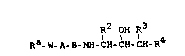Note: Descriptions are shown in the official language in which they were submitted.
HOECHST ARTIENGESELLSCHAFT HOE 89/F 128 Dr.MY/gm
;
De8cription 201~7~
DipQptide derivatives having enzy e-inhibitory action
The invention relates to dipeptide derivatives which
inhibit the action of the natural enzyme renin and to
processes for their preparation and use and to pharma-
ceutical preparations containing them. In ~P-A 0,255,082,
dipeptide derivatives are described which carry N-ter-
minal cycloalkylcarbonyl and cycloalkyl-alkylcarbonyl
substituents. In WO 88/05050, aminodiol derivatives
having renin-inhibitory action are described.
Surpri~ingly, it has now been found that substituted
cycloalkyl and cycloalkyl-alkyl derivatives contribute to
the considerable improvement of the action in vivo and to
the increased absorption of the compound.
The invention relates to compounds of the formula I
R2 OH R3
Ra-W-A-B-NH-CH-CH-CH-R4 (I)
in which
R' denotes ( C3-C~) -cycloalkyl, (C~-Cl0)-bicycloalkyl,
(C~-C~)-tricycloalkyl, ( C3-C~ ) -cycloalkyl-(cl-c6)-
alkyl, (C~-Cl0)-bicycloalkyl-(Cl-C6)-alkyl and
(C~-C~)-tricycloalkyl-(Cl-C6)-alkyl, in which in each
case the cycloalkyl, bicycloalkyl and tricycloalkyl
substituents can be substituted by one or two
identical or different radicals from the series
comprising F, Cl, Br, I, hydroxyl, (Cl-C6)-alkoxy,
(Cl-C6)-alkyl, carboxyl, (Cl-C6)-alkoxycarbonyl,
carbamoyl, carbo~ymethoxy, amino, (Cl-C0)-monoalkyl-
amino, (Cl-C6)-dialkylamino, amino-(Cl-C6)-alkyl, (Cl-
C6)-alkylamino-(Cl-C6)alkyl, di-(Cl-C6)-alkylamino-
(cl-c6)-alkyl~ amidino, hydroxamino, hydroximino,
hydrazono, imino, guanidino, (Cl-C6)-alkyloxy-
- 2 - 201 ~ 07 ~
sulfonyl, (C1-C~)-alkyloxysulfenyl, trifluoromethyl,
(Cl-C~)-alkoxycarbonylamino or (C6-Cl2)-aryl-(C1-C4)-
alkoxycarbonylamino;
W stands for -CO-, -O-CO- or -SO2-,
R2 denotes hydrogen, (C1-Cl0)-alkyl, (C~-C~)-cycloalkyl,
( C~-C7 ) -cycloalkyl-(C1-C~)-alkyl~ (C~-C1~)-aryl or
( CB_C1~ ) -aryl-(C1-C~)alkyl~
R3 denotes hydrogen, (Cl-C10)-alkyl, (C6-Cl~)-arYl-
(C6-C1~)-aryl-(Cl-Cj)-alkyl, hydroxyl or amino,
R4 denotes a radical of the formula II
(CH2)~-cHRs-D (II)
where
R5 stands for hydrogen, (C1-C,)-alkyl, (C1-C5)-
alkoxy, (Cl-C5)-alkylthio, (Cl-Cs)-alkylamino,
hydroxyl, azido, fluorine, chlorine, bromine or
iodine,
D denotes a radical Het, where
Het denotes a 5- to 7-membered heterocyclic
ring which can be fused to benzene, aromatic,
partially hydrogenated or completely hydro-
genated, which can contain one or two identical
or different radicals from the group N, O, S,
NO, SO or SO2 as heteroatoms and which can be
substituted by one or two identical or dif-
ferent radicals from the group comprising
(Cl-C~)-alkyl,(Cl-C~)-alkoxy,hydroxyl,halogen,
amino, mono- or di-(C1-C~)-alkyl~mino or CF3,
and
m denotes 0, l, 2, 3 or 4;
A and B independently of one another denote a radical of
an amino acid linked at the nitrogen terminus to R^-
~ 3 ~ 2 0 ~
W or A and at the carbon terminus to B or NH-CHR2-
CHoH-CHR9-R~ from the series comprising phenyl-
alanine, histidine, tyro~ine, tryptophan,
methionine, leucine, isoleucine, asparagine,
aspartic acid, ~-2-thienylalanine, ~-3-thienyl-
alanine, ~-2-furylalanine, ~-3-furylalanine, lysine,
ornithine, valine, alanine, 2,4-diaminobutyric acid,
arginine, 4-chlorophenylalanine, methionine sulfone,
methionine sulfoxide, 2-pyridylalanine, 3-pyridyl-
alanine, cyclohexylalanine, cyclohexylglycine, im-
methylhistidine,0-methyltyrosine,0-benzyltyrosine,
0-tert.-butyltyrosine, phenylglycine, l-naphthyl-
alanine, 2-naph*hylalanine, 4-nitrophenylalanine,
norvaline,0-2-benzotb]thienylalanine,~-3-benzo[b]-
thienylalanine, 2-fluorophenylalanine, 3-fluoro-
phenylalanine, 4-pyridylalanine, 4-fluorophenyl-
alanine, norleucine, cysteine, S-methylcysteine,
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid,
homophenylalanine, DOPA, 0-dimethyl-DOPA, N-methyl-
histidine, 2-amino-4-(2-thienyl)-butyric acid, 2-
amino-4-(3-thienyl)-butyric acid, 3-(2-thienyl)-
serine,(Z)-dehydrophenylalanine,(E)-dehydsophenyl-
alanine,l,3-dioxolan-2-ylalanine,N-pyrrolylalanine
and 1-, 3- or 4-pyrazolylalanine,
and their physiologically tolerable salts.
The chiral centers in the compounds of the formula I can
have the R-, S- or R,S-configuration.
Alkyl can be straight-chain or branched. The same applies
to r~dicals derived therefrom, such as, for example,
alkoxy, alkylthio, alkylamino, dialkylamino, alkanoyl and
aralkyl. (C3-C~)-Cycloalkyl is understood a~ meaning
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclo-
heptyl and cyclooctyl. (C~-C10)-Bicycloalkyl or (C~-C~)-
tricycloalkyl are understood as meaning an isocyclic
aliphatic, non-aromatic radical which can optionally
contain unsymmetrically distributed double bonds, and
_ 4 _ 2 01 5 0~
which can optionally also be substituted with open-chain
aliphatic ~ide chains. The two or three rings as com-
ponents of a radical of thi~ type are condensed or linked
in a spiro fashion and linked via a ring carbon atom or
a side chain carbon atom. Examples of these radicals are
bornyl, norbornyl, pinanyl, norpinanyl, caranyl,
norcaranyl, thu~anyl, adamantyl, bicyclo(3.3.0)octyl,
bicyclo(l.l.O)butyl and spiro(3.3)heptyl substituents.
If the cyclic compounds mentioned carry more than one
substituent, these can be both cis and trans to one
another.
