Note: Descriptions are shown in the official language in which they were submitted.
2~3~67 FP-ZP-5
~PY
DESCRIPTION
TITLE OF THE INVENTION
INDOLEACETIC ACID DERIVATIVES, PROCESS FOR THEIR
PREPARATION, AND MEDICINES CONTAINING SAME AS ACTIVE
INGREDIENTS
THCHNICAL FIELD
The present invention relates to an indoleacetic acid
derivative which has an anti-inflammatory action, an
antirheumatic action and an anti-ulcer action, and
particularly to an indoleace-tic acid derivative and a sal-t
thereof which have a reduced adverse effec-t than that of
conventional non-steroidal anti-inflammatory drugs, a
method of preparing them and an anti-inflammatory drug,
antirheumatic drug and anti-ulcer drug each of which
contains one of them as an active ingredient.
BACKGROUND ART
Anti-inflammatory drugs are classified into steroid
hormones, non-steroidal anti-inflammatory drugs,
antiphlogistic enzymes and immunosuppression agents. Of
these drugs, non-steroidal anti-inflammatory drugs are most
important drugs and, in recent, they ha~e been actively
developed all over the world. However, such non-s-teroidal
anti-inflammatory drugs have many problems in the clinical
field. The most critical problem is adverse effects such
203~6~7
as damages to the digestive organs and the kidneys.
Indomethacin, which is a typical indoleace-tic acid-type
compound having the strongest anti-inflammatory action
among non-steroidal anti-inflammatory drugs, is a first
choice drug as a remedy for rheumatism. Although
indomethacin has a great curative effect, i-t has adverse
effects such as injuries to the gastroin-testine and the
kidneys and thus has a critical problem in use. Such
adverse effects are serious since indomethacin must be
continuously administered to the rheumatics for a long
period and a large amoun-t of indomethacin must be used as
an anti-inflammatory drug. As -this type of anti-
inflammatory drugs manifest curative effects and adverse
effects which are much different depending upon patients,
it is necessary to use various types of drugs.
There is thus demand for an anti-i.nflammatory drug
which has reduced adverse effects such as injuries to the
gastrointestine and the kidneys and which is effective wi-th
excellent persistence.
On the other hand, a group of anticholinergic drugs
and histamine H2 receptor blockers such as cimetidine are
generally used as drugs effective for controlling the
gastric-acid secretion in the clinical field.
Anticholinergic drugs have adverse effec-ts of
controlling the gastric excretory movement and controlling
-the thirst, mydriasis and sudation, and also have a limi-t
2~3~
to the use for inhibi-ting the aggrava-tion of ulcer and
preventing -the recurrence thereof even if -the drugs are
used in amounts which allow the gastric-acid secre-tion to
be substantially inhibited. Cimetidine exhibits adverse
effects such as an undersirable central action and an
antiandrogen action, and particularly has a problem wi-th
respect to the lowering in defensive factors in gastric
mucosa, which is observed in curing treatmen-t for a long
period. It is said that this mainly causes the recurrence
of ulcer after the use of cime-tidine has been stopped.
There has been arisen a demand for a compound capable
of effec-tively controlling the secretion of the gastric
acid serving as an aggressive factor, capable of
reinforcing the defensive factor (cytoprotection ac-tion)
and also having an excellent anti-ulcer action.
Considering the above~mentioned situation, the
inventors energe-tically researched with a view to
developing an excellent anti-inflammatory drug,
antirheuma-tic drug, and anti-ulcerative drug. As a result,
the inventors found that novel indoleacetic acid
derivatives expressed by the formula (I) below and sal-ts
thereof have an excellent anti-inflammatory ac-tion,
antirheumatic action and anti-ulcer action and exhibit
extremely reduced adverse effects. This led to the
achievement of the present invention.
203~6~7
DISCLOSURE OF INVENTION
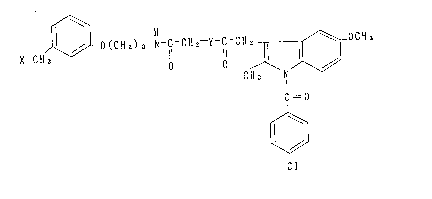
The present invention provides indoleacetic acid
derivatives expressed by the following formula (I) and salts
thereof: ~
X-C1~ ~ O(C11z)3-g-C-C1~z-Y-C-C1l ~ OC~13
C=O
( I )
[wherein X denotes a piperidino group, l-pyrrolidinyl group
or 3-hydroxy-l-pyrrolidinyl group; Y denotes
-(CH)l-- (wherein Rl denotes a hydrogen atom or a lower
Rl alkyl group, l denotes O , l or 2) or
-S - (CH)n~- (wherein R2 denotes a hydrogen atom, a lower
()m R2 alkyl group or a hydroxyl group; m denotes
O, l or 2; and n denotes l, 2, or 3)]. The present
invention also provides a method of producing these
compounds and an anti-inflammatory drug, anti-rheuma-tic drug
and anti-ulcerative drug each of which contains as an active
ingredient one of the compounds.
In tXe compounds (I) of the present invention, examples
of lower alkyl groups include straight chain, branched chain
or cyclic alkyl groups each having l to 6 carbon atoms such
5 2~3~6~7
as methyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-
butyl, isobutyl, 1-methylpropyl, tert-butyl,
cyclopropylmethyl, n-pentyl, cyclopentyl, n-hexyl,
cyclohexyl and the like.
