Note: Descriptions are shown in the official language in which they were submitted.
20~86'~6
- 1 -
Artificial promoter for the expression of proteins in
east
The present invention relates to a novel arti-
ficial promoter for the expression of proteins, in par-
05 ticular heterologous proteins, in yeast, to a vector
for the expression of said proteins which carries said
promoter, to the strains of yeast, and especially of
Saccharomyces cerevisiae, which are transformed by this
expression vector, and to a method of producing a re-
combinant protein with the aid of these strains.
Yeast, and in particular Saccharomyces cere-
visiae, a non-pathogenic microorganism whose genetics
has been studied in detail, is a preferred eukaryotic
host for the expression of proteins, especially hetero-
logous proteins. It is therefore important to discover
or construct novel promoters for the expression of said
proteins which are more advantageous than the known
promoters.
The structure of a yeast promoter, which is a
DNA sequence located upstream from a gene and res
ponsible for the transcription of said gene, is begin
ning to be partially known and understood. Said
promoter is known to comprise a TATA component located
in an AT-rich zone, a transcription initiation region
downstream from said component and, if appropriate, up-
stream from said component, sequences, called upstream
activation sequences (UAS) or upstream repression se-
quences (URS), which regulate the strength of the
promoter under the effect of an inducer or a repressor.
The Applicant constructed a novel hybrid pro-
moter from two known promoters: the promoter of the
GAL7 gene of Saccharomyces cerevisiae (TAJIMA et al.,
1986, Molecular Cellular Biology, 6, 246-256) and a
promoter with a sequence similar to that of the natural
ADH~ promoter (5'-flanking region of the ADH2 gene,
- 2 -
~~~6'~6
05
15
The invention therefore relates to a novel
artificial promoter for the expression of proteins in
yeast, which comprises:
- a sub-sequence upstream from the TATA component of
the sequence of the promoter of the GAL7 gene of Sac-
charomyces cerevisiae, which comprises the upstream
activation sequences UAS1 and UAS2; and.
- a sub-sequence of the sequence of an ADH2 promoter
comprising the TATA component and the transcription
initiation region.
Preferably, the sub-sequence upstream from the
TATA component of the promoter of the GAL7 gene of
Saccharomyces cerevisiae is the following sequence or
a sub-sequence thereof.:
described by RUSSEL et al. (1983); J. Biol. Chem. 258,
2674-2682), which is called an ADH2 promoter in the
- 3 -
M
L
a
I UAS1
CGCGTCTAT CTTCGGAGCACTGTTGAGCGAAG CTCATTAGATATATTTTCTGTCAT
__+______ __+_________+_________+ ________+_________+_____
AGATA GAAGCCTCGTGACAACTCGCTTC GAGTAATCTATATAAAAGACAGTA
05
UAS2
TTTCCTTAACCCAAAAATAAGGGAGAGGGTCCAAAAAGC CTCGGACAACTGTTGACCGT
____+_________+_________+_________+___ ____+_________+_____
AAAGGAATTGGGTTTTTATTCCCTCTCCCAGGTTTTTCG GAGCCTGTTGACAACTGGCA
G TCCGAAGGACTGGCTATACAGTGTTCACAAAATAGCCAAGCTGAAAATAATGTGTAGC
1 0 _ _+_________+_________+_________+_________+_________+_____
C GGCTTCCTGACCGATATGTCACAAGTGTTTTATCGGTTCGACTTTTATTACACATCG
S
P
h
15 I
CTTTAGCTATGTTCAGTTAGTTTGGCATG
____+_________+_________+____
GAAATCGATACAAGTCAATCAAACC
The sub-sequence of the sequence of an ADH2
promoter comprising the TATA component and the trans
20 cription initiation region is preferably selected from
the following sequence or a.sub-sequence thereof .
CCIATCACATATAAATAGA
.,_.._.. , .,....._
25 GTACGGATAGTGTATATTTATCT
GTGCCAG1AGCGACTTTTITCaCACTCGAGATACTC1TACTACTGC1CTCTTGTTGTTTT
...__.............,......___,__.~____.,_.......:,...._.
CACGGTCATCGCTGAAAAAAGTGIGAGCTCTATGAGAATGATGACGAGAGAACAACAAAA
TATCACTTCTTGTTTCT1C'1TGGTAAATAGAATATCAAGCTACAAAAAGCATACAATCAA
_._,.. _.....,.........,.........,_ ___--_~~........,._-.__.
30 ATAGTGRAGAACAAAGAAGAACCATTTAICT1ATAGTTCGpTGTTT1TCGTATGT1AGTT
C
1
a
I
CTATCAACTATTAACTA1A:~
35 __.,...._._..~....
GATAG1TGATAATTGAIA1AGC
20~486'~6
- 4 -
A particularly advantageous promoter is that
which comprises the following sequence:
05
1
a
I
CGCG1C1R1aC11CGGAGCaCTGIfGAGCCrAAGGC1CATTAGaIATAtIITCTGTCAT
..,_..._._._....._..._,...___...,.._......,_..__._..,_..__
AGatATGAAGCCTCGTGACAACTCGCTTCCGaGTAAICTATATAAAAGACaGTA
T1TCCTTRACCCAAAAATAAGGGAGAGGG1CCAAAaAGCGCTCGGACAAC1C11TGACCOT
_.._,....___._,... .,... .,._ .,. _
AAAGGAA11GGGT1ITTA1TCCC1C1CCCAGG111T1CGCGAGCCtGTTGACAACTGGCti
GATCCGAAGGACTGGCTATACAG1G1TCACAAAATRGCCAAGCTGRAAATAATGIGIAdC
_._.,...._._ .,__.__..._,._...._..,......___,.__.____.,.._..
CTAGGCTTCC1GACCGaTAIGTCACAAGTG1T1TATCGGTTCGAC111TATTACRCATCCi
S
n
I -
CTTTRGCTATG11CAG11RG1iTGGCATGCCTATCACAIAtAAAIAGA
_...,_.__...._,.._......,...._.,_.._._ ...___...
GAAATCGA1ACAAG1CAATCAAACCG-TACGGATAG1GIATAT1TATC1
GTGCCAGTRGCGRCTTI111CACiiCTCGaGATRCTCT7ACTRC1GCTCTCTTGTTGIT1T
.~. ..... ...~ _,..... ....... _,...
CpCGGTCATCGCTGaAAAaaGTGTGAGC1CIATGAGaATGAIGACGAGAGAaCAACaAAA
TA1CRCTTC1TGTTTCTTCTTGG1AAATAGAATR1CAAGCTHCAAAAAGCATACAATCAA
_... .,.. ..~...._........_.__.,.._....._,._..._
EiTAGTGARGAACaAAGAAGARCCRITTATC1TATAG1ICGGiIGTITTTCGTATGTTi~GTT
C
1
CIRTCAAG1A1TRACTATAtf
_...._._.__......,
GATAG11GATRRTTGATATAGC
The promoter of the invention may be obtained by the conven-
3 0 tional recombinant DNA techniques wellknawn to anyone skilled in the art.
The promoter of the invention has important
advantages over the known promoters and in particular
over the ADH2 promoter and the promoter of the GAL7
gene.
It permits a high maximum level of transcrip-
~0~~~~6
- 5 -
tion -and hence of expression, in particular for A.
flavus urate oxidase, and offers the possibility of
regulating said expression at three levels:
- zero level in the presence of glucose and in the
05 absence of galactose: no expression is detected,
which is an identical result to that published for
the promoter of the GAL7 gene in a strain of Sac
charomyces cerevisiae growing under these conditions
(TAJIMA _et al., 1986, Molecular Cellular Biology, 6,
246-256). The ADH2 promoter shows a low but detec-
table level of expression under these conditions.
- basic level in the absence of glucose and galactose:
there is weak expression, which is an intermediate
result between that observed for the ADH2 promoter
(maximum level) and that published for the promoter
of the GAL7 gene (zero level: TAJIMA et al., 1986,
Molecular Cellular Biology, 6, 246-256).
- maximum level of expression in the absence of glucose
and in the presence of galactose.
The advantage, for the expression of heterolo-
gous proteins in yeast, of having a promoter which
shows a zero level under certain conditions is that it
affords the possibility of avoiding any selection
pressure which would favor the least productive cells
during the propagation of the strain. This is particu-
larly important in the case where the protein is toxic
to the host cell.
The advantage of a promoter with two levels of
expression: the one a basic level (non-induced) and the
other a maximum level (induced), lies in the ability to
choose an intermediate level by varying the concentra-
tion of the inducer.
The invention further relates to an expression
vector for yeast which carries a gene of interest with
the means necessary for its expression, its replication
- 6 - ~04~6'~6
and the selection of the transformed cells, wherein the
gene of interest is under the control of the promoter
def fined above .
This gene of interest can be an endogenous gene
05 of yeast or a eukaryotic or prokaryotic exogenous gene.
Of particular value as eukaryotic exogenous genes are a
recombinant gene coding for Asperqillus flavu urate
oxidase, a recombinant gene coding for a human cyto
kinin and a recombinant gene coding for hirudin.
to zn the case where the protein coded for by the
exogenous gene is secreted naturally, the sequence
coding for this protein is preferably preceded by a
signal sequence. The function of this signal sequence,
which is chosen according to the host cell, is to
15 permit export of the recombinant protein out of the
cytoplasm, enabling the recombinant protein to adopt a
configuration similar to that of the natural protein
and considerably facilitating its purification. This
signal sequence can be cleaved either in a single step
2o by a signal peptidase which releases the mature pro-
tein, the eliminated sequence usually being called a
pre sequence or signal peptide, or in several steps
when this signal sequence comprises, in addition to the
sequence eliminated by the signal peptidase, called a
25 pre sequence, a sequence eliminated later in the course
of one or more proteolytic events, called a pro
sequence.
The invention further relates to the strains of
yeast, in particular of Sack aromyces cerevisiae, which
30 are transformed by the above expression vector, and to
a method of producing a pzotein of interest, which com
prises the culture of said strains in the presence of
galactose.
In particular, the invention relates to the strains
35 of Saccharomyces cerevisiae which have been deposited in the
depository authority named Collection Nationale de Culture
de Microorganismes -Intitut Pasteur - France under the
following numbers .
- 6 b i s - ~~~ti'~'6
I - 919 on December 28, 1989
I -1021 on December , 1990
I - 1022 on December , 1990
I - 1023 on December , 1990
05 The invention is illustrated, without impl;~~:,~ a
limitation, by means of the Examples below .
Many of the following tech.-:igues, which are well
known to those.skilled in the art, are described in
detail in the work by Maniatis et al.: "Molecular
cloning: a laboratory manual" published in 1984 by Cold
Spring Harbor Press in New York.
The synthesis of the oligonucleotides is ca~riec'
out by means of a DNA Synthetize Biosearch 4600.
ExAMPLE 1: nPrpYM~nation of the seguenrP ~r the coNA o'
A flavus urate ox
1 ) Isolation of the messe~gPr RNA's 'ron As~er~'_' ~~s
flavu~
The strain of A flavus which produces urate
oxidase was cultivated under conditions appropriate for
the production of urate oxidase, i.a. in a medium con
taining uric acid and having the following composition:
glucose 15 g/1, higS04.7H'O 1 g/1, KH~P04 0.75 g/1,
CaCO~ 1.2 g/l, uric acid 1.2 g/1, KOH 0.5 g/1, soy bean
oil 0.66 ml/1, FeS04.7H~0 10 mg/1, CuSO4.5H~0 1 mg/1,
ZnS04.7H~0 3 mg/1, rlnSO4.Ha0 1 mg/1. The medium is
adjusted to pH 7 with H~S04 1 M and sterilized at 120'C
for 80 min.
In a 5 1 Erlenmeyer flask, 1.5 1 of medium are
inoculated with about 1 to 3.10' spores. '
The culture is incubated for about 40 h at
30~C, with agitation (120 rpm). The mycelium is re-
covered by filtration on gauze, washed with water and
frozen in liquid nitrogen.
15 g of mycelium (wet weight) are thawed, re
suspended in 45 ml of lysis buffer and then taken up in
the same volume of beads (0.45 ~m in diameter). The
lysis buffer consists of guanidine thiocyanate 4 M,
_ 7 -
20~6'~~;
Tris-HC1 10 mM pH 7.6, EDTA 10 mM, l3-mercaptoethanol 50
ml/1. The mycelian suspension is ground in a Zell-
muhler mill (vibrogenic) for 5 min.
The ground material is recovered and the beads
05 are decanted. The supernatant is removed (about 45
ml), brought back to a final concentration of 3 t~i in
respect of lithium chloride and stored at 0°C.
After two days, it is centrifuged for 60 min at
10,000 rpm. The supernatant is discarded and the re
sidue is taken up in 40 ml of LiCl 3 M and centrifuged
again at 10,000 rpm for 1 h 30 min.
The following are added: proteinase K (SIGi~iA)
40 ug/ml, SDS (0.1% w/v) and EDTA 20 mM. The mixture
is incubated at 37°C for 3 h. Precipitation with 2
volumes of ethanol is followed by washing with 70%
ethanol. The residue is taken up in 0.5 ml of TE buf
fer (Tris-HC1 10 mM, EDTA 1 mM pH 7.5), the mixture is
extracted twice with chloroform and precipitation is
carried out with ethanol. The RNA's are stored at
-80°C in alcohol.
2) Purification of the poly A+ fraction of the RNA's
About 1 mg of RNA is precipitated for 20 min at
4°C (15,000 rpm) and then washed with 70o ethanol and
dried. The residue is taken up in 1 ml of TE buffer
and resuspended by agitation in a Vortex. Oligo dT-
cellulose type 3 (marketed by Collaborative Research
Inc., Biomedicals Product Division) is prepared accor-
ding to the manufacturer's recommendations. The RNA is
deposited on the oligo dT, agitated gently to resuspend
the beads and then heated for 1 min at 65°C.
The suspension is adjusted to 0.5 M NaCl and
then agitated gently for 10 min. It is then centri-
fuged for 1 min at 1000 rpm, the supernatant is removed
and the residue is washed twice with 1 ml of TE buf f er
containing 0.5 M NaCl. The supernatants are removed.
- g -
208676
The polyadenylated fraction of the RNA's (consisting of
the messenger RNA's) is eluted by suspending the beads
in 1 ml of TE buffer, then heating this suspension at
60°C for 1 min and subsequently agitating it for l0 min
05 on a tilting plate. It is then centrifuged for 1 min
at 1000 rpm, which makes it possible to recover on the
one hand the supernatant containing free mRNA's in
solution, and on the other hand the residue of cellu-
lose beads. The above series of operations (starting
from elution) is repeated. The supernatants obtained
in this way are pooled, the excess beads are removed by
centrifugation and the supernatant is precipitated with
ethanol containing NaCl in accordance with the usual
techniques (Maniatis: op. cit.).
3) Building of the cDNA librarv
The messenger RNA's isolated as described in
the previous section were used to build a cDNA library
in vector pTZl9R (marketed by PHARMACIA). This vector
is a plasmid comprising a polylinker containing unique
restriction sites.