(C6-C,4)-aryl is, for example, phenyl, naphthyl, bipheny-
lyl or fluorenyl; phenyl is preferred. ~he same applies
to radicals derived therefrom, such as, for example,
aryloxy, aroyl, aralkyl and aralkyloxy. Aralkyl is
under3tood as meaning an unsubstituted or substituted
(C6-C14)-aryl radical linked to (C,-C6)-alkyl, such as, for
example, benzyl, ~- and ~-naphthylmethyl, halobenzyl and
alkoxybenzyl, aralkyl, however, not being limited to the
radicals mentioned.
A radical Het in the sense of the preceding definition
is, for ex~mple, pyrrolyl, furyl, thienyl, imidazolyl,
pyrazolyl, oxazolyl, thiazolyl, pyridyl, pyrazinyl,
pyrimidinyl, indolyl, quinolyl, isoquinolyl, quinox-
alinyl, ~-carbolinyl or a derivative of these radicals
which is fused to benzene, cyclopentane, cyclohexane or
cycloheptane. This heterocycle may be substituted on a
nitrogen atom by oxido, (C~-C6)-alkyl, for example methyl
or ethyl, phenyl or phenyl-(C,-C~)-alkyl, for example
benzyl, and/or on one or more carbon atoms by (Cl-C~)-
alkyl, for example methyl, phenyl, phenyl-(Cl-C~)-alkyl,
for example benzyl, halogen, for example chlorine,
hydroxyl, (Cl-C~)-alkoxy, for example methoxy, phenyl-
(cl-c4)-alkoxy~ for example benzyloxy, or oxo and can be
partially ~aturated and i8 ~ for example, 2- or 3-pyr-
rolyl, phenylpyrrolyl, for example 4- or 5-phenyl-2-
- 5 - 2~
pyrrolyl, 2-furyl, 2-thienyl, 4-imidazolyl, methylimid-
azolyl, for example l-methyl-2-, 4- or 5-imidazolyl, 1,3-
thiazol-2-yl, 2-, 3- or 4-pyridyl, 1-oxido-2-, 3- or 4-
pyridino, 2-pyrazinyl, 2-, 4- or 5-pyrimidinyl, 2-, 3- or
5-indolyl, substituted 2-indolyl, for example l-methyl-,
5-methyl-, 5-methoxy, 5-benzyloxy-, 5-chloro- or 4,5-
dimethyl-2-indolyl, 1-benzyl-2- or 3-indolyl, 4,5,6,7-
tetrahydro-2-indolyl, cycloheptatb]-5-pyrrolyl, 2-, 3- or
4-quinolyl, 4-hydroxy-2-quinolyl, 1-, 3- or 4-isoquino-
lyl, 1-oxo-1,2-dihydro-3-isoquinolyl, 2-quinoxalinyl, 2-
benzofuranyl, 2-benzoxszolyl, 2-benzothiazolyl, benz[e]-
indol-2-yl or ~-carbolin-3-yl.
Partially hydrogenated or completely hydrogenated hetero-
cyclic rings are, for example, dihydropyridinyl, pyr-
rolidinyl, for example 2-, 3- or 4-N-methylpyrrolidinyl,
piperidinyl, piperazinyl, morpholino, thiomorpholino and
tetrahydrothiophenyl.
- Salts of compounds of the formula I are to be understood
as meaning, in particular, pharmaceutically utilizable or
non-toxic salts.
Such salts are formed, for example, from compounds of the
formula I which contain acidic groups, for example
carboxyl, with alkali metals or alkaline earth metals,
such as Na, K, Mg and Ca, and also with physiologically
tolernble organic amines, such as, for example, triethyl-
amine and tri-(2-hydroxyethyl)amine.
Compounds of the formula I which contain basic groups,
for example an amino group or a guanidino group, form
salts with inorganic acids, such as, for example, hydro-
chloric acid, sulfuric acid or pho~phoric acid and withorganic carboxylic or sulfonic acids, such as, for
example, acetic acid, citric acid, benzoic acid, maleic
acid, fumaric acid, tartaric acid and p-toluenesulfonic
acid.
- 6 - 2 0 ~ 5 0 ~ ~
Preferred compounds of the formula I are those in which
R' and W are as defined on page 1;
R2 denotes isobutyl, benzyl or cyclohexylmethyl;
R3 denotes hydrogen, ( C~-C5) -alkyl, (Cff-C10)-aryl,
S (C6-C1~)-aryl-(C,-C~j-alkyl or hydroxyl;
R~ denotes a radical of the formula II, in which
R5 stands for hydrogen, (C~-C~)-alkyl, (C1-C~)-
alkoxy, (Cl-C~)-alkylthio, (C1-C~)-alkylamino,
hydroxyl, azido, fluorine, chlorine, bromine or
iodine,
D is defined as the radical Het on page 2, and
m denotes 0, 1 or 2; and
A and B independently of one another denote a bivAlent
radical from the series comprising
; 15 phenylalanine, histidine, tyrosine, tryptophan,
; methionine, leucine, isoleucine, asparagine, aspar-
tic acid, ~-2-thienylalanine, ~-3-thienylalanine, ~-
2-furylalanine, ~-3-furylalanine, lysine, ornithine,
valine, alanine, 2,4-diaminobutyric acid, arginine,
4-chlorophenylalanine, methionine sulfone, methi-
onine sulfoxide, 2-pyridylalanine, 4-pyridylalanine,
3-pyridylalanine, cyclohexylalanine, cyclohexylgly-
cine, im-methylhistidine, 0-methyltyrosine, 0-
benzyltyrosine, 0-tert.-butyltyrosine, phenylgly-
cine, l-naphthylalanine, 2-naphthylalanine, 4-
nitrophenylalanine, norvaline, norleucine,cysteine,
S-methylcysteine, N-methylhistidine, 1,2,3,4-tetra-
hydroisoquinoline-3-carboxylicacid,homophenylalan-
ine, 2-amino-4-(2-thienyl)-butyric acid, 2-amino-4-
(3-thienyl)-butyric acid, 3-(2-thienyl)-serine, (Z)-
dehydrophenylalanine,(E)-dehydrophenylalanine,1,3-
dioxolan-2-ylalanine, N-pyrrolylalanine and 1-, 3-
or 4-pyrazolylalanine,
and their physiologically tolerable salts.
