Note: Descriptions are shown in the official language in which they were submitted.
2~
JAB 676
S YDRQXYALKYLFURANYL DERIVAllVES
10 BackgrQun~ of the invention
In US-4,556,660; 4,634,704; 4,695,569; 4,695,575; 4,588,722; 4,835,161;
4,897,401 and in EP-A-0,206,415 and 0,297,661 there are disclosed related furanyl
and/or aLkylfuranyl derivatives as antihistaminics and serotonin antagonists.
15 Descli~tiQ~Qf e invention:
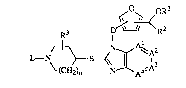
The present invention is concerned with compounds having the formula
D~Y~<
R3 I R2
L--N ~B <N3~ ~A2
~(CH~)n A4~A
20 the pharmaceutically acceptable acid addition salts and the stereochemically isomeric
forms thereof, wherein
-Al=A2-A3=A4- is a bivalent radical having the formula
-CH=CH-CH=CH- (a- 1),
-N=CH-CH=CH- (a-2),
-CH=N-~H=CH- (a-3), ~.
-CH=CH-N=CH- (a-4),
-CH=CH-CH=N- (a-S),
-N=CH-N=CH- (a-6) or :
-CH=N-CH=N- (a-7), ~ -
wherein one M two hydrogen atoms in said radicals (a-l) to (a-7) each independently
from one another may be rep]aced by halo, C1 6alkyl, C1 6alkyloxy, trifluoromethyl or
hydroxy;
:
:: :
: .
-2- . 2
- D is C1 4alkanediyl;
R1 is hydrogen, C1 6alkyl, arylCl 6alkyl or C1 6aLIcylcarbonyl;
R2 is hydrogen or C1 6alkyl;
S R3 is hydrogen or C1 6alkyl;
nisO, 1 or2;
BisNR4,0,S,SO,S02orCH2;
R4 is hydrogen, C1 6alkyl, C3 6cycloalkyl or arylC1 6alkyl;
L is hydrogen, Cl 12aL~cyl, C3 6cycloalkyl, C3 6aL~cenyl optionally substituted with
aryl, Cl 6alkylcarbonyl, Cl 6alkyloxycarbonyl, arylcarbonyl, arylCl 6alkyloxy-
carbonyl, C1 6alkylsulfonyl, arylsulfonyl, or a radical of forrnula
-Alk-R5 (b- I ),
-Alk-Y-R6 (b-2),
-ALIc-Zl-C(=X)-Z2-R7 (b-3), or
-CH2-CHOH-CH2-O-R8 (b-4), wherein
RS is halo, cyano, isocyanato, isothiocyanato, aryl, Het or arylsulfonyl;
R6 is hydrogen, aryl? Het or Cl 6alkyl optionally substituted with halo, aryl or Het;
R7 is hydrogen, aryl, Het or Cl 6alkyl optionally substituted with halo, aryl or Het;
20 R8 is aryl or naphthalenyl;
Y is 0, S, NR9, said R9 being hydrogen, Cl 6alkyl or Cl 6alkylcarbonyl;
zl and z2 each independently are 0, S, NR10 or a direct bond;
said R10 being hydrogen or C1 6alkyl;
X is 0, S or NR1 1; said Rl 1 being hydrogen, Cl 6alkyl or cyano;
25 each Alk independently is Cl 6alkanediyl;
each Het is a f1ve- or six-membered heterocyclic ring containing 1, 2, 3 or 4
hetero-atoms selected from oxygen, sulfilr and nitrogen, provided that no more than 2
oxygen and / or sulfur atoms are present, said five or six-membered ring being optionally
30 condensed with a five- or six-membered carbocyclic or heterocyclic ring also containing
1, 2, 3 or 4 heteroatoms selected from oxygen, sulfur and nitrogen, provided that the
latter ring does not contain more than 2 oxygen and/or sulfur atoms and that the total
number of heteroatoms in the bicyclic ring system is less than 6; and when Het is a
monocyclic ring system it may optionally be substituted with up to 4 substituents, when
35 Het is a bicyclic ring system it may optionally be substituted with up to 6 substituents,
said substituents being selected from a bivalent radical of fo~nula X; halo; isocyanato;
isothiocyanato; nitro; cyano; ~ifluoromethyl; a radical of formula -E; a radical of
forrnula -Y-E; or a radical of formula Zl-C(=X)-Z2-E; wherein X, y, z1 and z2 are as
previously def,ned hereinabove; and E is hydrogen, aryl or C1 6alkyl being optionally
substituted with aryl, Cl 6alkyloxy, aryloxy, hydroxy or Cl 6alkyloxycarbonyl; and
each aryl is phenyl optionally substituted with 1, 2 or 3 substituents each
independently selected from halo, hydroxy, nitro, cyano, trifluoromethyl, Cl 6alkyl,
Cl 6alkyloxy, Cl 6alkylthio, mercapto, amino, mono- and di(Cl 6alkyl)amino,
carboxyl, C1 6alkyloxycarbonyl and C1 6alkylcarbonyl;
provided that when R5 is Het, said Het is other than a 2-amino-3,4-dihydro-4-
oxo-5-pyrimidinyl group, wherein tne hydrogen on the 6-position may be replaced by a
Cl 6alkyl radical and wherein the nitrogen atom in the 3-position and the nitrogen atom
of the amino group optionally are substituted, or are linked by a bivalent radical of
formula-(cH2)2-~ -(CH2)3-, -CH=CH-, -CH=N-, -N=CH-, or-N=CH-CH2-,
wherein one or where possible two hydrogen atoms of said bivalent radicals each
independently from one another may be replaced by C1 6alkyl.
In ~he compounds of forrnula (I) where R5, R6 or R7 is Het, said Het may be partly
or completely saturated, or unsaturated. The compounds of formula (I) wherein Het is
partly saturated or unsaturated and is substituted with hydroxy, mercapto or amino, may
also exist in their tautomeric forms. Such forms although not explicitly indicated
hereinabove, are intended to be included within the scope of the invention.
As used in the foregoing definitions halo is generic to fluoro, chloro, bromo and
iodo; Cl 6allcyl defines straight and branch chained saturated hydrocarbon radicals
having from 1 to 6 carbon atoms such as, for example, methyl, ethyl, propyl,
I-methylethyl, butyl, l,1-dimethylethyl, 1-methylpropyl, 2-methylpropyl, pentyl, hexyl
and the like; C1 12alkyl defines C1 6alkyl radicals as defined hereinabove and the higher
homologs thereof having from 7 to 12 carbon atoms; C3 6cycloalkyl is generic to
;~ 30 cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl; C3 6alkenyl defines straight and
branch chained hydrocarbon radicals containing one double bond and having from 3 to 6
carbon atoms such as, for example, 2-propenyl, 3-butenyl, 2-butenyl, 2-pentenyl,3-pentenyl, 3-methyl-2-butenyl and the like, ~he carbon atom of said C3 6alkenylconnected to a heteroatom preferably being saturated; C1 4alkanediyl defines bivalent
straight and branch chained saturated hydrocarbon radicals having from l to 4 carbon
atoms such as, for example, methylene, 1,2-ethanediyl, 1,3-propanediyl, 1,4-butançdiyl
and the branched isomers thereof; C1 6alkanediyl defmes C1 4alkanediyl radicals as
4 2~3~9~
defined hereinabove and the higher homologs thereof having fr~m S to 6 carbon atoms
such as, for example, 1,5-pentanediyl, l,~hexanediyl and the branched isomers thereof.
Said pharmaceutically acceptable acid addition salts as mentioned hereinabove
5 comprise the therapeutically active non-toxic acid addition salt forms which the
compounds of formula (I) are able to form. Said salt forms can conveniently be obtained
by treating the base form of the compounds of formula (I) with appropriate acids such as
inorganic acids, for example, hydrohalic acid, e.g. hydrochloric, hydrobromic and the
like acids, sulfuric acid, nitric acid, phosphoric acid and the like; or organic acids, such
10 as, for example, acetic, propanoic, hydroxyacetic, 2-hydroxypropanoic, 2-oxopro-
panoic, ethanedioic, propanedioic, butanedioic, (Z)-2-butenedioic, (E)-2-butenedioic,
2-hydroxybutanedioic~ 2,3-dihydroxybutanedioic, 2-hydroxy-1,2,3-propanetricar-
boxylic, methanesulfonic, ethanesulfonic, benzenesulfonic, 4-rnethylbenzenesulfonic,
cyclohexanesulfamic, 2-hydroxybenzoic, 4-amino-2-hydroxybenzoic and the like acids.
15 Conversely the salt form can be converted by treatment with allcali into the free base
form.
The term acid addition salt also comprises the hydrates and solvent addition forrns
which the compounds of formula (I) are able to form. Examples of such forms are e.g.
hydrates, alcoholates and the like.
The compounds of this invention may have several asymmetric carbon atoms in their
s~ructure. Each of these chiral centers may be indicated by the stereochemical descriptors
R and S, this R and S notation corresponding to the rules described in Pwre Appl.
Chem., 1976, 45, 11-30.
25 Pure stereochemically isomeric fonns of the compounds of forrnula (I) may be
obtained by the application of art-known procedures. Diastereoisomers may be separated
by physical methods such as selective crystallization and chromatographic techniques,
e.g. counter current distribution, liquid chromatography and the like; and enantiomers
may be separated from each other following art-known resolution methods, for example,
30 by the selective crystallization of their diastereomeric salts with chiral acids. Pure
stereochemically isomeric forms may also be dçrived fiom the c~rresponding pure
stereochemically isomeric forms of the appropriate starting materials, provided that the
reactions occur stereospeci~lcally. Preferably, if a specific stereoisomer is desired, said
compound will be synthesized by stereoselective methods of preparation. These methods
35 will advantageously employ enantiomerically pure starting materials. S~ereochemically
isomeric forrns of the compounds of formula (I) are obviously intended to be included
within the scope of the invention.
-5- ~`D~%
The moiety _ D~ ~< in the compounds of formlula (I) as defined
4 3 R2
hereinabove, in particular is ~2-CHR2ORl-furan-5-yl)-C1 4alkyl-, (2-CHR20RI-furan-
5 4-yl)-Cl 4aLlcyl-, (2-CHR20Rl-furan-3-yl)-C1 4alkyl-, (3-CHR20Rl-furan-S-yl)-
Cl 4alkyl-, ~3-CHR20Rl-furan-4-yl)-C1 4aLtcyl- or (3-CHR20R1-furan-2-yl)-
Cl 4aLkyl-, wherein Rl and R2 are as defined hereinabove and Cl 4alkyl defines
straight and branch chained hydrocar'oon radicals having from 1 to 4 carbon atoms i.e.
methyl, ethyl, propyl, methylethyl, butyl, I-methylpropyl, 2-me~hylpropyl and
10 l ,l-dimethylethyl.
In particular, the radical Het as defined hereinabove may bç: . :
(i) an opdonally substituted five- or six-membered heterocyclic ring containing 1, 2, 3
or 4 heteroatoms selected from oxygen, sulfilr and nitrogen, provided that no more
than 2 oxygen and/or sulfur atoms are present;
(ii) an optionally substituted five- or six-membered hetçrocyclic ring containing 1 or 2
heteroatoms selected from oxygen, sulfur and nitrogen, being fused with an
optionally substituted five- or six-mem'oered ring through 2 carbon atoms or 1
carbon and 1 nitrogen atom, containing in the remainder of the fused ring only
car'oon atoms; or
(iii) an optionally subs~ituted five- or six-membered heterocyclic ring containing 1 or 2
heteroatoms selected from oxygen, sulfur and nitrogen, 'oeing fused with an
optionally substituted five- or six-membered heterocyclic ring through 2 carbon
atoms or I carbon and 1 nitrogen atom, containing in the remainder of the fused ring
1 or 2 heteroatoms selected ~om oxygen, sulfur and nitrogen;
wherein Het being a monocyclic ring system may be optionally substituted with up to 4
substituents; and wherein Het being a bicyclic ring system may be optionally substituted
with up to 6 substituents, said substituents being the sarne as defimed hereinabove.
More par~cularly Het is selected from p~idinyl, optionally substituted with one or
two substituents each independently selected from halo, amino, mono- and
di(Cl 6allyl)amino, arylCl 6alkylamino, nitro, cyano, aminocarbonyl, Cl 6alkyl,
C1 6alkyloxy, Cl 6alkylthio, Cl 6alkyloxycarbonyl, hydroxy, C1 6alkylcar'oonyloxy,
arylCl 6alkyl and carboxyl; pyridinyloxide, optionally substituted with nitro;
pylîmidinyl, optionally substituted with one or two substituents each independently
.:
.
. - , :
. . .