The cloning technique used is the one described
by Caput et al. (primer-adapter technique: Caput et
al., Proc. Natl. Acad. Sci. (U.S.A.) (1986) 83, 1670-
1674).
It consists firstly in digesting the vector
with Pstl, adding a polydC tail to the protuberant 3'
end and then digesting the resulting plasmids with
BamHI. The fragment corresponding to the vector is
purified on a column of Sepharose* CL4B (Pharmacia). It
therefore comprises a polydC tail at one end, the other
end being a sticky end of the BamHI type. Secondly,
the messenger RNA's are subjected to reverse trans-
cription starting from a primer having the sequence
5'<GATCCGGGCCCT~12~)<3. Thus the cDNA's have at their
5' end the sequence GATCC complementary to the BamHI
* - Trade-mark
A
_ g -
2048676
sticky end.
The RNA-DNA hybrids obtained by the action of
reverse transcriptase are subjected to alkaline hydro-
lysis, enabling the RNA to be removed. The single-
05 stranded cDNA's are then purified by 2 cycles on a
column of Sepharose CL4B and subjected to a treatment
with terminal transferase so as to add polydG's at the
3' end. The cDNA's are inserted in single-stranded
form into the vector prepared as described above. A
second oligonucleotide, the adapter, complementary to
the primer, is necessary in order to generate an "open"
BamHI site at the 5' end of the cDNA's. After hybridi-
zation of the vector, the cDNA and the adapter, the
recombinant molecules are circularized by the action of
the ligase of phage T4. The single-stranded regions
are then repaired by means of the DNA polymerase of
phage T4.
The plasmid pool obtained in this way is used
to transform the MC1061 strain for ampicillin resis
tance (Casabadan, Chou and Cohen, J. Bact. (1980) 143,
pages 971-980).
4) Determination of the partial sequence of urate
oxidase
An A . f lavus urate oxidase preparation ( SIGriA )
was repurified by chromatography on a column of Red
agarose 120 (SIGMA), this being followed by filtration
on Ultrogel Aca 44 (IBF), an acrylamide-agarose gel.
Direct amino-terminal sequencing of the protein
was attempted in order to obtain information on the
amino acid sequence of the purified urate oxidase,
making it possible to synthesize the probes necessary
for cloning the cDNA. This sequencing was not success-
ful, probably because of amino-terminal blocking of the
protein.
The following strategy was therefore developed
- 10 -
to obtain the partial sequence of urate oxidase:
- cleavage of the protein with proteolytic enzymes
(using the enzymes trypsin and protease V8 of staphylo-
coccus aureus)
05 - separation of the resulting polypeptides by reversed
phase HPLC
- sequencing of the purified peptides.
1) Hydrolysis of the urate oxidase with trypsin,
purification and seauencinct of the peptides:
The urate oxidase, at a concentration of 9
mg/ml in an ammonium carbonate buffer 100 mM pH 8.9,
was digested with trypsin (Worthington, TPCK), in a
ratio urate oxidase/trypsin of 30/1 by weight, at 30'C
for 24 h. After tryptic hydrolysis, 60 ~cg of digested
urate oxidase were directly injected on to a reversed
phase HPLC column of Brownlee G18 grafted silica
(column: 10 x 0.2 cm) equilibrated with acetonitrile 1%
(v/v) and trifluoroacetic acid 0.1% (v/v) in water.
The peptides were then eluted by a linear gradient of
acetonitrile in a solution of trifluoroacetic acid
( 0. 1% v/v) in water, varying from 1% to 60 0 of aceto-
nitrile in 60 min, at a rate of 150 ul/min. The pep-
tides leaving the column were detected by measurement
of the optical density at 218 nm.
The elution profile is shown in Figure 1, in
which the numbers following the letter T (trypsin) cor-
respond to the peaks identified.
Each peak was collected and stored at -20°C
until analyzed on a protein sequencer (model 470 A from
Applied Biosystems) equipped with a chromatograph
(model 430 A from Applied Biosystems), which con-
tinuously analyzes the phenylthiohydantoic derivatives
formed, after each degradation cycle.
Table (1) below shows the peptide sequences of
the 9 peaks identified.
- 11 - ic:0~~6~~
2) Hvdroly_sis of the urate oxidase with protease V8,
purification and sequencing of the peptides:
The urate oxidase, at a concentration of 2
mg/ml in an ammonium acetate buffer 100 mM pH 6.8, was
05 digested with the protease V8 of Staphylococcus aureus
(Boehringer-Mannheim), in a ratio urate oxidase/
protease V8 of 60/1, at 30 ° C for 72 h. 160 ~Cg of di-
gested urate oxidase were then injected on to a rever-
sed phase HPLC column of Brownlee G18 grafted silica
l0 (column: l0 x 0.2 cm: particles: 7 x 0.03 Vim), equili-
brated with acetonitrile 1% and trifluoroacetic acid
O.lo (v/v) in water. The peptides were then eluted by
a linear gradient of acetonitrile in a solution of tri-
fluoroacetic acid in water (O. to (v/v)), varying from
15 1 o to 60 0 of acetonitrile in 60 min, at a rate of 150
~1/min. The peptides leaving the column were detected
by measurement of the optical density at 218 nm.
The elution profile is shown in Figure 2, in
which the numbers following the letter V (protease V8)
20 correspond to the peaks identified.
Each peak was collected and stored at -20°C
until analyzed on the protein sequencer already men-
tioned.
Table (1) below shows the peptide sequences of
25 the 5 peaks identified.
35
i i
I I I 1 i
I I t ~ 1
N a~ a~ a s.. n. a c~
~ a a a
~
a .=. .. c I .-
~ n ~ w
I 1 I I I I ( 1 I I I
4 1r GL A ~ a. T I T
,
~
R a j V
V V
~
1 1 I I 1 1 ~ I I I
G N C~ T T d Ir ~ I Sr ~ i.r
~r T tr ...~ .-r ~ N ~
H
,_." v7
c~ a .s c~ V I N H
1 I I I , , I I I I I 1 I
N ~ a a~ a> a c .-. a~ a a
N T ~ ~ ~ ~ r1 t/) a~ O
rp ~ N '_'
T a E X a C~. C1. c7 r2 ~ .~ a. .-a
O 1 1 I I I I I 1 I I I I 1
w r. sa . a~ a a n> n. c.. 1... v..
s.. c
T ~-1 .C ~ ~ 'U d ~ V7 'U T QJ ~~ d
H H a. a a a. .c w H v~ c.7 cn
I I 1 I I I I I I I I I I
I i
a rr Lr rp Y..rT a a 1-n a C d I V 1r L
TJ ~I rp d --rU ~ .-~ ~-1 T ~ C7 .-r O O
~ d
O V ~ t/Wt V~ V V V H V n4 n--n ~ V~
N ~/7
t I I I 1 1 I I 1 1 1 I I I I I
I
~ ~r G a d. a O O N r0 L1. ~ ~ N N
~ C I
.t.1rD I d N d ~ L, Lr .-v n ~ T t/7 T
~r .-v
i
o ~ V a ~x a H w o.. z .s .s H , ,~ a
I .x I
N I I ~ 1 I I I 1 I I 1 , 1 I 1 I I
; I
_ .~
I U .-rC Q. ~1 D. O O O N 'U r0 C I ~ ~
~ 6'
~ ~ I
G ~O -i I N r0 N ~ Sr 1.a T ~.r r~ .-
' -n
w ~ c~ I .x ~ .x o. a. a. a c ~ .s .x ! .-.
.s .. .-r H
i I
~ I I ~ I 1 I I I I I I l I I i 1 n.
I ~ c s 1.. c L. I
N N ' n.
s.
O o a p. a m. a _ -. ~ N ~ T. T V7 Q1 V7
N Q7 .--~N .-a ~
I H ~ ft .~ H r- wt ~ V H E rC H ..7 J rC "C
v7
w
I 1 I I 1 1 1 1 I I I 1 1 I I 1
I
w
a I a 1 a lr ~ ~ O N lr ~ ~ d ~
N N N
. ~ .-1.-t.-1 ~ rp tp La T ~ .-r T T
~ T
v ~ v ~ t5 E-rc~ v> >. ~ a. a H ~ I .... ..,
H a .a
c_
I I I I I I , 1 1 1 I I 1 I 1 1
1 I
U
C C 1 d Lr d C O O a a L L i~ a T
S.-n N b a
O .--~-a ~ ~ Q7 .G N it ~ _ O a. O ~r d
O ~ ~ it d ~r
i
a V N I G..V7 C7. ~ D. LY ~7 ~7 H V) V .-7 CW
r4 H H S rC -7
D'
1 I I
1
d 1 1 I I I 1 I I I I 1 I 1
I I I I I
Q7 p, lr a L 1-aC C 1~ 1-~ L a. N ~ N
T ~. ~ C ~
N ~ G7 T O 7~ t N N N ~ O N I T n0 T
.--~ W ~~ Ir rp
7 Gl.I H w H H nC rat N H N fG ~ .-~ .-7
~2 V V H V 7 rtC 7
I t I I I I I 1 1 I i I 1 I I I I
1 I I I 1 1
C C ~ N d N 3-rC G it .~ L, N T L T
4J a 7 lr ~I a fr
N N _ O ~ .-1 T r0 ~ ~ --r .C
O d d t0 O
,Q ,Q i Z ~, Z N V C5 H ~. H H V H .-~ V
~y, .-.7 .~ t/~ ~ H
~ "
N I N N N f'1M M ~ N 1 l~1 ~D
N ~
H H I C-~E H E~ F H > > >
E
N
r 9 r p N
C r0
d O
G1 ~
O
a.
~,., ~ G~.
w w _
O ~ O
- 13 - ~~~86'i 6
5) Screening of the bacteria
1) Preparation of the labeled probes:
Two pools of probes deduced from amino acid
sequences of the protein were synthesized with the aid
05 of a Biosearch 4600 DNA synthesizer. The first pool
corresponds to the sequence of residues His-Tyr-Phe-
Glu-Ile-Asp (part of the sequence of T 27), i.e. from
5' to 3':
A T G G G
T C G A T T C A A T A T G
T C A A A
This pool in fact consists of 24 x 3 - 48 different
oligonucleotides, representing all the possible combi
nations.
The second pool corresponds to the sequence of
amino acid residues Gln-Phe-Trp-Gly-Phe-Leu (part of
the sequence of V5), i.e. from 5' to 3':
GG A G T
A AAGCCCCA AA TG
AA C A C
T
This pool consists of 24 x 4 = 64 combinations.
The probes are labeled with terminal deoxy-
nucleotide transferase (TdT) (marketed by IBI, Inc.).
The reaction is carried out on 100 ng of a
3o mixture of oligonucleotides in solution (100 mg/ml) in
"Cobalt" reaction buffer (supplied as a 10-fold concen-
trate by IBI, Inc.): 1.4 M potassium cacodylate - pH
7.2, 300 mM dithiothreitol, 1 ~1 of the enzyme terminal
deoxynucleotide transferase (IBI, Inc.) and 50 ~Ci of
deoxycytidyl triphosphate, dCTP, labeled with P32.
-14- 2048676
The reaction is carried out at 37°C for 10 min
and is then stopped by the addition of 1 ~,1 of EDTA
0.5 ri.
A phenol extraction is carried out and the
05 extract is dialyzed on a column of Biogel P10 poly-
acrylamide (Biorad: 150-1050).
2) --Hybridization and detection of the colonies
containinct,urate oxidase cDNA:
About 40,000 colonies are screened by the in
situ hybridization technique developed by Grunstein and
Hogness (1975, Proc. -Natl. Acad. Sci. (U.S.A.), 72,
3961). About 6000 bacteria are plated out in Petri
dishes to give isolated colonies. After incubation for
24 h at 37°C, each dish is replicated on 2 filters,
each filter being intended to be treated with one of
the 2 pools of probes, so that all the colonies ob-
tained are tested with the 2 pools of probes in paral-
lel.
The filters are hybridized with one of the 2
pools of probes in a buffer containing 6 x SSC, 10 x
Denhardt's solution and 100 ~g/ml of sonicated and de
natured salmon sperm DNA (SIGMA). The hybridization is
carried out at a temperature of 42°C for 16 h. The 6 x
SSC solution is obtained by diluting a 20 x SSC solu
tion. The preparation of the 20 x SSC buffer is des-
cribed by Maniatis, Fritsch and Sambrook (op. cit.).
In summary, this buffer contains 175.3 g/1 of NaCl and
88.2 g/1 of sodium citrate and is adjusted to pH 7 with
a few drops of NaOH 10 N. The 10 x Denhardt's solution
contains 1 g of Ficoll*, lg of polyvinylpyrrolidone and
1 g of human serum albumin per 500 ml of final volume.
After washing in the 6 x SSC solution at 42 ° C
(3 h with 5 changes of bath), the filters are wiped
with Joseph paper and subjected to autoradiography.
The filters are developed after 16 h. A fraction of
* - Trade-mark
- 15 - 20~86'~~
about 0.5% of the colonies was found to have hybridized
with the 2 pools of probes.
colonies from this fraction were taken up and
purified. The plasmid DNA was prepared from each of
05 these colonies and this DNA was analyzed by digestion
with either BamHI, or HindIII, or both BamHI and
HindIII.
After analysis on agarose gel, the 5 plasmids
obtained were found to have been linearized by BamHI
and by HindIII. The double digestions make it possible
to release a fragment corresponding to the whole of the
cloned cDNA. The size of this fragment is about 1.2 kb
in 3 cases and about 0.9 kb in the other 2 cases. For
the following determination, one of the 0.9 kb frag-
ments and one of the 1.2 kb fragments were selected and
recloned (see section 6 below).
6) Determination of the sequence of urate oxidase cDNA
On the one hand one of the 0.9 kb fragments
(clone 9A) and on the other hand one of the 1.2 kb
fragments (clone 9C) were recloned in the DNA of the
replicative form of single-stranded phage M13. The DNA
of the M13 clones, containing the 0.9 kb fragment on
the one hand and the 1.2 kb fragment on the other, was
digested with exonuclease so as to generate a series of
overlapping M13 clones (procedure: "Cyclone I Bio-
system" of IBI). Said clones were sequenced by the di-
deoxyribonucleotide method (Sanger et al., PNAS-U.S.A.
- 1977, 14, 5463-5467).
The nucleotide sequence of clone 9C is shown in
Figure 3, which also indicates, with an arrow, the
start of clone 9A and, with a nucleotide symbol fol
lowed by an asterisk ~, the sequenced nucleotides of
clone 9A which are not identical to those of clone 9C
(when matching the two sequences and the AccI and BamHI
restriction sites used in the subsequent constructions
- 16 -
2048676
(cf. 2)).
It is found that:
- the nucleotide sequence of the longer frag
ment (clone 9C) overlaps that of the shorter fragment
05 (clone 9A) but for two differences (see Figure 3). One
of the differences is quiescent and the other corres-
ponds to a change from a tryptophan residue to a gly-
cine residue. These differences may be due either to
differences in the messenger RNA's isolated (cf. 2)
above) or to errors in the reverse transcriptase used
when building the cDNA library (cf. 3) above).
