Note: Descriptions are shown in the official language in which they were submitted.
CAS~ 100-7683
:;
INDOLONAP~T~IRIDIN~S
.
The present invention relates ~o new 7a,8,9,10,11,11a-hexahydro
and 4,5,7a,8,9,10,11,11a-octahydro-7H-indololl,7-bcll2,6l-
naphthyridines.
These compounds, hereinafter referred to as new compounds, are
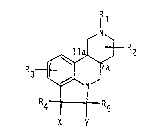
compounds of formula I
: 1 1
R2
R3 S~ I
R~ R5
X Y
: ~ wherein
~:; R1 is hydrogen, alkyl, alkylcarbonylalkyl, arylcarbonylalkyl,
aralkyl or carbamoylalkyl optionally mono- or disubstituted
by alkyl or aryl,
. ~
~:
; ~ "
: : :
.
- 2 - 100-7683
R2 is as defined for R1 and may additionally be trifluoromethyl,
allcoxy or alkylthio,
R3, R4 and Rs independently are hydrogen, halogen, alkyl,
alkoxy, alkylthio or trifluoromethyl and
X and Y are each hydrogen or form together a single bond~
in free base or acid addition salt form.
Depending on the substituents the new compounds may present two
(in position 7a and lla) or more asymmetrical carbon atoms. The
invention includes all resulting stereomers as well as their
mixtures, e.g. the racemic mixtures of the enantiomers.
In position 7a and lla the configuration of the new compounds may
be cis or trans.
Any alkyl, alkoxy or alkylthio group as well as any alkyl moiety
in an alkylcarbonylalkyl, arylcarbonylalkyl, aralkyl or
optionally substituted carbamoylalkyl group of a new compound
contains 1 to 4 carbon atoms.
An aryl moiety in an arylcarbonylakyl, aralkyl or arylcarbamoyl-
alkyl group is a five or six membered, saturated, unsaturated or
arom~tic carbocyclic ring optionally mono-9 di- or trisubstituted
by above-deiined alkyl, alkoxy or alkylthio, wherein one or two
carbon atoms may be replaced by nitrogen.
Halogen is fluorine, chlorine, bromine or iodine.
Insofar as above-defined alkyl, alkoxy or alkylthio groups are
present in the new compounds, these preferably have one or two
carbon atoms and especially signify methyl, methoxy or methyl-
thio.
.:
. ~ :
, , . j~
" '. ,
;
ri. !
- 3 - 100-76~3
An above-defined halogen is preferably fluorine or chlorine.
Rl is preferably alkyl, particularly methyl. R2, R3, R4 and Rs
are preferably hydrogen. Preferably X and Y each are hydrogen.
The invention includes for example a group of compounds of
formula I, wherein R1 is (C1_4)alkyl and R2, R3, R4, Rs? X and Y
are each hydrogen, in free base or acid addition salt form.
The invention also includes a group of compounds of formula I,
wherein R1 is (C1_4)alkyl, R2, R3, R4 and R5 are each hydrogen
and X and Y together form a single bond, in free base form or
acid addition salt form.
The preferred compound is the (~)-cis-4,5,7a,8,9,10,11,11a-
octahydro-7H-10-methyl-indolo[1,7-bcl[2,6lnaphthyridine in free
base or acid addition salt form.
The present invention also provldes a process for the production
of a compound of formula I or an acid addition salt thereof,
which includes the step of
a) for the production of a compound of formula Ia
~ N ~
R3 ~ Ia
R4 R5
,
~ ' ' ," :
'
: ~ ,.
~ 100~7683
wherein R1, R2, R3, R4 and R5 are as defined above, replacing
under reduction of the 7-oxo group the 10-ethoxycarbonyl
group of a compound of formula II
3 ~ R2 Il
R4 R5
wherein R2, R3, R4 and Rs are as defined above, by the group
R1, or
b) for the production of a compound of formula Ib
: R
~ R~ ~ R2 Ib
: R4 R5
~ wherein Rl, R2, R3, R4 and R5 are as defined above, oxidating
:~ ~ a compound of foFmula Ia,
and recovering the thus obtained compound of formula I in free
base or acid addition salt form.
:
' ` ' ! :
,
: ` 1 " ` ~ .
~ ' :
' i
- 5 - 100-7683
The reaction according to process a) may take place by known
methods.
