Note: Descriptions are shown in the official language in which they were submitted.
2 ~ 7 ,~
--1
MELT POLYMERIZATION PROCESS FOR MAKING POLYURETHANES
This invention relates to polyurethanes and is
more particularly concerned with a process for the
preparation of high f]exural modulus polyurethane
plastics by reactive extrusion.
U.S. Patents 4,376,834, 4,567,236, and
4,822,827 introduced novel classes of both thermoset and
thermoplastic polyurethane polymers to the plastics
molding art. These materials are characterized by high
impact resistance, stiffness, and other structural
strength properties similar to nylon and other engi-
neering thermoplastics. Additionally, some members of
thi~ group of new materials have exceptionally high Tg
values, as high as 165C. The advent of these materials
has provided the molding industry with excellent alter-
native engineering thermoplastic material choices.
Notably, some of these polyurethanes are thermoplastic
and contain little or no soft segment because of either
the very minor amount, or, complete absence, of high
molecular weight polyol in their formulations. Because
of their thermopl~etlc nature and, similarly, to the
well known softer thermoplastic polyurethanes, these
materials lend themselves to manufacture by the reactive
37,052-F _l_
2 ~ 7
extrusion process. This method is preferable because of
its economic advantages in terms of simply adding the
necessa~y ingredients into a sc~ew ~ruder in the
absence of solvents and continuously, but rapidly,
producing a finished product in the form of desirable
profi~e~ or pellets. Such continuous methods are
preferably carried out in multi-shaft screw extruders,
more preferably twin-screw extruders.
U.S. Patent 3,642,964 and its German counter-
part DE 2059570 were the first disclosures to the
continuous reactive extrusion of soft thermoplastic
polyurethanes in twin-screw extruders. This teaching
called for high shear mixing zones in the extruder
barrel with each zone having a series of kneading blocks
which can have various configurations. More than one
such mixing zone or series of blocks is employed on each
of the twin-screw shafts. The plurality of mixing zones
are either separated by conveying screws or else
connected as one long multi-zone depending on the
manufacturer's design of the extruder. It will be
obvious to one skilled in the art that such extruders
must also be equipped with the conventional conveyor
screws both in respect of a feed zone and metering zone
both before and/or after the mixing zones. Additional
types of screws may be employed such as, for example,
those which cause backward flow in order to keep mixing
zones filled. This process works admirably with soft
thermoplastic polyurethanes falling in the hardness
range of 60 Shore A to 75 Shore D~
U.S. Patent 3,963,679 dlsc'o3es a proces3 quite
similar to the one described above in employing the
extrusion apparatus set forth in Figure 1 of its
37,052-F -2-
~ ~ ~q ~
--3--
disclosure. Primarily, this method differs from that of
U.S. Patent ~,642,964 supra in requiring that the poly-
urethane forming mixture be subjected to its first hi~h
shear mixing during the time it has a viscosity of
10,000 to 100,000 centipoise (lO to lO0 Pa-s). This
limitation is paramount to the invention disclosed
because when only a single kneading zone is employed
after the viscosity has surpassed the upper viscosity
limit (Example lb, column 14) the product was
inhomogeneous. Similarly, when two zones are employed
(Example lc, lines 28 to 38) the product polyurethane is
inhomogeneous. Delaying the reaction until the first
zone is reached re~ults in the process of the invention
as described loccit, lines 40 to 45. This reference
inherently calls for more than one zone of high shear
mixing with the critical limitation being the placement
of the first zone to meet the reaction mixture viscosity
range set forth above.
Following the above art, there are a number of
disclosures to the reactive extrusion method for pre-
paring thermoplastic polyurethanes. U.S. Patent
4,245,081 discloses the continuous preparation using the
procedures described in U.S. Patents 3, 642,964 and
3,963,679 discussed supra with the inventive step being
the use of a mixture of glycol extenders. U.S. Patents
4,261, 946 and 4,342,847 each disclose essentially the
same process differing only in the component
30 proportions. The former is directed to modifying 70 to
98 parts of a thermoplastic polymer with 2 to 30 parts
of polyurethane forming components. The procedure is
accomplished by adding the thermoplastic polymer,
inclusive of preformed polyurethane, and the poly-
urethane forming ingredients to the first and second
37 ~ 052-F -3-
2 ~ 7 ~
--4--
inlets respectively of an extruder. The polyurethane
formed insltu in the extruder is inclusive of both those
with an~ w out soft segments. The latter has the
preformed thermoplastic polymer at 4 to 65 parts with
the polyurethane forming components at 35 to 96 parts.
Both patents disclose the use of the same twln-screw
technology described in
U.S. Patent 3,963,679 supra.
1~ U.S. Patent 4,595,709 discloses a method for
converting toluene diisocyanate distillation residues to
useful polyurethane polyaddition products containing
urethane groups by continuously reacting the residues
with low molecular weight compounds containing hydroxyl
groups. The reaction is carried out in multiple-screw
extruders using the extrusion technology of U.S. Patent
3,963,679 cited supra.