In the case of the longer fragment, an ATG
codon (in position 109 in Figure 3) opens an open
reading frame corresponding to a polypeptide of 302
amino acids, with a molecular weight of about 34,240
Da, whose sequence corresponds to the partial sequence
of purified A. flavus urate oxidase (cf. 4)).
Figure 4 shows the DNA sequence opened by the
ATG codon and the polypeptide coded for, and, with
arrows opposite the polypeptide coded for, the se
quenced peptides (cf. 4)) obtained by hydrolysis of A.
'avus urate oxidase with trypsin and protease V8.
It is found that the sequence of the poly
peptide terminates in the triplet Ser-Lys-Leu, which is
typical of peroxisomal location enzymes (could S.J. et
al., J. Cell. Biology 108 (1989) 1657-1664).
EXAMPLE 2: Construction of three expression vectors for
urate oxidase cDNA in breast: plasmid pEMR469
carrvinct an ADH2 promoter and plasmid
gE'~T'd73 and plasmid pEMR515 carrvina the
artificial promoter of the invention
The strategy employed uses fragments obtained
from pre-existing plasmids available to the public, and
fragments prepared synthetically by the techniques now
in common use. The cloning techniques employed are
2048676
- 17 -
those described by T. MANIATIS, E.F. FRITSCH and J.
SAMBROOK in "Molecular Cloning, a laboratory manual"
(Cold Spring Harbor Laboratory, 1984). The oligo
nucleotides are synthesized with the aid of a Biosearch
05 4600 DNA synthesizer.
The following description will be understood
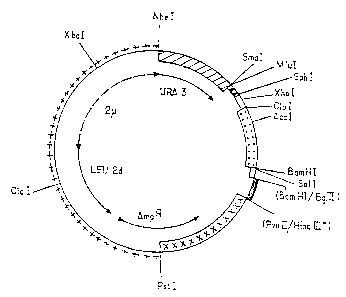
more clearly with reference to Figures 5, 6 and 7,
which respectively show restriction maps of plasmids
pEMR414, pEMR469 and pEMR473. The symbols used in
these Figures will be specified in the description
below. In the case where a site has been blunted by
Klenow polymerase, it carries the index "°"; where the
sites have been eliminated by ligation, they are indi-
cated in brackets.
1) Construction of~lasmid ~EMR469:
This plasmid was constructed from the shuttle
vector E. coli-yeast pEMR414, constructed by successive
ligations of the following components:
- the PstI-HindIII° fragment - symbolized by ,
++++ in Figure 5 - of plasmid pJDB207 (BEGGS, 1978:
Gene cloning in yeast - p. 175-203 in: Genetic Engi-
neering, vol. 2 - WILLIAMSON - Academic Press - London
UK), comprising the upstream part of the ampicillin
resistance gene AmpR of pBR322 (Sutcliffe, 1979, Cold
Spring Symp. Quart. Biol. 43, 779) and an endogenous 2~
fragment, B form, carrying the LEU2 gene of S. cere-
visiae partially modified by the deletion of its
promoter (called LEU2d), the locus STB (REP3) and the
origin of replication of the 2~ fragment (HARTLEY and
DONELSON, 1980, Nature, 286, 860-865). The HindIII end
of this fragment has been blunted by the action of
Klenow polymerase. It is denoted by HindIII° in Figure
5.
- the HindIII-SmaI fragment - represented by
~ in Figure 5 - of yeast chromosome V, containing
- is -
the URA3 gene with its promoter (ROSE et al., 1984,
Gene, 29, p. 113-124). This HindIII-SmaI fragment
originates from plasmid pFLl (CHEVALLIER et al., 1980,
Gene 11, 11-19). The HindIII end of this plasmid has
05 been blunted by the action of Klenow polymerase.
- an SamI-BamHI fragment - symbolized by
in Figure 5 - containing a synthetic version of the
promoter of the ADH2 gene which differs from the
natural version described by RUSSEL and SMITH (RUSSEL
et al. (1983) J. Biol. Chem. 258, 2674-2682) only by a
few base pairs intended for introducing restriction
sites. (The natural sequence could be used with only
slightly different results.) The sequence of this
fragment is given below:
20
30
- 19 - 2o~ss~s
S M
m 1
a
I I
~GGGACGCGTCTCCTCTGCCGGAACACCGGGCRTCTCCAAC~TRTAAGTTGGAG
.______,_________._________,_________,_________,______
05 . CCCTGCGCAGAGGAGRCGGCCT1GTGGCCCGTAGAGGTTGAATATTCAACCTC
AAATAAGAGAATTTCAGATTGAGAGAATGAAAAAAAAAAAAAAAAAAAAGGCAGAGuAGr'~-
___,_________,_________,_________,_________,_________,______
TTTATTCTCTTAAAGTCTAACTCTCTTACTTTTTTTTTTTTTTTTTTTTCCGTCTCCTCT
S
to p
h
I
GCATAGARATGGGGTTCACTTTTTGGTAAAGCTATAGCATGCCTATCACATATAAATAGA
___,_________,_________,_________,_________,_________,______
CGTATCTTTACCCCAAGTGAAAAACCATTTCGATATCGTACGGATAGTGTATATTTATCT
15 ~,TGCCAGTAGCGACTTT1TTCACACTCGAGATACTCTTACTACTGCTCTCTTGTTGTTTT
_ _ _ ~ _ _ _ _ _ _ _ _ _ , _ _ _ _ _ _ _ _ _ , _ _ _ _ _ _ _ _ _ , _ _ _ _ _
_ _ _ _ , _ _ _ _ _ _ _ _ _ i _ _ _ _ _ _
CACGGTCATCGCTGAAAAAAGTGTGAGCTCTATGAGAATGATGACGAGAGAACAACAAAA
TATCACTTCTTGTTTCTTCTTGGTAARTAGAATATCAAGCTACAAAAAGCATACAATCAR
___,_________,_________,_________~_________,_________,______
ATAGTGAAGAACAAAGAAGRACCATTTATCTTATAGTTCGATGTTTTTCGTATGTTAGTT
20 '
C a
1 m
a H
I
CTATCAACTATTAACTATATCGATACCATATGGATCCGTCGACTCTAGA~~;,TCGTC
_ _ _ , _ _ _ _ _ _ _ _ _ , _ _ _ _ _ _ _ _ _ , _ _ _ _ _ _ _ _ _ ; _ _ _ _ _
_ _ _ _ , _ _ _ _ _ _ _ _ _ , _ _ _
25 GATAGTTGATAATTGATATAGCTATGGTATACCTAGGCAGCTGAGATCTCCTAGCAG
B
a
n
H
GACTCTAGAG i
_ __ -_-
CTGAGATCTCCTAG
- the BgIII-HindIII fragment - symbolized by
in Figure 5 - carrying the 3' end of the yeast
PGK gene. This fragment originates from complete di-
gestion with BgIII of the HindIII fragment of the yeast
- 2 0 - ~U~6'76
chromosomal DNA, carrying the PGK gene described by
HITZEMAN et al. (1982, Nucleic Acids Res., 10, 7791-
7808), which has only one BgIII site. This digestion
makes it possible to obtain two HindIII-BgIII fragments
05 of which the smaller, of about 0.4 kb, which carries
the 3' end of the yeast PGK gene, is retained. The
sequence of the latter fragment is described by HITZE-
MANN et al. (op. cit.). The BgIII site is cloned in
the BamHI site of the previous fragment (the BamHI and
BgIII sites therefore disappearing), and the HindIII
site, blunted by the action of Klenow polymerase, is
cloned in the PvuII site of the PvuII-PstI fragment of
pBR322, described below.
- the PvuII-PstI fragment - symbolized by xxx
in Figure 5 - of pBR322, containing the origin of rep
lication and the downstream part of the ampicillin
resistance gene AmpR.
Plasmid pEl~t414 formed in this way therefore
contains the following components:
- an origin of replication and an ampicillin
resistance gene AmpR permitting the replication and
selection of the plasmid in E. coli cells. These com-
ponents permit transformation in E. coli cells.
- an origin of replication for the yeast (ARS),
the locus STB and the LEU2 gene of S. cerevisiae
without promoter and the URA3 gene of S. cerevisiae
with its promoter. These components permit the repli
cation and selection of the plasmid in S. cerevisiae
cells and a sufficient partition efficacy in cells
containing the endogenous 2~ plasmid.
Plasmid pEMR414 was completely digested with
the restriction enzymes NheI and ClaI. The small NheI-
ClaI fragment containing the URA3 gene, hereafter
called fragment A, was purified.
Plasmid pEMR414 was completely digested with
21 2~~6'~'Ci
the enzymes NheI and BamHI. The large NheI-BamHI
fragment containing especially the LEU2d gene and the
origin of replication of plasmid pBR322, hereafter
called fragment B, was purified.
05 The synthetic ClaI-AccI fragment, containing
the start of a gene coding for the protein deduced from
the urate oxidase cDNA sequence (clone 9C), was also
prepared. This fragment contains modifications, rela-
tive to clone 9C, introduced for the purpose of inser-
ting codons which are customary in yeast (q.v. SHARP et
al., 1986, Nucl. Ac. Res., vol. 14, 13, pp. 5125-5143)
without changing the amino acids coded for. The se-
quence of this fragment, hereafter called fragment C,
is as follows (the underlined nucleotides are those
modified relative to clone 9C):
C A
l c
a c
I I
CGATATACACAATGTCTGCTGTTAAGGCTGCTAGATACGGTAAGGACAACGTTAGAGT
___~_________+_ _ ._.+ _ .__._+.____ .__+____ ._.
TATATGTGTTACAGACGACAATTCCGACGATCTATGCCATTCCTGTTGCAATCTCAGA
The plasmid of clone 9C (cf. Figure 3) was
digested with the enzymes AccI and BamHI. The AccI-
BamHI fragment, which contains the end of urate oxidase
cDNA, hereafter called fragment D, was purified. This
fragment has the following sequence:
35
22 r2~'~~~~~
Accl.
~ CTACAAGGTTCACAAGG:\CGAGAAG
__+_________t_________,
~:cTTccA~c:c:~
vbv~VlW V
ACCGG~G.CCAGACGV~G~ACGAGATCACC G~C~GTGTGCTTCTGGAGGG~G:,G;.TTG:\G
_________+_________+_________~. _________+_________t_________t
TGGCCACnGGTCTGCC..~CA:GCTCTACTCG CAG.~CACnCGAAGACCTCCC:,CTCTAACTC
0 5 ACCTCTTACACCAAGGCCGACAACAGCGTC ATTGTCCCAACCGACTCCATTAAGAACACC
_________+_________~._________.f _____.___+_________+_________
TGV..C.,..~G~GG~~CCGVC~V~~G~CGC..G TA:,G.GCG~~GC~~G..GV~A...~C-.V~VV
ATTTAC:.TG1CCGCCA:.GCACAACCCCGTT ACTCCTCCCC:.GCTCTTCGG:.TCC:\TCCTG
_________.f_________.f_________+ _________+_________+_________.f
TAAATGTAGTGGCGGTTCGTCTTGGGGCAA TGAGGAGGGCTCGACAAGCCGAGGTAGGAC
GGCACACACT TCATTGAGAAGTACAACCAC ATCCA~GV.CG..TC:\CGTCAAC:.T:GTCTGC
1 0 _________+_________+_________+ _________+_________+_________+
CCGTGTGTGAAGTAACTCTTCATGTTCGTG TAGcTACGCCGAGTGCAGTTGTAACAGACG
C.\CCG\. ~ GGnCCCGG.,~ Gw\CA~ ~ G..CGV V. I1.,G\.G.CnCCC ~ C..C ~ CC ~ ~ C.. ~
........c.\C
_________+_________+_________; _________+_________+____.____+
G~uV\.GnCC~GGGCC~ACC~G~AAC~GCCv ~~CGV~GvGGVAG~G..GG/V1G~..GV..VV..V
AGCGAGGAGAAGCGGAATGTGCAGGTGGAC GTCGTCGAGCGG1AGGCCATCCATATCAAG
_________+___._____+_________+ _________+_________f_________+
TCGCTCCTCTTCGCCTTACACGTCCACCTG CACCAGCTCCCGTTCCCGTAGCTATAGTTC
TCGTCTCTGTCCCGCCTGACCGTGCTCAAG AGCACCa.ICTCGC:\GTTCTGGGVUTTCCTG
_________t_________.~._________.t _________+_________+_________
AGCACAGACAGGCCGGi.CT CGCACGACT T C T CG T GGT T CAGCG T C.1AC:\CCCCG:v\GGAC
CCTG.>CGACTACACCACACTTAAGC:.CaCC TGGGaCCCTATCCTGAGCACCGACGTCG:.T
_________*_________t_________+ _________i_________+_________+
GC.:.CTGCTCATGTGGTGTGAATTCCTCTGG ACCCTGGCATAGG:.CTCG:GGCTGC:\GCTa
GCCACTTGGCAGTGGAAGAATTTCAGTGGA CTCCAGGAGGTCCGCTCGCACGTGCCTAAG
2 0 _________+_________+_________+ _________t_________+_________+
CGV~GIV\CCGTCACCTTCTTNV1GTCACCT GAGGTCCTCCAGGCGAGCGTGCACGCaTTc
TTCGATGCTACCTGGGCCACTGCTCGCGAG GTCACTCTGF.AGACTTTTGCTGA.1G:\TAAC
AAGCTACGATGGACCCCGTGACGAGCGCTC CAGTG:.G:.CTTCTCAAA:.CG:.C::CTATTG
AGTGCG.CCGTGCAGCCCACTATGTACAAG ATGCCAGAGCAAATCCTGGCCCGCCAGCAG
2 5 Tc:.ccGTccc:.cc:cccc:cATACATcTTc TACCC:c:ccTTTACG~ccccGCGGTcGTc
CTGATCGAGACTGTCGAGTACTCG:TcCCT AACAAGCACTATTTCGAAATCG:.CCTGAGC
_________+_________+_________+ ._____.__+_______._+____.____+
GACTAGCTCTGACAGCTCATGAGCAACCGA TTGTTCGTGATAAAGCTTTAGCTCCACTCG
TGCCACAAGGCCCTCCAAAACACCCGCAAG AACGCCGACGTCTTCCCTCCTC:.GTCGG:.C
_________+_________+_________+ _________+_________+_________+
ACCGTGTTCCCGCAGGTTTTGTGCCCGTTC TTGCCGCTCCAGAAGCGAGG:.GTC:.GCCTG
3O CCCAACGGTCTGATCAAGTGTACCGTCCGC CGGTCCTCTCTCAAGTCTAAATTGTAA.ACC
_________+_________+_________+ _________+_________+_________+
GGGTTGCCAGACTAGTTCACATCGCAGCCC GCCACGAGAG:,CTTCAGATTT.1ACATTTCG
AACATGATTCTCACGTTCCGGAGTTTCCAA GGG'lllJICTGTATATAGTCTGCGATAGGGTA
____+_________+_________+ _________+______.__;_________+
TTGTACTAAGAGTGCAAGGCCTCA.1AGGTT CCGTTTGACATATATCAGACCCTATCCCAT
TAGC.~TTCATTCACTTGTTTTTT.1CTTCCA
3 5 _____ _t_________+_________+ _________+_________+_________+
ATCGTAAGTAAGTGAACAAAAAATCAACGT TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT
AAAGGGCCCC~ BamNl
_________+_________t________
TTTTTTTTTTTTTTTTTTTTTCCCCGGCCT AG
2 3 ~~,~~~6
Fragments A, B, C and D were ligated to give
plasmid pEMR469 shown in Figure 6, in which the symbols
have the same meanings as in Figure 5, the novel ClaI
AccI and AccI-BamHI fragments being symbolized by
05
Plasmid pEMR469 carries a sequence coding for
the protein deduced from the sequence of urate oxidase
cDNA, under the control of a promoter called an ADH
promoter, similar to the natural ADH2 promoter, com-
prising the sequence
H
1
a
_r
tGCGTCTCCTCTGCCGGAACACCGGGCATCTCCAACTTATAAGTTGGfaG
_._,.____....,._....__.,_..______,_________,._____
~AGAGGAGACGGCC1TGTGGCCCGTAGAGGTTGAATA1TCAACC7C
AAATAAGAGAATTTCAGATTGAGAGAA1GAAAAAAAAAAAAAAAAAAAAGGCAGAGGAGA
___,_....___.,_.... .._,......__.,.__._____,____..._.,_.____
TTTATTCTCTTAAAG1CTAAC1CIC1TACTT1TTTTTTTTTT7TTTTTTCCGTCTCCTCT
S
P
h TATA canw_ nent
I
GCATAGAAATGGGG1ICAC111TIGGTRAAGCTATAGCATGCCTATCACA1 AAATAGA
.,...____..,......................._.____,_.._.._ .,_.____
CGTATCTTTA;,CCCAAGTGAAAAACCA111CGATATCG~TACGGATAGIG1ATATTTATCT
GTGCCAGTAGCGACT1TTTTCACAC1CGAGATACTCITAC1ACTGCTCTC1TGTTGTTTT
._.,..___....,......_..,.........,.....___.,._.._..._,_..___
CACGGTCATCGCTGAaAAAAGIGTGAGCTCTATGAGaATGATGACGAGAGaACAACAAAA
TATCACTICTTGTTIC11CI1GGIAAATAGARTATCAAGCTACAAAAAGCATACAATCAA
.__,__.__.__.,....._.._,..__._._.,_________,_...._.._,____._
ATAGTGAAGAACAAAGRAGAACCA1TTA1~TTA1AGTTCGATGTTTTTCG1ATGTTAGTT
C Transcription
initiation region
a
1
CTATCAACTATTAACTA1AT~
-_.,.___...__,_._..__
GATAGTTGA1AATTGATATAGC
- 24 -
2 ) Construction of nl asmid oEt~iR473
Plasmid pEt~'IFt469 was completely digested with
the en2ymes MluI and Sphl. The large MluI-SphI frag-
ment, containing the urate oxidase gene, was then
05 ligated with the synthetic fragment, whose sequence is
given below, corresponding to a part (200 bp) of the
sequence upstream from the TATA component of promoter
GAL7 of S. cerevisiae, said part comprising two high-
affinity upstream activation sequences called UAS1 and
UAS2, which are boxed off below (q.v. R.J. BRAri et al.
