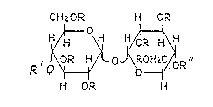Note: Descriptions are shown in the official language in which they were submitted.
2 ~
RAN 407 l /30
The present invention is concerned with novel sulphated
oligosaccharides of the formula
CH2 OR H O.;~
0 ~ H~ H
,~ 1 O~ O~R~oR
H O~ H
wherein R is hydrogen or a residue -SO3M; M is a cation; R is
a an equatorially or quasiequatorially linked sulphated
mono-, di- or tri- saccharide residue; or an axially linked
sulphated trisaccharide residue; and R is hydrogen or an
equatorially or quasiequatorially linked sulphated mono- or
disaccharide residue;
whereby the molecule contains a ma~imum of 6monosaccharide
units and on average at least one -SO3M group is present per
monosaccharide unit.
2s The invention is also concerned with a process for the
manufacture of the compounds of formula I, their use as
medicaments or as active ingredients for the manufacture of
pharmaceutical preparations, pharmaceutical preparations based
on compounds of formula I and a method of treating or
30 preventing arteriosclerotic disorders in man.
As the cation M there come into consideration all physio-
logically compatible cations, e.g. alkali metal cations such as ~a+
and K+; ammonium ions and substituted ammonium ions which
35 are derived from tertiary amines such as triethylamine, or
pyridine or imida;~ole; or quaternary ammonium ions such as
dodecyltri- methylammonium, ethylpyridinium and
Grn/7 . 8 . 9 l
... . . . . . . . . . . . . .. . . . . . . .. . . .. . .. . . . . . . .
. ~ .
2 ~5~
~enzethonium: as well as alkaline earth metal cations such as Ca++.
Compounds in which M is Na~ are prefe~ed.
The degree of sulphation means the number of -SO3M
s residues per monosaccharide unit w}lich are present in the
molecule on average. The degree of sulphation 1 therefore exists
e.g. when a hexasaccharide of formula I contains 6-SO3M
residues in the molecule. The degree of sulphation in the
compounds of formula I preferably amounts to 2-3.
Equatorially linked sulphated mono-, di- or trisaccharide
residues R' are preferably ,~-glycosidically linked residues; but an
equatorial linlcing, e.g. in the case of arabinose glycosides, can
accompany an a-glycosidically bonding of the residue R' to the
5 trehalose residue. The term "~uasiequatorial" relates to the
conformation of furanosides.
The compounds of formula I can be manufactured in
accordance with the invention by treating a corresponding tri-,
20 tetra-, penta- or hexasaccharide with a sulphating agent and
converting the reaction product into a salt or isolating it as such.
Examples of monosaccharide residues are glucopyranosyl, manno-
pyranosyl, arabinopyranosyl, galactofuranosyl, ar~binofuranosyl,
ribofuranosyl and rhamnofuranosyl. Examples of disaccharide
2s residues are maltosyl, cellobiosyl, lactosyl, melibiosyl, gentio-
biosyl and galactopyranosidoarabinopyranosyl. A trisaccharide
residue is, for example, maltotriosyl. The aforementioned
residues are sulphated as substituents R or R" in the scope of the
definition of formula I. Examples of compounds of formula I are
30 the compounds of formulae Ia and Ib in which R has the signifi-
cance given above and on average a Ieast one residue R per mono-
saccharide unit is -SO3M. Ill the compound Ia R is ,~-D-malto-
triosyl and R is hydrogen, in the compound Ib R' and R' are ,~-D-
maltosyl.
. .. _ . . . . . . . _ . _ . _ . . _ _ . . . . . . .
? ~ ~L
ClkOR CIIzOR CHZORCIIzOR 11 OR
R i\lo~ Il/Lo~\ol~ I~/l o,)\OR I~f ~J\OR 1~oJ\QIl2c/lo~ I~
H OR 11 01~ 1 I OR 11 OR 11
Cl120R CIIzOR CIIzOR 1~ 0~ CIIzOR CIIzOR
o ~ o ~0~ /OR 11\~ J I~ l Ib
R l~ ()J\oR ll/l ol~RO112l,~ OR
I 1 01~ 11 o~ 11 OR
The sulphation in accordance with the present invention can
be carried out using methods which are known per se for the
sulphation of hydroxy groups.
Examples of sulphating agents which can be used for the
20 manufacture of the compounds of formula I are S03-complexes
such as S03-pyridine, SO30trimethylamine, SO30dioxan and
S03-dimethylformamide. Other examples of sulphating agents are
chlorosulphonic acid, mixtures of chlorosulphonic acid and
sulphuric acid; and piperidine N-sulphate.
2~
The reaction is conYeniently effected in a suitable solvent,
especially a polar solvent, e.g. dimethylformamide, dimethyl
sulphoxide or hexamethylphosphortriamide. The reaction can be
carried out at room temperature or a higher temperature, e.g. at
30 20-70C, whereby the degree of sulphation can be influenced by
varying the reaction duration and reaction temperature. The
degree of sulphation achieved in each case can be assessed by
HPLC. The working-up of the reaction mixture and, respectively,
the isolation of the reaction product of formula I from the reaction
35 mixture can be effected according to methods known per se, e.g.
by gel filtration or ultrafiltration.
....... , .. , .. _ . . ..... .. . . .. _ _ . _ ~ _ _ _ ~ .. _ _ _ _ ~ _ . . . .. ...
2 ~
The free saccharides which are used as starting materials
are known or are accessible according to methods which are
known in principle. Enzymatic or synthetic chemical procedures
come into consideration for the preparation. The oligosaccha-rides
5 can be synthesized principally using sequential synthesis or block
synthesis. In this case glycosidic bonds are formed by reacting a
glycosyl acceptor with a glycosyl donor in the presence of a
suitable catalyst. Derivatized glycosyl compounds which are
activated at the anomeric centre, such as e.g. chlorides, bromides,
0 fluorides, acetates, trichloroacetimidates, alkylthio derivatives, etc,
are suitable as glycosyl donors.
Those saccharide derivatives in which the O~I groups to be
glycosylated are free and the remaining OH groups are completely
5 or partially protected are suitable as glycosyl acceptors. When the
remaining OH groups are only partially protected the glyco-
sylidation can be effected selectively or can be directed in a
particular direction by virtue of the hydroxyl groups having a
different environment.
The compounds of formula I inhibit the migration and
proliferation of cells of the vascular smooth musculature and
prevent proliferative arteriosclerotic lesions. Their blood
coagulation-inhibiting activity is lower than that of heparin. In
2s particular, the compounds have no in vitro anticoagulant activity,
i.e. they have no effect or only a very slight effect on the coagu-
lation factors thrombin (F.IIa) and F.Xa. The compounds of
formula I can therefore be used for the prophyla~cis of arterio-
sclerotic disorders, in man especially after bypass operations or
30 angioplasty, as well as for the treatment of patients having
progressive arteriosclerosis.
The blood coagulation-inhibiting activity was determined as
follows:
3s
aPTT factivated partial thromboplastin time! Test (see Walenga et
al., CRC Critical Reviews in Laboratory Sciences 22 (4) 361-389
(1986)): 100 ~,11 of citrated human plasma, which contains various
.. . _ _ _ , _ . . , .. . .. . _ _ _ . ~ ~ ~ _ _ _ . . _ _ _ _ _ _ . . ~ ~ _ j _
s
concentrations of test compound, is incubated at 37C for 8
minutes with 100 ~LI of Activated Thrombofax (Ortho Diagnostics,
Raritan, N.J., U.S.A.). 100 ~LI of pre-warmed 25 mM calcium
chloride solution are then added and the coagulation time is
5 measured in a Fibrometer Coagulation Timer (Becton, Diskinson
B asle).
anti-Xa Clotting Assay- 25 ~Ll of citrated plasma having
various concentrations of test compound are mixed with 75 ~,11 of
0 Factor Xa (Diagnostic Reagents, Thame, Oxon, Great Britain) diluted
1:100 in 0.63% citrate buffer (pH 7.3~ which contains 41 mM
imidazole, 82mM NaCl and 0.1% albumin. After warming to 37C
for 2 minutes 200 ~,11 of a 1:1 mixture of Factor X Deficient Plasma
(Diagnostic Reagents) and Platelet Substitute (Diagnostic Reagents)
5 are added and the mixture is incubated at 37C for 20 seconds.
