Note: Descriptions are shown in the official language in which they were submitted.
W09l/0072~ 2 ~ S 5 PCT/US90/03630
A~INO ACID ANALOG CCK ANTAGONISTS
This is a continuation-in-part of U.S. Patent
application Serial No. 376,778, filed July 7, 1989, which
is a continuation-in-part of PCT patent application Serial
No. PCT/US89/01412, filed April 4, 1989, which is a
continuation-in-part of U.S. patent application Serial No.
177,715, filed April 5, 1988.
Technical Field
The present invention relates to compounds and
compositions which antagonize cholecystokinin and gastrin,
processes for making such compounds, synthetic
intermediates employed in these processes and a method for
treating gastrointestinal disorders, central nervous
system disorders, cancers of the gastrointestinal system
- (i.e., pancreas, gall bladder, etc.), hypoinsulinemia, or
,; '.
- : ~ , , . :
. ..
WO9l/00725 PCT/US90/03630
., f~.
, . v
X~5 :;
potentiating analgesics, or regulating appetite disorders
with such compounds.
Backaround of the Invention
Cholecystokinins (CCK) are a family of polypeptide
hormones. CCK and a 33 amino acid fragment of CCK (CCK33)
were first isolated from hog intestine. (Mut and Jorpes,
Biochem. ~. 125, 628, 1981). Recently the CCK33 fragment
has been found in the brain, where it appears to be the
precursor of two smaller fragments, an octapeptide CCK8
and a tetrapeptide CCK4. (Dockray, Nat~re 264, 4022,
1979)-
CCK8, the carboxyl terminal octapeptide fragment ofCCK, is the smallest CCK fragment that remains fully
biologically active. (Larsson and Rehfeld, Brain ~
1~, 201-218, 1979). The localization of CCK fragments
in the cortex of the brain suggests that CCK may be an
important neuromodulator of memory, lea-ning and control
of primary sensory and motor functions. CCK and its
fragments are believed to play an impor-ant role in
appetite regulation and satiety. (Della-Fera, ~ ~s~
2Q6, 471, 1979; Gibbs et al., Nature ~, 599, 1981; and
Smith, Eating and Its Disorders, eds., ~aven Press, New
York, 67 ~984).
CCK antagonists (B.J. Gertz in Neu-oloay and
~eurobioloaY Vol 47, Cholecystokinin An-aaonists, Wang and
Schoenfeld eds. Alan R. Liss, Inc., New York, NY, 327-342,
1988; Sil~erman et al., a~ J Gastroent., 82(8), 703-8,
1987) are useful in the treatment and prevention of CC~-
related disorders of the gastrointestin~l (GI) (Lotti et
~ . . . - . .
. .
, . : - .
.
. . . .
WO91/00?25 PCT/US90/03630
~ ~ ~ ? 7 SS
~. .. .
al., J Pharm E~ IhsL~ 1), 103-9, 1987), central
nervous (CNS) ~Panerai et al Neuropharmacoloay, ~(9),
1285-87, 1987) and appetite regulatory systems of
animals, especially man. CCK antagonists are also useful
in potentiating and prolonging opiate induced analgesia
and thus have utility in the treatment of pain. ~Faris et
al., Science ~2~, 121S, 1984; Rovati et al., Clinical
Research, 34(2), 906A, 1986; Dourish et al., European J.
Pharmacology, 147, 469-72, 1988). Disease states that
may be treated with CCK antagonists are disorders of
gastric emptying, gastroesophageal reflux disease
~Setnikar et al ~z~ Forsch./Drua Research, 37(II) 10,
1168-71, 1987), pancreatitis, pancreatic and gastric
carcinomas (Douglas et al., Gas~roent. 2~, 4629, 1989;
Beauchamp et al., B~ Sura, 202, 313-9, 1985), disorders
of bowel motility, biliary dyskinesia, anorexia nervosa,
hypoglycemia (Rossetti, Diabetes, 36, 1212-15, 1987;
Reagan, ~uropean J. Pharmacoloav, 144, 241-3, 1987),
gallbladder disease, and the like.
Previously four distinct chemical classes of CCK
receptor antagonists have been reported. The first class
comprises derivatives of cyclic nucleotides as represented
by dibutyryl cyclic GMP (N. Barlos et al., ~m_ J.
Phvsiol~ 2, G161, 1982) and references sited therein)~
The second class is represented by the C-terminal
fragments of CCK tsee Jensen et al. Biochem Biophys.
~La, 757, 250 1983) and Spanarkel J. Biol. Chem. 258,
6746, 1983). The third class comprises amino acid
derivatives of glutamic acid and tryptophan as indicated
by proglumide (and its analogs) and benzotript (see Hahne
WO9l/~0725 PCT/US90/03630
SS
et al ~oc. Natl. ~cad. ~ci. U.S.A., 78, 6304, 1981 and
Jensen et al. ~i~chem. Biophys. B~S~_ 761, 269, 1983).