(1986) E2dB0 J., vol. 5, n~ 3, p. 603-608).
M
L
a
I UAS1
CGCGTCTAT CTTCGGAGCACTGTTGAGCGAAG CTCATTAGATATATTTTCTGTCAT
__+______ _+_________+_________+ ________+_________+_____
AGATA AAGCCTCGTGACAACTCGCTTC GAGTAATCTATATAAAAGACAGTA
UAS2
TTTCCTTAACCCAAAAATAAGGGAGAGGGTCCAAAAAGCGCTCGGACAACTGTTGACCGT
____+_________+_________+_________+___ ____+_________+_____
AAAGGAATTGGGTTTTTATTCCCTCTCCCAGGTTTTTC CGAGCCTGTTGACAACTGGCA
GA CCGAAGGACTGGCTATACAGTGTTCACAAAATAGCCAAGCTGAAAATAATGTGTAGC
_________+_________+_________+_________+_________+_____
. C AGGCTTCCTGACCGATATGTCACAAGTGTTTTATCGGTTCGACTTTTATTACACATCG
S
P
h
I
CTTTAGCTATGTTCAGTTAGTTTGGCATG
3 0 ____+_________+_________+____
GAAATCGATACAAGTCAATCAAACC
Plasmid pEMR473 obtained in this way is shown
in Figure 7, in which the symbols have the same
meanings as in Figure 6, the novel MluI-SphI fragment
introduced being symbolized by
Plasmid pEMR473 therefore carries a sequence
- 25 - 2~
coding for the protein deduced from the sequence of
urate oxidase cDNA, under the control of the artificial
promoter of the invention, which comprises the sequence
M
05
a
I UAS1
CGCGTCTAT CTTCGGAGCACTGTTGAGCGAAGGCTCATTAGATATATTTTCTGTCAT
AGATA GCCTCGTGACAACTCGCTTCCGAGTAATCTATATAAAAGACAGTA
UAS2
1 0 TTTCCTTAACCCAAAAATAAGGGAGAGGGTCCAAAAAGC,GCTCGGACAACTGTTGACCGT
I
AAAGGAATTGGGTTTTTATTCCCTCTCCCAGGTTTTTCC~CGAGCCTGTTGACAACTGGCA
GATCCGAAGGACTGGCTATACAGTGTTCACAAAATAGCCAAGCTGAAAATAATGTGTAGC
CTAGGCTTCCTGACCGATATGTCACAAGTGTTTTATCGGTTCGACTTTTATTACACATCG
15 -~ S
P
h
I TATA cannonent
CTTTAGCTATGTTCAGTTAGTTTGGCATGCCTATCACATATAAATAGA
GAAATCGATACAAGTCAATCAAACCGTACGGATAGTGTATATTTATCT
GTGCCAGTAGCGACTTTTTTCACACTCGAGATACTCTTACTACTGCTCTCTTGTTGTTTT
CACGGTCATCGCTGAAAAAAGTGTGAGCTCTATGAGAATGATGACGAGAGAACAACAAAA
TATCACTTCTTGTTTCTTCTTGGTAAATAGAATATCAAGCTACAAAAAGCATACAATCAA
' '
ATAGTGAAGAACAAAGAAGAACCATTTATCTTATAGTTCGATGTTTTTCGTATGTTAGTT
< >
C Transeriptian
l initiation region
a
I
CTATCAACTATTAACTATAT
GATAGTTGATAATTGATATAGC
3) Construction of plasmid ~EMR515:
Plasinid pEMR473 was partially digested with the
enzyme XbaI and totally digested with the enzyme MluI.
The large XbaI-MluI fragment was purified. This frag
ment contains especially the sequences of the origin of
replication and the locus STB of the 2~ fragment, the
LEU2d gene, the ampicillin resistance gene AmpR, the
- 26 -
origin of replication of pBR322 and the expression cas-
sette for urate oxidase. On the other hand, it con-
tains neither the URA3 gene nor that part of the 2~c
fragment which is between the XbaI and NheI sites.
05 The large XbaI-Mlul fragment was recircularized
via the following sequence adapter containing MluI and
modified XbaI sticky ends:
modified XbaI
1 0 ICTAGGCTAGCGGGCCCGCATGCA
CGATCGCCCGGGCGTACGTGCGC1
MIuI
Plasmid pEMR515 obtained in this way has only
15 one of the three components of the target FRT site of
the recombinase coded for by the FLP gene of the 2u
fragment.
Plasmid pEMR515 therefore carries a sequence
coding for the protein deduced from the sequence of
20 urate oxidase cDNA, under the control of the artificial
promoter of the invention.
EXAMPLE 3: Transformation of the EMY761 yeast strain by
plasmids pEMR469J, pEMR473 and p.EMR515 -
Transformation of the EMY500 and GRF18 yeast
25 strains by plasmid,~EMR515 - Transformation
with selection for the prototrophy of
leucine
Three non-isogenic strains of Saccharomyces
cerevisiae were used as recipient strains:
30 - the EMY761 strain (Mats, leu2, ura3, his3, gal)
- the EMY500 strain (Mats, leu2, ura3, pep4)
- the GRF18 strain (Mats, leu2, his3)
The GRF18 strain is well known to those skilled
in the art (terry FINK, MIT, USA). The EMY761 and
35 EMY500 strains are related to the GRF18 strain. They
- 27 -
were obtained by successively crossing the GRF18 strain
with a ura3 strain derived from the FL100 strain (depo-
sited in the ATCC under n° 28 383 ) and with the 20B12
strain (Mats, tspl, pep4) described by E.W. JONES (E.i9.
05 JONES et al. (1977) Genetics, 85, 23).
The GRF18 strain can be obtained by curing
plasmid pEMR515 of the GRF18 pEMR515 (leu~) strain
deposited in the CNCt~i under reference n° I-920 on 28
December 1989, and the Et~tY500 strain can be obtained by
curing plasmid pEMR515 of the EMY500 pEM.R515 (leu+)
strain deposited in the CNCM under reference n° I-919
on 28 December 1989.
These strains contain mutations (leu2 and ura3)
capable of being complemented by the LEU2d defective
selection marker and the URA3 selection marker, which
are present in each of plasmids pEriR469 and pEMR473.
The transformation technique used is a variant
of that described by Beggs et al. (Beggs et al. (1978),
Nature 275, 104-109). It consists in subjecting yeasts
to a protoplastization treatment in the presence of an
osmotic stabilizer, namely sorbitol at a concentration
of 1 M.
The precise transformation protocol is speci-
fied below:
a) 200 ml of liquid YPG medium (cf. Table I)
are inoculated with about 5 x 106 cells of a culture in
the stationary phase, and the culture inoculated in
this way is agitated overnight at 30°C.
b) When the density of the culture reaches
about 10' cells per ml, the cells are centrifuged at
4000 rpm for 5 min and the residue is washed with
sorbitol 1 M.
c) The cells are suspended in 5 ml of sorbitol
solution 1 M containing 25 mM EDTA and 50 mM dithio
threitol, and are incubated for 10 min at 30°C.
- 2 $ - ~Q~B~'~'~
d) The cells are washed once with 10 ml of
sorbitol 1 M and suspended in 20 ml of sorbitol. Zymo-
lase-100T (a preparation obtained by partial purifi-
cation of Arthobacter luteus culture supernatant on an
05 affinity column and containing 13-1,3-glucan laminari-
pentahydrolase, marketed by SEYKAGAKU KOGYO Co. Ltd.)
is added up to a final concentration of 20 ~g/ml and
the suspension is incubated at room temperature for
about 15 min.
e) The cells are resuspended in 20 ml of a
medium containing sorbitol, called sorbitol YPG medium
(cf. Table I below), and incubated for 20 min at 30°C,
with gentle agitation.
f) The cells are centrifuged for 3 min at 2500
rpm.
g) The cells are resuspended in 9 ml of trans-
formation buffer (sorbitol 1 M, Tris-HC1 10 mM pH 7.5
and CaCl2 l0 mM).
h) 0.1 ml of cells and 5 ~cl of DNA solution
(about 5 ~Cg) are added and the suspension obtained is
left for 10 to 15 min at room temperature.
i) 1 ml of the following solution is added:
polyethylene glycol PEG 4000 20%, Tris-HCl 10 mM pH 7.5
and CaCl2 10 mM.
j) 0.1 ml of the suspension obtained in i) is
poured into a tube containing leucine-free solid re-
generation medium (cf. Table I below) which has been
melted beforehand and kept liquid at about 45°C. The
suspension is poured into a Petri dish containing a
solidified layer of 15 ml of leucine-free solid re-
generation medium.
k) Step j) is repeated with the remainder of
the cell suspension obtained in h).
The transformed strains start to appear after
three days.
~o~s~~
- 29 -
A transformed strain EMY761 pEMR469 (leu~),
three transformed strains EMY761 pEhiR473 (leu~) (clones
1, 2 and 3 ) , a transformed strain EMY761 pEt-iR515
( leu+) , a transformed strain EMY500 pEMR515 ( leu+) and
05 a transformed strain GRF18 pEMR515 (leu~) were thus
retained.
TABLE I
Principal media used in Examples 3 4, Obis 6 and 7
- uracil-free solid medium
6.7 g of Yeast nitrogen base without Amino Acids
( from DIFCO )
5.0 g of casein hydrolyzate (Casamino acids from
DIFCO)
10 g of glucose
g of agar
Mix all the ingredients in distilled water and make
up the final volume to 1 1 with distilled water.
Autoclave for 15 min at 120°C.
- uracil-free liquid medium
Use the formulation of the uracil-free solid medium
20 without the agar. Autoclave for 15 min at 120°C.
- leucine-free solid medium
6.7 g of Yeast nitrogen base without Amino Acids
( from DIFCO )
20 mg of adenine
20 mg of uracil
20 mg of 1-tryptophan
20 mg of 1-histidine
20 mg of 1-arginine
20 mg of 1-methionine
mg of 1-tyrosine
30 mg of 1-isoleucine
30 mg of 1-lysine
50 mg of 1-phenylalanine
30 100 mg of 1-glutamic acid
150 mg of 1-valine
400 mg of 1-leucine
20 g of glucose
20 g of agar
Mix all the ingredients in distilled water. Make up
the final volume to 1 1 with distilled water. Auto
slave for 15 min at 120°C. After autoclaving, add
200 mg of 1-threonine and 100 mg of 1-aspartic acid.
- 30 -
- leucine-free solid regeneration medium
Use the formulation of the leucine-free solid medium,
mixing in 30~g of agar instead of 20 g and adding
182 g of sorbitol to the mixture.
- leucine-free liquid medium
05 Use the formulation of the leucine-free solid medium
without the agar. Autoclave for 15 min at 120°C.
After autoclaving, add 200 mg of 1-threonine and 100
mg of 1-aspartic acid.
- liquid YP medium
g of yeast extract (Bacto-yeast extract from
10 DIFCO)
g of peptone (Bacto-peptone from DIFCO)
Mix the ingredients in distilled water. Make up the
final volume to 1 1 with distilled water. Autoclave
for 15 min at 120'C.
- liquid YPG medium
Use the formulation of the liquid YP medium, adding,
15 after autoclaving, glucose at a concentration of 20
g/1.
- sorbitol YPG medium
Use the formulation of the liquid YPG medium, adding,
after autoclaving, sorbitol at a concentration of
1 M.
20 - ethanol-glycerol YP medium
Use the formulation of the liquid YP medium. After
autoclaving, add 10 ml of ethanol 100% (1% final con-
centration) and 30 g of glycerol.
