Note: Descriptions are shown in the official language in which they were submitted.
WO ~/14~05 PCT/US90~02736
2 ~
Açç~ul~ion o~ ~r~q~ lnt~ ~iDoso~e~ a~Pro~n Gr~ient
Fiçld Qf ~e~Inven~iqn
The pra~nt inv~ntion relates to ph~rmaceutlcal compoqi-
tions and methods of making liposome containing compositions
which exhibit chara~teristics o~ uptake which may be qreater than
expected by the relationship de~'ined by the Henderson-Hasselbach
equation.
The present invention al50 relates to liposomal composi-
tions wherein the liposome comprises in part a mem~rane-
stabilizing component, for example, cholesterol, which exhibits
favorable characteristics in preventing rapid release o~ a
pharmaceutical agent after it ha~ been ~or~ulated in liposomes.
The present invention also relates to novel sustained
release lipo~omal composition~ comprising the bronchodilators
metaproterenol, isoproterenol an~ terbutaline.
The pre~ent invention also relates to minimum ~ufferin~
capacity re~uired to achiPve liposomal encapsulation of
phrmaceutical agents with maintenance of a major portlon of the
initial pH gradient.
aackq~Qund o~' thç Invention
The th~rapeutic properties of many drugs may he dramati-
cally i~p~o~d ~y th~ ad~inistration in ~ lipo~omally encapsu-
lated fo~ ~S~Q, for ex~mple P.N. Shek and R.F. ~arber, Mod. ~ed.
~ , 41, 31~-332, (1986)]. In certain ca~es, for example, in
th~ ad~inistration of amphotericin B and doxorubicin ~Lopez-
Bernstein, et al., ~ Li_, 151, 70~-710, (198~) and ~ah-
man, et al., Ç3l~a~ ~, 40, 1532 (1980)] toxicity i5 reduced
while e~'~'icacy is maintained or even incr~as~d. The Senefit
wV~/1~
2 ~
obtain~d ~rom liposomally encapsulated ag~
and liXely results from the altered pharmc
biodistribution of th~ entrapp~d drug [Ost
o~. Phanml, in pr~q.
The pharmacokinetics and biodistr:
drug will largely depend on the character
Optimization of a lipsomal drug requires c
ber of variables, including vesiclQ SiZQ,
drug to lipid ratio. Most drug loading p2
not permit the independent variation o~ t~
which are passively trapped in liposomes
drug to lipid ratios as the liposome sizq
change~ in the trapped volu~e.
Several biogenic amines and antin~
been shown to acc~ulate in liposomes in
proton gradient known as "remote loading"
Mayer, et al., ~ :=SI~L .KLLL1L ~ S ~ ~5~
et al., ~iochemistrv, 27, 2053, ~l988) anc
C~e~ PhYs. LiPids, 4~, 97, ~l9~)]. Thi~
allows independent variation of any of the :
Much highQr drug to lipid ratioC can be ach:
conv~ntio~l t~chniqu~s [~ayer, et al- S~
333 (l986)~. In ~ddltion, the transr~e~brz
drug is generally d~tarmined by the protor
lates drug l~akag~ by ch~nges in the buff~
intrav~sicular ~ediu~. Th~ USQ 0~ pro1:on
to trap drugs which are non-zwitterionic
shown to b~ practic~l for adriamycin, thQ
WO90/14105 PCT/US90/027~
- 2 ~ ^3
dibucaine and dopamine and other drugs. Advantages o~ this
system include ef~icient drug trapping, slower rate~ o~ drug
releas~ than passively trapped drug, and higher drug to lipid
ratios than can otherwise be achieved. In addition, because the
liposomes can be prepared in the absence of the drug, problems
with drug release during storage, or drug degradation during
liposo~al preparation can be avoided.
Intraliposomal drug accumulation in respons~ to pH gradi-
ents is believed to occur in a manner similar to that of other
weak bases, for example, the pH gradient probe methylamine.
Methylamine equilibrates across liposomal membranes in the
uncharged foru, and re-ionizes according to the Henderson-
Hasselbach relationship of the pH o~ its environment. The equi-
librium distribution reflects the transmembrane pH gradient, and
its redistribution can be used to measure these gradients.
However, not all pharmaceutical agents which possess the
capacity to be ionized according to Hend~rson-Hasselbach rela-
tionships a~cumulate in liposomes according to this relationship.
In fact, certain agents do not seem to accumulate at all. In
addition, certain agents which may accumulate according to this
relationship immediately undergo release, resulting in unworkable
pharmaceutical formulations which must b~ used i~metiately after
production and ~hich are virtually unusable a~ ~ustained release
product~.
LiposomQs are completely clo~ed lipid bilayer membranes
which contain entrapped aqueous volume. Lipo omes are vesicles
which may be unilamellar (~ingle membrane) or multilammelar
(onion-liXe structures characterized by multiple me~brane
WO ~/141~ PCT/US~/027~
2 ~ ~ CJ .
bilay~r~, each separated ~rom the next by an aqu~ou~ layer). The
bilayer is co~posed o~ two lipid monolayers having a hydrophobic
"tail n region and a hydrophilic "head" rQgion. In the membrane
bilayer, the hydrophobic (nonpolar) "tails" o~ the lipid
~onolayers orient toward the c~nter o~ th~ bilayer, whereas th~
hydrophilic (polar) "heads" orient toward the aqu~ous phase. The
~asic structur~ of liposomes may b~ made by a variety o~ techni-
qu~ known in th2 art.
Liposomal encap~ulation could pot~ntially provid~
numerous beneficial e~f~cts ~or a wide variety o~ pharmaoeutical
agents and the remote loading technique should prov~ instrumental
in realizing th~ potential of liposomally en~apsulate~ agents.
In addition, a high trapping e~ficiency ~or loading liposomes
r2sults in v~ry little drug being lost during the encapsulation
process, an advantagu that prov~3 to be i~portant wh~n dealing
with expen~ive drug~. How~vQr, th~ us~ o~ liposom~ to
administer drug~ has rai~ed problem~ with r~g~rd both to drug
encapsulation and drug rQlQas~ during thQrapy. For example, even
with the present us~ of ~remote loading" syste~s, there is a con-
tinuing need to incr~ase trapping e~iciQncies so as to minimize
the lipid load pr*~nt~d to thG patient. S~condly, eY~n with
increa~d-trapping ~iciencie~, th~ro i8 no guarante~ that the
releas~ ch~ract~ri~tic~ o~ the load~d liposome wlll re~lect
acc~ptablQ sustain~d rQlease characteristics. Many drugs have
b~en ~ound to bc r~pidly rel~aY~d ~rom lipo~o~es a~t~r encapsula-
tion. Such rele~so r~duces th~ b~neSiclal a~octs of liposomal
encapsulation.
WO 90/1~05 PCr/US90/02736
2~ ?
Oblects Oe the P~esent In~ention
It is an object of the present invention to provide novel
liposomal compositions and general methods for maXing such co~-
positions which are designed to maximize the uptake of a
pharma~eutical agent into the liposome, thereby increasin~ the
amount of drug which can be loaded into liposomes and deoreasing
the lipid load presented to the patient du-ing administration o~
liposomes.
It is a further object o~ the present invention to pro-
vide novel liposomal compositions and methods for ma~ins such
compositions which prevent the rapid disadvantageous release of
pharmaceutical agent before administration of the liposomes.
It is a further object of the present invention to pro-
vide novel brochodilator liposomal compositions.
8rief Dçscri~tion o~ the FLqurç~
Figure 1 shows the accumulation of mitoxantrone by EPC
vesicles exhibiting a proton gradient with an internal aqueous
buffer system comprising 300 mM citrate, pH 4.0 and an external
buffer system comprising 300 mM NaCl, 20 mM HEPES, pH 7.5.
Accumulation wa~ rapid and complete and evidenced no release over
several hours.
Figure 2A shows the response o~ timolol uptake in EPC
vesicles (partial accumulation, uptaXe stable). ~he level of
uptake is about 100 nmoles/umole (about S0% of available drug).
Figu~e 29 shows thP response of quinamrine, which is
similar to ~i~olol in EPC vesicles. The level of uptake is about
80 nmoles/umole lipid after 30 minutes.
Figur~ 3A shows the response of quinidine uptake in EPC
WO ~l4l~ PCT/US~/027~
2~6~?~i
vesicles ~co~pl~te accumulatian, rapid r~ s~3. Within 30
minutes about 50% o~ the druy leak~ back out o~ the ve~icles.
Figure 3B shows the e~ect o~ added chole~t~rol to the
uptake of quinidins and the stability oX th~ pH gradient.
Figure 4 shows the ef~ect of phy~ostigmine tq transmem-
brane pH gradient. Under the conditions used to assess drug
uptake 5200 uM physostigmine) only a small decrease in the
measured pH is observed.
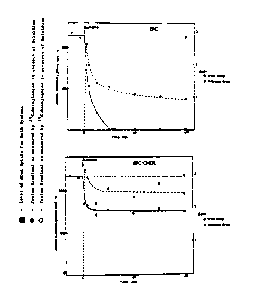
Figure 5 shows the entrapment o~ metaprot~renol, ter-
butaline and isoproterenol in response to p~ gradients using egg
phosphatidylcholine 200 nm extruded liposom~s.
