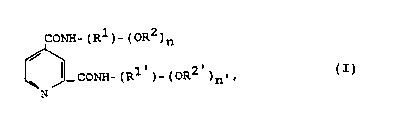Note: Descriptions are shown in the official language in which they were submitted.
HOECHST AXTIENGESELL5CHAFT Dr.SN/fe MOE 89/F 241
Description
N,N'-Bis(alkoxyalkyl)-pyridine-2,4-dicarbo~ylic acid di-
amides, preparation and their use
Compounds which inhiblt proline hydroxyla~e and ly~ine
hydroxylase cause a very selective inhibition of collagen
biosynthesis by influencing the collagen-specific
hydroxylation reactions. In the course of these, protein-
bound proline or ly~ine is hydroxylate~ by the enzymes
proline hydroxylase or lysine hydro~ylase. If this
reaction is sup~essed by inhibitor~, Zl non-functional,
hypohydroxylated collagen molecule iR formed, which can
be released by the cells into the extracellular space in
only a small amount. The hypohydroxylated collagen also
cannot be incorporated into the collagen matrix and is
very easily degraded by proteolysis. As a con~eguence of
these e~fect~, the amount of extracellularly deposited
collagen overall is reduced.
It is known that the inhibition of proline hydroxylase by
known inhibitors, such as ~ dipyridyl, leads to an
inhibition o~ the Clq biosynthesis of macrophages
(W. Muller et al., FEBS Lett. 90 (1978), 218; and Immun-
biolo~y 155 (1978) 47). This result~ in a failure of the
classical route of complement activation. Inhibitors of
proline hydroxylase therefore also act as i ~oSuppL~s-
sants, for example in cases of immune complex diseases.
It is known that proline hydlo~ylase is inhibited effec-
tively by pyridine-2,4- and -2,5-dicarboxylic acid
(K. Ma~amaa et al., Eur. J. Biochem. 138 (1984) 239-245).
However, in the cell culture the~e compounds are effec-
tive as inhibitors only in very high concentrations
(Tschank, G. et al., Biochem. J. 238, 625-633, 1987).
Ge -n Patent A-3,432,09A describes pyridine-2,4- and
-2,5-dicarboxylic acid diasters having 1-6 carbon atoms
-- 2 --
in the ester alXyl part as pharmaceutical& for inhibition
of proline hydroxylase and lys.ine hydroxylase.
Howeverl ~hese lower alkylated diesters have the dis-
advantage that they are split too rapidly in the organism
~o give the acids and do not arrive at their site of
action in the cell in a sufficiently high concentration,
and are therefore of little suitability fvr possible
a~ri n istra~ion as pharmaceu~icals.
G~rr-n Patent A-3,703,959, ~7e ~n Patent A-3,703,962 and
~erm~n Patent A-3,7~3,963 describe, in general form,
mi~ed ester/amides, higher alkylated diesters and di-
amides of pyridine-2,4- and -2,5-dicarboxylic acid which
effectively inhibit collagen biosynthe~is in the ~ni~l
model.
,.
German Patent A-3,703,959 thus describes, inter alia, the
synthesis o~ N,N'-bis(2-methoxyethyl)-pyridine-2,4-
dicarboxylic acid diamide (III)
CON~-CH2-CH2-O-C~3
~ CONH-CH~-CH2-O-CH3 (III)
and
N,N'-bis(3-isopropoxypropyl)-pyridine-2,4-dicarboxylic
acid diamide (IV)
~ CH3
CONH-CH2-CH2-CH~-O-CH
~H3", CH3 ~IV)
N ~ CONH CH2-CH2-C~2-~-CX ~
CH3
An improved proce~ for the preparation of N,N'-bis~2-
methoxyethyl)-pyridine-2,4-dicarboxylic acid diamide
~5 (III) is proposed in German Patent Applications
P 38 26 471.4 and P 38 28 140.6.