- 7 - 2 01 ~ 07 ~
Particularly preferred compounds of the formula I are
those in which
R' denotes (C~-C6)-cycloalkyl and tC~-C6)-cycloalkyl-
(Cl-C2)-alkyl, in which in each case the cycloalkyl
S substituent can be sub6tituted by one or two identi-
cal or different radicals from the series comprising
hydroxyl, (Cl-C~)-alkoxy, (Cl-C2)-alkyl, carboxyl,
(Cl-C2)-alkoxycarbonyl, carbamoyl, carboxymethoxy,
amino, (Cl-C2)-monoalkylamino, (cl-c2)-dialkyl~ino~
amino-(cl-c2)-alkyl~ (Cl-C2)-alkylamino-(Cl-C2)alkyl,
di-(Cl-C2)-alkylamino-(Cl-C2)-alkyl, amidino, tri-
fluoromethyl, (Cl-C~)-alkoxycarbonylamino, such as
tert.butoxycarbonylamino, (C6-Cl2)-aryl-(cl_c2)_
alkoxycarbonylamino Euch as benzyloxycarbonylamino
and methanesulfonylamino or trifluoromethane-
sulfonylamino;
W stands for -C0-;
R2 denotes isobutyl, benzyl or cyclohexylmethyl;
R3 denotes hydrogen or hydroxyl;
R~ denotes a radical of the formula II, in which
R5 stands for hydrogen or fluorine,
D stands for a 2-, 3- or 4-pyridine radical, a
2-, 4- or S-imidazole radical or a 2-oxazoline
radical, where the heterocycles mentioned can
in each case be substituted by one or two
radicals from the group comprising methyl,
ethyl, propyl, allyl, fluorine, chlorine,
bromine, CF3 and methoxy, and
m denotes 0, 1 or 2; and
0 A and B independently of one another denote a bivalent
radical from the series comprising
phenylalanine, histidine, tyrosine, tryptophan,
methionine, leucine, isoleucine, asparagine, aspar-
tic acid, ~-2-thienylalanine, ~-3-thienylalanine, ~-
- 8 - 2 0 ~ ~ ~ 7 ~
2-furylalanine, ly~ine, ornithine, valine, alanine,
2,4-diaminobutyric acid, arginine, 4-chlorophenyl-
alanine, methionine sulfone, methionine sulfoxide,
2-pyridylalanine, 3-pyridylalanine, 4-pyridylala-
nine, cyclohexylalanine, cyclohexylglycine, im-
methylhistidine,O-methyltyrosine,O-benzyltyrosine,
O-tert.-butyltyrosine,phenylglycine,l-naphthylala-
nine, 2-nsphthylalanine, 4-nitrophenylalanine,
norvaline, norleucine, 1,2,3,4-tetrahydroisoquino-
line-3-carboxylic acid, homophenylalanine, 2-amino-
4-(2-thienyl)-butyric acid and 1-, 3- and 4-pyrazol-
ylalanine,
and their physiologically tolerable salts.
The invention furthermore relates to a process for the
preparation of compounds of the formula I which comprises
coupling a fragment having a terminal carboxyl group or
its reactive derivative with a corresponding fragment
having a free amino group, optionally removing (a)
protective group(s) temporarily introduced for the
protection of other functional groups and optionally
converting the compound thus obtained into its physio-
logically tolerable salt.
Fragments of 8 compound of the formula I having a ter-
minal carboxyl group possess the formulae IIIa, IIIb or
IIIc below:
Ra-W-OH Ra W-A OH Ra-W-~-~-OH
IIIa IIIb IIIc
Fragments of a compound of the formula I having a ter-
minal amino group possess the formulae IVa, IVb or IVc
below:
9 20~7~
~2 ~ R3
H2N-A-B-NH-CH-CH-CH-R4 IVa
~2 ~R ~3
H2N-B-NH-CH-CH-CH-R4 IVb
~2 ~H ~3
H2N-CH-CH-CH-R4 IVc
Methods which are suitable for the preparation of an
S amide bond are describQd, for example, in Houben-Weyl,
Methoden der organischen Chemie (Methods of Organic
chemistry)~ volume 15/2; Bodanszky et al., Peptide syn-
thesis, 2nd ed. (Wiley & Sons, New York 1976) or Gross,
Meienhofer, The Peptides. Analy is, synthesis, biology
(Academic Press, New York 1979). The following methods
are preferably useds
active ester method using N-hydroxysuccinimide or 1-
hydroxybenzotriazole a8 the ester component, coupling
with a carbodiimide such ns dicyclohexylcarbodiimide or
with propanephosphonic anhydride snd the mixed anhydrlde
method with pivaloyl chloride.
The preparation of the optically active ~mines u~ed as
starting compounds of the for ula IVc
H2N-~H-CH- ~-R4 IVc
in which R2, R3 and R~ are as defined above, is carried
out starting from optically active ~-amino acids, the
asymmetric center of which is retained. For this purpose,
an N-protected amino acid aldehyde is prepared in a known
manner, coupled in an aldol-analogous addition to a
corresponding heteroarylalkyl building block and, after
removing the N-protective group, gives amino alcohols of
lo 2 0 ~
the formula IVc. Diastereomeric mixtures relative to the
OH-carrying center are obtained which can be separated
in a manner known per se, for example by fractional
crystallization or by chromatography. The checking of the
diastereomeric purity iE carried out by means of HPLC,
and the enantiomeric purity can be checked in a known
manner by converting into Mosher derivatives (H.S. Mosher
et al., J. Org. Chem. 34, 2543 (1969)).
The preparation of N-protected amino acid aldehydes is
carried out according to B. Castro et al. (Synthesis
1983, 676).
The aldol-analogou~ addition to N-protected amino acid
aldehydes (preferably N-tert.-butoxycarbonyl and benzyl-
oxycarbonyl protective groups) is carried out in a
solvent inert towards bases, such as ether, THF, toluene,
DMF, DMSO or dimethoxyethane.
Bases which can be used for the deprotonation of the
heteroarylalkyl component are alkali metal alcoholates,
such as potassium O-tert.-butylate, sodium methylate,
alkali metal hydrides, such as sodium hydride or potas-
sium hydride, organometallic bases, such as n-butyl-
lithium, s-butylIithium, methyllithium or phenyllithium,
sodium amide and alkali metal salts of organic nitrogen
bases, such as lithium dii~opropylamide.
The preceding and subsequent operations necessary for the
preparation of compounds of the formula I such as intro-
duction and removal of protective groups are known from
the literature and described, for example, in T.W.
Greene, "Protective Groups in Organic Synthesis n . Salts
of compounds of the formula I with salt-forming groups
are prepared in a manner known per se, for example by
reacting a compound of the formula I having a basic group
with a stoichiometric amount of a suit ble acid.
Mixtures of stereoisomers, in particular mixtures of
11- 20~7~
diastereomers, which are obtained using racemic amino
acids A or B, can be separated by fractional
crystallization or by chromatography in a manner known
per se.
The compounds of the formula I according to the invention
exhibit enzyme-inhibiting properties; in particular they
inhibit the action of the natural enzyme renin. Renin is
a proteolytic enzyme of the aspartylprotease class which,
as a result of various stimuli (volume depletion, sodium
deficiency, ~-receptor stimulation), iB secreted by the
~uxtaglomerular cells of the kidney into the blood
circulation. In the latter it cleaves the decapeptide
angiotensin I from the angiotensinogen liberated from the
liver. Thi~ is converted into angiotensin II by the
Nangiotensin converting enzyme~ (ACE). Angiotensin II
plays an essential role in the regulation of blood
pr~e~sure, since it directly increases the blood pressure
by means of vascular contraction. Additionally, it
stimulates the secretion of aldosterone from the adrenal
gland and in this manner increases the extracellular
fluid volume via the inhibition of sodium excretion,
which for its part contributes to an increase in blood
pressure. Inhibitors of the enzymatic activity of renin
cause a decreased formation of angiotensin I, which has
a decreased formation of angiotensin II as a consequence.
The lowering of the concentration of this active peptide
hormone is the direct cause of the hypotensive action of
renin inhibitors.
The activity of renin inhibitors can be checked by in
vitro tests. In this connection, the decrease in the
formation of sngiotensin I is measured in various systems
(human plasma, purified human renin).
1. Test principle
Human plasma, for example, which contains both renin and
angiotensinogen, is incubated at 37C with the compound
- 12 - 2~
to be tested. In the course of this, angiotensin I iB
released from angiotensinogen under the action of renin
and can subsequently be measured using a commercial
radioimmunoassay. This angiotensin release is inhibited
by renin inhibitors.
