Note: Descriptions are shown in the official language in which they were submitted.
~ 3 -
Novel aromatase inhibiting 4(5)~imidazoles
The present invention relates to substituted imidazole
derivatives and their non-toxic, pharmaceutically
acceptable acid addition salts, and their preparation,
to pharmaceutical compositions containing the same and
their use.
The imidazole derivatives of the present invention have
the general formulae (Ia) and (Ib):
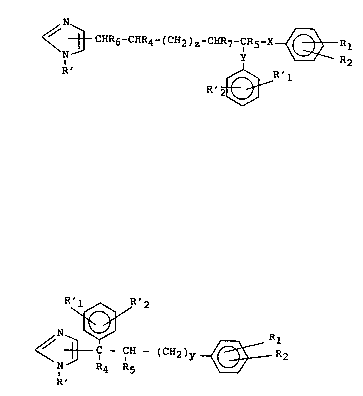
~ 3 CHR6-CHR4-(CH2, z-CHR7-CR5 x~RR2
R~ R'2 ~ (Ia~
wherein R1, R2, R'l and R'2, which can be the same or
different, are H, CH3, C2H5, C3H7~ OCH3, OH~ CH20H,
N02, NH2, CN, CF3, CHF2, CH2F or halogen; R' is H or
-CH2 ~ R3 where R3 is H, CH3 or halogen; R4 is H,
R5 is H or OH, R6 is H or OH and R7 is H or R4 and R6
together form a bond or R5 and R7 toyether form a bond;
X and Y, which can be the same or different, are a
bond, a straight Cl_2-alkyl or the corresponding alkenyl
and z is O to 2 and
~iJ )~J~3 ~
R'l R'2
3c CH - (CH2)y ~ R12
N R4 R5 (Ib)
R'
wherein Rl, R2, R'1 and R'2, which can be the same or
different, are H, CH3, C2Hs, C3H7, OCH3, OH, CH20H,
NO2, NH2, CN, CF3, CHF2, C~2F or halogen; R' is H or
-CH2 ~ R3 where R3 is H, C~3 or halogen; R4 is
H or OH and R5 is H or R4 and R5 together form a bond
and y is 0 to 4.
The non-toxic pharmaceutically acceptable acid addition
salts of these compounds are also within the scope of
the invention.
The compounds of the formulae (Ia) and (Ib) form acid
addition salts with both organic and inorganic acids.
They can thus form many pharmaceutically usable acid
addition salts, as, for instance, chlorides, bromides,
sulfates, nitrates, phosphates, sulfonates, formates,
tartrates, maleates, citrates, benzoates, salicylates,
ascorbates and the like.
The invention includes within its scope pharmaceutical
compositions comprising at least one of the compounds
of formula (Ia) or (Ib) or a non-~oxic, pharmaceutically
acceptable salt thereof, and a compatible
pharmaceutically acceptable carrier therefor.
The compounds of the present invention have been found,
depending on the substituents R', Rl, R2, R'l and R'2,
to possess varying degrees of aromatase and desmolase
t ~ ~
_f .J ~J ,J
inhibiting properties. Among them there are very
selective enzyme aromatase inhibiting compounds which
are valuable in the treatment of estrogen dependent
diseases, e.g. breast cancer.
Compounds of formula (Ia) can be prepared by the
following methods. Compounds of formula (Ia) wherein
the branches
-X ~ Rl and ~Y ~ RR'2
are identical, can be prepared by a successive sequence
of reactions comprising a Grignard reaction of 4(5)-
imidazole derivative of the formula (IIa)
o
N CH2CH2 ( CH2 ) ZCH2C-OR
N
H (IIa)
or its 1-benzyl derivative (IIIa) with an appropriate
aryl- or arylalkylmagnesium halide (IVa) following the
loss of water and hydrogenation.
N O
3 CH2CH2 ( CH2, zcH2 c o~
N (IIIa)
CH2 ~ - R3
Rl ~ (CH2)nMgHal (IVa)
In the formulae (IIa) to (IVa) R is alkyl, preferably
lower alkyl, n is O to 2 and Hal is halogen. The first
reaction step, the Grignard reaction, leads to the
following compounds of formula (Ia):
~ rs ~ s~ r~ r~
</ ~ CH2CH2(CH2)zcH2c~(cH2)n < ~ ~ R2
N (CH2)n
R' R2 ~ (Va)
In this reaction the aryl- or arylalkylmagnesium halide
derivative can be, for example, an aryl- or arylalkyl-
magnesiun~romide derivative, which is prepared by
reacting the corresponding aryl- or arylal~ylbromide
derivative with magnesium~ The Grignard reagent (IVa)
cannot be prepared if Rl and/or R2 are OH, CH20H or NH2.
Suitable solvents for the reaction include a variety of
ethers, preferably tetrahydrofuran.
The aryl- or arylalkylmagnesiumhalide derivative is
prepared in the usual way by adding the aryl~ or
arylalkylhalide derivative in a suitable solvent, e.g.
tetrahydxofuran, dropwise onto magnesium turnings covered
by tetrahydrofuran, at the boiling point of the reaction
mixture. When the magnesium turnings have reacted, the
mixture is cooled slightly and the 4t5)-imidazole
derivative (IIa) or its l-benzylsubstituted derivative
(IIIa) is added in solid form in small portions or
dropwise in tetrahydrofuran. After the addition, the
reaction mixture is refluxed until all of the 4~5~-
imidazole derivative has reacted. The reaction time
varies between one and five hours.
According to the feature of the invention, the compounds
of formula (Ia), wherein R7 and Rs both are hydrogen or
together form a bond, are prepared by dehydration of
the compounds of formula (Ia), where R5 is OH, and by
catalytic addition of hydrogen in the second step.
Water is eliminated by usual methods, i.e. by heating
with concentrated hydrochloric acid or by heating with
j v
s
dry potassium hydrogen sulfate. The unsaturated
compounds (VIa) (the compounds of formula (Ia) wherein
R7 and R5 together form a bond) are isolated and after
that hydrogenated. Alternatively they can be hydrogenated
directly in an acidic medium without previous isolation.
The hydrogenation is conveniently carried out at room
temperature with good stirring in alcohol, e.g. ethanol
in the presence of a catalyst in a hydrogen atmosphere.
Suitable catalysts are fox example platinium oxide,
palladium-on-carbon or Raney-nickel.
The reaction scheme for these steps can be illustrated
as follows:
CH2CH2(CH2)zcH2c~(cH2)n ~ RR2
R' R2 ~ (Ya)
wherein R', Rl, R2, z and n are as defined before
N ~ 2 2(CH2)zcH-c-(cH2) ~ RR2
R' R2 ~ (VIa)
</ ~ CH2CH2(C~2)zcH2~lcH~(cH2)n ~ RR2
N (CH2)n
R' R2 ~ (VIIa
If R' is a substituted or unsubstituted benzyl, this
group may be removed by hydrogenation as well. In this
case the hydrogenation is performed in an acidic medium
such as hydrochloric acid-ethanol mixture at elevated
temperature.
The reaction scheme of this hydrogenation which leads
to compounds of formula (Ia) wherein R', R7 and R5 each
are hydrogen can be illustrated as follows:
<, 3 CH2cH2(cH2)zcH~-cH-(cH2)n ~ R
R~ R2 ~ R1
~H2
< ~ CH2CH2(CH2)zcH2~cH~(cH2)n - ~ R
R2 ~ R1 (VIIIa)
wherein R1, R2, z and n are as defined before.
Another method to remove the benzylic R' group is a
hydrogen kransfer reaction in which the starting compound
(VIIa) is refluxed with ammonium formate and 10 ~ Pd/C
in an appropriate lower alcohol, such as methanol or
ethanol, or its aqueous solution. The compounds (VIIIa)
can also be prepared directly from the compounds (VIa)
by hydrogen transfer reaction with ammonium formate or
by hydrogenating both the double bond and the protecting
benzyl group at the same time. The compounds (VIIIa) can
also be prepared directly from the compounds (Va) by
hydrogen transfer reaction in which the starting compound
7 ~ ~ ?, "~, j
(Va) is refluxed with ammonium formate and 10 ~ Pd/C in
an acidic medium such as acetic acid.
The compounds of formula (VIIIa) can also be prepared
from ~he compounds of formula (Va) wherein R' is a
substituted or unsubstituted benzyl by removing first
the benzylic R' by a hydrogen transfer reaction as
described before to give the compounds of formula (IXa)
CH2CH2~CH~)zcH2c~(cH2)n { ~ RR2
R2 ~ Rl (IXa)
wherein Rl, R2, z and n are as defined before.
The benzylic R' can be removed by hydrogenation as
well. The compounds of formula (IXa) are further
dehydrated by the methods described before to form the
compounds of formula (Ia) where R5 and R7 together form
a bond (Xa).
</ ~ CH2CH2(CH2)zCH-C-(cH2)n ~ ~ ~ R2
R2 - ~ (Xa)
The compounds of formula (Xa) are further hydrogenated
by the methods described before to give the compounds
of formula (VIIIa).
When the compounds of formula (Ia3 wherein R1 andtor R2
are OH, CH20H or NH2 are wanted, they can be prepared by
the following reactions.
8 ~' '1 ~ ~, ~,~ ~,
~ J.,
The compounds of formula (Ia) wherein R1 andJor R2 are
OH can be prepared by reacting the 4(5)-imidazole
derivative (IIa) or (IIIa) with a Grignard reagent (IVa)
where R1 and/or R2 are OCH2Ph or OTHP (THP = tetrahydro-
pyranyl) and then hydrogenating catalytically by ~he
methods described to hydrogenate the benzylic R' group.
If R~ is a protecting benzyl group it will be removed
conveniently at the same time. Another method is to
dealkylate the compounds of formula (Ia) where R1 and/or
R2 are OCH3 by allowing them to react with BBr3, for
example.
The compounds o~ formula ~Ia) wherein Rl and/or R2 are
CH20H may be prepared ~rom the corresponding compounds
where Rl and/or R2 are CN by conventional methods, i.e.
by hydrolyzing the nitrile group and then catalytical
reduction of the acid group.
The compounds of formula (Ia) wherein R1 and/or R2 are
NH2 can be prepared by hydrogenating the corresponding
compounds where R1 and/or R2 are NO2. The protecting
benzyl group will be hydrogenated as well.
The compounds of formula (Ia) wherein R1 and/or R2 are
CN can be prepared from the corresponding compounds
where R1 and/or R2 are NH2 by diazotiæation. Compounds
of formula (Ia) wherein Rl and/or R2 are halogen may
also optionally be prepared by the same method.
Compounds o~ formula ~Ia) where R' is a benzyl group
can be prepared by benzylating the corresponding
compounds where R' is hydrogen. The starting compound
is first treated with a strong base such as sodium
hydroxide in water or sodium hydride in an appropriate
solvent, e.g. dimethyl formamide, to give the alkali
metal salt of the imidazole and then in the second step
9 ?J',fJ~
adding to this benzyl halide~ The reaction scheme c~n
be illustrated as follows:
~ 3 CHR6-cHR4-(cH2)z-cHR7-cR5-x ~ R
R'2 ~R'l
1)strong base > /N ~__~R
2) ~ CH2Hal ~ 3 CHR6-cHR4-(cH2)z-cHR7-cR5-x ~
R3 N Y R2
CH2 ~ R3 R~2 ~ R~1
The free OH, CH20H and NH2 substituenks must be protected
during the benzylation reaction.
Another process for the preparations of compounds of
formula (Ia) wherein the branches
-X ~ R1 and -Y - ~ R 1
are different, comprises in the first stage a series of
two successive Grignard reactions starting from 4(5)-
imidazole derivative (IIa) or its 1-benzyl substituted
derivative (IlIa) as previously. Now/ however, the amount
of the Grignard reagent is reduced as well as the reac-
tion temperature, to stop the reaction at the ketone
stage to give the ketone (XIa), which further is reacted
with another Grignard reagent (XIIa) to give a compound
of formula (Ia) where R5 is OH. The reactions are
illustrated as follows:
~.J J ~. -J ~) 3.,
N O R2 ~ (CH2)nMgHal
3 CH2cH2(cH2)zcH2-c-oR (IVa~>
N
R'
( CH2 )m-MgHal
N\O ~-~R1 R'2 (XIIa)
CH2CH2(CH2)zcH2~c~(cH2)n ~
NR2
R'(XIa)
~ 3 CH2CH2(CH2,~CH2 c (CH2)n < ~ lR2
R' R~2 ~ R'l (XIIIa)
In the reaction scheme above m and n, which can be the
same or different, are 0 to 2, z is 0 to 2 and R is an
alkyl group, preferably a lower alkyl. The compounds of
formula (XIIIa) are fuxther dehydrated and hydrogenated
by the methods described before to give the compounds
of formula
< ~ CH2CH2(CH2)zcH2-cH~(c~2)n ~ R
R' R'2 ~
wherein R', Rl, R2, R'1, R'2, z, n and m are as defined
before.
