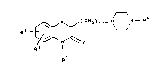Note: Descriptions are shown in the official language in which they were submitted.
2~ J~
HETEROCYCLICALLY SUBSTITUTED PIPERAZINOALKYLBENZOXAZINE
AND PIPERAZINOALKYLBENZOTHIAZINE COMPOUNDS, PROCESSES
FOR PREPARING THEM, AND MEDICAMENTS CONTAINING THEM
Backaround of the Invention
The present invention relates to novel benzoxazine-3-
one and benzothiazine-3-one derivatives which carry in the
2-position a piperazinoalkyl group substituted by a
heterocycle, and the corresponding 3-thione derivatives and
salts thereof; to processes for preparing such compounds,
and to pharmaceutical compositions containing such
compounds.
1,4-Benzoxazine-3-one derivatives which carry a
phenylpiperazinoalkyl group in the 2-position are known from
published European pate~t application No. EP 233,728. These
compounds have pronounced hypoten~ive and vasodilating
effects.
Summarv of the Invention
It is the object of the present invention to provide
new pharmaceutically active compounds which can be used as
anti-allergic agents.
Another object of the invention is to provide new
benzoxazine derivatives having valuable pharmacological
properties.
These and other objects of the invention are achieved
by providing a compound corresponding to the L ormula I
.
2 ~
wherein
X denotes oxygen or sulfur,
Y denotes oxygen or sulfur,
Rl denotes hydrogen or lower alkyl,
R2 denotes hydrogen, lower alkyl, halogen, lower alkoxy,
hydroxy, nitro or trifluoromethyl, and
R3 denotes hydrogen, lower alkyl, halogen or lower alkoxy,
or
R2 and R3 are bonded to adjacent carbon atoms and together
denote an alkylenedioxy group having 1-2 carbon atoms,
n is an integer from O to 4, and
R4 is a 6-membered unsaturated heterocycle containing 1 or
2 nitrogen atoms not directly bonded to the piperazine
2S ring, said heterocycle being substituted by O to 2
substituents bonded to carbon atoms selected from the
group consisting of lower alkyl, lower alkoxy and
halogen,
and physiologically acceptable acid addition salts thereof.
According to a further aspect of the invention, the
objects are achieved by providing a pharmaceutical
composition comprising an effective anti-inflammatory or
anti-allergic amount of a compound as described above and at
least one conventional pharmaceutical adjuvant.
In accordance with another aspect of the invention, the
objects are achieved by providing a process for preparing a
compound corresponding to the formula I
2~5~
~ 3 X~ C H2) n ~ N -- R
R
wherein
X denotes oxygen or sulfur,
Y denotes oxygen or sulfur,
Rl denotes hydrogen or lower alkyl,
R2 denotes hydrogen, lower alkyl, halogen, lower alkoxy,
hydroxy, nitro or trifluoromethyl, and
R3 denotes hydrogen, lower alkyl, halogen or lower alkoxy,
or
R2 and R3 are bonded to adjacent carbon atoms and together
denote an alkylenedioxy group having 1-2 carbon atoms,
n is an integer from 0 to 4, and
R4 denotes a 6-membered unsaturated heterocycle containing
1 or 2 nitrogen atoms not directly bonded to the
piperazine ring, said heterocycle being substituted by
0 to 2 substituents bonded to carbon atoms selected
from the group consisting of lower alkyl, lower alkoxy
and halogen,
and acid addition salts thereof, wherein said process5 comprises:
a) to prepare a compound corresponding to the formula
Ia
R~
2 ~ J ~ ~
wherein X, Rl, R2, R3, n and R~ have the above meanings,
reacting a compound corresponding to the formula II
R
R
wherein
X, Rl, R3 and n have the above meanings,
R2 has the meaning given for R2 except that any hydroxy
group is protected by a subsequently removable
protective group, and
L denotes an aminolytically cleavable radical,
with a piperazine derivative corresponding to the formula
III ~.
H-N N-~4
/
wherein R4 has the above meaning, or
b) reacting a compound corresponding to the formula IV
~ X~tt H~3n-- N N--H
wherein X, Rl, 2, R3 and n have the above meanings,
with a compound corresponding to the formula V
R4 - L'
wherein R4 has the above meaning and L' denotes halogen, and
then removing any hydroxy protective group, or
2 ~ 3
c) converting a compound corresponding to the formula
Ia to a compound corresponding to the formula Ib
~ ~ ~X~ ~ H ~ ~ n ~
Rl
wherein X, Rl, R2, R3, n and R4 have the above meanings.
Detailed Descri~tion of Preferred Embodiments
The novel heterocyclically substituted compounds of the
invention have ~een found to have valuable pharmacological
properties, to show anti-inflammatory and anti-allergic
effects, and to have an advantageous activity profile with
low toxicity and good tolerance. Due to- their activity
profile, the compounds of the invention are suitable for use
as anti-inflammatory active ingredients and anti-allergic
agents in the treatment of inflammatory and allergic
diseases.
2S The present invention therefore relates to novel
compounds of the general formula I
R2 ~CcN2~n - N 11~
R1
wherein
X denotes oxygen or sulfur,
Y denotes oxygen or sulfur,
Rl denotes hydrogen or lower alkyl,
R2 denotes hydrogen, lower alkyl, halogen, lower alkoxy,
hydroxy, nitro or trifluoromethyl, and~ R3 denotes hydrogen, lower alkyl, halogen or lower alkoxy,
or
- 5 -
'
.
2~;4~
R2 and R3 are bonded to adjacent carbon atoms and together
denote an alkylenedioxy group with 1-2 carbon atoms,
n is an integer from 0 to 4, and
R4 is a 6-membered unsaturated heterocycle containing 1 or
2 nitrogen atoms not directly bonded to the piperazine
ring, which heterocycle may optionally be substituted
by 1-2 substituents bonded to carbon atoms and selected
from the group consisting of lower alkyl, lower alkoxy
and halogen,
and physiologically acceptable acid addition salts thereof.
In so far as the substituents R2 and R3 in the compounds
of formula I and the substituents in the group R4 represent
or contain lower alkyl groups, these may be straight or
branched and contain in particular 1 - 4, preferably 1 - 2,
carbon atoms and represent in particular methyl or methoxy.
In so far as the substituents represent halogen, they may be
fluorine, chlorine or bromine, preferably chlorine. The
benzene ring of the ring structure may advantageously be
unsubstituted. In so far as the benzene ring is substituted
by a substituent R2, or by two substituents R2 and R3, lower
alkyl substituents, for example methyl substituents, are
considered especially suitable.
The substituent Rl advantageously represents hydrogen.
If Rl represents lower alkyl, it may be straight-chain or
branched and may contain 1 - 4, in particular 1 - 2, carbon
atoms.
In the compounds of the formula I, n represents 0 - 4.
An alkylene chain having 3 or 4 members has proved to be
particularly advantageous.