--6-- ;~L3~ ?
selected from halo, amino, Cl ~alkylamino, arylCl 6alkylamino, hydroxy, Cl salkyl,
Cl 6alkyloxy, Cl 6alkylthio and arylCl 6alkyl; pyridæinyl, optionally substituted with
C1 6aLkyl or halo; pyrazinyl, optionally substituted with halo, amino or Cl 6alkyl;
thienyl, optionally substituted with halo or Cl 6alkyl; furanyl, optionally substituted
5 with halo or Cl ~aLkyl; pyrrolyl, optionally substituted with Cl 6alkyl; thiazolyl,
optionally substituted wi~h Cl 6alkyl, Cl 6alkyloxycarbonyl, aryl or arylCl 6alkyl;
imidazolyl, optionally substituted with one or two substieuents each independently
selected from Cl 6alkyl, arylCl 6alkyl and nitro; tetrazolyl, optionally substituted with
Cl 6alkyl; 1,3?~thiadiazolyl, optionally substituted with Cl 6alkyl; 5,~dihydro-4H-
1() 1,3-thiazin-2-yl, optionally substituted with Cl 6alkyl; 4,5-dihydrothiazolyl, optionally
substituted with Cl 6alkyl; oxazolyl, optionally substituted with C1 6alkyl; 4,5-dihydro-
S-oxo-lH-tetrazolyl, optionally substituted with Cl 6alkyl; 1,4-dihydro-2,4-dioxo-
3~2O-pyrimidinyl, optionally substituted with Cl 6alkyl; 4,5-dihydro-
4-oxopyrimidinyl, optionally substituted with up to 3 substituents selected from Cl
15 6alkyl, amino, Cl 6allcylaminocarbonylamino, arylaminocarbonylamino, arylCl 6alkyl-
amino and Cl 6alkylamino; 2,3-dihydro-3-oxopyridazinyl; 2-oxo-3-oxazolidinyl;
pyrrolidinyl; piperidinyl; morfolinyl; thiornorfolinyl; dioxanyl, optionally substituted
with Cl 6alkyl; indolyl, optionally substituted with hydroxy or Cl 6alkyl; quinolinyl,
optionally substituted with hydroxy or Cl 6alkyl; quinazolinyl, optionally substituted
20 with hydroxy or Cl 6alkyl; quinoxalinyl, optionally substituted with Cl 6alkyl;
phthalazinyl, optionally substituted with halo; 1,3-dioxo-lH-isoindol-2(3O-yl; 2,3-
dihydro-3-oxo-4H-benzoxazinyl and 2,3-dihydro-1,4-benzodioxinyl, both being
optionally substituted with Cl 6alkyl or halo; 2-oxo-2H- l-benzopyranyl and 4-oxo-4H-
l-benzopyranyl, both being optionally substituted with Cl 6alkyl, and
a bicyclic heterocyclic radical of formula
R12
R l 3
x2 X
nl2
Rl3
3 ~ (C4)~
o
~ " '
-` ;2;C9~3~39~:
-7 -
G~ (C~s)' ~~ (c-6),
R12 R12
1~, (c-7), ~N~ (c-8),
wherein Xl and X2 each independently are O or S;
S each R12 independently is hydrogen, C1 6alkyl, arylC1 6alkyl, Cl 6alkyloxy-
C1 6alkyl, hydroxyCl 6alkyl or C1 6alkyloxycarbonyl;
each R13 independently is hydrogen, Cl 6alkyl, hydroxy, mercapto, C1 6alkyloxy,
C1 6alkylthio,haloorC1 6alkyloxycarbonylC1 6alkyl;
and the long dash in the radicals (c-1), (c-4), (c-S), (c-6) and (c-7) signifies that any
hydrogen atom of said radicals, including R12 and R13, may represent the bond linking
Het to respectively Alk, Y or z2 in the radicals of formula (b-1), (b-2) and (b-3);
G1 is-CH=CH-CH=CH~ or-S-CH=CH-;
G2 is -CH=CH-CH=CH-. -(CH2)4-~ -S-(CH2)2-~ -S-(CH2)3-~ -S-CH=CH-~
-CH=OEI-O-,-CH=C(CH3)-O-,-NH-(CH2)2-,-NH-(cH2)3-~ I-CH=CH-,
-NH-N=CH-CH2-, -NH-CH=N- or-NH-N=CH-;
G3 is -CH=CH-CH=CH-, -CH2-NH-~OEI2)2-, -S-CH=CH-, -S-(CH2)3-,
-N=CH-CH-CH-, -CH=N-CH=CH-, -CH=CH-N=CH-, -CH=CH-CH=N-,
-N=CH-N=CH- or-CH=N-CH=N-;
G4 is -CH=CH-CH=CH-, -CH2-NH-(CH2)2-, -N=CH-CH=CH-, -CH=N-CH=CH-,
-CH=CH-N=CH-, -CH=CH-CH=N-, -N=CH-N=CH- or -CH=N-CH=N-;
G5 is -CH=CH-CH=CH-, -N=CH-CH=CH-, -CH=N-CH=CH-, -CH=CH-N=CH-,
-CH=CH-CH=N-, -N=CH-N=CH- or-CH=N-CH=N-;
G6 is -CH=CH-CH=CH-, -N=CH-CH=C~l-, -CH=N-CH=CH-, -CH=CH-N=CH-,
-CH=CH-CH=N-, -N=CH-N=CH- or-CH=N-CH=N-;
wherein one or two hydrogen atoms in said radicals Gl, G2, G.3, G4, G5 or G6 or
in the benzene part of the radicals of formula ~c-2) M (c-3) may be replaced by
Cl 6alkyl, Cl 6alkylthio, C1 6alkyloxy or halo when connected to a carbon atom; or by
Cl 6alkyl, Cl 6alkyloxycarbonyl or arylCI 6alkyl when connected to a nitrogen atom.
Aryl as used in the definition of Rl, R5, R6 and R7, in particular is phenyl
optionally substituted with halo, C1~6alkyl or Cl 6alkyloxy; aryl as used in thedefinition of R4 and R8 iD particular is phenyl optionally substituted with halo.
.
. . . :
3L389
-8-
A particular subgroup among the compounds of formula (I) comprises those
compounds of folmula (I) wherein -Al=A2-A3--A4- is a bivalent radical of formula (a-1)
or (a-2); another particular subgroup among the compounds of formula (I) comprises
those compounds of formula (I) wherein -Al=A2-A3=A4- is a bivalent radical having a
5 formula (a-3) through (a-7~.
Particularly interesting compounds are those compounds of the former subgroups
wherein Rl is hydrogen or arylC1 6alkyl; or R3 is hydrogen; or B is NH or CH2; or n
is 1 or 2; or L is hydrogen, C1 6alkyl, C1 6alkylcarbonyl, C1 ,~alkyloxycarbonyl, or a
10 radical of formula (b-1), (b-2), (b-3) or (b-4); or the moiety
5~ ~<OR, is (2-CHR20RI-furan-5-yl)CI4alkyl or (3-CHR~ORl-furan-S-yl)-
--D ~ R2 Cl4alkyl; or several of these radicals have the meanings mentioned.
More particularly interesting compounds within the invention are those particularly
interesting compounds of formula (I) wherein R1 is hydrogen; or R2 is hydrogen; or n is
1 or 2; or L is methyl or a radical of formula (b-1), (b-2) or (b-3); or R5 is aryl or Het; or
R6 is Cl 6alkyl or Het; or R7 is aryl, Het or Cl 6alkyl; or Y is O or NH; or zl and z2
each independently are NR10 or a direct bond, R10 being hydrogen or Cl 6alkyl; or X
is O; or each Alk is C2 4alkanediyl; or Het is a more particular Het described
hereinabove; or several of the radicals have the meanings mentioned.
The most interesting compounds are those more particularly in~eresting compoundswherein
R5 is phenyl optionally subs~ituted with Cl 6aLIcyloxy; pyridinyl; 4,5-dihydro-5-oxo-
lH-tetrazolyl; 2-oxo-3-oxazolidinyl; 2,3-dihydro-2-oxo-lH-benzimidazl)lyl; or a
bicyclic radieal of formula
~ sN~C~I3
G2 N~ c4-a),
wherein G2 is -CH=CH-CH=CH-, -S-(CH2)3-~ -S-(CH2)2-~ -S-CH=CH- or
-CH=C(CH3)-O-; or
R6 is Cl 6 alkyl; pyTidinyl optionally substituted with nitro, pyrimidinyl; pyrazinyl;
pyridæinyl optionally substituted with halo; or 2,3-dihydro-3-ox~pyridazinyl; or
9 2~
the radical (b-3) is (arylcarbonyl)Cl 6alkyl, Cl 6alkylaminocarbonylC1 6alkyl or a
radical Hetl-C(=O)-NH-Cl 6alkyl wherein Hetl is l-methyl-lH-pyrrolyl,
furanyl, thienyl or aminopyrazinyl.
5 In order to simplify the structural representation of some of the eompounds and inter-
mediates in the following preparations the moiety containing the imidazole group fused to
a benzene, pyridine or pyrirnidine ring will hereinafter be represented by the symbol Q.
~0~ {)R
D '~ <R2
~ ~ A3 = ~Q
The compounds of formula (I) can generally be prepared by reacting an interrnediate
of formula (II) with an appropriately substituted diamine of formula (III).
~O~ORI
D ~1 R2
R3
rl~_B~ + N~ 1 3 (I)
~(CH2)r, W H2 A4
(lI) (III)
In this and the following reaction schemes W represents an appropriate reactive leaving
group such as, for example, halo, e.g. chloro, bromo or iodo; C1 6alkyloxy;
C1 6alkylthio, aryloxy or arylthio; and Xl denotes O, S or NH.
20 The derivatives of formula (II) wherein B is CH2 and W is halo may be generated in
situ, for example, by halogenating the corresponding carboxylic acid with thionyl
chloride, phosphorous trichloride, phosphoryl chloride, polyphosphoric acid and the like
reagents. The reaction of (II~ with (III) may be conducted in a suitable reaction-inert
solvent such as, for example, a hydrocarbon, e.g., benzene, hexane and the like; an
25 ether, e.g., 1,1'-oxybisethane, tetrahydrofuran and the like; a ketone, e.g., 2-propanone,
2-butanone and the like; an alcohol, e.g., methanol, ethanol, 2-propanol, l-butanol and
the like; a halogenated hydrocarbon, e.g., trichloromethane, dichloromethane and the
like; an organic acid, e.g., acetic acid, propanoic acid and the like; a dipolar aprotic
solvent e.g., N,N-dimethylforrnamide, N,N-dimethylacetamide and the like; or a mixture ;~
, ,
-10- ~ 3~Z
of such solvents. Depending upon the nature of the solvent and W it may ~e appropriate
to add to the reaction mixture a base such as is commonly employed in the art ofconducting N-alkylation reactions and/or a iodide salt such as an alkali metal iodide.
Elevated temperatures and stirring may enhance the reaction rate. ln some inst~nces the
S reaction of (II) with (III) may first yield an intermediate of formula (II-a~ which
subsequently may be cyclized to the desired compound of formula (I), either in situ or, if
desired, after isolation and purification.
O o
~ ~/
R3 D~ ~ 2
HN~ ~A ~ (I)
~(CH2),~ A4
(II-a)
10 The compounds of formula (I) can also be prepared by reacting an intermediate of
formula (IV) with an intermediate of formula (V) following art-known substitution
reaction procedures. In (IV) and hereinafter, M is hydrogen when B is other than CH2,
or M represents an alkali or earth alkaline metal such as, for example, lithium or
magnesium, when B represents CH2.
15 R3
rl~ W-Q
L--N )--B--M - V (I)
~(~H2)n (V)
Similarly, the compounds of forrnula (I) can also be prepared by reacting an
interrnediate of formula (VI) with an intermediate of forrnula (VII) wherein M has the
20 previously defined meaning. In formula (VI) and hereinafter wl represents an
appropriate leaving group such as, for example, halo, e.g., chloro, bromo and the like;
or a sulfonyloxy group such as, for example, methanesulfonyloxy, 4-methylbenzene-
sulfonyloxy and the like.
R3
rl~ M-B-Q
L--N ,~W~
~(CH2)l~ (VII)
25(VI)
The compounds of formula (I) wherein B is -CH2-, said compounds being
represented by forrnula (I-a), can also be prepared by reacting an interrnediate of formwla
(VIII) with an intermediate of formula (IX) or alternatively, by reacting an intermediate
of formula (X) with an intermediate of formula (XI).
R3
rl~ M-Q
L--N ~CH2W~
~(CH2)l, (IX)
(VIII) L N ~CH2Q
R3 ~(CH2)n
,1~ Wl-CH2-Q
L--N j--M ~I-a)
(CH2)n (XI)
(X)
The reactions of (IV), (VI), (VIII) and (X) with respectively (V), (VII), (IX) and (XI)
may conveniently be conducted in an appropriate reaction-inert solvent such as for
10 example, an aromatic hydrocarbon, e.g., benzene, methylbenzene and the like; an ether,
e.g. 1,4-dioxane, 1,1'-oxybisethane, tetrahydro~uran and the like; a halogenatedhydrocarbon, e.g. trichloromethane and the like; N,N-dimethylforrnamide;
N,~-dimethylacetamide; nitrobenzene; dimethylsulfoxide; l-methyl-2-pyrrolidinone and
the like; and when M is hydrogen, said solvent may also be a Cl 6alkanol, e.g.,
15 methanol, ethanol, 1-butanol and the like; a ketone, e.g., 2-propanone, 4-methyl-2-
pentanone and the like. In some instances, particularly when B is a heteroatom, the
addition of an appropriate base such as, for example, an alkali metal carbonate or
hydrogen carbonate, e.g., sodium carbonate, sodium hydrogen carbonate and the like;
sodium hydride; or an organic base such as, for example, N,N-diethylethanamine or
20 N-(1-methylethyl)-2-propanamine and/or the addition of an iodide salt, preferably an
alkali metal iodide, may be appropriate. Somewhat elevated temperatures and stirring
may enhance the rate of the reaction.