2&J~ 2~
-- 3 ~
The enteral absorbability of many of the compounds
described in German Patent A-3,703,959 i~ still un~atis-
factory, however, 80 ~hat there i~ a need to provide
compounds which effec~ively inhibit proline hydroxyla~e
S and lysine hydro~ylase after oral al' ini~tration even in
low dosagesr
It has been found that compounds of the formula I
CONH-(R~ O~ 3~
CO~H-(R~ oR2 ~n'~ (I)
N
wherein
R1 denotes linear or branched Cl-c4-alk~nediylt
R2 denotes unbranched Cl-C4-alkyl or hydrogen,
n denotes 1 or 2 and
R1, R~ and n~ have the ~ame ~anings as Rl, R2 and n, Rl
and R1, and RZ and R2 and n and n~ being
identical or different,
and physiologically tolerated 8alt8 thereof, excluding
N,N~-bis~2-methoxyethyl)-pyridine-2,4-dicarboxylic acid
diamide and N,N'-bi~ hydroxyethyl)-pyridine-2,4-dicar-
boxylic acid diamide, achieve the abov~- ~ntioned ob~e~t.
~0 In compari~on with the compounds N,N'-bis~2-methoxy-
ethyl)-pyridine-2,4-dicarboxylic aci~ diamide and N,N~-
bis(3-isopropoxypropyl)-pyridine-2,4-dicarboxylic acid
diamide de~cribed in Ge- -n Patent A-3,703,959, the
compounds of the formula I have both a better ph~rr-clo-
logical activity and a bekter enteral ab~orbability.
The invention furth~ Le relates to a process for thepreparation of N,N'-bi~-(alkoxyalkyl)-pyridine-2,4-
dicarboxylic acid diamides of the formula I
CONH- ~ R~ . ( OR2 ) n
~ CONH-~R~ (OR2 )n~ (I)
J ~ t)
wherein
R1 denotes linear ox branched C,-C4-~lk~ne~iyl,
R2 denotes unbranched Cl-C4-alkyl or hydrogen,
n denotes l or 2 and
Rl, RZ and n' have the same ~nin~s as Rl, R2 and n, R1
and R1, and R2 and R2 and n and n' being
identical or differant, excluding ~tN'-
bis(2-methoxyethyl~-pyridine-2,4-dicar-
boxylic acid diamide and N,N'-bis(2-
hydroxyethyl)-pyridine-2,4-dicarboxylic
acid ~ de,
which compri6es reacting a pyridine-2,4-dicarboxylic acid
halide with an alkoxyalkylamine or hydroxyalkylamine.
The invention likewise relates to a proce~s for the
preparation of N,N~-bis-(alkoxyalkyl)-pyridine-2,4-
dicarboxylic acid diamide~ of the formula I
CON~ Rl ) - ( oR2 ~ n
~ CONH-(R~ (OR~ )n~
wherein
R} denotes linear or branched Cl-c4-~kAnedi
R2 denotes unbr~nched Cl-C4-alkyl or hydrogen,
n denotes 1 or 2 and
Rl, R2 and n' have the same meanings as Rl, R2 and n, Rl
and Rl, and R2 and R2 and n and n' being
identical or different,
2~ which compri~es first
A)
~ i ng at least two equivalents of a halogenating
agent to pyridine-2,4-dicarboxylic acid and
2. dissolving at least 2 equivalent8 of a hydroxyalXyl-
amine or alkoxyalkylamine of the formula II or II'
H2N-(R )-(OR )n (II)
H2~ 1 )--( oR2 ) n ~
-- 5 --
ln which
Rl and R1 denote linear or branched Cl-C4-alkanediyl,
R2 and R2 denote unbranched C1-C4-alkyl or hydrogen,
n and n' denote l or 2 and
Rl and Rl, R2 and R2 and n and n~ are identical or
different, but II and II~ are differen ,
in a solvent
and then reacting the solution prepared according to l.
