Note: Descriptions are shown in the official language in which they were submitted.
2Q~~~°~7
-1-
4-18022/+
Parenterally administrable liposome formulation com~isino synthetic lipids
The invention relates to pharmaceutical compositions in the form of
parenterally
administrable liposome dispersions, or dry preparations that can be used
therefor,
comprising the zinc-phthalocyanine complex and synthetic, substantially pure
phospholipids, to a novel inventive process for the preparation of those
pharmaceutical
compositians and to the use of the dry preparations for the preparation of
intravenously
administrable liposome dispersions and to the use of the zinc-phthalocyanine
complex and
the synthetic, substantially pure phospholipids for the preparation of dry
preparations. The
invention relates also to the use of the pharmaceutical compositions in a
method for the
therapeutic treatment of the human or animal body.
Both the zinc-phthalocyanine complex itself and its therapeutic use in
photodynamic
chemotherapy for the treatment of tumours are known, see J.D. Spikes,
Photochem.
Photobiol. 43, 691 (1986). 'The zinc-phthalocyanine complex is administered
intraperitoneally to mice or rats in vivo in the form of an agueous suspension
and the
carcinoma induced in the experimental animals is irradiated 'with high-energy
light,
preferably with concentrated visible light (LASER).
The use of intraperitoneal dosage foams in human therapy generally gives rise
to
problems because of the pain caused by the piercing of the abdominal cavity
and the great
demands made of the skill of the physician. Attempts are therefore being made
to find an
alternative parenteral dosage form which is more acceptable to the patient,
but which is
also capable of ensuring systemic distribution of the zinc-phthalocyanine
complex to be
administered.
The intravenous dosage form offexs systemic distribution of the active
ingredient but
requires the active ingredient to be homogeneously distributed in the aqueous
injection
fluid.
For the preparation of suitable intravenous dosage forms it has therefore been
proposed to
use instead of the sparingly soluble zinc-phthalocyanine complex the water-
soluble
~039~'~~
-2-
derivatives thereof and to administer those derivatives intravenously, see J.
Rousseau et
al., Int. J. Appl. Radiat. Isot. 36, 709 (1985).
Although the introduction of hydrophilic groups, such as sulfonyl groups, into
the
porphyrin nucleus of the zinc-phthalocyanine complex increases the water-
solubility of
the camplex, the derivatives obtained are of a poor standard and are
unsuitable for
pharmaceutical use since they consist of non-uniform mixtures of the mono- to
tetra-substituted derivatives with several regioisomers, see F.H. Moser et
al., The
Phthalocyanines, CRC Press, 1983, Vol. II, page 20. The separation of the
numerous
isomeric derivatives would involve unrealistic expenditure.
As an alternative it has been proposed to solubilise the chemically pure,
water-insoluble
zinc-phthalocyanine complex in aqueous phase by the addition of a vehicle. For
example,
using suitable solubilisers in the form of phospholipids, for example di-
palmitoylphosphatidyl choline, the complex can be encapsulated in unilamellar
liposomes
which are substantially homogeneously dispersible in aqueous phase, see E.
Reddi et al.,
Br. J. Cancer (1987), SG, pages 597-600.
This homogeneous liposome dispersion is nevertheless still unsuitable for the
purposes of
intravenous administration to humans because the dispersion is prepared in
accordance
with the so-called injection method using relatively large amounts of toxic
pyridine, see G.
Valduga et al., J. Inorg. Biochem. 29, 59-65(1987). Pyridine is one of the few
solvents in
which the zinc-phthalocyanine complex is at all soluble. That solution is
diluted with
ethanol and the pyridine-containing ethanolic solution is injected at elevated
temperature
into water or buffer solution. In accordance with that method a residue of the
toxic
solvent pyridine will always remain in the aqueous phase as a result of the
formation of an
azeotropic mixture.
The preparation of liposome dispersions by other methods that do not use
organic solvents
also gives rise to problems. Even when solvent-free dry preparations, such as
lyophili-
sates or evaporation residues, are used for the formation of the aqueous
liposome
dispersions, the preparation of those dry preparations can in turn be effected
only from
solutions in selected organic solvents or solvent mixtures because of the
lipophilic nature
and the water-insolubility of the lipid components. Both the zinc-
phthalocyanine complex
and the phospholipids used have to be completely soluble in those solvents.
The
evaporation of the solvents must result in a homogeneous and pourable powder.
The
CA 02039477 2001-09-07
21489-8216
3
components must not be allowed to separate, as this would
result in the agglutination of the dry preparation and the
formation not of liposomes but only of poorly dispersible
aggregates, for example large micelles, which could cause
embolisms.
The problem underlying the present invention is to
prepare a parenterally, especially intravenously,
administrable liposome dispersion using those solvents in
which the zinc-phthalocyanine complex and the phospholipid
components are completely soluble and of which the residual
amount consisting of unremovable solvent residues is less
than 1 %, which amount is toxicologically harmless for
intravenous formulations.