2. Obtaining the plas ~
The blood is obtained from volunteer sub~ects (about
0.5 1 per person; Bluko sampling device from ASID Bonz
und Sohn, UnterschleiBheim) and collected in partially
evacuated bottles with ice cooling. The clotting is
prevented by adding EDTA (final concentration 10 mM).
After centrifuging (HS 4 rotor (sorvall)~ 3,500 rpm, O-
4C, 15 min; repeat if necessary), the plasma iB care-
fully pipetted off and frozen in suitable portions at
-30C. Only plasmas having a sufficiently high renin
activity are used for the test. Plasmas having low renin
activity are activated by cold treatment (-4C, 3 days)
(prorenin renin).
3. Carrying out the test
Angiotensin I is determined using the renin Maia kit
(Serono Diagnostics S.A., Coinsins, Switzerland). The
incubation of the plasma is carried out accordinq to the
instructions given theres
Incubation batch: 1000 ~1 of plasma (thawed at 0-4-C)
100 ~1 of phosphate buffer (pH 7.4)
(addition of 10-~ M
ramiprilate)
10 ~1 of PMSF solution
10 ~1 of 0.1 % Genapol PFIC
12 ~1 of DMSO or test preparation
The test preparations are in general dissolved at 10 2
in 100 ~ dimethyl sulfoxide (DNSO) and diluted correspon-
dingly with DMSO; the incubation batch contains a maximum
- 13 - 201~7~
of 1 ~ DMS0.
The batches are mixed in ice and placed in a water bath
(37C) ~or 1 hour for incubation. A total of 6 samples
(in each case 100 ~1) are taken without further incuba-
tion from an additional batch without inhibitor for the
determination of the starting angiotensin I content of
the plasma used.
The concentration of the test preparations are chosen 80
that approximately the range from 10-90 % enzyme inhibi-
tion i8 covered (at least five concentrations). At the
end of the incubation time, three 100 ~1 samples from
each batch are frozen on dry ice in precooled Eppendorf
vessels and stored at about -25C for the angiotensin I
determination (mean value of three individual samples).
Angiotensin I radioi~ unoassay (RIA)
The instructions for use of the RIA kit (renin Maiae kit,
Serono Diagnostics S.A., Coinsins, Switzerland) are
followed exactly.
The calibration curve include6 the region from 0.2 to
25.0 ng of àngiotensin I per ml. The base angiotensin I
content of the plasma is taken away from all measured
values. The plasma renin activity (PRA) is given as ng of
Ang I/ml x hour. PRA values in the presence of the test
substances are based on a batch without inhibitor
(= 100 %) and given as % residual activity. The IC50 value
is read off on the plot of ~ residual activity against
the concentration (M) of the test preparation (logarith-
mic scale).
The compounds of the general formula I described in the
pre~ent invention show inhibitory actions at concentra-
tions of about 10-5 to 10-1 mol/l in the in vitro test.
Renin inhibitors cause a lowering of blood pressure in
- 14 - 2015~7~
salt-depleted animals. Since human renin differs from the
- renin of other species, primates (marmosets, rhesus
monkeys) are used for the in vivo test of renin inhibi-
tors. Primates' renin and human renin are substantially
homologous in their sequence. An endogenous effusion of
renin is stimulated by i.v. in~ection of furosemide. The
test compounds are then administered and their action on
blood pressure and heart rate is measured. The compounds
of the present invention are in this case active in a
dose range from about 0.1 - 5 mg/kg i.v., and on intra-
duodenal administration by ga~troscope in the dose range
from about 1-50 mg/kg. The compound~ of the general
formula I described in the present invention can be used
as antihypertensives and also for the treatment of
cardiac insufficiency.
The HIV protease is excised autocatalytically from the
GAG-POL polypeptide and then cleaves the precursor
peptide p55 in the core antigens pl7, p24 and pl4. It is
thus an essential enzyme, whose inhibition interrupts the
life cycle of the virus and suppresses its reprocation.
In biological tests, it has been shown that the compounds
according to the invention have enzyme-inhibitory action
and also inhibit viral enzymes such as HIV protease. The
HIV protease-inhibiting action has a particular impor-
tance, which qualifies the compounds according to theinvention in particular for the therapy and prophylaxis
of disorders caused by infection with HIV. The compounds
of the general formula I according to the invention show
inhibitory actions at concentrations of about 10-~ to
10-~ mol/l in the in vitro tests used.
The invention furthermore relates to the use of compounds
of the formula I for the preparation of medicaments for
the therapy of high blood pressure and the treatment of
congestive cardiac insufficiency and for the therapy and
prophylaxis of viral disorders, in particular disorders
which are caused by HIV, and also to the medicaments
_ 15 - 2 ~1~ O l ~
mentioned.
Pharmaceutical preparations contain an effective amount
of the active compound of the formula I together with an
inorganic or organic pharmaceutically utilizable exci-
pient. Administration can be carried out intranasally,intravenously, subcutaneously or perorally. The dosage of
the active compounds depends on the warm-blooded animal
species, the body weight, the age and the manner of
administration.
The pharmaceutical preparations of the present invention
are produced in dissolving, mixing, granulating or
tablet-coating processes known per se.
For a form for oral administration, the active compounds
are mixed with the additives customary therefor such as
excipients, stabilizers or inert diluents and, by custo-
mary methods, brought into suitable forms for
administration, such as tablets, coated tablets, hard
gelatin capsules, aqueous, alcoholic or oily suspensions
or aqueous, alcoholic or oily solutions. Inert excipients
which can be used are, for example, gum arabic, magnesia,
magnesium carbonate, potassium pho~phate, lactose,
glucose, magnesium stearylfumarate or starch, in parti-
cular maize starch. The preparation can be carried out
both as dry and moist granules here. Suitable oily
excipients or solvents are, for example, vegetable or
animal oils, such as sunflower oil and cod liver oil.
For subcutaneous or intravenous administration, the
active compounds or their physiologically tolerable salts
are brought into solutions, ~uspensions or emulsion~, if
desired with the substances customary therefor such as
solubilizers, emulsifiers or other auxiliaries. Possible
solvents are, for example: water, physiological saline
solutions or alcohols, for example ethanol, propanediol
or glycerol, and in addition also sugar solutions such as
glucose or mannitol solutions, or else a mixture of the
- 16 - 20~507~
different ~olvents mentioned.
List of th~ abbreviations used:
ACC 4-aminocyclohexylcarbonyl
Boc tert.-butoxycarbonyl
BuLi n-butyllithium
DCC dicyclohexylcarbodiimide
DCI desorption chemical ionization
DNP 2,4-dinitrophenyl
DME dimethoxyethane
DMF dimethylformamide
EA ethyl acetate
EI electron impact
FAB fa~t atom bombardment
h hour
HOBt 1-hydroxybenzotriazole
M molecular peak
MS mass spectrum
MTBE methyl tert.butyl ether
NEM N-ethylmorpholine
20 n-PropPA n-propylphosphonic anhydride
THF tetrahydrofuran
The other abbreviations used for amino acid~ correspond
to the three letter code customary in peptide chemistry
as is described, for example, in Eur. J. Biochem. 138, 9-
37 (1984). If not expressly stated otherwise, the amino
acids are of the L-configuration.
