Note: Descriptions are shown in the official language in which they were submitted.
~3X~7~
BEElRINGWERRE A:~TI~NGESELLSC:HAFT HOE 89/B 050 - Ma 812
Dr. Ha/Sd
Descripkion
PRDl,ESS FOB~ T~E Pl~P~ID~ OF ~LYCOSYL~ YCLINONES
- _
~he pre~en~ invention relates to a proce~s, particularly
to a glyco~ylation process, for the preparation o 7-0-
~lycosylanthracyclinones which, owing to their cytostatic
activity, are ~uitable for the ~reatment of aarcino~e~.
The anthracycline clas~ of substance i6 descri~d in
depth in the technic~l litera~urb. Doxorubicin and it~
14-desoxy analog daunorubicin are named here a~ the most
successful representative~ of this clas~ of substance,
which can be employed in the clinic for ~he treatment of
a large number of solid tumor and leukemias. Other
analogs which have been modified both in the aglycone
moiety and in the carbohydrate unit have recently been
introduced in the clinic or are in clinical testing.
The chemical preparation of anthracyclines i8 carried
out, starting from a functionali~ed carbohydrat0 building
block as a donor and an anthracyclinone as an acceptor,
in the presence of a promoter. In thi~ case/ the hydrosyl
group on the C7 atom of the anthracyclinone acceptor is
glycosylated with a carbohydrate building blsck with the
formation of an ~-O-glyco~idic linkage.
~he following functionali~ carbohydrat~ building blocks
are u~ed for the glycosylation of anthxacyclinoness
1) Glycosyl halides u~ing ~Llver ~alts as promoters ~F.
Arcamone in hDoxorubicin", Academic Press, 1981, pp.
82-92 and pp. 194-258).
-- 2 --
2) Glycosyl halides using mercury ~alts a~ promoters
(T.M. Smith et ~1., J. Org. Chem. 42, 3653 (1977), F.
Axcamone et al., Cancer Treat Rep. 60, 829 (1976)).
3 ) Glycals using acid as a promoter (H. Umezawa et al.,
J. Antibiotic~ 33, 1581 (1980)).
4~ Glycals u~ing ~-iodosuccinimide ~D. Horton ~t al . in
"Anthracycline AntibioticQ", Editor H.S. ~l Khadem,
Academic Press, 1982, pp. 197 - 224).
5) l~O-Acyl-carbohydrate donor~ using trimethylsilyl
triflate as a promoter (EP O 143,323/1988).
~-Glycosides can be prepared ~electively by methods 1 and
2, but as the glycosyl halides are very un~table, their
use in glycoside synthe~i~, above all on the indus~rial
scale, is very problematical. ~oreover, expensivs ~ilver
salts or toxic mercury salts are used in thi~ c~se.
~-Glycosides can only be prepared as a mixture o tha ~-
- and ~-products by method 3. Only 2'-haloanthracyclines
can be prepared by method 4.
~-Glycosi~es can be pr~pared selectively by method 5, but
as the glycosyl donor is pre~ent a~ a mixture of the ~
and ~-O-acyl derivativ~s, the ~-O-acyl deriva~ives
preferably rsacting to completion in the glycosylation,
the ~-O-acyl component ufiually remain~ unre~cted during
the reaction. Expen~ive chro~atography i8 neCe~8ary i~
the purification of th* desired ~-glyco~ide~.
Surprisi~gly/ ~t has been ~hown in the glyc~ylation of
1 equivalent ~eq.) of ~-rhodomycinone ~th one eq. of l-
O-tert.-butyldimethyl8ilyld~unos~mine deriv~tive a~ donor
that ~-dauno~aminylrhodomycinone~ are ~electiv21y formed
in high yield. In this ~ynthesis of the ~-glyco5yl~nthra-
cyclinones, 3 e~. of glyco~ylation co~ponent, (4-O-p-
nitrobenzoyl-3-N-trifluoroacetyl~ dauno~minyl
_ 3 _ ~ 9
chloride) were formerly re~uired.