After the addition of 100 ~LI of pre-warmed 50 mM calcium
chloride solution the coagulation time is measured in a
Fibrometer.
The activity of the test compound is given as the ICso, which
is that concentration [~lg/ml] which leads to a coagulation time
which is double the control value.
Inhibition of Thrombin or Factor Xa in the Chromogenic
2s Substrate Assav (Teien et al., Thrombosis Research 10, 399-410
- (1977)): The determination was effect in a Cobas-Bio centrifugal
automatic spectrophotometer. The buffer solution used consisted
of 50mM Tris buffer, 18û mM NaCl, 7.~ mM EDTA Na2, 1% PEG
6000 and 0.02% Tween 80, pH 8.4. The test solution consisted of
50 ~Ll of buffer, 30111 of anti-thrombin III (1 U/ml), Kabi
Diagnostica) and 20 ~1 of plasma which contained vanous con-
centrations of test compounds. 30 ~11 of sample solution and
20 ~Ll of water with 18û 111 of thrombin were added to the test
cuvette in the automatic analyzer. After incubation at 37C for
3s 240 seconds 60 ~,11 of S-2238 (H-D-Phe-Pip-Arg-NH.pNA, Kabi
Diagnostica, Mondal, Sweden, 0.75 mM in water) and 20 111 of
water were added. The liberation of pNA (p-nitroaniline) was
followed during 60 seconds at 405 nm in 10 second intervals in
... .... , . . . . . .. . .. . ~ _ .
comparison to water as the blank. The inhibitory activity is given
as the ICso, which is the concentration [~,lg/ml] at which the
amidolytic activity of thrombin is reduced by 50% in comparison
to the plasma control value.
s
The inhibition of ~actor Xa was measured in the same
manner using a solution of Factor ~a (2.8 nkat/ml and 2nM S-
222 (Bz-CO-Ile-Glu-Arg-NH.pNA, Kabi Diagnostica) in water in
place of thrombin and, respectively, S-2238.
The anti-proliferative activity of the substances was
determined in cell cultures as follows: smooth muscle cells of rats
(cultivated in DMEM with 10% FCS at 37C and 5% CO2) were
applied to cell culture pla~es with a density of 8 x 103 cells/well.
15 After 4 hours the number of adhered cells was determined and
the substances to be tested (100 mg/mi) were added. Cells to
which test substance was not added served as a comparison and
heparin (100 mg/ml) served as a positive control. The cells were
incubated for 7 days and then the cell number was determined.
20 The antiproliferative activity of the individual substances was
calculated as the % inhibition in comparison to non-inhibited
growth:
cell number ( ) - cell number (inhib)
25 % Inhibition = x 100
cell number ( ) - cell number (d=o)
wherein cell number(d=o) = cell number after 4 h
cell number( ) = cell number to which test
substances was not added, a~ter 7 days.
cell number(inhib) = cell number with
100 ~LI/ml of the test substance
The results obtained in the experimental procedures
35 described above with compounds of formula I are listed in Table
1. Heparin served as the reference compound.
6~
Table I
Anti- Anti-coagulation activity IC50 [llg/ml]
Compoun proliferative Coagulation Amidolytic
d of activity % inhibitior activity
~xample inhibition aPI~ ~a Thrombin F.Xa
lG. 5 7 11 31 >1000960
2C 38 40 1 ]l~ >1000>1000
3C 5 9 1 2 33.5 >1000>1000
4C 64 1 1 32 >1000>1000
5C 39 33 69 >1000>1000
6D 6 7 14 . 5 28.5 > 1000900
7C 49 13.5 34 >1000~1000
Heparin 4 7 1.2 0.6 1.9_ 2.7
The test results show that the compounds in accordance
s with the invention have an anti-proliferative activity which, in
contrast to the likewise anti-proliferatively active heparin, is not
accompanied by or is accompanied to a very insignificant extent
by an anticoagu]ant activity.
o The medicaments based on the compounds in accordance
with the invention can be administered enterally, e.g orally in the
form of tablets, coated tablets, dragées, hard and soft gelatine
capsules, solutions, emulsions or suspensions, or rectally, e.g. in
the form of suppositories. However, the administration is
5 preferably effected parenterally, e.g. in the form of injection
solutions.
For the manufacture of tablets, coated tablets, dragées and
hard gelatine capsules the active ingredient can be mixed with
20 pharmaceutically inert, inorganic or organic excipients. As such
excipients for tablets, dragées and hard gelatine capsules there
can be used e.g. lactose, maize starch or derivatives thereof, talc,
stearic acid or its salts. Suitable excipients for soft gelatine
capsules are e.g. vegetable oils, waxes, fats, semi-solid and liquid
25 polyols; depending on the nature of the active ingredient no
excipients are, ho~ivever, usually required in the case of soft
... . . ... . . _ _ .. _ _ ... .. .. _ , ~.. _. _ . __ __ ._ ____ .. __ .. __ ._ . ... ,_ ....... .. ~ . , .
2 ~
gelatine capsules. Suitable excipients for the manufacture of
solutions and syrups are e.g. water, polyols, saccharose, invert
sugar and glwcose, suitable excipients for injection solutions are
e.g. water, alcohols, polyols, glycerine and vegetable oils and
5 s~itable excipients for s~ppositories are e.g. natural or hardened
oils, waxes, fats and semi-liquid or li4uid polyols.
The pharmaceutical preparations can contain, in addition,
preservatives, solubilizers, stabilizers, wetting agents, emulsifiers,
o sweeteners, colorants, flavorants, salts for varying ~he osmotic
pressure, buffers, coating agents or antioxidants. In the case of
enteral administration the resorption of the active ingredient can
be increased with the aid of liposomes.
The dosage of the active ingredient can vary within wide
limits and will, of course, be fitted to the individual requirements
in each particular case. In general, in the case of parenteral
administration a dosage of about 0.1 to 100 mg/lcg, preferrably of
about 1.5 to 15 mg/kg, per day should be appropriate for adults,
20 although the upper limit just given can also be exceeded when
this is shown to be indicated.
Example 1
25 A. A solution of 28.5 g of 2,2',3,3'-tetTa-O-benzyl-4,6-O-
benzylidene-~,oc-D-trehalose (Carbohydr. Res. ~, 51 (1978)) and
27 ml of bistributyltin oxide in 2.21 of toluene was heated under
reflex for 4.5 hours on a water separator and reduced to a volume
of 800 ml. Subsequently, 3.84 g of tetrabutylammonium bromide
30 and 42.8 ml of benzyl bromide were added and the mixture was
stirred at 100C. After 16 hours the solution was cooled and
worked-up by extraction in a sodium hydrogen
carbonate/methylene chloride system. The combined organic
phases were dried (MgSO4) and chromatographed on silica gel
3s with acetone/hexane 1:2 (containingl %o of triethylamine) as the
eluent. Product *actions were crystallized and gave 27.5 g (~6%)
of 2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,a-D-trehalose,
m.p. 122C.
. ... .. . ... . ... . . . . . _ . _ _ . .. _ . _ . _ _ _ _ _ _ _ ~ . _ _
2 ~ 3 ,j 3 ~ ~,
B. A solution of 32.7 g of deca-O-acetyl-a-D-maltotriosyl
bromide (K. Tabeo, K. Mine and T. Kuge, Carbohydr. Res 48, 197
(1976)), in 85 ml of allyl alcohol was stirred at 50-60~ for
5 90 minutes in the presence of 8.5 g of mercury-II cyanide,
concentrated and extracted in ethereal solution successively with
1 molar potassium iodide solution, sodium bicarbonate solution
and water. Chromatography of the crude product on silica gel
with ethyl acetate/hexane 1:1 as the eluent gave 25.3 g (79.6%)
10 of pure allyl deca-O-acetyl-,B-D-maltotrioside, [CC]2D=7S.OO (c= 0.5,
dioxan).
