Note: Descriptions are shown in the official language in which they were submitted.
- 203697~
4- 17972/+
Benzofurans
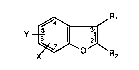
The invention relates to compounds of formula I
R~
wherein X is halogen, cyano, carbamoyl, N-lower alkylcarbamoyl, N,N-di-lower alkyl-
carbamoyl, N,N-lower alkylenecarbamoyl; N,N-lower alkylenecarbamoyl interrupted by
-O-, -S- or -NR"- wherein R" is hydrogen, lower alkyl or lower alkanoyl; N-cycloalkyl-
carbamoyl, N-(lower alkyl-substituted cycloalkyl)-carbamoyl, N-cycloalkyl-lower
alkylcarbamoyl, N-(lower alkyl-substituted cycloalkyl)-lower alkylcarbamoyl, N-aryl-
lower alkylcarbamoyl, N-arylcarbamoyl, N-hydroxycarbamoyl, hydroxy, lower alkoxy,
aryl-lower alkoxy or aryloxy, Y is a -CH2-A group in which A is imidazolyl, triazolyl or
tetrazolyl bonded by way of a ring nitrogen atom, or Y is hydrogen, each of Rl and R2
independently of the other is hydrogen, lower alkyl or a -CH2-A group as defined for Y, or
Rl and R2 together are lower alkylene, with the proviso that one of the radicals Y, Rl and
R2 is a -CH2-A group, with the further proviso that, in a -CH2-A group as the meaning of
Rl or R2, A is other than l-imidazolyl when X is bromine, cyano or carbamoyl, and with
the proviso that, in a -CH2-A group as the meaning of Y, A is other than l-imidazolyl
when X is halogen or lower alkoxy, Rl is hydrogen and R2 is hydrogen or lower alkyl,
and salts thereof, to a process for the preparation of those compounds, to pharrnaceutical
compositions containing those compounds, to the use of those compounds for the
therapeutic treatment of the human or animal body or for the preparation of pharma-
ceutical compositions.
Within the scope of this Application, the general definitions used hereinbefore and herein.-
after have preferably the following meanings:
The pre~1x "lower" denotes a radical having up to and including 7, and especially up to and
including 4, carbon atoms.
2036~7~
Lower alkyl is, for example, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl,
n-pentyl, neopentyl, n-hexyl or n-heptyl, preferably ethyl and especially methyl.
Halogen is especially chlorine and more especially bromine, but may also be fluorine or
iodine.
Halo-lower alkyl is, for example, trifluoromethyl.
Aryl is, for example, phenyl or naphthyl, such as 1- or 2-naphthyl. The phenyl and
naphthyl radicals may be unsubstituted or substituted, especially as indicated below for
phenyl. Aryl is preferably phenyl that is unsubstituted or substituted by from 1 to 4,
especially 1 or 2, substituents from the group comprising lower alkyl, lower alkenyl, lower
alkynyl, lower alkylene (attached to two adjacent carbon atoms), C3-Cgcycloalkyl,
phenyl-lower alkyl, phenyl, halo-lower alkyl, hydroxy, lower alkoxy, halo-lower aLkoxy,
phenyl-lower alkoxy, phenyloxy, lower alkenyloxy, halo-lower alkenyloxy, lower
alkynyloxy, lower alkylenedioxy (attached to two adjacent carbon atoms), lower
alkanoyloxy, phenyl-lower alkanoyloxy, phenylcarbonyloxy, mercapto, lower alkylthio,
phenyl-lower alkylthio, phenylthio, lower alkylsulfinyl, phenyl-lower alkylsulfinyl,
phenylsulfinyl, lower alkylsulfonyl, phenyl-lower alkylsulfonyl, phenylsulfonyl, halogen,
nitro, amino, lower alkylamino, C3- Cgcycloalkylamino, phenyl-lower alkylamino,
phenylamino, di-lower alkylamino, N-lower alkyl-N-phenylamino, N-lower alkyl-N-
phenyl-lower alkylamino; lower alkyleneamino or lower alkyleneamino interrupted by
-O-, -S- or -NR"- (wherein R" is hydrogen, lower alkyl or lower alkanoyl); loweralkanoylamino, phenyl-lower alkanoylamino, phenylcarbonylamino, lower alkanoyl,
phenyl-lower alkanoyl, phenylcarbonyl, carboxy, lower alkoxycarbonyl, carbamoyl, N-
lower alkylcarbamoyl, N,N-di-lower alkylcarbamoyl, N,N-lower alkylenecarbamoyl;
N,N-lower alkylenecarbamoyl interrupted by -O-, -S- or -NR"- wherein R" is hydrogen,
lower alkyl or lower alkanoyl; N-hydroxycarbamoyl, N-phenyl-lower alkylcarbamoyl,
N-phenylcarbamoyl, cyano, sulfo, lower alkoxysulfonyl, sulfamoyl, N-lower alkyl-sulfamoyl, N,N-di-lower alkylsulfamoyl and N-phenylsulfamoyl; wherein the phenylgroups occurring within the substituents are in each case unsubstituted or substituted in
their turn by lower alkyl, lower alkoxy, hydroxy, halogen and/or by trifluoromethyl.
Aryl is especially phenyl that is unsubsdtuted or substituted by lower alkyl, lower alkoxy,
hydroxy, halogen andlor by trifluoromethyl, and is most especially phenyl.
. . ' . ~.
.
,
'. - ~
,:-.
203697~
Substituted phenyl is preferably di-substituted and especially mono-substituted.
Aryl-lower alkoxy is, for example, phenyl-lower alkoxy and especially benzyloxy.
N-arylcarbamoyl is, for example, N-phenylcarbamoyl.
Imidazolyl bonded by way of a ring nitrogen atom is, for example, 1-imidazolyl.
Triazolyl bonded by way of a ring nitrogen atom is, for example, 1-(1,2,4-triazolyl),
1-(1,3,4-triazolyl), 1-(1,2,3-triazolyl) or 1-(1,2,5-triazolyl).
Tetrazolyl bonded by way of a ring nitrogen atom is, for example, 1-tetrazolyl or
2-tetrazolyl.
Lower alkylene formed by the groups R1 and R2 is preferably a -(CH2)n- radical wherein
n is 3, 4 or 5, especially 3 or 4, for example 1,3-propylene or especially 1,4-butylene, but
may also be substituted, for example by lower alkyl.
Cycloalkyl is preferably C3-Cg- and especially C3- or Cs-C6cycloalkyl, which is intended
to mean that it contains from 3 to 8 and 3, 5 or 6 ring carbon atoms, respectively.
Lower alkylene attached to two adjacent carbon atoms of a benzene ring is preferably
C3-C4alkylene, for example 1,3-propylene or 1,4-butylene.
Lower alkylenedioxy attached to two adjacent carbon atoms is preferably Cl-C2alkylene-
dioxy, for example methylenedioxy or 1,2-ethylenedioxy.
Lower alkyleneamino is, for example, C4-C7alkyleneamino and especially C4-Cs-
alkyleneamino, for example piperidino. Lower alkyleneamino interrupted by -O-, -S- or
-NR"- is, for example, such a C4-C7- and especially C4-Csalkyleneamino group in which
one ring carbon atom has been replaced by the corresponding hetero group, and isespecially morpholino, thiomorpholino, piperazino or 4-lower alkyl- or 4-acyl-piperazino.
Carbamoyl denotes the -CONH2 group. Accordingly, N,N-lower alkylenecarbamoyl, for
example, is lower alkyleneamino-carbonyl in which lower alkyleneamino is as defined
above.
-
203~975
- 4 -
Salts of compounds according to the invention are especially pharmaceutically acceptable
non-toxic salts. For example, compounds of forrnula I having basic groups can form acid
addition salts, for example with inorganic acids, such as hydrochloric acid, sulfuric acid or
phosphoric acid, or with suitable organic carboxylic or sulfonic acids, for example acetic
acid, fumaric acid or methanesulfonic acid, or with amino acids, such as arginine or
Iysine. Compounds of formula I having an acid group, for example l-tetrazolyl, form, for
example, metal salts or ammonium salts, such as alkali metal and alkaline earth metal
salts, for example sodium, potassium, magnesium or calcium salts, and ammonium salts
with ammonia or suitable organic amines, such as lower alkylamines, for example
triethylamine, hydroxy-lower alkylamines, for example 2-hydroxyethylamine, bis(2-
hydroxyethyl)amine or tris(2-hydroxyethyl)amine, basic aliphatic esters of carboxylic
acids, for example 4-aminobenzoic acid 2-diethylaminoethyl ester, lower alkyleneamines,
for example 1-ethylpiperidine, cycloalkylamines, for example dicyclohexylamine, or
benzylamines, for example N,N'-dibenzylethylenediamine, dibenzylamine or benzyl-b-
phenethylamine. Compounds of formula I having an acid group and a basic group may
also be in the form of internal salts, that is to say in zwitterionic form.