Building on this knowledge, the aim has been set for the
pre~ent invention of developing a novel process for the
glycosylation of anthracyclinone~ using 1-0-trialkylsilyl
susars.
This aim is achieved according to the invenkion by the
process for the preparation ~f an ~nthracycline of the
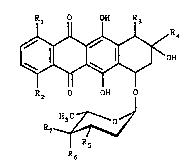
formula I
Rl OH R3
R2 OH O
1~,
in which
Rl is H or 9H
R2 is H, OH or OCH3
R3 is H, COOCH3, 0~ or an O-acyl protectin~ group
R4 i8 CH2CH3, COCH3, COCH20H or a CO-C~20-acyl protecting
group
R5 i NH2, an NH-acyl protecting group, OH or an O-acyl
pxotecting group
R~ is H, OH, an O-acyl protecting group, ~H2 or an ~H-
acyl protecting group
R7 is H, OH or an O-acyl protecting group.
An acyl prot~cting group i~ undsr~tood a~ meaning an ~cyl
group customary in carbohydrate chemi~try, which i8
derived fxom a Cl-C4-alkanoic acidt mvno-, di- or trihalo-
ac~tic acid or benzoic acid.
~ 4 ~ X~ Z9
An acyl protec ing ~roup is preferably acetyl, mono-, di-
or trihaloacetyl where halogen = fluorine or chlorine, or
benzoyl or p-nitrobenzoyl.
Preferred compounds of the fonnula I are those in which
R1, R2, R3, R4, R5, R~ and R7 retain their above-def ined
meaning, an acyl protecting group being acetyl, chloro-
acetyl, txifluoroacetyl or p-ni~robenzoyl.
The compounds of the formula I may optionally 1:~3 pre~ent
a~ ammonium ~alts.
The proce~s according to the invention for the prepara-
tion of a compound of the formula I compriY~s reacting an
anthracyclinone compound of the formula II
II
R2 OH OH
in which
R1 i~ H or OH
R2 i8 H, OH or OCH3
R3 is H, COOCH3 ~r an O-acyl protecting group and
R4 i~ CH2CH3, COCH3, or a CO-CH2-O-acyl protecting ~roup,
wi~h a functionalized carbohydrate building block of the
formula III
Ia
H3C~ o~ o.Si--~9
R'_~/ Rlo III
R ~5
~
in which
R5 is an NH-acyl pr~tecting group or an O-acyl protect-
ing group
R6 is H, an NH-acyl protecting group or an O-acyl
protecting group
R7 is H or an O-acyl protecting group and
RB, R~ and Rl are ~Cl-C4~-al~yl,
an acyl protecting gr~up for amino ~roups preferably
being trifluoroa~etyl and for hydroxyl groups preferably
being acetyl, trifluoroacetyl, chloroacetyl or p-nitro-
benzoyl, in the presence of a promoter such as a tri-
fluoromethanesulfonic acid tri-(Cl-C4)-alkylsilyl ester or
anhydride or BF3.ether, in an anhydrous organic ~olvent,
if desired in the pre3ence of a base or of an acid
entrainer and a drying agent at -50C to 25C to give a
compound of the formula I in which the radicals R1~ RZ,
R3, R4, R5, R6 and R7 retain the meaning defined above and
if appropriate deacylating these compounds preferably by
meanæ of an alkali liquor or an alcoholate to give
cytostatically active compounds of the formula I
in which
Rl is H or OH
R2 is H, OH or OCH3
R3 i~ H, COOCH3 or OH
R4 iB CH2CH3, CQC~I3 or COCH2OH
R5 i~ NH2 or OH
R6 is H, OH or NH2 and
R7 is H o~ OH.
In detail, the procedure in this ca~e i~ as iollows:
A glycosyl donor of the formula III i8 prepared ~tar~ing
from a carbohydrate pxecur~or, which contains one free
hydroxyl group on the Cl atom, and a ~ri- ~C~-C4) ~alkyl-
silyl halide in the pre~ence of an organic base such as
pyridi~e or imidazole and ~n oxg~nic ~olvent 8~ch as
dichloromethane or dichloroeth~ne at 20~-60C.