The Grignard reagents ~IVa) and (XIIa)cannot be prepared
when the substituents are OH, CH20H or NH2. When the
compounds of formula (Ia), wherein one or more of the
S~s ~
suhstituents Rl, R2, R~1 and R~2 are OH are wanted they
can be prepared by the following method.
The compounds of formula (Ia) wherein one or more of
the substituents R1, R2, R'l and R'2 are OH can be
prepared by reacting the 4(53-imidazole derivative (IIa)
or (IIIa) first with a Grignard reagent (IVa) and then
with reagent (XIIa) where the substituent/substituents
of either compound (IYa) or (XIIa) or both of them are
OCH2Ph or OTHP and hydrogenating catalytically by the
methods described to hydrogenate the benzylic R' group.
The protecting benzyl group will be remov~d conveniently
at the same time. Another method is to dealkylate the
compounds of formula (Ia) where the substituent/
substituents are OCH3 by allowing them to react with
BBr3, for example.
The compounds of formula (Ia) wherein one or more of
the substituents Rl, R2~ R'1 and R'2 are CH20H, NH2 or
CN can be prepared by the methods described before.
In order to achieve a better control of the reactions
above, the starting material may be an amide
N O
3 CH2CH2 ( CH2, zCH2C-NH2
N
R'
or a nitrile
N ~
CH2cH2(cH2)zC~CN
N
R'
as well.
Choosing appropriate conditions for the dehydration of
the compounds of formula (Ia) where R5 is OH results in
12 ~ J~ ? ~
the corresponding compounds of formula (Ia) where one of
the alkyl chains X or Y is transformed to the corre-
sponding alkenyl chain.
The starting compounds of the formulae (IIa) and (IIIa)
may be prepared for example from 4(5)-imidazole alkyl
acid of the formula
N ~ CH2cH2(cH2)zcH2cooH
H
and its 1-benzyl derivative of the formula
N
<~ ~ CH2CH2(CH2)ZCH2COOH
N
CH~ ~ R3
wherein z and R3 are as defined before, by esterifying
them according to the method described in US patent No.
3759944.
A further method of preparing the compounds of formula
(Ia) is the Wittig reaction where the starting compound
is an 4(5~-imidazole aldehyde (XIVa). In the formula
(XIVa) R' is as defined before.
N
CHO
N
R' (XIVa)
In the Wittig reaction the first step is to prepare a
phosphonium salt (XVa) from the corresponding halogenated
hydrocarbon ~XVIa) by reacting it with triphenyl-
13
phosphine. The reaction scheme can be illustrated asfollows:
Hal-CH2-(CH2)Z-cH2-cH-x ~ RR2
~ R'l
R 2 ~ (XVIa)
> Hal~ Ph P~_CH2_(CH2)ZCH2_CH_X ~ R
R'2 ~ (XVa)
in which Rl, R2, R'l, R'2, X, Y and z are as defined
before and Hal is halogen.
In the second step of the Wittig reaction the compound
(XVa) is treated with a strong base to form a phosphorus
ylide which is further allowed to react with the 4(5)-
imidazole aldehyde (XIVa) to achieve the compounds of
formula (Ia) wherein R4 and R6 together form a bond
(XVIIa). The strong base can be NaH or BuLi in a proper
solvent such as dimethoxyethane, tetrahydrofuran or
DMF. Further alkali metal alkoxides the corresponding
alcohols as solvent and NaH in DMSO can be used as
proton acceptors. The compounds (XVIIa) are isolated and
after that hydrogenated as has been described befoxe to
achieve the compounds of formula (Ia) wherein R4 and R6
both are hydrogen. The reaction scheme for these stsps
can be illustrated as follows:
3~
14
Hal~ Ph-P~-CH2-[CH2)zCH2~CH~X ~ l? strong base >
Ph y R2 2) ,N
< IN ~ CHO
R' (XIVa)
</ ~ CH=CH-(CH2)z~CH2~CH~X ~ Rl H2
R' ~ R'1
R'2 ~ (XYIIa)
- 3 CH2-CH2-(CH2)z~CH2-CH-X ~ 2
R' R'2 ~ (XVIIIa)
The Wittig reaction can be performed in another order
i.e. a phosphonium salt (XIXa) is prepared from the
corresponding halogenated 4(5)-imidazole derivative
(XXa) by reacting it with triphenylphosphine. In the
second step the compound (XIXa)is allowed to react with
a strong base and then with a substituted ketone of
the formula (XXIa) as described before to give the
compounds of formula (Ia) wherein R5 and R7 together
form a bond (XXIIa). The reaction scheme for these steps
can be illustrated ~s follows~
N
CH2cH2(cH2)zcH2Hal Ph3P
N (XXa)
R~
~ J.~
N Ph
~ ~ CH2cH2(cH2)zcH2p~-ph Hal~ 1) strong base
- N Ph 2) O=C-X ~ Rl
R' (XIXa) Y ~ R2
[~ R 1
3 CH2CH2(CH2)zCH=C-x ~ R12
R' R'2 ~ R'l (XXIIa)
wherein R', Rl, R2, R'1, R'2, z, X and Y are as defined
before.
The compounds of formula (Ia) can also be prepared by a
modified Wittig reaction, namely the Horner-Emmons or
Wadsworth-Emmons reaction where ~he phosphonate (XXIIIa)
which is prepared from the halogenated hydrocarbon (XVIa)
and a triester of phosphonic acid (e.g. (EtO)3P) by the
Arbuzow reaction reacts firstly with a base (e.g. NaH
in DMSO or in dimethoxyethane) and then with the aldehyde
(XlVa). The product (XVIIa) formed is a compound of
formula (Ia) where R4 and R6 together form a bond. The
reaction scheme can be illustrated as follows:
Hal CH2-(CH2)2-CH2-~H-x ~ R1 (RO)3P
R 2 ~ R~ l (XvIa)
~ff ," ~ , f
~6
Rl
(Ro)2-p-cH2-(cH2)z~cH2-cH-x ~ R2 1) base
- Y 2) N
R'2- ~ R'l < N 3
(XXIIIa) R' (XIVa)
=CH-(CH2)Z-CH2-CH-X ~ ~2
R' R~2 ~ (XVIIa)
In the formula ~XXIIIa) R is alkyl with 1-4 carbon atoms
and R1, R2, R'l, R'2, X, Y and z are as defined before.
The unsaturated compounds (XVIIa) are further
hydrogenated to form the compounds of formula (Ia)
wherein R4 and R6 both are hydrogen. The benzylic R'
group and the double bond of the compounds of formula
~XVIIa) can be removed at the same time by the methods
described before.
Another useful method to prepare compounds of forrnula
(Ia) is the Grignard reaction in which the 4(5)-imidazole
aldehyde (XIVa) is allowed to react with a Grignard
reagent (XXIVa) to give a compound of forrnula (Ia) where
R6 is OH (XXVa). The Grignard reagent is prepared by
reacting the corresponding halogenated hydrocarbon with
magnesium turnings in the usual way. The compound (XXVa)
is further dehydrated by heating ~ith KHS04 to achieve
the compounds of formula (Ia) where R4 and R6 together
form a bond (XVIIa).
The unsaturated derivatives are then hydrogenated to
forrn the compounds of formula (Ia) wherein R4 and R6
17 ~ 3 ~,J ~
both are hydrogen. The reaction scheme for these steps
can be illustrated as follows-
<, 3 CHO + HalNgcH2(cH2)zcH2-cH-x ~ RR2
R(XIVa) R'2 ~ (X~IVa
3--CH-CH2-(CH2~z-CH2-CH-X ~ = RR2
R~ R'2 ~ (XXVa)
H20~ ~ ~ CH=CH-(CH2)z-CH2-CH-X ~ R
R' R'2 ~ (XVIIa
~, // ~ CH2-cH2-(cH2)z-cH2-c~-x~RR2
R' R'2 ~ (XVIIIa)
In the formulae (XXVa), (~VIIa) and (XVIIIa) R', Rl,
R2, R'l, R'2, X, Y and z are as defined before.
If R' is substituted or unsubstituted benzyl, this
group may be removed by hydrogenation and hydrogen
transfer reaction as described before to give the
compounds of formula ~XXVIa).
18 )~ ?_S ' ~, ~,
3 CH2-CH2- ( CH2 ~ Z-CH2-CH-x C~RR2
H R~2 ~ (XXVIa)
The compounds of formula (XXVIa) can also be prepared
directly from the compounds (XVIIa) and (XXVa) by the
methods described before.
The Grignard reagent (XXIVa) cannot be prepared when the
substituents are OH, CH20H or NH2. The compounds of
formula (la) wherein one or more of the substituents
R1, R2, R'l and R'2 are OH, CH20H or NH2 can be prepared
according to the methods described before.
Further method to prepare the compounds of formula (Ia)
wherein one or more of the substituents Rl, R2, R'
and R'2 are NO2 is nitration of the corresponding
compounds wherein one or more of the substituen~s R1,
R2, R'l and R'2 are H.
Compounds of formula (Ib) can be prepared by the
following methods.
Compounds of formula ~Ib) can be prepared by a
successive se~uence of reactions comprising firstly a
Grignard reaction of 4(5)-imidazole aldehyde (IIb)
3 CHO
N
R' (~Ib)
with an appropriate arylalkylmagnesiumhalide (IIIb)
~ " ~ s-3 ~; ~f,
19
HalMgCH2~CH2)Y ~ R21
whicn leads to following compounds (IVb)
~ 3--C-C~(CH2)Y ~ lR2
R' (IVb~
In the reaction the arylalkylmagnesium halide derivative
can be, for example, an arylalkylmagnesiumbromide
derivative, which i5 prepared by reacting the corre-
sponding arylalkylbromide derivative with magnesium.
Suitable solvents for the reaction include a variety of
ethers, preferably tetrahydrofuran.
The arylalkylmagnesiumhalide derivative is prepared in
the usual way by adding the arylalkylhalide derivative
in a suitable solvent, e.g. tetrahydrofuran, dropwise
onto magnesium turnings covered by tetrahydrofuran, at
the boiling point of the reaction mixture. When the
magnesium turnings have reacted, the mixture is coolsd
slightly and the 4(5)-imidazole aldehyde (IIb) is added
in solid form in small portions or dropwise in tetra-
hydrofuran. After the addition, ~he reaction mixture
is refluxed until all of the 4(5)-imidazole aldehyde
(IIb) has reacted. The reaction time varies between one
and five hours.
The com~ounds (IVb) are further oxidized for example
with manganese dioxide to achieve compounds of formula
(Vb) which are allowed to react with another Grignard
reagent (VIb) to give the compounds of formula (Ib)
where R4 is OH (VIIb). The reaction scheme for these
steps can be illustrated as follows:
ZO ~ t,j '3f3~
<N3 OH ~ 12
R~ (IVb)
oxidation~ ~ 3 C-CH2(CH2)y ~ R12
R' (Vb)
R'l R'2
R'1 MgHal ~ R1
(VIb) > ~ C-OE12(CH2)y - ~ R2
R~ (VIIb)
The arylmagnesium halide (VIb) is prepared by reacting
the corresponding halogenated aromatic compound with
magnesium turnings in the usual way.
Compounds of formula (Ib) can also be prepared by
reacting an 4(5)-imidazole aldehyde (IIb) with an
arylmagnesiumhalide (VIb) which leads to following
compounds (VIIIb)
N~ ~EI 2
R' (VIIIb)
The compounds (VIIIb) are further oxidized for example
with manganese dioxide to achieve the compounds of
formula (IXb) which are further allowed to react with
the Grignard reagent (IIIb~ to give the compounds of
2 ~ J ~I SJ /~ ~j "~)
formula (Ib) where R4 is OH (VIIb). The reaction scheme
for these steps can ~e illustrated as follows:
N 3 ~ oxidatio ~ </ 3 C ~ R'
R' (VIIIb) R' (IXb)
R ~ R~2
HalMgCH2(C~2)Y ~ R2 N
~IIIb) ~ < 3 c CH2 ( CH2 j Y~RR2
R' (VIIb)
According to the feature of the in~ention, the compounds
of formula (Ib), wherein R4 and R5 both are hydrogen or
together form a bond, are prepared by dehydration of
the compounds of formula (Ib), where R4 is OH, and by
catalytic addition of hydrogen in the second step.
Water is eliminated by usual methods, i.e. by heating
with concentrated hydrochloric acid or by heating with
dry potassium hydrogen sulfate. The unsaturated
compounds (Xb) (the compounds of formula lIb) wherein R4
and R5 together form a bond) are isolated and after
that hydrogenated. Alternatively they can be hydrogenated
directly in an acidic medium without previous isolation.
The hydrogenation is conveniantly carried out at room
temperature with good stirring in alcohol, e.g. ethanol
in the presence of a catalyst in a hydrogen atmosphere.
Suitable catalysts are for example platinium oxide,
palladium-on-carbon or Raney-nickel.