To introduce a methyl group, the ethoxycarbonyl group can be
reduced in known manner, e.g. using aluminium hydride or complex
aluminium hydrides such as lithium aluminium hydride.
To introduce a higher alkyl or a subseituted allcyl group, the
ethoxycarbonyl group can be firstly cleaved and the amine
obtained can then be alkylated.
Oxidation of the compounds of ~ormula Ia according to process b)
may be carried out by known methods, for example using manganese
oxide. 02/cobalt salts in methanol, palladium dichloride in
triethylamine or benzene-selenic anhydride may also be used.
Working up of the obtained reaction mixtures and purification of
the compounds of formula I thus obtained may take place in
accordance with known methods.
Acid addition salts may be produced in known manner from the free
bases, and vice versa.
The process according to the invention may be effected using
starting products in the form of individual, optically active
isomers or mixtures thereof, especially the racemates thereof,
thus resultin~ in the corresponding end products.
The racemates can be split into individual, optically active
components, whereby known methods are employed, e.g. via
transient acid addition salt formation with optically active
acids, e.g. (~ resp. (-)]-di-O,O'-p-toluoyl-D-(-)-[resp.
L-(+)]-tartaric acid, and fractional crystallisation of the
diastereoisomeric acid addition salts.
,
,,
, ~ , , : , .. ,, ., .~,
:~ 1
~ 6 - 100-7683
The starting compounds of formula II may be produced by
commencing with isonicotinic acid chloride (known from Beilstein
4, vol. 22/1, page 526) and indoline! (known from Beilstein 4,
vol. 20/4, page 2896) or with deriatives thereof which can be
produced according to known methods, in accordance with the
following reaction scheme, e.g. as described in example 1 under
a) to e):
, :
' ~ '
,
7 100-7~3
, O
N ~ ~ R2
R4 R3 ~ R4 Vl
CH3 CH3I
R2 ~R2
R ~!, 4 R3 ~ 4 V
. ClC02Et
0~0\~
., I
~R2
~ 1. h ~
N ~ 0 2. Diaster.-
: ~ ~ R5 trennung II
R3__t_ 1 ~ . _
~/-- R4 [IT
: ', . ' :,
.. ' ,~ .
.
, j,, , ,f,
- 8 - 100-76~3
The 7a,8,9,10,11,11a-hexahydro- and 4,5,7a,8,9,10,11,11a-octa-
hydro-7H-indolol1,7-bc][2,6]naphthyridines and their physio-
logically acceptable acid addition salts, hereinafter referred to
as compounds according to the invention, exhibit interesting
pharmacological activities in animal tests and are, therefore,
useful as pharmaceuticals.
In particular, the compounds according to the invention have an
antagonist effect at the central 5HT-lC receptors.
The compounds according to the invention have a potent binding
affinity to central 5HT-lC receptors as e.g. measured according
to the method disclosed by D. Hoyer et al., Eur. J. Pharm., 118,
13 - 23 (198S). The compound of Examplc 5 has a pKD value of 7.8.
The compounds according to the invention antagonise the hypo-
locomotion induced in rats by administration of m-chlorophenyl-
piperazine (mCPP) according to the method disclosed by
G. A. Kennett and G. Curzon, Br. J. Pharmacol., 94, 137 - 147
(1988). In this test the compounds according to the invention
counteract the mCPP reduced locomotion after administration at
dosages of from about 0.5 to 30 mg/kg p.o.
The compounds according to the invention are therefore useful for
the prophylactic treatment of migraine or for the treatment of
disorders e.g. anxiety, depression, schizophrenia, autism,
obsessive compulsive disorders, panic attacks, priapism, bulimia
and situations of increased intracranial pressure.
An indicated daily dosage is in the range ~rom about 0.5 to about
300 mg of a compound according to the invention, together with
solid or liquid carriers or diluents.
.
~ .
.
"; ,, ,,, f,3
- 9 - 100-7683
In accordance with the foregoing, the present invention also
provides a compound according to the invention, for use as a
pharmaceutical, e.g. for the prophylactic treatment of migraine
or for the treatment of disorders e.g. anxiety, depression,
schizophrenia, autism, obsessive compulsive disorders, panic
attacks, priapism, bulimia and situations of increased intra-
cranial pressure.