- ethanol-glycerol-galactose YP medium
Use the formulation of the liquid YP mediu~a. After
autoclaving, add 10 ml of ethanol 100%, 30 g of
glycerol and 30 g of galactose.
EXAMPLE 4: Expression of urateoxidase by the EMY761
~EMR469 lleu;7 and EMY761 pEMR473 (leu~l
jclones 1, 2 and 3) strains - Immunodetec-
tion by Western blot - Assay of the urate
oxidase activity and the soluble proteins
1) Expression of urate oxidase:
a) Transformed strains
In a first stage, a colony of each of the
- 31 -
EI~iY761 pEriR469 ( leu+) and EZ~iY761 pEt~iR473 ( leu; ) ( clones
1, 2 and 3 ) strains was cultured in 25 ml of leucine-
free liquid medium (cf. Table I, Example 3). This made
it possible to obtain and maintain a large number of
05 copies of plasmids by carrying out the selection for
complementation of the leu2 mutation by the LEU2 gene
carried by plasmids pEt~t469 and pE2~473.
After 22 h at 30°C, with agitation, the two
cultures were centrifuged for 10 min at 7000 rpm. The
residues were taken up in 10 ml of sterile distilled
water and centrifuged again for l0 min at 7000 rpm.
Expression of the urate oxidase was induced by taking
up the cells in 20 ml of ethanol-glycerol YP medium for
the Et~iY761 pEMR469 (leu+) strain and in 20 ml of
ethanol-glycerol-galactose YP medium (cf. Table I,
Example 3 ) for the EMY761 pEMR473 ( leu~) strain. The
cultures were incubated again at 30°C for 27 h, with
agitation.
c) Control strain
The non-transformed EMY761 strain, i.e. the
EMY761 strain without plasmid, was cultivated as above
except that the first culture was carried out in liquid
YPG medium. It was subjected on the one hand to induc-
tion in 10 ml of ethanol-glycerol liquid YP medium and
on the other hand to induction in 10 ml of ethanol-
glycerol-galactose YP medium.
2) Preparation of the samples:
a) The cells cultivated in la), lb) and lc)
were centrifuged and the supernatant was removed. The
residues were taken up in 10 ml of distilled water and
centrifuged for 10 min at 7000 rpm. The residues
washed in this way were taken up in about 1 ml of tri-
ethyleneamine buffer, TEA, of pH 8.9. About 300 ~1 of
cells taken up in said buffer were lyzed in the pre-
sence of glass beads (from 400 to 500 ~m in diameter),
- 32 -
representing about half the final volume. This nixture
was agitated vigorously in a Vortex 4 times for 1 min,
the samples being placed in ice for 30 s between grin-
ding operations. The liquid was withdrawn from the
05 tubes with a Pasteur pipette and transferred to a
microtube. The glass beads were washed once with about
200 ~1 of TEA buffer of pH 8.9. The beads were agi-
tated in a Vortex once for 1 min and the liquid was
withdrawn with a Pasteur pipette and added to the above
lyzate. The lyzate was then centrifuged in a microtube
for 5 min at 7000 rpm. The supernatant was cautiously
withdrawn and stored at -20°C for Western blot, assay
of the urate oxidase activity and assay of the total
soluble proteins. The residue of the lyzed cells was
stored separately at -20°C for Western blot (cf. 3)
below).
Furthermore, samples of the cultures prepared
in la) and lb) were taken in the following manner
before induction: 2 ml of culture were centrifuged for
10 min at 7000 rpm. The residues were taken up in 500
ul of distilled water and centrifuged again for 5 min
at 7000 rpm. The residues were taken up in about 200
~cl of TEA buffer of pH 8.9 and lyzed as above in the
presence of glass beads. The supernatants and the
residues of the lyzed cells were stored separately at
-20°C. Assay of the oxidase activity and assay of the
total soluble proteins were performed on the super-
natants.
3) Immunodetection of the urate oxidase by Western
blot:
a) Procedure
The residues and the supernatants of the dif-
ferent samples were subjected to a Western blot - a
technique well known to those skilled in the art -
which comprises the following steps:
33
solubilization of the residue by boiling for 10 min
in a buffer, called a loading buffer, consisting of
Tris-HC1 0.125 M pH 6.8, SDS 40, bromophenol blue
0.0020, glycerol 20%, f3-mercaptoethanol 10% (accor-
05 ding to the protocol described by LAEt~iLI (U. K.
LAEMMLI, Nature, 227 (1970), 680-685)) (step perfor-
med solely for the residues);
- electrophoretic separation of the different proteins
contained in the solubilizate, according to the
protocol described by LAEMMLI ( U . K . LAEMriLI , Nature ,
227 (1970), 680-685); and
- transfer of said proteins contained in the gel on to
a nitrocellulose filter (according to the technique
of H. TOWBIN et al., Proc. Natl. Acad. Sci. USA 76
(1979) 4350-4354).
Immunodetection, performed according to the
technique of BURNETTE (W.W. BURNETTE, Ana. Biochem. 112
(1981) 195-203), involves the following successive
operations:
~ rinsing the nitrocellulose filter for 10 min with a
buffer A (Tris-HC1 10 mM, NaCl 170 mM, KC1 1 mri);
bringing the nitrocellulose filter into contact with
a buffer B (buffer A with bovine serum. albumin added
at a rate of 3 g per 100 ml) for 30 min at 37°C;
~ bringing the nitrocellulose filter into contact with
an immune serum (polyclonal antibodies recognizing A.
lavus urate oxidase) for 1 h at 37°C;
rinsing the nitrocellulose filter with buffer B;
bringing the nitrocellulose filter into contact with
a solution of protein G, labeled with iodine 125 at a
rate of 0.1 microcurie/ml, for 1 h at 37°C;
rinsing the filter with buffer A;
drying the filter between two absorbent sheets;
bringing the filter into contact with an X-ray film;
and
34 ~~~~r~~
developing the film.
b) Results
It is found that the EbiY761 pEMR469 (leu~) and
EMY761 pEMR473 (leu+) (clone 1) strains produce a pro
05 tein with an apparent molecular weight of about 33 kDa,
which is recognized by antibodies directed against A.
flavus urate oxidase (prepared in rabbits by techniques
well known to those skilled in the art: q.v. VAITU-
KAITIS et al. (1981) "Methods in enzymology", Academic
Press, New York, vol. 73, p. 46) and which is absent
from the control strain.
Comparison between the amounts of this protein
for the residues and the supernatants makes it possible
to deduce that about 800 of said protein is in soluble
form in the lyzate.
4) Assay of the urate oxidase activity:
The urate oxidase activity was measured on the
supernatants of the lyzed cells.
a) Principle
The conversion of uric acid to allantoin is
followed by the decrease in absorbance at 292 nm. The
reaction is as follows:
0
H H
~ N 0 H zNCONH N 0
HN~ 5 ~ Urate oxydase
2 4 ~ >
0~ N 0
H
H20 + 0~ H20 + C02
Uric acid Allantoin
(absorbs at 292 nm)
b) Rea eq nts
a) TEA 0.05 M pH 8.9/EDTA buffer
- 7.5 g of TEA (reagent for analysis - Prolabo ref.
287.46.266) are dissolved in 400 ml of distilled water;
35
- 0.372 g of Complexon III (Merck'- ref. 8418) is dis-
solved in 50 ml of distilled water;
- the two solutions are combined and made up to 500 ml
(solution 1);
05 - the pH of this solution is adjusted to 8.9 with HC1
0.2 N; and
- the volume is made up to 1000 ml with distilled water
(solution 2).
b) Uric acid stock solution
- 100 mg of uric acid (Carbiochem - ref. 6671) are dis-
solved in 50 ml of solution 1;
- the pH is adjusted to 8.9 with HC1 0.2 N; and
- the volume is made up to 100 ml with distilled water.
The solution obtained can be stored for one
week at 4°C.
c) Uric acid substrate solution
- 1.5 ml of uric acid stock solution (Carbiochem - ref.
6671) are taken and diluted to 100 ml with TEA buffer
(reagent for analysis - Prolabo ref. 287.46.266).
This solution must be used the same day.
c) Procedure
The following volumes are introduced into the
quartz cell of a spectrophotometer set to 292 nm and
thermostated at 30°C:
- 600 ~1 of uric acid substrate solution (preheated to
30°C); and
- 100 ul of the above supernatants to which 200 ~cl of
TEA pH 8.9 have been added (preheated to 30°C).
After mixing, the change in optical density
(sometimes abbreviated to OD hereafter) is read off
every 30 s for 5 min. Q E, the variation in optical
density per minute, is deduced from these readings.
d) Expression of the results
The urate oxidase enzymic activity A, expressed
in U/ml, is calculated from the aE measurement with the
- 36 -
aid of the formula
A = DE x Vr x d
I x VpE
05
in which the symbols Vr, d, I and Vp~ respectively
represent the reaction volume (0.9 ml), the dilution
factor (2), the extinction coefficient of uric acid at
292 nm (12.5) and the volume of the test sample (0.1
ml).
5) Assay of the total soluble proteins in the lvzates:
The protein assay kit from BIORAD was used for
assaying the total proteins present in the supernatant
of the lyzed cells. It is based on the observation
that the maximum absorbance of an acid solution of Coo-
massie brilliant blue g-250 changes from 465 nm to 595
nm when proteins become attached thereto (q. v. Reisner
et al., Anal. Biochem., 64, 509 (1975)).
procedure
The following volumes are introduced into the
cell of a spectrophotometer set to 595 nm:
- 10 ~1 of sample to which 790 ul of distilled water
have been added; and
- 200 ~C1 of concentrated Dye reagent (Biorad).
The ingredients are mixed and the optical den-
sity is read off at 595 nm. A calibration range with
increasing concentrations of BSA (bovine serum albumin)
was prepared in this way. The unknown concentration of
the total proteins in the lyzates is read off on the
calibration curve obtained.
6) Results:
The results obtained are collated in Table (II)
below, which specifies, for each strain, the culture
medium, the carbon and energy source of the culture,
the urate oxidase activity in U/ml, the amount of total
- 37 -
soluble proteins in mg/ml and the percentage of urate
oxidase in the total soluble proteins. This last para-
meter is calculated by assuming that the specific
activity of the recombinant protein is identical to
05 that of the urate oxidase obtained from A. f lavus : 30
U/mg.
15
25
35
2~~~'~
i
N
ro I
p
G
i
w .c
O
O +n M O O O O O ~ tf11f1 O
L
G1 O O O O O O O O O O N
d C tl'
tT.-~d0 0 0 0 0 0 .X 0 0 0 Ovl1 ppr.s
p .--I .r N rr .-.
G N
V ro
.-t
U ~
O
Yr
-,
N
d x
n.
O
i
N
~
G
_ O~ O~ o e~ tC1 N o .-..I.-~.-. tD ao ~ o ar
O
E
N d
w
E d' M \D tt1 ~ ~!'N M M M vf1 M ~f1 V N
r0
L
.~
w a.
O
I
d
N
roT
'~
+u
.--.
~x o 0 0 0 0 0 ~n o 0 0 o M
>
E
w O O O O O O O 00 O O O O t~ ~J N O
V U -1 M N
v
+~ v v v v v v
ro
S~
p
_ w ~ _w w
T .--n.-1 -r .-a
0, O O O O O O O O O O
L. La 1,. 1W . L 1r 1, ~. i'. 1..
p
v ~ Q~ U U p v O 'U V O J
G w U U U U U U U U U U
Q) fy ~ ~ ~ ~ T ?- T T T T T T T T
~
w tr N C7 V7 N N
'v ~ ~ ~ O ~ tn v~ v~ v~ v~ v. a7 tT O v~ a~
O v' d v~ a~
~
C y J U W ~ v W W W n v N v N W V7
W N
.-~ ro a ~ a ~ a .-,.-~ -. _ -. .~ o -. o -. o -.
a~ o .~ o
-.
CO U O --~ -~ --~--n -~ O O O O O O .-I O .~ O w O
w O y
C lr p~ D' p~ p~ p~ C C C C C G U G U G U C U C
~ U V
M a ro ro ro ro ro ro ro ro ro ro ro
g ro ro ~
ro
m o ~ s ~ t t _
. .c .~ ~ .~ .c --.
~ _.
,d ~.r r.r_ r-~ru r.., ..~ ro w ro w ro
N .u
E~ V p O1 d V N U D' U C~ CJ D' V
P O tT
~ ~ ~ ~ ~
d d O d CJ
_ _ _ _ _ N N V7 N N
'D 'D 'C7'~ '~ O
O
O
O
O
c ~ ~ ~ ~ ~ E ~ ~ ~ _
_
_
_
_
o ~ ro
w
w
ro
ro V V V V V _
_
_
..~ v v 'v w A. a. A. a. ro ro ~~ w
w ro > a ~ >. >. v, v. o. v. v.
O 1.. a a a a ~ v w
v w w
_ w ~ ~ w .--~-r .~ -. -i _
ro -. -. -.
.~70 0 0 0 O ~ O ~ O
.~. E a~ a~ a~ a~ ~ a~ O a~ O
~. a>
GY d d d d V V U U U U U U U U
d d ~ ~ T T T T T T T T T T
N O .-v 1-..1r L C7 .--wr ~ .-r.--m -a .-1
W w w w w 0. tT v~ U ~ v , ~ ~ v.
a ~ C~ ~ U
~ ,F, ~ ,F
w~L1 n v I I Y w w w ~ w ~
J
~
wwt
w
O O U p _ ~.r .--~~ .-r _
_
_
....~ ...-, ..--~
.-v
G G G G '~ O O O O O O '~ O 'O O '~J O
TJ O '~
N _ _ _ _ _ c m ~ ~
V U U U a r ~ ~ ro E ro E ro
a a a a o- v .c .c ro E ro_ E
t
a~ a~ a> a~ .-~~ .-~.-~.-~ .-~ o, .-. o, a .-~
o. ' a.
.-, ...,.~ .--~~ a> ai a> a~ ~ a> >. a> >. a> >.
a> >- am.
N M M ~ N ~ N M
d d d d d d d d d
C G C G C G C C G
O O O O O O O
O O
U U _ U V U _
. . U ~r . U U U
. . . . .
. . .
. .
- _ _ .-. - -
_
-.