The substituent R4 may represent an unsaturated
heterocycle containing one or two nitrogen atoms, for
example a heteroaryl group. Suitable R4 groups include, for
example, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl
residues, particularly pyridyl groups. If R4 represents a
pyridyl group, it is preferred to use a pyrid-2-yl group,
which optionally may be substituted. For example,
2~6~a~
unsubstituted pyridyl radicals or pyridyl radicals which are
substituted by lower alkyl, in particular methyl, are
suitable. A 4-methylpyrid-2~yl group is particularly
advantageous.
In accordance with the invention, the novel compounds
of formula I and their acid addition salts are obtained in
a known manner by
a) to prepare compounds of the general formula Ia
R2 ~CH2)n-- N N-- R4
R3
~1
in which X, Rl, R2, R3, n and R4 have the above meanings,
compounds of the general formula II
RZ ~ ~ ~ LCHzJn - L
R
R1
in which X, Rl, R3 and n have the above meanings, and R2 has
the meaning given for R2, but wherein a hydroxy group is
protected by a subsequently removable protective group, and
L is an aminolytically cleavable residue, especially
halogen,
are reacted with piperazine derivatives of the general
formula III
~ 4
H-N N-R
/
- 7 -
-,
.. ~ , .
; ~
' .
.:
,
9 ~ ~
in which R~ has the above meaning, or
b) compounds of the general formula IV
R 2 ~ ~X~ C H Z ) n-- N N-- H
R
in which X, Rl, R2, R3 and n have the a~ove meanings,
are reacted with compounds of the general formula V
q4 ~ I
in which R4 has the above meaning and L' denotes halogen, and0 any hydroxy protective group is subsequently removed, or
c) compounds of the general formula Ia are converted to
compounds of the general formula Ib
R2 ~XR~CH2)n-- N N-- R4
R1
in which X, Rl, R2, R3, n and R4 have the above meanings, and
optionally compounds of the general formula I which are
obtained in which Rl represents hydrogen, are alkylated to
form compounds of the general formula I in which R1
represents lower alkyl, and/or in compounds of the general
formula I which are obtained in which R2 represents methoxy,
the methoxy group is cleaved to form a hydroxy group, and
optionally free compounds of the formula I are converted to
their acid addition salts or the acid addition salts are
converted to the corresponding free compounds of formula I.
-- 8 --
2 ~ er~ J ~
The reaction of compounds of formula II with compounds
of formula III according to process variant a) may be
carried out by conventional methods for alkylating amines.
The reaction is advantageously carried out under basic
conditions in an organic solvent which is inert under the
reaction conditions.
Halogens, such as chlorine, bromine or iodine,
preferably bromine or chlorine, or also an acyloxy radical
O-Z, in which Z represents a lower alkanoyl group or an
organic sulfonic acid group, for example the radical of a
lower alkanesulfonic acid such as, for example, methane
sulfonic acid, or of an aromatic sulfonic acid such as
benzenesulfonic acid or benzenesulfonic acids substituted by
lower alkyl or by halogen, for example toluenesulfonic acids
or bromobenzenesulfonic acids, are suitable as
aminolytically cleavable residues L in the compounds of
formula II.
Aprotic solvents in particular, for example aromatic
hydrocarbons such as toluene, xylene or benzene; cyclic
ethers such as dioxane; dimethylformamide; or lower alkanols
such as ethanol; or mixtures of the foregoing solvents are
suitable as inert organic solvents.
The process is advantageously carried out at elevated
temperatures, for example temperatures between 50 and 150C,
preferably at the reflux temperature of the solvent.
The reaction is advantageously carried out with
addition of an organic or inorganic base. However, an
excess of the compound of the formula III may also be used,
and this may serve as an internal base. Examples of
suitable organic bases include tertiary organic amines, in
particular tertiary lower alkyl amines such as triethylamine
or tripropylamine, N-lower alkyl morpholines or N-lower
alkyl piperidines. Suitable inorganic bases include in
particular alkali metal carbonates or bicarbonates.
The reaction time may be between 2 and 8 hours
depending on the reaction conditions.
:,
. . . : : ~ :
.~,
.. , ':
2 Q o~ 3 3
Known ether protective groups, which can subsequently
be removed solvolytically or hydrogenolytically ln a known
manner, for example lower alkyl or benzyl groups, may be
selected as protective groups for any hydroxy group R2 which
may be present.
The reaction of compounds of formula IV with compounds
of formula V according to process variant b) may be carried
out by conventional methods for alkylating amines. For
example, it may be carried out in the manner described for
reacting compounds of formula II with compounds of formula
III.
The conversion of the 3-one group of the compounds of
formula Ia into the 3-thione group of the compounds of
formula Ib according to process varian$ c) may be carried
out by conventional methods for exchanging oxygen for sulfur
in oxo compounds. Thus the compounds of formula Ib may be
prepared in a known manner, for example by treating the
compounds of formula Ia with a phosphorus pentasulfide (for
example P4Slo) or also according to the method described by
20 Lawesson et al. (see Bull. Soc. Chim. Belg. 87, 525-534
(1978)) by reacting with 2,4-bis(methoxyphenyl)-1,3-dithia-
2,4-diphosphetane-2,4-disulfide of the formula VI
S s
2S CH30~ \s/ll OCH3
(= known as Lawesson,s reagent). The deoxosulfurization is
advantageously carried out in an organic solvent which is
inert under the reaction conditions, for example an aromatic
hydrocarbon such as xylene or toluene, at elevated
temperatures, for example temperatures between 50 and 150C,
advantageously at the reflux temperature of the reaction
mixture. When the reaction is completed, the sulfurized
-- 10 --
2 ~
compounds may be separated by filtration from the phosphorus
derivatives.
If desired, compounds of formula I which are obtained
in which Rl represents hydrogen may be subsequently alkylated
in a known manner to form the corresponding N-alkyl
compounds. Suitable alkylating agents include alkyl
halides, particularly iodides, alkyl sulfates or alkyl
sulfonates. A compound of formula I containing an amide or
thiamide group is advantageously reacted initially with a
strong base, such as for example an alkali metal hydride,
alkali metal amide or alkali metal alcoholate, in an inert
polar organic solvent and then further reacted with the
alkylating agent. The reaction may be carried out at a
temperature of 0C up to the reflux temperature of the
solvent. Dimethylformamide or cyclic ethers, such as
tetrahydrofuran or dioxane, are suitable as solvents
depending on the base used, or, if the base is a metal
alcoholate, also the corresponding alcohols. Thus, for
example, the reaction may advantageously be carried out in
dimethylformamide using sodium hydride.
In compounds of formula I in which R2 denotes methoxy,
the methoxy group may be cleaved to form the hydroxy group
in a known manner using methods suitable for cleaving
methoxyaryl ethers. For example, the ether may be cleaved
by treating with hydrogen iodide in a solvent which is inert
under the reaction conditions, for example acetanhydride, or
with iodotrimethylsilane or boron tribromide.