Figure 6 de~onstrates tXe ef~ect of drug uptake on the
residual pH gradi~nt as measured by mQthylamine redistribution in
the pre enc~ and absenco o~ isoproterznol. A~ shown, when the
inte~nal ~nd ext rn~l pH i9 7.4 or 4.0 (no gradient), the
methylamine doe3 not detect any pH gradiQnt.
Figure 7 shows the effect of temp~rature on the Entrap-
ment of Metaproterenol in response to pH gradients at 21'C, 37 C
and 60-C.
Figur~ 8 ~hows th~ influenc~ of choleqterol on the
accu~ulation Gf ~t~proterenol in respons~ to a p~ gradient.
Figur~ 9 -~ho~ ~ho influenco of varying the external druq
conc~ntration on thQ lQvel of ~etaproterenol uptake.
FigurQ 10 shows th~ ef~ect o~ intarnal buffering capacity
on drug uptake.
The present invention relates to liposo~al compositions
ha~ing a p~ gradient whic~ exhibit markedly increased accumula-
WO ~/14l0~ PCT/US90/02736
tion of pharmaceutical agents abov~ that expec~ed ~rom t~Henderson-Hasselbach relationship by ~ormulating the liposomes
utilizin~ a ~irst internal aqueoua burfer ~nd a sacond external
aqueous bu~fer wherein the conc~ntration of the pharmaceutical
agent exceeds its solubility product in the internal bu~fer fol-
lowing uptake. Therefore, pre~erably, the pharmaceutical agent
exhibits a solubility within th~ liposome which is less than the
final concentration of agent within the liposome. Preferably,
the solubility of the phar~aceutical agent is less than about 20
mM and most preferably less than about lO mM. In addition, the
internal buffer solution has a buffer strength of at least about
50 mMol, preferably about lO0 to about 300 mMol and ~ost
preferably about 300.
The present invention also relates to liposomal composi-
tions comprising in part, mem~rane-stabilizing components, for
exa~ple, cholesterol, among other lipids, to prevent the rapid
release of certain pharmaceutical agents from liposomes which do
not contain the membrane-stabilizin~ components. SUch liposomes
preferably comprise a miXtUrQ of phosphatidylcholine and
cholesterol in a molar weight ratio of about 55:45.
The present invention also relates to liposomal composi-
tions having a pH gradient containing bronchodilators selected
from tha group consisting of metaproterenol, terbutalinD and
i~oproteren~l. It has be~n shown that the above agents, which
heretofore hav~ not be~n formulated in such liposo~es, will
accumulate into liposomes to an apprecia~le extent to produce
effective, stable liposomal composition~ us~ful ~or trea~ing con-
dition~ re~uiring sustained releaso of bronchodilators. Such
WO ~/1410~ PcT/Us9o/o27
203~ ,.?.~
liposo~al compo~itions comprising bronchodilator ~or~ulations may
be usaful ~or treating a nu~ber o~ condition~, includin~ asthma.
Such compo~itions are exp~cted to hav~ a lonq~r re~idence tim~ in
the lung than the same ~ree drug, thus obtaining concentrations
of bronchodilator at the site o~ activity within the lung for a
period longer than for compositions pre~ently a~ailable. Such
compo~itions may be formulated a~ ae~osols within a pharmaceuti-
cally acceptable solution ~or ad~inistration o~ bronchodilators
dir~ctly into the lungs for tr~atment o~ acute asthma attacks.
The brochodilator compo~ieions o~ th~ present invention
have b~en shown to ef~ectively accumulate in lipo~omes comprisin~
egyphosphatidylcholine (EPC) as well a~ a ~ixtuse of
phosphatidylcholine ~nd cholesterol t55:45, w:w). The liposomes
comprising the mixture of phosphatidylcholine and cholesterol,
accu~ulate thQ bronchodilator to about th~ s~ma relativ~ extent
as the EPC liposom~s, although the time need ~or accu~ulation is
longer for ~he cholesterol containing lipo50~s.
In g~n~ral, the liposo~e composition~ o~ the present
invention have a drug to lipid molar ratio r~nging fro~ about
0.5~ up to about 50~. Tha liposome3 o~ th2 present in~ention may
compris~ phospholipids such as egg pho~phatidylcholine (EPC),
hydrog~t~d ~oy phoaphatidylcholine, dist~aroylphospha~idyl-
cholin~, di~yri~toylpho~phatldylcholinQ, or diarachidonoyl-
pho~ph~tidylcholins, a~ong oth~rs, and m~y additionally comprise
a nuDber o~ st~roidal compositions, as w~ll as other co~posi-
tion~.
In gen~ral, th~ liposo~e~ range in 5iZ8 fro~ about 0.05
to gr~at~r than 2 ~icron~, with a prefQrred ran~e being about
W~90/1~105 PCT/US90/0273~
O. 05 to about O. 3 microns. Mo~t pr~2~raoly, th~ lipo~om~3 ar~
unilamellar and range in size from about 0.1 to about 0.3
microns. These unilamellar llposomes may be homogeneous or
unimodal with re~ard to 5iZ~ distribution.
Tha liposomes of the present invention may be
administered via oral, pare~teral, buccal, topical, and trans-
dermal routes of administra~ion, among other routes of adminis-
tration.
Qetailed ~çscr~ption o~ ~he InventiQn
The presant inve~tion utilize~ ef~icient trapping of
phar~ac~utical agents in liposomes exhibiting a transmembran~
ionic gradient, preferably a trans~ambrane p~ gradient, which can
result in an accumulation o~ the agent in an amount signi~icantly
higher than otherwise expected ~rom the Hend~rson-Has~elbach
relationship. Liposom~ compositions Or the present in~ention
comprise at least one lipid, a phrmaceutical agent accumulated
therein, an internal buffer solution wherein the solubility of
the pharmaceutical agent within the bu~fer solution is less than
the concentration o~ the agen within the liposome and an
ex~ernal buffer solution wherein the solubility of th~
pharmaceutical ag~nt is pre~erably at least about 0.2 ~M. As
used throughout tho specification, the terms pharmaceutical agen
and drug are synonymous.
~ he liposom~s o~ th~ pre~en~ invention may be formed by
any o~ the ~ethod~ known in the art, but pre~erably they are
for~d according to t~e procedures disclosed in Ball~y, et
al., PCT Application No. US86/01102, published February 27, 1986
and Mayer, et al. PC~ Application No. US88/00646, published Sep-
WO ~/~4105 PCT/US90/0~7
1 0 ' ' --
2 ~
temb~r 7, 1988. These techniques allow the loading o~ liposomes
with ionizable pharmac~utical aqents to achieve intarior con-
ce~trations con~id~rably gr~ater than othorwise sxpected ~rom the
dru~s' solubility in aqueous solution at neutral pH and/or con~
centration~ greater than can he obtained by passive entrapment
techniquQs. In this technique, a transmembrane ion (pH) gradiens
is created between the internal and external membranes o~ the
liposomes and the pharmaceutical agent is loaded into the
liposomes by means o~ the ion (p~) gradient, which drives the
up~ake. The transmembrane gradient is generated by creating a
concentration gradi~nt for one or mors charged species, ~or exam-
pl~ Na+, C1-, R+, Li+, OH- and preferably H+, across the liposome
me~brane~, such that the ion gradi2nt drive~ t~a uptake of
ionizable phar~aceutical agQnts across the membranes. In the
present invention, tran~embrane ion (~+) gradients are
prefQrably employed to produce the ion gradient and load t~e
pharmaceutical agents, which tend to haYe weakly basic nitrogen
groups, into the liposomes.
In the present invention, liposo~ea are preferably -st
for~ed in an aquQous buffer solution. The first solution is
~ither acidic or basic, depending UpQn whether th~ pharmaceutical
agant to ~ load~d produces a charg~d sp~cie~ at ba~ic or acidic
pH~ For example, in the case of loading weakly basic
pharmac~utical ag~nt~, a charged specieC is produced at low pH,
i.e., a pH of about 2.0 to 5.0, preferably a pH o~ about 4Ø
After formatio~ o~ lipo~omes having an acidic int~rnal aqueous
buf~er solution, thQ bu~fer solution external to the liposomes is
thRn ~odi~ed to ~ pH significantly abov~ th~ pH o~ the internal
Wo ~/14105 PCT/US9~/02736
1 1
2 Q ~37~ 3
bu~fer solution, preferably at least about 3.0 to 4.0 pH units
abov~ the internal buffer solution. Th~ modification o~ the
axternal bu~f2r results in a pH gradient which drives the
accumulation of pharmaceutical agent within the liposome. The
internal buffer solution may differ from the external buffer
solution only in the difference in pH. In general, uncharged
pharmaceutical agent will pass through the lipid layer(s) of the
liposome much more readily than will charged (protonat~d, in the
case of weakly basic pharmaceutical aqents) agent. Thus,
uncharged phar~aceutical agent in the external buffer will
readily pass through the liposome into the internal buffer,
become protonatedt and remain wi~hin the liposome as a ~trapped~
protonated molecule which does not readily pass throush the
liposome layer(s). Pharmaceutical agent will thus concentrate in
the liposom~ as a function of the pH gradient b~tween the inter-
nal and external buf~er solutions.