The compounds of fonnula (I) wherein B is -NR4-, said compounds being
25 represented by formula (I-b), can also be prepared by reacting an inte~nediate of formula
(XII) with an intermediate of formula (VII) wherein B-M represents a radical -NR4-H,
said interrnediate being represented by formula (VII-a), following art-known reductive
N-alkylation procedures.
., , ~
.
-12- 2
R3 R3
rl~ R4-NH-Q rl~
L--N ~`CO --- L--N ~N--Q
~(CH2)r~ (VII-a) ~(CH2)n
(XII) (I-b)
The reaction of (XII) with (VII-a) can conveniendy be carried out by mixing the reactants
in a suitable reaction-inert solvent with an appropriate reductant. Preferably, the ketone
5 of formula tXII) is first reacted with the intermediate of foImula (VII-a) to form an
enamine, which optionally may be isolated and further purified, and subsequentlyreducing said enamine. Suitable solvents are, for example, water; C1 6 alkanols, e.g.,
methanol, ethanol, 2-propanol and the like; ethers, e.g., 1,4-dioxane and the like;
halogenated hydrocarbons, e.g., trichloromethane and the like; dipolar aprotic solvents,
lQ e.g., N,N-dimethylformamide, N,N-dimethylacetamide, dimethylsulfoxide and the like;
or a mixture of such solvents. Appropriate reductants are for example, metal or complex
metal hydrides, e.g., sodium borohydride, sodium cyanoborohydride, lithium aluminum
hydride and the like. Alternatively, hydrogen in the presence of a suitable catalyst such
as, for example, palladium-on-charcoal, platinum-on-charcoal and the like may be used
15 as reductant. In order to prevent the undesired further hydrogenation of certain functional
groups in the reactants and the reaction products it may be advantageous to add an
appropriate catalyst poison to the reaction mixture such as, for exarnple, thiophene and
the like.
20 The compounds of formula (I-b) wherein B is -NH-, said compounds being
represented by forrnula (I-b-1), can also be prepared by a cyclodesulfurization reaction of
an appropriate thiourea of formula (II-a) wherein X is S, said thiourea being represented
by formula (II-a-1), which may be forrned in situ by condensing an isothiocyanate of
formula (XIII) with a diamine of formula (III).
Said cyclodesulfurization reaction may be calTied out by reacting ~ a-1) with anappropriate alkyl halide, pref~rably iodomethane, in a suitable reaction-inert organic
solvent such as a C1 6alkanol, e.g., methanol, ethanol~ 2-propanol and the like.Alternatively, said cyclodesulfurization reaction may also be carried out by the reaction of
30 (II-a-l) with an appropnate metal oxide or salt such as, forexample, a Hg(lI) or Pb(ll)
oxide or salt, e.g., HgO, HgC12, Hg(OAc)2, PbO or Pb(OAc)2 in an appropriate solvent
following art-known procedures. In some instances it may be appropriale to supplement
the reaction mixture with a small amount of sulfur. Also methanediimides, especially
dicyclohexylcarbodiinnide may be used as cyclodesulfurizing agents.
-13- 2;~.3~
O ORI
R3 D'~l \ 2
rl~ HN~,A~A2
L--N >--N=C=S + ~ 3
2)n H2N~ A4
(XIII) (III)
O OR~
~ ~/
R3 D'~/ \ 2 ~ L--Nr ~NH Q
(CH2)n HN A4~ ~ (CH2)n
(II-a-l) (I-b-1)
The compounds of forrnula (I) can also be prepared by N-alkylating an intermecliate
S of formula (XV) with an appropriate alkylating reagent of formula (XIV).
O ORI
R3 ,H Wl-D~ /~( R3 D~
L--N ~--B~ A _ _ N3~ A
~(CH2)" A4~A (XIV) ~(CH2)n A4~
(XV) (I) :
Said N-alkylation reaction can conveniently 'oe conducted in a reaction-inert solvent such
10 as, for example, water; an aromatic hydrocalbon, e.g., benzene, methylbenzene,
dimethylbenzene and the like; an alkanol, e.g., methanol, ethanol, 1-butanol and the like;
a ketone, e.g., 2-propanone, 4-methyl-2-pentanone and the like; an ether, e.g.,
tetrahydrofuran, 1,4-dioxane, l,1'-oxybisethane and the like; a dipolar aprotic solvent,
e.g., N,N-dimethylformamide, N,N-dime~hylacetamide, dimet'nylsulfoxide,
15 nitrobenzenç, l-methyl-~-pyrrolidinone and the like; or a mixture of such solvents. The
addition of an appropriate base such as, for example, an allcali or an earth alkaline metal
car'oonate, hydrogen car'oonate, alkoxide, hydride, amide, hydroxide or oxide, e.g.,
sodium car'oonate, sodium hydrogen carbonate, potassium carbonate, sodium
methoxide, sodium ethoxide, potassium tert.-butoxide, sodium hydride, sodium amide,
20 sodium hydroxide, calcium car'oonate, calcium hydro~ide, calcium oxide and ~he like; or
- ~
:
.
-14-
an organic base, such as, for example, a tertiary amine, e.g., N,N-diethylethanamine,
~-(1-methylethyl)-2- propanamine, 4-ethylmorpholine, pyridine and the like may be
utilized to pick up the acid which is liberated during the course of the reaction. In some
instances the addition of an iodide salt, preferably an alkali metal iodide, is appropriate.
5 Somewhat elevated temperatures and stilTing may enhance the rate of the reaction.
Additionally, it may be advantageous to conduct said N-alkyla~on under an inert
atmosphere such as, for example, oxygen-free argon or nitrogen.
Alternatively, said N-alkylation may be carried out by applying art-known conditions
10 of phase transfer catalysis reactions. Said conditions comprise stirring the reactants with
an appropriate base and optionally under an inert atmosphere as described hereinabove,
in the presence of a suitable phase transfer catalyst such sas, for example, a
trialkylphenylmethylammonium, tetraalkylammonium, tetraalkylphosphonium,
tetraarylphosphonium halide, hydroxide, hydrogen sulfate and the like catalysts.
The compounds of formula (I) wherein Rl is hydrogen, said compounds being
represented by formula (I-c), can also be prepared by condensing a furan derivative of
formula (XVI) with an aldehyde R2-CHO (XVII) in the p~esence of a suitable acid or
base catalyst.
R3 D ~ R3 ~~<
,1~ N ~A:~A2 R2-CHO ,1~ N ~A~A2
1,--N >--B~\ ¦1 3 L--N ~B <~ ¦¦ I 3
~(~H2)n N--A4~A (XVII)~(CH2)n N~A4 A
(XVI) (I-c)
Said compounds of formula (I-c) can also be obtained by reducing a carboxylic acid
derivative of formula (XVTII) wherein R is hydrogen, alkyl or aryl, with a reducing s
25 agent such as, for example, lithium aluminum hydride, lithium borohydride, sodium
borohydride and the like in a reaction-inert solvent such as, for example, an ether, e.g.
tetrahydrofuran, 1, I '-oxybisethane, 1,4-dioxane and the like; or alternatively, by
reacting said carboxylic acid (XVIII) or a salt form thereof with an organometallic
reagent, in particular Cl 6alkyl lithium, and reducing the thus obtained intermediate
30 ketone with a reducing agent such as, for example, lithium aluminum hydride, lithium
borohydride, sodium borohydride and the like in a reaction-inert solvent such as, for
example, an ether, e.g. tetrahydrofuran, l,1'-oxybisethane, 1,4-dioxane and the like.
.
-15- 2(~
~ ~ COOR
R3
,1~ N ,~A~A2Reduction
~(CH2)n N~A4~A (I-c~
(XVIII)
The compounds of formula (I) wherein L is other than hydrogen, said 1, being
5 represented by L1, and said compounds being represented by formula (I-d) can also be
prepared by ~-al~ylatin~ a compound of formula tI) wherein L is hyd~ogen, said
compound being represented by (I-e), with an alkylating reagent of formula (XIX).
R3 R3
~1~ Ll-wl rl~
H--N ~B--Q _ Ll--N >--B--Q
(CH2)n ~(CH2)n
(I-e) ~XIX) (I-d)
Said N~alkylation is conveniently conducted following art-known N-alkylation
procedures as described hereinabove for the preparation of (I) from (XIV) and (XV).
The compounds of formu}a (I-d) wherein L is C3 6cycloalkyl, C1 12alkyl, a radical
15 of fonnula (b-1), ('~2) or (b-3), said radicals being represented by the radical L2H- and
said compounds by formula (I-d-1) can also be prepared by the reductive N-alkylation
reaction of (I-e) with an appropriate ketone or aldehyde of formula L2=o (XX), said
L2=o 'oeing an inter;nediate of forrnula L2H2 wherein two geminal hydrogen atoms are
replaced by =O, and L2 is a geminal bivalent radical comprising C3 6cycloalkylidene,
20 C1 l2alkylidene, R5-C1 6alkylidene, R6-Y-C1 6alkylidene and R7-Z2-C(-X)-Z1-
Cl 6alkylidene.
Reductive R3
(I-e) + L~=O~ -- ~ L2H~N ~Q
(XX) N-alkylation~CH~)n
(I-d- 1)
: . .
25 Said reductive N-alkylation can conveniently be carried out following the procedures
: ~
-16- ~
described herein~bove for the preparation of compounds of formula (I-b) from (VII-a)
and (XII), more particularly following the catalytic hydrogenation procedures.
The compounds of formula (I) wherein L is a radical of formula (b-2) and R6 is aryl
5 or Het, said R6 being represented by R6-a and said compounds by formula (1-d-2) may
also be prepared by alkylating a compound of formula (I) wherein L is a radical of
fonnula (b-2) and R6 is hydrogen, said compound being represented by formula (I-d-3),
with a reagent of formula (XXI).
R3 3
rl~ R~a Wl R
H-Y-Alk--N ~B~ R~a-Y-AL~--N ~B~
\~(CH2)n ~(CH2)n
~d-3) a~l-2)
Similarly, the compounds of formula (I-d-2) may also be prepared by treating a
compound of formula (I-d-4) with a reagent of forrnula (XXII).
YVl-Alk--N ~B4 ., R~a-Y-AL~c--N ~B4
~(CH23n ~(CH2)n
a~4) a~-v
The alkylation reactions of (I-d-3) with (XXI) and (XXII) with (I-d-4) may conveniently
be conducted in an inert organic solvent such as, for example, an aromatic hydrocarbon,
e.g., benzene, methylbenzene, dimethylbenzene; a ketone, e.g., 2-propanone, 4-methyl-
20 2-pentanone; an ether, e.g., 1,4-dioxane, 1,1'-oxybisethane, tetrahydrofuran; and a
dipolar aprotic solvent, e.g., N,N-dimethylfonnarnide; N,N-dimethylacetamide;
dimethyl sulfoxide; nitrobenzene; 1-methyl-2-pyrrolidinone; and the like. The addition of
an appropriate base such as, for exarnple, an alkali metal carbonate or hydrogencarbonate, sodium hydride or an organic base such as, for example,
25 N,N-diethylethanamine or N-(1-methylethyl)-2-propanamine may be utilized to pick up
the acid which is liberated during the course of the reaction. Somewhat elevatedtemperatures may enhance the rate of the reaction.
The compounds of formula (I) wherein L is a radical of formula (b-3), zl is NH, z2
30 is other than a direct bond and X is other than NRl 1, said z2 and X being represented
by z2-a and X2, and said compounds by (I-d-5), can be prepared by reacting an
:: , ' ' .. , ,. ' : -
.' . .' . .
- . -
-- ~ .
-17- ~ a~3
isocyanate (X~ = O) or isothiocyanate (X2 = S) of forrnula (I-d-6) with a reagent of
formula (XXIII).
R3 R3
~1~ R7 z2-a H rl
X2~N-Aik--N~ ~B Q R7-z2~a-c(=x2)-N~I-ALlc~N ~B-Q
(CH2)n ~m) ~ (CH2)n
a~ s~
s
The compounds of formula (I) wherein L is a radical of forrnula (b-3), z2 is NH, zl
is other than a direct bond and X is other than NRl 1, said zl and X being represented
by zl-a and X2, and said compounds by (I-d-7), can be prepared by reacting an
isocyanate (X2 = O) or isothiocyanate (X~ = S) of formula (XXIV) with a compound of
10 formula (I-d-8).
