Note: Descriptions are shown in the official language in which they were submitted.
RB87
~ .
6 7 "'J
PREPARATION OF BORONIC ACID DERIVATIVES
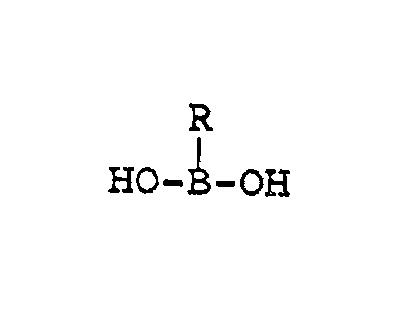
Compounds of the formula
HO-B-OH
or pharmaceutically accepta~le salts thereof,
wherein R is hydroxy, alkyl, alkenyl, cycloalkyl,
cycloalkenyl, alkoxy, carboxyalkyl, car~oxyalkenyl,
hydroxyalkyl, hydroxyalkenyl, alkoxyalkyl, alkoxy-
alkenyl, haloalkyl, haloalkenyl, aryl, arylalkyl or
(R1R2N)-alkyl, where Rl and R2 are each
independently hydrogen alkyl or arylalkyl or taken
together with the nitrogen to which they are
attached form a 5- or 6-membered nitrogen-containing
heterocycle are useful in the preparation of
pharmaceutically important agents.
For example, U.S. Patent No. 4,705,849
discloses boronic acid adducts of technetium-99m
dioxime complexes having the formula
II
99mTc X(Y)3Z
B
204~467
"
-2- RB87
wherein
X is an anion;
Y is a vicinal dioxime having the formula
5 (i) R'R"
HO-N-C-C=N-OH
wherein R' and R" àre each independently
hydrogen, halogen, alkyl, aryl, amino or a 5- or
6-membered nitrogen- or oxygen-containing
heterocycle, or together R' and R" are -(CR4R5)n
wherein n is 3, 4, 5, or 6 and R4 and Rs are each
independently hydrogen or alkyl;
and Z is a boron derivative of the formula
(ii) B-R.
These complexes are useful as imaging
agents.
To prepare.complexes of formula II,
pertechnetate ion (in the form of a salt) is
combined with a source of anion, a compound such
as that of formula I and a dioxime of
formula (i).
The pertechnetate ion can be obtained from
commercially available technetium-99m
parent-daughter generators; such technetium is in
the +7 oxidation state. The generation of the
pertechnetate ion using this type of generator is
well known in the art and is described in more
detail in U. S. Patent No. 3,369,121 and
3,920,995. These generators are usually eluted
with saline solution and the pertechnetate ion is
obtained as the sodium salt.
20~04~7
.....
_3_ RB87
The source of the anion moiety (X) can be
water or it can be an acid or salt which
dissociates to release an appropriate anion.
Exemplary anionic moieties are hydroxyl, halide,
isothiocyanato (N=C=Se) and thiocyanato (S-C=Ne).
The preferred anionic moieties are the halides,
and chloride is the most preferred halide.
Brown et al., J. Organometallics, 5, 2300
(1986) describe a process for the preparation
of methyl boronic acid which starts by reacting a
compound of the formula
(iii)
CH3Li
with a compound of the formula
(iv)
(ICH3)2
CH
o
B-O-CH-(CH3)2
CH
(CH3)2
in ether to provide the complex
(v) [CH3[(CH3)2CHO]3B ,Li ].
Treatment of complex (v) with an equivalent
of hydrogen chloride provides
20~04~i7
",,.
--4--
(vi) (IC 3)2
CH
o
CH3B-O-CH-(CH3)2
s
The byproduct LiCl is removed by a tedious
decantation. Hydrolysis of (vi) is then
accomplished by the addition of water to give
methyl boronic acid and the byproduct (CH3)2CHOH.