Such loading according to the above procedure, while
effective for certain pharmaceutical agents, often does not
result in maximum loading. ~ven if it is assumed that the
pharmaceutical agent is maximally soluble in the internal and
external buffer solutions and will readily pass through the
liposomal layer(s) (an assumption not always borne out by
reali~y), the maxi~u~ loading will generally reflect the rela-
tionship de ined by the Henderson-Hasselbach equation
[HA+]in/[HA+]out~ [H+]in/[H+]out- However, a number of fac~ors
are belie~ed to effect the ability o~ a pharmaceutical agent to
accumulate. These factors include the partitioning of the
unprotonated agent within the lipid layers(s), the difference in
WO ~/141~5 PcT/US9o/o2~
1 2
20~S.,~ 2~j
P~a b~twH~n protonated spoci~s that exist in the bu~er solution
and specie~ associatod wit~ th~ membrano, ~hs bu~er capacity o~
tha internal buffer solution and the solubility of the protonated
species within the internal buffer.
It ha~ now been deter~ined that th~ moct important ~actor
influencing th~ accumulation o~ phar~aceutical agent within a
liposome abovQ what is expected ~rom the ~enderson-Hacselbac~
equation is the solubility o~ th~ protonated species o~ the agent
within the internal bu~er solution. It has been determined that
th2 solubility o~ tha protonated species o~ thc pharmaceutical
agent to be accu~ulated will influence thP level o~ uptake and
accumulation w~ich may be substantially greater than that
predicted by tha Henderson-Hasselbach equation. ~iposo~e com-
positions which are formulated using an internal buf~er solution
in which an ioni~ed pharmacautical agant i~ minimally soluble and
which preferably praCipitatQs the ionizad ag~nt, will drive the
accumulation o~ the pharmaceutical ag~nt within the liposome
~eyond what would otherwisQ be expected to produc~ liposomes
which consistently should have high trapping efficiencies
approaching 100%.
In th~ prQ ent invention, it ha~ surprisingly been dis-
coY~red ~hat th- ~ol~bility o~ th~ pharuac~utical agent in th~
internal buffer ~ay ultimately control ~he ability of the
pharoac~utical agent to lo~d into th~ liposo~ to an extent
grea~er than that pr~dicted by the Henderson/Hasselbach relation-
ship. Th~s, in th~ present invention, liposome compositions are
pre~err~d which are ~ormed utilizing a first internal bu~fer
solution of either basic (pH about 8 to lO) or acidic (pH about
~ u~ PCT/US90/027~
13
2 ~ 3
3.0 to 5.0) charact~r and a second ext~rnal buffer solution, the
pH of which i~ preferably b~tween about 6.5 and A.0, preferably
7.4. The high or low pH of the int~rnal bu~rer relative to a
neutral pH of the ext~rnal bu~er produc~ a transmeMbrane gradi-
ent which acts to drive the accumulation of the agent in the
liposome. It has surprisingly been discovered that the most
i~portant factor in det~r~ining the ultimate amount of agent
which may be loaded into liposom2s using the transmembrane gradi-
ent to dri~e the accu~ulation of the~agent into the liposomes
above that expected by the H~nderso~-Hasselbach equation is the
solubility o~ the agent in the internal buffer solution.
In general, internal buffer solutions useful in embodi-
~ents of the present in~ention are chosen so that ths
phar~aceutical ag~nt to be accumulated has a solubility within
the internal bu~fer solution which is les~ than the total agent
to be accu~ulated in th~ liposo~e. Generally, the solubility o~
the phar~aceutical agent in the internal bu~fer solution is no
greater than about 65 ~M, pr~ferably no greater than about 20 mM
and most preferably no ~reater than about lO mM.
The internal buffer solution is also chosen to maximize
the buffer strangth of 'the internal solution. It is believed
that the bu~fer strength of the internal buffer solution is also
i~portant to the total accumulation o~ agent within the liposome
and internal bu~er solutions are chosen to maximize this
strength. 0~ course, the solubility of ~h~ a~ent within the
internal buffer solution is also a most important factor in
determining accumulation. Therefore, wherQ a bu~rer solution is
to be chosen, it is both the solubility ~actor and the buffer
W~ ~/l4l~ PCT/US90/02736
1 4
strength factor which should be maximizad in choo~ing useful
bu~fer solutions. In the present invention, it has been
determined that the bu~fer strength o~ the internal bu~r solu-
tion should be at least about S0 mM, preferably about lO0 mM to
about 300 mM and most preferably about 300 ~M.
Lipids which can be used in the liposome ~ormulations of
the prQSent inYention includ~ ~ynthetic or natural phospholipids
and ~ay include phosphatidylcholine ~PC),
phosphatidylethanolamine (PE?, phosphatidylserine (PS),
phosphatidylglycerol (PG), phosphatidic acid (PA),
phosphatidylinositol(PI), sphingomyelin (SPM) and cardiolipin,
among others, either alon~ or in combination. The phospholipids
useful in tho present inv~ntion may also include dimyristoyl-
phosphatidylcholine (DMPC) and dimyristoylphosphatidylglycerol
(DMPG). In other e~bodiments, distearylphosphatidylcholine
(DSPC), dipalmitoylphosphatidylcholine (DPPC), or hydrngenated.
soy phosphatidylcholine (HSPC) ~ay al90 be used. Dimyristoyl-
phosphatidylcholine (DMPC) and diarachidonoylphosphatidylcholine
(DAPC) may similarly be used. Due to the elevated transition
temperaturres (Tc) of lipida such as DSPC (Tc of about 65-C),
DPPC (Tc of abol~t 45-C) and DAPC (Tc ~ about 85-C), such lipids
are pref~r~bly h~ate~ to about their Tc or te~peratures slightly
higher, e.g., up to about 5'C higher than tho Tc, in order to
maXe theso lipo~om~. In pr~ferred ~bodim~nts, egg
phosphatidylcholin~ i~ uced.
In a ~umb~r o~ e~bodi~ents oP the pr~ont invention, a
steroidal componant may bQ added to th~ liposome. Any o~ the
a~ove-mentioned phospholipids may be used in combination with at
W~ ~/14l~ PCT/US~/027
205~'~?,5
least ona additional component selected ~rom ~he group consisting
o~ chole~tarol, cholestanol, coprostanol or cholestane. In addi-
tion, polyethylene glycol derivatives o~ cholesterol (PEG-
cholesterols), a~ well a organic acid d~rivativ~s o~ sterol-~,
fo~ example cholesterol hemisuccinate tC~S) may also be us~d in
combination with any o~ the above-mentioned phospholipids.
Organic acid derivatives of alpha-tocopherol hemisuccinate, (THS)
may also be used. CHS- and THS-containing liposomes and their
tris salt forms may generally be prepared by any method known in
the art ror preparing lipsomes containin~ sterols Any o~ the
above-mentioned sterols ~ay be used in liposomes, 50 long as the
rasultant phospholipid-sterol mixture yields stable liposomes.
In particular, sae the procedures of Janoff, et al., U.S. ~atent
No. 4,721,612, issued January 26, 1988, entitled "Steroidal
Liposomes", and Janoff, et al., PCT Pu~lication No. 87/02219,
published April 23, 1987, entitled "Alpha Tocopherol-8ased
Vehicles", relevant portions o~ which are incoporated by
reference herein.
In cPrtain embodiments in which the liposomes are
designed to prevent rapid release of the pharmaceutical agent,
cholesterol in an amount equal to about 30 mole% tu about 45
mole~ by waight o~ the lipid comprising the liposome is
preferably used in combination with any of the above named
phospholipids or phospholipid/steroid co~binations. Such com-
position should, in general, prev~nt the undesired rapid release
of accumulated ph~rmaceutica} agent f rom the liposome. Any com-
bination o~ ~embrane-stabilizing component and lipid may be used
which prevents rapid release o~ pharmaceutical agents ~rom the
WO ~tl4l05 PCT/US~/021~
2 o 5 fi L~ 3
liposome, and one o~ ordinary skill in the art will be able to
modify the membrane-~tabilizing component and th~ phospholipid to
~or~ulat~ liposomas which prev~nt rapid ralsas~ Or th~
pharmaceutical agent. Mos~ pre~erably, liposomes comprising a
mixture o~ about 45 mol~ ~ by wei~ht choles~erol and about 55
mole % by weight phosphatidylcholinQ are usQd in this aspect o~
the present invention. Althou~h a~y number of pha~maceutical
agents which show a proclivity to r~lease rapidly ~rom liposomes
may be used in thiC aspect of the present invention, it has beer
determined that th~ agents quinine, diphenhydramine and quinidir.a
are ~specially prone to rapidly release from liposomes and thus
liposomal formulations comprising these agents preferably com-
prise cholesterol in an amount equal to about 30 to 45 mole ~ and
preferably about 45 mol~ % of the lipid plu8 me~brane-stabilizing
componen~. Although it is di~icult to deter~lnQ, strictly on
th~ basis of chemical structur~, that a phar~aceutical agent will
rapidly release from a liposomal for~ulation, one o~ ordinary
skill in the art will be able to assess the degree of release of
the agent and formulate a liposomal product consistent with the
teachings of the present invention. As in other embodiments of
the present invention, any buffer solution may ~e used ~or the
internal a~d external bu~f er solutions in this a~pact of the
pr~ont inYention regardless of the solubility of the
phar~æc~utical ag~nt ther2in. However, tha pro~erred bu~er
solution~ are choc~n 80 that tha solubility of the pharmaceutical
agent is less ~han the concentration of t~ agent within th~ .
liposome, pre~erably is less than 20mM and ~o~t pre~erably is
less th~n 10 mM a~ is the case with other embodiments o~ th~
WO ~/14105 PCT~US90/02~
.t7
pr~sent invention.