For the purpose of isolation or purification it is also possible to use pharmaceutically
unsuitable salts, for example picrates or perchlorates. Only the pharmaceutically
acceptable non-toxic salts are used therapeutically and these are therefore preferred.
The compounds of formula I according to the invention have valuable, especially
pharmacologically useful, properties. In particular, they selectively inhibit the enzyme
aromatase in mammals, including humans. As a result, the metabolic conversion ofandrogens to oestrogens is inhibited. The compounds of formula I are therefore suitable,
for example, for the treatment of oestrogen-dependent diseases, including oestrogen-
dependent breast cancer, especially in post-menopausal women. They are also useful, for
example, in the treatment of gynaecomastia, i.e. breast development in males, since the
aromatisation of the steroids is inhibited.
These effects can be demonstrated by 1n vitro tests or in vivo tests, preferably on
mammals, for example guinea pigs, mice, rats, cats, dogs or apes. The dosage used is, for
example, within a range of approximately from 0.001 to 10 mg/kg, preferably from 0.001
to 1 mg/kg.
~ . :
203697~
The in vitro inhibition of aromatase activity can be demonstrated1 for example, using the
method described in J. Biol. Chem. 249, 5364 (1974). ICso values for aromatase
inhibition can furthermore be obtained, for example, in vitro from enzyme-kinetic studies
concerned with the inhibition of the conversion of 4-14C-androstenedione to 4-14C-
oestrone in human placental microsomes. The ICso values of the compounds according to
the invention are, at the minimum, about 10-9 M.
In v_, aromatase inhibition can be demonstrated, for example, by the suppression of the
ovarian oestrogen content of female rats that are injected first with mare's serum
gonadotrophin and, 2 days later, with human chorionic gonadotrophin, and treated p.o. the
next day with a compound of the invention and, 1 hour later, with androstenedione. A
further possible method of deterrnining aromatase inhibition in vivo is described hereafter:
androstenedione (30 mg/kg subcutaneously) is administered on its own or together with a
compound of the invention (orally or subcutaneously) for 4 days to sexually immature
female rats. After the fourth administration, the rats are sacrificed and the uteri are isolated
and weighed. The aromatase inhibition is determined by the extent to which the hyper-
trophy of the uterus caused by the administration of androstenedione on its own is
suppressed or reduced by the simultaneous administration of the compound according to
the invention. The minimum effective dose of the compounds of the invention in the in
vivo tests is approximately from 0.001 to 1 mg/kg.
The anti-tumoral activity, especially in the case of oestrogen-dependent tumours, can be
demonstrated in vivo, for example in DMBA-induced mammary tumours in female
Sprague-Dawley rats [cf. Proc. Soc. Exp. Biol. Med. 160, 296-301 (1979)]. The use of
compounds according to the invention brings about a regression of the tumours and
furthermore suppresses the occurrence of new tumours at daily doses of about 1 mg/lcg
and above p.o.
In addition, the compounds of formula I do not have an inhibiting effect on the cleavage of
the cholesterol side-chain and do not induce adrenal hypertrophy, as is demonstrated by
investigation of the endocrine system.
On account of their pharmacological properties as extremely selective inhibitors of the
enzyme aromatase, the compounds of formula I are suitable, for example, for the
treatment of oestrogen-dependent diseases, such as breast tumours (breast carcinoma),
endometriosis, premature labour or endometrial tumours in women, or of gynaecomastia
.
2036~7~
in men.
The invention relates especially to the compounds of formula I wherein X is halogen,
cyano, carbamoyl, N-lower alkylcarbamoyl, N-cycloalkyl-lower aLlcylcarbamoyl,
N,N-di-lower alkylcarbamoyl, N-arylcarbamoyl, hydroxy, lower alkoxy, aryl-lower
alkoxy or aryloxy, wherein aryl is phenyl or naphthyl each of which is unsubstituted or
substituted by lower alkyl, hydroxy, lower alkoxy, halogen and/or by trifluoromethyl; Y is
a -CH2-A group in which A is 1-imidazolyl, 1-(1,2,4-triazolyl), 1-(1,3,4-triazolyl), 1-
(1,2,3-triazolyl), 1-(1,2,5-tnazolyl), 1-tetrazolyl or 2-tetrazolyl, or Y is hydrogen; each of
R1 and R2 independently of the other is hydrogen, lower alkyl or a -CH2-A group as
defined for Y, or R1 and R2 together are -(CH2)n- wherein n is 3, 4 or 5; with the proviso
that one of the radicals Y, Rl and R2 is a -CH2-A group, with the further proviso that, in a
-CH2-A group as the meaning of R1 or R2, A is other than l-imidazolyl when X is
bromine, cyano or carbamoyl, and with the proviso that, in a -CH2-A group as themeaning of Y, A is other than 1-imidazolyl when X is halogen or lower alkoxy, R1 is
hydrogen and R2 is hydrogen or lower alkyl, and salts thereof.
The invention relates preferably to the compounds of formula I wherein the radical X is
attached in the 5- or 7-position and is halogen, cyano, carbamoyl OI phenyloxy; the radical
Y is attached in the 4- or 5-position and is a -CH2-A group in which A is 1-imidazolyl,
1-(1,2,4-triazolyl), 1-(1,2,3- triazolyl), 1-tetrazolyl or 2-tetrazolyl, or the radical Y is
hydrogen; Rl is hydrogen, lower alkyl or a -CH2-A group as defined for Y, R2 is
hydrogen or lower alkyl, or R1 and R2 together are -(CH2)4-; with the proviso that one of
the radicals Y and Rl is a -CH2-A group; with the further proviso that, in a -CH2-A group
as the meaning of R1, A is other than 1-imidazolyl when X is bromine, cyano or
carbamoyl, and with the proviso that, in a -CH2-A group as the meaning of Y, A is other
than 1-imidazolyl when X is halogen and Rl is hydrogen; and salts thereof.
Prominence is to be given to the compounds of formula I wherein the radical X is attached
in the 5- or 7-position and is halogen, cyano, carbamoyl or phenyloxy; the radical Y is
attached in the 4- or S-position and is a -CH2-A group in which A is 1-(1,2,4-triazolyl),
1-(1,2,3-triazolyl), l-tetrazolyl or 2-tetrazolyl, or the radical Y is hydrogen; Rl is
hydrogen, lower alkyl or a -CH2-A group as defined for Y, R2 is hydrogen or lower alkyl,
or Rl and R2 together are -(CH2)4-; with the proviso that one of the radicals Y and R1 is
a -CH2-A group; and salts thereof.
203~97~
The invention relates especially preferably to compounds of formula I wherein the radical
X is attached in the 5- or 7-position and is halogen, cyano, carbamoyl or phenyloxy; the
radical Y is attached in the 4- or 5-position and is a -CH2-A group in which A is
1-imidazolyl, 1-(1,2,4-triazolyl), 1-(1,2,3-triazolyl), I-tetrazolyl or 2-tetrazolyl; each of R
and R2 independently of the other is hydrogen or lower alkyl, or R1 and R2 together are
-(CH2)4-; with the proviso that, in a group Y = -CH2-A, A is other than 1-imidazolyl
when X is halogen and R1 is hydrogen; and salts thereof.
Preference is also given to the compounds of formula I wherein the radical X is attached
in the 5- or 7-position and is halogen, cyano, carbamoyl or phenyloxy; the radical Y is
hydrogen; R1 is a -CH2-A group in which A is 1-imidazolyl, 1-(1,2,4-triazolyl), 1-(1,2,3-
triazolyl), 1-tetrazolyl or 2- tetrazolyl, R2 is hydrogen or lower alkyl; with the proviso
that, in a group R1 = -CH2-A, A is other than 1-imidazolyl when X is bromine, cyano or
carbamoyl; and salts thereof.
Special preference is given to the compounds of formula I wherein the radical X is
attached in the 7-position and is bromine, cyano, carbamoyl or phenyloxy; the radical Y is
attached in the 4-position and is a -CH2-A group in which A is 1-imidazolyl, 1-(1,2,4-
triazolyl), l-tetrazolyl or 2-tetrazolyl; each of R1 and R2 independently of the other is
lower alkyl, or Rl and R2 together are -(CH2)4-; and salts thereof.