C A solution of 24.8 g of allyl deca-O-acetyl-,B-D-malto-
trioside in 150 ml of methanol was stirred at room temperature
5 for 18 hours in the presence of catalytic amounts of anhydrous
sodium carbonate, then filtered and neutralized with acidic ion
exchanger. After removing the solvent the residue was taken up
in 3~0 ml of dimethylformamide and treated at room temper-
ature with 8.0 g of sodium hydride (80% in Fefined oil). After
20 stirIing for 90 minutes the mixture was cooled to 1C, treated
with 32 ml of benzyl bromide and stirred while cooling with ice
for 30 minutes and at room temperature for 1 hour. Thereafter,
the mixture was added dropwise to 100 ml of methanol and
stirred for 1 hour. The reaction mixture was concentrated, taken
2s up in ethyl acetate and extracted with aqueous sodium
bicarbonate solution and with water. The organic phases were
dried and concentrated (38.5 g). 5 g of benzylated crude product
were suspended in 40 ml of acetic acidlwater 9:1 and treated in
an ul~asound bath for 3 hours in the presence of 2.46 g of
30 palladium chloride and 2.46 g of sodium acetate. Thereafter, the
mixture was suction filtered, washed, evaporated, the residue was
taken up in ethyl acetate and the solution was washed in
succession with aqueous sodium bicarbonate solution and water.
The ethyl acetate phases were dried (Na2~O4), concentrated and
35 chroma~ographed on silica gel with toluene/ethyl acetate 7:1 as
the eluent. There were obtained 3.99 g (90%) of syrupy deca-O-
benzyl-D-maltotriose.
. . .. .. _ _ . _ ____.__ ., _ .. .. _ _. , . . ._. .. .. _ __ _ .. _ ._ . . ..... __ _ _ _
2 ~
D. A solution of 0.25 ml of oxalyl chloride in 7 ml of absolute
dichlorome~hane was added dropwise at 3C during 45 minutes to
a solwtion of 3.69 g of deca-O-benzyl-D-maltotriose in 25 ml of
absolute dichloromethane and 0.1 ml of absolute dimethyl-
5 formamide. After stirring at room tempera~ure for 6 hours themixture was evaporated. The dried crude deca-O-benzyl-a- D -
maltotriosyl chloride was dissolved in 10 ml of absolute aceto-
nitrile and stirred at room temperatl~re for 3 hours in the
presence of 0.97 g of dry silver fluori~de. The reaction mixture
o was filtered and the precipitate was washed with ether. The
organic solutions were treated with aqueous saturated sodium
chloride solution and stirred vigorously for 15 minutes. After
suction filtration of the separated precipitate the filtrate was
concentrated, diluted with ether and washed in succession with
15 sodium chloride solution and water. The ethereal solutions were
evaporated. Chromatography on silica gel with ethyl acetate/
hexane 1:6 as the eluent gave 2.19 g (66%) of pure deca-O-
benzyl-~-D-maltotriosyl fluoride, [a]2D = 58.9 (c = 0.9, CHCl3).
20 E A solution of l l O ml of trifluoromethanesulphonic
anhydride in 5 ml of dry ether was added dropwise at -20C
within 1 hour in the presence of molecular sieve (4A) to a
solution of 1.69 g of well dried deca-~-benzyl-,~-D-maltotriosyl
fluoride and 53~ mg of 2,2',3,3',6'-penta-O-benzyl-4,6-O-
2s benzylidene-o~,a-D-trehalose in 20 ml of dry ether. The mixture
was warmed to room temperature and stirred for 48 hours. After
filtration over a filter aid, washing with ether and concentrating
the residue was chromatographed on silica gel using toluene/
ethyl acetate 14:1 as the eluent. There were obtained 414 mg
30 (30%) of 2,2',3,3',6'-penta-O-benzyl-4'-O-(deca-O-benzyl-oc-D-
maltotriosyl)-4,6-O-benzylidene-~,a-D-trehalose as a syrupy
product, [a]2D = +44 (c = 0.0846, Etr)H)
F. A solution of 295 mg of 2,2',3,3',6'-penta-O-benzyl-4'-O-(deca-
3s O-benzyl-a-D-matlotriosyl)-4,6-O-benzylidene-a~a-D-trehalose in
10 ml of ethanol and 5 ml of water was hydrogen- ated at room
temperature for 2 hours in the presence of 300 mg of 10%
palladium-on-charcoal. The reaction solution was filtered over a
.. . . ~
~ ~ r3 ~ .rJ3 '~.
1 1
filter aid, rinsed and freeze-dried. The crude product was gel-
chromatographed with water (on Sephadex6~ L1120) and the
product was lyophilized. There were obtained 65 mg o~ ~-O-(a-
D-maltotriosyl)-a,a-D-trehalose, [a]2D = +179.5 (c = 0.2, ~I2O).
C~ A solution of 30 mg of 4-O-(a-D-maltotriosyl)-oc,a-D-
trehalose in 1 ml of absolute dimethylformamide was stirred at
50C for 20 hours in the presence of 170 mg of sulphur trioxide-
trimethylamine complex, whereby a viscous syrup separated. The
0 solvent was decanted off, the residue was washed with methanol,
dissolved in 1.5 ml 10% sodium acetate solution and concen-
trated. The residue was taken up several times in water and
evaporated in order to remove trimethylamine. The residue was
gel-chromatographed (Sephadex~' LH 20) in order to remove salts.
s After ~reeze-drying there were obtained 67 mg of sulphated 4-O-
(a-D-maltotriosyl)-a,a-D-trehalose, S = 18.66%, AS (average
degree of sulphation) about 2.4.
Example 2
A. A solution of 4.2 g of a-D-acetobromoglucose in absolute
dichloromethane was added dropwise at -30C to a solution of
3.0 g of dry 2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,oc-D-
trehalose in 30 ml of absolute dichloromethane and 2.5 ml of
2s absolute tetramethylurea in the presence of 2.61 g of dry silver
trifluoromethanesulphonate. After stirring at room temperature
for 20hours a further addition of 3.44g of a-D-acetobromo-
glucose in 5 ml of dichloromethane, 2 ml of tetramethylurea and
1.75 g of silver trifluoromethanesulphonate was carried out at -
30 30~C. After stirring at room temperature for 18 hours themixture was filtered and washed with dichloromethane. The
organic solutions were washed with sodium bicarbonate solution
and water and dried over magnesium sulphate. Chromatography
on silica gel with toluene/ether 6:1 as the eluent gave 64% o~ pure
35 4'-0-(2,3,4,6-tetra-O-acetyl-~-D-glucopyranosyl)-2,2',3,3',6-penta-
O-benzylidene-a,a-D-trehalose as a syrup, [a]2D = +53.0 (c = 0.2,
dioxan~.
.. .... , . . .. . .. , . .. ... _ , . , .. . . . .. . _ _ . _ . .. _ . _ .. _ _ . _ _ _ ... . _ _ _ _ . _ . _
_ _
12 2~$~
B. A solution of 1.14 g of 4'-0-(2,3,4,6-tetra-O-acetyl-~-D-
glucopyranosyl)-2,2',3 ,3 ',6'-penta-O-ben~yl-4,6-O-ben~ylidene-
a,a-D-trehalose in 60 ml of absolute methanol and 20 ml of
absolute cycloh~xane (20 ml) was treated with 1.4 ml of 2%
5 sodium methylate solution. After 4 hours at room temperature
the mixture was neutralized with acidic ion exchanger, filtered,
concentrated and chromatographed on silica gel with e~hyl
acetate/methanol/water 95:1:1 as the eluent, whereby 840 mg of
product were obtained. A 370 mg a]liquot was hydrogenated at
10 room temperature in 60 ml ethanol/water 5:1 in the presence of
10% palladium-on-charcoal. After 90 minutes the mixture was
filtered, washed with ethanol and water and concentrated. There
was obtained a quantitative yield of 4-O-(~-D-glucopyranosyl)-
a,a-D-trehalose, [a]2D = +121.9 ~c = 0.2, H20).