with the solution prepared according to 2., or
B) converting the resulting N,N~-bis(alkoxyal~yl~-pyri-
dine-2,4-dicarboxylic acid ~ e into the bis(hydro~y-
alkyl) compound, or
C) reacting the pyridine-2,4-dicarboxylic acid halide
prepared according to A) 1. with a substituted or un~ub-
stituted benzyl alcohol to give the benzyl pyridine-2~4-
dicarboxylate, hydrolyzing the benzyl ester selectively
in the 2-position of the pyridine, converting the free
carboxylic acid in the 2-position into the acid halide
again in accordance with proces6 variant A) l. and adding
a solution prepared according to A~ 2. using a compound
of the formula II~ to the c~ -und thus obtAin~ a
pyridine-4-(carboxylic acid benzyl ester)-2-carboxylic
acid ~mide being formed, and then splitting off the
benzyl protective group in the 4-position by hydrogenoly-
25 ~i8 ~ converting the free carboxylic acid into the acidchloride again in accordance with process variant A) 1.,
and subsequently adding a solution prepared according to
A) 2. using a compound of the formula II, an unsymmetri-
cally substituted compcund of the formula I being formed,
and if appropriate then converting the resulting compound
of the formula I into its physiologically tolerated salt.
In the process for the preparation of the compound~ of
the formula I, the pyridine-2,4-dicarboxylic acid commer-
cially available 2S a starting subskance is suspended in
a solvent, such as toluene, and a halogenating agent,
preferably a chlorinating agent, ~uch as, for example,
SOCl2, i8 added at room temperature. 2-3 eguivalents,
preferably 2.5 equivalents, of a halogenating agent,
," ,i. ~ ~" ;,)
-- 6 --
based on the molar amount of pyridine-2,4-dicarbo~ylic
acid employed, are used. The resulting reaction mixture
is heated at 90-110~C, preferably at lOO~C, until no
further evolution of gas is tu be observed and a clear
solution has formed. 10% of the solution is then evapora-
ted off - preferably under a hlgh vacuum (down to about
10-3 mm Hg) - and the resulting carboxylic acid halide i8
reacted further.
2-4 times the molar amount of commercially available
lQ alkoxyalkylamine ox hydroxyalkylamine, based on ~he molar
amount of pyridine-2,4-dicarboxylic acid employ~d~ is now
dissolved in a solvent, such as toluene, and 2-4 times
the molar amount of a base, such as triethyli i n~ ~ is
pr~ferably added. The carbo~ylic acid halide is reacted
with the alkoxyalkylamine or the hydroxyalkylamine. ~his
i5 preferably done by ~in~ the solution of the alkyl-
amine mentioned dropwise to the dissol~ed pyridine-2,4-
dicarboxylic acid halide. However, it is also pos3ible to
add the solution of the carboxylic acid halide dropwi~e
to the solution of the alkoxyalkylamine or hydroxyalkyl-
amine~ The addition is carried out at a temperature of
-5 to ~5~C, pxeferably at 0~C. The reaction mixture ca~
then be after-reacted, for example, by warming it to room
temperature and subsequently stirring it fox a further 2-
5 hours, preferably 3 hours. The resulting product isthen acidified in order to ~ -ve excess hydroxy- or
alkoxyalkylamine from the desired product. The acidifica-
tion can be carried out, for example, with 0.2 molar
citric acid. The organic phase is then separated off and
washed with water. The or~anic phase i~ ~ub~equently
dried - pre~erably over magnesium sulfate - and finally
freed ~rom the solvent. On removal of the solvent, the
product is obt~;ne~ as a white solid or as an oil.
To prepare the N,N'-bis(hydroxyalkyl)-pyridine-2,4-
dicarboxylic acid diamides, a procedure i8 preferablyfollowed in which a correspo~ing bis(alkoxyalkyl)di-
amide, preferably bis(methoxyalkyl)~ ;de, is converted
~ 7 ~
into the corre~ponding bi~(hydroxyalkyl)diamide by
proce~es which are known from ~he litera~ure, for
example u~ing boron tribromide.