The invention relates to pharmaceutical
compositions in the form of parenterally administrable
liposome dispersions comprising
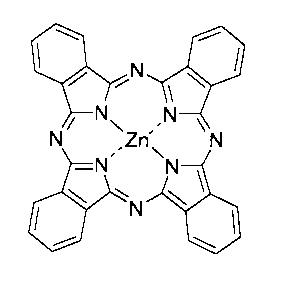
(a) a zinc-phthalocyanine complex of formula
~N 1
N N
N~ \Zri~ \N
-N~ N
~N
i
(b) a synthetic, substantially pure phospholipid
of formula
CA 02039477 2001-09-07
21489-8216
3a
1CH2 - O R1
R2 O 2CH O
Ra
+/
3CH2-O P (CnH2n) N Rb (I)
O- Rc
wherein R1 is Clo-Czoalkanoyl having an even number of carbon
atoms, Rz is Clo-Czoalkenoyl having an even number of carbon
atoms, Ra, Rb and R~ are hydrogen or Cl-C4alkyl and n is an
integer from two to four, optionally combined with a c)
synthetic, substantially pure phospholipid of formula
1CH2-O R3
R4 O 2CH O O
(S) (II)
3CH2- O p (CnH2n) CH C OH
O NH2 Y+
wherein R3 and R4 are each independently of the other Clo-
Czoalkenoyl having an even number of carbon atoms, n is an
integer from one to three and Y+ is the cation of a
~~9~~""~
_4_
pharmaceutically acceptable base, and
d) a pharmaceutically acceptable carrier liquid and, optionally, water-soluble
excipients
suitable for parenteral dosage forms.
The terms and definitions used hereinabove and hereinbelow preferably have the
following meanings in the context of the description of the invention:
The term "pharmaceutical composition" defines a mixture of substances that is
suitable for
parenteral administration, in the present case especially for intravenous
administration, to
humans and animals, preferably to humans, and that can be used for the
treatment of
various diseases, in the present case tumours.
The parenterally administrable liposome dispersion comprises liposomes in the
form of
unilamellar, multilamellar, large and small liposomes cansisting of a double-
layer
arrangement of phospholipids (I) and optionally (II) having an interior space
and a
spherical shape (unilamellar) or consisting of several concentric double-layer
arrangements of phospholipids (I) and optionally (II) having an interior space
and a
spherical shape ("onion-skin" structure of the double layers or membranes -
multilamellar). The size of the liposomes varies from approximately 1.0 x 10'8
to
approximately 1.0 x 10-5 m, depending upon the preparation process.
The therapeutic use of liposomes as carriers of active ingredients of
different kinds is
known. For example, liposomes have been proposed as carriers of proteins, for
example
antibodies or enzymes> hormones, vitamins or genes, or, for analytical
purposes, as
carriers of labelled compounds.
Pharmaceutical dosage forms based on liposomes are described in the synoptical
work by
Gregoriadis G. (Ed.) Liposome Technology, Col. II, Incorporation of Drugs,
Proteins and
Genetic Material, CRC Press 1984. In the synoptical work by Knight, C.G.
(Ed.),
Liposomes: From Physical Structure to Therapeutic Applications, Elsevier 1981,
the
advantages of a pharmaceutical dosage form based on liposomes are summarised
in
Chapter 16, page 166.
The liposome dispersion according to the present invention is free of solid
particles and
larger lipid aggregates, is stable to storage even at room temperature for
several days to
weeks, is reproducible as regards the proportion of the components, is
toxicologically
CA 02039477 2001-09-07
21489-8216
harmless as regards the lipid components used and the
residual amounts of organic solvents, and on the basis of
findings in vitro and in vivo is suitable for parenteral,
especially intravenous, administration to humans.
5 Unless expressly defined otherwise, the term
"lower" used in connection with organic radicals, for
example lower alkyl, lower alkylene, lower alkoxy, lower
alkanoyl etc., indicates that the organic radicals so
designated contain up to and including 7, and preferably up
to and including 4, carbon atoms.
The nomenclature for the phospholipids of formulae
I and II and the numbering of the carbon atoms is in
accordance with the recommendations made in the Eur. J. of
Biochem. 79, 11-21 (1977) "Nomenclature of Lipids" by the
IUPAC-IUB Commission on Biochemical Nomenclature (CBN) (sn-
nomenclature, stereospecific numbering).
The zinc-phthalocyanine complex a) corresponds to
the compound described on page 1 of the publication by G.
Valduga et al., Photochem. and Photobiology Vol. 48, No. 1
(1988), pages 1-5, which compound has been known for a long
time. The structure of this zinc-phthalocyanine complex is:
~N
N N
N~ \Zn~ \N
-N~ N
~N
i
CA 02039477 2001-09-07
21489-8216
5a
The purity of the synthetic phospholipids of
formulae I and II is more than 90 %, but preferably more
than 95 %, by weight.
This degree of purity can be demonstrated by known
analytical methods, for example by chromatography, such as
HPLC, gas chromatography or paper chromatography.
In a phospholipid of formula I, R1 as "Clo-
C2oalkanoyl having an even number of carbon atoms" is
preferably n-dodecanoyl, n-tetradecanoyl, n-hexadecanoyl, n-
octadecanoyl or n-icosanoyl.
In a phospholipid of formula I, R2 as "Clo-
C2oalkenoyl having an even number of carbon atoms" is
preferably 9-cis-dodecenoyl, 9-cis-tetradecenoyl, 9-cis-
hexadecenoyl, 6-cis-octadecenoyl, 6-trans-octadecenoyl, 9-
cis-octadecenoyl, 9-trans-octadecenoyl, 11-cis-octadecenoyl
or 9-cis-icosenoyl.
In a phospholipid of formula I, Ra, Rb and R~ are
preferably C1-C4alkyl, especially methyl.