The eluents used are abbreviated as followss
Ell CH2Cl2/CH30H 20sl
E12 CH2Cl2/CH30H 9sl
E13 CH2Cl2/CH30H 12sl
E14 CH2Cl2/CH30H/conc. NH3 10:1:0.1
E15 cyclohexane/ethyl acetate 9:1
E16 n-hexane/ethyl acetate 2:1
20~5~7~
- 17 -
The examples below serve to illustrate the present
invention without limiting it thereto.
Fxn ple 1
N-(N-Tert-butoxycarbonyl-cis-4-aminocyclohexylcarbonyl)-
L-phenylalanine-L-norvaline-(l-S-cyclohexylmethyl-2-S-
hydroxy-5-(2-pyridyl))pentylamide
98 mg of HOBt, 130 mg of DCC and 0.08 ml of NEM were
added to 235 mg of N-(N-tert-butoxycarbonyl-cis-4-amino-
cyclohexylcarbonyl)-L-phenylalanine diesolved in 3 ml of
DMF. 230 mg of L-norvaline-(l-S-cyclohexylmethyl-2-S-
hydroxy-5-(2-pyridyl))-pentylamide, dissolved in 3 ml of
DMF, were added to this solution and the mixture was
stirred overnight at RT. The DMF was distilled off in a
high vacuum, the solution remaininq was taken up in ethyl
acetate, precipitated urea was filtered off and the
filtrate was washed twice with satd. NaHCO3 and satd. NaCl
solution, and the organic phase was dried over NgSO~ and
concentrated in vacuo. 315 mg were isolated by column
chromatography (silica gel, Ell).
Melting point 100-103-C
MS (FAB) 748 (M+l)
a) N-(N-Tert-butoxycarbonyl-cis-4-aminocyclohexylcar-
bonyl)-L-phenylalanine
5.5 g of N-(N-tert-butoxycarbonyl-cis-4-aminocyclohexyl-
carbonyl) -L-phenylalanine benzyl ester were hydrogenated
in 230 ml of ethanol over 1 g of Pd/carbon under normal
pressure. After completion of the reaction, the cataly~t
was filtered off and the solvent was removed by distilla-
tion. 4.1 g of colorless product were obtained after
recrystallization from n-heptane/ethyl acetate.
Melting point 160-161C (decomposition)
MS (DCI) 391 (M+l)
_ 18 - 201~07~
b) N-(N-Tert-butoxycarbonyl-cis-4-aminocyclohexylcar-
bonyl)-L-phenylalanine benzyl ester
6.0 g of N-tert-butoxycarbonyl-cis-1,4-aminocyclohexane-
carboxylic acid and 6.3 g of L-phenylalanine benzyl ester
were dissolved in 75 ml of DMF and mixed with 24 ml of n-
prop PA and 15.7 ml of NEM at 0C and the mixture was
made to react overnight at RT. The solution was diluted
with CH2C12 and washed in each case with semi~aturated
NaHC03 solution, 10 ~ strength citric acid and water,
dried over NgS0~ and concentrated in vacuo. Flash chroma-
tography gave 5.7 g of pure prcduct.
: -14.6 (c = 1.1 in CH30H)
B~a ple 2
N-(cis-4-Aminocyclohexylcarbonyl)-L-phenylalanine-L-
norvaline-(1-S-cyclohexylmethyl-2-S-hydroxy-5-(2-pyri-
dyl))-pentylamide
77.7 mg of the compound described in Example 1 were
dissolved in 2 ml of CH2Clz and 2 ml of CF3COOH were added
at 0C under N2. The solution was concentrated after
30 min, the residue was taken up in ethyl acetate and
washed twice each time with satd. NaHC03 and NaCl solu-
tion.
Column chromatography (silica gel, ~11) gave 24.7 mg of
product.
NS (FAB) 648 (M+l)
19 -- 2 ~
X~ample 3
N-(N-Tert~butoxycarbonyl-~xans-4-aminocyclohexylcar-
bonyl)-L-phenylalanine-L-norvaline-(l-S-cyclohexylmethyl-
2-S-hydroxy-5-l2-pyridyl))pentylamide
Analogously to thP process described in Example 1, the
title compound was prepared from N-tN-tert-bu~oxycar-
bonyl-trans-4-aminocyclohexylcarbonyl)-L-phenylalanine
snd L-norvaline-~l-S-cyclQhexylmethyl-2-S-hydroxy 5-(2-
pyridyl))pentylamide. The yield was 270 mg.
Melting point: 219-220~C.
M5 lFAB) 748 (M+1)
a) N-(N-Tert-butoxycarbonyl-trans-4-aminocyclohexylcar
bonyl)-L-phenylalanine
4.45 g of N-~N-tert-butoxycarbonyl-tran~-4-aminocyclo-
hexylcarbonyl)-L-phenylalanine benzyl ester were cataly-
tically hydrogenated under normal pressuxe over 900 mg of
palladium-carbon in 200 ml of ethanol at RT. After
completion of the reaction, the catalyst was filtered off
and the solvent was removed by distillation in vacuo.
2.67 g of pure product were obtained after
recrystallization.
Melting point: decomposition from 160C
MS (DCI): 391 (M+)
b) N-(N-Tert-butoxycarbonyl-trans 4-aminocyclohexylcar-
bonyl)-L-phenylalanine benzyl ester
3 g of N-tert-butoxycarbonyl-trans-4-aminocyclohexane-
carboxylic acid and 3.2 g of L-phenylalanine benzyl e~ter
were di~solYed in 40 ml of DMF and 7.85 ml of NEM and
then 12 ml of n-propPA were added at 0C. The mixture was
made to react overnight at RT. After diluting with 200 ml
of ethyl acetate, the Eolution wa6 washed twice each with
H2O, 10 % strength citri.c acid, satd. NaHCO3 and satd.
NaCl solution. The remaining organic phase was dried
using MgSO4 and freed from the solvent in vacuo. ~he crude
- 20 - 2 0 ~ ~ ~7 ~
product was triturated with diethyl ether, and the
precipitated crystals were filtered off with suction and
dried. 4.45 g of colorless crystals were obtained.
Melting point: 176-177-C
MS (DCI) 481 (M+l)
~a ple 4
N-(N-trans-4-Aminocyclohexylcarbonyl)-L-phenylalanine-L-
norvaline-(l-S-cyclohexylmethyl-2-S-hydroxy-5-(2-pyri-
dyl))-pentylamide
The title compound was prepared by the proces6 described
in Example 2 from the compound de6cribed in Example 3.
Yield after chromatography (silica gel, E12) 27 mg.
MS (FAB) 648 (M+l)
RF 0,17 (CH2C12/CH3OH 8:2)
Exa ple 5
N-(N-Tert-butoxycarbonyl-tran~-4-aminocyclohexylcar-
bonyl)-L-phenylalanine-N(im)-trityl-L-histidine-(l-S-
cyclohexylmethyl-2-S-hydroxy-5-(2-pyridyl))pentylamide
The title compound was prepared from the phenylalanine
derivative described in Example 3a) and from FMOC-trityl-
His-(1-S-cyclohexylmethyl-2-S-hydroxy-5-(2-pyridyl))-
pentylamide by the process described in Example 1.
