Note: Descriptions are shown in the official language in which they were submitted.
-1-
A-176>6/=/CGM 341
Process for the preparation of tris-(2L4-diten-butvlnhenyl~phosphite
The invention relates to a process for the preparation of tris-(2,4-ditert-
butylphenyl)
phosphite from 2,4-ditert-butylphenol and phosphorus trichloride in the
presence of a
catalyst.
It is known from DE-A 2,007,070 to prepare triaryl phosphites from phenols and
phosphorus trichloride, the reaction taking place in three stages in
successive sections of
equipment. The process can be operated in the absence of solvent, but problems
then arise
with the formation of foam in the reaction mixture to the point where the
orderly course of
the reaction is interfered with. A process is known from EP-A 0,000,757 for
the
preparation of triaryl phosphites by reacting phosphorus trihalides with
hydroxyaromatic
compounds in the presence of catalysts. According to Example 4 of EP-A
0,000,757
tris-(2,4-ditert-butylphenyl) phosphite can be prepared with the concomitant
use of a
solvent. Working with a solvent has the advantage that the course of the
reaction and
hence also the undesirable foam formation can be controlled, but has, in turn,
the obvious
disadvantage that the solvent, by its volume, reduces the space/time yield and
has to be
heated and cooled with the reactants and, at the end of the process, removed
and worked
up.
The object of the present invention was to avoid the said disadvantages and to
provide a
process which makes it possible to prepare tris-(2,4-ditert-butylphenyl)
phosphite in a
simple manner and in high space/time yields.
In accordance with the invention this is achieved by means of a process which
comprises
carrying out the reaction in an at least three-stage process in which the
2,4-ditert-butylphenol and 40 - 100 % of the catalyst are combined in a
preliminary stage
and are brought together with the phosphorus trichloride in a first stage,
these being
allowed to react under normal pressure and at temperatures of 55 to
70°C for a dwell time
of 15 to 40 minutes, the reaction mixture is then reacted in a second stage
under normal
pressure and at temperatures of over 140°C, remaining amounts of
catalyst being added to
-2-
the first and/or second reaction stage, the reaction mixture is then kept
under reduced
pressure at temperatures of at least 186°C in a third reaction stage,
and the
tris-(2,4-ditert-butylphenyl) phosphite is then isolated from the reaction
mixture, the
process being carried out in the absence of solvents.
The following embodiments, independently of one another, are preferred: for
example,
that the reaction mixture is subjected to a fourth stage, the reaction mixture
being kept in
the fourth stage at at least 186°C and under reduced pressure,
preferably a pressure of 6 to
20 hPa; that a reaction time of 45 to 75 minutes is maintained in the second
stage; that a
reaction time of 1.5 to 2.5 hours is maintained in the third stage; that a
reaction time of 20
to 120 minutes is maintained in the fourth stage; and that a temperature of
190 to 195° is
maintained in the fourth stage.
In a preferred embodiment of the process the 2,4-ditert-butylphenol is added
in a 1-fold to
1.1-fold stoichiometric amount, relative to phosphorus trichloride.
Examples of catalysts available for the process according to the invention are
those
described in EP-A 0,000,757.
Examples of catalysts of this type are compounds belonging to the group
comprising
amines or ammoninium salts, amides of carboxylic acids or of carbonic acid,
non-aromatic
N-containing heterocyclic compounds and salts thereof, primary, secondary and
tertiary
phosphines and salts thereof or esters of phosphoric acids and phosphonic
acids.
The amines and ammonium salts, amides and nitrogen-containing heterocyclic
compounds
or phosphines can contain, as substituents, alkyl, cycloalkyl, aryl;
particularly phenyl,
alkaryl, particularly alkylated phenyl, aralkyl, particularly benzyl, or
alkaralkyl,
particularly alkylated benzyl, groups which preferably contain 1 to 18 C
atoms,
particularly 1 to 12 C atoms, and are interrupted, if appropriate, by oxygen
or sulfur atoms.
Alkyl contains especially 1 to 6 C atoms and cycloalkyl is especially
cyclopentyl and
cyclohexyl.