Now the new classes of polyurethanes set forth
in the three patents cited supra can be prepared using
the twin-screw extruder technology of the prior art
discussed aboveO However, there are problem~ encoun-
tered which appear to be unique to these new high melt-
ing materials. One problem is a tendency for gaseousformation or the appearance of small bubbles in the
extruded polymer. Another problem is the formation of
yellow coloration in what should be an essentially water
white polymer. Part of the problem lies with their high
melting characteristics and high melt visco ities. It
is a known phenomenon that the polymer chains of
thermoplastic polyurethanes can unzip, then ~ip back
together dur;ng thermal treatment. Generally speaklng,
this is not a problem with the softer polyurethanes made
by the prior art processes discussed above. However, in
37,052-F -4-
2 ~ r~ 7
--5--
the case of these newer materials, the higher
temperature required during processing may be the cause
of the difficulties. Wh le ~ wishing the present
invention to be limited by any theoretical
considerations but only by the claims appended herein-
b~low, it is believed that during the unzipping and
zipping of the polymer chains at the elevated processing
temperatures the reformed isocyanate groups can react
with each other to form carbodiimide and evolve carbon
dioxide thus resulting in bubble formation. The yellow
coloration appears to arise from some thermally
initiated side-reaction either connected to, or,
independent of, the above proposed carbon dioxide
formation.
Accordingly, it would be most desirable to have
a continuous reactive extrusion process available which
could ensure the preparation of these new polyurethane
products without the troublesome side ePfects noted
above.
The present invention is directed to an
improved reactive extrusion process for the continuous
preparation of a polyurethane having a Tg greater than
80C from a reaotion mixture comprising a polyisocyanate
and at least one polyol component by passing said reac-
tion mixture through a twin-screw extruder having
besides the feed zone, zones of high shear mixing and a
metering zone, wherein the improvement comprises limit-
ing the high shear mixing to a single zona.
The polyurethanes ?repared ln accordance with
the present invention are substantially free of bubble
formation and, surprisingly, are found to be much
37,052-F -5-
7 ~
--6--
lighter in color than those same chemically constituted
polyurethanes but prepared by the prior art method. The
difference in color is readily detected by COillpd~ir~g
Yellowness Index determinations for the respective
samples when measured in accordance with ASTM Test
Method D-1925.
Quite unexpectedly, the present reactive
extrusion process, if desired, can be carried out at
much lower L/D ratios than prior art extrusion methods.
The term L/D ratio refers to the overall length of the
twin-screw extruder barrel divided by the barrel
diameter, usually measured in millimeters (mm). In
running at lower L/D ratios which means shorter barrels,
this translates to an additional benefit of lowered
energy consumption compared with prior art methods.
Accordingly, the process in accordance with the
present invention meets the needs set forth above with
the added benefit of lower energy consumption.
The polyurethanes produced in accordance with
the present process are made up of either all hard
segments or hard segments with only a minor proportion
of soft segments arising from the small amount of high
molecular weight polyol uqed which is discussed in the
art cited supra. They are characterized by the
following properties: high impact resistance of at
least 1 ft. lb. per inch ~53 J/m), preferably at least 3
ft. lbs. per inch (160 J/m) of notch measured by the
notched Izod test in accordance with ASTM D256-56; high
heat deflection temperature when subJected to a 251l psl
(1.82 MPa) flexural load in accordance with ASTM D648-56
of at least 50C, preferably at least 70C; high flexural
37,052-F -6-
2 ~
--7--
modulus of at least about 150,000 psi (1034 MPa) as
measured by ASTM D790; Tg or secondary transition
tempcrat~.e greater than 80C; and hardl~ess values as
measured on the Rockwell hardness L ange R (as measured
by ASTM D785) of greater than 100~
Additionally, those polyurethanes prepared with
non aromatic polyisocyanates are characterized by having
essentially optical clarity and light stability.
The products produced in accordance with the
present invention find utility, for example, in the
molding of under the hood auto and truck parts such as
distributor covers, f;lter bowels, air-filter units and
covers, containers and covers for electronic circuitry,
medical devices requiring transparency and
autoclavability, surgical instrument trays and
containers for steam sterilization.
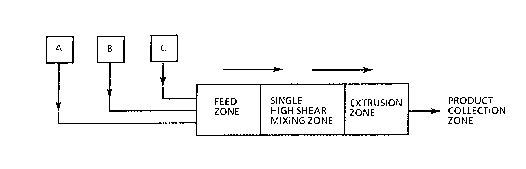
Figure l shows in schematic form one typical embodiment
of the process of the invention.
The reactive extrusion process in accordance
25 with the present invention is directed primarily to
certain specific classeq of hard, high temperature
resistant poly~lrethanes defined above. All of the
reactants, components, ingredients, and catalysts and
proportions therefor have already been set forth in
30 detail in U.S. Patents 4,376,834; 4,567,236; and
4,82Z,827. The compositions include both thermoplastic
injection-moldable resins and thermoset resins. The
latter are obtained when polyisocyanates, extenders and
polyols of functlonalities greater than two are employed
as taught in the previously mentioned patents.