Several methods may be used to ~or~ th~ liposomes of the
present invention. For exa~ple, ~ultilamQllar ve~icies (MLVs),
stable plurilamellar vesicles (SPI.Vs), or rev~rse phase evapora-
tion vesicles (REVs) may be used. Pref~rably, however, MLVs are
extruded through filter~ forming large unilamellar vesicle~
(LW s) of sizes dependent upon the filter size utilized. In gen-
eral, polycarb~nate filters of 30, 50, 60, lO0, 200 or 800 nm
por~s may b~ used. In this method, disclosed in Cullis, et al,,
PCT Publication ~o. W0 86~000238, ~anuary 15, 1986, relevant por-
tions of which are incorporated by reference herein, the liposome
suspension may be repeatedly passed through the extrusion device
resulting in a population of liposome~ o~ homog~neous size dis-
tribution. For example, the filtering may be performed through a
straight-throuqh msmbrane filter (a Nucleopore polycarbonate fil-
ter) or a tortuous path filter (e.g. a Nucleopore filter mem-
brafil filter (mix~d Gellulose esters) of O.l u~ size), or by
alternative size reduction techniques such as homogenization.
Tha size of the liposomes may vary from about 0.03 to above about
2 micron~ in dia~eter; pre~erably about O.OS to 0.3 microns and
most preferably abo~t O.l to about 0.2 microns. The size range
include~ liposom~ t~at are MLVs, SPLVs, or LW s. In the present
invention, the preferr2d liposomes are those which are unilam-
ellar liposom~s o~ about O.l to about 0.2 microns.
As described hereinahove, a nu~ber of lipid~ may ~ used
to form liposomes having a gel to li~uid crystalline Tc above
ambient te~perature. In such case~, an extruder having a heating
barrel or thermojacket may b~ employed. Such a device serves to
.
WO ~/1~105 PCT/US~/02~
1 ~3
2 ~ 5
increa~ the liposome suspension temperature allowing extrusion
of the LUVs. The lipid~ which are us~d with the thermoj~cXeted
extruder ars, for axample, DSPC, DPPC, DMPC and D~PC or mixtures
thereof, which may include cholasterol in certain embodi~ents ~or
preventing the rapid relea~a o~ pharmaceutical agents ~rom the
liposome. ~iposomos containing DSPC are generally extruded at
about 65-C, DPPC at about 45-C and DAPC at about 85-C (about S C
above the lipid Tc).
As indicated, the preferred liposome for us~ in the pres-
ent invention are LUYs o~ about 0.06 to about O.3 microns in
size. A~ defined in the present application, a homogeneous popu-
lation of vesicles is one comprising substantially the same size
liposome~, and ~ay ha~ a Gaus~ian distribution o~ particle
5iZ~. Such a population is said to be of uniSor~ size distribu-
tion, and may b~ unimodal with respect to -~iZQ. The term
"uni~odal~ referY to a population having a narrow polydisper~ity
of particle sizes, and the particles are of a single "mode".
A liposomal population is unimodal if, when measured by
quasi elastic light scattering methods, the population approxi-
mates to a Gaussian distribution, and if a second order poly-
nomial will fit the natural loga-ithm of the autocorrelation
function o~ a ~a~pl~ (Xoppel, 19~2, J~_~he~ P~., 57:4814).
~he clo3er this ~it, the better the measura of unimodality. The
clo~enes~ o~ thi~ ~it ~ay be deter~ined by how clo3e the chi
sguare (chi2) Yalu~ of the sample is to unity. A chi2 value of
2.l) or less is indicative of a unimodal population.
Other size reduction techniques may be employed in prac-
tiing the present in~ention. For eXampl2, ho~genization or mill-
W090/141~ PCT/US90/~t7~
~9
2 ~
ing technique5 may success~ully be employed. Such techni~ues mayyield liposome~ that are homogeneous or uni~odal with regard to
size distribution.
Liposomes ~ay be prepared which encapsulate the first
aqueous buf~er solution having the characteristics described
hereinabove. For a typical liposome preparation technique as
fully described hereina~ove, this ~irst aqueous buffer solution
will surround the liposomes as they are formed, resulting in the
buffer solution being internal and external to the liposomes. To
create the concentration gradien~, the original external buf~er
solution ~ay ~e acidified or basified so that the concentration
of charged species differs ~rom the internal bu~fer, or alterna-
tively, the external bu~fer may be replaced by a new external
mediu~ having different charge species. The replacement of the
external medium can be accomplished by various techniques, such
as, by passing tha liposome preparation through a gel filtration
column, e.g., a Sephadex column, which has been equilibrated with
the new medium, or by dialysis or related techniques.
During preparation of the liposo~es, oryanic solvents may
also be used to suspend the lipid~. Suitable organic solvents
for USQ in the present invention include those with a variety of
polaritie~ and di~lectric properties, which solubilize the
lipids, for exampl~, chloroform, methanol, et~anol, dimethylsul-
foxide (DMSO), u~thylene ~holo~ide, and solv~nt mixtures such as
benzene:methanol ~70:30), a~ong others. As a result, solutions
(mixture in which ths lipids and other components are uniformly
distri~u~ed throughout) containing the lipids are formed. Sol-
vents are generally chosen on the basis of their
W~ ~/l41~5 YCT/US90/027~
~3~ ~33
biocomp~tabllity, low toxicity, and solubilization abilities.
One pre~erred e~bodiment o~ th~ present inv~ntion is a 3
co~ponent liposomal-pharmaceutical agent tr~atment sy~te~ which
allows for highly ef~ici~nt entrap~ont Or the agent at the clini-
cal site. When the pharmaceutical agant i~ one that loads in
r~sponse to a transmambrane pH gradient wh~r~ the interior o~ the
liposome is acid, the ~irst component o~ thQ system (Vial 1) com-
prises liposo~es in an acidic buffer solution, in whi~h for exam-
ple, citric acid bu~fer (300 mmoi., pH about 3.8 to 4.2,
preferably 4.0) or another bu~fer in which the ionized ~orm of
the pharmaceutical agent to be trapped is only marginally soluble
(solubility less than the final concentration of agent within the
liposome, preferably no greater than about 20 mM and most
preferably no grnater than about 10 mM). ThOE sQcond component o~
the system (Vial 2) comprises a ba~ic buffer solution, ~or exam-
ple, a sodium carbonate or sodium bisphosphate solution at about
0.5 M, pH about 10 to 12, preferably abcut pH ll.S, which serves
to become part oS th~ external bu~fer solution of the liposome
formulation. For purposes of maximizing the loading of the
pharmaceutical agent within the liposomes it is preferable that
the phar~aceutical agent ha~ a solubility within the external
bu~Ser solution o~ at least about 0.2 ~ol. The t~ird component
(Vial 3) is the phar~aceutical agent. The above-described treat-
ment syste~ may bo provided as a 3-vial system, th~ ~irst vial
containing the liposomes in acidic medium, the secon~ vial con-
taining th~ bas~, and ths third vial containing the pharmaceuti-
cal agent as described hereinabove. A ~i~ilar treatment system
may be providPd for a pharmaceutical agent that loads in response
W090/14105 2 1 PCT/US~O/Ot736
2 ~
to a transmembrane gradient wheraln the internal bu~er of the
liposome~ i3 r~latively ba~ic i.e., has a pH about 8.5-l1.5. The
~irst component comprises liposome~ in a relatively basic bu~fer,
for example, sodium carbonate or sodium bisphosphate, at a pH o~
about 8.0-ll.O, proferably about lO. The second co~ponent com-
prises a relatively acidic or neutral solution as the external
buf~er for the liposomQs, ~or exampla, lSO mM NaCl bu~er/150 m~
HEPES buffer at a pH of about 7.4. The third component comprises
the phar~aceutical agant which i~ less ionized at the pH of the
external buf~er and is ionized at the p~ of the intPrnal bu~fer.
Following the formation of th~ pH gradient acro~s the
liposo~es (by admixing the first and second vials), the liposomes
may be heated prior to admixing with the drug. Under certain
circumstances, and in cases where the pharmaceutical agent is to
be loaded into liposomes comprising at least about 30 mole %
cholesterol to mini~ize the rapid release o~ the agent, it may be
advantageous to heat the llposomes up to about 60-C to facilitate
loading. To load the pharmaceutical agents into ~he liposomes
utilizing the above-described treatment systems, the methods
described in Mayer, et al. PCT Publication No. WO 88/06442, Sep-
te~ber 7, 1988, relvant portions of which are incorporated by
reSerance, h~r~in may b~ modi~ied for USQ with the ag~nts of the
present in~ention.
