Note: Descriptions are shown in the official language in which they were submitted.
2 ~ J.
A ~ROCESS FOR THE PREPARATION OF l-HALOGEN-l-OXOPHOSPHOLENES
BACKGROUND OF THE INVENTION
~ his invention relates to a process for the
preparation of l-halogen-l-oxophospholenes by the
continuous reaction of alkoxydihalogen phosphanes with
1,3-dienes.
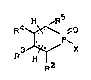
l-Halogen-l-oxophospholenes corresponding to the
following general foni,~la
~ x , ~ or ~ O
in which the groups R2, R3, R4 and R5 denote, independent-
ly of one another, hydrogen, chlorine, bromine or (Cl-C4)-
alkyl groups optionally substituted with chlorine or
bromine and X stands for chlorine or bromine are important
starting compounds, e.g. for substances used as flame-
retarding compounds in halogen-free synthetic resin
systems, as carbodiimidisation catalysts or as fungicides.
The increasing demand for l-halogen-1-oxophosphol-.
enes has made it essential to prepare these compounds in
~e A 27 569
industrial quantities. The methods of synthesis hitherto
known have, however, various disadvantages so that large
quantities could hitherto not be produced or only under
very disadvantageous conditions.
Thus DE-PS 1 191 204, for example, describes the
direct addition of phosphorus trihalides to 1,3-dienes to
produce trihalogenphospholenes which are then converted
into l-halogen-l-oxophospholenes by a reaction with
oxygen-donor substances such as water, alcohols, ketones,
orthocarbonyl compounds, etc. This process is, however,
very unsatisfactory on account of the rlaction times,
which extend over several days, the number of stages
required and the poor yields.
Another process uses phosphorous acid monoester
dichlorides as starting materials which are added to 1,3-
dienes in an autoclave and then split up into l-halogen-l-
oxophospholenes and alkyl halides or alkylene dihalides
[B.A. Arbusov, A.O. Vizel, Y.Y. Samitov and Y.F. Tarenko,
Izv, Akad. Nauk, SSSR, Ser. Khim (1967) (3), 648; N.A.
Razumova, L.J. Zubtsova and A.A. Petrov, Zh. Obsh. Khim.
40 (12), 2554, (1969)~. The reaction times may be reduced
within certain limits by the addition of phosphorus
trihalides tN.N. Bliznyuk, Z.N. Kvasha and A.F. Kolomiets,
Zh. Obschch. Khim. 37, 1811 (1967)] but even then,
economically acceptable short reaction times are not
obtained.
The problem therefore arose of providing a process
by which large quantities of l-halogen-l-oxophospholenes
could be produced economically without the above-described
disadvantages o~ the known processes, such as long
reaction times, low yields and polymerisation of the diene
components, ~nd which w~uld be simple to handle.
This problem has been solved by the process
according to the inYention.
Le A 27 569 2
DESC~IPTION OF THE PREFERRED EMBODIMENTS
This invention relates to a process for the
preparation of l-halogen-l-oxophospholenes by the reaction
of phosphorous acid monoalkyl ester dihalides with 1,3-
dienes which is carried out continuously in a single stageunder pressure and at elevated temperatures, using
phosphorus trihalides as solvents.
The reaction is illustrated by the following
reaction scheme:
R5
~ ~ R OPX2 3 ~ ~ ~ Rl-X
R3 1 as solvent R3 H R2
R2
in which R2, R3, R4 and R5 may denote, independently of
one another, hydrogen or (Cl-C4)-alkyl, preferably
(Cl-C2)-alkyl, optionally substituted with chlorine and/or
bromine, and the gr_up R1 is a ~Cl-C10)-alkyl group,
preferably a (Cl-C5)-alkyl group, optionally substituted
with chlorine and/or bromine and X may stand for chlorine
or bromine.
Particularly preferred is the continuous reaction
Df phosphorus trihalides, trialkylphosphites and 1,3-
dienes or phosphorus trihalides, 1,2-epoxides and 1,3-
dienes -in excess phosphorus trihalide. In this reaction,
the intermediate products (alkoxydihalogenphosphanes)
formed from the reaction of phosphorus trihalides with
trialkylphosphites or epoxides need not be icolated.
The phosphorus trihalide is preferably used in an
excess of from 75 to 125%.
Le A ~7 569 3
~ $s
The intermediate products talkoxydihalogenphos-
phanes~ are formed in accordance with the following
reaction scheme:
2 PX3 ~ p(oP~l) 3 PX3 ~, 3 RlOPX2
as solvent
or
R7 R8 R7 R8
PX3 1 ~C - C\ PX3 ~ R1OPX2, Rl = X-C -C
R6 o R9 as solvent R6 R9
in which X ctands for chlorine or bromine, Rl stands for a
(Cl-C10)-alkyl group, preferably a (Cl-C5)-alkyl group,
optionally substituted with chlorine and/or bromine and
R6, R7, R8 and R9 denote, independently of one another,
hydrogen, chlorine, bromine or (Cl-C~)-alkyl, preferably
(C1-C2)-alkyl, optionally substituted with chlorine and/or
10 bromine, or they denote (C5-C7)-cycloalkyl or phenyl.