C A solution of 840 mg of well-dried 4-O-(,~-D-gluco-
pyranosyl)-a,a-D-trehalose in 30 ml absolute dimethyl-
formamide was stirred at 50C for 20 hours in the presence of
5.08 g of sulphur trioxide-trimethylamine complex, whereby a
20 viscous syrup separated. The solvent was decanted off, the
residue was washed with methanol, dissolved in 30 ml of 10%
sodium acetate solution and concentrated. The residue was taken
up several times in water and evaporated in order to remove
trimethylamine. The residue was purified by gel chromatography
2s (Sephadex~' LH 20) in order to remove salts. After freeze-drying
there were obtained 2.0 g of sulphated 4-O-(,I~-D-glucopyranosyl)
-a,a-D-trehalose, S = 20.40%, AS about 3Ø
.
Example 3
A. A solution of 1.19 g of hepta-O-acetyl-a-D-maltosyl
bromide (J. Chem. Soc. 1962, 2823) in 5ml of absolute dichloro-
methane was added dropwise at 0C to a solution of 1.0 g of well-
dried 2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,a-D-
3s trehalose in 6 ml of absolute dichloromethane and 0.22 ml oftetramethylurea in the presence of 0.43 g of silver trifluoro-
methanesulphonate. After 8 hours at room temperature 109 mg
of silver triflate, 0.05 ml of tetramethylurea and 2g8 mg of
.. . .. . . .... . . _ _ _ _
2 ~ ~ ~ T3 ~3 ~
maltosyl bromide were added. After stiTring at room temperature
for 18 hours the mixture was filtered and washed with dichloro-
methane. The combined organic solutions were washed with
sodium bicarbonate solution and water and dried over magnesium
5 sulphate. Chromatography on silica gel with ethyl acetate/hexane
1:1 as the eluent gave 1.49 g (88%) of pure 4'-O-(hepta-O-acetyl-
~-D-maltosyl)-2,2',3,3 ',6'-penta-O-benzyl-4,6-O-beslzylidene-O-
a,a-D-trehalose as a syrup, [a]20= +84.0 (c = 0.2, dioxan).
o B. A solution of 1.43 g of 4'-(hepta-O-acetyl-~-D-maltosyl)-
2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,oc-D-trehalose in
15 ml of absolute methanol and 4 ml of absolute cyclohexane
was treated with 6 ml of 2% sodium methylate solution. After 1
minutes at room temperature the mixture was neutralized with
acidic ion exchanger, filtered and concentrated. The crude product
was dissolved in 20 ml of ethanol/water (3:1) and hydrogenated
at room temperature for 2 hours with 275 mg of 10% palladium-
on-charcoal. Filtration over a filter aid and rinsing gave pure 4-O-
(~-D-maltosyl)-a,oc-D-trehalose, (635 mg), [a]20= +150.0 (c = 0.2,
~0 H20)-
C A solution of 1.0 g of well-dried 4-O-(~-D-maltosyl)-a,a-D-
trehalose in 2~ ml of absolute dimethylformamide was stirred at
50C for 18 hours in the presence of 5.845 g of sulphur trioxide-
25 trimethylamine complex, whereby a viscous syrup separated.
Working-up as described in Example IG. gave 2.63 g of sulphated
4-O-~,B-D-maltosyl)-a,a-D-trehalose, S = 19.91%, AS about 2.8.
Example 4
A. A solution of 6.67 g of deca-O-acetyl-a-D-maltotriosyl
bromide (Carbohydr. Res 48, 197 (1976)) in 40ml of absolute of
dichloromethane was added dropwise at -30C to a solution of
4.0 g of well-dried 2,2',3,3',6'-penta-O-benzyl-4,6-O-benzyl-
35 idene-a,a-D-trehalose in 3~ ml of absolute dichloromethane and
3.1 ml of tetramethylurea in the presence of 1.74 g of silver
trifluoromethanesu lphonate. After 3 hours at room temperature
3.4 g of deca-O-acetyl-a-D-maltotriosyl bromide in 20 ml of
2 ~ 8 ~
14
absolute di~hloromethane and 1 g of 4A molecular sieve were
added at -30OC. The mixture was worked-up after 90 hours.
Chromatography on silica gel with ethyl acetate/hexane 3 :7 as the
eluent gave 4.03 g (50%) of 4'-(deca-O-acetyl-~-D-maltotriosyl)-
5 2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,a-D-trehalos~ as a
syrup, [a]20= +100.5 (c = 0.2, dioxan).
B. A solution of 3.46 g of 4'-deca-O-acetyl-,B-D-maltotriosyl)-
2,2',3,3',6'-penta-O-~enzyl-4,6-O-benzylidene-a,a-D-trehalose in
10 45 ml absolute methanol was stirred at room tempera~ure for 24
hours in the presence of a catalytic arnount of anhydrous sodium
carbonate. After filtration and neutralization with acidic ion
exchanger the mixture was concentrated and chromatographed on
silica gel with ethyl acetate/methanol/ water ~5:10:5 as the
15 eluent. The product fraction (2.29 g, 86%) was dissolved in
24 ml of ethanol/water (5:1) and hydrogenated at room
temperature for 3 hours in the presence of 10% pallidium-on-
charcoal. Filtration over a fil~er aid and rinsing gave pure 4-O-(,~-
~-maltotriosyl)-a,a-D-trehalose as a foam (1.21 g, 100%), [a]2D=
20 +172.5 (c=0.2, H2O).
C A solution of 500 mg of well-dried 4-O-(,~-D-maltotriosyl~-
a,a-D-trehalose in 10 ml of absolute dimethylformamide was
stirred at 50C for 18 hours in the presence of 2.85 g of sulphur
2s trioxide-trimethylamine complex, whereby a viscous syrup
separated. Working-up as described in Example lG. gave 1.32 g
of sulphated 4-O-(,I~-D-maltotriosyl)-a,a-D-trehalose, S =19.5%, AS
about 2.6.
Example 5
A. A solution of 1.19 g of dry hepta-O-acetyl-a-D-cellobiosyl
bromide (Ann. 435~ 1 (1923)) in 5 ml of absolute
dichloromethane was added dropwise at -30C to a solution of 1 g
of well-dried 2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,a-D-
3s trehalose in 1 ml of absolute dichloromethane and 0.3 mltetramethylurea in the presence of 442 mg of silver
trifluoromethanesulphonate. After stirring at room temperature
for 60 hours the mixture was filtered over a filter aid, rinsed with
,
. _ ... .. . _ _ . _ __ __. _ . _. .. . __ _ _ ~_ .. _ . .. __ ~ ,_ _ , . ..... ~ ~
2~fi~3~.'1.
1 5
dichloromethane, evaporated and chromatographed on silica gel
with dichloromethane/acetone 24: 1 as the eluent. There were
obtained 1.51 g (g8%) of pure 4'-O-(hepta-O-acetyl-~-D-
cellobiosyl)-2,2',3.3 ',6'-penta-O-benzyl-4~6-O-benzylidene-o~,oc-D -
s trehalose, [O~]2D= +35.0 (c = 0.3, CHC13)
B. A solution of 1.0 g of 4'-O-(hepta-O-acetyl-,B-D-cello-
biosyl)-2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-o~,a-D -
trehalose in 30 ml absolute methanol was stirred at room
o temperature in the presence of a catalytic amount of sodium (a
few mg). After 2 hours the mixture was neutralized with acidic
ion exchanger, evaporated and filtered over silica gel in chloro-
form/methanol 9:1. Evaporation gave a deacetylated crude
product (764 mg) of which 168 mg were hydrogenated at room
15 temperature in 10 ml of ethanol/water (4:1) in the presence of
10% palladium-on-charcoal. After 2.5 hours the mixture was
filtered over a filter aid, rinsed an evaporated. There were
obtained 93 mg (100%) of 4'-O-(,B-D-cellobiosyl)-oc,~-D-trehalose,
[a]2D= +97 (c = 0.3, H2O).
C A solution or 72 mg of 4'-O-(,B-D-cello~iosyl)-c~,a-D-
trehalose in 5 ml of anhydrous dimethylformamide was stirred at
5C for 20 hours in a presence of sulphur trioxide-trimethylamine
complex, whereby a viscous syrup separated. Working-up as
2s described in Example lG. gave 176 mg of sulphated 4-O-(,I~-D-
cellobiosyl)-(x,a-D-trehalose, S = 18.2%, AS about 2.7.