Prominence is also to be given to the compounds of formula I wherein X is halogen,
cyano, carbamoyl, hydroxy, lower alkoxy or phenyloxy9 wherein phenyl is unsubstituted
or substituted by lower alkyl, hydroxy, lower alkoxy, halogen and/or by trifluoromethyl,
Y is a-CH2-A group in which A is l-imidazolyl, 1-(1,2,4-triazolyl), 1-(1,3,4-triazolyl), 1-
(1,2,3-triazolyl), 1-(1,2,5-triazolyl), 1-tetrazolyl or 2-tetrazolyl, R1 is lower alkyl, R2 is
hydrogen or lower alkyl, or R1 and R2 together are -(CH2)n- wherein n is 3 or 4, and salts
thereof.
Special prominence is to be given to the compounds of formula I wherein the radical X is
attached in the 4- or 7-position and is halogen, cyano, carbamoyl or phenyloxy; the radical
Y is attached in the 4- or 5-position and is a -CH2-A group in which A is 1-imidazolyl,
1-(1,2,4-triazolyl), 1-(1,2,3- triazolyl), 1-tetrazolyl or 2-tetrazolyl, R1 is lower alkyl, R2 is
hydrogen or lower alkyl, or R1 and R2 together are -(CH2)4-; and salts thereof.
As sub-groups of a group of compounds of formula I, prominence is to be given to each of
203697~
the following: (a) compounds of formula I wherein the radical X is attached in the
7-position and the radical Y is attached in the 4-position; (b) compounds of formula I
wherein X is bromine or cyano; (c) compounds of formula I wherein X is carbamoyl; (d)
compounds of formula I wherein Y is a -CH2-A group in which A is 1-imidazolyl or1-(1 ,2,4-triazolyl).
The invention relates most especially to the specific compounds described in theExamples and to pharmaceutically acceptable salts thereof.
The compounds of forrnula I can be prepared in a manner known E~ se, for example by
(a) condensing a reactive esterified derivative of a hydroxymethyl compound of formula II
Y ~3 ~ (II)
wherein one of the radicals Y', R' 1 and R'2 is hydroxymethyl and the other two radicals
have the definitions given for Y, R1 and R2, respectively, under formula I, and X is as
defined under formula I, with a compound
A - H (III),
wherein A is as defined under formula 1, or with an N-protected derivative thereof, or .
(b) in a compound of formula IV
,~,
~OJl\R2
wherein X' is a radical that can be converted into a group X, and Y, R1 and R2 are as
defined under formula I, converting the radical X' into a group X, or
(c) for the preparation of compounds of formula I wherein A in ~he group -CH2-A is
`" 203697~
9-
l-tetrazolyl, reacting a compound of formula V
ya~3~ (V),
wherein one of the radicals ya, R 1 a and R2a is isocyanomethyl and the other two radicals
have the definitions given for Y, Rl and R2, respectively, under formula I, and X is as
defined under formula I, with hydrazoic acid or, especially, with a salt thereof; and/or, if
desired, converting a resulting compound of formula I into another compound of
formula I, and/or, if desired, converting a resulting salt into the free compound or into
another salt, and/or, if desired, converting a resulting free compound of formula I having
salt-forming properties into a salt, and/or separating a resulting mixture of isomeric
compounds of formula I into the individual isomers.
In the following, more detailed description of processes a), b) and c), unless indicated to
the contrary, each of the symbols X, Y, A, R1 and R2 has the meaning given underformula I.
Process (a): In a compound of formula n, reactive esteiified hydroxymethyl is hydroxy-
methyl that has been esterified by a leaving group, for example lower alkylsulfonyloxy-
methyl or arylsulfonyloxymethyl, such as methylsulfonyloxymethyl or p- toluenesulfonyl-
oxymethyl, or halomethyl, for example chloromethyl, bromomethyl or iodomethyl.
If, in the reaction according to process (a), 1,2,4-triazole is used as the compound of
formula Ill, then - depending on the reaction conditions chosen - mixtures of compounds
of formula I wherein A is 1-(1,2,4-triazolyl) and 1-(1,3,4-triazolyl) are norrnally obtained,
which can be separated, for example, by chromatography. Correspondingly, if 1,2,3-
triazole is used as the compound of formula III, then mixtures of compounds of formula I
wherein A is 1-(1,2,3-triazolyl) and 1-(1,2,5-triazolyl) are normally obtained, which
similarly can be separated, for example, by chromatography. Correspondingly, if tetrazole
is used as the compound of formula III, then mixtures of compounds of folmula I wherein
A is 1-tetrazolyl and 2-tetrazolyl are normally obtained, which similarly can readily be
separated, for example, by chromatography. In some cases it is possible, by using
compounds of formula In in which a specific ring nitrogen atom has been protected by a
2~3697~
- 10-
protecting group, to obtain selectively only one of the two compounds in question.
Suitable protecting groups for a ring nitrogen atom in a compound of formula III are, for
example, tri-lower alkylsilyl, for example trimethylsilyl, lower alkanoylt for example
acetyl, N,N-di-lower alkylcarbamoyl, for example N,N-dimethylcarbamoyl, or triaryl-
methyl, for example triphenylmethyl.
Another suitable protecting group is amino or ammonium, which is useful especially in
the selective preparation of 1-(1,2,4-triazolyl) compounds. For that purpose, in the
reaction according to process (a), 4H-1,2,4-triazole-4-amine (= 1-amino-1,3,4-triazole) is
used as the compound of formula III. A quaternary 1-benzofuranylmethyl-4-amino-
1,2,4-triazolium compound is initially obtained, which is converted into the desired
1-benzofuranylmethyl-1,2,4-triazolyl compound of formula I, for example by treatment
with hydrochloric acid, 50 % hypophosphorous acid (H3P02) and sodium nitrite.
The condensation reaction according to process (a) is known Per se and corresponds to a
conventional N-alkylation that is carried out, for example, without the addition of bases
or, preferably, in the presence of bases, such as, for example, potassium carbonate,
sodium, triethylamine or pyridine.
The starting compounds of formula II are preferably obtained in a manner known per se,
by esterification, from the corresponding hydroxymethyl compounds. The hydroxymethyl
compounds can be obtained, for example, by reduction, for example with LiAlH4 ordiisobutylalurninium hydride, from the corr~sponding carboxy or lower alkoxycarbonyl
compounds. The latter are known r se or can be prepared analogously to known
substituted benzofurancarboxylic acids and esters (cf. also Examples 1 and 6).
Process (b): Radicals X' that can be converted into a group X are, for example, amino that
can be converted, for example via diazotisation, for example into halogen, cyano or
hydroxy, or carboxy, lower alkoxycarbonyl, halocarbonyl, for example -COCI, or an acid
anhydride, which can be converted by reaction with ammonia or the corresponding
primary or secondary amine into carbamoyl or N-mono- or N,N-di-substituted carbamoyl,
respectively. The conversion of substituents on aromatic systems according to process (b)
is known ~ se.
The starting compounds of formula IV are prepared, for example, analogously to
203697~
- 11
process (a), there being used in the corresponding reactions, instead of the radical X, a
radical X'.
Process (c~: Isocyanomethyl is a -CH2-N=C radical. Salts of hydrazoic acid are especially
alkali metal azides, for example sodium azide.
The starting compounds of formula V are prepared, for example, from the analogous
compounds of formula 11 in which one of the radicals Y', R' 1 and R'2 is, for example,
bromomethyl. The latter compounds are first converted in a manner known ~ se, for
example by reaction with hexamethylenetetramine (urotropine), into the corresponding
aminomethyl compounds, and then converted in a manner known E~ se, for example by
reaction with dichlorocarbene (for example from chloroform and concentrated KOH), into
the desired isocyanomethyl compounds of formula V.
Compounds of formula I can be converted in a manner known Per se into other
compounds of formula I.
For example, compounds of formula I wherein X is halogen, especially bromine, can be
converted by reaction with a cyanating agent, for example copper(I) cyanide, into other
compounds of formula I wherein X is cyano.
It is also possible, for example, to convert compounds of formula I wherein X is halogen,
especially bromine, by reaction with hydroxyaryl compounds or corresponding alkali
metal salts thereof, for example potassium phenolate, into other compounds of formula I
wherein X is aryloxy, advantageously, for example, in the presence of copper.
Furthermore, for example, compounds of formula I wherein X is cyano can be converted
by partial hydrolysis, for example with potassium carbonate and aqueous H22 solution,
into other compounds of formula I wherein X is carbamoyl.
On the other hand, for example, compounds of formula I wherein X is carbamoyl orN-lower alkylcarbamoyl can also be converted, with the splitting-off of water or lower
alkanol, respectively, into compounds of formula I wherein X is cyano.
Finally, compounds of formula I wherein X is cyano can also be converted directly into
compounds of formula I wherein X is, for example, N-lower alkylcarbamoyl or N-cyclo-
,.. . .. .
~. : --.