The compounds of the formula I may be isolated from the
reaction mixture and purified in a known manner. Acid
addition salts may be converted to the free bases in a
conventional manner, and if desired, the free bases may be
converted in a known manner to pharmacologically acceptable
acid addition salts.
Suitable pharmacologically acceptable acid addition
salts of the compounds of formula I include, for example
their salts with inorganic acids, for example hydrohalic
'. ' -
,- :
2 ~
acids, particularly hydrochloric acid, sulfuric acid or
phosphoric acids, or with organic acids, for example lower
aliphatic monocarboxylic or dicarboxylic acids, such as
maleic acid, fumaric acid, lactic acid, tartaric acid or
acetic acid, or sulfonic acids, for example lower alkyl
sulfonic acids such as methanesulfonic acid, or
benzenesulfonic acids optionally substituted in the benzene
ring by halogen or lower alkyl, such as p-toluenesulfonic
acid, or cyclohexylaminesulfonic acid.
The compounds of the formula I contain an asymmetric
center in the 2-position of the benzoxazine or benzothiazine
structure and may exist in two optically active enantiomeric
forms or as a racemate. The present invention includes both
the racemic mixtures as well as the pure optical isomers of
the compounds of formula I.
If racemates of compounds of formula II or formula IV
are used in the synthesis, the resulting compounds of
formula I are obtained in the form of racemates. Optically
active compounds corresponding to formula I may be obtained
starting from optically active forms of compounds of formula
II or formula IV. Optically active compounds of formula I
may be isolated from racemic mixtures in a known manner, for
example by chromatographic separation on chiral separating
materials or by reacting with suitable optically active
acids, for example tartaric acid or 10-camphorsulfonic acid,
and then separating the resulting salts into their optically
active antipodes by fractional crystallization.
The starting compounds of formula II may be obtained
starting from 2-aminophenol derivatives corresponding to the
formula VII
R2'~R ~
. . -. , - ;
20~a9
in which Rl, R2, R3 and X have the above meanings.
Accordingly, the compounds of formula VII may be condensed
in a known manner wiin a B-bromo-acylbromide of formula VIII
8r
\CH _ ( CH) n -- L
ar_ C= O
in which L has the above meaning, and n' denotes 1 - 4, to
form compounds of the formula IIa
2 ~X~,CCHz~n _ L
R
R3
R
25 in which Rl, R2, R3, X, n' and L have the above meanings.
The condensation may be carried out in a solvent which is
inert under the reaction conditions, for example a
halogenated hydrocarbon such as chloroform, in the presence
of a base, for example an alkali metal hydrogen carbonate or
30 alkali metal carbonate, and is advantageously carried out in
the presence of a transfer catalyst, for example
benzyltrimethylammonium chloride. The compounds of the
formula VII can also be reacted in a known manner with
methyl B-bromo-alkanecarboxylates of the formula IX
R~._~XN~CCH7~n~ N N--H
R3 R1
-- 13 --
- :
~ ~ .
.
.' '', ~,,
' ~. ,; :. ' ' ' ` .:
': ~ -, ~' , .:-
,
2 ~
in which n' and L have the above meanings, to form compounds
of the formula IIa. The reaction may, for example, be
carried out in dimethylformamide in the presence of an
inorganic base, for example an alkali metal carbonate.
Compounds of the general formula VIIa
R2'~XH
NH
R 3
in which Rl, R2 and R3 have the above meanings, are known or
may be obtained in a known manner starting from compounds of
the general formula XIII
~ ~ / ~ 2
in which R2 and R3 have the above meanings. Compounds of the
formula XIII may be alkylated to introduce an alkyl radical
Rl in a known manner to form compounds of the general formula
XIV
2 ~C /~ NH
- 14 -
2 ~
in which R2 and R3 have the above meanings, and Rl denotes
lower alkyl.
~ ompounds of formula XIII or formula ~IV may be
converted to compounds of formula VIIa in a known manner by
thermal dissociation in an aqueous alkaline medium, for
example by heating in alkali metal hydroxide solution.
Compounds of the general formula VIIb
~, ~ `N H
R~ ¦
R
in which Rl, R2 and R3 have the above meanings, are known or
may be obtained by known methods or analogously to known
methods. 2-Alkylaminophenol compounds (R~ = lower alkyl) may
be obtained starting from the corresponding 2-aminophenol
compounds. For this purpose they are initially acylated,
whereby both the phenolic hydroxy group and the amino group
are provided with a protective acyl group. The amide group
in the resulting ester-amide compounds is alkylated in a
known manner by reacting the compounds with an alkylating
agent, for example a lower alkyl halide, alkyl sulfate or
alkyl sulfonate, in the presence of a strong base, for
example an alkali metal hydride or alkali metal hydroxide,
optionally in the presence of a transfer catalyst, for
example benzyltrimethylammonium chloride. The reaction may
take place, for example under the conditions given above for
the subsequent alkylation of compounds of the formula I.
After the alkylation is completed, the protective acyl
- 15 -
2 ~
groups may then be removed in a known manner by acid or
alkaline hydrolysis.
Compounds of formula II in which n denotes 0, may be
obtained starting from compounds of the formula X
R Z
R
R~
in which Rl, R2, R3 and X have the above meanings. Hence,
to prepare compounds of the formula IIb
2~ ~ X
R
R1
in which Rl, R2 and R3 have the above meanings, and L'
denotes halogen. A halogen substituent L', particularly
chlorine, can be introduced into compounds of the formula X
in a known manner by treatment with a halogenating agent,
for example sulfuryl chloride. The chlorination may be
carried out in a solvent which is inert under the reaction
conditions, for example a halogenated hydrocarbon such as
dichloromethane.
Compounds of formula X are known or may be obtained in
a known manner, for example by condensing compounds of
formula VII with chloroacetyl chloride. The condensation
- 16 -
2~5~9~3
may be carried out under the reaction conditions given above
for preparing compounds of formula IIa.
Compounds of the formula IV have not previously been
described in the literature and constitute novel, valuable
intermediate products for synthesizing pharmacologically
active compounds, for example the compounds of formula I.
Compounds of the formula IV may be obtained according
to known methods by reacting, for example, compounds of
formula II with an excess of piperazine. The reaction may
be carried out following conventional methods for alkylating
amines, for example under the conditions described above for
reacting compounds of the formula II with compounds of the
formula III.
Compounds of the formula IV may also be obtained from
compounds of the general formula XI
~ ~ ~ ~cu~ N - O
in which Rl, R2, R3 and X have the above meanings, and Q
denotes an amine protective group, by removing the amine
protective group in a known manner. Suitable amine
protective groups include the usual protective groups used
for protecting an amino function, for example hydrolytically
removable acyl groups or hydrogenolytically removable benzyl
groups. Suitable protective groups are known, for example
from E. McOmie, "Protective Groups in organic Chemistry";
Plenum Press, London (1971), page 44 ff. The formyl group
and lower carbalkoxy protective groups are particularly
suitable. They may be removed in a known manner by acid or
alkaline hydrolysis.