The present invention furthermore provides a pharmaceutical
composition comprising a compound according to the invention in
association with at least one pharmaceutical carrier or diluent.
Such compositlons may be manufactured in conventional manner.
In the following examples, all temperatures are uncorrected and
are in degrees Centigrade.
'~:
,~
. .
;- , .
- , : .
,
~ .
~ ! ~ .; , .,
- 10 - 100-7683
EXAMPLE 1: cis-4,597a,8,9,10,11,11a-octahydro--7~-10-methyl-
indolo[1,7-bc]~,6lnaphthyridine
2.03 g (53.4 mmols) of lithium aluminium hydride (aluminium
hydride can also be used) are placed at 0 in absolute tetra-
hydrofuran. A solution of 1.61 g (5.36 mmols) of cis-4,5,7a,8,9,-
lO,ll,lla-octahydro-7-oxo-7H-indolo[1,7-bcl~2,6]naphthyridine-10-
carbamic acid ethyl ester in absolute tetrahydrofuran is
subsequently added in drops. The reaction mixture is refluxed for
three hours and subsequently left to stand for 12 hours at room
temperature. Afterwards, the reaction solution is cooled to -20
and water is added in drops. The product is extracted with
toluene. The toluene phase is dried and concentrated by
evaporation. cis-4,5,7a,8,9,10,11,11a-octahydro-7H-10-methyl-
indololl,7-bc]l2,6]naphthyridine is obtained as a yellow oil. The
crude product is dissolved in acetone and mixed with the
equivalent quantity of malonic acid. After crystallisation from
methanol/acetone, the cis-4,5,7a,8,9,10,11,11a-octahydro-7H-10-
methyl-indolo[1,7-bc][2,61naphthyridine malonate is obtained with
the melting point 158 - 159.
The starting material may be prod~ced as follows:
a) 2,3-dihydro-1-(4-pyridinylcarbonyl)-1~-indole
327 g (2.05 mols) of isonicotinic acid chloride hydrochloride
are placed in methylene chloride (the hydrochloride is
prepared by reacting isonicotinic acid hydrochloride in
thionyl chloride according to a conventional process). To
the solution are added in drops at 20 532 ml (3.69 mols) of
triethylamine, followed by 206 ml (1.84 mols) of indoline
dissolved in methylene chloride. The mixture is stirred for
16 hours at 20. Afterwards, the preparation is washed with
water, the organic phase is dried and concentrated by
:
.
.
,
-:
~ 100-7683
evaporation. 2,3-dihydro-1-(4-pyridinylcarbonyl)-lH-indole is
obtained with a melting point of 127 - 128 (crystallisation
from methylene chloride/ethanol/hexane).
b) 4-(2,3-dihydro-1~-indol-1-yl-carbonyl~-1-methyl~yridinium-
iodide
224 g (l mol) of 2,3-dihydro-1-(4-pyridinylcarbonyl)-lH-
indole are suspended in acetone. The suspension is heated to
reflux, and 137.6 ml (2.21 mols) of methyl iodide are added
in drops. After 45 minutes, a further 68.8 ml (1.1 mols) of
methyl iodide are added. The reaction mixture is refluxed for
a further 2 hours. Afterwards, the crystallised product is
filtered off, washed with ether and dried. 4-(2,3-dihydro-lH-
indol-1-yl-carbonyl)-1-methylpyridinium-iodide is obtained in
the form of yellow crystals. It has a melting point of 242 -
243 (crystallisation acetone/ether).
c) 2,3-dihydro-1-(1,2,3,6-tetrahydro-1-methyl-4-pyridinyl-
carbonyl~ -indole
308 g (0.84 mols~ of 4-S2,3 dihydro-lH-indol-1-yl-carbonyl)-
1-methylpyridinium-iodide are suspended in ethanol. The
solution is cooled to +10. A mixture of 95.9 g (2.52 mols)
of sodium borohydride in 960 ml of water and 96 ml of 30%
caustic soda is added in drops whilst stirring intensely.
Stirring subsequently continues for 2 hours at room
temperature. Afterwards, the reaction mixture is concentrated
on a rotary evaporator. The residue is mixed ~ith ice water
and methylene chloride. The organic phase is separated and
the product is extracted therefrom with 2 n hydrochloric
acid. The aqueous solution containing hydrochloric acid is
rendered alkaline with 30% caustic soda, and the product is
extracted with methylene chloride. The organic phase is dried
:
.;, j ~:
,
. . .