37,052-F -7_
~ g~ h c Q 7
8--
Thermoplastic products are obtained by employing
substantially difunctional polyisocyanates and
difunctior.al extende.s alld, if used, polyols having
functionalities preferably not exceeding about 4. Since
the amount by weight of the polyol employed is
~ relatively small9 it is thus possible to employ such
components having functionalities greater than two
without detracting from the thermoplasticity of the
polymer. However, the thermoplastic materials are
greatly preferred and in this connection the following
sets forth some of the more preferred reactants which
can be employed.
Illustrative isocyanates but non-limiting
thereof are methylenebis(phenyl isocyanate) including
the 4,4'-isomer, the 2,4'-isomer and mixtures thereof,
m- and p-phenylene diisocyanates, chloro~henylene
diisocyanates, a,~'-xylylene diisocyanate, 2,4- and 2,6-
-toluene diisocyanate and the mixtures of these latter
two isomers which are available commercially, tolidine
diisocyanate, hexamethylene diisocyanate, 1,5-
-naphthalene diisocyanate and isophorone diisocyanate;
cycloaliphatic diisocyanates such as
methylenebis(cyclohexyl isocyanate) including the 4,4'-
-isomer, the 2,4'-isomer and mixtures thereof, and all
the geometric isomers thereof including trans/trans,
cis/trans, cis/cis and mixtures thereof, cyclohexylene
diisocyanate~ (1,2-; 1,3-; or 1,4-), 1-methyl-2,5-
-cyclohexylene diisocyanate, 1-methyl-2,4-cyclohexylene
diisocyanate, 1-methyl-2,6-cyclohexylene diisocyanate,
4,4'-isopropylidenebis(cyclohexyl isocyanate), 4,4'-
-diisocyanatodicy¢lohexyl, and all geometric isomers and
mixtures thereof. Also included are the modified forms
of methylenebis(phenyl isocyanate). By the latter are
37,052-F -8-
7 7
_9_
meant those forms of methylenebis(phenyl isocyanate)
which have been treated to render them stable liquids at
ambient temperat.lre (clrca 20C). Such piudu~ include
those which have been reacted with a minor amount (up to
about 0.2 equivalents per equivalent of polyisocyanate)
of an aliphat.c glycol or a mixture of aliphatic glycols
such as the modified methylenebis(phenyl isocyanates)
described in U.S. Patents 3,394,164; 3,644,457;
3,883,571; 4,031,026; 4,115,429; 4,118,411; and
4,299,347. The modified methylenebis(phenyl
isocyanates) also include those which have been treated
so as to convert a minor proportion of the diisocyanate
to the corresponding carbodiimide which then interacts
with further diisocyanate to form uretone-imine groups,
the resulting product being a stable liquid at ambient
temperatures as described, for example, in U.S. Patent
3,384,653. Mixtures of any of the above-named polyiso-
cyanates can be employed if desired.
A particularly preferred group of diisocyanates
includes aromatic and cycloaliphatic diisocyanates and
mixtures thereof as exemplified above. Most preferred
species within this group include methylenebis(phenyl
isocyanate) including both 4,4'- and 2,4'-isomers and
mixtures thereof with 4,4'- preferred, methylenebis-
(cyclohexyl isocyanate) including the 4,4'- and
2,4'-isomers and mixtures thereof including all of the
geometric isomers thereof with the 4,4'- preferred, and
4,4'-isopropyli~enebis(cyclohexyl isocyanate).
The at least one polyol component called for
above in its broadest scope will include the c~ain
extender and any polyol if it be employed. In respect
of the difunctional extenders, they are not strictly
37,052-F -9-
- l o -
limited to hydroxyl-containing extenders but can include
other active hydrogen materials such as amine groups or
...i:;'u.es of such extender types. r;owever, the preferred
extenders compri~e at least one diol having a molecular
weight of from 60 to 400. Included in this group are
the aliphatic diols having 2 to 1C carbon atoms,
inclusive of bis(hydroxyalkyl)cycloalkanes; and the
cycloalkane diols described in U.S. Patent 4,822,827 as
having 4 to 12 cycloaliphatic carbon atoms.
Illustrative of such diols are ethylene glycol, 1,3-
-propanediol, 1,4-butanediol. 1,5-pentanediol, 1,6-
-hexanediol, 1,2-propanediol, 1,3-butanediol, 2,3
-butanediol, 1,3-pentanediol, 1,2-hexanediol, 3-
-methylpentane-1,5-diol, 1,9-nonanediol, 2-methyloctane-
-1,8-diol, 1,4-cyclohexanedimethanol, neopentyl glycol,
hydroquinone bis(hydroxyethyl)ether, diethylene glycol,
dipropylene glycol and tripropylene glycol including
mixtures of two or more such diols; 1,2-cyclohexanediol,
1,3-cyclohexanediol, 1,4-cyclohexanediol, 2-cyclohexene-
-1,4-diol, 2-methyl-1,4-cyclohexanediol, 2-ethyl-1,4-
-cyclohexanediol, 1,3-cycloheptanediol, 1,4-
-cycloheptanediol, 2-methyl-1,4-cycloheptanediol and
4-methyl-1, 3-cycloheptanediol; 4,4'-
-methylenebis(cyclohexanol), 4,4'-methylenebis(2-
-methyicyclohexanol), 4,4'-methylenebis(3-
-methylcyclohexanol), 3,3'-methylenebis(cyclohexanol),
4,4'-ethylenebis(cyclohexanol), 4,4'-propylenebis-
(cyclohexanol), 4,4'-butylenebis(cyclohexanol), 4,4'-
-isopropylidenebis(cyclohexanol~, 4,4'-isobutylene-
bis(cyclohexanol), 4,4'-dihydroxydicyclohexyl, 4,4'-
-carbonylbis(cyclohexanol), 3,3'-carbonylbis(cyclo-
he~anol); ~'14'-sulfonylbis(cyclohexanol) and
4,4'-oxybis(cyclohexanol); and mixtures of any of the
above.