The reaction scheme for these steps can be illustrated
as follows:
~2
<~1 H~2 ( CH2 )y ~ Rl
R' (VIIb)
wherein R', Rl, R2, R'l, R'2 and y are as defined before
-~2 > ~ ~ R'2 RR21
R' (Xb)
< ~2(CH2)y <(~R2
R' (XIb)
wherein R , Rl, R2, R~l, R'2 and y are as defined
bafore.
If R' is a substituted or unsubstituted benzyl, this
group may be removed by hydrogenation as well. In this
case the hydrogenation is performed in an acidic medium
such as hydrochloric acid-ethanol mixture at elevated
temperature.
The reaction scheme of this hydrogenation which leads
to compounds of formula (Ib) wherein R', R4 and R5 each
are hydrogen can be illustrated as follows:
~JJ r~
23
~ } ~ (CH2)y ~ R1 (XIb)
R' ¦H2~ H+
\/
~z(CH2 )y ~=Rl
H (XIIb)
Another method to remove the benzylic R' group is a
hydrogen transfer reaction in which the starting compound
(XIb) is refluxed with ammonium formate and 10 % Pd/C in
an appropriate alcohol, such as methanol or ethanol, or
its aqueous solution. The compounds (XIIb) can also be
prepar~d directly from the compounds (Xb) by hydrogen
transfer reaction with ammonium formate or by
hydrogenating both the double bond and the protecting
benzyl group at the same time. The compounds (XIIb) can
also be prepared directly from the compounds (VIIb) by
hydrogen transfer reaction in which the starting compound
(VlIb) is refluxed with ammonium formate and 10 % Pd/C
in an acidic medium such as acetic acid.
The compounds of formula (XIIb) can also be prepared
from the compounds of formula (VIIb) wherein R' is a
substituted or unsubstitu~ed benzyl by removing first
the benzylic R~ by a hydrogen transfer reaction by the
method described before to give the compounds of formula
(XIIIb)
2 ~ ~ t3 ~
24
< I CH2 ( CH2 ) Y--~ Rl
N- OH R2
H (XIIIb)
wherein R1, R2, R'l, R'2 and y are as defined before.
The benzylic R' can be removed by hydrogenation as
well. The compounds of formula (XIIIb~ are further
dehydrated by the methods described before to form the
compounds of formula (Ib) where R4 and R5 together form
a bond (XIVb).
R ~ R 2
< ~ ~ C=CH(CH2)y ~ R
~ (XIVb)
The compounds of formula (XIVb) are further hydrogenated
by the methods described before to give the compounds
of formula (XIIb).
The benzylic R' can be removed already before the
oxidizing reaction by the methods described before. The
reaction scheme for this hydrogenation can be illustrated
as follows:
N 3 OH ~ RRl
R' (IVb)
~ r~, J i S~,i i'~ J
N OH R1
H2, H ~ </ 3 C-CH2(CH2)y ~ ~ = R2
The Grignard reagents (IIIb) and (VIb) cannotbe prepared
when the substituents are OH, CH20H or NH2. '~he compounds
of formula (Ib) wherein one or more of the substituents
Rl, R2, R~1 and R'2 are OH, CH20H or NH2 can be prepared
by the following methods.
The compounds of formula (Ib) wherein one or more of
the substituents Rl, R2, R'l and R'2 are OH can be
prepared by reacting the 4(5)-imidazole derivative (IIb)
first with a Grignard reagent (IIIb) and then with
reagent (VIb) or revise order where the
substituent/substituents of either compound (IIIb3 or
(VIb) or both of them are OCH2Ph or OTHP (THP =
tetrahydropyranyl) and then hydrogenating catalytically
by the methods described to hydrogenate the benzylic R~
group. If R~ is a protecting benzyl group it will be
removed conveniently at the same time. Another method
is to dealkylate the compounds of formula (Ib) where
the substituent/substituents are OCH3 by allowing them
to react with BBr3, for example.
The compounds of formula (Ib) wherein one or more of
the substituents R1, R2, R'l and R'2 are CH20H may be
prepared from the corresponding compounds where the
substituent/substituents are CN by conventional me~hods,
i.e. by hydrolyzing the nitrile group and then reducing
the acid group.
The compounds of formula (Ib) wherein one or more of
the substituents R1, R2~ R~1 and R~2 are NH2 can be
prepared by hydrogenating the corresponding compounds
'~; d~
26
where the substituent/substituents are N02. The
protecting benzyl group will be hydrogenated as well.
The compounds of formula (Ib) wherein one or more of
the substituents Rl, R2, R'l and R'2 are CN can be
prepared from the corresponding compounds where one or
more of the substituents are NH2 by diazotization.
Compounds of formula (Ib) wherein one or more of the
substituents Rl, R2, R'1 and R'2 are halogen may also
optionally be prepared by the s~me method.
Another method of preparing compounds of formula (Ib)
is a McMurry reaction which comprises a reducti~e
coupling of a benzoylimidazole (IXb)
~R ~ 2
R' (IXb~
and an appropriate aldehyde of the formula (XVb)
~I R
HC--( CH2 )y ~
R2 (XVb)
in an appropriate solvent, such as tetrahydrofuran or
dimethoxyethane, in the presence of a low valent titanium
reagent in an inert atmosphere, e.g. in nitrogen or
argon, to give the compounds of foxmula (Ib) where R4
and R~ together form a bond (Xb)
~J ~ J~
27
< ~ ~ R2 (Xb)
The unsaturated compounds (Xb) are further hydrogenated
as described before. The aldehyde (XVb) is prepared
from the corresponding alcohol in the usual way.
When the compounds of formula (Xb) where R'1 andtor R'2
are CH20H, NH2 or CN are wanted they can be prepared by
the methods described before.
The compounds of formula (Xb) where R'1 and/or R'2 are
OH and R1 and R2 are as defined before can be prepared
by a McMurry reaction from the compounds of formula
(IXb) where R'1 and/or R'2 are OH which can be prepared
by hydrogenating catalytically the compounds of formula
(IXb) wherein R'1 and/or R'2 are OCH2Ph or OTHP by the
methods described before. Alternatively the hydrogenation
can be done before the oxidizing reaction. If R' is a
protecting benzyl group it will be removed conveniently
at the same time. Another method is to dealkylate the
compounds of formula (IXb) where R'1 and/or R'2 are
OCH3 by allowing them to react with BBr3, for ~xample.
Compounds of formula (Ib) wherein R'l and/or R'2 are OH
and Rl and R2 are as defined before can be prepared by
a McMurry reaction in which the aldehyde (XVb) is allowed
to react with a ketone (IXb) wherein R'1 and/or R'2 are
OCH2Ph or OTHP to give a compound of formula (Xb~ where
R'1 and/or R'2 are OCH2Ph or OTHP and R1 and R2 are as
defined before which is further hydrogenated to give
compounds of formula (XIIb) where R'l and/or R'2 are OH.
2~3 c~ ~ "' s, r~ r ~
Compounds of formula (Ib3 where R' is a benzyl can be
prepared by benzylating the corresponding compounds where
R~ is hydrogen. The starting compound is first treated
with a strong base such as sodium hydroxide in water or
sodium hydride in an appropriate solvent, e.g. dimethyl
formamide, to give the alkali metal salt of the imidazole
and then in the second step adding to this ben~yl halide.
The reaction scheme can be illustrated as follows:
~CNR5-(CH2)Y~RR2
R'l R'2
l~stronq base > N ~ Rl
2)R3 ~ CH2Hal </ ~ CR4-CHRs(CH23y ~ R2
CH2 ~ R3
The free OH, CH20H and NH2 substituents must be protected
during the benzylation reaction.
Further method to prepare the compounds of formula (Ib)
wherein one or more of the substituents R1, R2, R'
and R~2 are NO2 is nitration of the corxesponding
compounds where one or more of the substituents are H.
The compounds of formula (Ia) and (Ib), their non-toxic,
pharmaceutically acceptable acid salts or mixtures
thereof may be administered parenterally, intravenously
or orally. Typically, an effective amount of the
2 9 ~ r; ~ s
compound is combined with a suitable pharmaceutical
carrier. As used herein, the term "effective amount'
encompasses those amounts ~hich yield the desired
activity without causing adverse side-effects. The
precise amount employed in a particular situation is
dependent upon numerous factors such as method of
administration, type o mammal, condition for which the
derivative is administered, etc., and of course the
structure of the compound.
The pharmaceutical carriers which are typically employed
with the compounds of the present invention may be
solid or liquid and are generally selected with the
planned manner of administration in mind. Thus, for
example, solid carriers include lactose, sucrose, gelatin
and agar, while liquid carriers include water, syrup,
peanut oil and olive oil. Other suitable carriers are
well-known to those skilled in the art of pharmaceutical
formulations. The combination of the compound and the
carrier may be fashioned into numerous acceptable forms,
such as tablets, capsules, suppositories, solutions,
emulsions and powders.
The compounds of the invention are especially valuable
as aromatase inhibiting agents and are therefore useful
in the treatment of estrogen dependent diseases, e.g.
breast cancer.
Estrogens are essential steroids in the physiology and
function of normal development of breast and sex organs
in women. On the other hand estrogens are known to
stimulate the growth of estrogen dependent cancers/
especially breast and endometrial cancers, and they may
increase the risk of development of breast cancer if
given at pharmacological doses for a long time. Excessive
production of estradiol may also cause other, benign
3 0 ~ rj ~ ~
disorders in hormone dependent organs. The importance
of estrogens as cancer growth stimulators and/o,r
regulators is clearly stressed by the fact that anti-
estrogens have reached a central position in the
treatment of estrogen receptor rich ~reast cancers.
Ar.tiestrogens act by binding to estrogen receptors and
thereby inhibiting the biological effects of estrogens.
Another approach for blocking estrogen effect is to
inhibit the synthesis of estro~ens. ThiS has been
achieved clinically by the unspecific steroid synthesis
inhibitor aminoglutethimide. The estro~en synthesis
could be blocked specifically by inhibiting the en~yme
aromatase, which is the key enzyme in biochemical
estrogen synthesis pathway. Aromatase inhibition is
important because several breast tumors synthesize
estradiol and estrone in situ and exhibit therefore
continuous growth stimulation ~Alan Lipton et al.,
Cancer 59:779-782, 1987).
The ability of the compounds of the invention to inhibit
the enzyme aromatase has been tested by in vitro assay
according to M. Pasanen (Biological Research in
Pregnancy, vol. 6, No. 2, 19~5, pp. 94-99). Human
aromatase enzyme was used. The enzyme was prepared from
human placenta, which is rich of ~he enzyme. Microsomal
fraction (100000 x g precipitate) was prepared by
centrifugation. The enzyme preparation was used without
further purification. Test compounds were added together
with 100000 dpm of 1,2[3H]-androstene-3,17-dione and
NADPH generating system. The concentrations of the test
compounds were 0,001; 0,01; 0,1 and 1,0 mM. The
incubation was carried out at 37C for 40 min.
Aromatization of 1,2[3H3-androstene-3,17-dione results
in the production of 3H20. The tritiated water and the
tritiated substrate are easily separated by a Sep-PakR
minicolumn, which absorbs the steroid but allows free
31
water elution. Radioactivity was counted by a liquid
scintillation counter. Aromatase inhibition was evaluated
by comparing the 3H20-radioactivity of inhibitor treated
samples to controls containing no inhibitor. IC-50
values were calculated as concentrations which inhibited
the enzyme activity 50 %. These concentrations are
presented in Table 2.
Cholesterol side chain cleavage (CSCC) activity
(desmolase) was measured according to the method of
Pasanen and Pelkonen (Steroids 43:517-527, 1984).
Incubations were carried out in 1,5 ml E~pendorf plastic
tubes, and an Eppendorf shaker, centrifuge and incubator
~ere used as a unit. In a 300 ~1 incubation volume, the
substrate (5 ~M) was prepared according to Hanukoglu
and Jefcoate (J. Chromatogr. 190:256-262, 1980), and
100000 dpm of radioactive 3H-4-cholesterol ~the purity
of the compound was checked by TLC) in 0,5 % ~een 20,
10 mM MgC12, 5 ~M cyanoketone and 2 mM NADPH was added.
Controls contained all the above substances but the
enzyme preparation was inactivated prior to the
incubation by the addition of 900 ~l of methanol. The
mitochondrial fraction (1 mg protein) from human placenta
or bovine adrenals was used as a source of enzyme.
After 30 min incubation at 37C, the reaction was
terminated by the addition of 900 ~l of methanol; 1500
dpm of marker 14C-4-pregnenolone was added to each
incubate and the tubes were vigorously shaken.
After 10 min equilibration, the methanol-precipitated
proteins were separated by centrifugation (8000 x g for
2 min) and th~ supernatant was sucked into 1 ml plastic
inj~ction syringe and loaded onto the pre-equili~rated
(7~ % methanol) minicolumn. The column was washed with
one ml of 75 ~ methanol and then with 3 ml of 80 ~
methanol. The 80 % methanol eluate was run into the
counting vial and 10 ml of scintillation liquid was
3 2
added. Radioactivity was counted using a double-label
program on a liquid scintillation counter (LKB RackBeta).