Compounds of the formula XI may be obtained in a known
manner, for example by reacting compounds of the formula II
with compounds of the formula XII
- 17 -
- . . .. . . . .
:
, ~ ,
:~ .
2 ~ $ ~
~
H-N N-Q
/
in which Q has the above meaning. The reaction may be
carried out following conventional methods for alkylating
amines, for example under the reaction conditions described
for the reaction of compounds of formula II with compounds
of formula III.
Compounds of the formula III are known or may be
prepared following known methods, for example by reacting
compounds of formula V with an excess of piperazine or with
a compound of formula XII and subsequently removing the
protective group Q.
The compounds of formula I and their pharmacologically
acceptable acid addition salts are characterized by
interesting pharmacological properties and have anti-
inflammatory and anti-allergic effects. In particular, the
compounds exhibit an advantageous activity profile for
treating asthmatic disorders with low toxicity and good
compatibility.
Asthma is a chronic inflammatory lung disease which is
characterized by an episodic, reversible obstruction of the
respiratory passages. It is generally assumed that the
initiation of asthmatic symptoms and attacks stems from a
parenchymal and interstitial cell type known as a mast cell.
These mast cells contain pre-formed inflammation mediators
and spasmogens, in particular histamine. They are also
capable of synthesizing a variety of mediators derived from
`35 membrane lipids. Mast cells also act in conjunction with a
number of associated cells which are all capable of
synthesizing inflammatory and pro-inflammatory mediators.
- 18 -
2 ~
As long as no allergy-inducing conditions are present,
the mast cells are in a quasi dormant waiting state. The
key to allergic reactions lies in the presence of high
concentrations of circulating IgE antibodies. When these
antibodies bind to a corresponding antigen, they activate
both the degranulation and release of pre-formed mediators
as well as the synthesis of other mediators.
Since asthma is an inflammatory obstructive lung
disease, the therapy is based on essentially two approaches:
alleviating the symptomatic complaints by administering
bronchodilators such as ~-sympathicomimetic agents, xanthine
derivatives and anti-cholinergic agents; administration of
anti-inflammatory active ingredients such as disodium
cromoglycinate and steroids; and targeted therapy directed
at specific mediators such as histamine, for example.
Treatment to alleviate the symptomatic complaints is
adequate in about 50 % of asthmatics, but does not
contribute anything to alleviating the cause, i.e. the
inflammation. Anti-inflammatory active ingredients may
control the inflammation, but often have undesirable side-
effects and are frequently administered simultaneously with
bronchodilators. Targeted therapy directed at a specific
mediator alone is totally inadequate, since there are
plethora of mediators.
The compounds of the invention are distinguished in
that they have an anti-inflammatory effect and act in
targeted fashion against one or more of the three types of
mediators: histamine, leucotrienes and blood platelet
aggregating factor, which contribute not only to acute
bronchospasms but also to maintenance of chronic
inflammation, or are also active against the respective
target cells via mediator-specific receptors.
The anti-inflammatory and anti-allergic properties of
the compounds of the invention can be demonstrated in vitro
and in vivo by standard pharmacological test methods.
-- 19 --
- ~
2~6~9
Descri~tion of the Test Methods
1. Determining inhibition of passive cutaneous anaphylaxis
(PC~) and histamine-induced anaphylactoid cutaneous reaction
The PCA reaction is carried out following the methods
5described by Goose et al. (J.N. Immunology 16 (1969), 749)
and by Martin et al. (Arch. Pharmacol. 316 (1981), 186).
The IgE-rich ovalbumin antiserum used in the test was
obtained from immunized Brown-Norway rats. The were
immunized with an intraperitoneal injection of a mixture of
100 ~q of ovalbumin with a Bordetella pertussis suspension
(VaxicoqTM, Manufactured by Institute Merieux, containing 5
X 109 organisms and 1.25 mg Al(OH)3). After 20 days the
animals were re-immunized with a further intraperitoneal
injection of a solution of 10 ~g ovalbumin in 0.5 ml
physiological saline solution. After a further four days
the blood was removed from the animals and centrifuged. The
resulting antiserum was stored at -20C until use.
The inhibition of the passive cutaneous anaphylaxis and
of the anaphylactoid cutaneous reaction induced by histamine
was determined as follows:
Sprague-Dawley rats having a body weight of 150 - 180
g were injected intradermally in one flank with 50 ~l of a
1:75 dilution of the IgE-rich ovalbumin antiserum in
physiological saline solution to passively sensitize them to
ovalbumin.
24 hours after sensitization a solution of 8.25 mg/kg
of ovalbumin and 26.4 mg/kg of a blue dye (Evans's Blue) was
administered to the rats intravenously according to Martin
et al. to trigger passive cutaneous anaphylaxis. The
ovalbumin challenge resulted in a local anaphylactic
reaction at the point where the antiserum was injected.
To determine the histamine-induced anaphylactoid skin
reaction, the animals are injected intradermally in the
flank opposite to the antiserum administration with 50 ~l of
a physiological saline solution containing 0.8 mg/ml
- 20 -
, ~
~. ,',,. . ,, ' ~
histamine directly before the intravenous injection of the
solution containing the ovalbumin and the blue dye.
On the day of the experiment the test substances were
dissolved in distilled water which contained 1 vol.%
dimethylformamide and 1 vol.% of TweenTM 20 (=
polyoxyethylene (20) sorbitan monolaurate). One hour before
administration of the ovalbumin challenge, each animal was
orally administered 2 x 105 mole/kg of test substance in 0.5
ml of solution. For comparison purposes a control group
received only the solvent.
The edematous anaphylactic (PCA) and anaphylactoid
(histamine-induced) reactions caused by the stimulation of
the intravenous ovalbumin injection, which manifested
themselves by edema formation, swelling and exudation of
blue dye, were evaluated 30 minutes after their initiation
by the intravenous ovalbumin injection. This was done by
visually determining the extent to which the blue dye
emerged at the sites of edema formation. The percentage
inhibition of anaphylactic and anaphylactoid reactions
induced by the test substances in comparison with the
reactions of the control animals not treated with any test
substance was determined using comparison scales.
The results obtained using compounds of for~ula I
according to the foregoing test methods are shown in the
following Table A. The example numbers given for the
compounds of formula I relate to the subsequent preparative
examples.