R3 ~j R3
,1~ R7 N=C=x2 ,1
H-ZI ~-Alk--N ~B Q _ ~ R7 NH C(=X2)-ZI a-Alk--N ~B4
(CH2)n ~ (CH2)n
~ 8) (1~-7)
The reaction of (XXIII) with (I-d-6), or (XXIY) with (I-d-8) can generally be conducted
15 in a suitable reaction-inert solvent such as, for example, an ether, e.g., tetrahydrofuran
and the like, a halogenated hydrocarbon, e.g., trichloromçthane and the like. Elevated
temperatures may be suitable to enhance the rate of the reaction.
The compounds of fiorrnula (I) wherein L is a radical of forrnula (b-3), z2 is a direct
20 bond, zl is other than a direc~ bond and X is other than NR1 1, said zl and X being
represented by zl-a and X2, said compounds being rçpresented by (I-d-9), can be
prepared by reacting a reagent of fonnula (XXV) or a reactive functional derivative
thereof wi~h a compound of fonnula (I-d-8).
R3 R3
~1~ R7 C~=X2)-oH ~1
HZI a Alk_N >--B-O~ . R7 C(=X2~Zl a-Alk N ~B-Q
(CH2)n ~ (CH2)n
25(~-8)
The reaction of (XXV) with lI-d-8) may generally be conducted following art-known
- . . ., , . .- .
. , .- . . ~ .
-18- ~ Z
esterification or amidation reaction procedures. For example, the carboxylic acid may be
converted into a reactive derivative, e.g., an anhydride or a carboxylic acid halide, which
subsequently is reacted with (I-d-8); or by reacting (XXV) and (I-d-8) with a suitable
reagent capable of fonning amides or esters, e.g., N,N-methanetetraylbis[cyclohexamine],
5 2-chloro-1-methylpyridinium iodide and the like. Said reactions may most conveniently be
conducted in a suitable solvent such as, for example, an ether, e.g., tetrahydrofuran, a
halogenated hydrocarbon, e.g., dichloromethane, trichloromethane, a dipolar aprotic
solvent and the like. The addition of a base such as, for example, N,N-diethylethanamine
and the like may be appropriate.
The compounds of formula (I) wherein L is a radical of formula L3-C2 6alkanediyl,
said L3 being aryl, Het, arylsulfonyl or a radical of formula R7-Z2-C(=X)-, and said
compounds ~eing represented by formula (I-d-10), may also be prepared by the addition
reaction of a compound of formula (I-e) to an appropriate alkene of forrnula (XXVI).
rl 3 L3-C2 6alkenediyl-Hrl 3
H--N ~Q ~ ~- L3-C2 6alkanediyl--N ~Q
(CH2)n ~ (CH2)n
(XXVI)
(I-e) (I-d-10)
The compounds of formula (I) wherein L is 2-hydroxy-C2 6alkyl or a radical of
formula (b-4), said compounds being represented by formula (I-d-11), can be prepared
20 by reacting a compound of formula (I-e) with an epoxide (XXVII) wherein R14 is
hydrogen, C1 4alkyl or a radical R8-O-CH2-.
R3 R ~ R3
,1~ ,1~
H--N ~B--Q ~Rl4-CHoH-CH2--N )--~Q
(CH~n ~(CH2)n
(I-e) (XXVII) (I-d-l 1)
25 The reaction of (I-e) with respectively (XXYI) and (XXVII) may be conducted by
stirring and, if desired, heating the reactants in a reaction-inert solvent such as, for
example, a ketone, e.g., 2-propanone, 4-methyl-2-pentanone, an ether, e.g., tetrahydro-
furan, 1,1'-oxybisethane, an alcohol, e.g., methanol, ethanol, 1-butanol, a dipolar
aprotic solvent, e.g., N,N-dimethylformamide, N,N-dimethylacetamide, and the like.
The compounds of forrnula (I) wherein R5, R6 or R7 are Het, may also be
~L3~39~
prepared following art-known procedures for preparing heterocyclic ring systems or
following analogous methods. A number of such cyclization procedures are described in
for example, US-4,695,575 and in the references cited therein, in particular
US-4,335,127; 4,342,870 and 4,443,451, all incorporated herein by reference.
The compounds of formula (I) can also be converted into each other following art-
known procedures of ~unctional group transformation. Some examples of such
procedures are cited hereinafter. The compounds of formula (I) containing a cyano
subsdtuent can be converted into the corresponding amines by stirring and, if desired,
10 heating the star~ng cyano compounds in a hydrogen containing medium in the presence
of an appropriate catalyst such as, for example, platinum-on-charcoal, Raney-nickel and
the like catalysts. Suitable solvents are, for example, methanol, ethanol and the like.
Amino groups may be converted into the corresponding isothiocyanato groups upon
treatrnent with CS2, optionally in the presence of N,N-methanetetraylbis-
15 lcyclohexamine]. Amino groups may be alkylated or acylated following art-known
procedures such as, for example, N-alkylation, N-acylation, reductive N-alkylation and
the like methods. The compounds of formula (I) containing an amino group substituted
with a radical arylCH2, may be hydrogenolyzed by treating the starting compound with
hydrogen in the presence of a suitable catalyst, e.g., palladium-on-charcoal, pladnum-
20 on-charcoal and the like, preferably in an alcoholic medium.
In all of the foregoing and in the following preparadons, the reaction products may
be isolated from the reacdon mixture and, if necessary, further purified according to
methodologies generally known in the art.
Some interrnediates and starting materials in the foregoing preparations are known
compounds which may be prepared according to art-known methodologies of preparing
said or similar compounds and others are new. A number of such preparation methods
will be described hereinafter in more detail.
3û
Starting materials such as the intermediates of forrnulae (II), (IV), (VI), (VIII), (X),
(XII), (XIII), (XV) and (XVI) can conveniently be prepared following procedures
similar to those described in for example, US-4,219,559; 4,556,660; 4,634,704;
4,695,569; 4,695,575, 4,588,722, 4,g35,161 and 4,897,401 and in EP-A-0,206,415;
35 0,282,133; 0,297,661 and 0,307,014.
The interrnediates of formula (III) can be prepared from an ar~matic starting material
.
-20- Z~
with vicinal halo and nitro substituents (XXVIII) by reaction with a suitable amine of
formula (XXIX), followed by art-known nitr~to-amine reduction.
D~\~/~ 1~\~/~
NH2 R2 , ¦ ~2
a~l~A~A2 (XXIX) ~ X 4~A3
~xvm
The interrnediates of formulae (V), (VII), (IX) and (XI) then, can be prepared from
the intermediates of formula (III) following art-known prc~cedures of converting aromatic
products with vicinal amino groups into benzimidazoles, imidazopyridines and/or
pur~nes.
The intermediates of formula (XVIII) can be conveniently prepared by N-alkylating
an intermediate of formula (XV) with an appropriately substituted furan-carboxylic acid
derivative of forrnula (XXX) wherein R is hydrogen, alkyl or aryl, following theprocedures described hereinabove for the preparation of the compounds of forrnula (I)
15 from the intermediates (XV) and (XIV).
~~
R3 H Wl-D~ COOR R3
rl~ N~A~A2 ~X A
~(CH2~n A4~(XXX) ~(CH2)n A4
(XV) (XVIII)
The compounds of formula (I), the pharmaceutically acceptable acid addition salts
20 and stereochemically isomeric forrns thereof possess useful pharmacological propcrties.
More particularly, they are active antihistaminics which can clearly be demonstrated by,
e.g., the results obtained in the test "Protection of Rats from Compound 48/80-induced
lethality", the test "Histamine antagonism in Guinea Pig" and the test "Ascaris Allergy
test in Dogs" described in Arch. Int. Pharmacodyn. Ther. 2~1, 39-51 (1981). Apart
25 ~rom their antihistaminic prvperties some of the the subject compounds generally also
show serotonin-antagonism, as can be demonstrated in the test "Gastric Lesions induced
by compound 48/80 in rats".
,
.
'' . ~ ' '
39~
-21 -
In view of their antihistaminic and serotoninergic properties, the compounds of
formula (I) and their acid addition salts are very useful in the treatment of allergic
diseases such as, for example, allergic rhinitis, allergic conjunctivities, chronic urticaria,
5 allergic astma and the like.
In view of their useful antiallergic properties the subject compounds may be
formulated into various pharmaceutical forms for administration purposes. To prepare
the antiallergic compositions of this invention, an effective amount of the particular
10 compound, in base or acid addition salt form, as the active ingredient is combined in
intimate adrnixture with a pharmaceutically acceptable carrier, which carrier may take a
wide variety of forms depending on the form of preparation desired for administration.
These pharmaceutical compositions are desirably in unitary dosage forrn suitable,
preferably, for administration orally, rectally, percutaneously, or by parenteral injection.
15 For example, in preparing the compositions in oral dosage forrn, any of the usual
pharmaceutical media may be employed such as, for example, water, glycols, oils,alcohols and the like in the case of oral liquid preparations such as suspensions, symps,
elixirs and solutions: or solid carriers such as starches, sugars, kaolin, lubricants,
binders, disintegrating agents and the like in the case of powders, pills, capsules and
20 tablets. Because of their ease in administration, tablets and capsules represent the most
advantageous oral dosage unit form, in which case solid pharmaceutica3 carriers are
obviously employed. For parenteral compositions, the carrier will usua]ly comprise
sterile water, at least in large part, though other ingredients, for example to aid solubility,
may be included. Injectable solutions, for example, may be prepared in which the carrler
25 comprises saline solution, glucose solution or a mixture of saline and glucose solution.
Injectable suspensions may also be prepared in which case appropriate liquid carriers,
suspending agents and the like may be employed. In the compositions suitable forpercutaneous administration, the canier optionally comprises a penetrat;on enhancing
agent and/or a suitable wetting agent, optionally combined with suitable additives of any
30 nature in minor proportions, which additives do not introduce a significant deleterious
ef~ect on the skin. Said additives may facilitate the administration to the skin and/or may
be helpful for preparing the desired compositions. These compositions may be
administered in various ways, e.g., as a transderrnal patch, as a spot-on or as an
oint~ent. Acid addition salts of (I) due to their increased water solubility over the
35 corresponding base f~rm, are obviously more suitable in the preparation of aqueous
compositions.
:.
-
-22- 2~ 8~2
It is especially advantageous to formulate the aforementioned pharrnaceutical
compositions in dosage unit forrn for ease of administration and uniforrnity of dosage.
Dosage unit form as used in the specification and claims herein refers to physically
discre~e units suitable as unitary dosages, each unit containing a predetermined quantity
5 of active ingredient calculated to produce ~he desired therapeutic effect in association with
the required pharrnaceutical carrier. Examples of such dosage unit forrns are tablets
(including scored or coated tablets), capsules, pills, powder packets, wafers, injectable
solutions or suspensions, teaspoonfuls, tablespoonfuls and the like, and segregated
multiples thereof.
The present invention also relates to a method of treating warm-blooded animals
suffering from said allergic diseases by administering to said warm-blooded animals an
antiallergically effective amount of a compound of forrnula (I) or a pharrnaceutically
acceptable acid addition salt form thereof.
15 Those of skil! in treating allergic diseases in warrn-blooded animals could easily
deterrnine the effective amount from the test results presented hereinafter. In general it is
contemplated that an antiallergically effective amount would be from about 0.001 mg~cg
to about 100 mg~cg body weight, and more preferably from about 0.01 mg/kg to about I
mg/kg body weight.
The following examples are intended to illustrate and not to limit the scope of the
present invention in all its aspects. Unless otherwise stated all parts therein are by
weight.
25 EXPERIMENTAL PART
A. ~paration of the interrnediate~
Exam~l
A mixture of 28.8 parts of ethyl 4-(lH-benzirnidazol-2-ylamino)-1-piperidinecarboxylate
(as prepared in Example XIV of U.S. patent 4,219,559), 33.9 parts of ethyl
30 5-chloromethyl-2-furancarboxylate, lS.9 parts of sodium carbonate and 282 parts of
[-dimethylforrnamide was stirred for 2 nights at 70C. The reaction mixture was
poured into water and the product was extracted with methylbenzene. The extract was
washed with water, dried, filtered and evaporated. The residue was purified by column
chromatography (silica gel; CHC13 / CH30H 97:3). The eluent of the desired fraction
35 was evaporated and the residue was stirred in I,1'-oxybisethane. The precipitate was
filtered off and dried, yielding 31.2 parts ~70.8%) of ethyl 4-[[1-~[5-(ethoxycar~onyl)-2-
-23- 20~ 38~2
furanyl]-methyl]- lH-benzimidazol-2-yl]amino]- 1 -piperidinecarboxylate; mp. 1 36.0C
(interm. 1).
In a similar manner ethyl 4-( l H-benzimidazol-2-ylamino)hexahydro- lH-azepine- 1-
carboxylate (as prepared in Example 9 of EP-0,297,661, published January 4, 1989)
5 was converted into ethyl 4-~1-[[5-(ethoxycarbonyl)-2-furanyl]methyl]-lH-
benzimidazol-2-yl]arnino]hexahydro-l~ azepine-1-carboxylate (interm. 2) and
ethyl 3-( lH-benzimidazol-2-ylamino)- 1 -pyrrolidinecarboxylate monohydrochloride (as
prepared in Exarnple 8 of EP-0,297,S61, published Januar~Y 4, 1989) into ethyl 3-[[1-
[[5-(ethoxycarbonyl)-2-furanyl]methyl]- lH-benzimidazol-2-yl]arnino]-1 -pyrrolidine-
10 carboxylate (interm. 3).