Typical activi~ies for placental and bovine adrenal
enæyme preparation were 0,5-3 and 50-100 pmol
pregnenolone formed/mg protein/min, respectively.
In inhibition experiments, the substance (final concen-
tration range from 1 to 1000 ~M) was added into incuba-
tion mixture in a volume of 10-20 ~1, usually as
methanol or ethanol solution. The same volume of the
solute was added into control incubation vial. The
IC-50 values (concentration causing a 50 % inhibition)
were determined graphically and are presented in Table
2.
Table 1: Compounds tested
No. Name
la. 4-[4-(4-methylphenyl)-4-phenylbutyl]-lH-imidazole
2a. 1-benzyl-5-[4-(4-methylphenyl)-4-phenylbutyl]-lH-
imidazole
3a. 1-benzyl-5-~4-(3-methylphenyl)-4-phenylbutyl]-lH-
imidazole
4a. 4-~4-(3-methylphenyl)-4-phenylbutyl]-lH-imidazole
5a. 4-[4-(2-methylphenyl)-4-phenylbutyl]-lH-imidazole
6a. 1-benzyl-S-[4-(2-methylphenyl)-4-phenylbutyl]-lH-
imidazole
7a. 1-benzyl-5-(4,5-diphenylpentyl)-lH-imidazole
~ i ~ s l ~ s~s ~
8a. 4-(4,5-diphenylpentyl)-lH-imidazole
9a. 4-(4,4-diphenylbutyl)-lH-imidazole
lOa. l-benzyl-5-(4,4-diphenylbutyl)-lH-imidazole
lla. 4-[4-(4-methoxyphenyl)-4-phenylbutyl]~lH-imidazole
12a. 4-[4,4-bis(4-methoxyphenyl)butyl]-lH-imidazole
13a. 4-[4,4-bis(3-methylphenyl)butyl]-lH-imidazole
14a. 4-[4-(3,5-dimethylphenyl)-4-phenylbutyl]-lH-
imidazole
15a. 4-[4-(3,4-dimethylphenyl)-4-phenylbutyl]-lH-
imidazole
16a. 4-[4-(3,5-dimethylphenyl)-4-(3-methylphenyl)butyl]-
lH-imidazole
17a. 4-[4,4-bis(4-methylphenyl)butyl]-lH-imidazole
18a. 4-~5,5-diphenylpentyl)-lH-imidazole
l9a. 4-(6,6-diphenylhexyl)-lH-imidazole
20a. 4-[4-(2-fluorophenyl)-4-phenylbutyl]-lH-imidazole
21a. 4-[4-(4-~luorophenyl)-4-phenylbutyl]-lH-imidazole
22a. 4-(4,4-diphenyl-1-butenyl)-lH-imidazole
23a. 4-[4,4-bis(4-fluorophenyl~butyl]-lH-imidazole
24a. 4-[4,4-bis(4-nitrophenyl)butyl]-lH-imidazole
34
25a. 4-[4,4-bis~4-aminophenyl)butyl]-lH-imidazole
26a. 4-[4-(4-ethylphenyl) 4-phenylbutyl]-lH-imidazole
lb. 4-[1-(4-fluorophenyl)-5-phenylpentyl]-lH-imidazole
2b. 4-[1-(4-fluorophenyl)-5-(2-methylphenyl)pentyl]-lH-
imidazole
3b. 4-[1-(4-fluorophenyl)-5-(3-methylphenyl)pentyl]-lH-
imidazole
4b. 4-[1-(4-fluorophenyl)-5-(4-methylphenyl)pentyl]-lH-
imidazole
5b. 4 [5-(3,5-dimethylphenyl)-1-(4-fluorophenyl)-
pentyl]-lH-imidazole
6b. 4-(1,5-diphenylpentyl)-lH-imidazole
7b. 4-(1,3-diphenylpropyl)-lH-imidazole
8b. 1-benzyl-5-(1,3-diphenylpropyl)-lH-imidazole
9b. 4-[1-(4-fluorophenyl)-3-phenylpropyl~-lH-imidazole
lOb. 4-[1-(4-fluorophenyl)-4-phenylbutyl]-lH-imidazole
llb. 4-[1-(4-fluorophenyl3-5-(3-methoxyphenyl)pentyl]-
lH-imidazole
12b. 4-[1-(4-fluorophenyl)-5-(4-methoxyphenyl)pentyl]-
lH-imidazole
13b. 4-[1-(4-fluorophenyl)-5-(2,6-dimethylphenyl)-
pentyl]-lH-imidazole
~ ,J
14b. 4-[1,3-bis(4-fluorophenyl)propyl]-lH-imidazole
15b. 4-tl,4-bis(4-fluorophenyl)butyl]~lH-imidazole
Table 2: Inhibition of human aromatase and desmolase
(CSCC) by test compounds. IC-50 represents the
concentration which inhibits the enzyme 50 %.
Compound IC-50IC-50 CSCC
No. ~mol/l~mol/l
la 2,9 96
2a 19 50
3a 13 38
4a 2,5 7,5
5a 3,4 29
6a 8,5 32
7a 15
8a 4,7 27
9a 2 320
lOa 7
lla 3,5 190
12a 10 46
13a 28 48
14a 7,5 65
15a 5,0 39
16a 125 95
17a 3,5 110
18a 1,7 68
l9a 14,5 61
20a 16 38
21a 2,8 80
22a 8,5 165
23a 3,3 175
24~ 20 37
36 ~ J ~~ J ~ rJ ~J
25a 26 210
26a 8,5 65
lb 2,8 19
2b 3,5 16
3b 3,8 57
4b 8,5 170
5b 7,5 80
6b 25
7b 4,5 7,5
8b 30
9b 0,72 22
lOb 0,75 31
llb 2,6 12
12b 3 22,5
13b 7,7 31
14b 2,2 22
15b 0,63 29
The anti-tumour effect was investigated in vivo against
DMBA-induced rat mammary adenocarcinomas by the following
method. Mammary adenocarcinoma was induced with DMBA in
50+2 days old female rats. Treatment with the compound
under test was started after palpable tumours had
appeared. Tumour size and amount of tumours were
evaluated once a week. Tumour sizes in the control
group, treated with solvent, were compared with the
test groups. Daily administration schedule was employed
for five weeks and animals were sacrificied. The change
in tumour sizes was evaluated.
Results were evaluated as changes in the sizes of tumours
and divided into three groups, namely increasing, stable
and decreasing sizes of tumours. The anti-tumour effect
of 4-(4,4-diphenylbutyl)-lH-imidazole (compound 9a in
~ J ~ ,~ ~3 ,J ~
37
Table 2) was tested and the results are presented in
Table 3.
Table 3: Number of different tumour types in control
and 4-(4,4-diphenylbutyl)-lH-imidazole treated
groups of DMBA-induced mammary tumour rats.
qroup decreasing stable increasin~
control 3 21 42
4-(4,4-diphenyl- 2 14 37
butyl)-lH-imidazole
3 mg/kg
4-(4,4-diphenyl- 21 3 15
butyl)-lH-imidazole
30 mg/kg
Acute toxicity, LD50, was determined by using young
adult female mice of NMRI-Strain. The administration of
the test compounds was oral. The LD50 values of the test
compounds of formula (Ia) were 350 mg/kg or more and of
formula (Ib) 400 mg/kg or more.
The daily dose for a patient varies from about 20 to
200 mg, administered orally.
The following examples illustrate the invention.
lH NMR spectra were determined with a Bruker WP 80 DS
apparatus (80 MHz). The reference substance was tetra-
methylsilane. MS spectra were determined with Kratos
MS80RF Autoconsole apparatus.
38 ~ i L~'J'.f'~
Example 1
4-(4,4-diphenylbutyl)-lH-imidazole
a) l-benzyl-5-(1-hydroxy-4,4-diphenylbutyl)-lH-imidazole
2,0 g of magnesium turnings are covered with 60 ml of
dry tetrahydrofuran. To the mixture is then added
dropwise a solution of 1-bromo~3,3-diphenylpropane
(22,9 g) in 20 ml of dry tetrahydrofuran at such a rate
that a smooth reaction is maintained. After the ~ddition
is complete, the reaction mixture is refluxed ~or one
additional ho~r and cooled to room temperature. The
reaction mixture is then added dropwise to a solution
of l-benzyl-5-imidazolecarbaldehyde (7,35 g) in 80 ml
of tetrahydrofuran at 60C. After the addition is
complete, the reaction mixture is refluxed for 2 hours,
cooled and poured into cold water. Tetrahydrofuran is
evaporated and to the solution is added conc. hydro-
chloric acid. The solution is cooled and the precipitate
which contains the product as hydrochloride salt is
removed by filtration, washed with water and dried.
Yield 14,1 g. M.p. 160-168C.
1H NMR (as HCl-salt, MeOH-d4):
1.50-2.30 (m, 4H), 3.82 (t, lH), 4.65 (t, lH), 5.43 (s,
2H), 7.05-7.50 (m, 16H), 8.62 (d, lH)
Using the same method ~or example the following compounds
included in the invention were pr~pared:
l-benzyl-5-[1-hydroxy-4-(2-methylphenyl)-4-phenylbutyl]-
lH-imidazole
~J .J ~ ~3 ~ ~;,
1H NMR (as HCl-salt, MeOH-d4):
1.50-2.40 (m, 4H), 2.21 and 2.15 (2s, 3H), 4.07 lt,
lH), 4.70 (m, lH), 5.49 and 5.46 (2s, 2H), 6.80-7.50
(m, l5H), 8-88 (s, lH)
l-benzyl-5-[1-hydroxy-4-(3-methylphenyl)-4-phenylbutyl]-
lH-imidazole
lH NNR (as HCl-salt, CDC13):
1.35-2.35 (m, 4H), 2.26 (s, 3H), 3.73 (t, lH), 4.62 (m,
lH), 5.38 (s, 2H), 6.80-7.40 (m, 15H), 8.51 (s, lH)
l-benzyl-5-[1-hydroxy-4-(4-methylphenyl)-4-phenylbutyl]-
lH-imidazole
lH NMR (as HC1-salt, CDC13~:
1.40-2.40 ~m, 4H), 2.23 and 2.24 (2s, 3H), 3.74 (t,
lH), 4.65 (broad t, 2H), 5.38 (s, 2H), 6.80-7.40 (m,
15H), 8.55 (s, lH)
l-benzyl-S-[l-hydroxy-4,4-bis(4-methylphenyl)butyl]-lH-
imidazole. M.p. of hydrochloride 155-158C.
l-benzyl-5-(1-hydroxy-4,5-diphenylpentyl)-lH-imidazole
H NMR (as hydrogen sulphate salt, MeOH-d4):
1.35-1.90 (m, 4H), 2.82 (broad s, 3H), 4.54 (t, lH),
5.42 (s, 2H), 6.85-7.50 (m, 16H), 8.80 (s, lH)
l-benzyl-5-[1-hydroxy-4-(4-methoxyphenyl)-4-phenylbutyl]-
lH-imidazole
lH NMR (as HCl-salt, MeOH-d4):
1.50-2.30 (m, 4H), 3.70 (s, 3H), 3.79 (t, lH), 4.69 (t,
lH), 5.46 (s, 2H), 6.79 (d, 2H), 7.0-7.55 (m, 13H),
8.85 (d, lH)
2 ~
l-benzyl-5-[1-hydroxy-4,4-bis(4-methoxyphenyl)butyl]-
lH-imidazole
lH NMR (as base, MeOH-d4):
1.50-2.20 (m, 4H), 3.66 (t, lH), 3.74 (s, 6H), 4.50 (t,
lH), 5.24 (s, 2H), 6.70-7.60 (m, 15H)
1-benzyl-5-[4-(2-fluorophenyl)-1-hydroxy-4-phenylbutyl]-
lH-imidazole. M.p. of hydrochloride 160-163C.
1H NMR (as HCl-salt, CDC13):
1.45-2.45 (m, 4H), 4.17 (t, lH), 4.67 (t, lH), 5.43 (s,
2H), 6.80-7.50 (m, 15H), 8.43 (s, lH)
1-benzyl-5-[4-(4-fluorophenyl)-1-hydroxy-4-phenylbutyl]-
lH-imidazole. M.p. of hydrochloride 168-171C.
lH NMR (as HCl-salt, MeOH-d4):
1.50-2.30 (m, 4H), 3.84 (t, lH), 4.68 (t, lH), 5.49 (s,
2H), 6.80-7.55 (m, 15H), 8.87 (d, lH)
l-benzyl-5-(1-hydroxy-5,5-diphenylpentyl)-lH-imidazole
1H NMR (as base, CDC13):
1.2-2.2 (m, 6H), 3.70 (t, lH), 4.54 (t, lH), 5.46 (s,
2H), 6.8-7.3 (m, 16H), 8.58 (s, lH)
1-benzyl-5-(1-hydroxy-6,6-diphenylhexyl)-lH-imidazole.