Example 6
30 A. 2.19 ml of bis-tributyltin oxide were added to a suspension
of 1.0 g of 2,2',3,3'-tetra-O-ben~yl-a,o~-D-trehalose (Chem. Pharm.
Bull. 3 0, 1169 (1982)) in 80 ml of toluene and the mixture was
heated under reflux on a water separator for 3.5 hours. Thereby,
the volume was reduced by 40 ml. Then, 157 mg of
3~ tetrabutylammonium bromide and 0.84 ml of benzyl bromide
were added and the mixture was stirred at 80C for 30 hours.
After the addition of the same amounts of tetrabutylammonium
bromide and benzyl bromide the mixture was stirred at 80C for a
, . . . . _ . .. .. .. . ..... .. . . .. _ _ _ _ _ . _ .. .. _ . .. _ _ . _ . .. _ _ _ _ . _ . _ . .
16
further 72 hours. The reaction mixture was poured into ice-
water/ methylene chloride and extracted with ~,vater and sodium
bicarbonate solution. After evaporating the organic phase the
residue was chromatographed on silica gel with ethyl acetate/
5 hexane 1 :2 as the eluent. There was obtained pure 2,2'3,3',6'-
hexa-O-benzyl-a,a-D-trehalose (1120 mg, 89%) as a syrup, [a]20 =
+111.5 (c = 0.5, dioxan).
B. A solution of 2.38 g of hepta-O-acetyl-a-maltosyl bromide
10 in 12 ml of anhydrous dichloromethane was added at -30C to a
suspension of 1.0 g of 2,2',3,3',6,6'-hexa-O-benzyl-oc,a-D-trehalose
and 0.88 g of silver triflate in 3 ml of anhydrous
dichloromethane and 0.61 ml of tetramethylurea. ~fter 10 days
at room temperature the mixture was filtered over a filter aid,
5 washed with dichloromethane, evaporated and the crude product
was chromatographed on silica gel with acetone/hexane 2:3 as the
eluent. There were obtained 1.96 g of pure 4,4'-bis-O-(hepta-O-
acetyl-~-D-maltosyl)-2,2',3,3',6,6'-hexa-O-benzyl-a,a-D-trehalose
(82%), melting point 85C, [a]2D= +84.5 (c = 0.2, chloroform).
C A catalytic amount of sodium methylate was added at room
temperature to a solution of 1.0 g of 4,4'-bis-O-(hepta-O-acetyl-~-
D-maltosyl)-2,2',3,3',6,6'-hexa-O-benzyl-a,a-D-trehalose in 40 ml
of methanol/dioxan (1:1). After 4 hours the mixture was
2s neutralized with acidic ion exchanger, filtered and evaporated.
The deacetylated compound was dissolved in 50 ml of ethanol/
water (1:1) and hydrogenated at room temperature in the
presence of 300 mg of 10% palladium-on-charcoal. After 1 hcur
the mixture was filtered over a filter aid, washed with ethanol/
30 water (1:1) and evaporated. There were obtained 465 mg of pure
4,4'-bis-O-(~-D-maltosyl)-a,oc-D-trehalose, [a]20= +148 (c = 1.3,
H20).
D. A solution of ~.3 g of 4,4'-bis-O-(~-D-maltosyl)-a,a-D- ;
35 trehalose in 15 ml of anhydrous dimethylformamide was stirred
at 5~C for 20 hours in the presence of 1.685 g of sulphur trioxide-
trimethylamine complex, whereby a viscous syrup separated.
Working-up as described in Example lG. gave 652 mg of
.. . . , . . . . . . .. . .. .. _ . _ . . . .. . .. , _ .... , . , . , _ . . , . . , _
2~5~-~3
1 7
sulphated 4,4'-bis-O-(,B-D-maltosyl)-a,a-D-trehalose, S - 18.93%,
AS = 2.5.
Example 7
A. 0.21 ml of trifluoromethanesulphonic anhydride was added
under argon to a cooled solution (-80C) of 618 mg of 2,3,4,6-
tetra-O-acetyl-D-glucopyranosyl trichloroacetimidate and 553 mg
of 2,2',3,3',6,6'-hexa-O-benzyl-a,a-D-trehalose in 5 ml of absolute
o dichloromethane. After 2.5 hours at -10C a further 123 mg of
2,3,4,6-tetra-O-acetyl-D-glucopyranosyl trichloro- acetimidate and
41 ml of trifluoromethanesulphonic anhydride were added at
-80C. The mixture was warmed to -10C, triethylamine was
added and the solvent was evaporated. After co-distillation with
15 toluene the residue was chromatographed on silica gel with
toluene/ethyl acetate 7:3 as the eluent and there were obtained
832 mg (86%) of pure 4,4'-bis-0-(2,3,4,6-tetra-O-acetyl-~-D-
glucopyranosyl)-2,2',3 ,3 ',6,6'-hexa-O-benzyl-a,a-D-trehalose [a]2D =
+ 42 (c = 0.3, chloroform).
~0
B. A solution of 696 mg of 4,4'-bis-0-(2,3,4,6-tetra-O-acetyl-,B-
D-glucopyranosyl)-2,2',3,3',6,6'-hexa-O-benzyl-a,oc-D-trehalose in
40 ml of anhydrous methanol was treated with a catalytic
amount of sodium. After 20 minutes at room temperature the
2s mixture was neutralized with acidic ion exchanger, filtered and
evaporated. Filtration over silica gel with chloroform/methanol
9:1 as the eluent gave 480 mg of deacetyl-ated compound which
was hydrogena~ed at room temperature in 50 ml of
ethanol/water (4:1) in the presence of 200 mg of 10% palladium-
30 on-charcoal. After 2 hours the mixture was filtered over a filter
aid, washed with ethanol/water (1:1) and evaporated. There were
obtained 250 mg of 4,4'-bis-O-(~B-D-glucopyranosyl)-a,a-D-
trehalose [a]20= ~108 (c = 0.2, H2O).
35 C A solution of 208 mg of 4~4'-bis-O-(,B-D-glucopyranosyl)-
a,a-D-trehalose in 5 ml absolute dimethylformamide was stirred
at 50C for 20 hours in the presence of 1.22 g of sulphur trioxide-
trimethylamine cornplex, whereby a viscous syrup separated.
.. ., . . . . ... .. .. . . .. . .. ... ~
.
2~'33~1
1 8
Working-up as described as in Example lG. gave 520 mg of
sulphated 4,4'-bis-O-(~-D-glucopyranosyl)-a,a-D-trehalose, S =
19.52%, AS about 2.7.
Example ~,
A. 0.52 g of silver trifluoromethanesulphonate and im~nedi-
ately thereafter 0.6 ml of absolute tetramethylurea were added
at 0-~C to a solution of 2.64 g of high vacuum-dried 2,2',3,3',6'-
0 penta-O-benzyl-4,6-O-benzylidene-a,lx-D-trehalose and 1.29 g of
2,3,4-tri-O-acetyl-6-0-(2,3,4-tri-O-acetyl-6-desoxy-a-L -
mannopyranosyl~-a-D-glucopyranosyl bromide [Ber. 70, 1098-
1101 (1938)] in 20 ml of absolute dichloromethane. After
stirring at 0-5C for a further 75 minutes the reaction mixture
5 was suction filtered over Dicalite and rinsed with dichloro-
methane, and the filtrate was washed twice with saturated
sodium bicarbonate solution, dried over magnesium sulphate and
concentrated. The residue was chromatographed on 85 g of silica
gel (70-230 mesh) with ethyl acetate/hexane 1 :4, 1 :2 and 1: 1 as
20 the eluent and gave 830 mg (29%) of 0-(2,3,~-tri-O-acetyl-6-
desoxy-a-~-mannopyranosyl)-( l ~ 6)-0-(2,3,4-tri-O-acetyl-~-D-
glucopyranosyl)-(l ~4)-2,2',3,3',6'-penta-O-benzyl-4,6-O-
benzylidene-a,a-D-trehalose as a foam, [a]20= + 32.2 (c = 0.5,
(~HC13).