The reaction solvent is then removed by
distillation followed by a tedious azeotropic
distillation with acetone of the excess water and
apparently also the (CH3)2CHOH. The desired
methyl boronic acid then remains as a residue.
Thus, any byproduct LiCl not removed in the
decantation process and any (CH3)2CHOH remaining
from the distillation are present as impurities in
the isolated methyl boronic acid. For the
preparation of methyl boronic acid and similar
compounds, i.e., compounds of fo-mula I, especially
on a manufacturing scale, an improved process would
be a very useful addition to the art.
In accordance with the present invention,
an improved process for preparation of compounds
of the formula
HO-B-OH
or pharmaceutically acceptable salts thereof, is
disclosed, wherein R is alkyl, alkenyl,
20~467
, ..
RB87
--5--
cycloalkyl, cycloalkenyl, alkoxyalkyl, alkoxy-
alkenyl, aryl, arylalkyl or (RlR2N)-alkyl, where
R1 and R2 are each independently alkyl or arylalkyl
or taken together with the nitrogen to which they
are attached form a 5- or 6-membered nitrogen
containing heterocycle. The present process
involves hydrolysis of a complex of the formula
III
[R(R3-0)3Be,Li~]
wherein R3 is alkyl to provide a complex of the
formula
IV
[R(OH)3B ,Li ]
which is thereafter treated with an acid to provide
compounds of formula I which are readily extracted
in high yields with an organic solvent.
The present invention provides a straight-
forward, high yield process for the preparation of
compounds of formula I. The present process is
therefore useful in preparation of various of the
the compounds described in U.S. Patent
No. 4,705,849 having the formula
II 9 mTc X(Y)3Z-
The present process is particularly useful in the
preparation of compounds of formula I wherein R is
2~ 1Q~ 7
~,,
RB87
--6--
methyl, i.e., methyl boronic acid. This is a key
intermediate in preparation of complexes of
formula II wherein X is chloro, Y is cyclohexane-
dione dioxime and Z is B-R where R is methyl (i.e.,
the boronic acid adduct, 99mTc (chlorine)(1,2-
cyclohexanedione-dioxane)3 methyl boron or
complexes where X is chloro, Y is
dimethylglyoxime, Z is B-R where R is 2-methyl-1
propane (i.e., the 2-methyl-1 propane boronic acid
adduct of chloro tris dimethylglyoxime technetium).
Listed below are definitions of the terms
used to describe the complexes of this invention.
These definitions apply to the terms as they are
used throughout the specification (unless they are
otherwise limited in specific instances) either
individually or as part of a larger group.
The terms "alkyl" and "alkoxy" refer to both
straight and branched chain groups. Those groups
having 1 to 10 carbon atoms are preferred.
The term "alkenyl" refers to both straight
and branched chain groups. Those groups having 2
to 10 carbon atoms are preferred.
The term "aryl", when used in the definition
of R, refers to phenyl and substituted phenyl
wherein the substituents can be any groups
compatible with the generation of the lithium
complexes or reagents of formula III, formula IV
and formula V, such as primary, secondary or
tertiary alkyl, dialkylaminoalkyl, alkoxy, or
alkoxyalkyl.
The term "aryl", when used in the definitions
of R ', R", Rl or R2, refers to phenyl and phenyl
substituted with primary, secondary or tertiary
2~~4~i7
",
_7_ RB87
alkyl, haloalkyl, aminoalkyl, alkylaminoalkyl,
dialkylaminoalkyl, alkoxy, alkoxyalkyl, halogen,
amino, hydroxy, or formyl groups.
Preferred "cycloalkyl" and "cycloalkenyl"
groups are those having 5, 6 or 7 carbon atoms.
The terms include those groups substituted with
alkyl, alkoxy, aryl, arylalkyl or (RlR2N)-alkyl
groups.
The terms "halide", "halo" and "halogen"
refer to fluorine, chlorine, bromine and iodine.