Most of the isomerically pure l-halogen-l-oxophos-
pholenes are in crystalline form at room temperature,
which normally renders them difficult to handle. Under the
conditions of synthesis according to the invention,
15 however, these compounds are always obtained as an
isomeric mixture of l-halogen-l-oxophospholene-3 and
l-halogen-1-oxophospholene-2, in most cases in liquid
form.
The phosphorus trihalides used for the process
20 according to the invention may be phosphorus trichloride
or phosphorus tribromide.
The alkyl dihalogenphosphanes to be used according
to the invention are ~ompounds corresponding to the
general formula
RlC)PX2
25 in which the group denoted by Rl is a (Cl-C10)-alkyl
Le A 27_569 4
L ~L~
group, preferably a (Cl-C5~-alkyl group, optionally
substituted with chlorine and/or bromine, and X may stand
for chlorine or bromine. These compounds may be prepared
by known methods, e.g. from phosphorus trihalides and
5 alcohols with removal of the resulting hydrogen chloride
in a vacuum. Other known processes for their preparation
are given in Houben-Weyl, Methoden der Organischen Chemie,
Volume 12/2, pages 12 et seq and Supplementary Volume E 1,
pages 352 et seq.
The following are examples of the trialkylphos-
phites of the general formula
i'
P(ORl)3,
wherein Rl has the meaning indicated above used for the
15 special embodiment of the process according to the
invention: ~rimethylphosphite, triethylphosphite, tri-
isopropylphosphite, tributylphosphite, tripentylphosphite,
tris-(2-ethyl-hexyl)-phosphite and tri-2-chloroethyl-
phosphite, the low boiling methyl and ethyl derivatives
20 being preferred. An important criterion for the choice for
the trialkylphosphites is the suitability for technical
utilization of the alkyl halides obtained as by-products
of the reaction with phosphorus trihalide. When methoxy-
dichlorophosphane prepared from phosphorus trichloride and25 methanol or by equilibration of phosphorus trichloride
with trimethylphosphite is used, chloromethane is obtained
as by-product in addition to l-chloro-l-oxophospholene and
may be used, for example, in the Rochow synthesis of
methylchlorosilanes.
3D The 1,2-epoxides to be used in the special embodiment of
the process according to the invention may be compounds
corresponding to the following formula
~e ~ 27 5~9 5
2 ~ ~ f ~
R7 R8
~6 ~ R9
wherein R6, R7, R8 and R9 denot2, independently of one
another, hydrogen, chlorine, bromine, (Cl-C4)-alXyl,
preferably (Cl-C2~-alkyl, optionally substituted with
chlorine and/or bromine, or (C5-C7)-cycloalkyl or phenyl.
Ethylene oxide, propylene oxide and epichlorohydrin are
preferred.
The 1,3-dienes used may in principle be any 1,3-
dienes corresponding to the following general formula
R4 ~H
R3~
in which R2, R3, R4 and R5 may denote, independently of
one another, hydrogen, chlorine, bromine or (Cl-C4)-alkyl,
preferably (C1-C2)-alkyl, optionally substituted with
chlorine and/or bromine. The following are examples of
particularly suitable 1,3-dienes: Butadiene, isoprene,
chloroprene and l-methyl-butadiene. Butadiene and isoprene
are particularly preferred.
Polymerisation inhibitors which are coluble in the
reaction mixture, e.g. phen~thiazine, hydroquinone,
p-tert.-butylpyrocatechol, etc. may be added to the
reaotion mixture in quantities of up to 1% by weiqht
(Houben-Weyl, Methoden der Orqanischen Chemie, Volume
14/1, pages 26 et seq).
~ç A 2? _569 6
2 ~ 3 ~'
The process according to the invention is prefer-
ably carried out in a temperature range of from 50 to
180~C, most preferably from 100 to 150~C.
The reaction times depend on the temperatures
employed. At the preferred temperature range, they are
generally from 15 minutes to 2 hours, in particular from
30 minutes to 1 hour.
The process according to the invention may, for
example, be carried out as follows:
The starting materials are introduced by dosing
pumps into a ~ixing vessel which is equipped for cooling
and also serves as receiver !for a pressure resistant
dosing pump used for conveying the reaction mixture into a
heatable pressure coil. A valve attached to the end of the
pressure coil maintains the required pressure inside the
coil. This pressure is equal to the vapour pressure of
the volatile components at the given reaction temperature.
Discharge of the reaction products also takes place
through the above-mentioned valve.
The l-halogen-l-oxophospholene is freed by known
methods, e.g. distillation at low pressure, from the
phosphorus trihalide used as solvent and the halogen or
dihalogenalkane produced as by-product.