In a phospholipid of formula I, n is an integer
from two to four, preferably two. The group
~~3~~"~~
-6-
of the formula -(Cnl-I2")- is unbranched or branched alkylene, for example 1,1-
ethylene>
1,1-, 1,2- or 1,3-propylene or 1,2-, 1,3- or 1,4-butylene. 1,2-ethylene (n=2)
is preferred.
In an especially preferred phospholipid of formula I, Rl is n-dodecanoyl, n-
tetradecanoyl,
n-hexadecanoyl ar n-octadecanoyl and RZ is 9-cis-dodecenoyl, 9-cis-
tetradecenoyl,
9-cis-hexadecenoyl, 9-cis-octadecenoyl or 9-cis-icosenayl, Ra, Rb and R~ are
methyl and n
is two.
A very especially preferred phospholipid of formula I is synthetic 1-n-
hexadecanoyl-2-
(9-cis-octadecenoyl)-3-sn-phosphatidyl choline having a purity of more than 90
%, but
preferably more than 95 %.
In a phospholipid of formula II, R3 and R4 are as defined for Rl and RZ under
formula I.
In a phospholipid of formula II, R3 and R4 as "Clo-CZOalkenoyl having an even
number of
carbon atoms" are preferably 9-ci.s-dodecenoyl, 9-cis-tetradecenoyl, 9-cis-
hexadecenoyl,
6-cis-actadecenoyl, 6-traps-octadecenoyl, 9-cis-octadecenoyl, 9-traps-
octadecenoyl,
11-cis-octadecenoyl or 9-cis-icosenoyl.
The cation Ym of a pharmaceutically acceptable base is, for example, an alkali
metal ion,
for example the lithium, sodium or potassium ion, the ammonium ion, a mono-,
di- or tri-
Ct-C4alkylammonium ion, for example the trimethyl-, ethyl-, diethyl- or
triethyl-ammonium ion, the tetramethylammonium ion, a 2-hydroxyethyi-tri-Ct-
C4alkyl-
ammonium ion, for example the choline cation, or the 2-hydroxyethylammonium
ion, or
the cation of a basic amino acid, for example lysine or arginine.
Ym is preferably the sodium ion.
In an especially preferred phospholipid of formula II, R3 and R4 are identical
and are, for
example, 9-cis-dodecenoyl, 9-cis-tetradecenoyl, 9-cis-hexadecenoyl, 9-cis-
octadecenoyl or
9-cis-icosenoyl, n is one and Y~ is the sodium ion.
A very especially preferred phospholipid of formula II is synthetic sodium 1,2-
di(9-cis-
octadecenoyl)-3-sn-phosphatidyl S-serine having a purity of more than 90 %,
but
preferably more than 95 %.
The names given in pwenthesis are also customary for the aryl radicals in the
phospholipids of formulae I and II:
9-cis-dodecenoyl (lauroleoyl), 9-cis-tetradecenoyl (myristoleoyl), 9-cis-
hexadecenoyl
(palmitoleoyl), 6-cis-octadecenoyl (petroseloyl), 6-traps-octadecenoyl
(petroselaidoyl), 9-
cis-octadecenoyl (oleoyl), 9-traps-octadecenoyl (elaidoyl), 11-cis-
octadecenoyl
(vaccenoyl), 9-cis-icosenoyl (gadoleoyl), n-dodecanoyl (lauroyl), n-
tetradecanoyl
(myristoyl), n-hexadecanoyl (palmitoyl), n-octadecanoyl (stearoyl), n-
icosanoyl
(arachidoyl).
In the pharmaceutically acceptable carrier liquid d) the components a) and b)
or a), b) and
c) are present in the form of liposomes, preferably multilamellar liposomes,
in such a
manner that for several days or weeks there is no re-formation of solids or
solid
aggregates, such as micelles, and the clear or in some cases slightly
opalescent liquid
comprising the said components can be administered, if necessary after
filtration,
parenterally, preferably intravenously.
The carrier liquid d) may comprise, for example, pharmaceutically acceptable,
non-toxic
water-soluble excipients that are necessary for the establishment of isotonic
conditions, for
example ionic additives, such as sodium chloride or non-ionic additives
(structure
formers), such as sorbitol, mannitol, glucose or lactose. In particular, the
dry preparation
comprises those additives, for example sodium chloride or mannitol, in the
prescribed
amounts, which are necessary for the establishment of isotonic conditions in
the
parenterally administrable solutions.
Suitable water-soluble excipients in the solution and in the dry preparation
are also
wetting agents or surfactants in the true sense that can be used for liquid
pharmaceutical
formulations, especially non-ionic surfactants of the fatty acid polyhydroxy
alcohol ester
type, such as sorbitan monolaurate, monooleate, monostearate or monopalmitate,
sorbitan
tristearate or trioleate, polyoxyethylene adducts of fatty acid polyhydroxy
alcohol esters,
such as polyoxyethylene sorbitan monolaurate, monooleate,~monostearate,
monopalmitate,
txistearate or trioleate, polyethylene glycol fatty acid esters, such as
polyoxyethyl stearate,
polyethylene glycol 400 stearate, polyethylene glycol X000 stearate,
especially ethylene
oxide/propylene oxide block polymers of the Pluronic~ type (Wyandotte Chem.
Corp.) or
Synperonic~ type (ICI).