2s
B. 1.5 ml of 1% Na in methanol were added at room temper-
ature to a solutiDn of 750 mg of 0-(2,3,4-~ri-O-acetyl-6-desoxy-a-
L-mannopyranosyl)-( l ~ 6)-0-(2,3,4-tri-O-acetyl-,~-D-gluco-
pyranosyl)-(l ~4)-2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene- '
30 oc,a-D-trehalose in 1.5 ml of diethyl ether and 7.5 ml of methanol
and the mixture was stirred for a further 3 hours. The reaction
solution was then neutralized with acidic ion exchanger
(Amberlite IR-120), stirred for 30 minutes, filtered off, rinsed
with methanol and the filtrate was evaporated. The residue was
3s chromatographed on 27 g of silica gel (70-230 mesh) with ethyl
acetate/methanol/water 98:1:1 as the eluent. 380 mg (62%) of
deacylated product were obtained, [a]2D= +7.0 (c =0.1, CHC13).
360 mg (0.303 mmol) of this in 27 ml of ethanol/water 2:1 were
.. ... _ . _ _
2~ ;3
I ~
hydrogenated at room temperature in the presence of 180 mg of
10% palladium-on charcoal. After 14 hours the reaction mixture
was suction filtered over Dicalite, rinsed with ethanol/water and
the filtrate was concentrated and dried in a high vacuum. There
were obtained 205 mg (100%) of 0-(6-desoxy-a-L-
mannopyranosyl)-(1~6)-O-~-D-glucopyranosyl-(1~4)-a,oc-D-
trehalose as an amorphous powder, [OC]ZD= + g.3 (c = 0.1, ~I2O).
C A solution of 200 mg of 0-(6-d~esoxy-a-L-mannopyranosyl)-
0 (1~6)-O-~-D-glucopyranosyl-(l~4)-a,a-D-trehalose in 7 ml of
absolute dimethylformamide was treated with 1.72 g of sulphur
trioxide-trimethylamine complex and stirred under argon at
60-65C for 20 hours. A resinous precipitate separated during
this time. Tbe solvent was evaporated and the residue was
5 dissolved in 11 ml of 10% sodium acetate solution and
concentrated in a waterjet vacuum. The residlle was tal~en up
several times (lOx) in 50 ml of water each time and evaporated
in order to remove triethylamine and was then gel-chroma-
tographed (Sephadex~' LH20, 150 g) in order to remove salts. The
20 fractions containing the sulphated tetrasaccharide were
evaporated and Iyophilized. There were obtained 320 mg (54%)
of sulphated 0-(6-desoxy-1~c-L-mannopyranosyl)-(1~6)-O-,B-D-
glucopyranosyl)-(1~4)-a,a-D-trehalose as an amorphous powder,
S = 19.62%, AS = about 2.7.
2s
Example 9
A. 0.52 g of silver trifluoromethanesulphonate, 1.40 g of 2,3,4-
tri-O-acetyl-6-0-(2,3,4,6-tetrta-O-acetyl-a-D-glucopyranosyl)-a-
30 D-glucopyranosyl bromide [Wolfram et al, J. Am. Chem. Soc., 71,
125-127, ~1949)] and 0.6 ml of absolute tetramethylurea
(5.0 mmol) were added in rapid sequence at 0-5C to a solution of
2.64 g of high vacuum-dried 2,2',3,3',6'-penta-O-benzyl-4,6-O-
benzylidene-a,a-D-trehalose in 20 ml of absolute
3s dichloromethane and the mixture was stirred at 0-5C for a
further 1 hour. The reaction mixture was suction filtered over
Dicalite, rinsed with dichloromethane, the filtrate was washed
twice with saturated sodium bicarbonate solution, dried over
.. .. . .. ... . . . . _ _ _ _ _ ... . .. .. .. .. _ . _. . .. .. ._ ~ _ . ~; ... .. _ . ~ _ _ _ _. .
. , _. _ _
2 i~
magnesium sulphate, filtered off and concentrated. The residue
was chromatographed on 85 g of silica gel (70-230 mesh) with
ethyl acetate/hexane 1:4, 1:2 and 1:1 as the eluent. 740 mg
(25%) of 0-(2,3,4,6-tetra-O-acetyl-a-D-gluco- pyranosyl)-(1~6)-
s 0-(2,3,4-tri-O-acetyl-,B-D-glucopyranosyl)-(1~4~-2,2',3,3',6'-
penta-O-benzyl-4,6-O-benzylidene-o~,a-D-trehalose were obtained
as a foam, [a]2D= + 78.2 (c = O.S, CHC13).
B. 1.32 ml of 1% Na in methanol were added at room temper-
o ature to a solution of 660 mg of 0-(2,3,4,6-tetra-O-acetyl-oc - D -
glucopyranosyl)-( l ~ 6)-0-(2,3,4-tri-O-acetyl-~-D-gluco-
pyranosyl)-(1 ~ 4)-2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-
a,a-D-trehalose in 1.5 ml of diethyl ether and 7.5 ml of methanol
and the mixture was stirred at room temperature for a further
15 3 hours. The reaction solution was then stirred with acidic ion
exchanger (Amberlite IR-120) for 10 minutes, filtered off and
rinsed with methanol. The filtrate was evaporated and chroma-
tographed on 27 g of silica gel (70-23û mesh) with ethyl
acetate/methanol/water 96:2:2 as the eluent. 420 mg (79%) of
20 deacylated product were obtained, [a]20= +103.0 (c = 0.1, CHC13).
400 mg (0.33 mmol) of this in 30 ml of ethanol/wa~er 2:1 were
hydrogenated at room temperature in the presence of 200 mg of
10% palladium-on charcoal. After 14 hours the mixture was ~'
suction filtered over Dicalite, rinsed with ethanol/ water and the `
2s filtrate was concentrated and dried in a high vacuum. There were
obtained 230 mg (100%) of O-(~-D-glucopyranosyl~-(1~6)-O~ -D-
glucopyranosyl)-(1~4)-o~,oc-D-trehalose as an amorphous powder,
[a]20= + 133.0 (c = 0.1, H2O).
30 C A solution ~f 213 mg of O-(a-D-glucopyranosyl)-(1~6)-O-(,B-
D-glucopyranosyl)-(1~4)-o~,a-D-trehalose in 8 ml of dry
dimethylformamide was stirred with 1.80 g of sulphur trioxide-
trimethylamine complex at 60-65C for 20 hours, whereby a
glassy, resinous precipitate separated. After decanting off the
35 solvent the residue was dissolved in 11 ml of 10% sodium acetate
solution and concentrated in a waterjet vacuum. The residue was
dissolved several times (12 x) in S0 ml of water each time and
evaporated in order to remove triethylamine. In order to remove
.
21 20.~3 ~ri~)$ I
salts, the residue was gel-chromatographed (Sephadex~ LH20, 150
g). Product fractions were evaporated and lyophilized. There
were obtained 390 mg (~58%) of sulphated O-a-D-
glucopyranosyl-(1~6)-0-,B-D-glucopyranosyl-(1~4)-oc,a-D-
trehalose as an amorphous powder, S = 20.36%, AS = about 3Ø
Example_ 10
A. 1.52 g of silver trifluoromethanlesulphonate, 4.13 g of
0 hepta-O-acetyl-oc-D-melibiosyl bromide [Jeanes at al, J. Am. Chem.
Soc., Z~, 3667-3672, (1953)] in 15 ml of absolute
dichloromethane and 0.76 ml of absolute tetramethylurea were
added successiYely and rapidly at 0-5C to a solution of 3.84 g of
2,2',3,3',4,6,6' -hepta-0-benzyl-4,S-O-benzylidene-a,a-D -trehalose
5 [S.Kato, K.Yogo, Bull. Chem. Soc. Jpn., 59, 411-14 (1986)] in 25 ml
of absolute dichloromethane. After stirring at 0-5C for a further
2 hours the reaction mixture was suction filtered over Dicalite and
rinsed with dichloromethane. The filtrate was washed twice with
saturated sodium bicarbonate solution, dried over magnesium
20 sulphate, filtered off, concentrated and chromatographed on
350 g of silica gel (70-230 mesh) with ethyl acetate/hexane 1 :4,
1:3 and 1:2 as the eluent. 2.~3 g (47%~ of pure 4-0-(hepta-0-
acetyl-,B-D-melibiosyl)-2,2',3,3',4,6,6'-hepta-0-benzyl-a,a-D -
trehalose were obtained as a foam, [a]2=+93.4(c=0.5,CHCl3).