-
' ~ ' :
2036~7~
alkyl-lower alkylcarbamoyl by first being treated with KOH/tert-butanol and then reacted
with a lower alkyl halide or a cycloalkyl-lower alkyl halide, respectively [S. Linke,
Synthesis 1978, 303].
Free compounds of forn~ula I having salt-forming properties that are obtainable according
to the process can be converted into their salts in a manner known ~r se: compounds
having basic properties, for example by treatment with acids or suitable derivatives
thereof, and compounds having acid properties, for example by treatment with bases or
suitable derivatives thereof.
Mixtures of isomers obtainable according to the invention can be separated into the
individual isomers in a manner known ~ se, racemates, for example, by forming salts
with optically pure salt-forming reagents and sepaMting the diastereoisomeric mixture so
obtainable, for example by means of fMctional crystallisation.
The reactions described above can be carried out under reaction conditions that are known
se, in the absence or, usually, in the presence of solvents or diluents, preferably those
which are inert towards the reagents used and are solvents thereof, in the absence or
presence of catalysts, condensing agents or neutMlising agents, and, depending on the
nature of the reaction and/or of the reactants, at reduced, normal or elevated temperature,
for example within a temperature range of from approximately -70C to approximately
200C, preferably from approximately -20C to approximately 150C, for example at the
boiling point of the solvent used, under atmospheric pressure or in a closed vessel, where
appropriate under pressure, and/or in an inert atmosphere, for example under a nitrogen
atmosphere.
In view of the close relationship between the compounds of formula I in free form and in
the form of salts, hereinbefore and hereinafter any reference to the free compounds or their
salts should be understood as including also the corresponding salts or free compounds,
respectively, where appropriate and expedient.
The compounds, including their salts, may also be obtained in the form of hydrates, or
their crystals may, for example, include the solvent used for crystallisation.
The starting materials used in the process of the present invention are preferably those
which result in the compounds described at the beginning as being especially valuable.
2~36~7a
- 13-
The invention relates also to those forms of the process in which a compound obtainable
as intermediate at any stage of the process is used as starting material and the remaining
process steps are carried out, or in which a starting material is formed under the reaction
conditions or is used in the form of a derivative, for example a salt thereof.
The present invention relates also to pharmaceutical compositions that contain one of the
pharmacologically active compounds of formula I as active ingredient. Compositions for
enteral, especially oral, administration and for parenteral administration are especially
preferred. The compositions contain the active ingredient on its own or, preferably,
together with a pharmaceutically acceptable carrier. The dosage of the active ingredient
depends upon the disease to be treated and upon the species, its age, weight and individual
condition, and also upon the mode of administration.
The pharmaceutical compositions contain from approximately 0.1 % to approximately
95 % active ingredient, forms of administration that are in single-dose form preferably
containing from approximately 1 % to approximately 90 % active ingredient, and forms of
administration that are not in single-dose form preferably containing from approximately
0.1 % to approximately 20 % active ingredient. Unit dose forms, such as drages, tablets or
capsules, contain from approximately 0.5 mg to approximately 100 mg of active
ingredient.
The pharmaceutical compositions of the present invention are prepared in a manner known
~ se, for example by means of conventional mixing, granulating, confectioning,
dissolving or Iyophilising processes. For example, pharmaceutical compositions for oral
administration can be obtained by combining the active ingredient with one or more solid
carriers, if desired granulating a resulting mixture and, if desired, processing the mixture
or granulate into tablets or dragee cores, where appropriate by addillg additional
excipients.
Suitable carriers are especially fillers, such as sugars, for example lactose, saccharose,
mannitol or sorbitol, cellulose preparations and/or calcium phosphates, for example
tricalcium phosphate or calcium hydrogen phosphate, and binders, such as starches, for
example corn, wheat, rice or potato starch, methylcellulose, hydroxypropylmethyl-
cellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone, and/or, if desired,
disintegrators, such as the above-mentioned starches, also carboxymethyl starch,
.
, . .
203~97~
- 14-
crosslinked polyvinylpyrrolidone, alginic acid or a salt thereof, such as sodium alginate.
Additional excipients are especially flow conditioners and lubricants, for example silica,
talc, stearic acid or salts thereof, such as magnesium or calcium stearate, andlor
polyethylene glycol, or derivatives thereof.
Dragee cores may be provided with suitable coatings which may be enteric coatings, there
being used, inter alia, concentrated sugar solutions which may contain gum arabic, talc,
polyvinylpyrrolidone, polyethylene glycol and/or titanium dioxide, or coating solutions in
suitable organic solvents or solvent mixtures, or, for the preparation of enteric coatings,
solutions of suitable cellulose preparations, such as acetylcellulose phthalate or hydroxy-
propylmethylcellulose phthalate. Dyes or pigments may be added to the tablets or drage
coatings, for example for identification purposes or to indicate different doses of active
ingredient.
Other orally administrable pharmaceutical compositions are dry-filled capsules consisting
of gelatin, and also soft sealed capsules consisting of gelatin and a plasticiser, such as
glycerol or sorbitol. The dry-filled capsules may contain the active ingredient in the form
of a granulate, for example in admixture with fillers, such as corn starch, binders andlor
glidants, such as talc or magnesium stearate, and, if desired, stabilisers. In soft capsules,
the active ingredient is preferably dissolved or suspended in suitable liquid excipients,
such as fatty oils, paraffin oil or liquid polyethylene glycols, to which stabilisers may also
be added.
Other oral dosage forms are, for example, syrups prepared in customary manner that
contain the active ingredient, for example, in suspended form and in a concentration of
approximately from 5 % to 20 %, preferably approximately 10 % or in a similar concen-
tration that provides a suitable single dose when the syrup is administered in quantities of
5 or 10 ml. Also suitable, for example, are powdered or liquid concentrates for the
preparation of shakes, for example in milk. Such concentrates may also be packaged in
single dose quantities.
Suitable rectally administrable pharmaceutical compositions are, for example,
suppositories that consist of a combination of the active ingredient with a suppository
base. Suitable suppository bases are, for example, natural or synthetic triglycerides,
paraffin hydrocarbons, polyethylene glycols or higher alkanols.
203697~
- 15-
For parenteral administration there are suitable, especially, aqueous solutions of an active
ingredlent ln water-soluble form, for example in the form of a water-soluble salt, or
aqueous injection suspensions that contain viscosity-increasing substances, for example
sodium carboxymethylcellulose, sorbitol and/or dextran and, if desired, also stabilisers. In
this case, the active ingredient, if desired together with excipients, may also be in the form
of a Iyophilisate and be dissolved by the addition of suitable solvents before parenteral
administration.
Solutions, such as are used, for example, for parenteral administration, may also be
administered in the form of infusion solutions.
The invention relates also to a method for the treatment of the pathological conditions
mentioned above. The compounds of the present invention can be administered
prophylactically or therapeutically, and are preferably used in the form of pharmaceutical
compositions. For a body weight of approximately 70 kg, a daily dose of from approxi-
mately 0.5 mg to approximately 100 mg, preferably from approximately 1 mg to approxi-
mately 20 mg, of a compound of the present invention will be administered.
The following Examples illustrate the present invention; temperatures are given in degrees
Celsius. The following abbreviations are used: ether = diethyl ether; THF = tetrahydro-
furan; hexane = n-hexane; DMF = dimethylformamide; DMSO = dimethyl sulfoxide;
TLC = thin-layer chromatography.
Example 1: 7-Bromo-4- r I -(1 ~214-triazolvl)methvll -2.3-dimethylbenzofuran
104 mg of 1,2,4-triazole, 139 mg of potassium carbonate and 10 mg of potassium iodide
are added in succession to a solution of 318 mg of 7-bromo-4-bromomethyl-2,3-dimethyl-
benzofuran in 10 ml of absolute acetone. After stirring for 2 hours at 55, the suspension is
cooled to room temperature and the acetone is evaporated. The reaction mixture is
partitioned between methylene chloride and water, and the organic phase is washed with
brine, dried over sodium sulfate and concentrated to dryness by evaporation. Theresulting oil is chromatographed with chloroform over silica gel to yield the title
compound. It is recrystallised from methylene chloride/ether/hexane; m.p. 108-110. IR
(CH2C12): 1631, 1605, 1504, 1404, 1199, 1140cm-1.
The starting compounds are prepared as follows:
203697S
- 16-
(a) 4-Bromo-3-(butan-3-on-2-yl)oxy-benzoic acid ethvl ester
5.17 g of potassium carbonate in 20.5 ml of acetone are added to 3.08 g of 4-bromo-
3-hydroxybenzoic acid ethyl ester (see DE-A-2 062 611) and 1.46 g of 3-chloro-2-butanone. After stirring under reflux for 16 hours, the sand-yellow suspension is cooled to
room temperature and filtered. The filtrate is concentrated by evaporation under reduced
pressure to yield the title compound in the form of a colourless oil that is used further
without further purification. TLC (silica gel; CH2C12/methanol 9S:S) Rf = 0.69. IR
(CH2C12): 1720, lS90, 1575, 1480, 1415, 1290 cm-1.