- 6 ~ 29~4
The preparation of trimethylsilyl and tert.butyl(di-
methyl~ 8ilyl derivative~ of the formula III i~ particu-
larly preferred in thi~ case.
1 eq. to 1.5 eq. of glyco~yl donor of the formula III are
as a rule required for the glycosylation of 1 eq. of
snthracyclinone. Trifluorome~hanesulfonic acid tri-(Cl-
C4)-alkylsilyl estPr, preferably tr~methyl~ilyl tri-
fluoromethanesulfonate or tert.butyl(dimethyl~ilyl
trifluoromethanesulfonate, or BF3-ether i~ preferably
employed as the pr~mot~r. The glycosylation i3 carried
out in an anhydrous organic solvent such as dichloro-
methane, dichloroethane, ether, toluene, acetonitrile or
mixtures thereof with acetone or ethyl acetate, as a rule
in the presence of a drying agent ~uch as a molecular
sieve or barium sulfate at -70aC to 25~C, preferably at -
50C to -20C. When using trifluoromethanesulfonic
anhydride as the promoter, an organic base such as
triethylamine or dimethylaminopyridine may be added.
The deacylation of the glycoside products is carried out
by means of an alkali metal ba~e such a~ NaOH or an
alcoholate such as sodium methylate.
The following examples illustrate the process ~ccordinq
to ~he invention without the latter being restricted ~o
the compounds mentioned in the examples.
- 7
~A~h~S
~ANPL~ 1
Preparation o~ carbohydrate building blocks
Methyl 4-0-p-ni~robenzoyl-2,3,6-tride~osy-3-trifluoro-
ace~amido-~-L-lyxohexopyrano3ide (~ompound 1)
61 g (237 mmol) of methyl 3-N-trifluoroace~yld~uno~amin-
ide were dissolved in 840 ml of dichlorome~hane/pyridine
(2:1~ and 33 g (177 ~ol) of p-ni~roben~oyl chloride were
added. The reaction mixture was stirred at 35~C for 3 h,
then evaporated in vacuo and rediRtilled with toluene.
The residue di~olved in 600 ml of dichloromethane was
washed with 0.1 N HCl, then with water. The organic phase
was dried over odium sulfat~ and evaporated. The crude
product was purified by column chromatography on 600 g of
silica gel (eluents chloroform/ethyl ~cetate 15~
Yield: ~2 g (95 %); Rf = 0.76 (6~1 CHC13/EtOAc; melting
point: 65-67C; [~]D -183C (c 1.03, chlorofo~m).
4-0-p-Nitrobenzoyl-2,3,6-tridesoxy-3-trifluoroacetamido-
L-lyxoh~xopyranose (Compound 2)
25 g (61.5 mmol) of ~e~hyl 4-0-p ni~robenzoyl-3-N-tri-
fluoroacetyl-~-L-daunosaminide were suspended in 250 ml
of 30 % 6trength trifluoroacetic acid. The reaction
mixture wa~ kept undex reflux for 10 min. The cooled
reaction solution was extracted twice ~ith ethyl ace~ate.
The organic ph ~e wa~ washed with pho~phate buffer
(O.1 mol of ~H~P04 adjusted to pH 7~5 with Q.l mol of
NaOH), then with water ~nd dried o~er sodium sulphate.
After evaporating the filtrate, the re~idue (21 g) ~a~
crystallized from ether. The mother liquor (6 g) was
purified by column chromatography on 100 g o~ ilica gel
(eluent: chloroform/ethyl a~etate 9:1). Yield: 17.4
(72.4 %); Rf = 0.21 (9sl CHCl3/Et~Ac); melting point:
213C; [~1 -192 (c 0.86, ~tOAc)~
- 8 ~ 29~
3,4~Bi6-(trifluoroacetylamino)-2,3,4,6-tetrade~oxy-1-
lyxohexopyranose (Compound 3)
Benzyl 3,4-bis-(trifluoroacetylamino)-2,3,4,6-tetrAdes-
oxy-~-L-lyxohexopyranoside (6.5 g) was dissolved in
S glacial acetic acid and hydrogenated in the pre~ence of
10 ~ strength Pd/C (5.5 g) for 48 hour6. The mixture wa~
then filtered and the filtrate was evaporated in vacuo.