Tne fourth and most recent class is comprised of 3-
substituted benzodiazepines, represented by L-364,718
(see: Evans et al. Proc. Natl. Acad. Sci. U.S.A., 83 4918,
lg86) .
With the exception of certain substituted
benzodiazepines and recently reported analogs of
proglumide (Makovec et al Arzneim.-Forsch./Druq Res.
36,(I), 98-102, 1986) , all of these compounds are
relatively weak antagonists of CCK usually demonstrating
IC50's between 10 9 and 10 6 M. The benzodiazepine CCK
antagonists or their metabolites may have undesireable
e~fects i~ v vo due to their interaction with
benzodiazepine or other receptors.
The C-terminal pentapeptide fragment of CCK is the
same as the C-terminal pentapeptide fragment of another
polypeptide hormone, gastrin. Gastrin, like CCK, exists
in the GI system. Gastrin antagonists are useful in the
treatment and prevention of gastrin related disorders of
the GI system such as ulcers, Zollinge--Ellison syndrome
and central G cell hyperplasia. There are no effective
receptor antagonists of the in vivo ef-ects of gastrin.
(Morely, Gut Pept. Ulcer Proc., Hiroshima Symp. 2nd, 1,
1983). A recent report (Bock J. ~. Chem., 32, 13-16,
1989) discloses potent n vitro gast- ~ antagonists.
: ', '. '. '
WOgl/00725 PCT/US90/03630
2~ S5
. ,. ,. `. .
~isclosure of the Tnvention
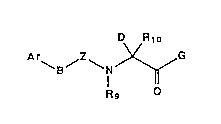
In accordance with the present invention, there are
cholecystokinin antagonists of the formula:
D R10
B ~ ~ r ~ G
Rg O
(I)
or a pharmaceutically acceptable salt thereof.
G is
(l) NH2 or
(2) substituted amino.
Rg is
(l) hydrogen,
(2) loweralkyl,
(3) carboxy-substituted alkyl or
(4) carboxyester-substituted alkyl.
Rlo is
(l) hydrogen,
(2) loweralkyl,
(3) functionalized alkyl or
(4) cycloalkyl.
D is
~) hydrogen,
(2) loweralkyl, ,
" , . . : - : . . ~ . .
... . : . : , . , . :,
W091tO0725 PCT~US90/03630
," ~,
7~5
(3) functionalized alkyl,
(4) cycloalkyl,
(5) aryl,
(6) functionalized oxyalkyl or
(7) heterocyclic;
or Rlo taken together with D is
(1) C4 to C6 alkylene,
(2) ~(CH2)q~V~(CH2)r~ wherein q is 1 to 3, r is 1
to 3 and
V is
(i) --O--,
( i i ) --S--,
(iii) -CH2- or
(iv) -N(R2s)- wherein R2s is hydrogen,
loweralkyl, haloalkyl, alkoxyalkyl, arylalkyl,
aryl or an N-protecting group;
or Rg taken together with D is
(1) C3 to Cs alkylene or
(2) -(CH2)p-V-(CH2)t- wherein p is 1 to 3, t is 1
to 3 and V is defined as above.
Z is
(1) --C (O)--,
(2) -C(S)- or
(3) -S(0)2-.
B is
(1) absent,
(2) alkylene,
(3) alkenylene,
.
~ : , : ,
WO 91/0072~ PCT/US90/03630
2C~7~5~
--7--
(4) substituted alkenylene,
~5) -R26-R27- wherein R26 is absent or -CH2- and
R27 is -O-, -S-, -NH- or -N(loweralkyl)- or
~6) -R27-CH2- wherein R27 is defined as above.
Ar is
~1) aryl or
~2) a heterocyclic group.
Compounds wherein D is indolylmethyl, indolinylmethyl
or oxindolylmethyl are disclosed in the copending parent
application PCT Patent Application Serial No.
PCT/US89/01412, filed April 4, 1989.
The term "loweralkyl" as used herein refers to
straight or branched chain alkyl radicals containing from
1 to 8 carbon atoms including, but not limited to, methyl,
ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, sec-
butyl, n-pentyl, 2-methylbutyl, 2,2-dimethylpropyl, n- -
hexyl, 2-methylpentyl, 2,2-dimethylbutyl and the like.
The term "functionalized alkyl" as used herein
includes
~1) haloalkyl, :-
~2) alkenyl,
(3) arylalkyl,
(4) arylalkyl wherein the alkyl group is
substituted by
(i) -OR16 wherein R16 is hydrogen or a hydroxyl
protecting group,
(ii) -NHRls wherein Rls is hydrogen or an
. . ~
- : . ,- . . ,................. :
:., . : ,' ~ :~. ,''. ', ' ::, ' '
: . :., , :, .