B. A solution of 2.75 g of 4-0-(hepta-0-acetyl-,B-D-melibiosyl)-
2,2',3,3',4,6,6'-hepta-0-benzyl-~,oc-D-trehalose in 5.5 ml of
diethyl ether and 27 ml of methanol was treated at room
temperature with 5.5 ml of 1% sodium in methanol and stirred
30 for 2 1/2 hours. The reaction solution was then stirred with acidic
ion exchanger (Amberlite IR-12û) for 10 minutes, filtered off
under suction and rinsed with methanol. The filtrate was
evaporated and the residue was chromatographed on 75 g of
silica gel (70-230 mesh) with ethyl acetate/methanol/water
35 96:2:2 as the eluent. 1.53 g (68%) of deacylated product were
obtained, [CC]20= +115 (c = 0.2, CHC13). 1.296g (lOmmol) of this
in 40 ml of ethanol/H20 3:1 were hydrogenated at room
temperature in the presence of 0.7 g of 10% palladium-on-
2 ~22
charcoal. After 8 hours the reaction mixture was suction filtered
over Dicalite and rinsed with ethanol/water. The filtrate was
concentrated and dried in a high vacuum and gave 0.679 g
(100%) of 4-O-(~-D-melibiosyl)-a,oc-D-trehalose as an amorphous
s powder, [a]2D= + 143.2 (c = ~.5, H2O).
C A solution of 660 mg of 4-O-(,B-D-melibiosyl)-a,a-D-
trehalose in 15 ml of dry dimethylformamide was treated with
4.17 g (30 mmol) of sulphur trioxidle-trimethylamine complex
o and stirred at 60-65C for 20 hours, whereby a viscous syrup
separated after 3 hours. The solvent was decanted off a~d the
residue was dissolved in 25 ml of 10% sodium ace~ate solution
and concentrated in a waterjet vacuum. The residue was
dissolved several times (12 x) in 50 ml of water each time and
5 concentrated in order to remove triethylamine. In order to
remove salts, the residue was gel-chromatographed (Sephadex~'
LH20, 150 g). Fractions containing sulphated tetrasaccharide were
evaporated and lyophilized. There were obtained 1.59 g (about
80%) of sulphated 4-O-(~-D-melibiosyl)-a,a--D-trehalose, [OC]20 = ,`~
20 +135.2 (c = 1.0, H2O), S = 20.58%, AS = about 3Ø
Example 1 1
A. 0.37 g of absolute N,N,N,N-tetramethylurea and 0.76 g of
25 dry silver trifluoromethanesulphonate were added at 0-5C under
argon and with the exclusion of light to a solution of 1.74 g of dry
2,2',3 ,3',6'-penta-O-benzyl-4,6-O-benzylidene-o~,a-trehalose
(Carbohydr. Res. 63, 51, (1978)) in 12.5 ml of absolute dichloro-
methane. A solution of 1.92 g of acetobromo-D-lactose (J. Am.
30 Chem. Soc. 37, 1270 (1915)) in 7.5 ml of dichloromethane was
added dropwise. After stirring at 0-5C for 1.5 hours the reaction
solution was filtered over silica gel and washed with
dichloromethane. The organic solutions were washed with sodium
bicarbonate solution and water and dried over magnesium
3s sulphate. Chromatography on silica gel with ethyl acetate/hexane
1:2 as the eluent gave 1.54 g of pure 4'-O-(hepta-O-acetyl-~-D-
lactosyl)-2,2',3,3 ',6'-penta-O-benzyl-4,6-O-benzylidene-a,cc-D -
trehalose as a white resin, FAB-MS (1521.2 (M+~a)+).
2~
23
B. A solution of 1.22 g of 4'-O-(hepta-O-acetyl-~-D-lactosyl)-
2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-oc,a-D-trehalose in
12.2 ml of absolute methanol and 2.5 ml of absolute ether was
s treated with 3.1 ml of 1 % sodium methylate solution. After
5.5 hours ar room temperature the mixture was neutralized with
acidic ion exchanger, filtered and concentra~ed. Chromatography
on silica gel with ethyl acetate/methanol/water 85:10:5 as the
eluent gave 0.91 g of pure 4'-O-(~-D-lactosyl)-2,2',3,3',6'-penta-O-
0 benzyl-4,6-O-benzylidene-a,a-D-trehalose as a white resin, FAB-
~IS (1227.3 (M+Na)+).
C A solution of 0.90 g of 4'-O-(,~-D-lactosyl)-2,2',3,3',6'-penta-
O-benzyl-4,6-O-benzylidene-a,cc-D-trehalose in 28 ml of
ethanol/water 3:1 was hydrogenated at room temperature for
6 hours with 0.5 g of 10% palladium-on-charcoal. Filtration over
a filter aid and rinsing gave, after drying in a high vacuum, 0.51 g
of pure 4'-O-(~-D-lactosyl)-a,a-D-trehalose as a white resin, FAB-
MS (689.0 (M+Na)+).
D. A solution of 0.49 g of well-dried 4'-O-(,B-D-lactosyl)-a,a-D-
trehalose in 11.2 ml of absolute dimethylformamide was stirred
at 65C for 22 hours in the presence of 3.094 g of sulphur
trioxide-trimethylamine complex, whereby a viscous syrup
2s separated. Working-up as described in Example lG gave ].1 g of
sulphated 4'-O-(,B-D-lactosyl)-a,a-D-trehalose, S = 19.93%, AS
about 2.9.
Example 12
A. 0.30 g of absolute N,N,N,N-tetramethylurea and 0.625 g of
dry silver trifluoromethanesulphonate were added at 0-5C under
argon and with the exclusion of light to a solution of 1.43 g of dry
2,2',3,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,a-D-trehalose
3s (Carbohydr. Res. 6~, 51, (1978)) in lO.Oml of absolute
dichloromethane. A solution of 1.70 g of acetobromo-D-
gentiobiose (J. Am. Chem. Soc. 49, 3170 (1927)) in 6.0ml of
methylene chloride was added dropwise. After stirring at room
,, . ... . . . . .. . . . . .. . . ....... . . . ... .. . .. ... .. . . _ . . . . . . .. ..... . _ _ _
2 ~ l3 ~r~
24
temperature for 20 hours the reactis)n solution was filtered over
silica gel and washed ~vith dichloromethane. The organic solutions
were washed with sodium bicarbonate solution and wate~ and
dried over magnesium sulphate. Chromatography on silica gel
s with ethyl acetate/toluene 1:2 as the eluent gave 1.22 g of pure
4'-O-(hepta-O-acetyl-~-D-gentiobiosyl)-2,2',3,3',- 6'-penta-O-
benzyl-4,6-O-benzylidene-a,a-trehalose as a white resin, ~AB-MS
(1521 .2 (M+Na)+).
B. A solution of 1.22 g of 4'-O-(hepta-O-acetyl-,~-D-gentio-
biosyl)-2,2',3 ,3',6'-penta-O-benzyl-4,6-O-benzylidene-a,a-D -
trehalose in 12.0 ml of absolute methanol and 2.5 ml of absolute
ether was treated with 2.44 ml of 1% sodium methylate solution.
After 5.5 hours ar room temperature the mixture was neutralized
l 5 with acidic ion exchanger, filtered and concentrated. Chroma-
tography on silica gel with ethyl acetate/methanol/water 85:10:5
as the eluent gave O.50g of pure 4'-O-(,B-D-gen~iobiosyl)-2,2',3,-
3',6'-penta-O-benzyl-4,6-O-benzylidene-oc,a-D-trehalose as a
white resin, FAB-MS (1227.4 (M+Na)+).
C A solution of 0.48 g of 4'-O-(~-D-gentiobiosyl)-2,2',3,3',6'-
penta-O-benzyl-4,6-O-benzylidene-a,a-D-trehalose in 11 ml of
ethanol and 3.7 ml of water 3:1 was hydrogenated at room
temperature for 6 hours with 0.27 g of 10% palladium-on-
25 charcoal. ~iltration over a filter aid and rinsing gave, after dryingin a high vacuum, 0.26 g of pure 4'-O-(,B-D-gentiobiosyl)-a,a-D-
trehalose, as a white resin, FAB-MS (689.0 ((M+Na)+).