In a lipo30m~-drug delivery system, the pharmaceutical
agent is entrapped in or associated with the liposome and then
administered to the patient to be treated. As used throughout
the specification, pharmaceutical agent, drug and agent are used
interchangably. For example, see RaAman et al., U.S. Patent No.
WO90/1~1~ PCT/US90/027~
22
2 ~ ~ ~ r c~ ..3
3,993,754; Sears, U.S. Pate~t No. 4,145,410: Papahadjopoulos et
al., U.S. Patent No. 4,235,871; Schneider, U.S. Patent No.
4,114,179; Lenk ~t al., u.S. Pat nt No. 4, 522, 803; a~d Fountain
et al., U.S. Patent No. 4,588,578. In ~he present invention, any
number of dif~er~nt pharmaceutical agents and different
pharmaceutical types may be entrapped in or associated with
liposomes. For example, pharmaceutical agents useful in the
present invention may includa any agont which readily passes
throush a liposomal layer(s) and ~xhibits limited solubility in a
buffer solutian internal to.the liposome at an ion concentration
or pH at which the pharmaceutical agent is in an ionized form.
Such agents may include antineoplastics: for example
mitoxantrone, local anaesthetics; for example, lidocain~,
dibucaine and chlorpromazina, ~ronchodilators; for example,
metaproterenol, ter~utaline and isoproterenol, beta-adrenergic
blockers, for example propanolol, timolol and labetolol;
antihypertensive agents, for example olonidine and hydralazine;
anti-depressants, for example, imipr.. ~e, amitryptyline and
doxepim, anti-convulsants, for exampl6, phenytoin, anti-emetics,
for example, procainamide and prochlorperazine; antihistamines,
for example, diphenhydramine, chlorpheniramlne and promethazine:
anti-arrhyth~lc agents, for example, quinidine and disopyramide,
anti-malarial agents, for example, chloroquine, quinacrine and
~uinine: and an~lqesics, among a number of additional
phar~ac~utical aqents.
In general, internal bufSers to be used in the liposomal
composition~ of th~ present inv~ntion are chosen using ~everal
criteria, the most important of which, after buf~er strength, is
W090~1410~ PCT/U~90/027~
23
the solubility characteristics of the pharmaceutical agent to be
loaded in the buffer solution, as described hereinabove. It is
preferrQd that th~ bu~r system usQd as the intornal bu~er has
a buffer stran~th of at least about 50 mM, preferably within the
range of about 100 mM to about 300 mM, and most preferably about
300 mM. The most preferred buf~er solution~ for use as the
internal buffer system o~ the present invention are there~ore
characterized by their inability to ~olubilize the ionized,
preferably protonated pharmaceutical agent, i.e., the ionized
pharmaceutical agent is generally soluble in the buffer solution
to an extent no greater than about 65 ~Mol, preferably no greater
than about 20mMol and most preferably no greater than about 10
mMol and which also have bu~fer strengths of at least about 50
mM, preferably about 100 to about 300 mM, most pre~erably about
300 mM. Mo~t preferably, the internal buf~er solution
precipitates the ionized species of the pharmaceutical agent out
of solution.
The choice of buffer to use as the internal buffer solu-
tion will vary depending upon the pharmaceutical agent chosen for
loading. One of ordinary skill in the art will be able to assess
the relative solubilities of ionized species of a phar~aceutical
agent ~nd the bu~fer strength to deter~ina the bu~er solution to
be used a~ the internal buffer solution.
Any buffer solution having the characteristics generally
described hereinaboYe may be used in the present invention, pro-
vided that ~h~ solution is pharmaceutically compatible, i.e., the
solution may be administered to the patient without deleterious
a~fects. Typical internal buffer solutions include citric acid,
WO ~/1~105 PCT/US~/02736
2 ~ '~ 6
oxalic acid, succinic acid and other organic acid salts being
preferred, among othsrs. Citric acid in a concentration ranging
from about lOo ~M to about 300 ~M iq preferred. Most preferably,
the citric acid buffer solution has a concentration ranging ~rom
about 100 mM to about 300 mM. Typical external bu~fer solutions
may include NaCl, XCl, potassium phosphate, sodium bicarbonate,
sodium carbonate, sodium bisphosphate, potassium sulfate and
~EPES, and mixturs~ the ~of, among othar~.
Loading efficiencies of p~ smaceutical ag~nts utiliziny
the present invention generally range from ~bo~t 20~ up to about
100%, preferably at least about 50S. In general, the loading
efficiencies for pharmaceutical agents according to the present
invention are greater than expected from the Henderson-Hasselbach
relationship. 0~ course, not all agents readily accumulate in
liposomes according to the Henderson-Hasselbach relationship, and
certain agents (see Table 1, Example 1) appear, in certain cases,
not to accumulate at all. This phenomenon may be the result of
the pharmaceutical agent being too polar for penetration of the
liposomes, or other factors. 0~ course, ona of ordinary skill in
the art will recognize that to maximize the loading o~ a
phar~aceutical agænt into liposo~es, it ~ay be n~cessary to
chango ths lipid constituents o~ the liposomes, or, in certain
cases, to utilize ionophor2s or other agents which may enhance
th~ p2n~tration o~ the liposome by the agent in practicing the
present invention.
The liposo~es ~ormed by the procedures o~ the present
invention may be lyophilized or dehydrated at variou~ stages of
formation. For example, the lipid film may be lyophilized after
WO90~l4l0~ PCr/US90/02736
removing the solvent and prior to addinq the drug. Alterna-
tively, the lipid-dru~ film may ~e lyophilized prior to hydrating
the liposome~. Such dehydration may bQ carried out by exposure
of the lipid or liposome to reduced pressure thereby removing all
suspending solvent. The liposomes may be dehydrated in the
presence of a hydrophilic agent accordinq to the procedures of
Bally et al, PCT Publication No. 86/01102, published February 27,
1985, entitled "Encapsulation of Antineoplastic Agents in
~iposomes", Janoff et al., PCT Publication No. 86/01103, pub-
lished February 27, 1986, entitled "Dehydrated Liposomes",
Schnelder et al., in U.S. Patent No. 4,229,360, issued October
29, 1980 and Mayer, et al. PCT Publication No. 88/06442, pub-
lished September 7, 1988, relevant portions of which are incor-
porated by reference herein. Alternatively or additionally, the
hydrated lipsome preparation may also be dehydrated by placing it
in surrounding medium in liquid nitrogen and freezing it prior to
the dehydration step. Dehydration with prior freezing may be
performed in the presence of one or more protective agents, such
as sugars in the preparation according to the techniques of
Bally, et al., PCT Application No. 8S/01103 published February
27, 1986, releYant portions of which ara also incorporated by
r~ference herein. Such techniques enhance the long-term storage
and stability of hte preparations. For example, the pharmaceuti-
cal agent may be ~ixed with a sugar solution in a suqar: lipid
weight/weight ratio ranging from about 0.5:1 to about 100:1,
preferably about ~0:1, without affecting ~he ability of the
liposome to retain loaded agent during rehydration. In this
aspect of the present invention, the liposomes preferably range
WO ~/1~1~ PCT/US9~/0~736
26
in size from about 0.1 to about 0.2 mlcrons.
In one pr~ferred ~bodiment, th~ sugar is mannitol, or
~annitol:glu~ose:lactose in a 2:1:1 w/w/w ratlo. Following
rehydration in distillod watsr, tho preparation is preferably
heated for ten minut~s at an elevated temperature, for example
60-C. Other suitabl~ methods may be used in the dehydration o~
the above- disclosed liposome preparations. The liposo~es may
also be dehydrat~d without prior freezlng.
once the liposomes have been dehydrated, they can be
stored for extended periods of ti~e until they are to be used.
The appropriate temperature for stora~a will depend on the lipid
for~ulation of the liposomes and the temperature sensitivity of
encapsulated ~aterials. For example, various antineoplastic
agents are heat labile, and thus dehydrated liposo~es containing
such agents should bQ stored under refrigerated conditions e.g.
a~ a~out 4'C, so that the pot0ncy o~ th~ agent is not lost.
~lso, for such agents, the dehydration process is preferably
carried out at reduced te~peratures, rather than at room
temperature.
When ~ dehydrated liposome~ are to be used, rehydrAtion
is accomplished by simply adding an aqueous solution, e.g., dis-
tilled water or an appropriate buf~er, to the liposomes and
allowing th~ to rehydrate. The liposo~es can be resuspended
into the aqueous solution by gentle swirling o~ th~ solution.