(b) 7-Bromo-2~3-dimethvlbenzofuran-4-carboxvlic acid
34 g of 4-bromo-3-(butan-3-on-2-yl)oxy-benzoic acid ethyl ester are added dropwise
within 20 minutes to 25 ml of ice-cooled concentrated sulfuric acid. The mixture is stirred
under nitrogen for 25 hours (bath temperature: 50). The reaction mixture is first poured
onto a mixture of ice-water and ethyl acetate and then partitioned between ethyl acetate
and saturated aqueous sodium hydrogen carbonate solution. The aqueous phase is
separated off and washed twice with ethyl acetate. The combined organic extracts are
extracted again with aqueous sodium hydrogen carbonate solution. The combined aqueous
extracts are adjusted to pH 1 with concentrated hydrochloric acid, a yellow solid
precipitating which is removed by filtration. After recrystallisation from ether, the title
compound is obtained. TLC (silica gel; toluene/ethyl acetate 9:1) Rf = 0.3. IR
(DMSO-d6): 1710, 1625, 1260, 1170cm-1.
(c) 7-Bromo-4-hvdroxvmethvl-2.3-dimethvlbenzofuran
1.58 g of 7-bromo-2,3-dimethylbenzofuran-4-carboxylic acid are added in portions to a
suspension, cooled to 0, of 226.3 mg of lithium aluminium hydride in 18 ml of absolute
THF. When the addition is complete, the solution is stirred further for 30 minutes at 0
and then for 19 hours at room temperature. The reaction mixture is then partitioned
between lN hydrochloric acid and ethyl acetate. The aqueous phase is separated off and
the organic phase is washed twice with aqueous sodium hydrogen carbonate solution and
twice with brine. The organic phase is dried over sodium sulfate and the solvent is
evaporated off under reduced pressure to yield the title compound in the form of a
light-yellow solid. TLC (CH2C12/methanol 9S:S) Rf = O.S. IR (CH2C12): 3597,2923,1632, 1602, 1404, 1206 cm-l.
(d) 7-Bromo-4-bromomethYl-2.3-dimethvlbenzofuran
1.98 g of 7-bromo-4-hydroxymethyl-2,3-dimethylbenzofuran are added at 0 within
203697~
- 17-
15 minutes to a solution of 0.8 ml of phosphorus tribromide in 20 ml of absolute ether.
After stirring for 2 hours at 0 and for a further 30 minutes at room temperature, the
mixture is partitioned between ethyl acetate and ice-cooled water. The organic phase is
washed in succession with water, aqueous sodium hydrogen carbonate solution and twice
with brine. After drying over sodium sulfate and evaporation of the solvent under reduced
pressure, the pure title compound is obtained. TLC (silica gel, chloroform/methanol 95:5)
Rf = 0.85. IR (CH2C12): 1630, 1601, 1487, 1444, 1402, 1203 cm-1.
Example 2: 7-Cvano-4-rl-(1,2 4-triazolyl)methvll-2.3-dimethYlbenzofuranA mixture of 306.2 mg of 7-bromo-4-[1-(1,2,4-triazolyl)methyl]-2,3-dimethylbenzofuran
(see Example 1) and 121.8 mg of copper(I) cyanide in 0.8 ml of pyridine is refluxed under
nitrogen for 18 hours. The black reaction mixture is treated with ethyl acetate and 37 %
aqueous ammonia solution and the organic phase is separated off. The organic phase is
washed again with aqueous ammonia solution, then twice with O.lN hydrochloric acid and
finally twice with water, and is dried and filtered. After evaporation of the solvent under
reduced pressure, the title compound is obtained. TLC (CH2C12/methanol 95:5) Rf = 0.38.
IR (CH2C12): 2233, 1615, 1504, 1408 cm-1.
Example 3: 7-Bromo-4-(1-imidazolvlmethyl)-2,3-dimethvlbenzofuran
1.76 g of sodium are added in portions to a solution of 22.3 g of imidazole in 350 ml of
absolute THF which is being maintained at 45. After stirring at 45 for 2.5 hours, the
orange-coloured reaction mixture is treated with a solution of 16.7 g of 7-bromo-4-bromo-
methyl-2,3-dimethylbenzofuran (see Example ld) in 250 ml of absolute THF. After 2
hours, the solvent is evaporated under reduced pressure and the reaction mixture is treated
with aqueous sodium chloride solution and 180 ml of lN sodium hydrogen carbonatesolution. The reaction mixture is extracted three times with ether and the extracts are
washed with brine, dried over sodium sulfate and concentrated to dryness by evaporation
to yield the title compound, m.p. 158- 160. TLC tchloroform/methanol 9: 1) Rf = 0.49.
IR (CH2C12): 1630, 1500,1400, 1385, 1230, 1209, 1150 cm-1.
Example 4: 4-(1-Im dazolylmethvl)-7-E~henoxv-2,3-dimethylbenzofuran hYdrochloride
A mixture of 25.4 g of 7-bromo-4-(1-imidazolylmethyl)-2,3-dimethylbenzofuran (see
Example 3), 11 g of potassium phenolate and 250 mg of copper powder is stirred for
18 hours at 150. The reaction mixture is cooled to room temperature and chromato-
graphed over silica gel (CH2C12/methanol 99.5:0.5), to yield the title compound in the
form of the free base. The latter, dissolved in methanol, is treated with ethereal HCI
20369~
- 18 -
solution to yield the title compound; m.p. 180- 181 (aRer crystallisation frommethanol/ether); IR (Nujol): 2550, 1470, 1380, 1220 cm-1.
Example 5: 7-Cyano-4-(1-imidazolvlmethyl)-2,3-dimethvlbenzofuran hvdrochloride
A mixture of 1 g of 7-bromo-4-(1-imidazolylmethyl)-2,3-dimethylbenzofuran (see
Example 3) and 0.32 g of copper(I)cyanide in 3 ml of N-methyl-2-pyrrolidone is stirred
for 20 hours at 200. The reaction mixture is cooled, poured into an ice-cooled 50 %
aqueous ethylenediarnine solution and extracted with methylene chloride. The organic
extracts are washed twice with 50 % aqueous ethylenediamine solution and twice with
water, dried over sodium sulfate and concentrated to dryness by evaporation to yield the
title compound in crude form. The latter is chromatographed over silica gel (chloro-
form/methanol 95:5) to yield the title compound in pure form. TLC (chloroform/methanol
95:5) Rf = 0.31. IR (CH2C12): 2250, 1625, 1505, 1235, 1210 cm-1. The hydrochloride of
the title compound is obtained in a manner analogous to that described in Example 4; m.p.
258-260 (with decomposition).
Example 6: 7-Bromo-3-(1-imidazolvlmethyl)-benzofuran hvdrochloride
0.63 g of sodium is added within 30 minutes at 20 to a solution of 4.35 g of imidazole in
45 ml of absolute THF. After stirring for 2.5 hours - once all the sodium has been
consumed - there is added dropwise to the reaction mixture, within 20 minutes, a solution
of 7.9 g of 7-bromo-3-bromomethyl-benzofuran in 25 ml of THF. After stirring for a
further 1.5 hours at room temperature,300 ml of water are added to the reaction mixture,
which is then extracted twice with 300 ml of ethyl acetate each time. The combined
organic extracts are washed S times more with 50 ml of water each time and once with 70
ml of brine, are dried over sodium sulfate and concentrated. Purification is effected by
column chromatography (silica gel, chloroform/methanol 9:1). The free base is dissolved
in ethanol/ether and converted into the title compound by treatment with 6N ethereal HCI
solution; m.p. 192-197 (with decomposition). IR (Nujol): 2960,1465, 1415, 1370, 1285,
1280 cm-1.
The starting compounds are prepared as follows:
(a) 2-(2-Bromophenoxy~-acetic acid ethvl ester
245.2 g of chloroacetic acid ethyl ester and 552 g of potash are added to 346 g of_-bromo-
phenol in 1 litre of acetone. The yellow suspension is heated to reflux temperature and
stirred at that temperature for 24 hours. The suspension is filtered with suction and the
-~ 203~7~
- 19-
residue is then washed repeatedly with acetone. The solution is concentrated under a high
vacuum and the resulting oil is dissolved in ether and washed in succession, while cooling
with ice, with 3 x 150 ml of 2N NaOH and 3 x 150 ml of brine. The ethereal phase is dried
over sodium sulfate, filtered and concentrated by evaporation. The pure title compound is
obtained by distillation under reduced pressure; b.p. 0.107 mbar 93-97. TLC (silica
geVchloroform): Rf = 0.69.