~D t~ tD
1 ~.. t~ 1 4 1 t~ i v 1
a a ~ a a. >..a a a a r a a a a
G
Q1 M M M Q1 M M M d M M M Q~
ro V ~ ~ r~ V r~ e~ e~ t~ ~ ~ V
~
L.1 ct' ~ Q' d' p' Q Q' d' V' ~T d' ~1'
w G CG tx G' O O GG p'..G: G'r O CC C
.' ~ P.' C~
_ ~3 ~ ~3 N N ~3 ~i ~i ~3 _
G3 Npl~f~~3
R G1 O..D. G C L1 G. GL GL C G1. L1 Q. Q.
w ro ro
_
.-- mr .r .~ H it ~ rr .--v.~ S-a
~D ~D tD ~D w w tD tD ~O ~D a~ ~D ~D ~O ~D
n n n I I f~ e~ A r~ I n n n t~
> a. >. ~. c G ~-. r ~- >. c a >. >.
c ~ ~ ~ c
c ~
2~~~'~~
- 39 -
This Table shows that:
a) in the presence of glucose, the ("repres-
sed") level of urate oxidase is not detectable in the
case of the artificial promoter of the invention
05 (strain EMY761 pEMR473 (leu+) (clone 1, 2 or 3)),
whereas it is detectable in the case of the ADH2
promoter (strain EMY761 pEMR469 (leu+)). In the
presence of glucose, therefore, the artificial promoter
permits better repression than the ADH2 promoter.
b) in the absence of glucose but in the pre-
sence of ethanol/glycerol, the level of urate oxidase
is high for the ADHZ promoter ( about 14% of the total
soluble proteins) and low but detectable for the arti-
ficial promoter.
c) in the absence of glucose but in the pre-
sence of ethanol/glycerol/galactose, the level of urate
oxidase retains a value little different from that of
the previous case for the ADH2 promoter (about 13.5% of
the total soluble proteins), but reaches a high value
(about 180 of the total soluble proteins) for the arti-
ficial promoter.
The artificial promoter of the invention there-
fore permits a high level of production of recombinant
protein and has three levels of expression:
- zero level a)
- basic level b)
- maximum level c)
EXAMPLE Obis: Expression, in an Erlenmeyer flask, of
urate oxidase cDNA by the EMY761 pEMR515
(leu~), EMY500 pEMR515 (leu+~ and GRF18
pEMR515 fleu+) strains
A colony of each of the above three strains was
cultured in 20 ml of leucine-free liquid medium.
After one night at 30°C, with agitation, the
three cultures were centrifuged for 10 min at 7000 rpm.
..~ 2s~s~s
- 40 -
The cell residues were taken up in 10 ml of sterile
distilled water and centrifuged again for 10 min. Ex-
pression of the urate oxidase was induced by taking up
the cells in 20 ml of ethanol-glycerol-galactose YP
05 medium (cf. Table I, Example 3). The cultures were
incubated again at 30°C for about 20 h, with agitation.
As a control, a culture of each non-transformed host
strain was prepared.
The cells of each of the six cultures are re
deposited by centrifugation and the supernatant is
removed. The residues were taken up in 10 ml of dis
tilled water and centrifuged for 10 min at 7000 rpm.
The residues washed in this way were taken up in about
1 ml of TEA buffer of pH 8.9 and the grinding and re
moval of the particles by centrifugation were carried
out in the manner described in Example 4, 2). The
supernatant of each culture is used, as before, for
assay of the urate oxidase and the total proteins. The
principal results obtained are collated in Table III.
below:
30
- 41 - 2o~s~s
T?.BLE III
Strain/culture conditionsUrate oxidaseTotal soluble% of orate
activity proteins obidase ~
(U/nl) (~g/al) in the sol~le
protei:a
05
GRF18 pEHR515 (leu')/a)< 0.1 2.2 < 0.05
EHY500 pEHR515 (leu')/a)< 0.1 0.9 < O.C5
EHY761 pEMR515 (leu')/a)< 0.1 1.8 < 0.05
GRF18 pEMR515 (leu')/b)38 5.4 23
1 0
EMY500 pEHR515 (leu')/b)20 2.5 26
EHY761 pEHR515 (leu')/b)33 4.2 26
a): the strains are cultivated in the presence of glucose (non-induction
conditions)
1 5 b): the strains are cultivated in the absence of glucose and in the
presence of
galactose (induction)
The promoter according to the invention there-
fore permits a high level of expression of orate
oxidase in three non-isogenic strains.
20 EXAMPLE 5: Construction of two expression vectors for
f3-qalactosidase in yeast: plasmid oEr1R429
carrying an ADH2 gromoter, and olasmid
pEMR437 carrying the artificial pronoter of
the invention
25 The strategy employed uses fragments obtained
from pre-existing plasmids available to the public, and
fragments prepared synthetically by the techniques now
in common use. The cloning techniques employed are
those described by T. MANIATIS, E.F. FRITSCH and J.
30 Sp~gROOK in "Molecular Cloning, a laboratory manual"
(Cold Spring Harbor Laboratory, 1984). The oligo-
nucleotides are synthesized with the aid of a Biosearch
4600 DNA synthesizer.
1) Construction of plasmid pEMR429:
35 Plasmid pEMR414 (cf. Figure 5) was completely
42
digested with the restriction enzyme BamHI. The BamHI
site, located between the promoter sequences (ADH~) and
terminator sequences (PGK), is unique. The linear DNA
of the plasmid is purified by elution from an agarose
05 gel after electrophoresis. Plasmid pt~iC1403 (ref. Casa-
daban et al. (1980), J. Bacteriol., 143, 971-980) was
completely digested with the enzymes BamHI and EcoRI,
which made it possible to release a BamHI-EcoRI DNA
fragment of about 3 kb containing the essential part
(upstream part) of the sequence coding for E. coli f3-
galactosidase. The complete sequence was reconstituted
with the aid of the synthetic fragment of the following
sequence:
EcoR I~ r,r n,.nrr~~~ ~r
5 ~ nATT T CAGE, i GnG~,v.~~uT~u
AGTCGACTCGCGGCCAGC
CTACCATTACCAGTTGGTCTGGT
GATGGTAATGGTCAACCAGACCA
GTCAAAAATAATAATAAG
CAGTTTTT:,TTATTATTCCTAGv
BamHl
The BamHI-EcoRI fragment originating from the
double digestion of pMC1403 is ligated at EcoRI with
the above synthetic fragment.
The BamHI-BamHI fragment obtained is then
ligated with the linear DNA of plasmid pEMR414 digested
with BamHI, to give plasmid pEMR429 shown in Figure 8,
in which the symbols have the same meanings as in
Figure 5, the BamHI-EcoRI and IcoRI-BamHI fragments
introduced being represented by .
In this plasmid, the sequence coding for B-
4 3 - ~~~b~! 6
galactosidase is under the control of the ADH2 promoter
described in Example 2.
2) Construction of plasmid pEMR461:
Plasmid pEMR429 was completely digested with
05 the enzymes MluI and SphI. The large MluI-SphI frag
ment, containing the 13-galactosidase gene, was then
ligated with the synthetic fragment whose sequence,
given below, comprises the upstream activation se
quences (UAS) of the promoter of the GAL7 gene of S.
cerevisiae and MluI and SphI sticky ends.
~A
U
CGCGTCTA1ACTTCGGAGCACTGTTGAGCGnAGGCTCA11AGATATATTITCTG1CAT
__,._.__._..,....._.__,__...___.,._.______,_______._,.____
AGATATGAAGCCTCGTGACAACTCGCTTCCGAGTAATCTA1ATAAFiGiG~CAG1A
TTTCCTTAACCCAAAAATAAGGGAGAGGGTCCAAAAAGCGCTCGGACAaCTGTTGACCGT
..._,___._____,.._._____,_____.___,____.____._________,_____
AAAGGAATTGGGTTTiTATTCCCTCTCCCRGGiTTITCGCGnGCCTGTTGt~CnACTGGCa
GaTCCGaaGGACTGGCTATACAGTGTTCACAar~ATAGCCAaGCTGAr~AATAATG1GTAGC
__._,,_____._._._..______,_._._..__,___..____,__._.____,_____
CTAGGCTTCCTGACCGATATGTCACAAGTGTTTTATCGGTTCGAC1TTTATTACACATCG
~ S
P
h
I
CTTTAGC1ATG1TCAGTTAGTT1GGCATG~
__..,_...__...,._.._._._,_....
GAAATCGATACAAGTCAATCAAAC G
Plasmid pEMR461 obtained in this way is shown
in Figure 9, in which the symbols have the same mea-
nings as in Figure 8, the novel MluI-SphI fragment
introduced being symbolized by ~.
In this plasmid, the sequence coding for B
galactosidase is under the control of the artificial
promoter of the invention, described in Example 2.
- 44 - ~iQ~ .~7~~~
EXAMPLE 6: Transformation of the DBY746 S. cerevisiae
strain by plasmids pEMR429 and pE2dR461
Transformation with selection for the proto-
05 trophy of uracil:
A colony of the DBY746 strain, which is (t~fata,
his3, leu2, ura3, trpl, cyhR) (ROSE et al. (1981), PNAS
USA, 78, 2460-2464), was used to inoculate 100 ml of a
medium called liquid YPG medium (cf. Table I of Example
3). When the cell density had reached 10' cells per
ml, the cells were treated with lithium acetate 0.2 i-i
for transformation by a technique well known to those
skilled in the art and described by ITO et al. (ITO et
al., 1983, J. Bacteriology 153, 163-168).
The DBY746 cells were transformed in parallel
with about 1 ~g of each of plasmids pEMR429 and
pEl~iR461. The transformed cells are selected for the
auxotrophic character of uracil (ura~) on a medium
called uracil-free solid medium (cf. Table I of Example
3). A transformed strain DBY746 pEMR429 (ura+) and a
transformed strain DBY746 pEMR461 (ura~) were thus
retained.
EXAMPLE 7: Production of fi-galactosidase with the aid
of the DBY746 pEMR429 (ura~l and DBY746
pEMR461 (uraW strains
1) ~~ression of f3-galactosidase:
A transformed colony DBY746 pEMR429 (ura~) and
a transformed colony DBY746 pEMR461 (ura+) were each
used to inoculate 20 ml of uracil-free liquid medium to
which tryptophan (10 mg/1) had been added beforehand.
After one night at 30°C, with agitation, 1% of glucose
is added and culture is allowed to continue for 4 h. A
check is then made to see that there is still some
glucose in the cultures. An aliquot is taken in order
to assay the B-galactosidase.
- 45 - 2~~~el~i
After one night at 30'C,~with agitation, the
two cultures were centrifuged for 10 min at 7000 rpm.
The residues were taken up in 10 ml of sterile distil-
led water and centrifuged again for 10 min at 7000 rpm.
05 Expression of !3-galactosidase was induced by taking up
the cells in 20 ml of ethanol-glycerol YP medium (cf.
Table I, Example 3) for the DBY746 pEMR429 (ura+)
strain and in 20 ml of ethanol-glycerol-galactose YP
medium (cf. Table I, Example 3) for the DBY746 pEMR461
(ura+) strain. The cultures were incubated again at
30°C overnight, with agitation.
2) Preparation of the samples and assay:
The cells cultivated above were centrifuged and
the supernatant was removed. The residues were taken
up in 10 ml of distilled water and centrifuged for 10
min at 7000 rpm. The residues washed in this way were
taken up in about 1 ml of B-galactosidase assay buffer
(EDTA 2 x 10-3 M; Na2HP04 7 x 10-2 M; NaH~P04 3 x
10-2 M; MgS04 10-3 M; MnS04 2 x 10-3 M). About 300 ~C1
of cells taken up in said buffer were lyzed in the pre-
sence of glass beads (from 400 to 500 ~Cm in diameter),
representing about half the final volume. This mixture
was agitated vigorously in a Vortex 4 times for 1 min,
the samples being placed in ice for 30 s between grin-
ding operations. The liquid was withdrawn from the
tubes with a Pasteur pipette and transferred to a
microtube. The glass beads were washed once with about
200 ~C1 of TEA buffer of pH 8.9. The beads were agita-
ted in a Vortex once for 1 min and the liquid was with-
drawn with a Pasteur pipette and added to the above
lyzate. The lyzate was then centrifuged in a microtube
for 5 min at 7000 rpm. The supernatant was cautiously
withdrawn and stored at -20°C for Western blot, assay
of the urate oxidase activity and assay of the total
soluble proteins. The residue of the lyzed cells was
- 46 -
stored separately at -20'C.
The f3-galactosidase activity was assayed by the
technique of PARDEE (PARDEE et al., J. Mol. B. (1959),
1, 1656-178).
05 Furthermore, the total soluble proteins were
assayed using the BIORAD protein assay kit, as des-
cribed in Example 4.
The results obtained are collated in Table IV
below:
TABLE IV
Strain Carbon and B-GalactosidaseTotal solubleCulture
energy ~ediu~
source of activity proteins
the
culture U/nl pg/nl
DBY746 glucose 59 375 liquid ~ediun
pEHR429
(ura~) without
uracil
+
tryptophan
(10
ng/1) +
glucose
(1%)
2 0 DBY746 ethanol/glycerol6500 1700 ethanol-glycerol
pE.u.R429 YP nediun
(ura~)
DBY746 glucose 0 350 liquid nediun
pEHR461
(ura~) without
uracil
+
tryptophan
(10
ng/1) glucose
(1%)
DBY746 ethanol/glycerol/400 520 ethanol-glycerol-
pEHR461
(uraa) galactose galactose
YP
aediun
This Table shows that:
_ in the presence of glucose, the EMY746 pEMR429 (ura+)
strain produces a small amount of f3-galactosidase,
whereas this protein is not detected for the DBY746
pEMR461 (ura+) strain. The artificial promoter of the
invention therefore permits better repression than the
ADH2 promoter.
CA 02048676 1999-12-22
- 47 -
- under induction conditions, the artificial promoter
leads to a high level of expression of ~3-galactosidase,
although under these conditions it is lower than the
level obtained with the ADH~ promoter.
S
M
In a
CGCGTCTATACTTCGGAGCACTGTTGAGCGAAGGCTCATTAGATATATTTTCTGTCAT
. . . _ _ _ . _ . .f _ _ _ _ _ . _ . _ ~. _ _ . . _ . _ _ _ y . . . . _ . . _
. .~. . . _ . . . . _ _ i . . . . .
AGATATGAAGCCTCGTGACAACTCGCTTCCGAGTAATCTATATAAAAGACAGTA
TTTCCTTAACCCAAAAATAAGGGAGAGGGTCCAAAAAGCGCTCGGACAACTGTTGACCGT
_ . . _ . _ . . _ i. . . . _ . _ . _ . + _ . . . . _ . _ _ + . _ _ _ _ . . _ _
~. . _ . . _ _ _ _ _ .~ . . _ _ _
rIAAGGAATTGGGTTTTCATTCCCTCTCCCAGGTTTTTCGCGAGCCTGTTGACAACTGGCA
GATCCGAAGGACTGGCTATACAGTGTTCACAAAATAGCCAAGCTGAAAATAATGTGTAGC
15 ._..+.._.__.__y....____.+________.+..._.....+______._.+.....
CTAGGCTTCCTGACCGATATGTCACAAGTGTTTTATCGGTTCGACTTTTATTACACATCG
P
h
1
CTTTAGCTATGTTCAGTTAGTTTGGCATGCCTATCACATATAAATAGA
_ . . _ ~. . . . . . . _ . . ~. _ . . . . . . _ _ + _ . . . _ . + . . . . . .
. . . ~. . . _ _ _ _
GAAATCGATACAAGTCAA-rCAAACCGTACGGATAGTGTATATTTATCT
GTGCCAGTAGCGACTTTTTTCACACTCGAGATACTCTTACTACTGCTCTCTTGTTGTTTT
. . . . _ . . . . + _ _ . . _ . _ . . + _ . . . . _ _ _ . .~ _ . . . . _ . . .