The catalysts to be used in the form of salts are preferably the halides and
particularly the
chlorides. The salts can also be formed in situ by means of the hydrogen
halide formed in
the course of the process: Nevertheless, it is advantageous in certain cases
to employ the
salts themselves as catalysts.
T
-3-
The amines and ammonium salts comprise one catalyst group. These can be
primary,
secondary and tertiary amines and also salts thereof. The salts also include
the quaternary
ammonium salts. The secondary amines, their salts and the quaternary ammonium
salts
are preferred. The alkyl-substituted and cycloalkyl-substituted amines or
ammonium salts
are preferred.
The following are examples: methylamine, ethylamine, propylamine, n-
butylamine,
t-butylamine, pentylamine, octylamine, dodecylamine, phenylamine, benzylamine,
dimethylamine, diethylamine, methylethylamine, methylbutylamine,
methyoctylamine,
methylphenylamine, ethylbenzylamine, trimethylamine, triethylamine,
tributylamine,
octyldirnethylamine and dimethylphenylamine and also tetramethylamonium,
trimethylethylamonium, triethylmethylamonium, tributylmethylamonium,
tetrabutylamonium, trimethyloctylamonium, triphenylmethylamonium and
tribenzylmethylammonium chloride, bromide or iodide. Examples of other
ammonium
salts are methylammonium, octylammonium, dimethylammonium,
methylcyclohexylammonium, dibenzylammonium, diphenylammonium,
trimethylammonium, tributylammonium, tribenzylammonium and triphenylammonium
chloride, bromide and iodide. The amines and ammonium salts can also contain
aromatic
N-heterocyclic radicals, for example pyridyl. These amines are more effective
than the
purely aromatic N-heterocyclic compounds.
The amides of carboxylic acids constitute another group of catalysts. This
group also
includes the areas and their bisurea derivatives. The amides can be derived
from
polyfunctional, preferably monofunctional, carboxylic acids containing, in
particular, 1 to
14 C atoms. The amides can also be derived from aromatic N-heterocyclic
compounds.
Cyclic amides, for example ~-caprolactam, are also suitable. The amides
derived from
carboxylic acids preferably have the formula
O R2
R1
~R3
n
in which, if n = l, Rt is phenyl, benzyl, naphthyl, cyclohexyl, cyclopentyl,
pyridyl,
hydrogen or alkyl having 1 to 13, preferably 1 to 6, C atoms, if n = 2, Rt is
phenylene,
naphthylene, cyclohexylene or alkylene having 1 to 12, preferably 1 to 6, C
atoms or a
d~'~ r~3W ~. id
-4-
direct bond, and R2 and R3 independently of one another are a hydrogen atom,
phenyl,
benzyl, cyclohexyl and alkyl having 1 to 12, preferably 1 to 6, C atoms or R2
and R3
together are alkylene which preferably has 4-7 C atoms and, if appropriate, is
interrupted
by O or S atoms. Examples are formamide, oxamide, dimethylfonnamide,
acetamide,
N,N-dimethylacetarnide, picoanilide, benzamide, terephthalamide and
trimellitamide.
Dimethylformamide is very particularly preferred as the catalyst in the
present process.
The .Following, besides urea, may be mentioned as examples of amides of
carbonic acid:
tetramethylurea, diphenylurea, dibenzylurea, diethylurea, di-n-octylurea and
bisurea
derivatives, for example ethylenebisurea. Examples of cyclic ureas are
hydantoin and
benzimidazolone.
Non-aromatic N-heterocyclic compounds constitute another group of catalysts
suitable for
the process according to the invention. These can contain 1 to 3 N atoms and,
if
appropriate, one or 2 O and S atoms. They can also be unsaturated. They can be
present in
the form of salts and also in the form of quaternary ammonium bases, and the N
atoms can
be substituted, preferably by alkyl groups having 1 to 12 C atoms. The
following are
examiples: pyrrolidine, D3-pyrroline, N-methylpyrrolidine, dihydroindole,
pyrazolidine,
imidazolidine, a2-pyrazoline, 1-phenylpyrazolidine, oxazolidine, thiazolidine,
oxazoline,
triazolidine, oxadiazolidine, thiadiazolidine, piperidine, morpholine, N-
methylmorpholine,
quinolidine, 1,2-dihydropurine, 8-aza-bicyclo-(3,2,1)-octane, piperazine and
N-methylpiperazine.