. , : ..
.
~ '' ' ~ ' ,
5l~
- ~2 - 100--7683
and concentrated by evaporation. 2,3-dihydro-1-(1,2,3,6-
~etrahydro--l-methyl~4-pyridinylcarbonyl)-lH-indole is
obtained with a melting point of 70 72 ~crystallisation
from ether/petroleum ether).
d) 4-(2,3-dihydro-l~-indol-1-yl-carbonyl)-1,2,3,6-tetrahydro-1-
pyridine-carbamic acid ethyl es~:er
145.4 g (0.6 mols) of 2,3-dihydro-1-(1,2,3,6-tetrahydro-1-
methyl-4-pyridinylcarbonyl)-lH~indole are placed in toluene.
156.6 ml of N-ethyldlisopropylamine are added and heated to
80~. At the same temperature, a solution of 196.8 ml
(2.1 mols) of chloroformic acid ethyl ester in toluene is
added to the reaction mixture in drops. The mixture is
stirred for 2 hours at 80 and subsequently cooled to 0. The
preparation is mixed with water-ice and washed with 2 n
hydrochloric acid. The toluene phase is separated, dried and
concentrated by evaporation. 4-(2,3-dihydro-lH-indol-l-yl-
carbonyl)-1,2,3,6-tetrahydro-1-pyridinecarbamic acid ethyl
ester is obtained wieh a melting point of 112 - 113
(crystallisation from methyl-tert.butylether/ether).
e) cis-4j5,7a,8,9,10,11,11a-octahydro-7-oxo-7~ dolo[1,7-bc]-
[2,6]naphthyridine-10-carba~ic acid ethyl ester
2.0 g (6.7 mmols) of 4-(2,3-dihydro-lH-indol-l-yl-carbonyl)-
1,2,3,6-tetrahydro-1-pyridinecarbamic acid ethyl ester in
toluene are irradiated under argon, whilst stirring, with a
400 watt mercury high-pressure lamp. The reaction vessel is
cooled under running waterO After an interval of 12 hours,
the dipping lamp is cleaned and the reaction solution is
treated with 3 mol X active charcoal and filtered over Hyflo.
After a total of 50 hours irradiation, the toluene is
evaporated. The crude product is purified by column chromato-
.
i--. ~ ' . '
- 13 - 100-7683
graphy (silica gel) and the cis/trans diastereoisomers are
thus separated. cis-4,5,7a,8,9,10,11,11a-octahydro-7-oxo-7H-
indolo[1,7-bc][2,6]naphthyridine-10-carbamic acid ethyl ester
is obtained as an oil.
IR(CH2Cl2) : r = 1665 cm [e-1] (clmide C=0); 1690 cm le-1
(carbamate C=0).
lH-NMR(D6-DMS0), 360 MHz : d (ppm) = 1.20 (t, J = 7.5 Hz; 3H,
carbamate-CH3); 4.08 (q, J Y 7.5 Hz; 2H, carbamate-CH2).
F,XAHPLE 2: erans-4,5,7a,8,9,10,11,11a-octahydro-7~-10-methyl-
indolo~l,7-bc][2~6]naphthy--dine
Production corresponds to that of the cis-diastereoisomer
(example 1). trans-4,5,7a,8,9,10,11,11a-octahydro-7H-10-mcthy].-
indololl,7-bc][2,6]naphthyridine is obtained with the melting
point 102 - 103 (ether/hexane). As the malonate with the melting
point 155 - 156 (acetone/methanol).
The starting material may be produced as follows:
trans-4,5,7a,8,9,10,11,11a-octahydro-7-oxo-?~-indolol1,7-bcl-
[2,6]naphthyridine-10-carbamic acid ethyl ester
Production corresponds to that of the cis-diastereoisomer
(example 1 e). trans-4,5,7a,8,9,10,11,11a-octahydro-7-oxo-7H-
indolol1,7-bc]l2,6lnaphthyridine-10-carbamic acid ethyl ester is
obtained with a melting point of 132 - 133 (crystallisation from
methyl-tert.butylether).