Examples of salts of the compounds (I) of the present
invention include salts, which are physiologically
allowable, for example, inorganic acid salts such as
hydrochlorides, sulfates, nitrates, and the like; and
organic acid salts such as acetates, oxalates, tartrates,
malates, citrates, maleates, fumarates and the like.
Typical examples of the compounds (I) of the present
invention include the following:
N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
N-[3-[3-(3-hydroxy-1-pyrrolidinylmethyl)phenoxy]-
propyl]carbamoylmethyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
2~[N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamoyl]-
ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
2~3:16~7
2--[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-rnethyl-3-
indolylacetate
2-[N-[3-[3-~3-hydroxy-1-pyrrolidinylmethyl)phenoxy]-
jpropyl]carbamoyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
l-methyl-2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]
carbamoyl]ethyl 1-(4-chlorobenzoyl3-5-methoxy-2-methyl-3-
indolylacetate
1-methyl-2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]
propyl]carbamoyl]ethyl 1-(9-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
3-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamoyl]-
propyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
3-[N-[3-[3-(1-pyrrolidinylmethyl)phehoxy]propyl]-
carbamoyl]propyl 1-~4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamoyl-
methylthio]ethyl l-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethylthio]ethyl. 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
2~3~6~
2-[N-[3-[3-(3-hydroxy-1-pyrrolidinylmethyl)phenoxy]-
propyl]carbamoylmethylthio]ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
2-~N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamoyl-
methylsulfinyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-
3-indolylacetate
2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethylsulfinyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylsulfonyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethylsulfonyl]ethyl l-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
2-[N-[3-[3-(3-hydroxy-1-pyrrolidinylmethyl)phenoxy]-
prcpyl]carbamoylmethylsulfonyl]ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
1-methyl-2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylthio]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
l-methyl-2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]-
propyl]carbamoylmethylthio]ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
8 2~3~7
1-methyl-2-[N-[3-[3-(3-hydroxy-1-pyrrolidinylmethyl)-
phenoxy]propyl]carbamoylmethylthio]ethyl 1-(9-
chlorobenzoyl)-5-methoxy-2-methyl-3-indolylacetate
1-methyl-2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylsulfinyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
1-methyl-2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]-
propyl]carbamoylmethylsulfinyl]ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
1-methyl-2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylsulfonyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
1-methyl-2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]-
propyl]carbamoylmethylsulfonyl]ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
1-methyl-2-[N-[3-[3-(3-hydroxy-1-pyrrolidinylmethyl)-
phenoxy]propyl]carbamoylmethylsulfonyl]ethyl 1-(4-
chlorobenzoyl)-5-methoxy-2-methyl-3-indolylacetate
3-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylthio]propyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
3-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl~-
carbamoylmethylthio]propyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
~03~667
3-[N-[3-[3-(piperi~inomethyl)phenoxy]propyl]-
carbamoylmethylsulfinyl]propyl 1-(9-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
3-[N-~3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethylsulfinyl]propyl 1-(4-chlorobenzoyl)-5-
methoxy 2-methyl-3-indolylacetate
3-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylsulfonyl]propyl 1-(4-chlorobenzoyl)-5-
me-thoxy-2-methyl-3-indolylacetate
3-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethylsulfonyl]propyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
2-hydroxy-3-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylthio]propyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
2-hydroxy-3-[N-[3-[3-(1-pyrrolidinylmethyl)-
phenoxy]propyl]carbamoylmethylthio]propyl 1-(4-
chlorobenzoyl)-5-methoxy-2-methyl-3-indolylacetate
2-hydroxy-3-~N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethylsulfinyl]propyl 1-(9-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate
2-hydroxy-3-[N-[3-[3-(1-pyrrolidinylmethyl)-
phenoxy]propyl]carbamoylmethylsulfinyl]propyl 1-~4-
chlorobenzoyl)-5-methoxy-2-methyl-3-indolylacetate
203~6~7
1 0
2-hydroxy-3-[N-[3-[3-~piperidinomethyl)phenoxy]propyl]-
carbamoylmethylsulfonyl]propyl 1-(4-chloroben~oyl)-5-
methoxy-2-methyl-3-indolylacetate
2-hydroxy-3-[N-[3-[3-(1-pyrrolidinylmethyl)-
phenoxy]propyl]carbamoylmethylsulfonyl]propyl 1-(4-
chlorobenzoyl)-5-methoxy-2-methyl-3-indolylacetate
For example, the compounds of the present invention can
be produced by the following method:
¢~L
X-CHz O(CH2) 3 - N - C - Cll z - Ya +
o
( Il ) o
H 3 C O ~,~C 1l ~ C - I'~
C = O ~ ( I )
( m )
Cl
[wherein Ya denotes a hydroxyl group, a halogen atom,
-(CH)l-z (wherein Z denotes a hydroxyl group or a
R1 halogen atom, R1 and 1 each denote the same as
that described above) or
-S - (CH)n-Z (wherein R2, m, n and Z each denote the
()m R2 same as that described above),
2~3~7
1 1
A denotes a hydroxyl group, a halogen atom or an active
ester residue, and X denotes the same as that described
above].
Namely, an acetamide compound expressed by the formula
(II) is reacted with an indoleacetic acid derivative
expressed by the formula (III) to form a compound (I) of the
present invention.
Examples of indole derivatives (III) include acid
halides, acid anhydrides, mixed acid anhydrides, 4-
nitrophenyl esters and 2,4-dinitrophenyl esters.
The reaction is preferably effected at -5 to 150C in
the presence or absence of a deoxidizer in a single or mixed
reaction solvent selected from the group consisting of
ether, tetrahydrofuran, dioxane, benzene, toluene,
chloroform, dichloromethane, acetonitrile and
dimethylformamide.