The expression "5 or 6-membered nitrogen
containing heterocycle" refers to all 5 and 6-
membered rings containing at least one nitrogen
atom. Exemplary aliphatic groups are dehydro
derivatives of a compound having a formula
CH2--( CH2 ,~
HN A
\ CH2 CH2
wherein m is 0 or 1 and A is 0, N-R6 or CH-R6
wherein R6 is alkyl, aryl or arylalkyl. Such
groups include pyrrolidinyl, piperidinyl, morpholinyl,
4-alkylpiperazinyl, 4-alkylpiperidinyl, and 3-alkyl-
pyrrolidinyl groups.
The expression "5 or 6-membered nitrogen or
oxygen containing heterocycle" refers to all 5 and
6-membered rings containing at least one nitrogen
or oxygen atom. Exemplary groups are those
described above under the definition of the
expression "5 or 6-membered nitrogen containing
heterocycle". Additional exemplary groups are
1,4-dioxanyl and furanyl.
2~!0467
.",.
-8- RB87
To carry out the present process, a
compound of the formula
R-Li
is reacted with a compound of the formula
VI
R3
o
B-O-R3
(wherein R3 can be alkyl, and is preferably
isopropyl, to provide a complex of the formula
III
[R(R3-O)3Be,Li~].
Preferably the above reaction is carried out in
diethyl ether cooled to between -60 and -80~C.
As opposed to the prior art process,
tri-ester complex III is thereafter hydrolyzed with
water or an aqueous solution to provide
IV
[R(oH)3Be,Li~].
One distinct advantage of the present process is
that complex IV can be readily isolated (as
opposed to the di-ester complex of the prior art),
~0167
....~ .
RB87
i.e., via evaporation and the like, so as to
remove any solvent and resultant R3OH by-product
while the desired intermediate of formula IV is in
a non-volatile form. This provides for much
easier isolation of the final product of formula I.
Thus, complex IV is concentrated to give
a solid residue. The so-treated complex is
thereafter treated with an acid to provide
compounds of formula I which are readily extracted
with an organic solvent using known techniques.
Preferably, the acid is an aqueous mineral
acid, such as hydrochloric, sulfuric, phosphoric
and the like, with hydrochloric acid being most
preferred. The solvent can be any convenient
organic solvent and preferably is a polar, low
boiling point solvent, such as an ether (e.g.,
diethyl ether and the like) or methyl acetate.
The present invention is further illustrated
by the following example.
20'~0~7
." .
RB87
--10--
Example
504 ml (21.4 mole) of triisopropyl borate
was added to 2200 ml of diethyl ether. This was
cooled in a dry ice/acetone bath and 1530 ml 1.4 M
(2.14 mole) methyl lithium in diethyl ether was
added slowly over two hours. When the addition
was complete, the cold bath was removed and the
reaction was allowed to warm to room temperature
over three hours. With vigorous stirring, 418 ml
of water was added, slowly. The resulting mixture
was stirred for 30 minutes. The water layer was
separated and the organic layer was extracted once
with 110 ml water. The combined water layer was
evaporated in vacuo at 50~. The resulting white,
solid residue was stirred with 2300 ml diethyl
ether and concentrated hydrochloric acid (201 ml,
2.40 mole) was added slowly until the pH of the
aqueous layer stayed at 2Ø The aqueous layer
was saturated with sodium chloride (~60 g) and the
ether layer was separated. The aqueous layer was
extracted with three 1000 ml portions of ether.
The combined organic layer was dried over magnesium
sulfate and evaporated at ~2 mm Hg/0-5~. Final
drying was accomplished with a vacuum pump at 20~
for ten minutes. The resulting granular solid was
suspended in 500 ml n-pentane and stirred for
fifteen minutes. After filtration, the solid was
washed with a little pentane and dried at 20 mm
Hg/room temperature for 45 minutes to give 118 g
(92 mole% yield) of the title compound.