The liposome dispersion can be prepared from a dry preparation to which the
present
_g_
invention likewise relates.
The present invention relates also to pharmaceutical compositions in the form
of a dry
preparation comprising
a) the zinc-phthalocyanine complex,
b) a synthetic, substantially pure phospholipid of formula I wherein Rl> R2,
Ra, Rb and R
and also n are as defined above, optionally combined with a
c) synthetic, substantially pure phospholipid of formula II wherein R3, R4, n
and Y~ are
as defined above, and optionally
d) water-soluble excipients suitable for intravenous dosage forms.
A dry preparation is the residue obtainable after any thermal drying process,
such as
evaporation at room temperature or elevated temperature or, preferably, freeze-
drying,
which residue contains less than 1 % (by weight), preferably less than 0.5 %,
organic
solvent residues, such as piperidine, tent-butanol or dimethyl sulfoxide.
In the dry preparation, the zinc-phthalocyanine complex is present in a ratio
of
approximately from 0.1 to 5.0 % by weight, preferably from 0.1 to 1.0 % by
weight, based
on the total amount of phospholipids of formulae I and optionally II -
components b) and
optionally c). In the dry preparation the mixing ratio of the phospholipids of
formula I -
the lecithin component - to the phospholipids of formula II - the serine
component - is
approximately 50:50 % by weight, preferably 70:30 % by weight.
The dry preparation of the present invention is distinguished by good filling
properties,
exact reproducibility of the weighed amounts introduced into the unit dose
forms and
storage stability.
The present invention relates also to a process far the preparation of a
pharmaceutical
composition in the form of a parenterally administrable liposome dispersion,
which
process comprises dispersing in aqueous phase a dry preparation comprising
a) the zinc-phthalocyanine complex,
b) a synthetic, substantially pure phospholipid of formula I, wherein Rl, R2,
Ra, Rb and R
and also n are as defined above, optionally combined with a
c) synthetic, substantially pure phospholipid of formula II, wherein R~, R4, n
and Y~ are
as defined above, and optionally
d) water-soluble excipients suitable for parenteral dosage forms and
optionally buffering
20394"~~
the resulting aqueous dispersion to pH 7.0-7.8 andlor isolating a liposome
fraction having
a desired diameter range,
The individual steps of the process are carried out in a manner known per se
by
reconstituting the dry preparation obtainable in accordance with the
invention, prior to
administration in the form of a liposome dispersion, in the prescribed amount
of liquid,
especially in sterile (pyrogen-free) water for injection.
Dispersion is effected, for example, by shaking (for example using a Vortex
mixer) or
stirring the aqueous phase to which the dry preparation has previously been
added. The
formation of liposomes, which may be large, small, unilamellar or
multilamellar, takes
place spontaneously, that is to say without the additional supply of external
energy and at
great speed. It is possible to disperse approximately from 0.1 to SO % by
weight (based on
the total weight of the aqueous dispersion), preferably from 2 to 20 % by
weight, of the
dry preparation in aqueous phase.
Aqueous dispersions having an acidic or basic reaction are preferably buffered
to pH
7.0-7.8, preferably pH 7.2-7.4. Optionally dispersion is effected in an
aqueous buffer
solution that has already been buffered to that pH value.
Dispersion is effected at temperatures of below approximately 36°C,
preferably at room
temperature. As appropriate, the process is carried out with cooling and/or
under an inert
gas atmosphere, for example a nitrogen or argon atmosphere. The resulting
liposomes are
stable in aqueous phase over a very long period (up to several weeks or
months).
The size of the liposomes formed depends inter alia upon the amount of active
ingredient
and the lipid components, the mixing ratio thereof and the concentration of
the
components in the aqueous dispersion and upon the method of preparation. For
example,
by increasing or reducing the concentration of lipid components it is possible
to produce
aqueous phases having a high proportion of small or large liposomes.
It is possible to obtain an especially uniform size distribution of the
liposomes by
aftertreatment of the liposome dispersion, for example by treatment with
ultrasound or
extrusion through straight-pored filters (for example Nucleopore~).
The separation and isolation of a fraction of large liposomes from a fraction
containing
- 10-
small liposomes, insofar as it is at all necessary, is effected by means of
conventional
separation methods, for example gel filtration, for example with Sepharose~ 4B
or
Sephacryl~ (Pharmacia SE) as carrier, or by sedimentation of the liposomes in
an
ultracentrifuge, for example with a gravitational field of 160,000 x g. For
example, after
centrifugation for several hours, for example about three hours, in that
gravitational
field,large liposomes are deposited, whereas small liposomes remain in
dispersion and can
be decanted. Repeated centrifugation results in complete separation of the
large
liposomes from the small liposomes.
Gel filtration especially can be used to separate off all the liposomes
present in the
aqueous phase having a diameter of more than about 6.0 x 10-s m and also
non-encapsulated components and excess, dispersed lipids that are present in
high
molecular weight aggregates and thus to produce an aqueous dispersion having a
fraction
of liposomes of relatively uniform size.
The completed formation of liposomes and their content in aqueous phase can be
demonstrated in a manner known Rer se by various physical measuring methods,
for
example with freeze fracture samples and thin sections under an electron
microscope or by
X-ray diffraction, by dynamic light scattering, by mass determination of the
filtrate in an
analytical ultracentrifuge and especially by spectroscopy, for example in the
nuclear
resonance spectrum (1H, 13C and 3tP).