The rehydration can b~ performed at room te~perature or at other
te~peratuses apprspriate to the composition o~ the liposomes and
their int~rnal contents. I~ the antineoplastic agent which is t_
be administered was incorporated into th~ high drug to lipid
WO90~14105 PCT/US90~27~
27
2 i~ 3
ratio liposomes prior to dehydration, and no further compositien
changes are desired, the reAydrated liposomes can be used
directly in the cancer therapy ~ollowing ~nown procedures for
administering liposome encapsulated drug5. Alternatively, using
the transmembrane pH gradient procedure~ described above,
ionizable antineoplastic agents can be incorporated into the
rehydrated liposomes just prior to administration. In connection
with this approach, the concentration gradient used to generate
the transmembrane pH gradient can be created either before
dehydration or after rehydration using the external medium
exchange techniques described above. For example, the high drug
to lipid ratio liposo~es may be dehydrated prior to establishing
the transmembrane pH gradient, for examplQ, dehydrated from their
first external medium. Upon rehydration, the pH gradient can be
established ~y adcixing the liposomes with the second external
medium of relatively acidic or basic pH. The antineoplastic
agent can be admixed with the liposomes simultaneously with or
following the establishment of the pH gradient.
In the case where the liposomes are dehydrated after
having a transm~brane pH gradient, the liposomes may be
rehydrated by ad~ixing them with an aqueous solution of neutral
pH.
For exa~ple, in the above-mentioned case where liposomes
containing citric acid buffer as the first medium are used, the
rehydration step would proceed by adding sodium carbonate and the
pharmaceutical agent, for example, propanolol. Where the
liposomes already contain the base (e.g. sodium carbonate), and
therefore already have ~he transmembrane pH gradient are
WO 90tl4105 PCT/US~/027~
28
2Q~ ?
rehydrated, water or another neutral a~ueous solutlon, and
doxaru~icin are adted. Finally, in the case where liposomes
having a transmem~rane pH gradient and contalning the
pharmac~utical aqent have been dehydrated, rehydration proceeds
using water or another a~leous solution. Alternatively, a second
phar~aceutical agent may be added, if d~sired.
Liposomes containing the pharmaceutical formulations of
the present invantion may be used therapeutically in mammals,
especially hurans, in the treatment of a number of disease states
or pharmacological conditions which require sustained release
formulations as well as repeated administration.
The mode of administration of the liposomes containing
the pharm~ceutical agents of the present invention may determine
th~ sites and cells in the organism to which the compound may be
d~livered. The liposomes of the present invention may be
administered alone but will generally be ad~inistered in admix-
ture with a pharmaceutical carrier selected with regard to the
intended route of administration and standard pharmaceutical
practice. The preparations may be injected parenterally, for
example, intravenou~ly. For parenteral administration, they can
be used, for exampl~, in the form of a sterile aqueous solution
which may contain other solutes, for example, enough salts or
gluco3Q to maXQ the solution isotonic. The liposo~es of the
present invention may also be employed subcutaneously or
intra~uscularly. Other uses, depending upon the particular
properties of tha preparation, may be envisioned by those skilled
in the art.
For the oral mode of administration, the liposomal for-
W0 ~14105 2 9 PCT/US90/02736
2~5~3~5
mulations of the present invention ca~ he used in the ~orm Oetablets, capsules, losenges, troches, powders, syrups, elixirs,
aqu~ous solution~ and suspensions, and ths liXe. In the case of
tablets, carriers which can be used includa lacto~e, sodium
citrate and salts o~ phosphoric acid. Various disintegrants such
as starch, lubricating agents, and talc are commonly used i~
tablets. For oral administration in capsule form, useful
diluents are lactosa and high mol~cular weight polyethylene
glycols. When aqueous suspensions are required for oral use, the
active ingredient is co~bined with emulsifying and suspending
agents. If desired, certain swestening and~or ~lavoring agents
can be added.
For the topical mode of ad~inistration, the liposomal
formulations of the present invention may bç incorporated into
dosage form~ such as g~ls, oils, emulsions, and the like. These
formulations may be administered by direct application as a
cream, paste, ointment, qel, lotion or the like.
For ad~inistration to humans in the treatment of disease
stat~s or pharmacological conditions, the prescribing physician
will ultimately deter~ine the appropriate dosage of the
neoplastic drug ~or a giYen hu~an subject, and this c~n be
expQcted to ~ary according to th~ age, weight and re~ponse of the
individual as w~ll as the phaxmacokinetics o~ the agent used.
Also the nature and severity of the patient' 5 disease stat~ or
pharmacological condition will influence the dosage regimen.
While it is e~pected that, in general, the dosage of the drug in
liposomal for~ will be about that employed for ~he free drug, in
som~ cases, it may ~ necessary to administer dosages outside
WO ~14105 PC~/US9~/027~
2~5~ 5
these limits.
The following examples are givan for purposes of
illustration only and are not to he viewed aq a limitation o~ the
scope o~ the invention.
PreParation o~ a~d Li~osomes
T~e following pharmaceutical agents were loaded or
attempted to be loaded into liposomes comprising egg
phosphatidylcholine (EPC, from Avanti Polar Lipids, lnc.,
Birmingham, Alabama) or EPC and cholesterol 55:45 (molar ratio)
(cholesterol from Sigma Chemicals, St. Louis, MO.): propanolol,
timolol, di~ucaine, chlorpromazine, lidocaine, quinidine,
pilocarpine, phy~ostigmine, dopamine, imipramine, diphen-
hydramine, quinine, chloroquine, quinacrine, dauorubicin, vin-
cristine and vinblastine (obtained from Sigma Chemicals, St.
Louis, MO.), doxorubicin, epirubicin (o~tained fro~ Adria
Laboratories, ~ississauga, Ont. Canada), mitoxantrone (obtained
from Cyanamid Canada Inc., Montreal Que. (Canada), codeine and
pethidine (obtained ~rou Abbott Laboratories, Ltd. Downsview,
Ont. Canada). Th~ radiolabels, 3~-
dipal~ oylpho~;phatidylcholinP, 14C-
dipaluitoylphosphatidylcholine, l4C-dopamine and l4C-Imipramine
were obtained fro~ Amersham while 3H-c~lorpromazine, 3H-
propranolol, l4C-pilocarpine, 14C-chlorpromazine, 14C-methylamine
and l4C-lidocaine were obtained ~rom ~ew England Nuclear. The
Liposome Company, Inc. ~Princeton, N.J.) kindly pravid~d l4C-
timolol. Salts and rea~ents used were of analytical grade.
All loading o~ pharmaceutical agents were into EPC
WO ~/141~ PC~/US~/~t7~
3 1
2 ~ 3 ,~
vesicles Icontaining 3H-Dipalmitoylphosphatidylcholine) or
EPC:cholesterol mixtures (55:45 molar ratio). EPC:cholesterol
~ixture3 were prepared by colyophilization from benzene:methanol
(95:5 v/v). The dry lipid waq hydrated with 300 mM citrate p~
4.0 as internal bu~fer solution and the resultant MLVs were sub-
jected to five freeze-thaw cycles employing liquid nitrogen to
enhance solute distribution according to Mayer, et al. ~iochim.
3io~hyal_~ç~, 817, 193 (1986). ~arg~ unilamellar vesicles were
then prepared using an Extruder (Lipex 3iomembranes, vancouver,
Canada) employing the LUVET procedure as described by Hope, et
al. ~iohim. ~iophy~, Acta, 812, 55, (1935) with 100 nm pore size
polycarbonate filters (Nucleopore, Inc.). To establish a pH gra-
dient the vesicle~ were then pa sed down a Sephadex G-50 (fine)
colume ~1.5 X lOcm) preequilibrated with 300 ~M NaCl, 20 mM
HEPES, pH 7.5.
Large unilammelar vesicles (approximately 1 mM lipid)
were incubated with the agent (0.2 mM) in 300 ~M NaCl, Z0 mM
HEPES, pH 7.5 at 25-C. At various times up to 2 hours, aliquots
(100 ul) of the mixture were taken and vesicles separated from
unentrapped drug by centrifugation thxough a 1 ml "uinicolumn" o~
Sephadex G-50 (mQdium) a~ described by Pick, AL~h~ 3iochem.
~io~h~. 212, 186, 1981. Lipid and drug were quantiÇied by the
following procedure.
Lipid concentrations were deter~ined ~y liquid scintilla-
tion counting o~ 3H-DPPC or 14C-DPPC using a Pac~ard 2000 CA
instrument. Si~ilarlyl pilocarpine, chlorpromazine, timolol,
propranolol, imipramine, lidocaine and dopamine were quanti~ied
~sing tracer quantities of 3H- or 14C-radiolabel.
WO90~l43~ PCT/USgO~02~
32
2 8 ~
Physostigmine was assayed by ~luorescenc~ spectroscopy
following solubilization of vesicles in 60% ethanol (v/v). The
exc~tation and emission of wavelengths used were 305 and 350 nm,
respectively. Quinacrine, chloroquine and quinine were also
quantified from their ~luorescence using excitation and emission
wavelenqths of 420 nm, 505 nm; 335 nm, 375 nm; and 335 nm, 365
nm; respectiv~ly.
Vinblastine and ~incristine were assayed ~y U.V. spec-
troscopy from their absorbances at 262 nm and 297 nm, respec~
tively, following solubilization of the vesicles in 80% ethanol.
Codeine was also ~easured by U~V. ~pectroscopy at 220 n~ in this
case after solubilization in 40 mM octyl-beta-D-glucopyr~noside.