Unsymmetrically substituted compounds of the formula I
can be synthe~ized, for example, as followss reaction of
a pyrldine-2,4-dicarboxylic acld halide, preferably the
chlor~de, with ~ub~tituted or unsub~tituted benzyl
alcohol to give benzyl pyridine-2,4-clicarboxylate,
subsequent sslective hydroly~i~ of the ester in the 2-
position (for example in the presence of a copper cata-
lyst, Acta Helv. 44, 1963, page 637), conver~ion of the
free acid in the 2-position into the acid halide, reac-
tion with a compound of the formula (II') ~o give the
pyxidine~4-(carboxylic acid benzyl ester)-2-car~o~ylic
acid amide, splitting off of the remain~ng benzyl pxotec-
tive group by hydrogenolysi~ (for example with H~Pd, see
Houben-Wey} Volume IV/lc ~1980), pages 381-82) and
subsequent conversion of the free acid in the 4-po~ition
of the pyridine ring into the acid halide.
~he acid halide can now be converted into the mixed
diamide ~I) with an amine II (see Figure 1).
If appropriate, the product~ can be worked up, for
example, by extraction or by chromatography, for example
over silica gel. The product isolated can be Lecly~tal-
lized and if appropriate reacted with a suitable acid togive a physiologically tolerated salt. Example6 of
possible suitable acid~ are: mineral acids, such as
hydrochloric and hydrobromic acid as well a~ ~ulfuric,
phosphoric, nitric or perchloric acidt or organic acid~,
such as formic, acetic, propionic, succinic, glycolic,
lactic, malic, tartaric, citric, maleic, fumaric, phenyl-
acetic, benzoic, methanesulfonic~ toluene~ulfonic,
oxalic, 4-aminobenzoic, naphthalene-1,4-di~ulfonic or
ascorbic acid.
l'he compounds o~ the formula I can be used ae medicaments
in the form of phA -ceutical preparations which contain
-- 8 ~
them, if appropriate toge~her with tolerated pharmaceuti-
cal excipients. The compound~ can be used as medicines,
for example in the form o~ pharmaceutical preparations
which contain these compounds as a mixture with a
pharmaceutical/ organic or inorganic excipient suitable
for enteral, percutaneous or parenteral admini~tration,
such as, for example, water, gum axabic, gelatin, lac-
tose, starch, ma~nesium stearate, talc, vegetable oils,
polyalkylene glycols, vaseline and the :Like.
The ph~ ~?utical preparation~ can be in solid form, for
example as tablets, coated tablets, suppositorie~ or
capsules; in semi-solid form, for e~ample as ointments,
or in liquid form, for example as solutions, ~uspensions
or emulsions. If a~loyliate, they are ~terilized and/or
contain auxiliaries, such as pre~ervatives, s$abilizers,
wetting agents or emulsifiers/ salts for modifying the
osmotic pressure or buffers. They can al~o additionally
contain other ther~peutically active substances.
It has been found that the c~ ~)ounds of the formula T
have exceptionally good enteral ~bsorbabilities. ~he
absorbability was inve~tigated on Wis~ar ra~s to whi~h
the compounds according to the invention were ~ ni~ter-
ed intragastrally. ~he serum level dropped in the first
hours after administration of the substance, and after
about 5 hours ~eached a plateau which 5till fell only
slightly. The good absorbability of the substances can be
conclude~ from the initially very high serum level
directly after ~-' i n~ stration of the substances.
The invention i~ illustrated in more detail below with
the aid o~ examples.
rJ ~ 5; ~ éJJ
Exampl~ 1
Pyridine-2,4-dicarboxylic acid bis-N,N~-(metho~ opyl)-
amide
3 g of pyridine-2,4-dicarboxylic acid are initially
S introduced into 50 ml of toluene and 1 ml of dimethyl-
formamide, and 2.7 ml of thionyl chloride are added
dropwi~e to the solution. The mixture i~ heated until no
further evolution of gas is to be ob~erved (about 2.5
hours~. It i5 cooled, 5 ml of toluene are di~tillsd off
and 4.6 ml of 3-methoxypropylamine and S ml of triethyl-
amine are added dropwise to the solution. ~fter the
solution has been 6tirred at room temperature for 4
hours, it is evaporated, the residue is taken up in water
and the mixture is extracted 4 tLmes with methylene
lS chloride. The combined organic phase~ are dried over
magne~ium sulfate and evaporated. The crude product i~
chromatographed with silica gel ~solvent: ethyl acetate).