Exam~le 2
To 470 parts of ~,~-dimethylformamide were added portionwise 17.3 parts of a
dispersion of sodium hydride in mineral oil (50%) and 91.6 parts of 2-~[1-(phenyl-
15 methyl)-4-piperidinyl]methyl]-lH-benzimidazole (as prepared in Example 16 of U.S.
patent 4,695,575) while stirring under a nitrogen atmosphere. After stirring .for 1 hour,
67.9 parts of ethyl 5-chloromethyl-2-furancarboxylate were added dropwise while
cooling. Stirring was continued for 1 hour and then water was added tO the reaction
mixture. The product was extracted with methylbenzene and the extract was washed with
20 water, dried, filtered and evaporated. Tlle residue was dried azeotropically with
methylbenzene (2x), yielding 119 parts (86.7%) of ethyl 5-[[2-[[1-(phenylmethyl)-4-
piperidinyl]methyl]-lH-benzimidazol-l-yl]methyl]-2-furancarboxylate (intelm. 4).Following the same procedure and starting from the appropriate starting materials, there
were als~ prepared:
25 methyl 5-[[2-[[1-[2-(4-me~hoxyphenyl)ethyl]-4-piperidinyl]amino]-lH-benzimidazol-l-
yl]methyl]-2-furancarboxylate; mp. 1?4.4C (interm. 5),
ethyl 5-[[2-[[1-[2-(4-methoxyphenyl)ethyl]-4-piperidinyl]amino]- lH-benzimidazol- 1-
yl]methyl]-3-furancarboxylate; mp. 121.9C (interm. 6),
methyl 5-[[2-r( 1 -methyl-4-piperidinyl)amino]- 1 H-benzimidazol- 1 -yl]rnethyl]-2-
30 furancarboxylate; mp. 169.5C (interm. 7),ethyl 5-[[2-~(1-methyl-4-piperidinyl)amino]-lH-benzimidazol-1-yl]methyl]-3-
furancarboxylate (E)-2-butenedioate~1:2); mp. 200.9C (interm. 8),
ethyl 2-[[2-[[1-[2-(4-methoxyphenyl)ethyl]-4-piperidinyl]amino]-lH-benzimidazol- 1- -
yl]me~hyl]-3-furancarboxylate (intenn. 9),
35 ethyl 4-[~1-[[3-(ethoxycarbonyl)-2-furanyl]methyl]-lH-benzimidazol-2-yl~amino]-1-
piperidinecarboxylate; mp. 162.3(: (intelm. 10),
38~:
-24-
ethyl ~[[1-[[2-(methoxycarbonyl)-3-furanyl]methyl]-lH-benzimidazol-2-yl]amino]-1-
piperidinecarboxylate (interm. 11), and
methyl 3-[[2-[[1-[2-(~methoxyphenyl)ethyl]-4-piperidinyl]amino]-lH-benzimidazol-1-
yl]methyl]-2-furancarboxylate ~interm. 12).
E~ple 3
a) A mixture of 55 parts of N-(2-furanylmethyl)-3-nitr~2-pyridinamine, 2 parts of a
solution of thiophene in methanol (4%) and 400 parts of methanol saturated with
ammonia was hydrogenated at normal pressure and at room temperature with 4 parts of a
platinum-on-charcoal catalyst 5%. After the calculated amount of hydrogen was taken
up, the catalyst was filtered off and the filtrate was evaporated, yielding 48 parts of
;~2-(2-furanylmethyl)-2,3-pyridinediamine as a residue (intenn. 13).
b) A mixture of 54 parts of ethyl 4-isothiocyanato- 1-piperidinecarboxylate, 48 parts of
intermediate (13) and 450 parts of tetrahydrofuran was stirred overnight at reflux
temperature. The reaction mixture was evaporated and the residue was crystallized from a
mixture of 2-propanone and 2,2'-oxybispropane, yielding 76 parts (75%) of ethyl
4-[[[2-[(2-furanylmethyl)amino]-3-pyridinyl]aminothioxomethyl]aminol- 1 -piperidine-
carboxylate; mp. 132.7C (interm. 14).
In a similar manner ethyl hexahydro-4-isothiocyanato-1~-azepine- 1-carboxylate ~as
prepared in Example 9 of EP-0,297,661, published January 4, 1989) was converted into
ethyl hexahydro-4-[[[[2-[[[5-(hydroxymethyl)-2-furanyl]methyl~amino]-3-
pyridinyl]amino]thioxomethyl]an~ino]-l~-azepine-1-carboxylate (interrn. 15).
c) A mixture of 74 parts of intermediate (14), 96 parts of mercury{ll)oxide, 0.1 parts of
sulfur and 800 parts of ethanol was stirred and refluxed for 3 hours. The reaction mixture
was filtered over diatomaceous earth and the filtrate was evaporated. The residue was
crystallized from acetonitrile, yielding 52.5 parts (79%) of ethyl 4-[[3-(2-furanyl-
methyl)-3H-imidazo~4,5-b~pyridin-2-yl]amino]- 1 -piyeridinecarboxylate; mp. 149.2C
(interm. 16).
Example 4
a) A mixture of 16.3 parts of 4~6-dichloro-5-pyrimidinamine, 14 parts of
5-(aminomethyl)-2-furanmethanol, 12 parts of ~I.N-diethylethanamine and 200 parts of
water was stirred for 10 hours at reflux temperature. The reaction mixture was
evapora~ed and the residue was purified by column chromatography ~silica gel; (~H2CI~ /
CH30H(NH3) 95:5). The eluent of the desired ~raction was evaporated and the residue
was crystallized from acetonitrile. The pr~duct was filtered off and dried, yielding 19.5
.. ... ~
';' ~' '
'
389
-25-
parts (76.6%) of S-[r(S-amino 6-chloro~pyrimidinyl)amino]methyl]-2-fi~ranmethanol;
mp. 136.7C (interm. 17).
b) A mixture of 18.5 parts of intermediate (17), 1 part of a solution of thiophene in
S methanol 4%, 119 parts of methanol and lO parts of calciumoxide was hydrogenated at
normal pressure and room temperature with 4 parts of palladium-on-charcoal catalyst
10%. After the calculated amount of hydrogen was taken up, the catalyst was filtered off
and the filtrate was evaporated, yielding 15.9 parts (100%) of 5-~[(5-amino-4-
pyrimidinyl)amino}methyl]-2-filranmethanol (interm. 18).
Ex~
a) A mixture of 15.9 parts of 4-chloro-~nitropyndine, 12.7 parts of 5-(arninomethyl)-2-
furanrnethanol, 13.3 parts of ~ -diethylethanamine and 745 parts of trichloromethane
was stirred for 3 hours at reflux temperature. After cooling, the reaction mixture was
15 washed with K2C03 taq.), dried, filtered and evaporated. The residue was crystallized
from acetonitrile. The product was filtered off and dried, yielding 17.76 parts (71.3%) of
S-[t(3-nitro~pYndinyl)amino]methyl]-2-furanmethanol; mp. 134.9C (inteIm. 19).
b) A rnixture of 17.3 parts of intermediate (19), 1 part of a solution of thiophene in
methanol 4% and 158 parts of methanol was hydrogenated at normal pressure and room
20 temperature with l part of platinum-on-charcoal catalyst 5%. After the calculated amount
~ of hydrogen was taken up, the catalyst was filtered off and the filtra~e was evaporated.
- The residue was crystallized f~om acetonitrile. The product was filtered off and dIied,
yielding 12.7 paras (83.9%) of 5-~(3-amino-~pyIidinyl)amino]methyl]-2-furanmethanol
(interm. 20~.
25 c) A mL~ rre of 14.3 parts of ethyl 4-isothiocyanato-1-piperidinec~rboxylate, 12.7 parts
of intermediate (20) and 188 parts of ~,~-dimethylformarnide was s~r~ overnight at
60C. The reaction mixture was evaporated,yielding 25.1 parts (100%) of ethyl 4-[[[[4-
[[[S-(hydr~xymethyl)-2-furanyl~methyl]amino]-3-pyridinyl]-amino]~hioxomethyl~-
amino]-l-pipendinecar~oxylate (interm. 21).
30 Following the same procedure, inteImediate (18) was eonverted into ethyl ~[[r[4-~[[~-
(hydr~xymethyl)-2-furanyUmethyl]amino]-S-pylimidinyu-amino]thioxomethyl}amino]-
l-piperidinecarboxylate ~intenn. 22).
~nple 6
35 a) To a solution of 25 parts of 1-(phenylmethyl)4-piperidineacetonit~ile in 178 parts of
tetrahydrofilran were added dropwise 37.97 parts of ethyl chloroformate at room
. ,. ~
-26- 2~ 9
temperature. The reaction rnixture was stirred for 15 hours at room temperature and was
then evaporated. The residue was taken up in 90 parts of ethyl acetate and the whole was
washed successively with HCl 3N, NaHCO3 and NaCl (sat.). The solvent was
evaporated and the residue was distilled (100-1 lO~C / 6.7 Pa), yielding 18.7 parts
(81.4%) of ethyl 4-~cyanomethyl)-1-piperidinecarboxylate (interm. 24).
b) A mixture of 18.32 parts of intennediate (24), 4.30 parts of ethanol and 74.5 parts of
trichloromethane was cooled in an ice-bath while hydrochloric acid was bubbled through
for 30 minutes. The reaction mixture was left in a refrigerator for 48 hours and was then
evaporated. The residue was triturated with 142 parts of 1,1'-oxybisethane. The product
was dried in vacuo, yielding 14.2 parts (54.1 %) of ethyl 4-(2-ethoxy-2-iminoethyl)- 1-
piperidinecarboxylate monohydrochloride (interm. 25).
Following the same procedure, 1-(phenylmethyl)-4-piperidineacetonitrile was converted
into ~ethyl 1-(phenylmethyl)-4-piperidineethanimidate dihydrochloride (interm. 26).
B. Pr~thefinalcom~ounds
Examp~e 7
a) To a stirred mixture of 4.4 parts of interrnediate (1) and 133.5 parts of tetrahydrofuran
were added dropwise S ml. of a solution of lithium tetrahydroborate in tetrahydrofuran
2M under a nitrogen atmosphere. Stirring was continued overnight at reflux temperature
and then there were added successively 2-propanone and acetic acid. The whole was
evaporated. The residue was taken up in water and basified with K2CO3 The product
was extracted with dichloromethane. The extract was dried, filtered and evaporated. The
residue was crystallized from 4-methyl-2-pentanone, yielding 2.08 parts (52.2%) of
ethyl ~[[1-l[S-(hydroxymethyl)-2-furanyl]methyl]-lH-benzimi-dazol-2-yl}amino]-1-piperidinecarboxylate; mp. 141.6C (comp. 3.05).
b) A mixture of 75.7 parts of compound (3.(35), 106.5 parts of potassium hydroxide and
390 parts of 2-propanol was stirred overnight at reflux temperature. After cooling, the
reaction rnixture was evaporated and the residue was taken up in water. The product was
extracted with dichloromethane and the extract was filtered over diatomaceous earth. The
~lltrate was evaporated and the residue was crystallized from 2-propanone, yielding 46.5
parts (75.0%) of 5-[[2-(4-piperidinylamino)-lH-benzimidazol-1-yl]m~.thyl]-2-
furanmethanol; mp. 1~6.3C (comp. 3.11).
c) A mixture of 4.53 parts of chloroacetonitrile, 16.26 parts of compound (3.11), 8 parts
of sodium carbonate and 141 parts of N~N-dimethylformamide was stirred for 2 hours at
room temperatur~. The reaction mixture was poured into water and the product wasextracted with trichloromethane. The extract was dried, filtered and evaporated. The
residue was stirred in 2,2'-oxybispropane and the product was filtered off, yielding
. . ~ , . .
':
-27- 26~ 392
17.71 parts (96;.9%) of 4-[[1-[[5-(hydroxymethyl)-2-furanyl~-methyl]-lH-benz-
imidazol-2-yl]amino]-1-piperidineacetonitrile; mp. 209.8C (comp. 3.22).
d) A mixture of 16.6 parts of compound (3.22) and 790 parts of methanol saturated with
ammonia was hydrogenated at norrnal pressure and at room temperature with 6 parts of
S Raney nickel. A~ter the calculated amount of hydrogen was taken up, the catalyst was
filtered off and the filtrate was evaporated. The residue was crystallized successively
from acetonitlile and 2-propanol, yielding 7.4 parts (435%) of 5-[[2-[[1-(2-amino-
ethyl)-4-piperidinyl]amino]- lH-benzimidazol- 1 -yl]methyl]-2-furanmethanol;
mp. 164.6C (comp. 3.23).