; :
The following compounds of formula I are produced analogously to
example 1:
. .
: " , : ,! , ,.
- 14 - 100-7683
~ ..... _
Example R config. m.p. of dihydrochloride
. _ - ...... _ --- - I
3 ethyl (~)-cis 280 - 281
4 n-propyl (+)-cis 294 - 296
EXAHPL~ 5~ c;s-4 5,7a,8,9,10,11?11a-octahydro-7~-10-methyl-
. . ? __
indolo[l,7-bc][2,6]naphthyridine
13.57 g (59 mmols) o (+/-)-cis-4,5,7a,8,9,10,11,11a-octahydro-
7H-10-methyl-indololl,7-bc]l2,6lnaphthyridine (for preparation
see example 1) are dissolved in acetone. This solution is mixed
with 23.92 g (59 mmols) of (-)-di-0,0'-p-toluyl-L-tartaric acid
monohydrate i~ acetone. The solution is concentrated, mixed with
ether and the crystallised salt is filtered off and washed with
acetone/ether. The salt is recrystallised from ethanol/acetone
until reaching a constant rotatory value [to obtain the
(-)-enantiomer (example 6), the mother liquors are collected
separatelyl. (-)-di-090'-p-toluyl-L-tartrate is obtained with a
melting point of 158 - 159 and the rotatory value of -46 (c =
0.5, methanol). The base is released from the crystallisate
having constant rotatory value by adding ice/conc. ammonia
solution and methylene chloride. (+)-cis-4,5,7a,8,9,10,11,11a-
octahydro-7H-10-methyl-indolol1,7-bcl[2,6]naphthyridine is
obtained with the rotatory value + 132 (c = 0.5; methanol). The
base crystallises as the malonate with the melting point 129 -
130 (crystallisation from acetone/ether) and the rotatory value
~71 (c = 0.5; methanol).
.
' ~ ' ~ ' , :
s~
- 15 - 100-7683
EXAMPLE 6~ cis-4,5,7a,8,9,10,11,11a-o _ ahydro-7~-10-methyl-
indololl,7-bcll2,61naphthyridine
Preparation is from (~ cis-4,5,7a,8,9,10,11,11a-octahydro-7H-
10-methyl-indololl,7-bcl[2,6]naphthyridine (example 1) or from
the collected mother liquors of (~)-enantiomer production, using
(-~)-di-0,0'-p-toluy'l-D-tartaric acid. The process is analogous to
the preparation of the (+)-enantiomler (example 5). (~)-di-0,0'-p-
toluyl-D-tartrate is obtained with the melting point 160 - 161
(methanol/acetone) and the rotatory value ~46 (c = O.S,
methanol). After release of the base, (-)-cis-4,5,7a,8,9,10,11,-
- lla-octahydro-7H-10-methyl-indolo[1,7-bcl[2,61naphthyridine is
obtained with the rotatory value -130 (c , 0.5, methanol). The
base crystallises as the malonate with the melting point 129 -
130 (acetone/ether) and the rotatory value -75 (c = 0.5,
methanol).
E~AMPLE 7. cis-7a,8,9,10,11,11a-hexah~d___10-methyl-7H-indolo-
[1,7-bcll2,63naphthyridine
200 mg (0.9 mmols) of cis-4,5,7a,8,9,10,11,11a-octahydro-7H-10-
methyl-indolo[lt7-bc][2,6lnaphthyridlne (axample 1) are dissolved
in methylene chloride. 2.00 g of manganese(IV)oxide (precipitated
active) are added whilst stirring at 20. Stirring is effected
for 4 hours at room temperature, the reaction mixture is
subsequently filtered over Hyflo and the methylene chloride
j filtrate is concentrated by evaporation. After crystallisation
from ether, cis-7a,8,9,10,11,11a-hexahydro-10-methyl-7H-indolo-
[1,7-bc][2,63naphthyridine is obtained with the melting point
125 - 126.
lH-NMR, 360 MHz (CDCl3): d (ppm) = 2.31 (s; 3H, 10-N-CH3).
As the hydrogen fumarate, the compound has the melting point 127
(decomp.).
.-: , ,
.
.. .
, : . .