The present invention relates also to a novel, inventive process for the
preparation of a
pharmaceutical composition in the form of a dry preparation, which process
comprises
dissolving
a) the zinc-phthalocyanine complex and
b) a synthetic, substantially pure phospholipid of formula I, whereim Rt, R2,
Ra, Rb and R~
and also n are as defined above, optionally combined with a
c) synthetic, substantially pure phospholipid of formula II, wherein R3, R4, n
and 'lr~ are
as defined above, in a pharmaceutically acceptable organic solvent, the
residual amount of
which in a dry preparation is toxicologically harmless, and optionally
d) adding carrier liquid and water-soluble excipients suitable for parenteral
dosage forms,
and removing the solvent or solvent mixture.
A pharmaceutically acceptable organic solvent, the residual amount of which in
a dry
preptrration is toxicologically harmless, is suitable for the preparation of a
clear solution of
-11-
the zinc-phthalocyanine complex and if possible the phospholipid components
(I) and (II).
If the phospholipids are insoluble in the organic solvent in which the zinc-
phthalocyanine
complex is soluble, they are dissolved in a second solvent, for example in
tort-butanol, and
that solution is combined with the solution of the zinc-phthalocyanine
complex.
Preferred organic solvents that meet the toxicological requirements as regards
the residual
amounts present in the dry preparation and in which the zinc-phthalocyanine
complex is
soluble are dimethyl sulfoxide, N-methyl-2-pyrrolidone and piperidine, and
mixtures
thereof.
The zinc-phthalocyanine complex is soluble in dimethyl sulfoxide and
N-methyl-2-pyrrolidone (NMP) but the phospholipids (I) and (II) are not. Both
the zinc-
phthalocyanine complex and the phospholipids (I) and (II) are soluble in
piperidine. All
solvents are toxicologically harmless in the residual amounts of less than 1
%, preferably
less than 0.5 °lo, present in the dry preparation.
When dimethyl sulfoxide or NMP is used, the zinc-phthalocyanine complex is
dissolved
in the minimum necessary amount of that solvent and the solution is combined
with a
second solution of the phospholipids that is miscible with the first solution
of the zinc-
phthalocyanine complex. In addition to the requirement of being miscible with
the
dimethyl sulfoxide solution, this second solvent is subject to the same
toxicological
requirements as regards the residual amounts present in the dry preparation.
Such a
suitable solvent in which the phospholipids (I) and (1I) are completely
soluble is,,for
example, tert-butanol.
The preparation of the dry preparation, preferably a lyophilisate, can be
effected by
modifying known methods, by first dissolving the introduced amount of the
phospholipid,
for example 1-hexadecanoyl-2-(9-cis-octadecenoyl)-3-sn-phosphatidyl choline,
if required
with gentle heating, in the amount of tert-butanol required for the
dissolution process and
combining that clear solution with a second solution comprising a defined
amount of the
zinc-phthalocyanine complex in an amount of dimethyl sulfoxide or NMP
appropriate for
the dissolution process. Alternatively, it is possible to dissolve the
phospholipids (I) and
(II) at approximately from 0°C to roam temperature, preferably at
0°C, and the
zinc-phthalocyanine complex at from room temperature to approximately
40°C, preferably
room temperature, in the minimum necessary amount of piperidine or in
piperidine
containing up to about 10 %, preferably abaut 2 to 3 %, water and to remove
the solvent
- 12-
from that clear solution in order to produce the dry preparation. The clear
solution in the
said organic solvents can then be combined with the carrier liquid d)
comprising
water-soluble excipients, especially non-ionic additives such as lactose. The
dry
preparation can be prepared by removing the solvent mixture at low temperature
below
about 0°C (lyophilisation) or at normal or elevated temperature (film
formation). The
solvent mixture used for the preparation of the dry preparation can also be
dialysed for the
purpose of purification and the dialysed aqueous solution, which contains no
organic
solvent, concentrated by ultrafiltration. The dry preparation is then prepared
by removing
water, preferably by lyophilisation. Measured amounts of the solution to be
lyophilised
can be introduced into suitable containers for a unit dose, such as ampoules,
for example
glass phials. The filled containers can then be frozen at about -40° to
-50°C, especially at
-45°C, and then lyophilised at a pressure of about 0.2-0.6 mbar by
slowly heating to a final
temperature of about 25°-35°C.
The solvent or solvent mixture comprising components a), b) and c) can be
converted into
a dry preparation also directly, without cooling, thermally in a larger vessel
(film
formation method). Freeze-drying from small vessels containing a unit dose is
preferred,
however, as this method avoids the operation of filling the dry preparation
itself and
therefore allows the accurate apportionment of unit doses from liquids.
Surprisingly, it has been possible using the said processes to prepare,
reproducibly, dry
preparations, especially lyophilisates, and liposome dispersions
reconstitutable therefrom,
that are st<lble and suitable for injection.
The present invention relates also to the use of the dry preparation
obtainable in
accordance with the said processes for the preparation of intravenously
administrable
liposome dispersions.
The present invention relates also to the use of the zinc-phthalocyanine
complex and the
phospholipid components of formula I and optionally of formula II for the
preparation of
dry preparations, especially lyophilisates, in accordance with the methods
described
hereinabove. The invention relates also to the use of the pharmaceutical
compositions in a
method for the therapeutic treatment of the human or animal body, especially
in
chemotherapy for the treatment of tumours. The liposome dispersion is
administered
parenterally, especially intravenously, and the carcinoma is irradiated with
high-energy
light, preferably with concentrated visible light (LASER).