10 e) To a stirred solution of 1.38 parts of 1-methyl-lH-2-pyrrolecarboxylic acid, 2.81 parts
of 2-chloro-1-methylpyridinium iodide, 2.2 parts of N.N-diethylethanamine and 199.5
parts of dichloromethane was added a solution of 3.7 parts of compound ~3.23) in a
mixture of dichloromethane and N.N-dimethylacetamide. After stirring for 3 hours, the
reaction mixture was poured into water. The product was extracted with dichloro-
15 me~hane and ~he extract was dried, filtered and evaporated. The residue was purified bycolumn chromatography (silica gel; CHC13 / CH30H (NH3) 95:5). The eluent of the
desired fraction was evaporated and the residue was crystallized from acetonitrile,
yielding 2.25 parts (47.2%) of N-[2-[4-[[1-[[5-~hydroxymethyl)-2-furanyl]methyl]-lH-
benzimidazol-2-yl]amino]- 1 -piperidinyl]-ethyl]- l-methyl- 1 -pyrrole-2-carboxarnide;
20 mp. 182.2C (comp. 3.24).
Example 8
a) To a stirred and refiuxing mixture of 12 parts of lithium alum;num hydride in 445 parts
of tetrahydro~uran, was added dropwise a solution of 137 parts of intermediate (4) in
25 tetrahydrofuran under a nitrogen atmosphere. Refluxing was continued for 1 hour. After
cooling, there were added successively ethyl acetate, 42 parts of NaOH 15% (dropwise)
and 36 parts of water. The whole was stirred and f~iltered. The filtrate was evaporated and
the residue was taken up in water. The product was extracted with dichloromethane and
the extract was dried, filtered and evaporated. The residue was crystallized from
30 acetonitrile, yielding 64.1 parts (51.4%) of 5-[[2-[[1-~phenylmethyl)-4-piperidinyl]-
methyl3-1}~-benzimidazol-1-yl]methyl]-2-furanrnethanol; mp. 143.8C (comp. 1.01).
b) To a stirred mixture of 14.1 parts of 2-chloro-1-methylpyridinium iodide, 11.5 parts
of N~N-diethylethanamine and 282 parts of ~-dimethylformamide were added
dropwise 3.3 parts of acetic acid at room temperature. After stirring for I hour, 41.5
35 parts of compound (1.01) were added. Stirring was continued ove}night and then the
reaction rnixture was poured into water. The product was extracted with dichloromethane
and the extract was dried, filtered and evaporated, yielding 40 parts (87.4%) of 5-[[2-[[1-
.
-: .
-28- 2(~389~
(phenylmethyl)-4-piperidinyl]methyl]-lH-benzimidazol-1-yl]methyl]-2-furanmethanol
acetate ~ester) (comp. 1.02).
c) To a stirred and refluxing mixture of 45.8 parts of compound (1.02) and 2~1 parts of
methylbenzene, were added dropwise 12 parts of ethyl chloroformate. Refluxing was
5 continued for 1 hour. After cooling, water was added to the reaction mixture and the
product was extracted with methylbenzene. The extract was dried, filtered and evapo-
rated, yielding 44.0 parts (100%) of ethyl 4-[[1-[[5-[(acetyloxy)-methyl}-2-furanyl]-
methyl]- lH-benzimidazol-2-yl]methyl]- 1 -piperidinecarboxylate (comp. 1.03).
This compound also contained an amount of a side product, namely ethyl 4-[[1-[[5-
10 [[(phenyl)msthoxy]methyl]-2-furanyl]methyl]- IH-benzimidazol-2-yl]methyl]- 1- piperidinecarboxylate.
d) A mixture of 44.0 parts of compound (1.03), 56 parts of potassium hydroxide
and 234 parts of 2-propanol was stirred overnight at reflux temperature. The reaction
mixture was evaporated and the residue was taken up in water. The product was
15 extracted with dichloromethane and the extract was dried, filtered and evaporated. The
residue was purified by column chromatography (silica gel; CHC13 / CH30H (NH3)
95:5 ~ $0:20). The eluent of the second fraction was evaporated, yielding 27.5 parts
(84.5%) of 5-[12-(4-piperidinylmethyl)-1~-benzimidazol-1-yl]methyl]-2-furanmethanol
(comp. 1.û4).
20 Evaporation of the first fraction yielded 9 parts of 2-[[5-(phenylmethoxy)methyl]-2-
furanyl]methyl]-2-(4-piperidinylmethyl)-lH-benzimidazole (comp. 1.05).
e) A mixture of 3.25 parts of compound (1.04), 2 parts of polyoxymethylene, 2 parts of
a solution of thiophene in methanol 4% and 119 parts of methanol was hydrogenated at
no~nal pressure and at room temperature with 2 parts of palladium-on-charcoal catalyst
25 10%. After the calculated amount of hydrogen was taken up, the catalyst was filtered off
and the filtrate was evaporated. The residue was taken up in dichloromethane and the
whole was washed with NH4OH (aq.). The organic layer was dried, filtered and
evaporated. The residue was crystallized successively from 4-methyl-2-pentanone and
acetonitrile, yielding 1.72 parts (50.7%) of 5-[[2-[(1-methyl-~piperidinyl)methyl]-lH-
30 benzimidazol-1-yl]methyl]-2-furanmethanol; mp. 158.1~C (comp. 1.17).
Example 9
Through a stirred mixture of 8.4 par~s of intermediate (16), 5.05 parts of a fo~naldehyde
solution 40% and 4.25 parts of piperidine was bubbled hydrochloric acid till all solid
35 en~ered the solution. The whole was stirred over weekend and treated with ammonia. The
product was extracted with trichloromethane. The extract was dried, filtered andevaporated. The residue was purified by column chromatography (silica gel; CHC13 /
, . ,:
"
.:
-29- X~iL3892
CH3OH 97:3 ~ 98:2). The eluent of the desired fractions was evaporated and the
residue was crystallized from a mixture of 4-methyl-2-pentanone and 2,2'-oxybis-propane, yielding 1.0 part (12.5%) of ethyl ~[[3-l[S-(hydroxymethyl)-2-furanyl]-methyl]-3~-imidazo[4,5-b]pyridin-2-yl]arnino]-1-piperidinecarboxylate; mp. 148.3C
S (comp. 2.01).
Exam~le 1Q
A mixture of 105.9 parts of intermediate (25), 197.5 parts of 5-[[(3-amino-2-pyridinyl)-
amino]methyl]-2-furanmethanol and 470 parts of ~,~-dimethylformarnide was stirred
10 for 3 days at room temperature. The reaction mixture was poured into water. The whole
was basified with K2CO3 and extracted with dichloromethane. The exlract was dried,
filtered and evaporated and the residue was taken up in 435 parts of methylbenzene. A
few parts of 4-methylbenzenesulfonic acid were added and the whole was stirred for 2
hours at reflux temperature. After cooling and basifying with K2C03 (aq.), the product
15 was extracted with dichloromethane and the extract was dried, filtered and evaporated,
yielding 156 parts (100%) of ethyl 4-[[3-[[S-(hydroxymethyl)-2-furanyllmethyl]-3H-
imidazol4,5-b]pyridin-2-yl]methyl]-1-piperidinecarboxylate (comp. 2.13).
Exam~ç~
20 A mixture of 20 parts of compound (3.05) and 237 parts of methanol was acidified with
sulfuric acid (pH 1) while stirring. Stirring was continued overnight at reflux
temperature. After cooling, the reaction rnixture was basified with methanol saturated
with ammonia and was then evaporated. The residue was taken up in water and the
product was extracted with dichloromethane. The extract was dried, filtered and
25 evaporated. The residue was purified by column chromatography (silica gel; CHC13 /
CH30H ~7:3). The eluent of the desired ~raction was evaporated, yielding 29 parts
(10Q%)ofethyl 4[[1-[[5-(methoxy~nethyl)-2-furanyl]methyl]-lH-benzimidazol-2-
yl]amino]-l-piperidinecarboxylate (comp. 3.06).
30 ~me!~2
A mixture o~ 2 parts of [(4-fluorophenoxy)methyl]oxirane, 3.26 parts of cornpound
(3.11) and 39 parts of 2-propanol was stirred for 48 hours at reflux temperature. The
reaction mixture was evaporated and the residue was purified by column chromatography
(silica gel; CHC13 / CH30H (NH3) 97:3). The eluent of the desired fraction was
35 evaporated and the residue was crystallized ~rom a mixture of acetonitrile and
2,2'-oxybispropane, yielding 2.80 parls (56.6%) of a-[(4-fluorophenoxy)methyl~-4-[[1-
~389~:
-30-
[[5-thydroxymethyl)-2-furanyl}methyl]-lH-benzim~dazol-2-yl]amino)-1-
piperidineethanol; mp. 136.8C ~comp. 3.20).
Example 13
S To a stirred and cooled (0C) ;nixture of 3.3 parts of compound (3.11), 1.01 parts of
1,1'-oxybisethane and 94 parts of N~N-dimethylformamide was added dropwise a
solution of 0.8 parts of acetylchloride in N~N-dimethylformamide. The reaction mixture
was allowed to walm up to room temperature and was then evaporated. The residue was
boiled 4 times in trichloromethane and decanted. The combined liquid phases were dried,
10 filtered and evaporated. The residue was purified by column chromatography (silica gel;
CH2C12 / CH30H 95:5). The eluent of the desired fraction was evaporated and the
residue was crystallized from methanol, yielding 0.8 parts (21.7%) of l-acetyl-N-[1-[[5-
(hydroxymethyl)-2-furanyl]methyl]- lH-benzimidazol-2-yl]4-piperidinamine;
mp. 213.9C (comp. 3.21).
Exam~le 14
A mixture of 3.7 parts of 7-(2-bromoethyl)-3,4-dihydro-8-methyl-2-H,6H-pyrimido-[2,1-b][1,3]thiazin-6-one monohydrobromide, 3.2 parts of compound (3.11), 2.1 parts
of sodium carbonate and 160 parts of 4-methyl-2-pentanone was stirred overnight at
20 reflux temperature. The reaction mixture was evaporated and the residue was taken up in
water. The product was extracted with dichloromethane and the extract was dried, filtered
and evaporated. The residue was purified by column chromatography (silica gel;
CH2C12 / CH30H(NH3) 95:5). The eluent of the desired fraction was evaporated andthe residue was crystallized frorn acetonitrile. The product was filtered off and dried,
25 yielding 1.5 parts (28.0%) of 3,4-dihydro-7-[2-[4-[[1-[[5-(hydroxymethyl)-2-
furanyl]methyl]-lH-benzirnidazol-2-yl]amino]-1-piperidinyl]ethyl]-8-methyl-2H,~_-
pyrimidor2,1-b][1,3]thiazin-6-one; mp. 226.9C (comp. 3.31).
Exarnpl~l S
30 A mixture of 3.3 parts of 2-chloroacetonitrile, 13 parts of compound (2.03), 5 parts of
~,~-diethylethanamine and 94 parts of ~-dimethylforrnamide was stirred for 4 hours
at room temperature. After the addihon of potassium carbonate, the reaction rnixture was
diluted with water~ The whole was stirred briefly and the product was extracted with
dichloromethane. The extract was dried, filtered and evapo~ted. The residue was stirred
35 in 1,1'-oxybisethane, filtered off and dried, yielding 10.85 parts ~74%) of 4-[[3-L[s-
(hydroxymethyl)-2-furanyl]methyl]-3H-imidazo[4,5-b]pyridin-2-yl]amino]- 1 -piperidine-
acetonitrile (comp. 2.26).
- , '
~,
~3~39;~
-31-
ExamDle 11~
A mixture of 3 parts of 2-ethenylpyridine, 3.7 parts of compound (2.28) and 122 parts of
1-butanol was stirred overnight at reflux temperature. The reaction mixture was
5 evaporated and the residue was purified by column chromatography (silica gel; CH2cl2 /
CH30H / CH30H(NH3) 90: 10:0 ~ 90:8:2). The eluent of the desired fraction was
evaporated and the residue was converted into the (E)-2-butenedioate (2:3) salt in
ethanol. The salt was recrystallized from ethanol (2x), yielding 2.14 parts (35.3%) of S-
[~2-[[1-[2-~2-pyridinyl)ethyl]-4-piperidinyl]methyl~-3H-imidazo[4,5-b]-pyridin-3-
10 yl]methyl]-2-furanmethanol (E)-2-butenedioate(2:3); mp. 159.8C (comp. 2.35).
Example 17
A mixture of 4.5 parts of 3,6-dichloropyridazine, 11.1 parts of compound (1.14) and 3.2
parts of sodium carbonate was stirred for 1/2 hour at 150C. After cooling, the reaction
lS mixture was diluted with water. The product was extracted with trichloromethane and the
extract was dried, filtered and evaporated. The residue was boiled in acetonitrile. After
cooling, the precipitate was filtered off and dried, yielding 9.72 parts (67.4%) of 5-[[2-
[[1 -[2-[t6-chloro-3-pyridazinyl)amino]ethyl]-4-piperidinyl]-methyl l-lH-ben7.imidazol- 1-
yl]methyl]-2-~uranmethanol; mp. 188.4C (comp. 1.21).