M.p. of hydrochloride 147-149C.
H NMR (as HCl-salt, MeOH-d4):
1.6-2.2 (m, 8H), 3.85 (t, lH), 4.60 (t, lH), 5.53 (s,
2H), 7.0-7.4 (m, 15H), 7.47 (s, lH), 8.90 (s, lH)
l-benzyl-5-[4-(4-ethylphenyl)-1-hydroxy-4-phenylbutyl~-
lH-imidazole
41
lH NMR (as HCl-salt, MeOH-d4):
1.71 (t, 3H), 1.50-2.35 (m, 4H), 2.57 (q, 2H), 3.78 (t,
lH), 4.68 (t, lH), 5.46 ts, 2H), 6.9-7.5 (m, 15H),
8.82 (d, lH)
l-benzyl-5-[4,4-bis(4-fluorophenyl)-1-hydroxybu~yl]-lH-
imidazole
lH NMR (as HCl-salt, MeOH-d4):
1.50-2.00 (m, 4H), 3.86 (t, lH), 4.70 (t, lH), 5.52 (s,
2H), 6.80-7.50 ~m, 14 H), 8.89 (d, lH)
b) 1-benzyl-5-(4,4-diphenyl-1-butenyl)-lH-imidazole
1-benzyl-5-(1-hydroxy-4,4-diphenylbutyl)-lH-imidazole
hydrochloride (5,0 g) and 30,0 g of anhydrous potassium
hydrogen sulphate are heated at 150C for 4 hours. The
mixture is cooled, 90 ml of ethanol is added to dissolve
the product. The mixture is then filtered and the
filtrate is evaporated to minor volume. Water is added
and the mixture is made alkaline with sodium hydroxide.
The product is extracted in methylene chloride, washed
with water and evaporated to dryness. The product is
then made to hydrochloride salt with dry hydrochloric
acid in dry ethylacetate. Yield is 2,9 g. M.p. 204-206C.
lH NMR (as HCl-salt, MeOH-d4):
2.88-3.05 (m, 2H), 4.08 (t, lH), 5.29 (s, 2H3, 6.22-
6.32 (m, 2H), 7.00-7.50 (m, 16H), 8.87 (d, lH)
Using the same method for example the following compounds
included in the invention were prepared:
1-benzyl-5-[4,4-bis(3-methylphenyl~-1-butenyl]-lH-
imidazole. M.p. of hydrochloride 152-156C.
~,2 7~ J J .~
H NMR (as HCl-salt, MeOH-d4):
2.26 (s, 6H), 2.85-3.05 (m, 2H), 3.99 (t, lH), 5.27 (s,
2H), 6.21-6.31 (m, 2H), 6.80-7.50 (m, 14H), 8.86 ~d, lH)
1-benzyl-5-[4-(3,5-dimethylphenyl)-4-(3-methylphenyl)-
l-butenyl]-lH-imidazole
H NMR (as HCl-salt, MeOH-d4):
2.22 (s, 3H), 2.23 (s, 3H), 2.26 (s, 3H), 2.85-3.05 (m,
2H), 3.94 (t, lH), 5.26 (s, 2H), 6.20-6.30 (m, 2H),
6.75-7.50 (m, 13H), 8.84 (d, lH)
1-benzyl-5-[4-(3,5-dimethylphenyl)-4-phenyl-1-butenyl]-
lH-imidazole. M.p. 110-112C.
H NMR (as HCl-salt, MeOH-d4):
2.22 (s, 6~), 2.85-3.05 (m, 2H), 3.98 (t, lH), 5.27 (s,
2H), 6.20-6.30 (m, 2H), 6.75-7.5 (m, 14H), 8.87 (d, lH)
1-benzyl-5-[4-(2-methylpheny~)-4-phenyl-1-butenyl]-lH-
imidazole
1H NMR (as hydrogen sulphate, MeOH-d4):
2.19 (s, 3H), 2.80-3.05 (m, 2H), 4.28 (~, lH), 5.29 (s,
2H), 6.0-6.60 (m, 2H), 7.0-7.5 (m, 14H), 7.52 (d, lH),
8.85 (d, lH)
l-benzyl-5-[4-(3-methylphenyl)-4-phenyl-1-butenyl]-lH-
imidazole
1H NMR (as base, C~Cl3):
2.27 (s, 3H), 2.75-3.95 (m, 2H~, 3.93 (t, lH), 4.89 (s,
2H), 5.70-6.10 (m, 2H), 6.80-7.40 (m, 16H)
1-benzyl-5-[4-(4-methylphenyl)-4-phanyl-1-butenyl]-lH-
imidazole
43 ~ ~ r~
lH NMR (as base, CDC13):
2.27 (s, 3H), 2.70~2.95 (m, 2H), 3.93 (t, lH), 4.89 (s,
2H), 5.60-6.20 (m, 2H), 6.80-7.50 (m, 16H)
l-benzyl-5-[4,4-bis(4-methylphenyl)-1-butenyl]-lH-
imidazole. M.p. of hydrochloride 128-132C.
l-benzyl-5-[4-(4-methoxyphenyl)-4-phenyl-1-butenyl]-lH-
imidazole
lH NMR (as HCl-salt, CDC13):
2.70-2.95 ~m, 2H), 3.95 (t, lH~, 5.16 (s, 2H), 5.70-
6.20 (m, 2H), 6.70-7.40 (m, 15H), 8.99 (s, lH)
l-benzyl-5-[4,4-bis(4-methoxyphenyl)-1-butenyl]-lH-
imidazole
lH NMR (as base, CDC13):
2.60-2.90 (m, 2H), 3.74 (s, 6H), 3.78 (t, lH), 4.92 (s,
2H), 5.80-6.00 (m, 2H), 6.65-7.50 (m, 15H)
l-benzyl-5-(4,5-diphenyl-1-pentenyl)-lH-imidazole
H NMR (as base, CDC13):
2.30-2.55 (m, 2H), 2.88 (m, 3H), 4.93 (s, 2H), 5.5-6.1
(m, 2H), 6.8-7.5 tm, 17H)
l-benzyl-5-[4-(2-fluorophenyl)-4-phenyl-1-butenyl]-lH-
imidazole. M.p. of hydrochloride 195-201C.
lH NMR tas base, CDC13):
2.75-3.00 (m, 2H), 4.34 (t, lH), 5.97 (s, 2H), 5.80-
6.10 (m, 2H), 6O75-7.50 (m, 16H)
l-benzyl-5-[4-(4-fluorophenyl)-4-phenyl-1-butenyl]-lH-
imidazole
~-v ~'~ J~J
44
lH NMR (as base, CDCl3):
2.60-2.95 (m, 2H), 3.96 (t, lH), 4.93 (s, 2H), 5.80-
6.05 (m, 2H), 6.75-7.50 tm, 16H)
1-benzyl-5-(6,6-diphenyl-1-hexenyl)-lH-imidazole. M.p.
of hydrochloride 184-186C.
1-benzyl-5-[4-(4-ethylphenyl)-4-phenyl-1-butenyl]-lH-
imidazole
1H NMR (as HCl-salt, MeOH-d4):
1.17 (t, 3H), 2.56 (q, 2H), 2.93-2.98 (m, 2H), 4.03 (t,
lH), 5.28 (s, 2H), 6.20-6.34 (m, 2H), 7.08-7.41 (m,
14H), 7-52 (d, lH), 8.87 (d, lH)
1-benzyl-5-[~,4-bis(4-fluorophenyl)-1-butenyl]-lH-
imidazole
HNMR (as HCl-salt, CDC13):
2.78-2.94 (m, 2H), 4.01 (t, lH), 5.27 (s, 2H), 5.82-
6.34 (m, 2H), 6.83-7.40 (m, 14H), 9.21 (d, lH)
c) 1-benzyl-5-(4,4-diphenylbutyl)-lH-imidazole
1-benzyl-5-(4,4-diphenyl-l-butenyl)-lH-imidazole
hydrochloride (2,0 g) is dissolved in ethanol and a
catalytic amount of Pd/C (10 ~) is added. The reaction
mixture is a~itated vigorously at room temperature in a
hydrogen atmosphere until the uptake of hydrogen ceases.
The mixture is filtered and the filtrate is evaporated
to dryness. The residue which is the product is purified
by flash chromatography eluting with methylene chloride-
methanol mixture. Yield 1,3 g. M.p. of the hydrochloride
salt is 200-202C.
4 5 ~ ~ ~ c r~
1H NMR (as HCl-salt, MeOH-d4):
1.30-1.70 (m, 2H), 1.85-2.20 (m, 2H), 2.61 (t, 2H),
3.83 (t, lH), 5.35 (s, 2H), 7.05-7.50 (m, 16H), 8.89
(d, lH)
Using the same method for example the following compounds
included in the invention were prepared:
.
1-benzyl-5-[4-(2-methylphenyl)-4-phenylbutyl]-lH-
imidazole. Mp. of hydrochloride 200-205C.
1H NMR (as HCl-salt, CDCl3):
1.30-1.80 (m, 2H), 1.80-2.15 (m, 2H), 2.22 (s, 3H),
2.48 (t, 2H), 4.02 (t, lH), 5.31 (s, 2H), 6.96 (s, lH),
7.0~7.5 (m, 14H), 9.28 (s, lH)
1-benzyl-5-[4-(3-methylphenyl)-4-phenylbutyl]-lH-
imidazole. M.p. of hydrochloride 148-158C.
H NMR (as HCl-salt, CDCl3):
1.30-1.80 (m, 2H), l.B0-2.25 (m, 2H), 2.30 (s, 3H),
2.49 (t, 2H), 3.78 (t, lH), 5.29 (s, 2~), 6.90-7.50 (m,
15H), 9.24 (s, lH)
l-benzyl-5-[4-(4-methylphenyl)-4-phenylbutyl]-lH-
imidazole. M.p. of hydrochloride 164-170C.
H NMR (as HCl~salt, CDCl3):
1.25-1.75 (m, 2H), 1.80-2.25 (m, 2H), 2.29 (s, 3H;,
2.48 (t, 2H), 3.78 (t, lH), 5.31 (s, 2H), 6.80-7.50 ~m,
15H~, 9.35 (s, lH)
1-benzyl-5-(4,5-diphenylpentyl)-lH-imidazole. M.p. of
hydrochloride 166-170C.
46
lH NMR ~as HCl-salt, MeOH-d4):
1.15-1.90 (m, 4H), 2.~9 (t, 2H), 2.81 (m, 3H), 5.30 (s,
2H), 6.90-7.50 (m, 16H), 8.82 (s, lH)
l-benzyl-5-[4-(4-methoxyphenyl)-4-phenylbutyl]-lH-
imidazole. M.p. of hydrochloride 180-187C.
lH NMR (as HCl-salt, MeOH-d4):
1.25-1.70 (m, 2H), 1.85-2.20 (m, 2H), 2.62 (t, 2H),
3.74 (s, 3H), 3.78 (t, lH), 5.34 (s, 2H), 6.81 (d, 2H),
7.0-7.5 (m, 13H), 8.84 (d, lH)
1-benzyl-5-[4-(2-fluorophenyl)-4-phenylbutyl]-lH-
imidazole. M.p. of hydrochloride 185-196C.
lH NMR (as HCl-salt, MeOH-d4):
1.25-1.75 (m, 2H), 1.80-2.25 (m, 2H), 2.65 (t, 2H),
4.18 (t, lH), 5.37 (s, 2H), 6.80-7.5 (m, l5H), 8.89 (d,
lH)
l-benzyl-5-[4-(4-fluorophenyl)-4-phenylbutyl]-lH-
imidazole. M.p. of hydrochloride 172-174C.
H NMR (as HCl-salt, MeOH-d4):
1.25-1.70 (m, 2H), 1.80-2.25 (m, 2H), 2.64 (t, 2H),
3.85 (t, lH), 5.37 (s, 2H), 6.80-7.50 (m, 15H), 8.90
(d, lH3
l-benzyl-5-(5,5-diphenylpentyl)-lH-imidazole
lH NMR (as base, MeOH-d~):
1.1-1.6 (m, 4H), 1.8-2.1 (m, 2H), 2.37 (t, 2H~, 3.72
(t, lH), 5.11 (s, 2H), 6.67 (s, lH), 6.9-7.3 (m, 15H),
7.59 (s, lH)
~ ~3 ~ .~
47
l-benzyl-5-[4-(4-ethylphenyl)-4-phenylbutyl]-lH-imidazole
H NMR (as HCl-salt, MeOH-d4):
1.17 (t, 3H), 1.40-1.70 (m, 2H), 1.90-2.20 (m, 2H),
2.57 (q, 2H), 3.80 (t, lH), 5.34 (s, 2H), 7.00-7.50 (m,
15 H), 8-87 (d, lH)
1-benzyl-5-[4,4-bis(4-fluorophenyl)butyl]-lH-imidazole
1H NMR (as HCl-salt, MeOH-d4):
1.30-1.70 (m, 2H), 1.80-2.25 (m, 2H), 2.64 (t, 2H),
3.87 (t, lH), 5.40 (s, 2H), 6.80-7.50 (m, 14 H), 8.92
(d, lH~
d) 4-(4,4-diphenylbutyl)-lH-imidazole
l-benzyl-5-(4,4-diphenylbutyl)~lH-imidazole hydrochloride
(0,6 g) is hydrogenated in the mixture of 20 ml of 2 N
hydrochloric acid and 10 ml of ethanol at 80C Pd/C
(10 ~) as catalyst. When the uptake of the hydrogen
ceases, the reaction mixture is cooled, filtered and
evaporated to dryness. Water is added and the mixture
is made alkaline with sodium hydroxide. The product i5
then extracted to methylene chloride which is washed with
water, dried with sodium sulphate and evaporated to
dryness. The residue is the product as base and it is
made to its hydrochloride in ethyl acetate using dry
hydrochloric acid. Yield 0,2 g. M.p. 204-206C.