- 21 -
2 9 ~
Table A
Test Inhibiting effect on cutaneous anaphylactic
Substance and anaphylactoid reactions in Rats
Example % inhibition by 2 x 105 mole/kg dose per os
5 No. Passive cutaneous ~ Histamine-induced
anaphylaxis (PCA) ' ana~hylactoid reaction
22 69 45
7 70 40
9 60 30
10 1 88 55
8 73 60
12 80 50
13 55 30
1516 50 45
17 70 35
11 55 20
3 80 55
2018 80 53
19 80 60
21 78 55
24 80 60
2527 60 35
28 65 40
2. Determination of minimum toxic dose.
Maximum doses of 300 mg/kg of the test substance were
administered orally to male mice weighing 20 - 25 g. The
animals were observed carefully for 3 hours for toxicity
symptoms. In addition, all symptoms and deaths were
recorded over a period of 24 hours after administration.
Associated symptoms were also observed and recorded. If
death or severe toxic symptoms were observed, additional
mice were administered increasingly lower doses. The lowest
- 22 -
2 ~
dose which produced death or severe toxic symptoms is given
in the following Table B as the minimum toxic dose.
Table B
5 Test Substance Minimum toxic dose mg/kg mouse
Example No. per os
1 300
22 300
8 100
9 300
2 > 300
11 > 3
12 300
13 100
16 300
';:
3. Investigation of anti-histamine-(H~) effect based on
histamine-(H~)-receptor antagonism in vitro.
To investigate the histamine-(HI)-receptor antagonism of
the test substances, the inhibiting effect of the substances
on histamine-induced contractions of the smooth muscle was ~-
determined in vitro on the isolated organ. Isolated strips
of organ from the ileum are suitable for this purpose. In
an organ bath of physiological saline solution the smooth
muscle strips react to the addition of histamine by
contracting. Addition of the compounds of the invention
decreased the histamine-induced contraction of the smooth
muscle of the ileum strips. The extent of regression of the
contraction is an indication of the anti-histamine-(H~)
activity of the compounds. The investigation was carried
out analogously to the method originally described by Magnus
(Pfluegers Arch. 102, 123 (1904)).
Procedure for determining the smooth muscle contraction
inhibiting effect on isolated guinea pig ileum induced by a
5 x 10~ molar histamine concentration
.
.. . .
.,.
' ' '' ' , ' ' : .~ .~
-
2~5~
Segments of the ileum 1.5 cm long from Dunkin Hartley
guinea pigs having a body weight of 300 500 g were used for
the test. Each strip was placed in an organ bath of 10 ml
of physiological saline solution according to Krebs-
Henseleit and attached to a conventional apparatus forisotonic measurement of changes in length of the ileum
strips (automated Celaster measuring apparatus), so that the
tissue was under 1 g of tension. The bath was kept at a pH
of 7.4 and gassed with a mixture of 5 % CO2 and 95 ~ 2-
After an equilibration phase, an isotonic contraction of thetissue was induced by adding histamine in a final
concentration of 5 x 104 mole/liter and washing it out again
after a contact time of 30 seconds. Only tissues samples
from which three reproducible contractions were obtained at
10 minute intervals were used in the subsequent test. The
test substances were then added in a final concentration of
104 mole/liter, and after 30 seconds contact time histamine
was again added up to a concentration of 5 x 104 mole/liter.
The resulting contractions were measured over 30 seconds.
The tissue was then washed several times over a period of 10
minutes. Histamine was then added again to stimulate a
contraction. The resulting contractions were again measured
over 30 seconds. The difference between the amplitude of
the contraction obtained by histamine addition alone and the
amplitude of the contraction obtained in the presence of the
test substance was determined and expressed in terms of %
inhibition.
The following Table C shows the results obtained with
the test substances according to the method described above.
The inhibiting effects on the contractions induced by
histamine 30 seconds after administration of the test
substance and on the contractions induced by the addition of
histamine 10 minutes later a~e listed in the table.
~'
-:
2~5~
Table c
Test in vitro (Hl)-receptor antagonism, ~ inhibition
~ubstance of histamine-induced ileum contractions at a
Example histamine concentration of 5 x 10~ mole/l and
5 No. a test substance concentration of 104 mole/l
after 30 seconds 'after 10 minutes
7 85 74
8 42 5
13 42 15
16 30 27
11 32 35
2 51 60
42 36
3 66 28
18 78 83
19 60 42
21 50 89
23 70 83
24 70 86
32
27 16 72
28 ~38 45
4. Determination of anti-PAF activity in vitro.
PAF (= Platelet Activatlng Factor) is a phospholipid
mediator which has many effects. Activation of platelet
aggregation induces protracted broncho-contraction and
hyper-reactivity of the air paths.
In this test the effect of the test substances on
platelet aggregation induced by adding PAF to a suspension
of platelets obtained from rabbit blood is investigated
using the method described by Mikashima et al. (Jap. J.
Pharmacol. 44 (1987) 387-391).
A suspension of platelets obtained from rabbit blood
3S containing 4 x 109 platelets/ml in a modified Tyrode buffer
solution (= Tyrode solution with 1.3 mM/l CaCl2 and 2.5 g/l
- 25 -
- ,
: . - -
.. .. ~ . : - , :. ~ .
, - - .-: . :
-. : ' -~;
2 ~
gelatine added) adjusted to pH 7.4 was used. Tyrode
solution is an aqueous solution containing 136.9 mmoles
NaCl, 2.68 mmoles KCl, 2.31 mmoles CaCl2, 1.0 mmole MgCl2,
11.9 mmoles NaHC03, 1.45 mmoles NaH2P04 and 5.55 mmoles
glucose per liter. The platelets were obtained from 10 ml
blood samples from each of three rabbits (New Zealand
hybrids, body weight 3 - 4 kg). For this the blood samples
were treated with ethylenediamine tetraacetic acid and
washed according to the method of Artley et al. (Brit. J.
Hematol. 19 (1970), 7-17). A platelet-rich plasma was then
initially separated by centrifuging (20 minutes at 400 x g).
The blood platelets were separated from the plasma by re-
centrifuging for 15 minutes at 1,400 x g. The platelets
remaining as sediment after centrifuging were resuspended in
a Tyrode buffer solution (but without calcium). 0.4 mmole
of lysine acetylsalicylate was then added, and after 15
minutes the platelets were sedimented again. The sediment
was resuspended in the aforementioned modified Tyrode buffer
solution, and the number of platelets in the res~lting
suspension was adjusted to the desired content.
A 40 x 109 molar PAF solution was used as a reagent.
This solution was made from a 1.8 x 10-3 molar stock solution
in chloroform. For this purpose a 10 ~1 sample of the stock
solution was evaporated to dryness and redissolved in 180 ~1
of the modified Tyrode solution to which 0.25 % of lipid-
free bovine serum albumin had been added. From this 10-5
molar working solutions were then prepared and stored
frozen. Samples of these solutions were appropriately
diluted for the tests.
To carry out the test 50 ~1 of the platelet suspension
and 10 ~1 of a 40 x 1o-5 molar solution of the compound being
investigated were added with stirring (1,000 rpm) to 330 ~1
of the modified Tyrode buffer solution in an aggreqation
tube provided with a small magnetic stirrer. This
corresponds to a final test substance concentration of 1o-5
mole/l. After 90 seconds preincubation time, 10 ~1 of the
- 26 -
- .................... -. - ~ -
--
2 ~ 3
PAF preparation were added. The agqregation occurring in
the aggregation tubes was measured over 4 - 5 minutes with
the aid of a computerized aggregometer.