A tertiary amine such as pyridine, trimethylamine,
triethylamine or the like, an alkali carbonate, alkali
hydroxide, alkali hydride or basic resin can be
appropriately selected and used as a deoxidizer.
When A in the formula (III) is a hydroxyl group, it is
preferable to use a condensing agent such as 1,3-
dicyclohexylcarbodiimide (DCC), 1,1'-carbonyldiimidazole
(CDI) or the like.
2~3~667
12
An acetamide compound (II) which is a raw material in
the above-described method can be produced by, for example,
the following reaction:
X-CHz ~ O(CH2)3- Nllz ~ RO - C- CH2 - Y -H
o
(IV) (V)
( 11 )
[wherein R denotes a lower alkyl group or a phenyl group
which may be substituted, and X and Y each denote the same
as that described above].
In other words, a phenoxypropylamine compound (IV) is
reacted with an acetate compound (V) in a reaction solvent
to form an acetamide compound (II). As occasion demands,
the hydroxyl group of the compound (II) is then halogenated
by a usual halogenation method to form a compound (II) in
which Ya is a halogen atom.
The thus-formed compound (I) of the present invention
can be changed to a desired salt by a general method.
A description will now be given on the pharmacological
action, adverse effects and toxicity of the compounds (I)
of the present invention.
(a) Pharmacological action~
[Test of carrageenin plantar edema]
2~3~7
13
Male Wistar rats each having a body weight of about
100 to 200 g were divided into groups each consisting of 10
rats, and were fasted under water feeding for 24 hours.
After the body weight of each rat had been measured, the
volume of the right hind-leg of the rat was measured by
using an apparatus for measuring plantar volumes, The ra-ts
were then divided into groups so that the groups have the
same body weight average and plantar volume average. A
test compound was suspended in an aqueous solution of 5%
tragacanth gum and then orally administered -to each of the
rats. 0.1 ml of a physiological saline of 1% carrageenin
was hypodarmically injected into the sole of the right
hind-leg of each rat. The volume was measured for each rat
every 1 hour for 5 hours. An edema rate was determined by
using a rate of increase in the volume measured after
treatment to the volume measured before treatment. The
results are shown in Table 1.
-
`
14 2~3~67
Table 1
Compound Molecular Dose Carrageenin e~ema
weight ~mol/kg mg/kg Inhibition ED50
rate at peak ~mol/k~ ~ ma/kg
Example 8.4 6.4 34.9
2 762.24 25.2 19.2 50.427.8 21.1
84.0 64.0 62.6
Example 8.4 6.9 19.1
822.36 28.0 23.0 41.247.8 39.3
84.0 69.0 68.4
280.0 230.0 72.7
Indo- 2.8 1.0 24.6
methacin357.81 8.4 3.0 49.820.6 7.4
(Control 28.0 10.0 59.4
compound _ 4 0 30.0 55.9
As seen from Table 1, the compounds in Examples 2 and
5 showed ED50 values which were substantially -the same as
and about 2 times, respectively, that of indomethacin.
Concerning the inhibi-tion rate at the peak during
administration of 84.0 ~mol/kg, both compounds exhibited
slightly higher values than that of indomethacin.
[Test of injury to gastric mucosa]
Male Wistar rats each having a body weight of abou-t
180 to 200 g were divided into groups consis-ting o~ 10 rats
and were fasted for 24 hours. ~f-ter body weight of each
2 ~ 7
rat has been measured, a test compound was suspended in an
aqueous solution of 5% tragacanth gum and then orally
administered to each rat. The rats were fasted again, and
after 5 hours had passed, they were killed by cervical
dislocation. A 1% formalin solution was injected into the
gastric cavity of each of the rats so tha-t it was semi-
fixed. The gaster of each rat was cut along -the greater
curvature of the stomach, and the ulcer index of each rat
was measured under a stereoscopic microscope and determined
by the Munchow method. The Mann-Whitney U-test was applied
to the statistic data processing. The results are shown in
-
\\
%~3~ ~67
16
Table 2
Compound Molecular _ Dose __ Ulcer Ulcer .
weight ~mol/kg mg/kg Index occurrence
_ _ (%) .
Control _ _ _ _ 0.0+0~00 . 0
Example 25.2 19.21.3+0.63 0
2 762.2484.0 64.05.9+4.92 14
_ 251.9 1~2.017.5~+15.84* 40
Example 28.0 23.00.1+0.10 0
822.3684.0 69.00.0+0.00 0
280.0 230,0 ~ 0
Indo- 28.010.046.5i15.00** 70
methacin 357.81 56.0 20.0 52.8+17.47** 70
(control 84.0 30.0 62.1+16.50** 80
compQund .
* p<0.05 ** p<0.01
As can be seen from Table 2, compounds of Examples 2
and 5 show an extremely low degree of injuiry to the
gastriG mucosa, as compared with indomethacin.
Particularly, indomethacin shows 70% ulcer occurrence while
the compound of Example 5 shows no occurrence of ulcer even
when a dose by mole was 10 -times -tha-t of indomethacin.