(b) 3-(2-Bromophenoxv)-2-oxo-succinic acid diethvl ester
99 ml of ethanol are added dropwise within 20 minutes to a suspension of 86.6 g of 50 %
sodium hydride in 2 litres of ether. During that procedure, the temperature rises to 25. To
the resulting suspension there are added dropwise within 25 minutes 245 ml of oxalic acid
diethyl ester. The reaction mixture is heated to reflux temperature and then a solution of
425.3 g of 2-(2-bromophenoxy)-acetic acid ethyl ester in 500 ml of ether is added thereto.
After 30 minutes - when everything has dissolved - the reaction mixture is cooled and is
poured, with stirring, onto 1.5 kg of ice. A pH of 3 is established by the addition of 2N
HCI and the phases are subsequently separated. The organic phase is washed with water
and then with brine and is dried with sodium sulfate. After concentration under reduced
pressure, the resulting title compound [IR (CH2C12): 1750, 1670, 1590 cm-l] is further
reacted without additional purification.
(c) 7-Bromo-2,3-dlethoxycarbonvl)benzofuran
366.4 g of 3-(2-bromophenoxy)-2-oxo-succinic acid diethyl ester are added dropwise
within 1 hour with stirring and at room temperature to 1.135 litres of 90 % sulfuric acid.
The dark-brown reaction mixture is then stirred at 55 for 1 day. After cooling, the
reaction mixture is poured, with stirring, onto 2 kg of ice. After extraction with ether and
separation of the aqueous layer, the batch is washed in succession with brine, lN aqueous
sodium hydrogen carbonate solution and brine until neutral. After drying over sodium
sulfate, filtration and concentradon under reduced pressure, the crude product so obtained
is purified by chromatography (silica geVchloroform). TLC (chloroform) Rf = 0.67.
IR (CH2C12): 1740, 1600, 1475, 1370 cm-l.
(d) 7-Bromo-3-ethoxycarbonv!-benzofuran
A mixture of 87.7 g of 7-bromo-2,3-di(ethoxycarbonyl)benzofuran, 30 g of sodium
chloride and 9.2 ml of water is stirred in 530 ml of DMSO for 3 hours at 150 (CO2
evolution). After cooling the reaction mixture, the undissolved material is removed by
filtration over Hyflo Super Cel(g) (kieselguhr). The filtrate is freed of DMSO at 40 under
203697~
- 20 -
reduced pressure. The residue is dissolved in ethyl acetate and washed with 3 x 300 ml of
water and 1 x 300 ml of brine. The organic phase is dried over sodium sulfate and
concentrated under reduced pressure. The black residue is purified by column chromato-
graphy (silica geVchloroform). The title compound is recrystallised from petroleum ether;
m.p. 54-56. IR (CH2C12): 1725, 1590, 1585, 1480 cm-1.
(e) 7-Bromo-3-hyd oxvmethvl-benzofuran
28 g of 7-bromo-3-ethoxycarbonyl-benzofuran are added within 60 minutes at 0 under
nitrogen to 182.6 ml of a 20 % solution of diisobutylaluminium hydride in toluene. After
stirring for a further 60 minutes at 0, the reaction mixture is poured onto a mixture of
400 ml of ice and 50 ml of 70 % sulfuric acid. After stirring and separation of the organic
phase, extraction is carried out three times more with 50 ml of toluene each time. The
combined extracts are washed with 4 x 35 ml of water and 1 x 30 ml of brine, dried over
sodium sulfate and concentrated under reduced pressure. The residue is crystallised from
ethyl acetate/petroleum ether; m.p. l l l- 112. IR (CH2C12): 3600, 1615, 1595,
1470 cm-1.
(f) 7-Bromo-3-bromomethyl-benzofuran
6.25 g of 7-bromo-3-hydroxymethyl-benzofuran are added within 30 minutes at 3 to a
solution of 2.73 g of phosphorus tribromide in S0 ml of ether. After the temperature has
risen to 15, the reaction mixture is stirred for a further I hour at 5 and is then poured
onto 100 ml of ice. After the addition of ether, the phases are separated. The organic
solution is washed with 3 x 40 ml of water and 1 x 40 ml of brine, is dried over sodium
sulfate and concentrated. The title compound is crystallised from ethyl acetatelpetroleum
ether; m.p. 123-124.5; IR (CH2C12): 1590,1480, 1415 cm-1.
Example 7: 7-Cvano-3-(1-imidazolvlmethvl)-benzofuran hvdrochloride
Analogously to Example S,5.98 g of 7-bromo-3-(1-imidazo1ylmethyl)-benzofuran (see
Example 6) are converted with 2.13 g of copper(I) cyanide in 19.5 ml of N-methyl-2-
pyrrolidone into the title compound which is crystallised from ethanoUether; m.p.
237-239; IR (KBr): 3090,3000, 2800, 2236, 1575,1425, 1280 cm-1.
Example 8: 7-Bromo-S-(1-imidazolylmethvl)-2,3-dimethyl-benzofuran hydrochloride
Analogously to Example 6,31.8 g of 7-bromo-S-bromomethyl-2,3-dimethyl-benzofuranare converted into the title compound; m.p. (after crystallisation from methanol/ether)
272-274. lR (Nujol): 2962,1464, 1416, 1370, 1287, 1281 cm- 1.
2036~7~
The starting compound is prepared as follows: -
(a) 7-Bromo-5-bromomethvl-23-dimethyl-benzofuran
Analogously to Example Id, 76.5 g of 7-bromo-5-hydroxymethyl-2,3-dimethylbenzofuran
are converted into the title compound; m.p. 122-124. IR (CH2C12): 1629, 1600,1488,
1443, 1404, 1203 cm-l.
Example 9: 7-Cyano-5-(1-imidazolvlmethyl)-2.3-dimethvl-benzofuran hvdrochloride
Analogously to Example 5, 12.2 g of 7-bromo-5-(1-imidazolylmethyl)-2,3-dimethyl-benzofuran (see Example 8) are converted with 3.9 g of copper(I) cyanide into the title
compound; m.p. (after crystallisation from ethanol/ether) 258-259. IR (KBr): 3092, 2238,
1470, 1285 cm-l.
Example 10: 5-Bromo-3-(1-imidazolvlmethvl)-benzofuran hvdrochloride
Analogously to Example 6, 33.05 g of 5-bromo-3-bromomethyl-benzofuran are converted
into the title compound; m.p. (after crystallisation from ethanoVether) 190.5-191.5;
IR (KBr): 2750, 1450, 1270, 1110 cm-l.
The starting compounds are prepared as follows:
(a) 2-(4-Bromophenoxv)-acetic acid ethyl ester
Analogously to Example 6a, 364.04 g of 4-bromophenol are converted into the title
compound; m.p. (after crystallisation from hexane) 56-58; IR (CH2C12): 1760, 1580,
1490, 1210 cm-l.
(b) 3-(4-Bromophenoxv)-2-oxo-succinic acid diethvl ester
Analogously to Example 6b,356.2 g of 2-(4-bromophenoxy)-acetic acid ethyl ester are
converted into the title compound; IR (CH2C12): 1745, 1665, 1585, 1475, 1230 cm-1.
(c) 5-Bromo-2.3-di(ethoxvcarbonyl)benzofuran
Analogously to Example 6c,465 g of 3-(4-bromophenoxy)-2-oxo-succinic acid diethyl
ester are converted into the title compound; m.p. (after crystallisation from
ether/petroleum ether) 45-48; IR (CH2C12): 1725, 1580, 1300 cm-1.
203697~
- 22 -
(d) 5-Bromo-3-ethoxvcarbonyl-benzofuran
Analogously to Example 6d, 87.2 g of 5-bromo-2,3-di(ethoxycarbonyl)benzofuran are
converted into the tide compound; m.p. (after crystallisation from petroleum ether)
81-83; IR (CH2C12): 1725, 1560, 1440 cm-1.
(e) 5-Bromo-3-hydroxymethvl-benzofuran
Analogously to Example 6e, 84.5 g of 5-bromo-3-ethoxycarbonyl-benzofuran are
converted into the title compound; m.p. (after crystallisation from ethyl acetate/petroleum
ether) 72-75; IR (CH2C12): 3555, 1440, 1190 cm-1.
(f) S-Bromo-3-bromomethyl-benzofuran
Analogously to Example 6f, 52.3 g of 5-bromo-3-hydroxymethyl-benzofuran are
converted into the title compound; m.p. (after crystallisation from hexane) 75-78;
IR (CH2C12): 1445, 1180,1105 cm-1.