Further purification oP the l-halogen-1-oxophos-
2j pholene may also be carried out by distillation at reduced
pressure.
The invention will now be described in more detail
with the aid of the following Examples.
Le A 27 569 7
EXAMPLES
Example l
Continuous preparation of an isomeric mixture of 1-
chloro-3-methyl-1-oxophospholenes
275 g (4 mol) of isoprene, gl7 g (6,67 mol) of
phosphorus trichloride and 165 g (1.33 mol) of trimethyl-
phosphite are measured per hour into a water cooled mixing
vessel. The tempe ature in the mixing vessel is 18~C.
A pressure resistant pump delivers the homogeneous
reaction mixture (1.2 l/hour) into a reaction coil of
700 ml capacity situated in an oil bath heated to 130~C.
The average dwe~l time is 35 minutes. The pressure inside
the reaction coil is 15 bar. As the reaction mixture
leaves the reaction coil, the pressure in the mixture is
released and the mixture cools and is collected in a crude
product container by way of a pressure retaining valve.
Most of the chloromethane escapes at this stage and is
condensed in a brine condenser and collected in another
container also cooled with brine. The yields are deter-
mined by measuring the hourly rates of throughput. The
crude product is heated to 80~C at normal pressure to
drive out any remaining chloromethane. Excess phosphorus
trichloride is drawn off at 20 mbar up to a sump tempera-
ture of 70~C and is condensed in a brine condenser.
Distillation of the residue (110 - 120~C at 3 mbar) yields
568 g (94.4% of the theoretical yield) of 1-chloro-3-
methyl-l-oxophospholene. The isomeric mixture is composed
of 58% of 1-chloro-3-methyl-1-oxophospholene-3 and 42% of
l-chloro-3-methyl-1-oxophospholene-2. The yield of
chloromethane is 196 g (97% of theoretical yield).
Ex~
The same procedure is carried out in Example 2 as
in Example 1. Methoxydichlorophosphane is in this case
used as starting material which has previously been
prepared in a separate apparatus by a known method from
phosphor~s trichloride and methanol and already contains
Le A 27 569 8
3~
the excess phosphorus trichloride required as solvent for
the subsequent reaction.
The following are introduced per hour into the
cooled mixing vessel:
S 275 g (4 mol) of isoprene and 1082 g of the
previously prepared methoxydichlorophosphane/phosphorus
trichloride mixture containing 4 mol of methoxydichloro-
phosphane.
Reaction and working up are carried out as
described in Example 1.
539 g (89.5% of theoretical yield) of 1-chloro-3-
methyl-l-oxophospholene purified by distillation are
obtained per hour.
Example 3
The following are reacted per hour under conditions
analogous to those of Example 1:
275 g (4 mol) of isoprene, 917 g (6.67 mol) of phosphorus
trichloride and 221 g (1.33 mol) of triethylphosphite.
The pressure maintained during the continuous
process is 10 bar.
The quantity of reaction mixture collected per hour
is used for determining the yield.
After the product has been worked up by distilla-
tion, 500 g (83.0% of the theoretical amount) of an
isomeric mixture of 1-chloro-3-methyl-1-oxophospholene are
obtained. It consists of 21.8% of 1-chloro-3-methyl-1-
oxophospholene-3 and 78.2% of 1-chloro-3-methyl-1-
oxophospholene-2.
238 g (92.2% of the theoretical amount) of ethyl
chloride are obtained.
~xam~le 4
The following are continuously reacted per hour
under the conditions described in Example 1:
275 g (4 mol) of isoprene, 917 g (6.67 mol) of phosphorus
trichloride and 388 g (1.33 mol) of tri-n-pentylphosphite.
Le A 2? 569 9
The pressure in the reaction coil i~ 3 bar.
The quantitv of reaction mixture collected per hour
for distillative working up is used for ~etermining the
yield. The following are obtained:
358 g ~84% of theoretical amount) of n-pentyl
chloride and 495 g ~.2% o~ theoretical amount) of an
isomeric mixture of 18.1% of 1-chloro-3-methyl-1-oxophos-
pholene-3 and 81.9% of 1-chloro~3-methyl-1-oxophos-
pholene-2.
Example 5
The procedure is the same as in Example 1.
But~diene, phosphorus trichloride and trimethylphosphite i
are put into the process.
The following were put through per hour:
216 g (4 mol) of butadiene, 917 g (6.67 mol) of phosphorus
trichloride and 165 g (1.33 mol) of trimethylphosphite.
Determination of the yield is based on the quantity
of reaction mixture obtained per hour, which after
distillative working up of provides 494 g (90. 5~) of an
isomeric mixture of 56% of 1-chloro-1-oxophospholene-3 and
44% of 1-chloro-1-oxophospholene-2.
It is undexstood that the specification and examples
are illustrative but not limitative of the present invention
and that other embodiments within the spirit and scope of the
invention will suggest themselves to those skilled in the art.
e A 27 ~69 10