37,052-F -10-
2 ~ 7 ~
--1 1
Preferred for use in the present process are
1,4-butanediol, ',,-;.e~anediol, 1,6-hexanediol, 1,4-
-cyclohexanedimethanol, the cyclohexanediols including
the 1,2-, 1,3-, and 1,4-isomers, and ll,4'-isopropyli-
denebis~cyclohexanols), and mixtures c,f the above inaccordance with the teachings of the previously
mentione~ patents.
In respect of the polyol component, its use is
optional but, even if employed, it will not be present
in proportions which would lower the flexural modulus to
values below 150,000 psi (1034 MPa) as measured in
accordance with ASTM Test Method-D790. Accordingly, the
expedient concentration for the polyol falls in the
range of from 0 to 25 parts by weight per 100 parts of
total urethane reactants ba ed on (i) organic
polyisocyanate, (ii) at least one chain extender and
said polyol (iii). If it is selected for incorporation
for whatever reason, for example, to increase the impact
strength, elongation and tensile strength propertie~ of
the resulting plastics, then an advantageous range is
from 1 to 15 parts per 100 parts of reactants, and
preferably from 1 to 5 parts.
Minimum requirements for the polyol component
are a molecular weight of at least 500 and functionality
of at least 2. Advantageously, the molecular weight
falls within a range of from 500 to 12,000 with a
functionality of from 2 to not greater than 6;
preferably the molecular weight and functionality are
from 500 tc 6,000 and from 2 to 4 respectively; most
preferably the functionality is from 2 to 3.
37,052-F -11-
2 ~ 7 ~
-12-
Exemplary of the classes of polyols which can
be optiorally employed are: polyether polyols, poly-
ester polyols, hydroxy-terminated p~iy~arbonates~
hydroxy-terminated polybutadienes, hydroxy-terminated
polyhutadiene-acrylonitrile copolymers, hydroxy-
-terminate1 copolymers of dialkyl siloxane and alkylene
oxides such as, for example, ethylene oxide and
propylene oxide, and mixtures in which any of the above
polyols are employed as major component (greater than 50
percent w/w) with amine-terminated polyethers and amino-
-terminated polybutadiene-acrylonitrile copolymers.
Illustrative of polyether polyols are polyoxy-
ethylene glycols, polyoxypropylene glycols which,
optionally, have been capped with ethylene oxide resi-
dues, random and block copolymers of ethylene oxide and
propylene oxide, propoxylated tri- and tetrahydric
alcohols such as glycerine, trimethylolpropane and
pentaerythritol, which propoxylated compounds have been
capped with ethylene oxide, polytetramethylene glycol,
random and block copolymers of tetrahydrofuran and
ethylene oxide and or propylene oxide, and products
derived from any of the above reaction with di- or
higher functional carboxylic acids or esters derived
from said acids in which latter case ester interchange
occurs and the esterifying radicals are replaced by
polyether polyol radicals. The preferred polyether
polyols are random and block copolymers of ethylene and
propylene oxide of functionality fro~ 2 to 4,
preferably, 2 to 3 and polytetramethylene glycol
polymers of functionality greater than or equal to 2Ø
Generally speaking, the overall proportions of
the components (i), (ii), and (iii) are such that the
37,052-F -12-
7 77
-13-
active hydrogen-oontaining components (ii) and (iii)
balance the isocyanate component (i) so that stoichio-
metric equivalency of the rea~tants is maintained.
However, for various reasons, it is not always possible
nor desirable to meet the 1:1 equivalency. Advanta-
geously, the proportions are quch that the overall ratioof isocyanate groups to active hydrogen groups is in the
range of from 0.90:1 to 1.10:1, preferably, from 0.95:1
to 1.05:1 and, more pre~erably, from 0.98:1 to 1.02:1.
It is frequently desirable, but not essential,
to include a catalyst in the process. Any of the cata-
lysts conventionally employed in the art to catalyze the
reaction of an isocyanate with a reactive hydrogen-
-containing compound can be employed for this purpose;
see, for example, Saunders et al., Polyurethanes,
Chemistry and Technology, Part I, Interscience,
New York, 1963, pages 228-232; see also Britain et al.,
J. Applied Polymer Science, 4, 207-211, 1960. Such
catalysts include organic and inorganic acid salts of,
and organometallic derivatives of, bismuth, lead, tin,
iron, antimony, uranium, cadmium, cobalt, thorium,
aluminum, mercury, zinc, nickel cerium, molybdenum,
vanadium, copper, manganese and zirconium, as well as
phosphines and tertiary organic amines. Representative
organotin catalysts are stannous octoate 7 stannous
oleate, dibutyltin dioctoate and dibutyltin dilaurate.