The aggregation which occurred in test tubes containing
only platelet suspension was evaluated as 0 %, whereas the
aggregation which occurred in test tubes containing platelet
suspension and PAF preparation was evaluated as 100 %. The
aggregation which still occurred during the inhibition of
the PAF induced platelet aggregation due to the addition of
the test substances was measured, and the resulting
aggregation inhibition was calculated therefrom in %.
The results obtained using the compounds of formula I
according to the foregoing method are shown in the following
Table D.
Table D
Test Anti-PAF activity in vitro
Substance % inhibition of the PAF-induced aggregation
Example of rabbit blood platelets at a test
20 No. substance concentration of lo-5 mole/l
22 42
9 51
8 48
11 96
2S 2 60
18 76
21 37
74
27 35
28 45
5. In vitro determination of cyclooxygenase inhibition and
5-lipoxygenase inhibition.
After a cell is activated, arachidonic acid contained
in cell membranes is metabolized in two ways. Leucotrienes,
inter alia leucotriene C4, are formed due to the action of
- 27 -
.
.
2 ~
the enzyme 5-lipoxygenase (a 5-L0), and prostanoids are
formed due to the action of the enzyme cyclooxygenase (=
C0). In in vitro systems these metabolites are secreted
from the cell.
To evaluate the cyclooxygenase-inhibiting and 5-
lipoxygenase-inhibiting properties of the test substances,
their inhibitory activity on the biosynthesis of the
arachidonic acid derivatives leucotriene C4 (= LTC4) and 6-
keto-prostaglandin F~ (= 6-keto-PGF~I) was determined ln
vitro on mouse peritoneal macrophage cells. For this
purpose the LTC4 and 6-keto-PGFI contents of a mouse
peritoneal macrophage cell culture medium were determined by
zymosan stimulation as described by Scott et al. (J. Exp.
Med. 152 (1980), 324-335) and by Fradin et al.,
15 (Prostaglandins, 33 (1987), 579-589).
A cell suspension containing peritoneal cells from male
mice 8 - 10 weeks old was obtained in a known manner. A
solution marketed under the designation RPMI 1640
(Manufactured by Gibco) was used as the cell culture medium,
to which heparin (10 international units/ml) and antibiotics
were added according to the procedure of Bonney et al.
(Biochem. J. 176 (1978) 422-433). The cell suspension was
adjusted to a cell ~concentration of lo6 cells per ml and
distributed uniformly on titer dishes containing 24 l-ml
titer cells (wells). These were kept for two hours in a
humidified incubator filled with air enriched with 7% C02.
Cells not adhering to the titer well walls were then removed
by washing. The remaining macrophage cells adhering to the
walls were incubated for about 12 hours in a suspension
medium to which 0.1 % of bovine serum albumin (BSA = Bovine
Serum Albumin) was added. The suspension medium was then
replaced by a Hanks salt solution (= Hanks Balanced Salt
Solution = HBSS) with 10 mmoles of HEPES (= hydroxyethyl-
piperazinoethanesulfonic acid) to which a 0.1 % strength
solution of the test substances in aqueous, 1 % strength
dimethylformamide or only the solvent had been added. After
- 28 -
2 ~
15 minutes the arachidonic acid metabolism was stimulated by
adding 10 particles of zymosan (= glycoprotein mixture
isolated from cell walls of beer yeast, Saccharomyces
cerevisiae, Manufactured by Sigma Chemical Co., Munich) per
titre cell. After 2 hours samples of the respective
supernatant liquid in each cell were examined for their 6-
keto-PGF~ and LTC4 contents by an enzyme immunoassay (= EIA).
This EIA is carried out following the method of Pradelles et
al. (Analytical Chem. 57 (1985), 1170-1173). The
determination of LTC4 and the determination of 6-keto-PGF~
were each carried out on suitable dilutions of the samples
(1 : 50 to 1 : 250 for the LTC4 determination and 1 : 2S0 to
1 : 1,250 for the 6-keto-PGF~I determination) in comparison
with a comparative scale. To determine the inhibiting
effect of a 105 molar concentration of the compounds, the
amounts of reference eicosanoid were determined, and the
inhibiting effect was calculated therefrom in % inhibition
compared to the measurements of the zymosan controls. The
results obtained in this test are shown in the following
20 Table E.
Table E
Test In vitro % inhibition in zymosan-stimulated
Substance mouse peritoneal macrophage cells at a
25 Example concentration of lo-5 mole/l upon release of
No. 6-keto-PGF~ LTC~
7 17 46
9 32 65
1 27 48
36 72
46 9
11 0 42
Due to their activities described above, the compounds
of formula I are useful as anti-inflammatory and anti-
allergic medicaments for larger mammals, in particular
- 29 -
,
'
2 ~
humans, for treating inflammatory and allergic diseases.
The orally active compounds of the invention may act in
several ways, since they are active against several of the
principal mediators implicated in inflammatory processes and
asthmatic complaints. As a result of this activity profile
it can be assumed that in the treatment of allergy-based and
non-allergy-based asthma svmptoms, the compounds of the
invention will not only alleviate the symptomatic complaints
associated with asthmatic diseases, but also may reduce the
associated inflammation.
The doses to be used may vary from individual to
individual and of course will vary depending on the nature
of the condition to be treated, the substance used, and the
manner of administration. For example, parenteral
formulations will generally contain less active substance
than oral preparations. However, medicament forms having an
active ingredient content of 10 to 250 mg per individual
dose are generally suitable for administration to larger
mammals, in particular humans.
As medicaments, the compounds of formula I may be
contained with conventional pharmaceutical auxiliaries in
galenic formulations such as, for example, tablets,
capsules, suppositories, or solutions. These galenic
formulations may be produced by known methods using
conventional solid carriers such as, for example, lactose,
starch or talcum, or liquid paraffins, and using
conventional pharmaceutical auxiliaries, for example, tablet
disintegrating agents, solubility promoters or
preservatives.
The following examples are intended to illustrate the
invention in further detail without limiting its scope.
Exam~le 1: 2-{4-[4-(4-methylpyridin-2-yl)-piperazin-1-yl]-
butyl}-2H-1,4-benzothiazine-3(4H)-one.
A) 45.3 g (= 0.23 m) of 6-bromocaproic acid were
treated with 63.6 g (= 0.235 m) of phosphorus tribromide
- 30 -
, .i
2 ~ 9
with stirring, and then 75.1 g (= 0.047 m) of bromine were
added, the first half being added dropwise and the remainder
more rapidly. The mixture was then heated at a temperature
of 85-90C and maintained at this temperature for 1.5 hours.