Yield: 4.3 g; oil
1H-NMR ~CDCl3): ~ = 1.6-2.3 (4H, m); 3.2-3.8 (14H, m);
7.8-8.0 (lH, m); 8.3-8.5 (lH, m); 8.6-
8.8 (lH, m).
~xample 2
Pyridine-2,4-dicarboxylic acid bis-N,N'-(etho~y~Lo~yl)-
amide
Yor instructions see Example l; amine component: ethoxy-
propylamine
Yield: 4.5 g, melting point: 46-48~C.
H-NMR (CDCl3): 6 - 1.3 (6H, tr); 1.7-2.1 (4H, m); 3.3-3.8
(12H, m); 7.8-B.0 (lH, m); 8.4-8.5
(lH, m); B . 5-8 . 8 ( lH, m) .
2~3~ r~
-- 10 --
~xample 3
Pyridine-2/4-dicarboxylic acid bi~-M,N'~(2-dimetho~y~
ethyl)amide
For instructions see Example 1 amine component: 2-
S dimethoxyethylamine
Yield: 1.6 g (~rom 3 g of pyridine~-2,4-dicarboxylic
acid), oil
H-NMR (CDC13): ~ = 3.4 (12H, ~); 3.7 (4H, m); 4.5 (2H,
m); 7.9-8.0 (lH, m); 8~4-8.5 (lH, m);
8.7-8.8 (lH, m).
r - ~le 4
Pyridine-2,4-dicarboxylic acid bis-N,N'-(2-m0thoxy-
isoprop~l)a~ide
For instructions ~ee Example 1; amine component: 2-
methoxyisopropylamine;
Yield: 3.3 g tfrom 3 g of pyridine-2,4-dicarboxylic
acid), oil
H-NMR (CDCl3): 6 = 1.3 (6H, d); 3-2 (6H~ ~); 3-5 (4H~
4.4 (2H, m~; 7.9 8.0 (lH, m); 8.4-8.5
(lH, m); 8.7~8.8 (lH, m).
Rxample 5
Pyridine-2,4-dicarboxylic acid bis -N, N ' - ( 2-ethoxyethyl)-
amide
For in~truction6 ~ee Example 1; ~mine c~ nent: etho~y-
ethyli- Ine~
Yields 7.8 g (from 10 g of pyridine-2,4-dicarboxylic
acid), melting point: 42-44~C
H-NMR (CDCl3): ~ = 1.2 (3H, tr); 3.3-3.8 (12H, qu. and
m); 7.9 (lH, m); 8.4-8.S (lH, m); 8.7-
8.8 (lH, m).
Example 6
Pyridine-2,4~dicarboxylicacidbi~-N,N'-(3-hydroxyethyl)-
amide
0.5 g of pyridine-2,4-dicarboxylic acid bis-N,N'-(3-
methoxyethyl)~mide are dis~olved in lO ml of methylene
chloride, and boron tribromide (11 ml~ 1 molar .501ution
in methylene chloride) is added dropwisle at ~78~C. When
the addition ha~ ended, the mixture i8 allowed to come to
room temperature and is subsequen~ly stirxed for 3 hours.
It is poured onto 109 ml of saturated bicarbonate solu-
tion and extracted 3 times with ethyl acetate. The
combined organic phases are dried with magnesium ~ulfate
and evaporated. The crude product is chromatographed on
silica gel.
Yield: 0.45 g; oil
H-NMR (CDCl3): ~ - 1.5-2.2 t4H/ m); 3.4 (4H, m); 3.6 (4H,
m); 7.9-8.0 (lH, m); 8.4-B.5 (lH, m);
8.7-8.8 (lH, m).