17 2 ~3~ ~ 67
[Test of inhibition of gas~ric-acid secretiOn by histamine
stimulation]
Male SD rats (Japan Charles River Co., Ltd.) each
weighing 1~0 to 200 g were ~asted for 24 hours before
celiotomy with etherrization. A feeding tube (fr. 3.5) for
injecting test medicines was inserted into the duodenum,
and the pylorus was ligatured. A polyethylene tube (inner
diameter: 7mm) was used in the anterior stomach and gastric
fistula was performed. Af-ter washing the inside of the
stomach with heated physiological salt solution (37C)
several times, the body was closed. A tube with a fin was
inserted into the caudal vein and was fastened with a tape
and it was connected to a infusion pump (Harvard). The
rats were`enclosed in a KN Ballman II cage (Natsume
Seisakusho). Gastric juice flowed from the fistula was
collected with a messcylinder every one hour. Starting
for~ one hour after the operation, histamine was
continuously injected at a rate of 1. 4 ml/hour through the
caudal vein. Then, test compounds suspended in 0.5% sodium
caboxy methyl cellulose (Na-CMC) were administered one hour
after the start of the histamine injection. The gastric
juice thus extracted in a period from one hour after the
administration of the chemical to four hours after that was
titrated by using an automatic titration aparatus (Kyoto
Electronic) with 0.lN NaOH to adjust pH 7, and the quantity
18 2~31L~7
of the gastric juice was measured to obtain the total acid
output.
The inhibition rate (%) of the gastric acid secretion
was calculated form the following e~uation, which results
are shown in Table 3.
Inhibition Rate (~) of Gastric Acid secretion
/total acid out~ut of test com~ound ~rou~t \ x 1 oo
total acid output of control group J
Tabl 3 .
Test compound Dose Gastric-acid secre~ion
(mg/kg) inhibition rate (%)
Example 5 22 47.4
t. 44 74.1 _
Cimetidinè
(Control 30 32.3
compound) .
[Acute toxicity test]
A test compound was suspended in an aqueous solution of
5% gum arabic and then orally administered to each of 4 to
5-week old ICR male mice (Charles River Co., Ltd.) each
having a body weight of 22 to 30 g in each group consisting
of 3 to 4 mice, once in each of doses 25, 50, 75, 200 and
500 mg/kg. They were observed for 1 week.
2~3~67
1 9
As a result, all the mice in the groups, to which 25,
50 and 75 mg/kg of indomethacin were respectively
administered, died within 48 to 96 hours. Meanwhile,ns death
example was observed in all the groups, to which the
compounds of Examples 2 and 5 were respectively
administered, 1 week after the administration. As
described above, each of the compounds of the present
invention has an extremely low degree of toxicity as
compared with indome-thacin.
Each of the compounds of the present invention is
administered in an oral, intramuscular, subcutaneous, or
intravenous manner or a parenteral manner used for
suppositories. Although the dose of each compound depends
upon the disease conditions, age and sexuality of a patient,
it is generally 1 to 1000 mg per day for one adult, and it
is particularly preferable to administer 5 to 300 mg of a
compound 1 to 3 times a day.
The compounds of the present invention are formed into
preparations such as tablets, granules, powder medicines,
capsules, injection drugs, solutions, eye drops, nasal
drops, suppositories, ointments, plasters, cream drugs,
suspensions, aqueous liniments, or the like by normal
methods used in the technical field of preparation.
Examples of additives that are used in preparation include
cellu ose, lactose, sucrose, mannitol, sorbitol, starch
~316~7
Ipotato, cone, rice, wheat and the like), gelatin,
tragacanth rubber, polyvinyl pyrrolidone, carboxymethyl
cellulose, sodium carboxymethyl cellulose, calcium
carboxymethyl cellulose, hydroxypropyl cellulose, talc,
magnesium stearate, calcium stearate, synthetic aluminum
silicate, polyethylene glycol, polyoxyethylene hardened
castor oil, polysorbate, propylene glycol, glycerin, boric
acid, benzalconium chloride, chlorobutanol, sodium
dehydroacetate, wittepsole, macrogol, cacao butter, sodium
chloride, potassium chloride, alcohol, water and the like.
Some of these additives are appropriately selected and used.
Examples
The present invention is described in detail below with
reference to examples.
(Example 1)
N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethyl 1-~4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
_ _ _ _ . _ _ _
\\~
.~
2 1 2 0 3 ~ 6 ~ 7
N-CHz ~ 0 (CH2) 3-N-C-Cllz
O
Cl,
C=O
~C I
3.6 g of 2-hydroxy-N-[3-[3-tpiperidinomethyl)phenoxy]-
propyl] acetamide and 1.5 ml of pyridine were dissolved in
50 ml of dry acetonitrile, and 4 g of 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetyl chloride was gradually
added to the resultant solution at room temperature under
agitation over a time of 20 minutes. ~fter agitation at
room temperature for 1 hour, the solvent was distilled off
under reduced pressure to obtain an oily substance. 30 ml
of a saturated aqueous solution of sodium hydrogencarbonate
was then added to the oily substance, followed by 3
extractions with 50 ml of chloroform. After the thus-
obtained extract had been then dried over anhydrous sodium
sulfate, chloroform was distilled off under reduced
pressure. The thus-obtained residue was purified by using
silica gel column chromatography (developing solvent;
chloroform : methanol = 20 : 1) to obtain as an oily
22 ~3~7
substance 6.9 g of N-[3-[3-(piperidinomethyl)phenoxy]
propyl]carbamoylmethyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate.