Representative tertiary organic amine catalysts are
triethylamine, triethylenediamine, N,N,N',N'-
-tetramethylethylenediamine, N,N,N',N'-tetraethyl-
ethylenediamine, N-methylmorpholine, N-ethylmorpholine,
N,N,N',N'-tetramethylguanidine, N,N,N',N'-tetramethyl-
-1,3-butanediamine, N,N-dimethylethanolamine and N~N-
-diethylethanolamine. The amount of catalyst employed
37,352-F -13-
éj 7
-14-
is generally within the range of from 0.02 to 2.0
percent by weight based on the total weight of the
reactants.
Included in the process is the one-shot
procedure, wherein all the reactants are brou@h~L
together all at once in the extrusion apparatus, and the
prepolymer or quasi-prepolymer techniques. The use of a
prepolymer technique would, of course, be limited
primarily to those formulations employing the polyol
component (iii) wherein part or all of the polyol is
first reacted with isocyanate and the isocyanate
prepolymer, then fed to the extrusion apparatus along
with the extender. However, the preferred method is the
one-shot reaction.
Additionally, the process can also include
various additives such as impact modifiers, fillers and
fiber glass; antioxidants, pigments, fire retardants,
plasticizers, reinforcing agents and W2X lubricants
commonly employed in the art in such compositions.
These may be added along with the reactants or at a
later stage through a downstream feed port, or at a post
reactor compounding step.
The process in accordance with the present
invention is carried out by feeding the above ingre-
dients into a commercial multi-screw extruder which is,
generally speaking, a twin-screw extruder. The screws
can be co- or counter-rotating. Preferably, a co-
-rotating and self-cleaning twin-screw extruder is
employed. The general procedures and extrusion
equipmer.t described in U.SO Patents 3,642,964 and
3,963,67g can be employed herein except for the novel
37,052-F -14-
¢; ~ ~
-15-
exceptions discussed hereinbelow. Generally speaking,
the reactive extrusion process is carried out within an
overall temperature ran~e of f~ " , 5aoc tG 280C. That
is to say, individual zones may not all be at the same
temperatures but their individual values will fall
with n this range.
The reactants can be measured out using known
means such as, for example, gear pumps, membrane or
piston pumps for delivery to the extruder feed zone.
Figure 1 shows one schematic embodiment of the process
wherein (A), (B), and (C) represent three separate
reactant feed lines for the isocyanate component, polyol
component~ and catalyst component respectively. It is
not essential that all three lines be utilized. For
example, catalyst may be included in the (B) feed line;
alternatively, and, if desired, the reactants may be
mixed in a suitable mixing head before they are
introduced into the extruder. In that case, (A)
represents a mixing head which includes all the
reactants with catalyst or else the catalyst may be
added separately as (B). One of the advantages of the
present process i5 that viscosity build-up of the
reaction mixture in the FEED ZONE is not in any way a
particular problem. In fact, one of the key features of
the present process i3 that the reaction mixture
viscosity should exceed 100,000 cps (~00 Pas) before
reaching the critical SINGLE HIGH SHEAR MIXING ZONE
(hereinafter MIXING ZONE). In this regard, the gel time
for the reactant systems should be less than about 10
seconds, preferably less than about 6 seconds and FEED
ZONE temperatures can fall wlthi~. a range of from 150C
to 250C. he FEED ZONE length and conveying screws are
in no way critical to the present process and can be
37,052-F -15-
2 ~
-16-
configured as in the previously mentioned U.S. patents.
Actual barrel lengths for the FEED ZONE will be defined
by ~he overall L/D (lengt;l/diameter) of the ext~uder
which will be discussed in detail below.
Besides the diff~rences in the reaction mix
viscosities in the FEED ZONES between the present
process and the prior art cited supra particularly U.S.
Patent 3,~63,679, the limitation o~ the present MIXING
ZONE to one single zone o~ high shear mixing is a very
critical and novel ~eature which results in polymer
improvements that otherwise cannot be obtained with the
hard polyurethanes. Such multiple zones of high shear
mixing are described in detail in the previously
mentioned patents, particularly the '679 reference.
Generally speaking, such high shear mixing zones employ
two series of broad edged multiple kneading elements or
blocks mounted in intermeshing relationship on the pair
of parallel mounted screws in the twin-screw extruder in
each zone. The individual kneading elements or blocks
can be triangular, circular or elliptical as typically
disclosed in the figures 2 through 7 of the '679 patent.
The dimensions are such that there is minimal radial
clearance between the inner surface of the barrel and
the perimeter edges of the blocks. Hence, rotation of
the screws subjects the reaction mixture to high shear
force~. Additional mixing and application of high shear
forces can be imparted, if desired, by mounting
appropriate baffles but more usually reverse pitch
kneading blocks. Generally speaking, conveying screws
are present in the same extruder barrel sections which
contain the zone of kneading blocks.