A further 18.4 g (= 0.115 m) of bromine were added at this
temperature, and the reaction mixture was maintained at a
temperature of 85-90C for a further 18 hours. The reaction
mixture was worked up by cooling it to 20C and adding it to
a mixture of 700 ml cooled water and S00 ml of cold hexane.
The organic phase was separated, and the aqueous phase was
washed two more times with 1oo ml of hexane each time. The
organic phases were combined and dried over sodium sulfate.
The solvent was then distilled off. The 2,6-dibromocaproic
acid bromide which remained as an oily residue was
characterized by IR and NMR spectroscopy and further
processed without additional purification.
B) 9.25 g (= 0.07 m) of 2-aminothiophenol, 27.9 g of
benzyltrimethylammonium chloride, and 25 g of sodium
hydrogen carbonate were introduced into 150 ml of chloroform
with stirring. The resulting suspension was cooled to -5C,
and a solution of 25.3 g of 2,6-dibromocaproic acid bromide
in 70 ml of chloroform was added slowly with stirring to the
cooled suspension. The addition took 30 minutes, and the
temperature varied between -5C and +5C. The mixture was
maintained in this temperature range for a further hour.
The mixture was then heated under reflux for 7 hours.
Afterward it was cooled, filtered and the chloroform was
distilled off from the filtrate. The remaining residue was
treated with water and toluene. The organic phase was
separated, dried, and fractionated by column chromatography.
The fraction containing 2-(4-bromobutyl)-2H-1,4-
benzothiazine-3(4H)-one was separated. After
crystallization in the presence of diethyl ether, 5.9 g of
2-(4-bromobutyl)-2H-1,4-benzothiazine-3(4H)-one were
obtained from this fraction as slightly beige-colored
crystals having a melting point of 100C.
2 0 ~
C) A mixture of 3.6 g (= 0.012 m) of 2-(4-bromobutyl)-
2H-1,4-benzothiazine-3(4H)-one, 2.12 g (= 0.012 m) of N-(4-
methylpyridin-2-yl)-piperazine, and 1.83 g (= 0.018 m) of
triethylamine in 100 ml of toluene was heated at reflux for
9 hours with stirring, during which a further 0.46 g of
triethylamine was added after 3 hours, and again a further
0.9 g of triethylamine was added after 6 hours.
The reaction mixture was worked up by cooling it to
20C and adding 200 ml of water. The organic phase was
separated, and the toluene was distilled off. ~he remaining
residue was dissolved in 50 ml of 20 ~ strength aqueous
hydrochloric acid solution, and the solution was washed with
toluene. The aqueous phase was made alkaline to pH 9 and
then extracted with dichloromethane. The dichloromethane
extract was washed, dried over calcium chloride, and the
dichloromethane was distilled off. The crude title compound
which remained as a residue was purified by column
chromatography and then recrystallized from a 50/50 v/v
mixture of toluene and isopropanol. 2.4 g of 2-{4-[4-(4-
20 methylpyridin-2-yl)-piperazin-1-yl]-butyl}-2H-1,4-
benzothiazine-3(4H)-one were obtained as colorless crystals
having a melting point of 119C.
Example 2: 2-{4-[4-(4-methylpyridin-2-yl)-piperazin-1-yl]-
25 butyl)-2H-1,4-benzothiazine-3(4H)-thione.
5.95 g (= 0.015 m) of 2-{4-[4-(4-methylpyridin-2-yl)-
piperazin-1-yl]-butyl}-2H-1,4-benzothiazine-3(4H)-one
prepared according to Example 1 were dissolved in 60 ml of
xylene at a temperature of 60C. 6.9 g of P4Slo were added to
the solution, and the reaction mixture was heated at reflux
for 1 hour with stirring.
The reaction mixture was cooled, and the resulting
precipitate was separated and added to a mixture of 160 ml
of 2N aqueous sodium hydroxide solution and 200 ml of
dichloromethane. Further addition of 50 ml of water and S0
ml of dichloromethane yielded a reaction solution with a
- 32 -
., ~ :, ...
. ... , ~. .
r , .
' ' 1 ' .,
21[~ ~ r~
slight precipitate, which was removed by filtration. The
organic phase then was separated, dried, and the solvent was
distilled off. 5.2 g of a residue were obtained and
fractionated by column chromatography. The title compound
was crystallized from the fraction which contained it in a
50/So v/v mixture of diethyl ether and ethyl acetate. 3.1
g of 2-{4-~4-(4-methylpyridin-2-yl)-piperazin-1-yl]-butyl}-
2H-1,4-benzothiazine-3(4H)-thione were obtained in the form
of beige-colored crystals having a melting point of 119C.
Exam~le 3: 4-methyl-2-{4-~4-(4-methylpyridin-2-yl)-
piperazin-1-yl]butyl}-2H-1,4-benzothiazine-3(4H)-one.
2 g (= O.oos m) of 2-{4-[4-(4-methylpyridin-2-yl)-
piperazin-l-yl]-butyl}-2H-1,4-benzothiazine-3(4H)-one
lS prepared according to Example 1 were dissolved in 20 ml of
anhydrous dimethylformamide with stirring. 0.151 g of
sodium hydride were added under a nitrogen atmosphere, and
the resulting suspension was maintained at room temperature
for 10 minutes. 0.86 g of methyl iodide was then added all
at once, and the reaction mixture was stirred for 2.5 hours
at room temperature. The reaction mixture~was worked up by
distilling off the solvent, adding 100 ml of water to the
residue, and extracting the mixture with 100 ml of ethyl
acetate. The organic phase was concentrated by evaporating
the solvent, and the crude title compound which remained as
a residue was purified by column chromatography. The
resulting base was dissolved in isopropanolic 2.3N
hydrochloric acid. The hydrochloride of the title compound
which precipitated as a white precipitate was filtered out.
0.7 g of4-methyl-2-{4-t4-(4methylpyridin-2-yl)-piperazin-1-
yl]-butyl}-2H-1,4-benzothiazine-3(4H)-one dihydrochloride
having the empirical formula C23H3~40S, 2HCl, 2.5 H2O and a
melting point of 163C were obtained.
Example 4: 4-methyl-2-{4-[4-(4-methylpyridin-2-yl)-
piperazin-l-yl]butyl}-2H-1,4-benzothiazine-3(4H)-one.
-
2~5~
A) 0.1 mole of 2-aminobenzothiazole and 0.1 mole of
methyl iodide were heated under reflux in 50 ml of absolute
ethanol for 12-15 hours. The 2-imino-3-methylbenzothiazole
hydroiodide which precipitated was separated and dissolved
in hot water. The solution was rendered alkaline by adding
a saturated aqueous sodium carbonate solution. The 2-imino-
3-methylbenzothiazole which precipitated was separated,
washed with water and dried under reduced pressure.
B) The product obtained above was heated under reflux
for 36 hours in 50 % strenqth potassium hydroxide solution.