Yield after chromatography (silica gel, Ell) 0.4 g
MS (PAB) 1028 (M+l), 1034 (M+7).
RF 0~5 (toluene/ethanol 8:2)
- 21 -
Example 6
N-~N-Tert-butoxycarbonyl-trans-4-aminocyclohexylcar-
bonyl)-L-phenylalanine-L-histidine-(1-S-cyclohexylmethyl-
2-S-hydroxy-5-(2-pyridyl))pentylamide
40 mg of the substance obtained in Example 5 were dis-
solved in 5 ml of 90 % strength acetic acid and the
mixture was heated to 60-C for two hours. The solution
was ad~usted to pH 8 using Na2CO3 and extracted three
times using ethyl acetate. The organic phase was washed
twice each with satd. NaHCO3 solution and with satd. NaCl
solution, then dried over MgSO~ and concentrated in vacuo.
Chromatography (silica gel, Ell) gave 16 mg of pure
product. 155C decomposition
MS (FAB) 786 (M+l)
Esa ple 7
N-(trans-4-Aminocyclohexylcarbonyl)-L-phenylalanine-L-
histidine-(l-S-cyclohexylmethyl-2-S-hydroxy-5-(2-pyri-
dyl))pentylamide
300 mg of the substance described in Example 5 were
stirred for 15 min at O-C and for 2 h at RT with 5 ml of
CF3COOH and 0.25 ml of H20. The solution was concentrated
in vacuo, and the residue was rendered alkaline with
NaHCO3 solution and extracted using ethyl acetate. The
organic phase WaB concentrated in vacuo and the residue
was chromatographed (~ilica gel, E12). Melting point 114-116C.
MS (FAB) 686 (M+l)
~x~ ple 8
N-(N-Tert-butoxycarbonyl-cis-4-aminocyclohexylcarbonyl)-
L-phenylalanine-N(im)-trityl-L-histidine-(l-S-cyclohexyl-
methyl-2-S-hydroxy-5-(2-pyridyl))pentylamide
The title compound was prepared from the phenylalanine
. - 22 - 2~
derivative described in Example la) and from FMOC-trityl-
His-(l-S-cyclohexylmethyl-2-S-hydroxy-5-(2-pyridyl)-
- pentylamide by the process described in Example 1. Yield
after chromatography (silica gel, Ell) 0.5 g.
MS (FAB) 1028 (M+l), 1034 (M+7) RF 0,26 (El 1)
~xa ple g
N-(N-Tert-butoxycarbonyl-cis-4-aminocyclohexylcarbonyl)-
L-phenylalanine-L-histidine-(l-S-cyclohexylmethyl-2-S-
hydroxy-5-(2-pyridyl))pentylamide
The title compound was prepared by the process described
in Example 6 from the compound described in Example 8.
MS (FAB) 786 (M~l)
Yield: 61 mg. Melting point 127-130C. RF 0,21 (El 2)
Es~mple 10
N-(cis-4-Aminocyclohexylcarbonyl)-L-phenylalanine-L-
histidine-(1-S-cyclohexylmethyl-2-S-hydroxy-5-(2-pyri-
dyl))pentylamide
The title compound was prepared by the process described
in Example 7 from the compound described in Example 8.
Yield 180 mg. Melting point 177C(decomp.)
MS (FA~) 686 (M+l)
Bs~ ple 11
N-(N-Tert-butoxycarbonyl-trans-4-aminocyclohexylcar-
bonyl)-O-methyl-L-tyrosine-N-(im)-trityl-L-histidine-(l-
S-cyclohexylmethyl-2-S-hydroxy-5-(2-pyridyl))pentylamide
The title compound was prepared from N-(N-tert-butoxycar-
bonyl-trans-4-aminocyclohexylcarbonyl)-methyl-L-tyrosine
and trityl-L-His-(l-S-cyclohexylmethyl-2-S-hydroxy-5-(2-
pyridyl))pentylamide by the process described in Example
l. R 0,54 (toluene/ethanol 8:2)
MS (~AB) 1058 (M+l)
~ 2~ _ 2~
~ampl~ 12
N-(N-Tert-butoxycarbonyl-trans~4-aminocyclohexylcar-
bonyl)-0-methyl-L-tyrosine-L-histidine (l-S-cyclohexyl-
methyl-2-S-hydroxy-5-(2-pyridyl))pen~ylamide
The title compound was prepared by the process described
in Example 6 from the compo~nd described in Example 11.
MS (FA~) 816 tM+l) 180C decomposition, RF 0~2 (toluene/
ethanol 8:2)
Example 13
N-(trans-4-Aminocyclohexylcarbonyl)-0-methyl-L-tyrosine-
L-histidine (l-S-cyclohexylmethyl-2-S-hydroxy-5-(2-
pyridyl))pentylamide
The title compound was prepared by the process described
in Example 7 from the compound de~cribed in Example 11.
MS (FAL) 716 ~M+l)
E~ample 14
N-(N Tert-butoxycarbonyl-ci~-4-aminocyclohexylcarbonyl)-
0-methyl-L-tyrosine-N-(im)-trityl-L-histidine (l-S-cyclo-
hexylmethyl-2-S-hydroxy-5-(2-pyridyl))pentylamide
The title compound Wa8 prepared from N-(N-tert-butoxycar-
bonyl-cis-4-aminocyclohexylcarbonyl~-0-methyl-L-tyrosine
and trityl-L-Hi~-(l-S-cyclohexyLmethyl-2~S-hydroxy-5-(2-
pyridyl))pentylamide by the proces~ described in Example
1. RF 0,75 (El 2).
MS (FAB) 105~ (M~l)
- 24 - 201507~
B~aple 15
N-(N-Tert-butoxycarbonyl-cis-4-aminocyclohexylcarbonyl)-
O-methyl-L-tyrosine-L-histidine (1-S-cyclohexylmethyl-2-
S-hydroxy-5-(2-pyridyl))pentylamide
The title compound wa8 prepared by the process described
in Example 6 from the compound described in Example 14.
MS (FAB) 816 (N+l) RF 0,80 (El 4)
~ ple 16
N-(cis-4-Aminocyclohexylcarbonyl)-O-methyl-L-tyrosine-L-
histidine (1-S-cyclohexylmethyl-2-S-hydroxy-5-(2-
pyridyl))pentylamide
The title compound was prepared by the process described
in Example 7 from the compound described in Example 14.
MS (FAB) 716 (M+l) RF 0~50 (El 4)
~ pl~ 17
N-(N-(N-(eis-4-Tert.butoxycarbonyl~mino-cyclohexylcar-
bonylphenylalaninyl)-h$~tidinyl)-2-(5-S-amino-6-cyclo-
hexyl-3S,4R-dihydroxy-n-hexyl)-pyridine
60 mg of the compound from Example 17 a) are stirred with
40 mg of thiophenol in 2 ml of acetonitrile for 2 h.
After concentrating, the residue is chromatographed
(silica gel, E14) and 45 mg are obtained as an amorphous
powder.