NMR (CDC13) ~: 1.33 - 1.67 (6H, br), 1.76 (2H, q), 2.20
- 2.50 (4H, br), 2.36 (3H, s), 3.25 (2H, s), 3.38 (2H, 5),
3.72 (2H, s), 3.79 (3H, s), 3.88 (2H, t), 4.55 (2H, s), 6.13
(lH, br), 6.66 - 7.25 (7H, m), 7.38 t2H, d), 7.60 (2H, d)
(Example 2)
N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamoyl-
methyl 1-(9-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate maleate
6.4 g of the indoleacetic acid compound obtained in
Example 1 was dissolved in 50 ml of benzene, and a solution
obtained by dissolving 1.4 g of maleic acid in 50 ml of
ether was added to the resultant solution. After excess
ether had been further added to the resultant mixture and
then cooled, the solvent layer was removed by decantation.
50 ml of benzene was then added to the oily residue,
followed by agitation under cooling, to obtain 6.5 g of
slightly yellow crystals. The thus-obtained crystals were
recrystallized from benzene to obtain N-[3-[3-
(piperidinomethyl)phenoxy]propyl]carbamoylmethyl 1-(4-
chlorobenzoyl)-5-methoxy-2-methyl-3-indolylaceta-te maleate.
Melting point: 92 to 95C
23 2~31~7
IR (KBr) cm~1: 3400 - 3800, 2940, 1745, 1663, 1580,
1480, 1360, 1165
NMR (CDCl3) ~: 1.5 - 2.1 (8H, br), 2.30 (3H, s), 3.0 -
3.7 (6H, br), 3.76 ~3H, s), 3.7 - 4.1 (2H, m), 4.05 (2H,
br), 4.55 (2H, s), 6.26 (2H, s), 6.50 - 7.26 (7H, m), 7.36
(2H, d), 7.58 (2H, d), 13.5 - 14.7 (2H, br)
(Example 3)
N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamoyl-
methyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate maleate
366 mg of sodium hydride (55%) was suspended in 6 ml of
anhydrous dimethylformamide, and 3 g of indomethacin was
added to the resultant suspension under agitation. 2 g of
potassium iodide and 2.6 g of N-[3-[3-(piperidinomethyl)-
phenoxy]propyl]-2-chloroacetamide were added to the
resultant mixture, and the mixture was then heated at 80 to
90C for 3 hours under agitation. The solvent was
distilled off under reduced pressure, and the resultant
residue was dissolved in 80 ml of water. The resultant
aqueous solution was then subjected to 2 extractions with 60
ml of ethyl acetate. The obtained organic solvent layer
was washed with a saturated salt solution and then dried
over magnesium sulfate. The solvent was distilled off
under reduced pressure, and the resultant residue was
purified by separation using silica gel column
24 2~31~7
chromatography (developing solvent; chloroform : methanol =
97 : 3) to obtain 1.4 g of an oily substance.
The thus-obtained oily substance was dissolved in 20 ml
of benzene, and an saturated ether solution of maleic acid
was then added to the resultant solution until oily
precipitates were no longer produced. After the solvent
layer had been then removed by decantation, 20 ml of benzene
was added to the thus-obtained oily substance, followed by
agitation under cooling, to obtain 1.6 g slightly yellow
crystals of N-[3-[3-(piperidinomethyl)phenoxy]propyl]-
carbamoylmethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate maleate.
Melting point: 92 - 95C
(Example 4)
2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamoyl-
methylthio]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate
CN-CII 2 -¢~ O (Cll z) 3-N-C-CH 2
S CH ZCH Z O C CH 1 ~ ~I OCI 3
c =o
2~3~6~7
4.2 g of 2-(2-hydroxyethyl~1-thio~-N-~3-[3-
(piperidinomethyl)phenoxy]propyl] acetamide and 5 ml of
anhydrous pyridine were dissolved in 70 ml of dry
acetonitrile, and 4.8 g of 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetyl chloride was gradually added to the
resultant solution over a time of 20 minutes under agitation
at room temperature. The resultant mixture was then
agitated for 1 hour at the same temperature and further at
40 to 50C for 1 hour. The solvent was distilled off under
reduced pressure, and 50 ml of a saturated aqueous solution
of sodium hydrogencarbonate was then added to the resultant
residue. The resultant mixture was then subjected to 2
extractions with 50 ml and 30 ml of chloroform, and the
chloroform solution was dried over anhydrous sodium sulfate.
After the solvent had been distilled off under reduced
pressure, the resultant residue was purified by silica gel
column chromatography (developing solvent; chloroform :
methanol = 42 : 3) to obtain as an oily substance 7.2 g of
2-[N-[3-[3-(piperidinomethyl)phenoxy]-
propyl]carbamoylmethylthio]ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate.
IR ~liquid film) cm~1: 3500 - 3400, 2950, 1730, 1655,
1590, 1530, 1488, 1452, 1262, 1220, 1160, 1103, 1038, 752
- .
~3~6~7
26
NMR (CDC13) ~: 1.50 (6H, br), 1.99 (2H, q), 2.35 (3H,
s), 2.25 - 2.55 (4H, br), 2.79 (2H, t), 3.20 (2H, s), 3.41
(2H, t), 3.45 (2H, s), 3.67 (2H, br), 3.81 (3H, s), 4.02
(2H, t), 4.28 (2H, t), 6.52 - 7.25 (7H~ m), 7.42 (2H, d),
7.67 (2H, d)
The thus-obtained oily substance was dissolved in
isopropyl ether and then crystallized by being allowed to
stand at 5~C for 12 hours.