The primary, secondary and tertiary phosphines and salts thereof constitute
another group
of catalysts to be used in accordance with the invention. The tertiary
phosphines and their
salts are preferred, amongst the salts the hydrohalides, preferably the
hydrochlorides,
hydrobromides and hydriodides. The phosphorus atom can be unsubstituted or
substituted
by phenyl, benzyl, cyclohexyl and/or alkyl having 1 to l2, preferably 1 to 6,
C atoms. The
following are examples:
methylphosphine, ethylphosphine, hexylphosphine, dadecylphosphine,
dimethylphosphine, ethylmethylphosphine, diphenylphosphine,
dicyclohexylphosphine,
dibenzylphosphine, phenylmethylphosphine, triphenylphosphine,
tribenzylphosphine,
tricyclohexylphosphine, trimethylphosphine, triethylphosphine,
tripropylphosphine,
tributylphosphine, triisobutylphosphine, tripentylphosphine, trihexylphosphine
and
dimethylphenylphosphine and hydrochlorides, hydrobromides and hydiodides
thereof.
-5-
In the case of the esters of phosphoric acid and phosphonic acid the alcohol
radicals are
preferably derived from phenols, and especially, Ct-CtBalkanols and
cycloalkanols, for
example from phenol, 2-methylphenol, cyclohexanol, methanol, ethanol,
propanol,
butanol, hexanol, octanol, i-octanol, dodecanol and octadecanol. The following
are
examples of phosphonic acids: phenylphosphonic, benzylphosphonic,
cyclohexylphosphonic, methylphosphonic, ethylphosphonic, propylphosphonic,
butylphosphonic, pentylphosphonic and hexylphos,phonic acid.
The catalyst can be employed in amounts of, for example, 0.005 to 10 mol-%,
appropriately in amounts of 0.05 to 8 mol % and preferably of 4 to 6 mol %, in
each case
relative to the phosphorus trichloride.
The process according to the invention is carried out in apparatus known per
se. Reaction
vessels in the form of stirred kettles are particularly appropriate and
reaction vessels in the
form of reaction columns, particularly with inserts which force intimate
mixture on the
reactants by means of circuitous paths are also possible.
A heatalile reaction vessel eqiupped with at least two inlets, one outlet and
a stirring
device are appropriately envisaged for carrying out the first stage of the
process. The
phosphorus trichloride is preferably introduced directly into the reaction
vessel via one
inlet, the 2,4-ditert-butylphenol through a column, for example a packed
column, and
through the second inlet. Depending on its nature, the catalyst can be
introduced into the
reaction vessel in part separately via a third inlet. 40 - 100 % of the
catalyst are put into
the second inlet, also via the packed column. The streams of starting
materials are
appropriately controlled in such a way that the phosphorus trichloride is
taken initially and
the catalyst and the phenol compound reach the reaction vessel via the packed
column.
The HCl formed during the reaction which sets in escapes through the packed
column
countercurrent to the phenol compound. Entrained phosphorus trichloride is
absorbed by
the phenol compound and recycled into the reaction vessel. The reaction is
already
catalysed by the catalyst which is present in the column at the same time, and
the escaping
phosphorus trichloride reacts with the phenol compound which flows through the
column.