Mitoxantrone was quantified fro~ its absorbance at 670 nm follow-
ing solubilization of the vesicles in 2% Triton-X100.
Diphenhydramine was assayed by gas-liquid chromatography
using a HP 9850 gas chromatoqraph fitted with a Chromatographic
Specialties DB-225 (25% cyanopropylphenyl) capillary column. The
helium carrier flow rate was 1 ml/min and detection was by flame
ionization. An internal standard, methylpentadecanoate, was used
to quantify diphenhydramin~ following its extraction from the
aqueous 5a~pl~ in di~thylether and its separation fro~ EPC by
thin lay~r chromatography. Transbilayer pH gradients were
quantified e~ploying the weak base methyla~ine (14C-labelled) as
previously described by Bally, et al-, 5~Y~L 3~Y .~ 47
97, (1988).
The results o the loading experiment appear in Table l,
below. 3asically, the loading of liposo~es with the agents
described above may ~ efined on the basis of their uptaXe char-
WO ~/l4105 P~T/US90/02736
33
2l~5~ i~ 3~;
acteris~ics. Four drug categorles may be defined based upontheir uptake characteristics.
The rirst cat~gory o~ phar~ac~utical agents exhlbLted
completR, stablo uptake. Propanolol, dopamine, dauonorubicin,
epirubicin, dibucaine, imipramine and doxorubicin exhibited the
characteristics of this drug category. All o~ the drugs within
this categary exhibitud uptakQ greater than predicted from the
Henderson/Hasselbach equation. The accumulation o~ mitoxantrone
by EPC liposome~ exhibiting a proton gradient i~ shown in Figure
1. .
The second category showed partial, but s~able up~ake.
Timolol, lidocaine, chlorpromazine, serotonin and chloroquine
exhikited the characteristics of this drug category. Timolol was
loaded to the extent o~ about 100 nmoles/umole lipid (about 50~
o~ available drug, see ~iqure 2A) and quinacrine was loaded to a
level of about 80 nmoles/umole lipid after 30 ~inutes (see figure
2B). While accumulation is lower than in the first group of
agents, nevertheless uptake is quite substantial. In the case of
timolol, an internal concentration of about 65 m~ is achieved
against an external concentration of 109 uM.
Tha third category shows a partial uptake followed by a
rapid relea~e of agent rom the lipusom~. Figure 3 indi~ates a
rapid virtually complete accumulation o~ quinidine into the
vesicles and within 30 ~inutes about 50~ of the agent has leaked
back out of the vesicles (Figure 3A). Other agents whi~h leak
back out of EPC vesicles include quinine, diphenhydramine, vin-
blastine and vincristine. The leakage rates vary considerably
with vincristine and vinblastine loaded vesicl~s losing only 27%
WO9~/14l05 PCT/US90/027~
34
2 0 ~
o~ initially seyuestered d~g over two-hours. This loss is asso-
cia~ed ~ith a corresponding reduction in re~idual change i~ pH as
detQrmined using ~thylamina. A simllar d~crease in proton gra-
dient is obs~rv~d a~ quinin~ and diph~nhydrami~ ar~ released
from EPC v~sicles.
The fourth category of phar~aceutical agents, physostig-
mine, codeinR and pilocarpine exhibited no ~asurabl~ r~sponse t~
the transmembrane pH gradient. Tha suggestion that these agents
causQ a ma~or increa ~ in me~brane permeability resulting in loss
of ion gradient is not borne by the data from physostlgmine (Fig-
ure 4). Und~r th~ conditions us~d to as~ess uptake of physostig-
~ine (200 u~) only a~small d~creasQ in m~asured change in pH was
o~servad.
~kL~L
Extent and Stability of accumulatian of Various Drugs
Ve~icl2J Exhibiting a p~ Gradient
,,
Drug Class Uptake 15 Minutes Uptake 2 Hours
(nmoles/umoles lipid) ~nm/um lipid)
S~
Mitoxantrone Antineoplastic 200 198
Epirubicin Antin~oplastic 201 200
Daunorubicin AntinRoplastic 200 204
Doxorubi~ln ~ntinQopla~tic 202 203
Dibucaine LoG~l Ana~sthetic~ 194 176
Propanalol Adr6n~rgic 198 187
Dopa~inQ Biogenic Amine 190l 177
cateqo~t Z
Timolol Adrenergic gs 97
Lidocain~ Lccal Anaesthetic 87 87
WO 91)/11105 PCI/US90/02736
Chlorpromazine Local Anaesthetic 98 96
Serotonin Biogenic Amine 802 78
Chloroquine Antimalarial 1043 88
Quinacrine Antiprotozoal 731 71
Cateqory ~
Quinidine Antiarrhythmic 203 74
Quinine Antimalarial 1483 81
Diphenhydramine Antihistamine 1~63 87
Vinblastine Antineoplastic 1753 127
Vincristine Antineoplastic 178 130
categor~ 4
Codeille Analgesic <1 ~1 -
Pilocarpine Cholinergic <1 ~1
Physostigmine Cholinergic ~2 <1
, ~
Footnote: L ~ maxlmum uptake at 30 mlnutes, ~, maxlmum uptake at
90 minutes, 3, .maximum uptake at 5 minutes
Examplç ~
Com~arisQn ~e~ween I~evel of ~rua U~take and
th~D~ua's Octanol: Water Partition Coefficient
Levels of drug uptake were co~pared to their
octanol/water partition coefficients to determine the extent that
partition coefficient and the possibility that an agent was
partitioning into the liposome bilayer raight deter;nine the extent
of uptake o~ a pharmaceutical agent. Fros~ Table 2, below, it
appears that no clear relat onship exists between drug uptake and
its partition coe~ficient. Although the values ~iven may not
accurately reflect mem~rane/water partition coefficients, they
are ~erely being used for a comparative basis. Chlorpromazine
and doxorubicin, for example, have si~ilar partition coefficients
yet display very di~fer~ont uptake levels (98 vs 202). On the
WO ~/14105 PCT/US90/027~
20~S~
other hand, timolol and chlorpromazin~ are accumulated by
vesicles to a similar extent despite a large di~ference in their
partition coe~icients. While partition coef~icient for a
ionized drug may in~luence dru~ up~ake, it can not be taken to
explain the di~erences between the agents studied.
A comparison Bet~een the L~vel of Drug Uptake and
Its Octanol: Water Partition Coef~icient
Drug Maximum Uptake Log Octanol:Water
nmoles/umole lipid Partition Coefficient
Daunorubicin 2Q0 3.5
Doxorubicin202 . l.l
Vincristine178 2.8
Chlorpromazine 98 l.5
Dibucaine 194 4.4
Propranolol2 198 l.3
Timolol2 95 . -0.l
Physostigmine 0 O.2
Imipramine 182 4.6
Diphenhydramine 176 3.4
Quinine l48 l.7
Codein~ 0 l.2
FootnoteY ~- All data taken from Leo, A., e~ al. Chem. Rev., 7l,
525~ (l971) except for propranolol and timolol.
- Fro~ Merbate, L. 5., et al-, ~igLhY~ 49, 9l,
(1986).
WO ~141~ PCT/US~/027~
37
2 ~ 3
~xam~le 3
cQm~ri~Q~ ~e~e~ ~vql Q~ IL. -
th~ prya'~ ~o~ t~ i~ 9u~ o1y~Q~
~ esidea bu~er strength, the factor that in~luences the
level o~ druq uptake to the greatest extent is the solubility 5
the protonated spRcies in the internal bu~er. Wh~n the con-
cQntration of protonated drug inside the vQsiclQ exceeds its
solubility product and precipitation occur3 thie will e~fectively
reduc~ the transm~mbrane concentration gradiQnt ~or the remaining
solube fraction thus allowing further accumulation by the
vesicles. In table 3 is show~ the maximum apparent solubilities
in 300mM citrate buffer, p~ 5.O for most o~ the drugs whose
proton gradient dependent uptake was examined. Drugs such as
mitoxantrone, epirubicin, doxorubicin and daunorubicin which
exhibit complete and stable uptak~ are relatively insoluble in
the intravesicular medium. This indicates that most of the
accumulated drug is in the form of a precipitata and does not
contribute to the concentration gradient of the soluble
protonated species, thu~ accounting for the high levels o~ uptake
observed. In addition, if most Or the intravesicular drug is
precipitat~d the concentration of free dru~ available to parti-
tion into the mc~brans is correspondingly reduced which will con-
tribute to the obsQrv~d stability o~ th~ transme~brane proton
gradisnt. As ~xpect~d, agents such as timolol, lidocaine,
quinacrin~ and chloroquine which exhibit uptake in good agreement
with the Hender~on-Hasselbach equation have apparent solubilities
which are in excess of the intravesicular concentrations achieved
(See table 3, below). Without being bound by any theory, the
solubility data may explain most of the observed di~ferences in
Wo ~1410~ RCT/US~/02736
38
2 ~
uptaX~ characteristic~ ~or the various druqs examined. The data
indicate~ that solubility data is most important in determinin~
upta~e o~ drugs into liposomes.
We note that dibucaine, propranolol and dopamine ~ay also
~e loaded in liposomes in an amount significantly greater tha~
predicted by the ~enderson-Hasselbach equation. T~is is a sur-
prising result considering that the three agents' apparent
solubility is greater than the final internal concentration of
the agent in the liposome.