-13-
The invention relates preferably to pharmaceutical compositions in the form of
intravenously administrable liposome dispersions comprising
a) the zinc-phthalocyanine complex,
b) a synthetic, substantially pure phospholipid of formula I, wherein R1 is n-
dodecanoyl,
n-tetradecanoyl, n-hexadecanoyl or n-octadecanoyl and R2 is 9-cis-dodecenoyl,
9-cis-
tetradecenoyl, 9-cis-hexadecenoyl, 9-cis-octadecenoyl or 9-cis-icosenoyl, Ra,
Rb and R
are methyl and n is two, optionally combined with a
c) synthetic, substantially pure phospholipid of formula II, wherein R3 and R4
are identical
and are 9-cis-dodecenoyl, 9-cis-tetradecenoyl, 9-cis-hexadecenoyl, 9-cis-
octadecenoyl or
9-cis-icosenoyl, n is one and Y~ is the sodium ion, and
d) a pharmaceutically acceptable carrier liquid and, optionally, water-soluble
excipients
suitable for intravenous dosage forms.
The invention relates preferably also to pharmaceutical compositions in the
form of dry
preparations comprising the said preferred components a), b), optionally
combined with
c), and d) water-soluble excipients suitable for intravenous dosage forms.
The invention relates especially to pharmaceutical compositions in the form of
intravenously administrable liposome dispersions comprising
a) the zinc-phthalocyanine complex,
b) synthetic, substantially pure 1-n-hexadecanoyl-2-(9-cis-octadecenoyl)-3-sn-
phosphatidyl choline (I), optionally combined with
c) synthetic, substantially pure sodium 1,2-di(9-cis-octadecenoyl)-3-sn-
phosphatidyl
S-serine (II), and
d) a pharmaceutically acceptable carrier liquid and, optionally, water-soluble
excipients
suitable for intravenous dosage forms.
The invention relates especially preferably to pharmaceutical compositions in
the form of
lyophilisates comprising
a) the zinc-phthalocyanine complex,
b) synthetic, substantially pure 1-n-hexadecanoyl-2-(9-cis-octadecenoyl)-3-sn-
phosphatidyl choline (I), optionally combined with
c) synthetic, substantially pure sodium 1,2-di(9-cis-octadecenoyl)-3-sn-
phosphatidyl
S-serine (II), and
d) water-soluble excipients suitable for intravenous dosage forms.
~03~4~'~
- 14-
The invention relates especially to pharmaceutical compositions in the form of
intravenously administrable liposome dispersions comprising
a) the zinc-phthalocyanine complex,
b) approximately 70 °l° by weight (based on component c))
synthetic, substantially pure
1-n-hexadecanoyl-2-(9-cis-octadecenoyl)-3-sn-phosphatidyl choline (I),
combined with
c) approximately 30 % by weight (based on component b)) synthetic,
substantially pure
sodium 1,2-di(9-cis-octadecenoyl)-3-sn-phosphatidyl S-serine (II), and
d) a pharmaceutically acceptable carrier liquid and, optionally, water-soluble
excipients
suitable for intravenous dosage forms.
The invention .relates especially also to pharmaceutical compositions in the
form of
lyophilisates comprising:
a) the zinc-phthalocyanine complex,
b) approximately 70 °l° by weight (based on component c))
synthetic, substantially pure
1-n-hexadecanoyl-2-(9-cis-octadecenoyl)-3-sn-phosphatidyl choline (I),
combined with
c) approximately 30 °l° by weight (based on component b))
synthetic, substantially pure
sodium 1,2-di(9-cis-actadecenoyl)-3-sn-phosphatidyl S-serine (II), and
d) water-soluble excipients suitable for intravenous dosage forms.
The following Examples illustrate the invention.
Exam-ale 1: 1 mg of zinc-phthalocyanine, 70 mg of at least 95 °lo pure
1-n-hexadecanoyl-
2-(9-cis-octadecenoyl)-3-sn-phosphatidyl choline and 30 mg of at least 95
°l° pure 1,2-di-
(9-cis-octadecenoyl)-3-sn-phasphatidyl serine are dissolved in 2 ml of
piperidine in a
round-bottomed flask. The solution is sterile-filtered using an ACRODISC
membrane
filter (2.2 x 10-~ m) and the clear solution is introduced into phials.
After the solution has been frozen at -40°C, the phials are dried in
vacuo until a
temperature of 25°C has been reached and are sealed under an argon
atmosphere.
Before use, 1.5 ml of sterile, calcium- and magnesium-free, phosphate-buffered
(pI-I
7.2-7.4) sodium chloride solution (Dulbecco) are introduced into the above-
mentioned dry
preparation (lyophilisate) at room temperature using a sterile syringe and the
phials are
shaken for one minute on a standard laboratory shaker (Vortex, stage 7). The
resulting
liposome dispersion can be stored at 4°C and is suitable for
intravenous administration.