MS (FAB) 802 (M+l)
- 25 - 2 0 ~ ~ a ~ ~
a) Boc-cis-ACC-Phe-DNP-His-2-(5-amino-6-cyclohexyl-
3S,4R-dihydroxy-n-hexyl)pyridine
60 mg of BOC-DNP-His-(3S,4R,5S)-2-(5-amino-6-cyclohexyl-
3,4-dihydroxyhexyl)pyridine are stirred in 5 ml of
DME/HCl for 2 h. After concentrating, the crude product
is dissolved in 3 ml of DMF together with 50 mg of N-(N-
tert.butoxycarbonyl-cis-4-aminocyclohexylcarbonyl)-L-
phenylalanine, 120 mg of DCC and 80 mg of HOBt. The
solution is ad~usted to pH 9 using NEM and stirred for
16 h. The solution is filtered, diluted with ethyl
acetate and washed once each with 3 % strength NaHCO3
solution, H20 and satd. NaCl solution, dried using MgSO~,
concentrated and chromatographed on silica gel (E13).
60 mg of the title compound are obtained as a yellow
foam.
MS(FAB) 968 (N+l)
b) BOC-DNP-His-(3S,4R,5S)-2-(5-amino-6-cyclohexyl-3,4-
dihydroxyhexyl)pyridine
0.5 mmol of the (3S,4R,5S)isomer from Example 17 c are
stirred with 5 ml of HC~ in DME (saturated) for 2 h.
After concentrating in vacuo, the re6idue is dissolved in
3 ml of absolute DMF. O.5 mmol each of BOC-DNP-His-OH,
dicyclohexylcarbodiimide and HOBt are added. The solution
is ad~usted to pH 9 using NEM and stirred for 24 h. After
filtration, it is diluted with EA and washed with each 3
% strength NaKCO3 solution, dried using MgSO~ and
concentrated. Chromatography on silica gel (Ell) gives
the title compound a8 a yellow resin.
NS (FAB) 696 (N+l)
c) 2-((3S,4R,5S)-5-tert.butoxycarbonylamino-3,4-di-
hydroxy-6-cyclohexyl-n-hexyl)pyridine
1.4 ml (1 mmol) of n-BuLi are added at -78C to 93 mg
(1 mmol) of 2-picoline in 10 ml of THF. After warming to
RT, the mixture is stirred for 30 min, then cooled to
- 26 - 2Q~
-40'C. 1 mmol of (2RS,3R,4S)-3-tert.-butyldimethylsilyl-
oxy-4-tert.-butoxycarbonylamino-5-cyclohexyl-1,2-oxopen-
- tane (known from EP-A 189,203, Example 6) are added
(dissolved in 5 ml of THF). After 10 hours at RT, the
mixture is diluted with water and extracted using MTBE.
The crude product (0.4 g) is dissolved in THF and stirred
at 0-C with 5 ml of a lM solution of tetrabutylammonium
fluoride in THF for 1 h. After diluting with water and
extracting with EA, 0.15 q of the (3S,4R,5S)-isomer (MS
(FAB)s 391 (M+1) and 0.12 g of the (3S,4S,5S)-isomer (NS
(FAB): 391 (M+1) are obtained.
Fxample 18
cis-4-Aminocyclohexylcarbonyl-L-Phe-L-His-(1-S-cyclo-
hexylmethyl-2R,3S-dihydroxy-5-(2-pyridyl))-n-pentylamide
tristrifluoroacetate
25 mg of the compound described in Example 17 are stirred
in 1 ml of TFA/1 ml of CH2Cl2 for 2 h. After concentrat-
ing, the residue is taken up in H2O and lyophilized. 20 mg
of product are obtained as a colorless powder.
MS (FAB) 702 (M+l)
~sa ple 19
BOC-trans-ACC-L-Phe-L-Hi~-(lS-cyclohexylmethyl-2R,3S-
dihydroxy-5-(2-pyridyl))-n-pentylamide
The title compound was prepared by the process described
in Example 17 from the compound described in Example l9a.
MS (FAB) 802 (M+l)yield 79,5 mg. RF 0,19 (El 2)
a) BOC-trans-ACC-L-Phe-L-DNP-His-(lS-cyclohexylmethyl-
2R,3S-dihydroxy-5-(2-pyridyl))-n-pentylamide
The title compound was prepared by the process described
~s~3'~;
- 27 -
in Example 17 a) from the compound~ de~cribed in Examples
3 a) and 19 b). RF 0~40 (El 2)
MS ~FAB) g68 (M+1)
b) DNP H-His-(3S,4R,5S)-2-(5-amino-6-cyclohexyl-3,4-
dihydroxy-n-hexyl)-pyridine hydrochloride
400 mg of the compound from ~xampl2 17 b) are stirred in
10 ml of DME/HCl (satd.) for 2 h. Af~er concentrating,
the residue is taken up in toluene twice and in each ca~e
concentrated ayainr The title compound i8 obtained as a
yellow foam.
MS (FAB) 596 (M+l)
~ample 20
trans-ACC-L-Phe-L-His-(lS-cyclohexylmethyl-2R,3S-di-
hydroxy-5-(2-pyridyl))-n-pentylamide
The title compound was prepared by the proces~ described
in Example 18 from the compound de~cribed in Example 19.
MS (FAB) 702 (~+1) Yield: 49 mg
~ampl~ 21
BOC-trans-ACC'-O-methyl-L-Tyr-L-His-(lS-cyclohexylmethyl-
2R,3S-dihydroxy-5-[2-pyridyl))-n-pentylamide
The title compound was prepared by the proces~ described
in Example 17 from the compound described in ~xample 21
a~.
MS (FAB) 832 ~M~l)
5 a) BOC trans-ACC-O-methyl-L-Tyr-DNP L-Hi~-(lS-cyclo-
hexylmethyl-2R~3S dihydroxy-5-(2-pyridyl~)-n-pentyl-
amide
The title compound wae prepared by the proce~s de~cribed
in Example 17 a) fro~ the compound~ de~cribed in ~xamples
- 28 - 2 ~ ~ ~ S~ ~
21 b) and 19 b).
MS (FAB) 998 (M+l)
b) BOC-trans-ACC-Tyr(OCH3)-OH
0.05 mol of BOC-trans-ACC-Tyr(OCH3)-OCH3 are dissolved in
250 ml of DNE and the mixture is stirred with an equi-
molar amount of lN NaOH solution for 5 h. The solution is
then concentrated and the residue is taken up with water,
acidified to pH 4 using RHSO4 and extracted with EA. The
crude product is recry~tallized from n-heptane/EA snd the
title compound is obtained as a white powder.
NS (DCI) 421 (M+l) Melting point: 181C(decomp.)
RF 0,20 (toluene/ethanol ~:2)
c) BOC-trans-ACC-Tyr(OCH3)-OCH3
0.1 mol of BOC-trans-ACC-OH and 0.1 mol of L-tyrosine-
(OCH3) methyl ester are dissolved in 500 ml of DNF and
the mixture is stirred at O-C under N2 with 100 ml of n-
propPA (50 % strength in CH2Cl2) and 0.5 mol of triethyl-
amine. The solution is made to react overnight. After
completion of the reaction (TLC checking)~ the solvent i8
removed by distillation in a high vacuum and the residue
is taken up in EA. The organic phase is washed twice each
with 10 % strength citric acid, water, semisaturated
NaHCO3 solution and satd. NaCl solution, dried over NgSO~
and concentrated in vacuo, and the residue is
chromatographed (silica gel, E15).
Melting point: 150-155 C.