1H NMR !as HCl-salt, MeOH-d4):
1.40-1.90 (m, 2H), 1.90-2.30 (m, 2H), 2.75 ~t, 2H),
3.95 (t, lH), 7.00-7.40 tm, llH), 8.72 (d, lH)
Using the same method for example the following compounds
included in the invention were prepared:
4-[4,4-bis(3-methylphenyl)butyl]-lH-imidazole. M.p. ~f
hydrochloride 122-129C.
H NMR (as HCl-salt, MeOH-d4):
1.40-1.90 (m, 2H), 1.90-2.30 (m, 2H), 2.26 (s, 6H),
2.73 (t, 2H), 3.85 (t, lH), 6.80-7.25 (m, 9H), 8.73 (d,
lH)
4-~4-(3,5-dimethylphenyl)-4-(3-methylphenyl)butyl]-lH-
imidazole. M.p. of hydrochloride 75-82C.
lH NMR (as HCl-salt, MeOH-d4):
1.40-1.90 (m, 2H), 1.90-2.30 (m, 2H), 2.22 (s, 3H),
2.23 (s, 3H), 2.27 (s, 3H), 2.74 (t, 2H), 3.81 (t, lH),
6.75-7.30 (m, 8H), 8.72 (d, lH)
4-[4-(3,5-dimethylphenyl)-4-phenylbutyl]-lH-imidazole.
M.p. of hydrochloride 104-106C.
lH NMR (HCl-salt, MeOH-d4):
1.40-1.90 (m, 2H), 1.90-2.30 (m, 2H), 2.23 (s, 6H),
2.74 (t, 2H), 3.85 (t, lH), 6.84 (m, 3H), 7.22 (m, 6H),
8.72 (d, lH)
4-[4-(3,4-dimethylphenyl)-4-phenylbutyl]-lH-imidazole.
M.p. of hydrochloride 118-121 C.
4-[4-(2-methylphenyl)-4-phenylbutyl]-lH-imidazole. M.p.
of hydrochloride 151-154,5C.
lH ~R (as HC1-salt, CDC13):
1.50-2.20 (m, 4H), 2.22 (s, 3H), 2.71 (t, 2H), 4.09 (t,
lH), 6.81 (s, lH), 7.0-7.4 (m, 9H), 9.04 (s, lH)
5-[4-(3-methylphenyl)-4-phenylbutyl]-lH-imidazole. M.p.
of hydrochloride 140-153C.
49
lH NM~ (as HCl-salt~ MeOH-d4):
1.40-1.85 (m, 2H), 1.85-2.25 (m, 2H), 2.27 (s, 3H),
2.74 (t, 2H), 3.90 (t, lH), 6.80-7.30 (m, lOH), 8.69
(d, lH)
4-[4-(4 methylphenyl)-4-phenylbutyl]-lH-imidazole. M.p.
of hydrochloride 173-177C.
lH NMR ~as HCl-salt, CDC13 + 2 drops of MeOH-d4):
1.40-1.80 (m, 2H), 1.80-2.25 (m, 2H), 2.28 (s, 3H),
2.71 (t, 2H), 3.87 (t, lH), 5.86 (d, lH), 7.09 (s, 4H),
7.21 (s, 5H), 8.71 (d, lH)
4-[4-(4-methoxyphenyl)-4-phenylbutyl]-lH-imidazole.
M.p. of hydrochloride 156-159C.
lH NMR (as HCl-salt, CDC13):
1.40-1.90 (m, 2H), 1.90-2.30 (m, 2H), 2.71 (t, 2H),
3.76 (s, 3H), 3.87 (t, lH), 6.82 (d, 2H), 6.90 (s, lH),
7.13 (d, 2H), 7.21 (m, 5H), 8.68 (s, lH)
4-[4,4-bis(4-methoxyphenyl)butyl]-lH-imidazole. M.p. of
hydrochloride 138-142C.
H NMR (as HCl-salt, CDC13):
1.40-2.25 (m, 4H), 2.71 (t, 2H), 3.75 (s, 6H) under
which there is (t, lH), 6.78 (d, 4H), 6.83 (s, lH),
7.08 (d, 4H), 9.02 (s, lH)
4-(4,5-diphenylpentyl)-lH-imidazole
lH NMR ( as HCl-salt, CDC13):
1.20-1.90 (m, 4H), 2.57 (t, 2H), 2.83 (m, 3H), 6.71 (s,
lH), 6.80-7.40 (m, lOH), 8.84 (s, lH)
4-(5,5-diphenylpentyl)-lH-imidazole
1H NMR (as HCl-salt, MeOH-d4):
1.3-1.5 (m, 2H), 1.5-1.7 tm, 2H), 1.8-2.3 (m, 2H),
2.656 (t, 2H), 3.746 (t, lH), 7.06-7.2 (m, llH), 8.716
(d, lH)
4-(6,6-diphenylhexyl)-lH-imidazole
lH NMR (as base, CDCl3):
1.1-1.7 (m, 6H), 1.8-2.2 (m, 2H), 2.530 (t, 2H), 3.847
(t, lH), 6.685 (s, lH), 7.2 (s, lOH), 7.470 (s, lH),
9.6 (broad s, lH)
4-[4,4-bis(4-methylphenyl)butyl]-lH-imidazole. M.p. of
hydrochloride 176-179C.
4-[4-(4-fluorophenyl)-4-phenylbutyl]-lH-imidazole. M.p.
of hydrochloride 175-182C.
4-[4-(2-fluorophenyl)-4-phenylbutyl]-lH-imidazole. M.p.
of hydrochlo~ide 182-190C.
4-[4-(4-ethylphenyl)-4-phenylbutyl]-lH-imidazole
1H NMR (as HCl-salt, MeOH-d4):
1.18 (t, 3H), 1.40-1.90 (m, ~H), 1.90-2.30 (m, 2H),
2.57 (q, 2H~, 2.75 (t, 2H), 3.91 (t, lH), 6.95-7.30 (m,
lOH), 8.73 (d, lH)
4-[4,4-bis(4-fluorophenyl)butyl]-lH-imidazole
lH NMR (as HCl-salt, MeOH-d4):
1.40-1.85 (m, 2H), 1.90-2.30 (m, 2H), 2.77 (t, 2H),
3.98 (t, lH), 6.80-7.40 (m, 9H), 8.72 (d, lH)
5~ ;J~
Example 2
4-[4-(4-fluorophenyl)-4-phenylbutyl]-lH-imidazole
A concentrated water solution of ammon~umformate (0,98
g, 15,6 mmol) is added dropwise to the boiling mixture
of l-benzyl-5-[4-(4-fluorophenyl)-4-phenylbutyl]-lH-
imidazole (1,5 g, 3,9 mmol) and 10 % Pd/C (0,156 g) in
16 ml of 50 % ethanol. The mixture is refluxed for 2
hours. The catalyst is filtrated off and the solvent is
evaporated. 2 M NaOH is added and the product is
ext~acted into ethyl acetate. The ethyl acetate phase
is dried and evaporated to dryness to give the produc~.
Yield 1,02 g. Nelting point of the hydrochloride salt
(from ethyl acetate) is 175-182C.
lH NMR (as HCl-salt, MeOH-d4):
1.40-1.90 (m, 2H), 1.90-2.30 (m, 2H), 2.75 (t, 2H),
3.96 (t, lH), 6.85-7.36 (m, 10H), 8.74 (d, lH)
According to the same procedure as the example the
following substituted dexivative was prepared:
4-[4-(2-fluorophenyl)-4-phenylbutyl]-lH-imidazole. M.p.
of hydrochloride 182-190C.
H NMR (as HCl-salt, MeOH-d4):
1.45-1.95 (m, 2H), 1.95-2.30 (m, 2H), 2.77 (t, 2H),
4.29 (t, lH), 6.85-7.45 ~m, 10H), 8.74 (d, lH)
Example 3
4-(4,4-diphenylbutyl3-lH-imidaz~le
a) 1-benzyl-5-(4-hydroxy-4,4-diphenylbutyl)-lH-imidazole
2 ~
52
~agnesium turnings (0,49 g) are covered with 4 ml of
dry tetrahydrofuran. Brombenzene (3,18 g) i~ 7 ml o~
dry tetrahydrofuran is added dropwise to the mixture at
such a rate that a smooth reaction is maintained. The
reaction mixture is refluxed for an additional hour.
Ethyl 4-(1-benzyl-lH-imidazol-5-yl)butyrate (1,10 g) in
15 ml of dry tetrahydrofuran is then added dropwise to
the Grignard reagent and the reaction mixture is refluxed
for 2 hours. Saturated ammonium chloride is added to
the cooled reaction mixture. Tetrahydrofuran is
evaporated and the precipitated product is collected.
Yield 1,41 g. Melting point of the hydrochloride salt
is 197-201C.
lH MMR (as HCl-salt, MeOH-d4):
1.35-1.80 (m, 2H), 2.20-2.45 (m, 2H), 2.61 (t, 2H),
5.33 (s, 2H), 7.0-7.5 (m, 16~), 8.81 (d, lH)
b) l-benzyl-5-(4,4-diphenyl-3-butenyl)-lH-imidazole
1-benzyl-5-(4-hydroxy-4,4-diphenylbutyl)-lH-imidazole
(1,3 g) is refluxed in 20 ml of ethanol containing 5 %
(W/w) hydrogen chloride for 1 hour. The solvent is
evaporated and the HCl-salt of the product is
precipitated with ethyl acetate. Yield 1,1 g, m.p. 161-
168C.
lH NMR (as HCl-salt, MeOH-d4):
2.22-2.60 (m, 2H), 2.60-2.90 (m, 2H), 5.29 (s, 2H),
6.06 (t, lH), 6.90-7.60 (m, 16H~, 8.88 (d, lH)
c) 1-benzyl-5-(4,4-diphenylbutyl)-lH-imidazole
1-benzyl-5-(4,4-diphenyl-3-butenyl)-lH-imidazole
hydrochloride salt is hydrogenated in ethanol as
~ J~
53
described in Example 1 c). M.p. of the product as
hydrochloride is 200-202C.
d) 4-(4,4-diphenylbutyl)-lH-imidazole
The benzyl group of l-benzyl-5-(4,4-diphenylbutyl)-lH-
imidazole is hydrogenated as is described in Example
1 d).
Example 4
4-[4,4-bis(4-nitrophenyl)butyl]-lH-imidazole
1,8 g (14,6 mmol) of urea nitrate is added in small
portions to a mixture of 2,0 g (7,3 mmol) of 4-(4,4-
diphenylbutyl)-lH-imidazole in 6,4 ml of concentrated
sulphuric acid under 10 C. The reaction mixture is
stirred for 2 hours at room temperature. The mixture is
made alkalin~ with 2 M sodium hydroxide and the product
is extracted into ethyl acetate. The product is purified
by flash chromatography using methylene chloride-methanol
(95:5) as eluent.
H NMR (as HCl-salt, MeOH-d4):
1.4-1.95 (m, 2H), 2.0-2.45 (m, 2H), 2.81 (t, 2H), 4.34
(t, lH~, 7.27 (broad s, 1~), 7.5 (d, 4H), B.17 (d, 4H),
8.73 (d, lH)
Example 5
4-t4,4-bis(4-aminophenyl)butyl]-lH-imidazole
4-[4,4-bis(4-nitrophenyl)butyl]-lH-imidazole is
hydrogenated in ethanol using 10 ~ palladium on carbon
(Pd/C) as a catalyst.
54
1H NMR (as HCl-salt, MeOH-d4):
1.4-2.25 (m, 4H), 2.69 (t, 2H), 3.70 (t, lH), 6.77 ~d,
4H), 6-95 (d, 4H)t 7.08 (broad s, lH), 8.57 (d, lH)
Example 6
4-(4,4-diphenyl-1-butenyl)-lH-imidazole
a) 4-(1-hydroxy-4,4-diphenylbutyl)-lH-imidazole
A concentrated water solution of ammonium formate (4,0
g) is added dropwise to the boiling mixture of l-benzyl-
5-(1-hydroxy-4,4-diphenylbutyl)-lH-imidazole (4,5 g)
and 10 % Pd/C (0,5 g) in 50 ml of 50 % ~thanol. The
mixture is refluxed for 2 hours. The catalyst is
filtrated and the solvent is evaporated. 2 M NaOH is
added and the product is extracted into ethyl acetate.