Example 7a
Pyridine-2,4-dicarboxylic acid dibenzyl e~ter
30g of pyridine-2,4-dicarboxylic acid are coJ-v~lLed illtO
the acid chloride using 30 ml of thionyl chloride analo-
gously to Example 1 and the acid chloride is reacted with
43.8 g of benzyl alcohol. The product is recrystallîzed
from diisopropyl ether.
Yield: 42.1 g Melting point 63-65~C
Rxample 7b
Pyridine-2-(carboxylic acid)-4-carboxylic acid benzyl
ester
40 g of pyridine-2,4-dicarboxylic acid dibenzyl ester
from Example 7a are added to a suspension of 27.8 g of
12 -
copper-II nitrate in 700 ml of methanol. The mixture i~
boiled under reflux for one hour and, after cooling, the
copper complex is ~iltered off. The comple~ is su~pended
in dioxane and carbon disulfide is pa~sed in. The copper
sulfide which has precipitated is filtered off and ths
organic phase is concentrated. The product i~ ~tirred
with petroleum ether.
~ield: 25.3 g Melting point 113-115~C
~ample 7c
Pyridine-2-[(3-methoxypropyl)-carboxylic acid amide]-4-
carboxylic acid benzyl ester
3.9 g of pyridine-2-tcarboxylic acid)-4-carboxylic acid
benzyl ester from Example 7b are converted into the acid
chloride using 1.2 ml of thionyl chloride analogously to
Example 1 and the acid chloride is reacted with 3-meth-
oxypropylamine to give the amide. For purification, the
product is chromatographed over silica gel using a
mi~ture of cyclohe~A~/ethyl acetate (1:1).
Yield. 4.3 g Oil
r _le 7d
Pyridine-4-(carboxylic acid)-2-(3-methoxypropyl)-car-
boxylic acid amide
4.3 g of the e, ~ur.d from ~xample 7c are dissolved in
100 ml of dio~ane and hydrogenated using 50n mg of
palladium/charcoa]. (10% strength) catalyst under normal
pressure for 4 hours. When the uptake of hydrogen has
ended, the catalyst i~ filtered off with suction and the
solvent i3 stripped of~.
Yield: 3.5 g Melting point 124-126~C
2 ~ s~ Ji
- 13
~xample 7e
Pyridine~4-[carboxylic acid-(2-me~hoxyethyl)~amide]-2-
carboxylic acid t3-metho~ypropyl)-amide
1.8 g of the compound from Example 7d are converted into
S ~he acid chloride using 0.B ml of thionyl chloride in
accordance with ~xample 1 and the acid chloride i~ then
reacted with 2-me~hoxyethyl, inP. For purification, the
product is chromatographed over silica gel using a
mixture of methylene chloride/methanol (20:1).
Yield: 1.0 g Oil
~-NMR ~CDCl3)~ 9-2.0 (2H~ qui); 3.4 (6H, ~); 3.5-
3.7 (8H, m); 6.9 ~lH, s, br); 8.0 (lH,
dd); 8.4 (lH, s, br); 8.5 (lH, s); 8.7
(lHt d)-
~xa~ple 8
Pyridine-2-[carboxylic acid-(2-metho~yethyl)-amide]-4-
carboxylic acid (3-methoxypropyl)-amide
The compound according to Example 8 is prepared analo-
gously to Examples 7a-e by using 2-methoxyethy~ n~ in
the reaction step of Example 7c and 3-metho~y~ o~ylamine
in the reaction step of Example 7e.
Melting point: 69-72~C
H-NMR ~CDCl3): ~ = 1.9-2.0 (2H, qui); 3.4 (3H, s); 3.45
(3H, s); 3.6-3.7 (~H~ m); 7.4 (lH,
br); 7.9 (lH, ddl; 8.3 (lH, br); 8.4
(lH, d); 8.7 ~lH, d).
E~ampl~ 9
~nteral absorbability
Female Wistar rat~ of about 150 g body weight are given
an intragastral administration of 50 mg/kg of the sub~
stance under investigation by means of a stomach probe.