Example 11: 5-Cvano-3-(1-imidazolylmethvl)-benzofuran hvdrochloride
Analogously to Example 5, 19.2 g of 5-bromo-3-(1-imidazolylmethyl)-benzofuran (see
Example 10) are converted with 6.8 g of copper(I) cyanide in 60.6 ml of N-methyl-2-
pyrrolidone into the title compound; m.p. (after crystallisation from ethanol/ether)
222-224; IR: 2240, 1500,1470 cm-1.
Example 12: The following compounds can be prepared in a manner analogous to that
described in the Examples:
2~3~97~
- 23 -
CH2
~ R~
_ A X Rl R2 IR (CH2C12) [cm~l]
(a) l-Tmidazolyl Cl CH3 CH3 1629,1502, 1231
(b) l-Imidazolyl Br . H H 1630, 1501,1232
(c) l-Imidazolyl Br H CH3 1628, 1501, 1231
(d): 1-Imidazolyl Br CH3 H 1630, lS00 1231
(e) 1-Imidazolyl Br -(CH2)4- 1630, 1503 1230
(f) 1-~nidazolyl Br C2Hs C2Hs 1630, lS01, 1228
(g) l-Imidazolyl CN H CH3 2248, 1625, 1504, 1234
(h) l-Imidazolyl CN CH3 H 2249, 1627, lS01, 1233
(i) l-Imidazolyl CN H H 2247, 1628, 1500, 1235
(i) :: 1-Imidazolyl CN -(CH2)4- 2250, 1629, 1502, 1231
(k) 1-(1,2,4-Triazolyl) Cl CH3 CH3 1631, 1605, 1504, 1199
(1) 1-(1,2,4-Triazolyl) Br H CH3 1628, 1607, 1502 1200
(m) 1-(1,2,4-Triazolyl) Br CH3 H 1629, 1605 1502 1200
(n) 1-(1,2,4-Triazolyl) Br -(CH2)4- 1630, 1604 1504, 1198
(o) 1-(1,2,4-Triazolyl) Br H H 1631, 1605, 1503, 1199
(P) 1-(1,2,4-Triazolyl) CN H H 2233, 1615, 1504
(q) 1-(1,2,4-Triazolyl) CN H CH3 2235, 1617, 1503
(r) 1-(1,2,4-Triazolyl) CN : CH3 H 2232, 1615, 1503
(s) 1-(1,2,4-Triazolyl) CN -(CH2)4- 2233, 1614, 1504
,~
(t): 1-(1,2,4-Triazolyl) ~ ~ CH3 CH3 1602, 1505, 1227
\=/
(u) 1-(1,2,3-Triazolyl) Br CH3 CH3 1631, 1605, 1504, 1199
(v) 1-(1,2,3-Triazolyl) CN H CH3 æ36, 1618, 1504
(w) 1-(1,2,3-Triazolyl) CN CH3 H 2235, 1616, 1506
(x) 1-(1,2,3-Triazolyl) CN CH3 CH3 2236, 1617, lSOS
(Y) 1-(1,2,3-Triazolyl) CN -(CH2)4- 2235, 1617, 1505
(Z) 1-(1,2,3-Triazolyl) ~~9 CH3 CH3 1602, 1506, 1228
(aa) 1-Tetrazolyl Br CH3 CH3 1630, 1606, 1504, 1200
(ab) 1-Tetrazolyl CN H CH3 2234, 1616 1505
(ac) 1-Tetrazolyl CN CH3 H 2232, 1616 1506
(ad) l-Tetrazolyl CN CH3 CH3 2232, 1618, 1505
. . , ,. ~ ~.
, :
.
. ~
,
~$~
- 24-
~ .... _ _
A X Rl R2 IR (CH2CI2) [cm~l]
. _ . .__ _ .. ._ _ _ .
(ae) 1-Tetrazolyl CN ~(CH2)a,- 2235,1617, 1503
(a~ 1 -Tetrazolyl ~ ~3 CH3 CH3 1602, 1505,1225
(ag) 2-Tetrazolyl Br CH3 CH3 1628, 1606, 1504, 1201
(ah) -Tetrazolyl CN H CH3 2234, 161S, 1505
(ai) 2-Tetrazolyl CN CH3 H 2232, 1614, 1505
(aj) 2-Tetrazolyl CN -(CH2)4- 2234, 1616, 1507
(ak) 2-Tetrazolyl ~~=~ CH3 C~3 1602, 1507, 1227
. _ _ ,
Example 13: The following compounds can be prepared in a manner analogous to that
described in the preceding Examples:
A-CH2 \~ i CH3
~ CH3
X
.
A X IR [cm~~
.
(a) 1 -(1,2,4-Triazolyl) E3r 1635,1604, 1507
(b) 1-(1,2,4-Triazolyl) CN 2238, 1617, 1505
(c) l-Tetrazolyl Br 1633,1604, lSOS
(d) l-Tetrazolyl CN 2237, 1619, 1504
(e) 2-Tetrazolyl Br 1634, 1606, lSOS
(f) 2-Tetrazolyl CN 2236, 1618,15()4
. _ . _
Example 14: The following compounds can be prepared in a manner analogous to that
described in the preceding Examples:
203~7~
- 25 -
~CH2-A
~ O
X
_ IR [cm~l]
(a) 1-Imidazolyl -O~ 1602, 1506, 1495,
(b) 1-(1,2,4-Triazolyl) Br 1632, 1605, 1504,1198
(c) 1-(1,2,4-Triazolyl) CN 2236, 1618, 1505
(d) 1-(1,2,4-Triazolyl) -O~ 1601, 1507, 1496,
(e) l-Tetrazolyl Br 1630, 1606, 1505, 1197
(f) l-Tetrazolyl CN 2235, 1615, 1505
(g) l-Tetrazolyl -O~ 1600, 1507, 1494,
(h) 2-Tetrazolyl Br 1631,1606, 1504, 1199
(i) 2-Tetrazolyl CN 2235, 1617, 1508
(i) 2-Tetrazolyl ~3 1602, 1506, 1495,
Example 15: The following compounds can be prepared in a manner analogous to that
described in the preceding Examples:
X ~ ,CH2-A
~oJI
2~36~7~
- 26 -
A X IR [cm-l]
(a) 1-(1,2,4-Triazolyl) Br 1632, 1606, 1503, 1198
(b) 1-(1,2,4-Triazolyl) CN 2237, 1614, 1502
(c) 1-(1,2,3-Triazolyl) Br 1630, 1605, 1502, 1200
(d) 1-(1,2,3-Triazolyl) CN 2234, 1615, 1504
(e) 1-Tetrazolyl Br 1631, 1606, 1502, 1197
(f) 1-Tetrazolyl CN 2235, 1615, 1503
(g) 2-Tetrazolyl Br 1632, 1605, 1504,1202
(h) 2-Tetrazolyl CN 2237, 1614, 1504
Example 16: (a) 7-Bromo-4-(2-tetrazolvlmethvl)-2~3-dimethvlbenzofuran and
(b) 7-bromo-4-(1-tetrazolvlmethvl)-2~3-dimethvlbenzofuran
315 mg of dry tetrazole, 417 mg of potassium carbonate and 30 mg of potassium iodide
are added in succession to a solution of 955 mg of 7-bromo-4-bromomethyl-2,3-dimethyl-
benzofuran in 15 ml of acetone. After stirring at 55 for 1.17 hours, the reaction mixture is
cooled and concentrated. The residue is taken up in CH2C12/water. The organic phase is
separated off, washed with brine, dried over sodium sulfate and concentrated. Column
chromatography (SiO2, CH2C12) yields first 7-bromo-4-(2-tetrazolylmethyl)-2,3-
dimethylbenzofuran; m.p. (after recrystallisation from ether/hexane): 117-120; 1H-NMR
(DMSO-d6): w = 2.25 (s,3H), 2.42 (s, 3H), 6.27 (s, 2H),7.07 and 7.47 (arom. H,2H), 9.0
(s, lH) ppm; and then 7-bromo-4-(1-tetrazolylmethyl)-2,3-dimethylbenzofuran; m.p. (after
recrystallisation from ether): 168-170; lH-NMR (DMSO- d6): w = 2.25 (s, 3H), 2.44 (s,
3H), 6.03 (s, 2H), 6.97 and 7.47 (arom. H,2H),9.47 (s, lH) ppm.