The ethyl acetate phase is dried and evaporated to
dryness to give the product which is used in the
following step b).
b) 4-(4,4-diphenyl-1-butenyl)-lH-imidazole
4-(1-hydroxy-4,4 diphenylbutyl)-lH-imidazole (3,0 g)
and 20 g of anhydrous potassium hydrogen sulfate are
heated at 150 C for 4 hours. The mixture is cooled and
90 ml ethanol is added to dissolve the product. The
mixture is made alkaline with sodium hydroxide. The
product is extracted into methylene chloride, washed
with water and evaporated to dryness. The product is
then made ~o hydrochloride salt with dry hydrogen
chloride in ethyl acetate. M.p. above 240 C.
lH NMR (as HCl-salt, CDC13):
2.904-3.068 (m, 2H), 4.116 (t, lH), 6.05-6.35 (mr 2H),
6.g98 (d, lH), 7.22-7.25 (m, 10H3, 8.719 (d, lH)
~ ~J ~
Exam~le 7
4-[1-(4~fluorophenyl)-5-phenylpentyl]-lH-imidazole
a) 1-benzyl-5-(1-hydroxy-5-phenylpentyl)-lH-imidazole
2,1 g of magnesium turnings are covered with 60 ml of
dry tetrahydrofuran. A solution of 4-phenylbutylbromide
(18,8 g) in 20 ml of dry tetrah~drofuran is then added
dropwise to the mixture at such a rate that a smooth
reaction is maintained. After the addition is complete,
~he reaction mixture is refluxed for one additional
hour and cooled to room temperature. The reaction mixture
is then added dropwise to a solution of l-benzyl-5-
imidazolecarbaldehyde (6,5 g) in ~0 ml of tetrahydrofuran
at 60C. After the addition is complete, the reaction
mixture is refluxed for 2 hours, cooled and poured into
cold water. Tetrahydrofuran is evaporated and conc.
hydrochloric acid is added to the solution. The product
which is separated as an oil, is extracted with methylene
chloride and evaporated to dryness.
b) 4~ hydroxy-5-phenylpentyl)-lH-imidazole
1-benzyl-~ hydroxy-5-phenylpentyl)-lH-imidazole
hydrochloride (8,5 g), prepared in the Step a, i5
hydrogenated in the mixture of 100 ml of 2 N hydrochloric
acid and 10 ml of ethanol at 60C Pd/C (10 ~) as
catalyst. When the uptake of the hydrogen ceases, the
reaction mixture is cooled, filtered and evaporated to
dryness. Water is added and the mixture is made alkaline
with sodium hydroxide. The product is then extracted to
methylene chloride which i8 washed with water, dried
with sodium sulphate and evaporated to dryness. The
residue is the product as base, and it is used as such
in Step c.
56
lH NMR (as base, MeOH-d4 + a drop of CDCl3):
1.2-2.0 ~m, 6H), 2.61 (distorted t, 2~), 4.65 (t, lH),
6.91 (dd, lH), 7.0-7.3 (m, 5H), 7.56 (d, lH)
c) 4-(1-oxo-5-phenylpentyl)-lH-imidazole
5,5 g of 4-~1-hydroxy-5-phenylpentyl)-lH-imidazole and
7,0 g of manganese dioxide are refluxed stirring in
tetrachloroethylene for four hours. The reaction mixture
is filtered and the filtrate is evaporated to dryness.
Water is added and the product is extracted into
methylene chloride. The com~ined extracts are washed
with water and evaporated to dr~ness.
d) 4-[1-(4-fluorophenyl)-l-hydroxy-5-phenylpentyl]-lH-
imidazole
0,52 g of magnesiurn turnings are covered with 60 ml of
dry tetrahydrofuran. Then a solution of l-~romo-4-
fluorobenzene (3,8 g) in 60 ml of dry tetrahydrofuran is
added dropwise to the mixture at such a rate that a
smooth reaction is maintained. ~fter the addition is
complete~ the reaction mixture is refluxed for one
additional hour and cooled to room temperature. The
reaction mixture is then added dropwise to a solution
of 4-(1-oxo-5-phenylpentyl)-lH-imidazole t3,8 g) in 40
ml of tetrahydrofuran at ~0C. After the addition is
complete, the reaction mixture is refluxed for 3 hours,
cooled and poured into cold water. Tetrahydrofuran is
evaporat~d and conc. hydrochloric acid is added to the
sol~tion. The product is extracted as hydrochloric salt
into methylene chloride. Combined methylene chloride
extracts are then evaporated to dryness.
~ J3
57
e) 4-[1-(4-fluorophenyl)-5-phenyl-1-pentenyl]-lH-
imidazole
4-[1-(4-fluorophenyl)-l-hydroxy-5-phenylpentyl]-lH-
imidazole hydrochloride (5,0 g) and 30,0 g of anhydrous
potassium hydrogen sulphate are heated at 150C for 4
hours. The mixture is cooled and 90 ml of ethanol is
added to dissolve the product. The mixture is then
filtered and the filtrate is evaporated to minor volume.
Water is added and the mixture is made alkaline with
sodium hydroxide. The product is extracted into methylene
chloride, washed with water and evaporated to dryness.
The product is then made to hydrochloride salt with dry
hydrogen chloride in dry ethylaceta~e.
lH NMR (as base, CDCl3):
1.5-2.7 (m, 6H), 4.8 (broad s, lH), 6.34 (t, lH), 6.48
(broad s, lH), 6.9-7.4 (m, 9H), 7.52 (broad s, lH)
Using the same method for example the following compounds
included in the invention were prepared:
4-[1-(4-fluorophenyl)-5-(3-methylphenyl)-l-pentenyl]-
lH-imidazole
4-[l-(4-fluorophenyl)-5-(4-methylphenyl)-1-pentenyl]-
lH-imidazole
4-[1-(4-fluorophenyl)-5-(2-methylphenyl)-1-pentenyl]-
lH-imidazole
4-[5-(3,5-dimethylphenyl)-1-(4-~luorophenyl)-l-pentenyl]-
lH-imidazole
4-[1-(4-fluorophenyl)-5-(3-methoxyphenyl)-l-pentenyl]-
lH-imidazole
?, ~
4-[5-(3/5-dimetho~ypheny~ (4-fluorophen
pentenyl]-lH-imidazole
f) 4-[1-(4-fluorophenyl)-5-phenylpentyl]-lH-imidazole
4-[1-(4-fluorophenyl)-5~phenyl-1-pentenyl]-lH-imidazole
hydrochloride (2,0 g) is dissolved in ethanol and a
catalytic amount 10 % Pd/C is added. The reaction mixture
is agitated vigorously at room temperature in a hydrogen
atmosphere until the uptake of hydrogen ceases. The
mixture is filtered and the filtrate is evaporated to
dryness. The residue which is the product is purified
by flash chromatography eluting with methylene chloride-
methanol mixture. Yield 82 ~.
1H NMR (as base, CDCl3):
1.1-2.7 (m, 8H), 3.84 (t, lH), 6.71 (broad s, lH),
6.80-7.38 (m, 9H), 7.47 (broad s, lH), 9.22 (broad s, lH)
Using the same method for example the following compounds
included in the invention were prepared:
4-[1-(4-~luorophenyl)-5-(3-methylphenyl)pentyl]-lH-
imidazole
MS: 322 (20, M+-), 189 (28), 176 (38), 175 (72), 149
(100), 125 (20), 121 (14), 109 (42), 105 (16), 97 (21)
1H NMR (as HCl-salt, MeOH-d~):
1.1-2.7 (m, 8H), 2.27 (s, 3H), 4.06 (t, lH), 6.7-7.5
(m, 8H), 7.37 (d, lH), 8.77 (d, lH)
4-tl-(4-fluorophen~1)-5-(4-methylphenyl)pentyl]-lH-
imidazole
59 ~ J~
H NMR (as H~l-salt, MeOH-d4):
1.1-2.7 (m, 8H), 2.26 (s, 3H), 4.05 (t, lH), 6 8-7.6
(m, 9H), 8-78 (d, lH)
4-[1-(4-fluorophenyl)-5-(2-methylphenyl)pentyl]-lH-
imidazole
MS: 322 (53, M~-), 189 (30), 176 (55), 175 (100), 148
(18), 121 (12), 105 (42), 101 (11), 79 (12), 77 (13)
lH NMR (as HCl-salt, MeOH-d4):
1.1-2.7 (m, 8H), 2.24 (s, 3H), 4.11 (t, lH), 6.9-7.5
(m, 9H), 8.79 (d, lH)
4-t5-(3,5-dimethylphenyl)-1-(4-fluorophenyl~pentyl]-lH-
imidazole
MS: 336 (47, M+-), 189 (90), 176 (67), 175 (100), 166
(13), 148 (16), 121 (12), 119 (16), 91 (14)
H NMR (~s HCl-salt, MeOH-d4):
1.1-2.6 (m, 8H), 2.22 (s, 6H), 4~09 (t, lH), 6.6-7.4
(m, 7H), 7.40 (broad s, lH), 8.80 (broad s, lH)
4-[1-(4-fluorophenyl)-5-(3-methoxyphenyl)pentyl~-lH-
imidazole
4-[5-(3,5-dimethoxyphenyl)-1-(4-fluorophenyl)pentyl]-lH-
imidazole
Example 8
4-(1,3-diphenylpropyl)-lH-imidazole
a) l-benzyl-5-(1-oxo-3-phenylpropyl)-lH-imidazole
7i ~ ~J
l-benzyl-5-(l-hydroxy-3-phenylpropyl)-lH-imidazole is
oxidized with manganese dioxide in tetrachloroethylene,
as it is described in Example 7 c).
lH NMR (as base, CDCl3):
3.03 (m, 4H), 5.53 (s, 2H), 7.07-7.4 (m, lOH), 7.60 (s,
lH), 7.77 (s, lH)
Using the same method for example the following compounds
included in the invention were prepared:
l-benzyl-5-[5-(2,6-dimethylphenyl)-1-oxopentyl]-lH-
imidazole. M.p. of hydrochloride 175-180C.
1-benzyl-5-(l-oxo-5-phenylpentyl)-lH-imidazole. M.p. of
hydrochloride 185-189C.
b) l-benzyl-5-(1-hydroxy-1,3-diphenylpropyl)-lH-imidazole
Grignard reagent is prepared from 5,0 g of bromobenzene
and 0,76 g of Mg turnings in tetrahydrofuran. This
solution is then added to 3,1 g of 1-benzyl-5-(l-oxo-3-
phenylpropyl)-lH-imidazole in tetrahydrofuran and the
reaction mixture is refluxed for 3 hours. The mixture
is then poured into cold water, tetrahydrofuran is
evaporated and the solution is made acidic with
hydrochloric acid. The hydrochloride of the product is
filtered, washed with toluene and dried. M.p. 196-198~C.
Using the same method for example the following compounds
included in the invention were prepared:
1-benzyl-5-(1-hydroxy-1,5-diphenylpentyl)-lH-imidazole.
M p. of hydrochloride 193-196~C.
?3 7) ,3
61
l-benzyl-5-[5-(2,6-dimethylphenyl)-1-hydroxy-1-
phenylpentyl]-lH-imidazole. M.p. of hydrochloride 190-
192C.
c) l-benzyl-5-tl,3-diphenylpropyl)-lH-imidazole
1-benzyl-5-(1-hydroxy-1,3-diphenylpropyl)-lH-imidazole
is treated with anhydrous potassium hydrogen sulphate
at 150C as described in Example 7 e). The double bond
of the obtained intermPdiate, l-benzyl-5-(1,3-diphenyl-
1-propenyl)-lH-imidazole, is hydrogenated as described
in Example 7 f). M.p. of the hydrochloride salt is 154-
174C (from diethylether).
1H NMR (as HCl-salt, MeOH-d4):
2.2-2.7 (m, 4H), 3.90 (t, lH), 5.12 (AB q, the middle
of the quartet, 2H), 6.90-7.37 (m, 15H), 7.70 (broad s,
lH), 8.88 (d, lH)
Using the same method for example the following compounds
were prepared:
l-benzyl-5-(1,5-diphenylpentyl)-lH-imidazole
lH NMR (as base, CDCl3~:
1.18-2.61 (m, 8H), 3.56 (t, lH), 4.59 and 4.81 (AB q,
2H), 6.76-7.40 (m, 17H)
1-benzyl-5-[5-(2,6-dimethylphenyl) l-phenylpentyl]-lH-
imidazole
d) 4-(1,3 diphenylpropyl)-lH-imidazole
1-benzyl-5-(1,3-diphenylpropyl)-lH-imidazole is
hydrogenated in the mixture o~ 2N hydrochloric acid and
ethanol at 60C 10 % Pd/C as catalyst. The product is
~2
isolated as in Example 7 b) and is purified by flash
chromatography methylene chloride-methanol (9,5:0,5) as
eluent. Yield 73 %.