- 15-
Example 2: Depending upon the batch, from 0.1 to 1 mg of zinc-phthalocyanine
and from
15 to 400 mg of a 7:3 mixture by weight of 95 to 100 % pure 1-n-hexadecanoyl-2-
(9-cis-
octadecenoyl)-3-sn-phosphatidyl choline and 95 to 100 % pure 1,2-di(9-cis-
octadecenoyl)-3-sn-phosphatidyl serine are dissolved in 2 ml of piperidine in
a
round-bottomed flask. The solution is sterile-filtered using an ACRODISC
membrane
filter (2.2 x 10-~) and the clear solution is introduced into phials.
The phials are set in rotation at 150 rpm and the solvent is blown off in a
stream of
purified nitrogen filtered at 1 bar.
The phials are then evacuated in a vacuum of 6.0 x 10-2 bar. The phials are
sealed under
an argon atmosphere. Before use, 1.5 ml of sterile, calcium- and magnesium-
free,
phosphate-buffered (pH 7.2-7.4) sodium chloride solution (Dulbecco) are added
to the
above-prepared film at room temperature using a syringe and the phials are
shaken for 10
minutes on a standard laboratory shaker (stage 7). The resulting liposome
dispersion can
be stored at 4°C and is suitable for intravenous administration.
Example 3: In a manner analogous to Example 1, after being frozen the
solutions
mentioned in Example 2 are dried in vacuo and lyophilisates are prepared.
Example 4: In a manner analogous to Examples 2 and 3 it is also possible to
prepare
solutions comprising from 5:5 to 9:1 mixtures by weight of 1-n-hexadecanoyl-2-
(9-cis-
octadecenoyl)-3-sn-phosphatidyl choline and 1,2-di(9-cis-octadecenoyl)-
phosphatidyl
serine.
Example 5: 125 ml of 95 % pure 1-n-hexadecanoyl-2-(9-cis-
octadecenoyl)-3-sn-phosphatidyl choline (Avanti, Polar Lipids) are dissolved
in 0.5 ml of
tert-butanol in a round-bottomed flask. 0.05 mg of zinc-phthalocyanine is
dissolved
separately in 0.5 ml of sterile dimethyl sulfoxide in a second round-bottomed
flask.
The two solutions are combined and sterile-filtered through an ACRODISC
membrane
filter (2.2 x 10-~ m). After the introduction of the clear solution into a
phial, it is frozen at
-80°C. The phials are dried ip vacuo until a temperature of 25°C
has been reached and are
sealed under an argon atmosphere.
- 16-
The further process steps are carried out analogously to Example 1.
Example 6: 12.5 to 25 mg of 95 °lo pure 1-n-hexadecanoyl-2-(9-cis-
octadecenoyl)-3-sn-
phosphatidyl choline (Avanti, Folar Lipids) are dissolved in 0.5 ml of tert-
butanol in a
round-bottomed flask. 0.05 mg of zinc-phthalocyanine is dissolved separately
in 0.5 ml of
sterile dimethyl sulfoxide in a second round-bottomed flask.
Analogously to Example S the two solutions are combined, sterile-filtered
using an
ACRODISC and introduced into phials. The phials are set in rotation at 150 rpm
and the
solvent is blown aff in a stream of purified nitrogen filtered at 1 bar. The
phials are then
evacuated under a pure vacuum of 6.0 x 10-2 bar. The phials are sealed under a
protective
argon atmosphere.
The further process steps are carried out analogously to Example 2.
Example 7: 0.5 mg of zinc-phthalocyanine and 250 mg of 95 °lo pure 1-n-
hexadecenoyl-
2-(9-cis-octadecanoyl)-3-sn-phosphatidyl choline are dissolved in 1 ml of
sterile
piperidine in a round-bottomed flask. The solution is sterile-filtered using
an ACRODISC
membrane filter (2.2 x 10-7 m) and the clear solution is introduced into
phials.
Analogously to Example 5 it is possible to produce a lyophilisate or, if
desired, a film
residue analogously to Example 6, which is then converted into an
intravenously
administrable liposome dispersion analogously to Example 1 or 2.
Example $: In a manner analogous to Example 7 it is possible to produce a
lyophilisate or
a film residue by dissolving 3 mg of zinc-phthalocyanine in 1 ml of sterile
piperidine.
From these dry preparations there is then produced an intravenously
administrable
lipasome dispersion analogously to Example 1 or 2.
Example 9: In a measuring flask at room temperature, depending upon the batch
200-400 mg of zinc-phthalocyanine are dissolved in 400 ml of piperidine
puriss. and the
solution is made up to 500 ml. In a second flask at room temperature,
depending upon the
batch 120-240 mg of a 7:3 mixture by weight of 95-100 °!o pure 1-n-
hexadecanoyl-
2-(9-cis-octadecenoyl)-3-sn-phosphatidyl choline and 95-100 % pure 1,2-di(9-
cis-
octadecenoyl)-3-sn-phosphatidyl S-serine are dissolved in 6 ml of piperidine
puriss., and
6-12 ml of the above zinc-phthalocyanine solution are added. The resulting
solution is
~~3~4'~~
-17-
sterile-filtered using an ACRODISC membrane filter (2.2 x 10-~) and the clear
solution is
introduced into phials.
The further process steps are carried aut analogously to Example 1.