Melting point: 81 - 83~C
IR (KBr) cm~1: 3300, 1730, 1679, 1640
NMR (CDC13) ~: 1.34 - 1.48 (2H, m), 1.48 - 1.62 (4H,
m), 1.99 (2H, m), 2.24 - 2.42 (4H, m), 2.37 (3H, s), 2.77
(2H, t), 3.21 (2H, s), 3.42 (2H, s), 3.48 (2H, q), 3.67 (2H,
s), 3.83 (3H, s), 4.04 (2H, t), 4.26 (2H, t), 6.64 - 7.22
(8H, m), 7.46 (2H, d), 7.66 (2H, d)
Elementary analysis value (as C38H44N3o6scl):
C H N
Experimental value 64.70 6.36 5.81
Calculated value 64.62 6.28 5.95
(Example 5)
2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbomoyl-
methylthio]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate maleate
The same procedure as in Example 2 was followed with
the exception that 6 g of the indoleacetic acid compound
27 ~3~6~7
obtained in Example 4 was used to obtain as an amorphous
substance 6.5 g of 2-[N-[3-[3-~piperidinomethyl)phenoxy]
propyl]carbamoylmethylthio] ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate maleate.
NMR (CDCl3) ~: 1.53 - 2.25 (8H, br), 2.33 (3H, s), 2.82
~2H, t), 3.21 (2H, s), 3.33 - 3.70 (6H, m), 3.67 (2H, s),
3.80 (3H, s), 3.99 (2H, t), 4.26 (2H, t), 6.30 (2H, s), 6.65
- 7.30 (7H, m), 7.37 (2H, d), 7.63 (2H, d), 12.6 - 13.2 (2H,
br)
(Example 6)
6.5 g of the indoleacetic acid compound obtained in
~xample 4 was dissolved in 20 ml of acetonitrile, and a
solution obtained by dissolving 1.3 g of oxalic acid
dihydrate in 80 ml of acetonitrile was then added to the
resultant solution. After the resultant mixture had been
allowed to stand at room temperature for 5 hours and then at
4C for 20 hours, the crystals separated were filtered off
and then recrystallized from acetonitrile to obtain 5.5 g of
2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]carbamolymethyl-
thio]ethyl 1-(4-chlorobenzoyl)-3-methoxy-2-methyl-3-
indolylacetate oxalate.
Melting point: 102 - 103C
IR (KBr) cm~1: 3290, 2600 - 2200, 1735, 1675, 600
NMR (CDC13) ~: 1.35 (2H, m), 1.83 (4H, m), 1.98 (2~,
m), 2.36 (3H, s), 2.58 (2H, br), 2.82 (2H, t), 3.25 (2H, s),
28 2~3~67
3.44 (2H, m), 3.59 (2H, br), 3.68 (2H, s), 3.83 (3H, s),
3.98 (2H, t), 4.12 (2H, s), 4.28 (2H, t), 6.64 - 7.48 (8~,
m), 7.47 (2H, d), 7.66 (2H, d)
Elementary analysis value (as C3~H44N306SCl C2H4o4~2H2o)
C H N
Experimental value 64.70 6.36 5.81
Calculated value 64.62 6.28 5.95
(Example 7)
2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethylthio]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate
~N-CHz O(CHZ) 3-N-C-CHZl
S--C H 2 C H 2--O--C--C H z ~ O C 11 3
C=O
'
530 mg of N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]
propyl~-2-(2-hydroxyethyl-1-thio) acetamide was dissolved in
a mixture solvent containing 7 ml of dry acetonitrile and
0.5 ml of pyridine, and 775 mg of 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetyl chloride was then added to
the resultant solution at a stroke under ice-cooling.
29 20316~7
After addition, the resultant mixture was agitated for 10
minutes in an iced bath, 30 minutes at room temperature, and
then for 1 hour at 50 to 60C, and the reaction solution was
then concentrated under reduced pressure. 16 ml of a
saturated aqueous solution of sodium hydrogencarbonate was
poured on the thus-obtained residue, followed by extraction
with chloroform. The extract was dried over magnesium
sulfate, and chloroform was then distilled off under reduced
pressure to obtain a brown oily brown substance. The thus-
obtained oily substance was subjected to silica gel column
chromatography (developing solvent: chloroform : methanol =
6 : 1) to obtain as a slightly yellow oily substance 332 mg
of 2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamolymethylthio]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-
methyl-3-indolylacetate.
IR (liquid film) cm~1: 1735, 1675
NMR (CDCl3) ~: 1.60 - 2.20 (6H, m), 2.37 (3H, s), 2.40
- 3.10 (6H, m), 3.22 (2H, s), 3.25 - 4.45 (lOH, m), 3.85
(3H, s), 6.60 - 7.80 (12H, m)
(Example 8)
2-[N-[3-[3-(1-py-rrolidlnylmethyl)phenoxy]propyl]
carbamoylmethylsulfonyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
30 2~3~67
~N-CII.~LO(CU,),- -C-CII,~
S--C H 2 C H z - O - ICI - C il ~
C =O
(a) 1.3 g of N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl-
2-(2-hydroxyethyl-1-thio) acetamide was dissolved in 8 ml of
acetic acid, and 3.1 g of an aqueous solution of 31%
hydrogen peroxide was then added to the resultant solution,
followed by agitation under heating at an external
temperature of 65C. After 1.5 hours had passed, 10 ml of
water was added to the mixture which was then concentrated
under reduced pressure. 16 ml of a saturated aqueous
solution of sodium hydrogencarbonate was then added to the
remaining solution, and the resultant mixture was then
subjected to extraction with chloroform. After the extract
had been dried over magnesium sulfate, chloroform was
distilled off under reduced pressllre to obtain a brown oily
substance. The thus-obtained substance was then subjected
to silica gel column chromatography (developing solvent:
chloroform : methanol = 4 : 1) to obtain as a colorless oily
31 2~3~6~7
substance 377 mg of N-[3-[3-(1-
pyrrolidinylmethyl)phenoxy]propyl]-2-(2-hydroxyethyl-1-
sulfonyl) acetamide.