1H NMR (as base, CDC13):
2.1-2.7 (m, 4H), 3.88 (t, lH), 6.71 (broad s, lH),
7.01-7.26 (m, 10H), 7.30 (d, lH), 10.5 (broad s, lH)
Using the same method for example the following compound
was prepared:
4-~1,5-diphenylpentyl)-lH-imidazole
lH NMR (as HCl-salt, MeOH-d4):
1.2-2.3 (m, 6H), 2.57 (distorted t, 2H), 4.05 (t, lH),
7.05-7.40 (m, llH), 8.73 (d, lH)
Example 9
l-benzyl-4-(1,3-diphenylpropyl)-lH-imidazole
2,0 g of benzylbromide in 5 ml of toluene is added
dropwise to the mixture of 4-(1,3-diphenylpropyl)-lH-
imidazole (2,6 g), 48 % NaOH (10 ml), toluene (20 ml)
and tetrabutylammoniumbromide (O,2 g) at room
temperature. After addition the reaction mixture is
stirred at room temperature for 3 hours. Water is added
and the toluene layer is separated. The toluene phase
is then washed with water and evaporated to dryness.
The residue contains the isomers l-benzyl-4-(1,3-
diphenylpropyl)-lH-imidazole and 1-benzyl-5-(1,3-
diphenylpropyl)-lH-imidazole and the former is separated
and purified by flash chromatography ~methylene chloride-
methanol 9,5;0,5).
63
Example 10
4-[1,4-bist4-fluorophenyl)butyl]-lH-imidazole
a) l-benzyl-5-[1-(4-fluorophenyl)-l-hydroxymethyl]-lH-
imidazole
Grignard reaction of 4-bromofluorobenzene and l-benzyl-
5-imidazolecarbaldehyde is performed analogously to
Example 7 a). The product is crystallized as
hydrochloride salt from ethylacetate. Yield 94 %.
lH NMR (as base, CDC13 + MeOH-d4):
5.05 and 5.21 (AB q, 2H), 5.64 (s, lH), 6.61 (5, lH),
6.97-7.1 (m, 4H), 7.26-7.33 (m, 5H), 7.41 (s, lH)
MS: 282 (22, M+-), 265 (5), 191 (18), 91 (100)
b) l-benzyl-5-[1-(4-fluorophenyl)-1-oxomethyl]-lH-
imidazole
Oxidation of l-benzyl-5-[1-(4-fluorophenyl)-1-
hydroxymethyl]-lH-imidazole is performed analogously to
Example 7 c). The crude product is recrystallized as a
base from methanol. Yield 72 %.
lH NMR (as base, CDCl3):
5.62 (s, 2H), 7.1-7.4 (m, 7H), 7.5-8.0 (m, 4H)
MS: 280 (46, M+-), 123 (13), 107 (4), 95 (19), 91 (100)
c) 1-benzyl-5-[1,4-bis(4-fluorophenyl)-1-hydroxybutyl]-
lH-imidazole
l-benzyl-5- L 1, 4-bis(4-fluorophenyl)-1-hydroxybutyl]-lH-
imidazole is prepared analogously to Example 7 a)
~ S~3 1 ,~
64
starting from 3-(4-fluorophenyl)-1-bromopropane and 1-
benzyl-5-[1-(4-fluorophenyl)-1-oxomethyl]-lH-imidazole.
The product is p~rified by flash chromatography.
lH NMR (as base, CDCl3):
1.2-1.4 (m, lH), 1.65-1.85 (m, lH), 2.1-2.25 (m, 2H),
2.52 (t, 2H), 4.73 and 4.81 (AB q, 2H), 6.75-7.3 (m, 15H)
Using the same method for example the following compounds
included in the invention were prepared:
l-benzyl-5-[1-(4-fluorophenyl)-1-hydroxy-3-phenylpropyl]-
lH-imidazole
MS: 386 (M+-, 8), 368 (22), 281 (lO0), 159 (26), 91
(99), 65 (34)
l-benzyl-5-[1,3-bis(4-fluorophenyl)-1-hydroxypropyl]-
lH-imidazole
MS: 404 (M+ , 2), 386 ~18), 195 (16), 281 (33), 123
(34), 91 (100), 65 (20)
l-benzyl-5-[l-(4-fluorophenyl)-1-hydroxy-4-phenylbutyl]
lH-imidazole
MS: 400 (M+ , 2), 382 (2), 281 (38), l91 (3), 91 (100),
65 (9)
l-benzyl-5-[1-(4-fluorophenyl)-1-hydroxy-5-(3-
methoxyphenyl)pentyl]-lH-imidazole
lH NMR (as HCl-salt, MeOH-d4):
1.0-1.8 (m, 4H), 2.26 (t, 2H), 2.52 (t, 2H), 4.71 (s,
3H), 5.06 and 5.31 (ABq, 2H), 6.6-7.5 (m, 13H), 7.7 (s,
lH), 8.6 (s, lH)
~ 3
l-benzyl-5-[5-(2,6-dimethylphenyl)-1-(4-fluorophenyl~-
l-hydroxypentyl]-lH-imidazole
MS: 442 (M+ , 19), 281 (71), 256 (5), 191 (7), 119
(21), 91 (100)
d) 4-[1,4-bis(4-fluorophenyl)butyl]-lH-imidazole
1-benzyl-5-[1,4-bis(4-fluorophenyl)-1-hydroxybutyl]-lH-
imidazole (10 g) is dissolved in conc. acetic acid (100
ml). Into the solution is added 0,1 g of palladium on
carbon and 0,8 g of ammoniumformate. The mixture is
refluxed for an hour and cooled to room temperature.
The solution is filtered through silicous earth. The
acetic acid filtrate is evaporated, the residue is
dissolved in methylene chloride and washed once with 2
M aqueous sodium hydroxide solution and once with water.
The methylene chloride solution is dried with sodium
sulphate and evaporated under raduced pressure. The
product is then made to hydrochloride salt with dry
hydrogen chloride in diethylether. M.p. ~35-137 C.
lH NMR (as base, CDCl3):
1.4-1.65 (m, 2H), 1.8-1.95 (m, lH), 2.05-2.2 (m, lH),
2.5-2.65 (m, 2H)~ 3.87 (t, lH), 6.7 (s, lH~, 6.8-7.2
(m, 8H), 7.5 (s, lH)
Using the same method for example the following
compounds included in the invention were prepared:
4-tl-(4-fluorophenyl)-3-phenylpropyl]-lH-imidazole
1H NMR (as base, CDCl3):
2.0-2.7 (m, 4H), 3.7-4.0 (m, lH), 6.7 (s, lH), 6.75-
7.45 (m, 9H), 7.57 (s, lH)
66 2 Jf~v ~J
MS: 280 (4, M~-), 189 (9), 176 (100), 148 (15), 121 (9),
91 (12)
4-[1,3-bis(4-fluorophenyl)propyl]-lH-imidazole
lH NMR (as base, CDC13):
2.1-2.25 (m, lH), 2.3-2.6 (m, 3H), 3.88 (t, lH), 6.76
(s, lH), 6.9-7.2 (m, 8H~, 7.63 (s, lH)
MS: 298 (5, M+-), 189 (8), 176 (100), 122 (22), 109 (42)
4-[1-(4-fluorophenyl)-4-phenylbutyl3-lH-imidazole
lH NMR (as base, CDC13):
1.3-2.3 (m, 4H), 2.6 (t, 2H), 3.88 (t, lH), 6.67 (s,
lH), 6.7-7.3 (m, 9H), 7.39 (s, lH)
MS: 294 (31, M+ ), 189 (24), 175 (100), 91 (31)
4-[1-(4-fluorophenyl)-5-(3-methoxyphenyl)pentyl]-lH-
imidazole
H NMR (as base, CDC13):
1.1-2.3 (m, 6H), 2.5 (t, 2H), 3.76 (s, 3H), 3.85
(distorted t, lH), 6.6-7.6 (m, lOH)
MS: 338 (19, M+ ), 189 (43), 175 (70), 148 (12), 121
(20), 36 (100)
4-[1-(4-fluorophenyl)-5-(4-methoxyphenyl)pentyl]-lH-
imidazole
lH NMR (as base, CDC13):
1.1-2.3 (m, 6H), 2.49 (~, 2H), 3.75 (s, 3H), 3.8
(distorted t, lH), 6.6-7.6 (m, lOH)
67 ~3 ~3. ~ ~ r~ ~-t ''
MS: 338 (10, M+ ), 189 ~20), 175 (22), 148 (6), 121
(22), 36 (100)
4-[5-(2,6-dimethylphenyl)-1-(4-fluorophenyl)pentyl]-lH-
imidazole
lH NMR (as base, CDC13):
1.25-1.5 (m, 4H), 1.8-1.95 (m, lH), 2.05-2.2 (m, lH),
2.25 (s, 6H), 2.52 (t, 2H), 3.86 (t, lH~, 6.71 (s, lH),
6.9-7.0 (m, 5H), 7.1-7 2 (m, 2H), 7.4 (s, lH)
MS: 336 (50, M+-), 217 (20), 189 (75), 175 (100), 148
(13), 119 (23)
Example 11
l-benzyl-5-~1-(4-fluorophenyl)-3-phenyl-1-propenyl~-
lH-imidazole
Titanium tetrachloride (0,03 mol) is added dropwise to
a stirred suspension of zink powder (0,06 mol) in
tetrahydrofuran (30 ml) at -10 C under dry nitrogen.
The mixture is heated to reflux and refluxing is
continued for 1 hour. The solution is cooled to 0 C
and 1-benzyl-5-[1-(4-fluorophenyl)-1-
oxomethyl]-lH-imidazole (0,005 mol) in tetrahydrofuran
(10 ml~ and phenylacetaldehyde (0,006 mol) in
tetrahydrofuran (10 ml) are added into the mixture,
respectively. The mixture is refluxed with stirring for
1 hour. The dark mixture is poured into water (60 ml),
tetrahydrofuran is evaporated and the mixture is
extracted with methylene chloride (2 x 100 ml). The
methylene chloride solution is washed with 2 N sodium
hydroxîde and water, dried with sodium sulphate and
evaporated to dryness. The residue is purified by flash
chromatography.
- 6~ -
H NMR (as base, C~C13):
3.35 and 3.45 (2d, 2H), 4.62 and 4.65 (2s, 2H), 6.03 and 6.2
(2t, lH), 6,8-7.3 (m, 15H), 7.50 and 7.64 (2s, lH).
Example 12
4-[1,3-bis(4-nitrophenyl)propyl]-lH-imidazole
1,8 g (14,6 mmol) of urea nitrate is added in small portions
to a mixture of 2,7 g (7,3 mmol) of 4-(1,3-diphenylpropyl)-
lH-imidazole in 6,4 ml of concentrated sulphuric acid under
10C. The reaction mixture is stirred for 2 hours at room
temperature. The mixture is made alkaline with 2 M sodium
hydroxide and the product is purified by flash chromatography
using methylene chloride-methanol (95:5) as eluent.
In formula Ia, preferably Rl, R2, R'l and R'2 are
each H and R4, R5, R6 and R7 are each H. If one phenyl group
in formula Ia is mono-substituted then preferably Rl, R'2 and
R2 are each H and Rl is not hydrogen and is in the ortho,
meta or para position of the phenyl group, preferably the
para position. When one phenyl group is mono-substituted in
the para position and the other group is unsubstituted,
preferably the substituent is OCH3.
If both phenyl groups in formula Ia are mono-
substituted, preferably R2 and R'2 are both H, Rl and R'l are
both not H and are each in the para position on the
respective phenyl groups. Rl and R'l are preferably both F.
If a phenyl group is di-substituted, preferably R'l
and R'2 are both H, Rl and R2 are both not H and are in the 3
and 5 or 3 and 4 positions on the phenyl group.
,- " s~ f~ ~ ~
- 69 -
Preferred compounds of formula Ia also include
those in which R4 and R6 together form a bond, and those in
which R' is H.
In formula Ib, preferably R4, R5 and R' are each H.
If one phenyl group in formula Ib is mono-substituted then
preferably Rl, R2 and R'2 are each H and R'l is not hydrogen
and is in the para position of the phenyl group. Preferably
R'l is F.
If both phenyl groups in formula Ib are
mono-substituted, preferably R2 and R'2 are both H, Rl and
R'l are both not H, Rl is in the ortho, meta or para position
of the phenyl group and R'l is the para position of the
phenyl group. Preferably Rl is in the para position.
Preferred substituents are CH3 and F, in particular it is
prferred that Rl and R'l are both F or Rl is CH3 and R'l is
F.
If one phenyl group is di-substituted and the other
is mono-substituted, preferably R'2 is H, Rl, R2 and R'l are
each not H and R'l is the para position of the phenyl group
and Rl and R2 are in the 3 and 5 or 2 and 6 positions of the
phenyl group.