,
- 16 - 100-7683
EXAMPLE 8: trans-7a,8,9,10,11,11a-hexahydro-10-methyl-7H-
indololl?7-bc][2~6]naphthyridine
Production corresponds to that of the cis-diastereoisomer
(example 7). trans-7a,8,9,10,11,11a-hexahydro-10-methyl-7H-
indololl,7-bc]l2,6]naphthyridine is obtained with the melting
point 143 - 145 (crystallisation from ethanol).
lH-NMR, 360 MHz(CDC13): d (ppm) = 2.45 (s; 3~, 10-N-CH3); 6.44
(m; lH, C-4-H).
The starting material may be produced as follows:
a) trans-4,5,7a,8,9,10,11,11a-octahydro-7-oxo-7~-indolo[1,7-bcL-
l2,6]naphthyridine-10-carbamic acid ethyl ester
-
Production corresponds to that of the cis-diastereoisomer
(example 1 e). trans-4,5,7a,8,9,10,11,1la-octahydro-7-oxo-7H-
indololl,7-bc][2,6]naphthyridine-10-carbamlc acid ethyl ester
is obtained with a melting point of 132 - 133 (crystalli-
sation from methyl-tert.butylether).
b) trans-4,5,7a,8,9,10911,11a-octahydro-7~-10-methyl-indolo-
jl,7-bc][2,6]naphehyr~ e
Production corresponds to that of the cis-diastereoisomer
(example 1). trans-4,5,7a,8,9,10,11,11a-oceahydro-7H-10-
methyl-indolo[1,7-bc][2,6~naphthyridine is obtained with a
melting point of 102 - 103 ~ether/hexane). As the malonate
with a melting point of 155 - 156 (acetone/methanol).
The following compounds of formula I, wherein R2, R4, R5, X and Y
are hydrogen, are produced analogously to example 7:
`
. -
, `
~, ,
- 17 - 100-7683
_. , .. __ . . ._ ._~ I
Example Rl R3 config. m.p.
. __ __ . __ __
9 ethyl H (~)-cis 156-158 (hydrogen maleinate)
10 n-propyl H (i)-cis 105-107 (hydrogen maleinate)
11 methyl Z-Cl (~ rans 324 (decomp.) (hydrochloride)
EXAMPLe 12~ cis-7a,8,9~10,11l11a-hexahydro-10-methyl-7~-
indolo[l,7-bc][2,~naphthyridine
16.10 g (71 mmols) of (+/-)-cis-7a,8,9,10,11,11a-hexahydro-10-
methyl-7H-indolo[1,7-bc]12,6]naphthyridine (for production see
example 7) are dlssolved in acetone. This solution is mixed with
28.80 g (71 mmols) of (-)-di-0,0'-p-toluyl-L-tartaric acid mono-
hydrate (in acetone). The solution is concentrated, mixed with
ether and the crystallised salt is filtered off and washed with
acetone/methanol. The salt is recrystallised from methylene
chloride/methanol until reaching a constant rotatory value. (The
mother liquors are collected separately, see example 13). The
base is released from the crystallisate having constant rotatory
value by adding ice/conc. ammonia solution and methylene
chloride.
(-)-cis-7a,8,9,10,11,11a-hexahydro-10-methyl-7H-indolo[1,7 bc]-
[2,6]naphthyridine is obtained with the melting point 99 - 100
and the rotatory value l]20 = -50 (c = 0,25, CH2C12).
.
: .. . . .
.
- 18 - 100-7683
~AHPLE 13: ~+)-cis-7a,8,~?,10,11,11a-hexahydro-10=methyl-7~-
indololl,7-bcll2t61naphthyridirle
Preparation is from (+/-)-cis-7a,8,9,10,11,11a-hexahydro-10-
methyl-7H-indolo[1,7-bc][2,6]naphthyridine (example 7) or from
the collected mother liquors of ~-)-enantiomer production, using
(~)-di-0,0'-p-toluyl-D-tartaric acid. The process is analogous to
the preparation of the (-)-enantiomer (example 12). After release
of the base, (+)-cis-7a,8,9,10,11,11a-hexahydro-10-methyl-7H-
indololl,7-bc]l2,6]naphthyridine is obtained with the melting
point 101 - 102 (ether/hexane) and wlth the rotatory value
1~]2 = +49 ~c , 0,25, CH2C12)-
: .: : : . ;
.`~ '
. ~ ` ` ,,