Thus a preliminary step already takes place in the column. The reaction
mixture then
flows into the first reaction vessel. The reactants can be intimately mixed in
the reaction
vessel by means of a stirring device. At the same time the reaction mixture is
heated to
temperatures of 55° to 70°C. Temperatures of 60 to 70°C
are preferred. The HCl formed
in the reaction is removed via the packed column and is disposed of. The first
stage of the
-6-
reaction is complete after 15 to 40 minutes. It is then possible to carry out
the second stage
of the reaction in the first reaction vessel, but preferably the contents of
the reaction vessel
from the first stage are transferred into a second reaction vessel, preferably
a heatable
stirred kettle. This stirred kettle preferably has feed inlets for the
reaction mixture from the
vessel of the first stage and, if appropriate, for the addition of a second
part amount of the
catalyst, and also has a gas outlet equipped with a reflex condenser for
discharging the
I-ICl formed during second reaction stage. The second stirred kettle is also
preferably
equipped with a stirring device. When the second stage starts the reaction
mixture is
immediately heated to over 140°C, for example 145 to 200°C,
preferably 150 to 170°C
and particularly preferably about 160°C, and is preferably kept at
these elevated
temperatures for 45 minutes to 75 minutes, preferably 60 minutes. When the
second stage
is complete, the reaction mixture is subjected to the third stage either in
the same reaction
vessel or, preferably, in another reaction vessel. The heatable reaction
vessel for the third
stage is preferably equipped with an inlet for the reaction mixture, an outlet
for reactants
which escape in the form of gas, an outlet for the reaction mixture and,
preferably, a
stirring device. The outlet for the reactants which are evolved, in particular
HCI, can have
a reflex condenser and, if appropriate, also a desublimer in order to separate
the escaping
reactants. Furthermore, since the third stage is carried out under reduced
pressure,
appropriate arrangements are provided, such as a vacuum pump and appropriate
seals and
valves. The third stage of the reaction is carried out at at least
186°C, appropriately at
186°C to 210°C and preferably at 190 to 195°C, under a
reduced pressure of,
appropriately, 10 to 60 hPa, in particular 10 to 20 hPa and preferably 15 hPa.
The reaction
time for the third stage is, for example, 1 to 2.5 hours, preferably 2 hours.
After the expiry
of the reaction time the reaction mixture is, if desired, subjected to the
fourth stage. This is
effected either by bringing the reaction mixture into the reaction vessel of
the third stage
or, preferably, bringing it into another reaction vessel and subjecting it to
the conditions of
the fourth stage in this other xeaction vessel, which can be heated and
evacuated. The
reaction mixture is also preferably stirred in the fourth stage. The xeaction
vessel of the
fourth stage should therefore contain not only an inlet for the reaction
mixture but also a
stirring device and a suitable outlet fox removing, in particular, the gaseous
products of the
reaction, preferably an oulet containing a reflex condenser and a desublimer.
Finally, a
suitable outlet should be provided at this reaction vessel for discharging the
reaction
material. Since the fourth stage is to be carried out under reduced pressure,
appropriate
means of producing a vacuum and seals should be provided. The reaction mixture
is
appropriately subjected to the fourth stage for 20 to 120 minutes, preferably
60 minutes.
Meanwhile the pressure can be, for example, 6 to 20 hPa, appropriately 10 to
15 hPa and
_7_
preferably 10 hPa. The temperature in this fourth stage is at least
186°C, appropriately
186°C to 210°C and especially 190 to 195°C.
It is possible to distill off the 2,4-ditert-butylphenol which may be used in
excess from the
reaction vessel of the third stage or, if used, the fourth stage, and, if
desired, to recycle it to
the reaction. The excess 2,4-ditert-butylphenol can also be disposed of from
the reaction
mixture of the third stage or, if used, the fourth stage.
A preferred embodiment consists in carrying out the process of the present
invention in a
three-stage kettle cascade.
The particularly preferred embodiments of the present invention include
carrying out the
process in a four-stage kettle cascade.
Another preferred embodiment is to stir the reaction mixture in at least one
stage.
It is very particularly preferable to stir in all four stages.
It is also preferable in the present process to maintain a temperature of 170
to 190°C in the
second stage.
Another preference in the process of the invention is to maintain a
temperature of 190 to
195°C in the third and the fourth stage, independently of one another.
In the process according to the invention the amount of catalyst envisaged is
added to the
reaction to the exent of 40 to 100 % through the packed column in which the
preliminary
stage described above is earned out. It is appropriate to add 50 to 100 %,
preferably 70 to
100 % and especially 90 to 100 %, of the catalyst to the reaction via the
packed column.
Remaining amounts of catalyst, insofar as not fed in via the packed column,
are added in
the first and/or second stage of the reaction.
Remaining amounts of catalyst, unless 100 % of the catalyst are added to the
reaction via
the packed column, are preferably added to the first reaction stage.