~a~le 3
Appar~nt Maximum Drug Solubility in 300 mM
Citrat~ Buffer, pH 5.0
Drug Apparent ~axlmu~ Solubility
(mM)
Mltoxantrone <O.01
Epirubicin 0.26
Daunorubicin .9.10
Doxorubicin 0.24
Vincristine >35
Vinbla~tine 19.1
Lidocaine 240
Dibucaine >700
Propanolol
Timolol 135
Quinidin~
Dopa~ine 1400
Quinine 1.05
Chloroquine 585
Quinacrine 90
.
Wo ~/1~1~ PCT/U~/027~
2~ 3
Exa~DlQ ~
Isopr~terenol. MetaDroterenol and Ter~utaline
E~g phosphatidylcholine (EPC) purohased from Avanti Polar
Lipids (8irmingham, Alabama), and 14C-methylamine was purchas2d
from N~w England Nuclear. All other chemicals and buf ~ers were
purchased from Sigma (St. ~ouis, M0.) and were used without
puri~ication.
Large Unilamellar Vesicles (LW s) were produced by extru-
sion according to the method of Hope, et al., 3i9C.LU~_.3~D~h~_
Ac~a, 817, 193 (1985) Briefly, LUVs were pro~uced by extrusion
o~ ~rozen and ~hawed lipid dispersions prepared in 300 ~M
citrate, pH 4.0, through 0.1 or 0.2 um polycarbonate filters
(Nucleopore) employing an extrusion device (Lipex 8iomembranes,
Vancouver, Canada~. Vesicles prepared by this t~chnique employ-
ing 0.~ um filt~rs have trapped volum~s of 1.5 uL/umole
phospholipid as deter~ined using 14C or 22Na and have an averag~
diameter of 90 n~. Phospholipid concentrations wzre determined
by assay of lipid phosphorous as previously described by Fiske
and Subbarrou, 2, Biol. Ch~., 66, 375, tl925). Transmem~rane pH
gradient~ wer~ a~tablishQd according to Exampla 1, and untrapped
int~rnal bu~er re~o~ed by passing tha LW s down a Sephadex G-S0
column equilibrated with tha external bu~er ~150 mM NaCl, 20 mM
HEPES, p~ 7.4]. Induced p~ gradients w~re detQr~ined by measur-
ing the transme~brane distributions of 14C-methylamine as
described by HOPQ , et al., ~EE~. In short, methylamine was
added to the ve3icle system to a final concentration of 0.5
uCi/~L. At appropriate time~, aliquots (100 uL~wer~ removed and
WO ~/14105 P~T/U~90/027
4 0
2 ~ C~
passed down 1 mL sephadex G-50 mini-columns as previously
described. The trapp~d probe was determined by liquid scintilla-
tion counting employing a Packard 2000CA liquid scintillation
counter, and phospholipid concentrations w~ra determined. Trans-
membrane pH gradients were calculatad according to the equation
change in pH~ log[MeAm]i/~MeA~]O.
The bronchodilators, isoproterenol, metaproterenol and
terbutaline were incubated with liposomes with a transmem~ranepH
gradient prepared as above at the indicted temperatures in a 150
mM NaCl, 20 mM HEPES, pH 7.4 buffer containing 500 uM o~ the
bronchodila~or and 6 mM of the phospholipid. Control samples
without a trans~embrane pH gradient were incubated at pH 4.0 or
7.4 (both insida and outside the vesicles) to determine the
degree of gradient-indepemdent membrane binding. The pH 4.0 con-
trol consisted o~ tha vesicles prepared as above, but the
external buffer was 150 mM NaCl, 20 m~ citrate, pH 4Ø For the
pH 7.4 control, the vesicles were prepared with 150 ~M NaCl, 20
mM HE~ES pH 7.4, both internally and externally.
Free drug was separated from vesicles using 1 mL Sephadex
G-50 minicolumns as described above and assayed ~photometri-
cally). Figur~ 5 ~hows the entrapment of tho bronchodilators
metaproterenol, terbutalin~ and isoproterenol in response to pH
gradients using ( PC) 200 nm extruded liposomes. Vesicles con-
taining 300 m~ citrate, pH 4.0 were incubated with a 500 uM drug
solution at pH 7.4. Th~ liposomes accu~ulate the drug to levels
o2 greater than 60 nmoles/umole lipid. This is roughly 70~ trap-
ping ef~iciency, and corresponds to an inside:outside drug con-
centration ratio oS about 190:1, which assume~ an internal volume
.
wo 90/l~lûS PCr/US90~01736
41
2 ~ 3 S ~ 3 3
o~ 2.2 uL/umole lipid. I~ all drug i5. in the aqueous space of
the vesicles, an internal drug concentration of about 30 mM is
calculated. Uptake i5 stable for at least 4 hours and is com
plete within ~inutes. In the absence o~ the transmembrane pH
gradient, background binding of these drugs is less than 15
nmoles/umole lipid for both pH 4.0 (inside and outside the
vesicles) and pH 7.4 (in and out), indicating that non-specific
binding to the llpid is not responsible for the ;sociation o~
thesa drugs with the liposomes, and that the associ~tion is a
function of the proton gradi~nt rather than the absolute pH.
Exa~ 5
Effect of Dru~ U~ake on ResiduaL ~H Gradie~t
As M~surç~k~MethYL3~nç ûi$t~ib~lt~on
The loqarithm of the ratios of the internal and external
concentrations ~f the radioactive methylamine can be used to
measure the transmembrane change in p~, because the methylamine
probe does not dissipate the internal proton poool at these con-
centrations. When the internal and external pH is 7.4 or 4.0 (no
gradient) the methylamine does not datect any gradient (Figure .
6). When vesicle~ with an internal pH of 4.0 are incubated in a
pH sf 7.4, th~ m~thylamine distribution indicates a 3.0 unit pH
gradient, in good agreem~nt with th~ 3.4 pH unit gradient. When
~etaproter~nol is added to the ~xternal buffer to a final con-
centration of 500 u~, the gradient dissipates to about 2.3 pH
units as the drug i5 accumulated. The date indicates that the
190 fold achieved by the drug approximates the residual proton
gradient (pH o~ 2.3 units).
W090/141U~ PCT/US90/027~
42
2 ~ 3 ~
~R~ 6-
E~ec~ o~.H~at o.~ E~r~m~nt o~ ~e~a~rQt~ereno1
The rate of metaproterenol entrapment is increased by
increased temperature (See Fiqure 7), reachiny steady-state
levels arter 2 hours at 21-C, but ~aster than 15 minutes when
incubatQd at 60 C. The extent of drug uptake is not dramatically
affected by the temperature o~ incubation.
Exam~Q 7
Effeçt~ o~ Choles~rol on ~ntraD~ent o~ 8ronchodilators
The inclusion of cholesterol in liposomes on the.uptake
of bronchodilators was in~estigated. Cholesterol decreased both
the rate o~ uptaXe, and ths total am~unt o~ trapped drug per
u~le of lipid. 5inc~ the internal trapped volu~e o~
EPC:cholesterol (55:45 mole %) determined using l~C inulin and
22Na is about 30% lower than vesicles compos~d of EPCalone, the
actual cancentration gradients of metapro~erenol achiaved is
similar in both ca~es. See Figure 8.
xa~ple_8
Effect of Dru~ to Li~id Ratio on the Amount o~ T~a~Ped Druq
The amount of entrapped drug also depends on the initial
drug to lipid ratio (see Figure 9). At low drug to lipid ratios,.
the a~ount oS drug entrapped by the vesicle~ reflects the imposed
~ransmembrane pH gradient greater than 3 units. At sufficiently
high drug concentration, the reionization of the drug in th~
vesicle interior causes a decrease in the internal pH (Figure 6).
Equilibrium levels o~ drug entrapment would there~ore be expected
to relect the final change in pH rather than the initial chan~e
in pH as the ionized drug overwhelms the internal buffering capa-
city o the vesicle~.
WO ~/~410~ P~/US90tO27~
43
Example g
E~ec~ o2 ~5fer S~ren~t~ Qn E~traDment
Extent o~ the buf~er capacity (buf~er strength) is a fac-
tor which affect~ the ability of the liposome to trap ionized
drug . Low internal buf~r capacity affects the extent of drug
accumulation. ~elow 100 mM citrate, the extent of drug accumula-
tion is indeed af~ected by the internal citrate concentration
(Figure 10). Under theqe conditions, re-ionization of the drug
ov~rwhelms the internal bu~fering capacity of the vesicle inte-
rior, raising the pH of the vesioles and dissipating the change
in pH (inset). Including greater levels o~ citrate than 300 mM
in the vesicles does not dramatically increa~e the levels of
trapped drug, since there is still a large residual pH gradient
in these cases and the internal ~ufferin~ capacity does not limit
drug uptake.
It will be understood by those skilled in the art that the
foregoing description and examples are illustrative o~ practicing
the present invention, but are in no way limiting. Variations of
the detail presonted herein may be made without departing fro~
the splri~ and scop~ o~ the pres2nt invention.