~. . _ _ . . . . . . ~. . _ . _ . .
CACGGTCATCGCTGAAAAAAGTGTGAGCTCTATGAGAATGATGACGAGAGAACAACAAAA
TATCACTTCTTGTTTCTTCTTGGTAAATAGAATATCAAGCTACAAAAAGCATACAATCAA
_.__._.._.F..._._... r..._...._~.._._____.~......__...H__..._
ATAGTGAAGAACAAAGAAGAACCATTTATCTTATAGTTCGATGTTTTTCGTATGTTAGTT
C
1
~S 1
C'T'ATCAACTATTAACTATAT
_....._..~.....__
GATAGTTGATAATTGATATAGC
3~
CA 02048676 1999-12-22
- 47a -
EXAMPLE 8 . Construction of two vectors for the
expression and secretion of the human
cytokinin gro-(3 in yeast plasmids
pEMR575
and pEMR583 carrying the artificial
promoter of the invention
The examples described above concern proteins
whose localisation is intracellular. Now, it is known
that yeast can secrete recombinant proteins in the
culture medium. The use of the metabolic pathway
leading to secretion of the protein has several
important advantages .
1 - It enables a reasonably pure and correctly matured
product to be recovered from the culture
supernatant.
2 - It enables the protein to benefit from the
modifications associated with the secretion
pathway, such as the formation of disulfide
bridges, glycosylation etc.
There are several proteins or polypeptides
naturally secreted by yeast. In the majority of known
cases, these proteins are synthesized in the form of a
longer precursor whose NH2-terminal sequence is
decisive for entry into the metabolic pathway leading
to secretion. In certain cases, these NHS-terminal
sequences can be used for the secretion of heterologous
proteins. Among these sequences, it is known to use the
pre-pro system of the alpha pheromone. The alpha sex
pheromone of yeast is a peptide of 13 amino acids which
is secreted in the culture medium by S. cerevisiae
yeasts of the Mata sex type.
The alpha factor arrests the cells of the oppo-
- H ~ -
20,~0,~~
site sex type (t~fata) in the G 1 phase and induces the
biochemical and morphological changes necessary fcr
conjugation of the 2 types of cells. Kurjan, J. a.::d
Herskowitz, I. (1982), Cell, 30, 933-943, cloned the
05 structural gene of the alpha factor and deduced from
the sequence of this gene that this actor of 13 a::,ino
acids is synthesized in the for:a of a pre-pro prec~.:rsc=
protein of 165 amino acids. The precursor contains a
hydrophobic amino-terminal sequence of 22 amino acids
followed by a sequence of 61 amino acids containing 3
glycosylation sites, followed finally by 4 copies of
the a factor. These 4 copies are separated by space=
sequences and the mature protein is released fron the
precursor by virtue of the ~ollowing enzymic activi
ties:
1 - an endopeptidase of the cathepsin B type (product
of the KEx2 gene, called yscF) which cleaves Lys-
Arg dipeptides at the carboxy terminal end.
2 - an exopeptidase of the carboxypeptidase type
(product of the KEX1 gene) which cleaves the basic
residues at the carboxy terminal end of the excised peptides.
3 - a dipeptidylaminopeptidase (called A) {prcduct of
the STE13 gene) which removes the Glu-Ala and Asp-
Ala doublets.
A first example of a protein which is secreted
by this system and uses the promoter of the invention
is the human cytokinin gro-f3. The cDNA of this pro-
tein, which is called either gro-a (S. Haskill et al.,
1990, Proc. Natl. Acad. Sci. USA, 87, 7732-7736) or
MIP-2a {P. Tekamp-Olson et al., 1990, J. Exp. tiled.,
172, 911-919) was recently cloned and sequenced, gro-f3
belongs to a family of cytokinins whose members appear
to be involved in modulation of the inflammatory res-
ponse and in activities of the growth factor type.
The Applicant tested the use of the promoter
~s,~s,~s
- 49 -
for the secretion of gro-B by ~. cerevisiae. To do
this, it replaced the natural signal sequence of gro-!3
with the pre-pro sequence of the alpha pheromone and
placed the precursor of gro-!3 behind the promoter of
05 the invention.
1 - Construction of plasmid pEMR530 (cloning vector):
Plasmid pEMR4~3 described above (ExaMple 2 2) -
Figure 7) was digested with the enzymes Xhoz and Sall
and the large fragment, hereafter called fragnent E,
was isolated. This large fragment comprises the
sequences of the URA3 gene, the origin of replication
and the locus STB of the 2~ fragment, the arapicillin
resistance gene AmpR, the origin of replication of
plasmid pBR322 and the UA5 of the promoter of the GAL7
gene of ~. cereyisiae, as well as the terminator of the
PGK gene.
A double-stranded oligonucleotide sequence of
about 400 base pairs, called fragment F, was synthe-
sized in the form of an Xhol-SalI fragment. This
sequence brings the TATA region and the initiation
region of the ADH~ promoter, which is extended by a
synthetic sequence preceding the start of the pre-pro
region of the alpha pheromone. The sequence of this
pre-pro region of the alpha pheromone differs from that
described by Kurjan and Herskowitz, 1982, Cell, 30,
933-943, in the introduction of a HindIII site by the
silent mutation of the TCT codon - corresponding to
serine 81 of the precursor of the pheromone - to AGC.
The whole sequence of the fragment is given below:
35
-50-
x
h
0
I
CGAGATACTCTTACTACTGCTCTCTTGTTGTTTTTATC~CTTCTTGTT~C
_ i ~ _ ~ _ _ _ . ~ . . _ _ ~ _ _ ~ _ i ~ _ ~ _ ~ _ _ _ _ y ~ ~ _ _ _ _ _ _ _
a ~ _ _ _ _ _ _ _ _
TATGAGAATGATGACGAGAGAACAACAAAAATAGTGAAGAACRAAG
05
TTCTTGGTAAATAGAATATCAAGCTACAAAAAGCATACAATCAACTaTCAATCAGATCTA
y.......__~___~__~~~a~~_~~~~~~a~________y_________~_________
AAGAACCATTTATCTTATAGTTCGATGTTTTTCGTATGTTAGTTGr7TAt;TTAGTCTAGr=.T
ATATTAATAAAAAATGAGATTTCCTTCFiATTTTTACTGCAGTTTTATTCGCA~;CATCCTC
y____~____a__~~~_...y.........~_~_~__~_~~~~_______._________
TATAATTATTTTTTACTCTAAAGGAAGTTAAAAATGACGTCAAAATr'iAI,~CGTCGTAGGr'aG
CGCATTAGCTGCTCCAGTCAACACTACAACArAAGATGAAACGGCIaCAC,ATTCCGGCTGG~
y _ _ _ ~ ~ _ _ _ _ ~ _ _ _ ~ _ ~ _ _ _ ~ _ ~ _ ~ ~ _ _ . . y . . . _ _ _ _ ~
~ a _ ~ _ _ _ _ _ _ _ a ~ _ _ _ _ _ _ _ _
GCGTAATCGACGAGGTCAGTTGTGATGTTGTCTTCTACTTTGCCGTGTTTAAGGCCGr~C~
AGCTGTCATCGGTTACTTAGATTTAGAAGGGGATTTCGATGTTGCTGTTTTGCCaTTTTC
y.....____~__~~_____y~_____~~~~_~_______~._____.__~_________
TCGACAGTAGCCAATGAATCTAAATCTTCCCCTAAAGCTACAACGACAAAACGGTAAaAG
CAACAGCACAAATAACGGGTTATTGTTTATAAATACTACTATTGCCAGCATTGCTGCTAA
GTTGTCGTGTTTATTGCCCAATAACAAATATTTATGATGATAAC~GTCGTAACGaCGaTT
H
i
n
d S
I a
I 1
I I
AGAAGAAGGGGTAAGCTTGCATGCCTGCAGG ~
a_._______i_________.._______~-i__ ___~
TCTTCTTCCCCAT TCGAACGTACGGaCGTCCpGC~
Fragments E and F were ligated to give plasmid
pEMR530 shown in Figure 10, in which the symbols have
the same meanings as in Figure 7, the novel Xhol-Sall
_fragment (fragment F) introduced being represented by:
This plasmid comprises the artificial promoter
of the invention, the sequence of which was given in
51 ,~'~'~6"7S
Example 2.
2 - Construction of plasmid pEMR583 (expression plasmid
for the human Cytokinin gro-f~)
Plasmid pEMR530 was completely digested with
05 the enzymes Nhel and HindIII. The small NheI-HindIIT_
fragment, containing the artificial promoter and the
pre-pro region of the a pheromone, as well as the URA3
gene, was purified (hereafter called fragment G).
Plasmid pEMR473 was completely digested with
the restriction enzymes NheI and BainI~iI. The large
fragment (hereafter called fragment H), comprising the
origin of replication and the locus STB op the 2u frag
ment, the LEU2d gene, the ampicillin resistance gene,
the origin of pBR322 and the terminator of the PGK
gene, was purified.
The cDNA of gro-13 was cloned and sequenced
according to the method described by Tekamp-Olson et
al., op. cit. The sequence of the cDNA of gro-B
(described in detail in said reference - in particular
Figure 2) has an EcoRI site (sequence: 5'-GAATTC) which
covers the ATT codon (isoleucine in position +18 of the
mature sequence of gro-f~, Tekamp-Olson et al., op.
cit.) and overlaps the two flanking codons GGA and CAC.
The cloned cDNA of gro-I3 terminates at the 3~ end in a
polyA tail flanked by a BamHI restriction site.
The EooRI-BamHI fragment, comprising the major
part of the coding sequence of gro-13, followed by the
3' sequence corresponding to the non-translated end of
the mRNA flanked by the polyadenylated tail, was iso-
lated by the customary techniques described in Maniatis
et al. (op. cit.). The fragment is hereafter called
fragment I.
A synthetic HindIII-IrcoRI fragment, containing
the end og the pre-pro region of the a pheromone (cor
responding to the residues Ser Leu Asp Lys Arg) and the
- 52 -
start of the sequence coding for the mature protein of
gro-f~, was also prepared. The sequence of this frag-
ment, called fragment J, is given below. It will be
noted that the 'sequence Corresponding to the start or
05 the cDNA op gro-13 has been modified relative to the
sequence of the cDNA described by Tekamp-olson et al.,
op. cit., so that the codons used are among those most
frequently used by S. cerPvisiae (q. v. Sharp et al.,
1.986, Nucleic Acids Research, vol. 14, 13, 5125-5143).
Hind III
AGCTTGGATAAAAGAGCGCCTTTGGCTACTGAATTGAGATGTCAATGTTTGCAAACCTTGCAAGG
1 5 ACCTATTTTCTCGCGGAAACCGATGACTTAACTCTACAGTTACAAACGTTTGGAACGTTCCTTAA
EcoRI
Fragments G, H, I and J were ligated to give
plasmid pEMR583 shown in Figure 11, in which the
2p symbols have the same meanings as in Figures 7 and lo,
fragment E being represented by ~ and fragment F by
The nucleotide arid peptide sequences of the
start of the mature protein of gro-f3, and of the end of
25 the pre-pro region v~ the a pheromone, are as follows:
35
- 53 -
G T A.A G C.T T G.G A T.A A A.A G A.~G C G.C C T.T T G.
Val. Ser. Leu. Asp. Lys. Arg. (Ala. Pro. Leu.
05 End of the pre-pro region of the Start of the
a pheromone (mature protein
Cleavage site of the
endopeptidase of cathep-
lo sin B type: yscF (product
of the KEX2 gene)
EXAMPLE 9: Secretion of the cvtokinin aro-l3 by lea=
1 Transformation of the EMY751 yeast strain by plasmid
15 pEMR583 and expression of gro-I3 by the transformed
strain. The EMY761 strain (Mata, leu2, ura3, hi~3)
described in Example 3 was transformed by plasmid
pEMR583 far the prototrophy of leucine by the tech
nique already described 1n Example 3). Two trans
2o formed strains, hereafter called EMY767. pEMR583 (1)
and EMY761 p~NIR583 (2), were retained.
2 Expression, in an Erlenmeyer flask, of the cDNA og
gro-B by the EMY761 pEMR583 (1) and EMY761 pEMR583
(2) strains. Detection of the protein in the culture
25 medium on pol.yacrylamide gel/sodium dodecylsulfate
SDS ) according to the protocol described by LAEMtdLz
(U. K. LAEMMLI, Nature, 22~ [1970] 680-685).
culture
a/ A colony of each of the EMY761 pEMR583 (1) arid
30 EMY761 pEMR583 (2) strains was cultured in 50 ml
of uracil-free liquid medium. This medium con-
tains the following per liter:
6.7 g of Yeast Nitrogen base without amino acids
(from DIFCO)
35 5.0 g of casein hydrolyzate (Casamino acids from
.M. - 5 4
DIFCO)
g of glucose.
After one night at 30'C, with agitation, the two
cultures were centrifuged for 10 min at 7000 rpr~.
05 The residues were taken up in 10 ml of sterile
distilled water and centrifuged again for 10 min
at 7000 rpm. Expression of gro-D was induced by
taking up the cells in 50 ml of ethanol-glycerol-
galactose YNB medium. The ethanol-glycerol-
10 galactose YNB medium contains the following per
liter:
6.7 g of Yeast Nitrogen base without Amino acids
(from DIFCO)
5.0 g of casein hydrolyzate (casamlno acids fron
DIFCO)
30 g of glycerol
30 g of galactose
10 ml of ethanol
The cultures were incubated again at 30'C for 24 h,
with agitation.
b/ Control strain:
The non-transformed EMY761 strain, i.e. the EMY761
strain without plasmid, was cultivated as above.
It was subjected on the one hand to preculture in
50 ml of uracil-free liquid medium to which uracil
had been added (20 ug/ml), and on the other hand
to induction in 50 m1 of ethanol-glycerol-
galactose YNB medium to which uracil had been
added (20 ug/ml).
Preparation of the samples:
The cells cultivated in 1 a/ and 1 b/ were centri
fuged for 20 min at 10,000 rpm and the supernatant
was collected. 5 ml of 50$ trichloroaCetic acid
containing 2 mg/ml of deoxycholate were added to
10 ml of supernatant.
- 55 -
The mixture was cooled at *4°C for 3o min and then
centrifuged for 30 min at 10,000 rpm. The residue
was taken up in about 1 ml of cold acetone (T4'C)
and centrifuged again for 30 min at 10,000 rpr~.
05 After having been dried, the residue is taken up
in about 20 ul of a so-called loading buffer con-
sisting of Tris-HC1 0.125 M pH 6.8, SDS 4%, brono-
phenol blue 0.002%, glycerol 20%, B-mercapto-
ethanol 10% (according to the protocol described
by LAEMMLI [1970 0 . The residue is solubilized by
boiling for 15 min and then neutralized by the
addition of 10 N sodium hydroxide solution until
the bromophenol blue turns blue.