It is very particularly preferable to add the whole amount of catalyst, i.e.
100 % of the
_g-
catalyst, to the reaction via the packed column. All percentage figures relate
to weight.
The continuous mode of operation is particularly preferred for the process
according to the
invention.
The present process according to the invention has the advantage that
tris-(2,4-ditert-butylphenyl) phosphite, which, in terms of processes, can
only be obtained
with difficulty, can be obtained in a high space/time yield. As a result of
dispensing with a
solvent more reaction volume is available in the present process, which makes
higher
reaction temperatures and hence higher reaction rates possible, and the
heating up times
and the time needed for distilling off the solvent prior to the further
working up, in
particular the crystallization, of the product are omitted.
The processes of the state of the art quoted initially are incapable of giving
satisfaction in
this respect, since it is absolutely necessary to use a solvent for the
compound of present
interest, tris-(2,4-ditert-butylphenyl) phosphite, by virtue of its high
melting point.
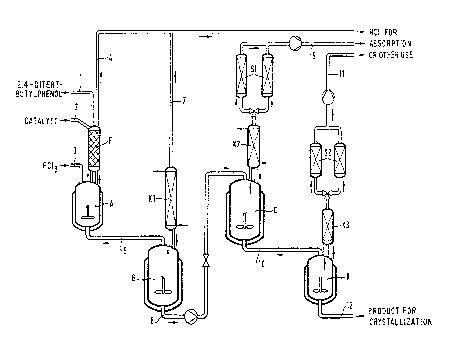
Example: The following flows of materials:
Addition of ~ g/hour ~ Addition to
2,4-ditert-butylphenol, 994 via packed column (F) into
98.9% pure [1] i I reactor (A)
Phosphorus trichloride (3] I 216.2 ~ Reactor (A)
Dimethylformamide (2] ~ ( ~ via packed column (F) into
(catalyst) reactor (A)
are set up in a four-stage stirred kettle cascade such as can be seen from the
diagram
(Figure 1), composed of a) a reactor (A) having a packed column F for the
first stage, b) a
main reactor (B) equipped with a reflux condenser (Kl) for the second stage,
c) a second
main reactor (C) equipped with a reflux condenser (K2) and after that a
desublimer (S1)
for the third stage, and finally d) an after-reactor (D) equipped with a
reflux condenser
(K3) and a desublimer (S2).
a) Conditions in reactor (A):
-9-
Normal pressure, temperature 65°C, dwell time 30 minutes, discharge of
89.8 g/hour of
HCI (4] via the packed column (F) to waste disposal and discharge of 1126.4
g/hour of
reaction mixture [5] via the bottom valve to the first main reactor (B).
b) Conditions in the first main reactor (B):
Normal pressure, temperature 16S°C, dwell time 1 hour, discharge of 64
g/hour of I-ICl [7]
via a reflex condensder (K1) and discharge of 1062.4 g/hour of reaction
mixture [8] via
the bottom valve to the second main reactor (C).
c) Conditions in the second main reactor (C):
Reduced pressure 15 hPa, temperature 190°C, dwell time 2 hours,
discharge of I3 g/hour
of HCl [9] via a reflex condenser (K2) and a desublimer (S 1) and discharge of
1049.4
g/hour of reaction mixture [ 10] via the bottom valve into the after-reactor
(D).
d) Conditions in the after-reactor (D):
Reduced pressure 10 hPa, temperature 190°C, dwell time 60 minutes,
discharge of 5.4
g/hour of HCl [I 1] via a reflex condensder (K3) and a desublimer (S2) and
discharge of
1045 g/hour of reaction mixture [12], containing 1003.2 g/hour of
tris-(2,4-ditert-butylphenyl) phosphite, to be crystallized.
The end product is of high purity, and virtually no content of monochloro and
dichloro
compounds or unreacted PC13 could be detected.
Figure 2 shows an alternative embodiment differing from the embodiment
described
above in that the reflex condenser (K3) also constitutes a distillation device
in which the
excess 2,4-ditert-butylphenol can be removed. This 2,4-ditert-butylphenol can
be recycled
to the reaction via the feed inlet [I].