The residue wa~ redistilled with 2sl methanol/toluene.
The re~ulting product was purified by column chromato
graphy on Bilica gel ( 120 g) u~ing 3: 1 dichloromethane-
methanol. Yield: 4.39 g; 1~D -63.3 (c 1, EtOAc).
2,6-Didesoxy-3,4-di-O-p-nitrobenzoyl-L-lyxohexopyrano6e
(Compound 4)
2,3-Didesoxy-1,3,4-tri-O-p-nitrobenzoyl-~ and ~-L-lyxo-
hexopyranose (2.5 g, 4.2 mmol) was dissolved in methanol
(60 ml) and aminated silica gel (~erck, 3 g) was added.
The reaction mixture was ~tirred at xoom temperature for
3 hours and then filtered. Af~er evaporating the filt-
rate, the residue wa~ dissol~ed in dichloromethane
(120 ml) and washed with phosphate buffer (pH 8, 60 ml x
2), then wi~h water. The organic phase was dried over
magnesium sulfate and evaporated in vacuo. ~he residue
was purified by column chromatography on silica gel
~130 g) using 7:1 dichloromethane-acetone. Yield: 1.42 g
(76 ~).
~xample 2
Prepara~ion of l-O-trialkyl~ilylcarbohydxate derivativQ~
4-O-p-Nitroben~oyl--2,3,6-tride~o~y 3-trifluoroacetamido-
l-O-trimethylsilyl-~-L-lyxohexopyrano~e (comp~und 5)
4.6 g (11,7 mmol) of 4 O-p-~itrobenzoyl-2,3,6-tridesoxy
3-trifluoroacetamido-~-lyxohexopyrano~e were dissolved in
80 ml of pyridine/dichloromethane (1:13 and 4.46 ml
(35.1 mmol) of trL~ethyl~ilyl chloride were added at 0~C.
After 16 h, 100 ml of dichloromethane were added ~o the
reaction mixture, which was washed twice ~ith 200 ml of
Z03~97~
_ g
phospha~e buffer (pH 7.5) each tLme. The organic phase
was dried over ~odium sulfate and eYaporated in vacuo.
The re~idue was additionally red~stilled with toluene.
The crude product, dried in a high vacuum, wa~ purified
by column chromatography on 100 g of silica gel ~eluent:
dichloromethane/pe~roleum ether/acetone 5:5:0.5). ~ield:
5.07 g (93-3 ~)
l-O-t-Butyl-dimethylsilyl-4-O-p-nitrobenzoyl-2,3,6-
tride~oxy-3-trifluoroacetamido-~ lyxohexopyranose
(Compound 6)
4.05 g (10.3 mmol) of 4-O-p-Nitrobenzoyl-3-N-trifluoro-
acetyl-L-daunosamine were dis6clved in 160 ml o~ pyrid-
ine/1,2-dichloroethane and 7.7 g (51.5 mmol) of t-butyl-
dimethylsilyl chloride were added. After stirr~ng at 60C
for 16 h, the reaction mixture wa~ dilu~ed with 200 ml of
dichloromethane ~nd washed twice with pho~phate buf~er
(O.1 mol of KHzPO4 ad~usted to pH 7.5 with 0.1 mol o~
NaOH) with re-extraction of the aqueou6 phase. The
organic phase was dried over sodium ~ulfate and evapora-
ted in vacuo. The re~idue was redi6tilled ~everal times
with toluene until pyridine was no lon~er presen~, then
purified by column chromatography on 150 g of ~ilica gel
(eluent:petroleumether/dichloromathane/acetone 10:10:1).
Yields 4.7 g ~90.3 %). Rf = 0.43 (10.10:1 chloroform/pet-
roleum ether/acetone); melting point: 72-74C; 1~]D -91
(c 1, chloroform).