IR (liquid film) cm~1: 3300, 1650, 1310, 1115
NMR (CDC13) ~: 1.75 - 2.30 (6H, m) r 2.65 - 3.25 (4H,
m), 3.25 - 3.70 (m), 3.80 - 4.50 (m), 6.65 - 7.75 (5H, m)
(b) 370 mg of N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]-
propyl]-2-(2-hydroxyethyl-1-sulfonyl) acetamide was
dissolved in a solvent containing 5 ml of dry acetonitrile
and 0.3 ml of pyridine, and 966 mg of 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetyl chloride was then added to
the resultant solution at room temperature, followed by
agitation at an external temperature of 50C. After 1.5
hours had passed, the reaction solution was poured into a
mixture containing 20 ml of a saturated aqueous solution of
sodium hydrogencarbonate and 20 ml of a saturated aqueous
solution of sodium chloride. After extraction with
chloroform, the extract was dried over magnesium sulfate,
and chloroform was then distilled off under reduced pressure
to obtain a brown oily substance. The thus-obtained
residue was subjected to silica gel column chromato~raphy
(developing solvent: chloroform : methanol = 7 : 1) to
obtain a yellow oily substance. The thus-obtained yellow
oily substance was then subjected to alumina column
chromatography (developing solventj chloroform : methanol =
32 2~3~7
7 : 1~ to obtain as a yellow oily substance 401 mg of 2-[N-
[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]carbamoylmethyl-
sulfonyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetate.
IR (KBr) cm~1: 1735, 1670, 1320, 1150 - 1110
NMR (CDCl3) ~: 1.55 - 2.15 (6H, m), 2.25 - 2.75 (4H,
m~, 2.35 (3H, s), 3.15 - 4.20 (15Hr m), 4.55 (2H, t), 6.50 -
7.80 (12H, m)
(Examp1e 9)
2-[N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
carbamoylmethylsulfinyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
~N- Cl12--¢~L O (Cl12) :J- N-C-CH 2
0
l O
C H .
C=O
(a) 918 mg of N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]-
propyl]-2-(2-hydroxyethyl-1-thio) acetamide was dissolved in
33 2~31~7
3 ml of acetic acid, and 549 mg o~ an aqueous solution of
31% hydrogen peroxide was then dropwisely added to the
resultant solution at room temperature, followed by
agitation at the same temperature. After 1.5 hours had
passed, the resultant mixture was poured into a saturated
aqueous solution of sodium hydrogencarbonate, and sodium
chloride was then added to the mixture to saturate it.
After extraction with butanol, the extract was dried over
magnesium sulfate, and n-butanol was then distilled off
under reduced pressure to obtain 804 mg of a slightly brown
oily substance. The thus-obtained oily substance was then
subjected to alumina column chromatography (developing
solvent: chloroform : methanol = 7 : 1) to obtain as an oily
substance of N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]propyl]-
2-(2-hydroxyethyl-1-sulfinyl) acetamide.
IR (liquid film) cm~1: 3275, 1650, 1025
NMR (CDCl3) ~: 1.60 - 2.20 (6H, m), 2.30 - 2.75 (4H,
m), 3.03 (2H, t), 3.20 - 4.25 (llH, m), 6.65 - 7.65 (5H, m)
(b) 637 mg of N-[3-[3-(1-pyrrolidinylmethyl)phenoxy]
propyl]-2-(2-hydroxyethyl-1-sulfinyl) acetamide was
dissolved in a mixture containing 7 ml of acetonitrile and 1
ml of pyridine, and 1.3 g of 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetyl chloride was then added to the
resultant solution at room temperature, followed by
agitation at an external temperature of 50C. After 1 hour
34 ~3:~67
had passed, the reaction solution was concentrated under
reduced pressure, and 15 ml of a saturated aqueous solution
of sodium hydrogencarbonate was poured on the concentrated
solution. The resulting solution was then subjected to
extraction with chloroform, and the extract was dried over
magnesium sulfate. Chloroform was then distilled off under
reduced pressure to obtain a brown oily substance. The
thus-obtained oily substance was then subjected to alumina
column chromatography (developing solvent; chloroform :
methanol = 7 : 1) to obtain 1.6 g of a brown oily substance.
The thus-obtained oily substance was then subjected to
silica gel column chromatography (developing solvent;
chloroform : methanol = 7 : 1) to obtain as a slightly
yellow oily substance 280 mg of 2-[N-[3-[3-(1-pyrrolidinyl-
methyl)phenoxy]propyl]carbamoylmethylsulfinyl]ethyl 1-(4-
chlorobenzoyl)-5-methoxy-2-methyl-3-indolylacetate.
IR (CHCl3) cm~1: 1730, 1665, 1030
NMR (CDCl3) ~: 1.68 - 2.20 (6H, m), 2.38 (3H, s), 2.48
- 2.90 (4H, m), 2.90 - 4.28 (15H, m), 4.38 - 4.68 (2H, m),
6.70 - 7.88 (12H, m)
(Example 10)
2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]
carbamoylmethylsulfonyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate
35 203:~6~
C~N- C 11 2 ~L 0 ( C ~1 2 ) 3- N - C - C 11 2 1
r~
L S C l12 C H z--O--C - C 112 ,~ 0 C 113
C=O
¢~,
(a) The same proc.edure as in the method (aj in Example 8
was followed with the exception that N-[3-~3-(piperidino-
methyl)phenoxy]propyl]-2-(2-hydroxyethyl-1-thio) acetamide
was used to obtain N-[3-[3-(piperidinomethyl)phenoxy]
propyl]-2-(2-hydroxyethyl-1-sulfonyl) acetamide.