37,052-F -16-
What distinguishes the present process is the
fact that the high shear mixing is carried out in just a
single ~ uf these multiple kneading elements and not
a series of repetitive zones. Advantageously, the
actual length of the kneading elements within the single
zone will fall within a range of from 60 ~n. to 240 mm,
preferably from 80 to 180, and more preferably from 120
to 180 mm.
In order to provide sufficient shearing forces
in the polymer forming mixture, a sufficiently high
shear rate or velocity gradient must be achieved.
Generally speaking, this is related to the combination
of the minimal radial clearances between the barrel
(extruder wall) and shearing edges of the blocks
referred to above and the speed of screw rotation.
Advantageously, this clearance will fall within a range
of from 0.05 nm to 0.6 mm, preferably from 0.1 to 0.4
mm. Screw rotation expressed in rotations per minute
(r.p.m.) will fall within a range of from 140 to 500
r.p.m., preferably 150 to 300 r.p.m.
Temperatures in the MIXING ZONE will be
controlled within a range similar to the FEED ZONE with
optionally a slightly higher range, that is to say, from
150C to 280C. Capability of cooling this zone should
be provided in the event that exotherm could exceed the
upper limit of 280C.
The EXTRUSION ZONE, sometimes called the
METERING ZONE, can be configured similarly to the prior
art w th onl~ conveying screws. Temperatures are
advantageously controlled to a range of from 180C to
280C. The reacted melt product is simply extruded at
37,052-F -17-
-18-
the end through any desired molding tool or die known to
the art such as, for example, slot dies, profile-forming
ules and rods.
There is no particular limit on overall barrel
ength other than what is dictated by practical con-
siderations. However, a particularly attractive feature
of the present process are the short barrel lengths
which are possible. Advantageously, the L/D ratio can
fall within a range of from 10/1 to 44/l, preferably
from 15/1 to 25/1, most preferably from 15/l to 20/l.
Overall temperature control throughout the above L/D
ranges, whether it be through heating and/or cooling in
particular zones, will fall within the previously stated
range of from 150C to 280C. Overall residence times in
the extruder when operating under these L/D,
temperature, and r.p.m. conditions set forth above will
fall within a range of from 5 to 45 seconds, preferably
from 6 to 30 seconds, more preferably from 6 to 25
seconds.
The extruded polyurethanes can be in finally
desired shape or else comminuted or pelletized for
further molding or injection molding into desired
article~. The polymers a3 obtained need no further
treatment, havin~ attained their superior physical and
mechanical properties as set forth in the three patents
cited supra and already incorporated herein.
The following examples describe the manner and
process of making and using the invention and set forth
the best mode cor.templated by the inventors of carrying
37,052-F -18-
_19_
out the invention but are not to be construed as limit-
ing.
Exam~le 1
The following experiment sets forth a com-
parison of two redc~ive extrusion methods, one in
accordance with the invention (run 1) and the other
(comparison 1? in accordance with the prior art.
A Werner and Pfleiderer ZSK-53 self-cleaning,
co-rotating twin-screw extruder was fitted with five
barrels of which four had high shear mixing zones. Each
of the latter zones were 240 mm in length but only 120
mm of this length was formed of the actual high shear
kneading blocks themselves with the remainder given over
to conveying screws. Radial clearance between extruder
wall and outer diameter of the shearing edges of each
kneading block was about 0.2 mm. Screw speed was con-
trolled to 460 r.p.m. Extruder temperatures were con-
trolled by five independent barrel heating or cooling
zones. Feed zone temperature was 225 + 5C, with
extrusion zone being 215 ~ 5C and the intermediate
zones including the four high shear mixing zones being
190 to 210C. ~ sheeting die, 200 mm width and 3 mm gap
was flanged to the end of the extrusion zone.
For comparison 1, a mixture in the proportional
pa~ts by weight of 100 part~ of 1,4-cyclohexanedi-
3 methanol, 0.82 parts of trisnonylphenyl phosphite and0.69 parts of Irganox 1010 (antioxidant supplied by Ciba
Geigy Corp.) was degassed and dehydrated under vacuum at
99C for 2 hours. Using a gear pump, this mixture was
delivered to the front feed port of the extruder. This
means that the length/diameter ~L/D) ratio for this feed
37,052-F -19_
-20-
passing through the 2344 mm of length for the five zones
of the barrel was 44/1. A second gear pump delivered to
''-.e s~ e ront feed port 175.2 parts of~ melted 4,4'-
-methylenebis(phenyl isocyanate) per 100 parts of the
above diol,-while a third pump delivered at the same
proportional rate 0.33 part of a polyurethane catalyst
Fomrez UL-22 which is dimethyltindidodecyl mercaptide
supplied by Witco Corporation. Gel time of this
reacting mixture determined by hand mixing the
components rapidly in a beaker was less than 10 seconds.