3,4-Bis-(trifluoroacetylamino)-l-O-ter~.-bu~yl(dime~hyl)-
silyl-2,3,~,6-tetra-de~oxy-L-lyxohexopyranose
(Compound 7)
Starting from 3,4-bi~-(trifluoroacetylamino)-2,3,4,6-
tetradesvxy-L-lyxohexopyTano~eandtert.-butyl(diemthyl~-
~ilyl chloride, the ti~le compound was ~ynthe~ized by the
procedure for the preparation of compound 6.
2,6-Didesoxy-3,4-di-O-p-nitrobenzoyl-l;~O-tert.-butyl(di-
methyl)silyl-L--lyxohexopyrano~e (Compound 8)
The title compound wa~ prepared by the procedure for the
- lo- ~ 2974
synthesis of compound 6 starting from 2,6-didesoxy-3,4-
di-O-p-nitroben~oyl-L-lyx~hexop~ranose and tert.-butyl-
(dLmethyl)silyl chloride~
Example 3
S Preparation of 7-O-trimethyl~ilyl~ o)rhodomycinones
7-o-TrLmethyl~ilyl-e-rhodomycinone (Compound 9)
200 mg (0.429 mmol) of ~-rhodomycinone (92 ~ by HPLC)
were dissolved in lO ml of pyridine/dichloromethane and
0.180 ml (1.4~ mmol) of trimethyl6ilyl chloride was added
at 0C. After stirring for 30 min, the r~action mixture
was diluted with 20 ml of dichloromethane and wa~hed
twice with phosphate buffer (pH 7.5). ~he organic phase
was dried over sodium ~ulfate and then evaporated, and
the residue was redistilled with toluene to remove
pyridine xesidues. The acid-sensitive product was puri-
fied by column chromatography on 20 g of ~ilica g~1
[eluent: chloroform/triethylamine 200:1~. Yield: 180.6 mg
(84 ~); t~]D +279~ (c 0.037, ~hloroform).
7-O-Trimethylsilyl-~-isorhodomycinone (Compound lO~
200 mg (O.45 mmol) of ~-isorhodomycinone were dissolYed
in 10 ml of pyridine/dichloromethane (1:13 ~nd 0.180 ml
(1.398 mmol) of trimethylsilyl chloride was added at 0C.
After ~tirring for 30 min, ~h~ reaction mixture was
diluted with 20 ml of dichlorometha~e and washed twice
wi h phosphate buffer SpH 7.5). ~he organic pha~e w~s
dried over ~odium ulfate, ~hen evaporated in vacuo. The
residue was redistill~d with toluene to ramove pyridine
residues. The acid-~ensitive product was purified ~y
column chromatography on 20 g of ~ilica gel (elue~ts
chlorofo~/triethylamine (200~ Yields 180 ~g (82 %);
[3~D ~3~5 (C 0-0061, chloroform~.
~ample 4
Glycosy}ation of anthracyclinones with functionalized
3~74
carbohydrate building blocks
7-0-~4-O-p-Nitrobenzoyl-2,3,~-tridesoxy-3-trifluoroacet-
ylamino-~-L-lyxohexopyrano~yl)-~-isorhodomycinone
(Compound 11)
~-I60rhodomycinone (6.0 ~, 11.2 mmol), daunosamine donor
(compound 6, 8.51 g, 16.8 mmol) and molecular ~ieve 4 A
(6.0 g) were suspended in 5:1 dichloromethane-ethyl
acetate (7~Q ml) undex protective ga~. After the addition
of trimethylsilyl ~rifluorome~h~ne~ulfona~e (4.97 ~,
22.4 mmol) at -35~C, the reaction mixture ~as stirred for
5 hours. Triethylamine (10 ml) was added to the cooled
mixture, which was filtered. The filtra~e was wa~hed
twice with citrate buffer (0.1 molar citric acid solution
adjusted t~ pH 5 with 0.1 molar NaOH), then with ice-
water. The organic ph~se was dried over sodium sulfate
and evaporated in vacuo. The residue (12.6 g) wa~ puri-
fied ~y column chromatography on silica gel (450 g) uRing
200:10:1 chloroform-ethyl acetate-formic acid. Yield:
7.15 g t78 %); Rf = 0.33 (solvent - see above).