(b) The same procedure as in Example 4 was performed with
the exception that 477 mg of the acetamide compound obtained
in (a), 540 mg of 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
indolylacetyl chloride, 119 mg of anhydrous pyridine and 10
ml of dry acetonitrile were used to obtain as an oily
substance 320 mg of 2-[N-[3-[3-(piperidinomethyl)phenoxy]
propyl]carbamoylmethylsulfonyl]ethyl 1-(4-chlorobenzoyl)-5-
methoxy-2-methyl-3-indolylacetate.
IR (liquid film) cm~1: 1735, 1660, 1325
2~316~7
NMR (CDC13) ~: 1.34 - 1.79 (6H, m), 2.10 - 2.33 (2H,
m), 2.48 - 2.94 (7H, m), 3.16 - 4.40 (17H, m), 6.43 - 7.75
(llH, m)
(Example 11)
2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]
carbamoylmethylsulfinyl]ethyl 1-(4-chloroben~oyl)-5-methoxy-
2-methyl-3-indolylacetate
C~N-CI12- ) O(CIIZ) 3-N-C-C112
l O
c~
C=O
(a) The same procedure as in the method (a) in Example 8
was followed with the exception that N-[3-[3-(piperidino-
methyl)phenoxy]propyl]-2-(2-hydroxyethyl-1-thio) acetamide
was used to obtain N-[3-[3-(piperidinomethyl)phenoxy]
propyl]-2-(2-hydroxyethyl-1-sulfinyl) acetamide.
(b) The same procedure as in Example 4 was followed with
the exception that 262 mg oî the acetamide compound obtained
in (a), 309 mg of 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-3-
37 2~3:1~67
indolyacetyl chloride, 70 mg of anhydrous pyridine and 5 mlof dry acetonitrile were used to obtain as an oily substance
168 mg of 2-[N-[3-[3-(piperidinomethyl)phenoxy]propyl]
carbamoylmethylsulfinyl]ethyl 1-(4-chlorobenzoyl)-5-methoxy-
2-methyl-3-indolylacetate.
IR (CHC13) cm~1: 1730, 1670, 1030
NMR (CDC13) ~: 1.37 - 1.74 (6H, m), 1.84 - 2.19 (2H,
m), 2.19 - 2.55 (7H, m), 3.13 (2H, t), 3.30 - 4.21 (15H, m),
4.40 - 4.67 (2H, m), 6.65 - 7.84 (llH, m)
Drug preparation examples according-to the invention
will be described below.
(Tablet)
100 g of the compound of Example 5, 126 g of lactose,
12 g of calcium carboxymethyl cellulose and 2 g of magnesium
stearate were mixed well and compression-molded by a tablet
machine to form tablets.
(Granule)
25 g of the compound of Example 5 was dissolved in 170
ml of ethanol. 200 g of lactose, 250 g of cone starch and
25 g of hydroxypropyl cellulose were mixed in a high-speed
mixer, and the resulting mixture was then added to the
solution separately prepared and kneaded well. The thus-
obtained kneaded material was granulated by an extrusion
granulating machine and -then air-dried at 40C for 2 hours
to form granules.
` 38 ~31~7
(Capsule)
100 g of the compound of Example 2, 192 g of lactose
and 6 g of hydroxypropyl cellulose were well mixed, and 2 g
of talc was added to the thus-obtained mixture. The
mixture was then charged in each capsule to form a capsule
medicine.
(Injection)
lO g of the compound of Example 5 was dissolved in
polysor.~ate 80, and the thus-obtained solution and 8 g of
sodium chloride were dissolved in distilled water used for
injection so that the total volume was 1 1. The thus-
formed solution was filtered and sterilized and then charged
in 1-ml ampoules to form injections.
(Suppository)
100 g of the compound of Example 7 was dissolved in
l900 g of Wittepsole, and the resultant solution was then
charged in a suppository container and then cooled to form
suppositories.
(Eye drops)
0.3 g of the compound of Example 5, 0.4 g of
chlorobutanol, 602.0 g of polyoxyethylene hardened castor
oil and 0.2 ml of ethanol were agitated on a water bath at
about 80C to form a solution. Purified water at about
80C was then added to the thus-formed solution. After
dissolution had been confirmed, the resultant mixture was
39 2~3~667
cooled, and 1.0 g of boric acid was added to the mixture.
Purified water was added to the mixture so that the total
volume was 100 ml, and an appropriate amount of borax was
added to the resultant solution. After the pH value of the
solution had been adjusted to 7.0, the solution was filtered
by 0.22 ~m membrane filter, charged in a 10-ml eye dropper,
capped and then sterilized by heating to form eye drops.
INDUSTRIAL APPLICABILITY
Each of the compounds ~I) of the present invention has
an extremely reduced degree of adverse effects such as
damage to the alimentary canal, which are great problems of
conventional non-steroidal anti-flamma-tory drugs represented
by indomethacin, a low degree of toxicity and an effect of
inhibiting carrageenin edema to an extent which is
substantially the same as indomethacin. The compounds also
exhibit excellent anti-ulcer activities. They are therefore
very useful as anti-inflammatory, anti-rheumatic and anti-ulcer
drugs.