D. A solution of 0.20 g of well-dried 4'-O-(~-D-gentiobiosyl)-
30 c~,a-D-trehalose in 5.0 ml of absolute dimethylformamide was
stiIred at 65C for 22 hours in the presence vf 1.26 g of sulphur
trioxide-trimethylamine complex, whereby a viscous syrup
separated. Working-up as described in Example lG gave 0.50 g
of sulphated 4'-O-(,B-D-gentiobiosyl)-a,a-D-trehalose, S = 20.25%,
3s AS about 3.3.
, . . .. . . . . .. . .... . . . . . .. .... .. . . .. . ~ . . . , . . . ~ . . . .
2 '') ~ ~ ~ ? 1
E~xample 13
A. 50 ml of acetic anhydride were added to a suspension of
S g of 3-O-~B-D-galactopyranosyl-D-arabinose in 75 ml of
5 pyridine and the mixture was stirred at room temperature for
6 honrs. The reaction solution was concentrated in a waterjet
vacuu n at 140C bath temperature. The syrupy residue was
treated with 200 ml of ice-wa~er andl extracted twice with 200 ml
of e~hyl acetate each time. The extracts were washed twice with
0 cold 5% H2SO4, twice with saturated sodium bicarbonate solution
and once with saturated sodium chloride solution, dried over
magnesium sulphate, filtered of~ and evaporated and dried in a
high vacuum overnight. There were obtained 8.84 g (91~o) of
1 ,2,4-tri-O-acetyl-3 -0-(2,3,4,6-tetra-O-acetyl-,B-D -
5 galactopyranosyl)-D-arabinose (according to NMR the mixture also
contained furanose derivative). 7.6 g thereof were dissolved in
15 ml of dichloromethane, cooled to 0C and treated within lS'
- with 45 ml of 33% hydrobromic acid in acetic acid and stiTred at
0C for a further 2hours. The reaction mixture was poured on to
20 250 ml of ice-water and extracted three times with 100 ml of
dichloromethane each time. The extracts were washed twice with
100 ml of ice-water each time and twice with 100 ml of cold
saturated sodium bicarbonate solution each time, dried over
magnesium sulphate, filtered off and evaporated at <25C. The
2s residue was chromatographed on 200 g of silica gel (70-230
mesh) with dichloromethane/diethyl ether 9:1 and 4:1 as the
eluent. There were obtained 4.15 g (53%) of 2,4-di-O-acetyl-3-O-
(2,3,4,6-tetra-O-acetyl-,i~-D-galacto- pyranosyl)-,l~-D-
arabinopyranosyl bromide in the form of a foam, [a]20= -148.8 (c
30 = 0.5, CHC13).
B. 2.0 ml of absolute tetramethylurea and 1.55 g of silver
trifluoromethanesulphonate were added in succession at 0-5C to
a solution of 3.52 g of high vacuum-dried 2,2',3,3',6'-penta-O-
35 benzyl-4,6-O-benzylidene-a,a-D-trehalose and 3.76 g of 2,4-di-O
acetyl-3 -0-(2,3,4,6-tetra-O-acetyl-~-D-galactopyranosyl)-,B-D -
arabinopyranosyl bromide in 30 ml of absolute dichloro-
methane. After ome hour at 0-~C the reaction mixture was
3 ~
26
sucti~n filtered over Celite, rinsed with dichloromethane, the
f;ltrate was washed twice with saturated sodium bicarbonate
solution, dried over magnesium sulphate, filtered off and evapor-
ated. The residue was chromatographed on 190 g of silica gel (70-
s 230 mesh) with ethyl acetate/hexane 1 :2 and 1: ] as the eluent.There were obtained 3.33 g (58%) of 0-(2,3,4,6-tetra-0-acetyl-~-
D-galactopyranosyl)-(1 ~ 3)-0-(2,4-di-0-acetyl-a-D -
arabinopyran osyl)- (1 ~ 4) -2,2' ,3,3 ' ,6'-penta-0- ben~yl-4,6- 0 -
benzylidene-a,a-D-trehalose as a foam, [a]20= +42.0 (c = 0.5,
I o CHC13).
C 3.4ml of 1% Na in methanol were added to a solution of
3.4 g of 0-(2,3,4,6-tetra-0-acetyl-~-D-galactopyranosyl)-(1~3)-0-
(2,4-di-0-acetyl-a-D-arabinopyranosyl)-(1 ~4)-2,2',3,3',6'-penta-
S 0-benzyl-4,6-0-benzylidene-a,a-D-trehalose in 7 ml of diethyl
ether and 35 ml of methanol and the mixture was stirred at room
temperature for 1.5 hours. The reaction solution was neutralized
with ion exchanger (Amberlite IR-120), stirred for 15 minutes,
filtered 03ff, rinsed with methanol and the filtra~e was evaporated.
20 The residue was chromatographed on 90 g of silica gel (70-230
mesh) with ethyl acetate/methanol/ water 93 :5 :2 as the eluent.
1.93 g (69%) of deacylated product were obtained as a foam, ~a]23
= +52.0 (c = 0.5, CHC13). 1.80 g (1.53 mmol) of this in 60 ml of
ethanol/water 3: 1 were hydrogenated at room temperature in the
2s presence of 1.0 g of 10% palladium-on charcoal. After 16 hours
the reaction mixture was suction filtered over 1:3icalite, rinsed with
ethanol/water and ~he filtrate was evaporated and dried. There
were obtained 1.12 g of a product which, for ~urther purification,
was acetylated using pyridine (22 ml) and acetic anhydride (11
30 ml). After 22 hours at room temperatule the reaction mixture
was evaporated and chromatographed on 85 g of silica gel
(70-230 mesh) with ethyl acetate/hexane 1: 1 and 2: 1 as the
eluent. There were obtained 1.06 g (59%) of 0-(2,3,4,6-tetra-0-
acetyl-,l~-D-galactopyranosyl)-(1 ~3)-0-(2,4-di-0-acetyl-a-D-
35 arabinopyranosyl)-(1~4)-hepta-0-acetyl-a,a-trehalose as a foam,
[a] D = + 68.00 (c = 0.4, CHC13). 1.02 g (0.86 m3mol) of this were
dissolved in 30 ml of methanol/ dimethoxyethane/water l:l:1
and treated dropw3ise with 1 % sodium methylate solution (pH
. ... ., . . .... . . .. . _ . _ .. . . . .. ... _ _ _ _ _ _ _ _
.
2 ~ 3 :1
27
1~-13) until saponification was complete. After 6 hours the
reaction solution was neutralized with ion exchanger (Amberlite
IR-120), stirred for 15 minutes, suction filtered, rinsed with
methanol/water and the filtrate was evaporated and dried in a
s high vacuum. There were obtained 480 mg (88%) of O-~-D-
galactopyranosyl-(1~3)-O-a-D-arabino- pyranosyl-(1~4)-a,a-
trehalose as an amorphous powder, [a]20= + 9.4O (c = 0.1, H2O).
D. A solution of 440 mg (0.69 mmol) of O-,B-D-galacto-
o pyranos yl- ( 1 ~ 3 ) - O-a -D- arabinopyran osyl -(1~ 4)-a, a-trehalose in
15 ml of dry dimethylformamide was treated with 2.87 g
(20.7 mmol) of sulphur trioxide-trimethylamine complex and
stirred at 60-65C under argon for 20 hours. During this time a
viscous precipitate separated. Working-up as described in
5 Example lG gave 960 mg (about 75%) of sulphated O-~-D-
galactopyranosyl-(1~3)-O-a-D-arabinopyranosyl-(1~4)-a,a-
trehalose as an amorphous powder, [a]2D= +158.8 (c = 0.5, H2O), S
= 19.24%, AS about 3Ø
Example 14
For the production of an injection solution, 5 mg of a
compound of formula I and 9 mg of sodium chloride are dissolved
in water ad 1 ml. The solution is treated with ascorbic acid
2s (0.5 mg/ml) and benzyl alcohol (0.1 mg/ml) and then filtered
s~erile.