7-0-(4-O-p-Nitrobenzoyl-~,3,6-tridesoxy-3-trifluoroacet~
ylamino-~-L-lyxohexopyranosyl)-~-rhodomycinone
(Compound 12)
The title compound 12 was synthesized by the procedure
for the prep~ration of compound 11 starting from
e -rhodomycinone and daunos~mine donor (~ompound 6).
7-0-(4-O-p-Nitrobenzoyl-2,3,6-tride~oxy-3-trif~uoroace~-
ylamino-~-L-lyxohexopyranosyl~-daunomycinone
(Compound 13)
The title compound 13 wa~ prepared by the procedure for
th~ ~ynthesis of c~mpound 11 starting from daunomycinone
and daunosamine donox (compoulld 6).
7-0-(3,4-Bi~-(trifluoroacetylamino)-2,3,4,6 te~ra-de~oxy-
~-L-lyxohexopyranosyl)-4-O-methyl-10-O-p-nitrobenzoylw~-
rhodomycinone (Compound 14)
4-O-Methyl-10-O p-nitrobenzoyl-~-rhodomycinone (2.64 g!
4.80 mmol), 3,4-bis-tri1uoroa~etylamino sugar (compound
12 ~ 3Z97~
7, 2.17 g, 4.80 mmol) and 4 A molecular ~ieve (3.0 g)
were dissolved in dichloromethane (220 ml) and trimethyl-
silyl triflate (1.2 ml3 were added at -35~Co The reaction
mixture was stirred at -35C for 3 hours, then triethyl-
S amine (2.5 ml) wa~ added and the mixture wa~ filtered.
The filtrate was wa~hed with phosphate buffer ~pH 7,
60 ml x 2), then with water, dried (~odium sulfate) ~nd
evaporated in vacuo. The residue was purified by colu~n
chroma~ograp~y on silica gel (220 g) u~ing 15:1 chloro-
form-acetone. Yield: 3.3S g (81 ~), Rf = 0.37 (15:1
chloro~oxm-a~etone); ~elting point: 222~C; alpha D + 250
(c O.05, chloroform~.
7-~-(3,4-Bis-O-p~nitrobenzoyl-2,6-didesoxy~alpha-L-lyxo-
hexopyranosyl)-4-0-methyl-10-0-p-nitrobenzoyl-~-rhodo-
mycinone (Compound 15)
The title compound was synthesized by the procedure forthe preparation of ~ompound 14 starting from 2-de~oxy-
fucose donor (compound 8) and 4-0-methyl-10-0-p-nitro-
benzoyl-~-rhodomycinone.
~xample 5
Deblocking of anthxacyclines
The anthracycline derivatives described in Example 4 were
deblocked by known processes, a~ de6cribed in the follow-
ing general procedure:
Protected anthracycline compound was dissolved ~n metha-
nol or chloroform-methanol and 1 N NaOH was added at O~C.
After the reaction had taken place, the reaction mixture
was neutralized with 1 N HCl. After th~ cu~tomary work-
up, the deblocked compound was purified on RP-~8 silica
gel or amino 8ilica gel.
The following c~mpounds were prepared by thi~ procedure:
O~'Z~374
_ 13 -
7-0-(3-Amino-2,3,6-tridesoxy-alpha-L-lyxohexopyrano~yl)-
~-isorhodomycinone (Compound 16)
7-0-(3-Amino-2,3,6-tridesoxy-alpha-L-lyxohexopyranosyl)-
~-rhodomycinone (Compound 17)
7-0-(3-Amino-2,3,6-~ride~oxy~ lyxohexopyranosyl)-
daunomycinone (Compound 18)
7-0-(3,4-Diamino-2,3,4,6-tetra-deso~y-~-L-lyxohexopyrano-
syl)-4-0-methyl-~-rhodomycinone (Compound 19)
7-0-(2,6-Didesoxy-~-L-lyxohexopyranosyl)-4 O-methyl-~-
rhodomycinone (Compound 20)