The solution was worked up by diluting it with water and
adjusted to pH 6 by adding aqueous 5N acetic acid solution.
The aqueous solution was then extracted several times with
ethyl acetate. The solvent was distilled off from the
combined organic phases under reduced pressure. The 2-
(methylamino)-thiophenol which remained as a residue was
used in the subsequent reaction step without further
purification.
C) 4.5 g (= 0.032 m) of 2-(methylamino)-thiophenol, 6
g of triethylbenzylammonium chloride, and 11.20 g of sodium
hydrogen carbonate were added to 120 ml of chloroform. The
resulting suspension was cooled to 5C, and then a solution
of 10.88 g of 2,6-dibromohexanoyl bromide in 20 ml of
chloroform was added dropwise so slowly that the temperature
did not exceed 5C. 20 minutes were required for the
addition. The reaction mixture was then allowed to warm to
room temperature and was subsequently heated under reflux
for 4 hours. The react~on mixture was worked up by cooling
and filtering it. The chloroform phase was washed, dried
over sodium sulfate, and the solvent was distilled off. The
2-(4-bromobutyl)-4-methyl-2H-1,4-benzothiazine-3(4H)-one
which formed was isolated from the remaining residue by
fractional column chromatography. 2.78 g of white crystals
having a melting point of 116C were obtained.
3S D) The product obtained above was reacted analogously
to Example 1 C) with N-(4-methylpyridin-2-yl)-piperazine in
- 34 -
. . .
.. . .
- , 1 .. . .... - . ~
,
- . . .:
., . , ,. , -
.,
,: ~
2 ~
toluene with addition of triethylamine. The resultiny 4-
methyl-2-{4-[4-(4-methylpyridin-2-yl)-piperazin-1-yl]-
butyl}-2H-1,4-benzothiazine-3(4H)-one was converted as
described in Example 3 to the corresponding dihydrochloride
having the empirical formula C~3H3~40S, 2HCl, 2.5 H20 and a
melting point of 163C.
Example 5: 2-[4-(4-methylpyridin-2-yl)-piperazin-1-yl]-2H-
1,4-benzothiazine-3(4H)-one.
A) 0.8 ml (= 1 equivalent) of sulfonyl chloride was
added dropwise with stirring at room temperature to a
suspension of 10.0 mmoles of 2H-1,4-benzothiazine-3(4H)-one
in 10 ml of dichloromethane, and the reaction mixture was
stirred for a further 5 hours at room temperature. The
mixture was worked up by evaporating it to dryness under
reduced pressure. The 2-chloro-2H-1,4-benzothiazine-3(4H)-
one which remained as a residue (melting point 189-210C
with decomposition) was processed further in the next step
without additional purification.
B) 7 g (= 0.035 m) of 2-chloro-2H-1,4-benzothiazine-
3(4H)-one, 6.2 g of N-(4-methylpyridin-2-yl)-piperazine, and
7 g of triethylamine were heated at reflux with stirring for
5 hours in 100 ml of toluene. The suspension was worked up
by diluting it with 200 ml of toluene, and the resulting
precipitate was filtered out and dissolved in 20 % strength
aqueous hydrochloric acid solution. The solution was
rendered alkaline by adding an ammonium hydroxide solution,
and the resulting precipitate was filtered out, washed with
water and dried. 2 g of beige-colored crystalline 2-t4-(4-
methylpyridin-2-yl)-piperazin-1-yl]-2H-1,4-benzothiazine-
3(4H)-one having a melting point of 245C were obtained.
Example 6: 7-hydroxy-2-{4-[4-(4-methylpyridin-2-yl)
piperazin-1-yl]butyl}-2H-1,4-benzothiazine-3(4H)-one.
360 mg of 7-methoxy-2-{4-[4-(4-methylpyridin-2-yl)-
piperazin-l-yl]-butyl}-2H-1,4-benzothiazine-3(4H)-one (see
- 35 -
2 ~
Example 19) prepared analogously to Example 1 were added to
5 ml of dichloromethane with exclusion of moisture. After
cooling to -5C, a solution of 0.73 g of boron tribromide in
1 ml of methylene chloride was added dropwise with stirring.
Stirring was then continued for a further 30 minutes at room
temperature. The reaction mixture was worked up by adding
it with stirring to a mixture of ice and aqueous sodium
hydrogen carbonate solution. 200 ml of chloroform were then
added. The resulting organic solution was washed with
water, dried over sodium sulfate, filtered and concentrated
under reduced pressure. The title compound was obtained
from the remaining residue by fractional column
chromatography. The fractions containing the title compound
were concentrated and treated with ether. 30 mg of 7-
hydroxy-2-{4-[4-(4-methylpyridin-2-yl)-piperazin-1-yl]-
butyl}-2H-1,4-benzothiazine-3(4H)-one were obtained as white
crystals having a melting point of 212C.
The compounds of formula I listed in the followin~
Table I were also obtained following the procedures
described in the foregoing examples.
- 36 -
-
'- . ' :.
. . ' ,
2 0 ~ 9
, ~ q ~ ~ ~ . t ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ . ~ t
,~ o o o o U C o ~ o o o ~o o o o o o o o o o o
X o o o o o o 0
~L +~t~
_ I I T T I I I II _ I I~ _ _ ~ _ _ _ _ I 1~ T _ O
~1: I I I I T I I I I T I I I I I I I T T T T C I ~ _ ¦
_ _ _ _ O _ _ _ e ~ -- ~ w O N _ N _ _ _ C = ~C O
-- 37 --
2~5 ~ ~J~
Exam~le I: Tablets containing 2-{4-[4-(4-methylpyridin-2-
yl)-piperazin-l-yl]-butyl}-2H-1,4-benzothiazine-3(4H)-one
Tablets were produced having the following composition
per tablet:
2-{4-[4-(4-methylpyridin-2-yl)-piperazin-1-
yl]-butyl}-2H-1,4-benzothiazine-3(4H)-one 20 mg
Corn starch 60 mg
Lactose 135 mg
Gelatine (10 % strength solution)6 mg
The active ingredient, the corn starch and the lactose were
mixed with the 10 % strength gelatine solution to form a
paste. The paste was comminuted and the resulting granules
were placed on a suitable plate and dried at 45C. The
dried granules were passed through a comminuting machine and
mixed with the following further adjuvants in a mixer:
Talcum 5 mg
Magnesium stearate 5 mg
Corn starch 9 mg
and then pressed to form 240 mg tablets.
The foregoing description and examples have been set
forth merely to illustrate the invention and are not
intended to be limiting. Since modifications of the
described embodiments incorporating the spirit and substance
of the invention may occur to persons skilled in the art,
the invention should be construed broadly to include all
variations falling within the scope of the appended claims
and equivalents thereof.
- 38 -
. - - . ~ .
- ~ ~. ~ , . .... .
., . - - -
- ~ : ' ' ' '
,,
-,, ~:
,~ . : . . -; ~ ....