Example 17: 7-Cvano-4-(2-tetrazolvlmethvl)-23-dimethvlbenzofuran
A solution of 154 mg of 7-bromo-4-(2-tetrazolylmethyl)-2,3-dimethylbenzofuran and
50 mg of copper(I) cyanide in 0.9 ml of N-methyl-2-pyrrolidone is stirred at 190-200 for
2.5 hours. After cooling, the reaction mixture is diluted with CH2C12, washed twice with
aqueous ethylenediamine solution (50 %), twice with water and twice with brine and, after
being dried over sodium sulfate, is concentrated. The title compound is purified by column
chromatography (SiO2, toluene to toluene/ethyl acetate 95:5) and subsequently
crys~allised from CH2C:12/ether/hexane; m.p. 134-135; TLC (silica gel, methylene
chloride): Rf = 0.3. IR (CH2C12): 2234, 1631, 1616, 1407 cm-1.
Example 18: 7-Carbamoyl-4-(1-imidazolylmethvl)-2,3-dimethylbenzofuran hydrochloride
18 mg of potassium carbonate and 48 ml of a 30 % aqueous H22 solution are added at
203~97~
room temperature to a suspension of 9S mg of 7-cyano-4-(1-imidazolylmethyl)-2,3-dimethylbenzofuran hydrochloride in O.S ml of DMSO and I ml of CH2C12. After thefurther addition of 20 mg of potassium carbonate and 0.2 ml of H22 solution, the bath
temperature is adjusted to 50-60 and, after the further addition of 0.2 ml of H22
solution, the reaction mixture is stirred at room temperature for 19 hours to complete the
reaction. 3 ml of water are added to the reaction mixture which is then stirred for I hour
while cooling with ice. The solid is removed by filtration, washed with water and dried
over P2Os in a hot desiccator. The solid is crystallised from methylene
chloride/methanol/ether. After dissolving in methylene chloride/methanol, there are added
to the solid 40 ml of 9N methanolic HCI solution; after the addition of ether, the title
compound crystallises out. After filtering off, washing with ether and drying in a
desiccator, the title compound is obtained. TLC (silica gel, methylene chloride/methanol
9:1) Rf = 0.31; IR (KBr): 3420, 1660, 1610, 1575,1410 cm-1.
Example 19: 7-Carbamovl-4-rl-(1.2.4-tnazolyl)methYll-2.3-dimethY1benzofuran
20 mg of potassium carbonate and 48 ml of a 30 % hydrogen peroxide solution are added
to a solution of lOO.S mg of 7-cyano-4-[1-(1,2,4-triazolyl)methyl]-2,3-dimethylbenzofuran
(Example 2) in O.S ml of DMSO and a small amount of CH2C12. After stirring at room
temperature for 17.5 hours, a further 48 ml of a 30 % hydrogen peroxide solution are
added and the reaction mixture is stirred again for 3 hours. While cooling with ice,3 ml of
water are then added to the beige suspension, which is then stirred for 45 rninutes and
filtered. After being washed, the filtration residue is dried over phosphorus pentoxide.
After stirring with CH2C12, the pure title compound is obtained; IR (KBr): 3427,3188,
1694, 1616, 1503, 1407,1272 cm-1.
Example 20: 7-N-(cyclohexylmethyl)carbamoyl-4-rl-(1~2~4-triazolYI)methyll-
2~3-dimethvlbenzofuran
69 ml of aminomethylcyclohexane, 114 mg of N,N'-dicyclohexylcarbodiimide, 3 mg of
4-N,N-dimethylaminopyridine and 8 mg of N-hydroxybenzotriazole are added to an ice-
cooled suspension of 136 mg of 7-carboxy-4-[1-(1,2,4-triazolyl)methyl]-2,3-dimethyl-
benzofuran in 4 ml of CH2C12 and 1 ml of DMF. After 10 minutes, the cooling bath is
removed and the reaction mixture is stirred at room temperature for 7 hours. A further
57 mg of N,N'-dicyclohexylcarbodiimide, 35 ml of aminomethylcyclohexane and 2 mg of
4-N,N-dimethylaminopyridine are then added to the reaction mixture. After 6 hours, the
solid is removed by filtration. The filtrate is diluted with CH2C12 and washed in
succession with aqueous sodium hydrogen carbonate solution, water and brine. After
2~3~7~
- 28 -
drying and concentration, the crude product is purified by column chromatography (SiO2,
CH2Cl2 to CH2C12/methanol 95:5) and subsequent digestion in hexane; IR (CH2C12):3439, 2925, 1661, 1610, 1540, 1504, 1449 cm-l.
The starting compound is prepared as follows:
(a) 7-Carboxv-4-r l -~1 2,4-triazolyl~methvll-2.3-dimethylbenzofuran
A solution of 252 mg of 7-cyano-4-[1-(1,2,4-triazolyl)methyl]-2,3-dimethylbenzofuran
(Example 2) in 4 ml of ethanol and 4 ml of 4N NaOH is stirred under reflux conditions for
20.25 hours. After cooling by means of cooling with ice-water, a pH of 3 is established
with 2N H2SO4. The ethanol is evaporated off and the resulting white suspension is
cooled in a refrigerator for 1.5 hours. The crude product is obtained by filtration and is
purified by being stirred in CH2C12; IR (KBr): 3429, 3140, 1693, 1610, 1512, 1405,
1292 cm-l.
Example 21: 7-N-(cvclohexvlmethvl)carbamovl-4-(1-imidazolylmethvll-2.3-dimethyl-benzofuran
109 mg of potassium hydroxide are added to a solution of 100 mg of 7-cyano-4-(1-imidazolylmethyl)-2,3-dimethylbenzofuran (Example 5) in 0.3 ml of tert-butanol and the
mixture is stirred at 80 for 20 minutes. After cooling, 0.3 ml of bromomethylcyclohexane
are added dropwise to the mixture which is then stirred under reflux for 30 minutes. After
cooling, the mixture is poured onto water and extracted with CH2Cl2. The organic phase
is dried and concentrated. The crude product is purified by column chromatography (SiO2,
CH2C12 to CH2C12/methanol 95:5); IR (CH2Cl2): 3439, 2924, 1661, 1610, 1541, 1505,
1449, 1389 cm-l.
Example 22: 7-N-(n-propvl)carbamovl-4-(1-imidazolvlmethvl)-2~3-dimethvlbenzofuran
Analogously to Example 21, 100 mg of 7-cyano-4-(1-imidazolylmethyl)-2,3-dimethyl-
benzofuran in 0.3 ml of tert-butanol are converted with 109 mg of potassium hydroxide
and 178 ml of n-propyl bromide into the title compound. The latter is purified by column
chromatography (SiO2, CH2C12 to CH2C12/methanol 98:2) and stirring in hexane; IR(CH2C12): 3435, 3040, 1660, 1610, 1539, 1505, 1457, 1389 cm-l.
Example 23: ?-N-(2-propyl)carbamovl-4-(1-imidazolylmethvl)-2,3-dimethvlbenzofuran
Analogously to Example 21, 135 mg of 7-cyano-4-(1-imidazolylmethyl)-2,3-dimethyl-
benzofuran in 0.4 ml of tert-butanol are converted with 148 mg of potassium hydroxide
., . , . . ~ ~ , ..... .
2~3697~
- 29 -
and a total of 515 ml of isopropyl iodide within 8 hours into the title compound. The latter
is purified by column chromatography (SiO2, CH2C12 to CH2C12/methanol 98:2);
IR (CH2C12): 4325, 2966,1658, 1610, 1535, 1505, 1457, 1387 cm- 1.
Example 24: 10 000 tablets are prepared, each containing 5 mg of active ingredient, for
example one of the compounds prepared in Examples 1-23:
Composition:
active ingredient 50.00 g
lactose 2535.00 g
corn starch 125.00 g
polyethylene glycol 6000 150.00 g
magnesium stearate 40.00 g
purified water quantum satis
ocedure: All the pulverulent constituents are sieved through a sieve of 0.6 mm mesh
width. Then the active ingredient, the lactose, the magnesium stearate and half of the
starch are mixed in a suitable mixer. The other half of the starch is suspended in 65 ml of
water and the resulting suspension is added to a boiling solution of the polyethylene glycol
in 260 ml of water. The paste formed is added to the powder mixture and the resulting
mixture is granulated, if desired or necessary with the addition of more water. The
granulate is dried overnight at 35C, forced through a sieve of 1.2 mm mesh width and
pressed into tablets having a breaking notch.
Example 25: 1000 capsules are prepared, each containing 10 mg of active ingredient, for
example one of the compounds prepared in Examples 1-23:
Composition:
active ingredient 10.00 g
lactose 207.00 g
modified starch 80.00 g
magnesium stearate 3.00 g
Procedure: All the pulverulent constituents are sieved through a sieve of 0.6 mm mesh
width. Then, in a suitable mixer, the active ingredient is mixed first with the magnesium
stearate and then with the lactose and the starch until homogeneous. No. 2 hard gelatin
2~13697~
- 30-
capsules are each filled with 300 mg of the resulting mixture using a capsule-filling
machine.