MS (DCI) 435 (N+1)
E~ ple 22
.
trans-ACC-O-methyl-L-Tyr-L-His-(lS-cyclohexylmethyl-
2R,3S-dihydroxy-5-(2-pyridyl))-n-pentylamide
The title compound was prepared by the process described
in Example 18 from the compound described in Example 21.
MS (FAB) 732 (M+1)
20~ ~Q7~
2g
Bxa ple 23
Boc-cis-Acc-o-methyl-L-Tyr-L-His-(ls-cyclohexylmeth
2R,3S-dihydroxy-5-(2-pyridyl))-n-pentylamide
The title compound was prepared by the proce~s described
in Example 17 from the compound described in Example
23 a). RF 0,21 (El 2)
MS (FAB) 832 (M+1)
a) BOC-cis-ACC-O-methyl-L-Tyr-DNP-L-His-(lS-cyclo-
hexylmethyl-2R,3S-dihydroxy-5-(2-pyridyl))-n-pentyl-
amide
The title compound was prepared by the process describedin Example 17 a) from the compounds described in Examples
23 b) and 19 b).
MS (FAB) 998 (M+l)
b) BOC-cis-ACC-Tyr(OCH3)-OH
The title compound is obtained analogously to the process
Example 21 b). d25= + 18,6 (c=1, CH30H)
MS (DCI) 421 (M+l)
c) BOC-cis-ACC-Tyr(OCH3) methyl ester
The title compound was prepared analogously to the
process in Example 21 c).
MS (DCI) 435 (M+l)
~x~ ple 24
cis-ACC-O-mothyl-L-Tyr-L-Hi~-(lS-cyclohexylmethyl-2R,3S-
2S dihydroxy-5-(2-pyridyl))-n-pentylamide
The title compound was prepared by the process described
in Example 18 from the compound described in Example 23.
~ 30 - 20~7~
MS (FA~) ~32 (M+1) RF 0,56 (El 4)
~xa ple 25
BOC-cis-ACC-Phe-His-(lS-cyclohexylmethyl-2S-hydroxy-5-(N-
propyl-2-imidazolyl))pentylamide
The title compound was prepared by the process described
in Example 17 from the compound described in Example 25.
MS (FAB) 817 (M+l)
a) BOC-cis-ACC-Phe-DNP-His-(lS-cyclohexylmethyl-2S-
hydroxy-5-(N-propyl-2-imidazolyl))pentylamide
80 mg of the compound from Example b are stirred with
4 ml of HCl in DME (satd.) for 2 h. After concentrating
in vacuo, the residue is dissolved in 3 ml of absolute
DMF. 44 mg of BOC-cis-ACC-Phe-OH and 0.11 mmol each of
DCC and HOBt are added. The solution is ad~usted to pH 9
using NEN snd ~tirred for 24 h. After filtering, it i~
diluted with EA and washed twice each with 3 % strength
NaHCO3 solution, H20 and satd. NaCl solution, dried using
MgSO~ and concentrated. Chromatography on silica gel gives
the title compound as a yellow resin.
MS (FAB) 9B3 (M+l)
b~ BOC-DNP-His-(lS-cyclohexylmethyl-2S-hydroxy-5-(N-
propyl-2-imidazolyl))pentylamide
100 mg of the compound from Example c are stirred with
5 ml of HCl in DNE (satd.) for 2 h. After concentrating
in vacuo, the residue is dissolved in 3 ml of absolute
DMF. 105 mg o$ BOC-DNP-His-OH and O.3 mmol each of DCC
and HOBt are added. The solution i8 ad~usted to pH 9
using NEM and ~tirred for 24 h. After filtration, it is
diluted with EA and washed twice each with 3 % strength
NaHCO3 solution, H20 and satd. NaCl solution, dried using
MgSO~ and concentrated. Chromatography on silica gel gives
the title compound as a yellow resin.
- 31 - 2~ ~ ~ 07 ~
MS (FAB) 711 (M+l)
c) 3-BOC-4S-cyclohexylmethyl-2,2-dimethyl-5-(3-(2-N-
propylimidazolyl)-propyl)oxazolidine
500 mg of the compound from Example d are dissolved in
5 ml of absolute THF. Under an argon atmosphere, 1.9 ml
of a 1.5 molar solution of n-butyllithium in n-hexane are
added at -60-C. After 15 min, 500 mg of the compound from
Example e dissolved in 5 ml of absolute rn~ are added.
After 1 h at ~60C, 10 ml of a satd. NaHCO3 solution are
added. After warming to RT, the mixture iB extracted
three times with EA, dried using Na2SO4 and concentrated.
The title compound is obtained by chromatography on
silica gel (El EA).
R(f) 0.3 (EA); MS (PAB) M+l)
d) 2-Methyl-N-n-propylimidazole
10 g of 2-methylimidazole are heated to boiling in 40 ml
of n-propylbromide for 2 h. The solution i8 then poured
into 150 ml of a 5 % strength NaHSO~ solution, and the
mixture is extracted three times using EA, dried using
Na2SO4 and concentrated. The residue i8 taken up in ether
and separated from insoluble starting material by filtra-
tion. In this manner, the title compound is obtained as
a colorless oil.
R(f) 0.2 (EA/CH30H lOsl)
e) 3-Boc-cyclohexylmethyl-2,2-dimethyl-5-(2-bromo-
ethyl)oxazolidine
1.6 ml of diathyl azodicsrboxylate are added dropwise at
20-C under argon to 690 mg of 3-BOC-lS-cyclohexylmethyl-
2,2-dimethyl-5-(2-hydroxyethyl)oxazolidine, 2.6 g of
triphenylphosphine and 1.6 g of pyridinium bromide in
15 ml of CH2Cl2. After 16 h at RT, water is added and the
mixture i8 diluted using 100 ml of CH2C12. The organic
phase is washed twice with satd. NaHCO3 solution and once
20~507~
- 32 -
with NaCl solution. The organic phase dried with Na2SO4 i~
concentrated, and the residue is taken up in a little EA
and filtered to separate PPh3. Purification on silica gel
yields the title compound (E16).
R~f) 0.3 (E16); NS 404 (M+l).
f) 3-BOC-4S-cyclohexylmethyl-2,2-dimethyl-5-(2-hydroxy-
ethyl~oxazolidine
10 g of BOC-ACHPA-OC2H5 (preparation according to J. Ned.
Chem. 28, 1779 (1985)), 500 mg of p-toluenesulfonic acid
and 7.2 ml of dimethoxypropane are heated at 80-C in
160 ml of toluene under argon for 2 h. The mixture i8
then concentrated. The residue i~ added dropwise at 0-C
to a suspension of 2 g of LiAlH4 in 200 ml of THF. After
2.5 h at 0-C, 100 ml of 5 ~ strength NaHSO~ solution are
added and the mixture is extracted three times with EA.
The combined organic phases are washed with satd. NaHCO3
solution. After drying using Na2SO~, the mixture is
concentrated and chromatographed on silica gel (E16).
R(f) 0.1 (E16); MS (DCI) 342 (M+l).
E~ample 26
cis-ACC-Phe-His-(lS-cyclohexylmethyl-2S-hydroxy-5-(N-
propyl-2-imidazolyl))pentylamide
The title compound was prepared by the method de~cribed
in Example 18 from the compound in Example 25.
NS (FAB) 717 (N+l)