The polymer was extruded onto a retal conveyor belt at
26C, cooled and diced. At this point the solidified
polymer was observed for the formation of bubbles.
After drying at 115C in a dehumidifying hopper dryer
with a dew point below -28C, the pellets were injection
molded into test specimens. Their physical properties
were determined according to ASTM test procedures with
the results set forth in Table I.
Run 1 in accordance with the invention was
carried out identically to the above comparison run in
every respect except the reactant~ were fed into a
downstream feed port. The second feed port was located
in the extruder barrel which was just prior to the la~t
(i.e. fourth) high shear mixing zone. Feeding the
reactants at this point shortened the barrel to 919 mm
and reduced the L/D to 17.3/1. More importantly, the
reaction mixture passed through a single zone of high
shear mixing wherein the actual length of kneading
blocks was 120 mm.
Comp2riscn of the two extrudates showed run 1
to be essentially clear with the presence of very few
minute bubbles, essentially no more than what are to be
37,052-F -20-
2 ~
expected in the extrusion of transparent type thermo-
plastics. Contrastingly~ Comparison 1 showed copious
bubbles in the sol~dLl . Gd eAtrudate. In respect of
color formation as measured by Yellowness Index, run 1
was much lower in color compared with comparison 1. As
for molecular weights and other physical properties, ru~
1 appeared to be superior in most respects.
37,052-F -21-
~ :?~
-22-
TA8LE I
Run Comp. 1
Polymer M.W.l 232,432 286,654
5 Flex Modulus, psi ~- 103 (GPa) 311.3 (2.15) 305.0 (2.1)
Flex strength, psi (MPa) 14,714 (101.4) 14,898 (102.7)
Tensile strength, psi (MPa)
Yield 11,624 (80.1) 11,634 (80.2)
10 8reak 8,634 (59.5) 9,086 (62.6)
Tensile moduius, psi x 103 (GPa) 293.4 (2.02) 289.2 (1.99)
Elongation (~)
Yield 8.2 8.3
8reak 34.6 31.0
HDT2 ( C )
66 psi (455 kPa) 133 135
264 psi (1820 kPa) 123 123
Notched Izod3 ft-lbs/in (J~m) notch
1/8" (3.175 mm) thick sample 1.10 (58.7) 1.24 (66.2)
1/4" (6.35 mm)thick sample 0.74 (39.5~ 1.40 (74.7)
Yellowness IndeY4 3.05 5.75
~ubble Formation5 copious very few
bubbles bubbles
Footnotes to TALLL I
-
1 Polymer weight average molecular weight determined by gel
permeation chromatography using a polystyrene reference as
standard.
30 2 ~DT: Heat deflectiun temperature measured at the specified
pressures in accordan~e with AST~ Test Method D-648.
3 Notched Izod: Izod impact strength measured on 1/8" and 1~4"
(3.175 mm and 6.35 mm) thick samples in accordance with ASTM
D256-56.
37, 052-F -22-
r~
4 Yellowness Index: determined usinq ASTM D-1925 standard at a two
degree observer angle using a Pacific Scientific Spectrogard
Color Syste~, Silve~ Spring, Marylan~ 20910 with the illuminant
being simul~ted average daylight.
5 Bubble Formation: refers to the qualitative visual observation
of the extruded polymer for th~ formation o^ bubbles; presence of
a very few minute b~bbles can be tole~ated ~Ind the polymers
classified as essentially a ~lear plastic, ~hereas the formation
of a copio~s number of bubbles is not accep`:able.
Example 2
Similarly to Example 1~ this experiment
describes comparison 2 and run 2 reactive extrusions
wherein the same apparatus and conditions were employed
herein with the exception of different reactant com-
ponents.
The mixture pumped to the first feed port for
comparison 2 which ran through the four mixing zones was
as follows: a degassed and dehydrated mixture of 100
parts of an 85/15 mole ratio of 1,4-cyclohexane-
dimethanol and hydrogenated bisphenol-A, 0.77 parts
trisnonylphenyl phosphite, and 0.64 parts of Irganox
1010; 158.35 parts of melted 4,4'-methylenebis(phenyl
isocyanate); and 0.31 part of Fomrez UL-22. For run 2
which ran through only the ~ingle zone of high shear
mixing, the same three part ingredient mixture was fed
into the downstream feed port for the L/D ratio of
17.3i1.
The properties of the injection molded samples
from the two runs are set forth in Table II. Signifi-
c~rtly less gassing in the initial extrudate and lower
color for molded run 2 specimens compared to comparison
37,052-F -23-
2 0 ~ 7 ~
-24-
2 was observed. Their physical properties were
essentially comparable.