2 ~ 5 ~ ~)
- 14 -
In each case 4 rats are anesthetized after 5; lO; 15; 30;
60; 120; 180 and 240 minutes and exsanguinated via the
Vena Cava. The blood i8 cen~ri~uged immediately and the
compound a.~ stered i8 extracted from the serum using
ether. A~ter the ether ha~ been evaporatRd, the residue
is taken up in 100 ml of mobile phase. ~he mobile phase
consists of 0.05 ~ phosphoric acid and acetonitrile
(4:1). 50 ~l ~f thi~ sample are in~ected into a high
performance liquid chromatography column. Detection is
performed under W of 200 nm with a retention time of 2.2
minutes. The results are documented in Table l.
Table 1: Serum levels of the compounds according to the
invention from Examples 1-3 after a~inistra
tion of 50 mg/kg perorally
1~
Tlme Substance from Substance from Substance ~rom
(minutes) Example 1Example 2 Example 3
x SD x SD x SD
~.3 + 15.451.4 ~ 11.2 8.9 t 3.1
~9.8 + 3.639.2 + 4.0 11.5 + 0.6
39.9 + 11.029.4 ~ ~.71~.7 ~ 1.9
28.1 ~ 3.215.2 + 5.6 10.7 ~ 1.9
9.~ ~ 5.51.4 + 1.0 11.3 ~ 1.5
120 0.3 + 0.3<DL 5.5 ~ 0.9
180 <DL <DL 2.9 + 0.5
240 ~DL <DL 1.7 i 0.4
x = mean value of 4 measurements
SD = stAn~rd deviation
<DL = below the detection lLmit
~xample 10
ph~ ~cological activity
To demonstrAte the effective inhibition of proline
hydroxyla~e and lysine hydroxylase by the compounds
- 15 ~
according to the invention, the hydroxyproline concentra-
tion~ in the liv2r and the 7s 5IV)-collagen concentra-
tions in the serum o~
a) untreated rats (control)
b~ rats to which carbon tetrachloride had been ~ini~-
tered ~ CC14 control)
c) rat~ to which first Cc14 and then a compound according
to the invention had been ~r 1 n i ~tered
were measured (this ~ast method is described by
1~ ~ouiller, C., experimental toxic injury of the liver; in
The Liver, C. Rouiller, Volume 2, pages 335-476, New
York, Academic Press, 1964)o
The action potency of the compounds according to the
invention was det~rrined as the percentage inhibition of
the liver hydroxyproline synthesis and serum 7s-(IV)-
collagen synthesis following oral administration in
comparison with control ~ni 9l B to which only carbon
tetrachloride had been A~' i n i Rtered ~ CC14 control). ~he
result~ are shown in Table 2. The compounds from E~amples
2 and 3 of Ge ~n Patent ~~3r703~559 ~,N'-bis(2-methoxy-
ethyl)-pyri~ine-2,4-dicarboxylic acid diamide and N,N'-
bis(3-isopropox~propyl)-pyridine-2,4-dicarboxylic acid
diamide) are likewise also ~hown as comparison ~ubstan-
ces. Surprisingly, the compounds according to the inven-
tion show a better activity, even after oral ~-' ini~t:ra-
tion, than the intraperitoneally ~l inistered compound
from Example 2 of Germ~n Patent A-3~703,959.
2 ~ 1 .J i~ ~ i f J r
-- 16 ~
Table 2:
SubstancQ Dosage Liver Serum 7B- Admini~-
from hydxo~y~ (IV)-collagen tration
Example proline ~% [% inhibition~
inhibition~
1 2x2 mg 62 28 p.o.
2xlO m~ 90 67 p.o.
2 2x2 mg 25 2 p.o.
2xlO mg 60 35 p.o.
2 (from 2x25 mg 55 48 i.p.
DE-A-3,703,959
3 (from 2x25 mg 49 11 p.o.
DE-A-3,703,~59'
p.o. = peroral
i.p. = intraperitone~l