Example 10: In a measuring flask at room temperature, depending upon the batch
200-400 mg of zinc-phthalocyanine are dissolved in 400 ml of piperidine
puriss. and the
solution is made up to 500 ml. In a second flask, depending upon the batch 120-
240 mg' of
a 7:3 mixture by weight.of 95-100 % pure 1-n-hexadecanoyl-2-(9-cis-
octadecenoyl)-
3-sn-phosphatidyl choline and 95-100 % pure 1,2-di(9-cis-octadecenoyl)-3-sn-
phosphatidyl S-serine are dissolved in 6 ml of a piperidine/water mixture (2-
10 %) at
0-4°C and at that temperature 6-12 ml of the above zinc-phthalocyanine
solution are added
dropwise thereto. The resulting solution is sterile-filtered using an ACRODISC
membrane filter (2.2 x 10- ) and the clear solution is introduced into phials.
The further process steps are carried out analogously to Example 2.
Example 11: After sterile-filtration, a solution of zinc-phthalocyanine and
lipid mixture
prepared according to Example 9 or Example 10 is not introduced into phials
but is
lyophilised in a batch process. Aliquot amounts of the resulting lyophilisate
are converted
into a liposome dispersion analogously to Example 1.
Example 12: 100 mg of zinc-phthalocyanine, 7000 mg of 95 % pure 1-n-
hexadecanoyl-2-
(9-cis-octadecanoyl)-3-sn-phosphatidyl choline (POPC) and 3000 mg of 95 % pure
sodium 1,2-di(9-cis-octadecanoyl)-3-sn-phosphatidyl S-serine (OOPS) are
dissolved in
205 ml of freshly distilled piperidine in a round-bottomed flask.
To that solution there are added, with stirring, 2000 ml of 9.75 % W/V aqueous
lactose
solution. After the pII value has been adjusted to 7.0 by the addition of
dilute HCl, the
resulting dispersion is concentrated to a concentration of 0.5 mg of
zinc-phthalocyanine/ml and dialysed against 2000 ml of 9.75 % lactose using a
Millipore
Minitan~ tangential dialysis apparatus.
The resulting lactose-containing dispersion is then sterile-filtered using an
ACRODISC
membrane filter (2.2 x 10- ) and introduced into phials (2 ml per phial
containing 1 mg of
zinc-phthalocyanine). After the dispersion has been frozen at -4U°C,
the phials are dried
-18-
in vacuo until a temperature of 25°C has been reached and are sealed
under an argon
atmosphere.
The resulting dry preparations can be stored at 4°C for several
years.
Before administration, 2.0 ml of water are injected using a sterile syringe
into a dry
preparation (lyophilisate) in a phial at room temperature. After a maximum
dissolution
time of one minute there is formed a liposome dispersion suitable fox
intravenous
administration.
Example 13: In a manner analogous to Example 12, it is possible to carry out
the process
with 10 000 mg of POPC.
Example 14: In a manner analogous to Example 13, it is possible to carry out
the process
with 300 mg of POPC or 210 mg of POPC and 90 mg of OOPS.
Example 15: In a flask, 35 g of POPC and 15 g of OOPS are dissolved in 500 ml
of
tertiary butanol, with stirring, at 40°C. In a further flask, 0.'i g of
zinc-phthalocyanine is
dissolved in 125 ml of NMP (ultrasonic bath).
The two solutions are mixed together, the dye solution being poured into the
tertiary
butanol solution. The mixture is heated to 40°C in order to obtain a
clear solution. Using
a dynamic mixer, that solution is mixed with 10 litres of lactose medium that
has been
cooled to 4°C and that contains per litre 94.9 g of lactose for
injection and 24 g of sodium
chloride.
The organic phase is pumped at a rate of 107 ml/min and the lactose solution
at a rate of
1714 ml/min into the dynamic mixer, which is operated at a pressure of 3 bar.
The resulting blue, slightly opalescent dispersion (10625 ml) is concentrated
to 1 litre and
then dialysed against 10 litres of lactose medium using a Millipore tmgential
dialysis .
apparatus.
The further procedure is described in Example 12.
Example 16: In a manner analogous to Example 15, 3.5 g of POPC and 1.5 g of
OOPS are
- 19-
dissolved in 100 ml of tort-butanol and mixed with 25 ml of NMP and 100 mg of
zinc-
phthalocyanine. The organic phase is then mixed with 2 litres of lactose
medium (25 g of
lactose and 0.064 g of NaCI per litre). The dialysis medium has the same
composition.
Example 17: In a manner analogous to Example 16; the organic phase is mixed
with
2 litres of lactose medium containing 50 g of lactose and 0.127 g of NaCl per
litre.
Example 18: In a manner analogous to Example 15, I0.5 g of POPC and 4.5 g of
OOPS'
are dissolved in 200 ml of tort-butanol and mixed with 50 ml of NMP containing
200 mg
of zinc-phthalocyanine. The organic phase is mixed with 4 litres of lactose
medium
containing 37.5 g of lactose and 0.095 g of NaCI per litre. The dialysis
medium has the
same composition.
Exam 1p a 19: In a manner analogous to Example 18, the organic phase is mixed
with
4 litres of lactose solution containing 75 g of lactose and 0.1905 g of NaCI
per litre. The
dialysis medium has the same composition.
Example 20: In a manner analogous to Example 15, 3.5 g of POPC and 1.5 g of
OOPS are
dissolved in 200 ml of tert-butanol and mixed with 50 m1 of NMP containing 200
mg of
zinc-phthalocyanine. The organic phase is mixed with 4 litres of lactose
medium
containing 25 g of lactose and 0.064 g of NaCI per litre. The dialysis medium
has the
same composition.