Example 18
A mixtu3~ of 1.1 parts of 4-chloro-3-nitropyndine, 2.5 parts of compound (3.23), 1 part
of sodium carbonate and 39.5 parts of ethanol was sti~ed overnight at room temperature.
Water was added and the product was extracted with dichloromethane. The extract was
25 dried, filtered and evaporated. The residue was crystallized from a mixture of acetonitrile
and ethanol, yielding 1.9 parts (56.~%) of 5-[[2-[[1-[2-[(3-nitro-4-pyridinyl)amino]-
ethyl]-~piperidinyllamino]- lH-benzimidazol- 1 -yl]methyl]-2-furanmethanol;
mp. 191.0C (comp. 3.26).
30 ~m~çl2
A mixture of I .74 parts of 4-chloro-3-nitropyridine, 4.22 parts of compound (4.11),
1.11 parts of N.N-diethylethanamine and 149 parts of trichloromethane was stirred
overnight at room temperature. The reaction mixture was washed with K2CO3 (aq.). The
aqueous layer was separa~ed and extracted with dichloromethane. The combined organic
35 layers were dried, ~lltered and evaporated. The residue was purified by column
chromatography (silica gel; CH2C12 / CH30H / CE13OH(NH3) 90: 10: 1). The eluent of
the desired fraction was evaporated and the residue was crystalliæd from a mix~ure of 4-
-32- ~ 8
methyl-2-pentanone and ethanol. The product was filtered off and dried, yielding 1.2
parts (21.2~o) of 5-[[2-[[hexahydro-1-[2-[(3-nitro-4-pyridinyl)amino]ethyl]-lH-azepin-
4-yl]arnino]-lEI-ben7imidazol-1-y}]methyl]-2-furanmethanol hemihydrate; mp. lS5. 1 C
(comp. 4.14).
_xam~le 20
To a soludon of 1.3 parts of lithium aluminum hydride in 89 parts of tetrahydrofuran was
added a solution of 4.6 parts of intermediate (3) in tetrahydrofuran. After refluxing for 2
hours, the reaction mixture was treated with ethyl acetate. Then there were added
10 dropwise 7.9 parts of NaOH 15% and 4.8 parts of water, while stirnng. The whole was
filtered over diatomaceous earth and the filtrate was evaporated. The residue was purified
by column chromatography (silica gel; CH2C12 / CH30H I CH30H (NH3) 90:5:5). The
eluent of the desired fraction was evaporated and the residue was crystallized from a
mixture of acetonitrile and 2,2'-oxybispropane (2x), yielding 0.65 parts (18.4%) of 5-
15 [[2-[(hexahydro-1-methyl-lH-azepin-4-yl)amino3-1~-benzimidazol-1-yl]methyl]-2-
furanmethanol (comp. 4.02).
Example 21
A mixture of 2.5 parts of 3-bromo-N-(1-methylethyl)propanamide, 3.26 parts of
20 compound (3.11), 1.26 parts of sodium hydrogen carbonate and 39.5 parts of ethanol
was stirred overnight at reflux temperature. The reaction mixture was evaporated and the
residue was taken up in water. The product was extracted with a mixture of
trichloromethane and ethanol and the extract was dried, filtered and evaporated. The
residue was crystallized from acetonitrile, yielding 3.20 parts (72.8%) of 4-~[1-[[5-
25 (hydroxymethyl)-2-furanyl]methyl]- lH-benzimidazol-2-yl]amino]-N-(1 -methylethyl)- I -
piperidinepropanamide; mp. 139.7C (comp. 3.12).
Exarnple 22
A mixture of 4.36 parts of compound (1.21~, I part of a solution of thiophene in30 methanol 4%, 198 par~s of methanol and 2 parts of calciumoxide was hydrogenated at
norrnal pressure and room temperature with 2 parts of palladium-on-charcoal catalyst
10%. After the calculated amount of hydrogen was taken up, the catalyst was filtered off
and the filtrate was evaporated. The residwe was taken up in water and the product was
extracted with dichloromethane. The extract was dried, filtered and evaporaeed. The
35 residue was purified by colurnn chromatography (silica gel; CH2C12 / CH30H(NH3)
9S:S). The eluent of the desired fraction was evaporated and the residue was converted
into the edlanedioate (1:3) salt in a mixture of methanol and ethanol. The product was
33 ~ 392
~lltered off and dried, yielding 4.51 parts (69.9%) of 5-[[2-~[1-E2-(3-
pyridazinylamino)ethyl]-4-piperidinyl]methyl]- lH-benzimidazol- 1 -yl]methyl]-2-furanmethanol ethanedioate (1:3); mp. 203.5C (comp. 1.24).
S E~xarn~le 2~
A mixture of 3.4 parts of compound (1~21), 0.82 parts of sodium acetate and 73.5 parts
of acetic acid was stirred for 4 hours at reflux temperature. The reaction mixture was
evaporated and the residue was taken up in water. After basifying with K2CO3, the
solution was extracted with dichloromethane and the extract was dried, filtered and
10 evaporated. To the residue there were added 3.5 parts of potassium hydroxide and 39
parts of 2-propanol and the whole was stirred for 3 hours at reflux temperature. The
solvent was evaporated and water was added to the residue. The product was extracted
with l-butanol and the extract was dried, filtered and evaporated. The residue was
purified by column chromatography (silica gel; CH2C12 / CH3OH(NE13) 90:10). The
lS eluent of the desired fraction was evaporated and the residue was converted into the
ethanedioate (2:5) salt in ethanol. The product was filtered off and dried, yielding 2.20
parts (45.7%) of 6-[[2-[4-[[1-[[S-(hydroxymethyl)-2-furanyl]methyl]-lH-benzimidazol-
2-yl]methyl]-1-piperidinyl]ethyl]arnino]-3(2O-pyridazinone ethanedioate (2:5); mp.
210.2C (comp. 1.25).
~0
Example 24
A rnixture of 22.4 parts of interrnediate (15), 13 parts of mercury(II)oxide, a spoonful of
sulfur and 178 parts of tetrahydrofuran was stirred for 3 hours at reflux temperature. The
reaction mixture was filtered and the filtrate was evaporated. The residue was partitioned
25 between H2SO4 and dichloromethane. The organic layer was dried, filtered and
evaporated and the residue was purified by column chromatography (silica gel; CH2C12 /
C2HsOH 95:5). The eluent of the desired fraction was evaporated, yielding 9.9 parts
(47.9%) of ethyl hexahydro-4-[[3-[[5-(hydroxymethyl) 2-furanyl]methyl]-3H-
imidazo[4,5-b]pyridin-2-yl]amino]-lH-azepine-1-carboxylate (comp. 4.04).
All new compounds listed in Tables 1 to 6 were prepared following the proceduresdescribed hereinabove. For each compound the actually used procedure is referred to in
the column captioned by "Ex. No.".
-34- Z~38
Ta~lç 1 IcH2~cH2-oR
L--N~}CH2~\ ~
N
, . .
S No Ex. R1 L Physical data- mp.
. . . ..... ~ .
1.01 8a H- C6Hs-CH2- 143.8C
1.02 7e CH3-CO- CSHs-cH2
1.03 & ~H3-C~ C2H5OOC-
1.04 8d H- H-
1.05 8d C6Hs-CH2- H-
S ~N~,,CH3 : .
1.06 7c C6H5-CH2- ~--N~I`CH2-cH2-- 180.5C / 1/2H2O / 3/2* :
oO
1.07 7c H- C2H5--NJ\N--(CH2)2-- 124.8C
N=N
1.08 7c H- ~ ~CH2-CH2-- 159.7C
: 15 o
1.09 7c H- ~CH2-CH2- 145.0C/H2O
O
S ~N CH3
1.10 7c H- <--N~ CH2--CH2-- 1 83.0C
O
1.1 1 7c H- 4-CH3O-~6H4-(CH2)2- 1 89.7C / 1/2 C2HsOH / *
1.12 7c H- iC3H7-NH-CO-(~H2)2- 1 35.0C
1.13 7c H- NC-CH2-
25 1 14 7d _ H2N-(CH2)2- _ . _ ?
:: , . . , . - :
' , : ~ ,. : ~ ,;
1' '~
' '
`
Z~ 392
-35-
, . _
Co Nxb. Rl L Physical data - mp.
-- CH3 O
1.15 7e H- 6~C-NH--(cH2)2-- 176.3C/2(COOH)2/ 1/2 H20
1.16 7e H- 6~C--NH-(CH2)2-- 137.4C / 2(COOH)2 / H2O
1.17 8e H- CH3- 158.1C
1.18 7c H- 4F-C6H4-cO-(cH2)3- 120.0C
CH3~ 2 (c-c6Hll-NHso3H)
1.19 14 H- O'N~CH2-CH2- 87.3C
1.20 12 H- N=N 1/2 * / 227.8C
1.21 17 H- CI~NH--(CH2)2-- 1 88.4C
1.22 17 H- <=~NH--(CH2)2-- * /184.2C
/=N
1.23 18 H- ~ /~NH--(CH2)2-- 111.5C
1.24 22 H- ~--NH--(CH2)2-- 3 (COOH)2/203.5C
N--N
1.25 23 H- O~-NH--(CH2)2-- 5/2 (COOH)2 / 210.2C
o
= (E)-2-butenedloate
Table 2 I H2~CH2-OR1
~ N~N
L--N~B~\ ~
.
-36- ~ ~38~;~
_ . _ . _ __ ... ----` -I
CNoO ENxo. B R1 L Physical data - mp.
~ _ .. .
S 2.01 9 NH H- C2Hs-OOC- 148.3C
2.02 7b NH H- H- 220.2C / 3l2*
2.03 7b NH H- H- 124.8C / 1/2 H2O
2.04 7c NH H- 4-CH3O-C6H4-(CH2)2- 111 .0C / H2O
2.05 7c NH H- C2Hs-O-(CH2)2- 105.8C I 112 H2O
2.V6 7c NH H- 4-F-C6H4-CO-(cH2)3- 200.5C I (COOH)2
J~
2.07 7c NH H- HN N--(CH2)2- 231.3C
S ~N CH3
2.08 7c NH H- <--N~CH2-CH2- 250.0C / H20
o
S _~N CH3
2.09 7c NH H- ~CH2-CH2- 239.0C
2.10 15 NH H- ~CH2-CH2- 177.7C l 2 H2O / S/2*
2 .11 1 0 ~H2 H- C6Hs-CH2- 11 5.4C
2.12 10 CH2 CH3- C6H5-CH2- 201.5C / (COOH)2
2.13 10 CH2 H- C2H5OOC-
2.14 7b CH2 H- ~g 3 151 .9C 1 H20 12*
2.15 lS CH2 H- <--N~CH2-CH2- 203.3C I *
O
J~
2.16 14 NH H- C2H5--N~ N--(CH2)2-- 173.1C
2.17 14 NH H- i-C3H7-NHcO-c2H4- 175.3C I 3l2(COOH)2
2.18 8e NH H- CH3- 1 83.3C
_ _ . .. ~
.. .: -
37 20~B92
~ ,. . ~
Co Ex. B R1 L Physical data - mp.
_ . _
2.19 12 NH H- 4F-C6H4-OCH2-CHOH-cH2- 161.6C
2.20 7e CH2 H- ~NH - (CH2~2 - 174.7C
2.21 7e NH H- ~LNH - (CH2)2 - 175.9C
S~N CH3
2.22 14 CH2 H- ~N~CH2-cH2- 163.7C
2.23 7c CH2 H- 4F-C6H4-cO-(cH2)3- 174.6C /
(C-C6Hl l-NHso3H)
2.24 7c CH2 H- NC-CH2-
2.25 7d CH2 H- H2N-(CH2)2-
2.26 15 NH- H- NC-CH2-
lS 2.27 7d NH- H- H2N-(CH2)2-
2.28 7b CH2 H- H- 137.6C
2.29 14 CH~ H- O ~CH2 - cH2 - H20 / 91.2C
2.30 7e NH- H- ~N L IC, _NH_(CH2)2 - 177.5C
CH3
~N~CH3
2.31 14 CH2 H- Y~(CH~2_ 178.4C
~N CH3
2.32 14 NH- H- ~CH2-CH2_ 202.5C
0
~S YNll'CH3
2.33 14 CH2 H- ~N~(C~2)2- H2O/ 159.5C
o
- . _ .. ._ .
-38- ;~
. _ ~
Co Ex. B R1 L Physical data - mp.
NH - (CH2)2-- .... ____ .
2.34 17 CH2 H- CH3 N) 1/2 H2O / 145.3C
2.35 16 CH2 H- ~(CH2)2-- 3/2 * /159.8C
2.36 7e CH2 H- ~ CH2)2-- 174.7C
N~C ~NH--(CH2)2--
2.37 7e CH2 H- ¢N~\N~2 1 33.0C
2.38 21 CH2 H- C2Hs-NH-CO-NH-tCH2)2- 119.7C
* = tE)-2-butenedioate
~abl~ 3 ~3
4 R
/~ N ~,
L--N~s~\ _!l J
N ~
15 . .. I
Co Ex. B R L Physical data mp
3.01 8a NH- 2-CH2OH 4-CH3O-C6H4-(CH2)2- 110.1C
3 .02 8a NH- 3-CH20H 4-CH3O-C6H4-(CH2)2- 136.1 C
3.03 8a NH- 2-CH2OH CH3- 124.1C/ 1/2H20
3.04 8a NH- 3-CH2OH CH3- 133.3C/ 1/2 H2O
3.05 7a NH- 2-CH20H C2Hs-OOC- 141 .6C
3.06 11 NH- 2-CH2OCH3 C2Hs-OOC- -
3.07 7b NH- 2-CH20CH3 H- 240.8C / 2*
_ _ . ._
,
. , . .: : '
39 2~3L3~
~ .. ._ .......................... .. I
Co ENo. B R L Physical data - mp. .