The samples are deposited on polyacrylamide gel/
SDS:
1/ Size marker
2/ Non-induced non-transformed
EMY761 : 20 ul deposited
3/ Non-induced EMY761 pEMR583
( 1 ) . 20 ~tldeposited
4/ EMY761 pEMR583 (1) induced
for 24 h . 15 ~C1deposited
5/ EtdY~61 pEMR583 induced
(1)
for 24 h : 5 ul deposited
6/ Size marker
7/ EMY761 pEMR583 (2) induced
for 24 h : 5 ~1 deposited
8/ EMY761 pEMR583 (2) induced
for 24 h . 15 ~Cldeposited
9/ Non-induced EMY761 pEMR583
(2) . 20 ~1
10/ Induced non-transformed
EMY761 : 20 ~1 deposited
- 56 -
After electrophoresis, the proteins are stained
with Coomassie blue.
$~SULTS:
Analysis of the geI obtained shows that a
05 supernumerary protein with an apparent molecular weigrt
of about 8 kDa is produced by the EldY761 pEMR583 (1)
strain (lanes 4 and 5) and EMY761 pEMR583 (?.) strain
(lanes 7 and 8) and is not produced in the culture
supernatants of the non-transformed EMY761 strain
(lanes 2 and 10). It is also apparent that the syn-
thesis of this supernumerary protein is associated with
induction of the promoter by growth on ethanol-
glycerol-galactose (bands absent in lanes 3 and 9).
Analysis of the amino-terminal sequence of this
protein purified by HPLC made it possible to verify
that it was the mature gro-B protein described by
Tekamp-Olson et al., op. cit.
It is therefore apparent that the cytokinin
gro-f3 can be secreted under the control of the promoter
of the invention.
The EMY761 pEMR583 (1) strain has been depo-
sited at the Institut Pasteur under the number I - 1021.
EXAMPLE 10: Construction of a vector fob the expression
and secretion of IL-8 '~ veast~ plasmid
pEMR611 carry~~ the artificial pror.!o er og
the invention
A second example of a secreted protein whose
expression can be regulated by the promoter of the in-
vention is the human cytokinin IL-8. This cytokinin of
about 8000 Da, produced by monocytes, has been des-
cribed by several teams: Yoshimura et aI. (1987), J.
Immunol., 139, 788-793, ShrtSder et al. (1987), J.
Immunol., 139, 3474-3483, and Walz et al. (1987), ~io-
chem-Hiophys. Res. Gommun., 149, 755-761. IL-8 acts as
a chemical attractant of neutrophils. IL-8 has remar-
- 57 - 2fl'~~'~~
kable structural similarities with l3-thromboglobin (Van
Damme et al. (1989), Eur. J. Biochem. 181, 337-344).
The cytokinin IL-8 exists in several forms which differ
from one another in their NHS-terminal end. The major
05 form is composed of 72 amino acids but 5 other minor
forms are also produced, 3 of which have a truncated
NHa-terminal end compared with the major form, and 2 of
which have respective extensions of 5 and 1 amino acids
compared with this major form.
The cDNA of IL-8 was cloned and sequenced
according to the method described by ?datsushima et al.,
1988, J. Exp. Med. 167, 1883-1993. The sequence of
this cDNA contains a single HindIII site (5'-AAGCTT)
which covers the 42nd and 43rd codons of the mature
part (form 72 aa: q.v. Matsushima et al., op. cit.).
Furthermore, the clone of the cDNA of~IL-8 used
in this cloning has a single BamHI site directly
flanking the end (3') of the polyA tail. The BamHI-
HindIII fragment, carrying the 3' end of the cDNA of
IL-8, was purified.
A cloning vector was prepared in the following
manner:
Plasmid pEMR583, described in Example 8, was
digested with HindIII and BamHI.
This double digestion releases 5 fragments: the
heaviest fragment, IiiridIII-BamHI, corresponds to the
cloning vector (about 7760 base pairs). The other 4,
HindIII-HindIII (with sizes of 169 base pairs, 92 base
pairs, 529 base pairs and 187 base pairs), correspond
to the sequence of the cDNA of gro-J3. The HindIII site
of the cloning vector is located slightly upstream from
the insertion site at the end of the pro sequence of
the alpha pheromone (and covers the sequence of the
codon - serine 81 - of the precursor). The BamHI site
is located upstream from the terminator of the PGK.
-5a- 2~,
The DNA of the HindTII-BamHI fragment was
purified. The HindIII-BamHI fragment, containing the
3' part of the sequence of the cDNA, was ligated with a
synthetic HindIII-HindIIZ DNA fragment. The sequence
05 of this fragment, which is given below, is intended for
reconstituting the sequence of the major mature form of
IL-8 (72 amino acids), preceded by the sequence of the
Cleavage site (Ser-Leu-Asp-Lys-Arg). The novel
HindIII-BamHI fragment was ligated with the HindIII-
l0 BamHI fragment corresponding to the cloning vector.
The sequence of the synthetic HindIII-HindIII fragment
is as follows:
5'-AGCTTGGaTAAAAG~TCTGCT~GG~ATTG~GATGTCAATGT~TC~G~.CTTACTC:~GCC~TTCC
1 5 ACCTAT:'TTCTAG~CG~TTCCTTAACTCTACAGTTAC~T~GT:C:G~TG~G~TTCCGT~1GG
dCCCrI~G:T. CnTC~CGc'lciTTGaG~GTTc'~TCG~ATCTGGTCC~ICaCTGTGCTr~CaCTGr~c'~r~TTA~
T CG G : ii T C~rIGTr'~G TTC~, T'"lAAC T CTCc~ATc'~G CTTc'1G?~C
Cc~GGTGTGr~Cc~CGc~T i GTGnC i ~TdAT~1
20 CGTTA
GC~TTCGa- 5'
The plasmid obtained in this way, carrying the
cDNA of IL-8 preceded by the pre-pro sequence of the a
25 pheromone and the promoter of the invention, whose
sequence was specified in Example 2, is called pEt~R611.
~,~,I~PLE 11 : Tr ansformation of the EMY761 yeast strain
by plasm ~,gEMR611, and secretio of IL-8
The EMY76I strain (Mata, ura3, leu2, his3),
30 described in Example 3, was transformed into the (leu-~)
strain by the DNA of plasmid pEMR611 according to the
technique already described. Of the (leu~) colonies
obtained, one was removed at random and cultivated in
order to study the secretion of rL-e. This strain is
35 hereafter called EMY761 pEMR611.
89
The protocol for analysis of the proteins sec-
reted by the yeast is that described in Example 9. The
proteins secreted by EMY761 pEMR611 were compared with
those secreted by EMY761. This revealed a superriu-
05 merary mayor band corresponding to a protein of 8000 Da
secreted by the EMY761 pEMR611 cells induced by galac-
tose. This protein is specifically recognized by
rabbit antibodies directed against human IL-8 (supplied
by Endogen: Anti human IL-8 polyvalent P801) according
to the results of analysis by Western blot - a method
which is well known to those skilled in the art and
whose protocol is given in detail in Example 4.
Expression plasmid pEMR611 therefore per:aits a
high level of expression of IL-8 in transformed yer~sts.
This expression is under the control of the crt~~icial
promoter of the invention. '
The EMY761 pEMR611 strain has been deposited in
the Institut Pasteur collection under the number I-1023.
EXP.MPLE, 12: Cons ruction of yector for the expr~on
and secretion of iruain: pl, ~ml4c~pE2~T?5~c
-~; ncr the ar~~ fic~ aI p r~motcr F the
invention
Naturally produced by the leech, rirudin
is a very specific and very effective inhibitor of
thrombin. A number of variants have been identified
and designated by HV1, HVz and HV~ (Dodt J. et al.
(1986), FEBS Lett. ~, 373, 377). Some of these
natural variants and other analogs have subsequently
been prepared by genetic engineering in a variety of
host cells. The present Example concerns the variant
rHV~-Lys47 described in the patent publication EP-A-
0273800. More particularly, the expressed sequence
codes for a precursor of this variant rHVs-Lys47, which
contains a signal sequence: Met - Arg - Phe - Ser -
Thr - Thr - Val - Ala - Thr - Ala - Ala - Tyr - Ala -
- 60 -
20~~'~~
Leu - Phe - Phe - Thr - Ala - Ser - Gln - val - Ser -
Ala, directly preceding the start of the sequence of
mature hirudin. .
The construction and the structure of this pre
05 cursor are described in the patent publication FR
2646437. The expression of this precursor permits the
release of the variant rHV2-Lys47 in the culture super
natant of the transformed cells.
Plasmid pTG3867, whose construction and
to description are given in detail in the patent
application FR 2 646 437, is a secretion vector for
hirudin. In this construction, the hirudin is
synthesized in the form of a precursor containing a
signal sequence. The hirudin is placed behind the
15 promoter of the MFal gene, which is a constitutive
promoter in yeast strains or the a conjugation type.
Construction of p~a5mid pEMR~47
Plasmid pEt~IR547 is derived fzom plasmid pt;MR515
(cf. Example 2) by deletion of the small sequence ori
20 ginating from plasmid 2~ and located between the end of
the LEU2d gene and the EcoRI site bordering the se-
quence of plasmid pBR322. Plasmid pEMR515 was digested
partially with BsptdI and totally with EcoRI. The large
BspMI-EcoRI fragment, corresponding to plasmid pEtrLR515
25 from which the 3' part of LEU2 and the small adjoining
sequence of the 2u fragment have been deleted, was
ligated with a synthetic Bspbtl-EcoRI° sequence intended
for reconstituting the 3' region Of the LEU2d gene.
This synthetic sequence is as follows:
35
- sl - 2U~6"~6
EcoRI° AATTGCCCGGGACGTCTTRTGTACAAATATCATAAAAAAAGAGAATCTTTTT
+_________+_________+_________+_________+_________
~CGGGCCCTGCRGAATACATGTTTATRGTATTTTTTTCTCTTAGaAaaA
AAGCAAGGATTTTCTTRACTTCTTCGGCGACRGCATCRCCGACTTCGGTGGTACTGTTGG
+_________+_________+_________+_________+_________+_________
05 TTCGTTCCTAAAAGAATTGAAGAAGCCGCTGTCGTAGTGGCTGAAGCCACCATGr'1CAACC
B
s
P
M
I
AACCACCTRAATCACCAGTTCTGATACCTGCRTCC
+_________+_________+_________.________
TTGGTGGATTTAGTGGTCAAGACTATGGACGTAGGTTTT
The reconstituted plasmid is called pEt~iR547.
Co struction of g],.asr~id pEMR5~8
Plasmid pEMR568 is derived from pEMR547 in the
following manner:
The DNA of plasmid pEMR547 was digested with
Mlul and Nhel. This double digestion makes it possible
to linearize plasmid pEMR547 (6.6 kb), the 2 sites MluI
and NheI being situated within a few base pairs of one
another. A double-stranded synthetic oligonucleotide
of the sequence
2 5 CTAGCGAGCTCAAGCTTA
GCTCGAGTTCGAATGCGC
was inserted between these 2 sites.
The resulting plasmid is called pEMR568.
Construe ion of ,plasm'~pEMR576, an a nressi~Q, asmid
for hirudin
The sequence of the variant rI-iv2-Lys47 of
hirudin was obtained from plasmid pTG3867, whose con-
struction and description are given in detail in the
patent application FR 2 646 437. In this construction,
62
the hirudin is synthesized in the form of a precursor
composed of a signal peptide of 23 amino acids (inclu-
ding the methionine corresponding to the initiation
codon). The messenger RNA coding for the precursor is
05 transcribed with the aid of the MFal promoter, which is
a constitutive promoter in yeast strains of the alpha
conjugation type.
AccI-SalI double digestion of plasmid pTG3867
releases several fragments, the shortest of which,
numbering about 200 base pairs, is readily puri~iable
on 2~ agarose gel. The AccI site which borders this
fragment is located a few base pairs from the star of
the mature sequence, according to the sequence
AccI
ATTACGITA TACAGAC...
T.yATGC ATlATGTCTG
~0 Mature
sequence
The SalI site is located downstream from the
stop codon of the coding sequence of hirudin.
The AccI-SalI fragment of 200 base pairs
carries the information for the greater part of the
mature sequence of hirudin. The complementary infor-
mation (signal sequence and start of the mature
sequence) is provided by a synthetic sequence o~ about
90 nucleotides, which is specified below:
- 63 - ~~
CGATATACACAATGCGTTTCTCTACTACAGTCGCTACTGCAGCTACTGCGCTATTTTTCACAGCCTCC
TATATGTGTTACGCAAAGAGATGATGTCAGCGATGACGTCGATGACGCGnTAAAAAGTGTCCGr~CG
CCAAGTTTCAGCTATTACGT
0 5 GGTTCAAAGTCGATAATGCATA
Vector pEMR468 was linearized with Sall and
partially digested with ClaZ. The Clal-SalI fragment
of about 5.6 kb, corresponding to this vector from
which the sequence of urate oxidase has been deleted,
was ligated with the AccI-Sall fragment of about 200
base pairs, which carries the information for the
sequence of mature hirudin, and with the small syn-
thetic ClaI-ACCI sequence intended for reconstituting
the sequence of the precursor of hirudin. The resul-
ting plasmid is called pEMR576.
EXAMPLE 13: Cor'rPYion of h'_ruc~in
1) Transformation of the EMY761 strain by
plasmid pEMR576
2p The EMY761 strain (Mats, ura3, his3, leu2) was
transformed into the (leu+) strain by plasmid pEMR576
according to the technique already described.
A transformed strain, hereafter called EMY761
pEMR576, was isolated.
2) Expression of hirudin
As a negative control, a (leu~) strain derived
from EMY761, hereafter called EMY761 (leu~), was con-
structed. The technique used is described in detail in
the patent application FR 2 646 437. The EMY761
pEMR576 and EMY761 (leu+) strains were Cultivated in
parallel in the manner described below:
The precultures take place in medium of the
following composition: Yeast Nitrogen base (Difco)
0.7~, histidine 50 ~1g/ml and uracil 50 ~g/ml. After
24 h, the cultuxes are inoculated with l0a cells in
- 64 -
medium having the following composition: Yeast nitrogen
Base (Difco) 0.7$, ethanol 1~, casamino acids 0.5~,
uracil 100 ug/ml_, glycerol 3~ and galactose 1% . After
culture for 72 h in the latter medium, the supernatant
05 is separated from the cells by filtration on 0.2 u.
The inhibitory activity of the supernatant on t!~.ro.::bin
is measured by using the colorimetric test described in
FR 2 646 437 (proteolytic activity of thrombin on a
synthetic substrate: chromozyme TH marketed by
Boehringer Mannheim).
The Table below shows the results of the assays
in ~g of hirudin per ml of supernatant, at an optical
density of 1, i.e. 0.3 x 10' cells/ml.
1~
Strain I ug/ml
EMY761 (leu~) non-detectable
EMY761 pF..MR576 0.5
It is therefore apparent that hirudin can be
secreted under the control of tha promoter of the
invention.
The EMY761 pEMR576 strain has been deposited in
y5 the CNC2-i under the number I - 1022.
35