TA8LE II
Run Comp. 2 2
Polymer M.W. 203,761 212,193
Fles Modulus, psi x 103 (GPa) 320.6 (2.Zl) 311.6 (2.14)
Flex strength, psi (MPa) 12,918 (89~ 12,812 (88)
10 Tensile strength, psi (MPa)
Yield 11,887 (82) 11,825 (81.5)
Break 8,729 (60.2) 8,664 (59.7)
Tensile modulus, (psi x 103) 295.8 (2.04) 291.4 (2.01)
Elongation (~)
Yield 8.3 8.2
Break 22.1 24.9
~DT (~C)
66 psi (455 kPa) 145 145
264 psi (1820 kPa) 135 139
Notched Izod ft-lbs/in (J/m) notch
1/8" (3.175 mm) thick sample 1.15 ~61.4) 1.02 (54.4)
1/4" (6.35 mm) thick sample 0.82 (43.8) 0.93 (49.6)
25 Yellowness Index 8.13 5.64
Subble Formation copious very few
bubbles bubbles
3 Example 3
This experiment similar to those above compared
the properties of the extrudates and the injection
molded samples ~rom comparison 3 and run 3. Using the
same extruder configurations and conditions set forth in
37,052-F -24-
2 ~
-25-
Example 1, the comparison 3 ingredients were fed into
the front feed port of the extruder barrel while the
same ingredients were fed into tile uownsiream feed port
for run 3 with the latter having only the single zone of
high shear mixing. The ingredients were as follows:
degassed and dehydrated mixture of 100 parts of 1,6-
-hexanediol, 0.94 part trisnonylphenyl phosphite, and
0.78 part of Irganox 1010; 213.8 part of melted 4,4'-
-methylenebis(phenyl isocyanate); and 0.19 part of
Fomrez UL-22.
The properties of the two product runs are set
forth in Table III. The data show significantly less
gassing and lower color for run 3 compared with its
comparison sample.
3o
37,052-F -25-
`2~
-26-
TABLE III
Run Comp. 3 3
-- , , .
PolymPr K.W. 583,000 642,500
5 Tensile strength, psi (MPa)
Yield 8960 (61.8) 7650 (52.7)
Break 6600 (45.5) 9865 (68)
Tensile modulus, psi x 103 (GPa) 256 (1.77) 273 (1.88)
Elongation (~)
Yield 7
Break 27 166
HDT (C)
66 psi (455 kPa) 88 86
264 psi (1820 kPa) 75 76
Notched Izod ft-lbs~in (J/m) notch
1/8" (3.175 mm) thick sample 1.747 (93.3) 2.399 (128)
1/4" (6.35 mm) thi~k sample 1.294 (69.1) 1.782 (95.1)
Bubble Formation COPIOUS VERY FEW
- BU~BLES ~UB8LES
EXample 4
This example describes the preparation of a
hard thermoplastic polyurethane polymer in accordance
with the present invention ~run 4) and comparison 4.
In run 4, a self-cleaning twin-screw Werner and
Pfleiderer ZSK-120V was employed with a single zone of
high shear mixing at an L/D ratio of 9.5/1 from the feed
port and with an overall extruder L/D of 18.8/1. The
actual length of kneading blocks in the high shear
37,052-F -26-
-27-
mixing zone was 180 mm. Screw speed was 250 r.p.m. and
with about 0.2 mm radial clearance between extruder wall
and outei~ dia,l-eLe} of the shearing edges of each
kneading block. Zone temperatures were feed, 188C;
mixing, 177 to 199C; and extrusion, 2300.
The reaction mixture ~hich was metered into the
feed port in three streams in the following proportions
in parts by weight was as follows: degassed and
dehydrated mixture of 43.2 parts of 1,4-cyclohexane-
dimethanol, 23.6 parts of 1,6-hexanediol 10.4 parts of
a 650 molecular weight polytetramethylene glycol, 0.52
part of Irganox 1010, and 0.31 part of triphenyl
phosphite; 130.79 parts of 4,4-methylenebis(phenyl
isocyanate); and 0.10 part of a 50/50 weight mixture of
stannous octoate and dioctyl phthalate. The extruded
product was fed onto a cool conveyor as described in the
previous examples and dicedO The extruded material was
clear transparent with only a few minute bubbles being
detectable. The injection molded product had the
following properties set forth in Table IV.
Comparison 4 was carried out under essentially
the same temperature conditions with identical
ingredients to run 4 but using a twin-screw Werner and
Pfleiderer extruder having an 83 mm diameter, equipped
with three zones of high shear mixing and an L/D of
30~7/1.
3o
A comparison of the two runs shows that run 4
provided a product with much superior molecular weight
to the compar-son run ~ znd a measurably better yellow
index and transmission. Notably, this was accomplished
37,052-F -27-
20~ 9~rlJ7
--28--
with much less power consumption as compared with
co~parison 4.
TABLE IV
Run Comp. 4 4
.
Polymer M.W. 266,302 359,352
Tensile Strength, psi (MPa)
Yield 10,305 9,896
Break 7,867 7,703
Tensile Modulus, psi x 103 (GPa) 274 (1.89) 274.8 (1.9)
Elongation (~)
Yield 6.4 5.7
Break 42 45
Yellowness Index 5.52 4.74
Transmission at 380 NMl 58.05~ 61.97
Footnote to Table IV
1 Light transmittance in percent measured on injection molded disk
measuring 2 inches (51 mm) diameter x 1/8" (3.2 mm) thick at 380
nanometers using Pacific Scientific Spectrogard Color Systems,
Silver Spring, Maryland, 20910.
37, 052-F -28-