__
3.08 7c NH- 2-CH2OCH3 4-CH3O-C6H4-(CH2)2- 201.3C / 2*
3.09 7c NH- 2-CH2OCH3 ~ CH2-CH2- 181.7C/3/2H20/2*
S ~ N CH3
3.10 7c NH- 2-CH2OCH3 ~--N~ ~ cH2-cH2- 1 59.6C/H20/2*
3 .11 7b NH- 2-CH2OH H- 1 56.3C
3.12 21 NH- 2-CH2OH iC3H7-NH-cO-(cH2)2- 139.7C
3 . 13 7c NH - 2-CH2OH H3C-CH2--N~ ,N--(CH2)~-- l 63 .1 C
3 .1 4 7c NH - 2- CH2OH ~ N~ ~ CH2-CH2- 208 . 8 C
3.15 7c NH- 2-CH2OH <~ N~ CH2-CH2- 210.4C / H2O
3.16 7c NH- 2-CH2OH C2Hs-O-(CH2)2- 1 77 .0C/2(COOH)2
~ N CH3
3.17 7c NH- 2-CH20H ~N~CH2-CH2- 237.0C/ 1/2 H20
3.18 7c NH- 2-CH2OH 4F-C6H4-cO-(cH2)3- 1 34.8C
3 .1 9 7c NH- 2-CH20H HN N--~CH2)2- 206.8C
~
3.20 12 NH- 2-CH20H 4F-c6H4-o-cH2-cHoH-cH2 1 36.8C
3.21 13 NH- 2-CH2OH CH3-CO- 213.9C
3.22 7c NH- 2-CH20H NC-CH2- 209.8C
-._ .. _ , ~ .
'
.
~ L3892
_ . _
Co Ex. B R L Physical data - mp.
~ _ _
3.23 7d NH-2-C~H20H H2N-(CH2)2- 1 64.6C
CH3 o
3.24 7e NH-2-CH20H ~C~ (CH2)2- 1 82.2C
3 .25 7e NH-2-~H20H ~c--NH--(CH2)2-- 200.2C
NO2
3.26 18 NH-2-CH20H N~NH--(CH2)2- 191.0C
H3C~pN C~13
3.27 7c NH--C H2OH ~CH2--CH2-- 206.2C / 1/2 H20
3.28 &L NH-3-CH20H HsC2o(cH232- 118.0C / H2O
3.29 8aL NH-4-CH20H (~-CH3Oc6H4)-c~I2-cH2- 137.7C
3~3 ¦ 20 ¦ NH- ¦ -CH2OH ¦CH3 ¦ 205.4OC
3.31 14 NH- 2-OEI20H b~((cH2)2-- 226.9C
3.32 24 NH- 2-CH(OH)~H3 CH3 187.6C / 3/2*
3.34 20 NCH3 2-CH20H CH3 185.1C / 3~2*
3.35 O 2-CH20H CM3
3.36 S 2-CH20H CH3
. ~ j:~ Itenedio Lte ~- . ___
Table 4 Cl H2~CH20H
L--N/~
~(~H2)n N
`
;. , . . . : ::
-41-
. _ ._
Co. Ex. n Al L Physical data- mp.
No. No. _ _
4.01 7a 2 CH- C2H5C~C 176.8C
4.02 20 2 CH- CH3- 119.7C
4.03 7b 2 CH- H- 203.4CI 2*
4.04 24 2 N HsC2OOC-
4.05 7b 2 N H- 167.6C / 2*
~ 1~
4.06 14 2 CH- ~ ~(CH2~ - 169.0 C
1~ o
4.07 14 2 CH- (cH3)2cH-~nH-c(o)-(cH2)2- 184.9C / 1/2 H20
o 2(c.C6Hl lNHS03H)
4.08 14 2 CH- o N - (CH~ - 174.8 C /2*1112 H20
4.09 14 2 CH- (4-CH3O-C6H4)-(C~l2)2- 148.6C / 2*
4.10 7c 2 CH- NC-CH2- 157.3C
4.11 7d 2 CH- H2N-(CH2)2-
4.12 7e 2 CH- ~c - NH - ~CH~2 - 167.3C
~Nll~CH3
4.13 14 2 CH- ~(CHi~ - 185.2C / 1/2 H2O
2(c.C6Hl lNHS03H)
~NO2
4.14 19 2 CH- N~ 155.1C / 1/2 H2O
NH--(CH~2--
L~Nll'CH3
4.15 14 2 CH- ~(CH~2 - 177.1 C
. o
4.16 8e 2 N CH3 175.9C/2*/ 1/2 ~2
4.17 20 0 CH- CH3 133.1C
_ _ _ . , .__ . . .~
,:
:, '
-42
TableS O
f~2~CH20H
L--N~} ~N3~ A~A2
~3~4~ ~ ¦ Physical~ ¦
5.01 24 -N=CH-N=CH- ~5c2OoC- 1~.7C
5.02 7b -N=CH-N=CH- s ~N CH3 1 60.1 C / H20
5.03 14 -N=CH-N=CH- ~N ~ 160.8C/H~O
(CH~ -
5.04 8e -N=CH-N=CH- CH3- 191.3C
5.05 24 -CH=CH-N=CH- H5C2OOC- 176.1C
5.06 7b -CH=CH-N=CH- H-
5.07 8e -CH=CH-N=CH- CH3- 188.4C
5.08 8e -CH=N-CH=CH^ CH3-
5.09 7b -CH=CH-CH=N- H- 207.7C
5.10 8e -CH=CH-CH=N- CH~ 172.5C/H20
5 Tab~6
HOCH2 0
CH2
L-N ~ NH ~\ ~
_~ . . . . ~
Comp. Ex. L Physic~
No. No.
. ~ . . ,:-
6.01 20 CH3- 142.8C
6.02 8a(4-CH3O-C6H4~-(CH2k- 141.1C
., . . ,:
.
L3
-43-
C. PhalmaçolQg~Example
ExamplQ25
The useful antihistaminic properties of the compounds of formula (I) can be
5 demonstrated in the test "Protection of rats frorn compound 48/80-induced lethality",
which is described in US-4,556,660, incorporated herein by reference, and the res~llts of
which are given in Table 7 below. The term (sc) signifies subcutaneous administration.
Tabl~
CQ. ED50 (mg/lcg)
No. compound 48/8
induced lethality
in rats
1.07 0.04 (sc)
1.08 0.01 (sc)
1.09 0.02 (sc)
1.10 0.01 (sc)
1.1 1 O.t)4 (sc)
1.12 0.01 (sc)
1.15 0.04 (sc)
1.17 0.04 (sc)
1.22 0.04 (sc)
1.23 0.02 (sc)
1.~4 0.02 (sc)
1.25 0.01 (SC)
2.09 0.01 (sc)
2.10 0.04 (sc)
2. 1 1 0.04 (sc)
2.16 0.04 (sc)
2.18 0.01 (sc)
2.20 0.04 (sc)
2.22 0.(~4 (sc)
2.23 0.04 (sc)
2.32 0.02 (sc)
44 ~ 3
Co. EDso (mg~g):
No. compound 48/8C
induced lethality
in rats.
. _ _
2.33 0 04 (sc)
2.35 0.02 (sc)
3.03 0.Q2 (sc)
3.11 0.04 (sc)
3.12 0.04 (sc)
3.13 0.04 (sc)
3.15 0.04 (sc)
3.17 0.04 (sc)
3.19 0.02 (sc)
3.22 0.04 tsc)
3.25 0.02 (sc)
3.26 0.04 (sc)
~.31 0.04 (sc)
4.05 0.02 (sc)
4.07 0.04 (sc)
4.12 0.04 (sc)
4.14 0.04 (sc)
4.16 0.02 (sc)
5.03 0.04 (sc)
5.04 0.02 (sc)
:~
D. Composition Examples
Example 26: ORAL DROPS
500 Parts of the A.I. was dissolved in 0.5 1 of 2-hydroxypropanoic acid and 1.5 1 of the
polyethylene glycol at 60~80C. After cooling to 30~4ûC there were added 35 l of
polyethylene glycol and the mixture was stirred well. Then there was added a solution of
1750 parts of sodium saccharin in 2.5 1 of purif1ed water and while sti~ing there were
35 added 2.5 1 of cocoa flavor and polyethylene glycol q.s. to a volume of 50 l, providing
,
: .
-45- ~ 2~89
an oral drop solution comprising 10 mg/ml of A.I.. The resulting solution was filled into
suitable containers.
Exarnple 27: ORAL ~,OLUTION
S 9 Parts of methyl 4-hydroxybenzoate and 1 part of propyl 4-hydroxybenzoate were
dissolved in 41 of boiling purified water. In 31 of this solution were dissolved first 10
parts of 2,3-dihydroxybutanedioic acid and thereafter 20 parts of the A.I. The latter
solution was combined with the remaining part of the forrner solution and 12 l
1,2,3-propanetriol and 31 of sorbitol 70% solution were added thereto. 40 Parts of
sodium saccharin were dissolved in 0.51 of water and 2 ml of raspberry and 2 ml of
gooseberry essence were added. The latter solution was combined with the former, water
was added q.s. to a volume of 201 providing an oral solution comprising 5 mg of the
active ingredient per teaspoonful (S ml). The resulting solution was filled in suitable
containers.
Example 28: CAPS~ILES
20 Parts of the A.I., 6 parts sodiurn lauryl sul~ate, 56 parts starch, 56 parts lactose, 0.8
parts colloidal silicon dioxide, and 1.2 parts magnesiurn stearate were vigorously s~irred
together. The resulting rnixture was subsequently filled into 1000 suitable hardened
gelatin capsules, comprising each 20 mg of the active ingredient.
Example 29: FILM-Ç(;;)ATED TA~BLETS
A mixture of 100 parts of the A.I., 570 parts lactose and 200 parts starch was mixed well
and thereafter humidified with a solution of 5 parts sodium dodecyl sulfate and 10 parts
polyvinylpyrrolidone (Kollidon-K 90 (~ in about 200 ml of water. The wet powder
mixture was sieved, dried and sieved again. Then there was added 100 parts micro-
crystalline cellulose (Avicel @~)) and 15 parts hydrogenated vegetable oil (Sterotex ~). The
whole was mixed well and compressed into ~ablets, giving 10.000 tablets, each con-
taining 10 mg of the active ingredient.
~Qat ~
To a solution of 10 parts methyl cellulose (Methocel 60 HG~)) in 75 ml of denaturated
ethanol there was added a solution of S parts of ethyl cellulose (Ethocel 22 cps ~) in 150
ml of dichloromethane. Then there were added 75 ml of dichloromethane and 2.5 ml1,2,3-propanetriol. 10 Parts of polyethylene glycol was molten and dissolved in 75 ml of
dichloromethane. The latter solu~ion was added to the forrner and then there were added
2.5 parts of magnesium octadecanoate, 5 parts of polyvinylpyrrolidone and 30 ml of
-46- 2~3~
concentrated colour suspension (Opaspray K-1-2109~) and the whole was
homogenated. The tablet cores were coated with the thus obtained mixture in a coating
apparatus.
S Examplç 3Q INIECIABLE SOLUTION
1.8 Parts methyl 4-hydroxybenzoate and 0.2 parts propyl 4-hydroxybenzoate were
dissolved in about 0.5 1 of boiling water for injection. After cooling to about 50C there
were added while stirring 4 parts lactic acid, 0.05 parts propylene glycol and 4 parts of
the A.I.. The solution was cooled to room temperature and supplemented with water for
10 injection q.s. ad I 1, giving a solution comprising 4 mg/ml of A.I.. The solution was
sterilized by filtration (U.S.P. XVII p. 811) and filled in sterile containers.
Example 31: ~PPOSITORIES
3 Parts A.I. was dissolved in a solution of 3 parts 2,3-dihydroxybutanedioic acid in 25
15 ml polyethylene glycol 400. 12 Parts surfactant (SPAN(~) and triglycerides (Witepsol
555 ~) q.s. ad 300 parts were molten together. The latter mixture was mixed well with
the former solution. The thus obtained mixture wàs poured into moulds at a temperature
of 37-38(: to form 100 suppositories each containing 30 mg/ml of the A.I.
20 Example 32: IN3ECI'ABLE SOLUTION
60 Parts of A.I. and 12 parts of benzylalcohol were mixed well and sesame oil was
added q.s. ad I 1, giving a solution cornprising 60 mg/ml of A.I. The solution was
steriliæd and filled in sterile containers.
`