- . , . .: . .
:: ~ . , :
WO gltO0725 Pcr/ussnto3630
2~ 755 ~
N-protecting group,
(iii) -OR13 wherein R13 is loweralkyl,
(iv) -OR14 wherein R14 is an aryl group or
(v) -SR13 wherein R13 is loweralkyl,
(5) heterocyclicalkyl,
(6) heterocyclicalkyl wherein the alkyl group is
substituted by
(i) -OR16 wherein R16 is hydrogen or a hydroxyl
protecting group,
(ii) -NHRls wherein Rls is hydrogen or an
N-protecting group,
(iii) -OR13 wherein R13 is loweralkyl,
(iv) -OR14 wherein R14 is an aryl group or
(v) -SR13 wherein R13 is loweralkyl,
(7) loweralkyl substituted by -S-loweralkyl,
-S(O)-loweralkyl or -S(0)2-loweralkyl,
(8) loweralkyl substituted by -S-aryl,
-S(O)-aryl or -S(0)2- aryl and
(9) loweralkyl substituted by -NHR12 wherein R12
lS
(i) hydrogen,
(ii) -C(O)R4 wherein R4 is ir.dependently
selected from loweralkyl, alkenyl, aryl,
arylalkyl, heteroaryl and heteroarylalkyl,
~iii) -CO2R4 wherein R4 is independently defined
as above,
(iv) an N-protecting group,
(v) -C(O)-A-aryl wherein A is alkenylene,
substituted alkenylene, -OCH~-, -SCH2-, -NH-,
-N(loweralkyl)-, -S- or -O-.
.::. , . :: :
.
.
WO91/00725 PCT/US90/03630
Z~-?755
9 ; . ., ~
The term "functionalized oxyalkyl" as used herein
includes -T-OR11 wherein
T is
(1) alkylene or
(2) arylalkylene and
Rll iS
(1) hydrogen,
(2) loweralkyl,
(3) haloalkyl,
(4) alkenyl,
(5) arylalkyl,
~6) hydroxyl protecting group,
(7) -C(O)-(L)~-R4 wherein R4 is independently
defined as above, s ~s 0 or 1 and
L is
(i) o,
(ii) S or
(iii) NH or
(8) -C(O)-A-aryl wherein A is independently
defined as above.
The term "haloalkyl" as used here-n refers to a
loweralkyl radical in which one or more hydrogen atoms
have been substituted by halo groups i~cluding, but not
limited to, fluoromethyl, trifluoromet~yl, chloroethyl,
2,2-difluorethyl, 2,3-dibromopropyl and the like.
The term "alkoxyalkyl" as used he-ein refers to an
alkoxy group appended to a loweralkyl =adical.
The term "cyanoalkyl" as used herein refers to a
cyano group (-CN) appended to a lowerarkyl radical.
._ .... ... ~ ................. - . ,
.-
.,, .. : ~ - . :
.. .. . . .
WO91/00725 PCT/US90/03630
~=~
' ' ' ,
Z(~ 755 - 1 o -
The term "hydroxyalkyl" as used herein refers to a
hydroxy group (-OH) appended to a loweralkyl radical.
The term "cycloalkyl" as used herein refers to an
alicyclic ring having 3 to 7 carbon atoms including, but
not limited to, cyclopropyl, cyclopentyl, cyclohexyl and
the like.
The term "cycloalkylalkyl" as used herein refers to a
cycloalkyl group appended to a loweralkyl radical
including, but not limited to, cyclopropylmethyl,
cyclohexylethyl and the like.
The term "carboxy-substituted alkyl" as used herein
refers to a carboxy group ~-COOH) appended to a loweralkyl
radical.
The term "carboxyester-substituted alkyl" as used
herein refers to a carboxyester group ~-COOR' wherein R'
is loweralkyl, cycloalkyl, aryl or arylalkyl) appended to
a loweralkyl radical.
The term "alkenyl" as used herein refers to a
straight or branched chain of 2 to 8 carbon atoms
containing a carbon-carbon double bond including, but not
limited to, vinyl, allyl, butenyl and the like.
The term "alkylene group" as used herein refers to a
straight or branched chain spacer group containing 1 to 8
carbon atoms including, but not limited to, -CH2-,
-CH(CH3)-, -CH(CH3~CH2-, -(CH2)3- and the like.
The term "alkenylene group" as used herein refers to
a straight or branched chain spacer group of 2 to 8 carbon
atoms containing a carbon-carbon double bond including,
but not limited to, -CH=CH-, -C(CH3)=C:i-, -CH2-CH=CH-,
-CH(CH3)-CH2-CH=CH-CH2- and the like.
,
.. . . .
:- . .: .
.
. :.,
WO9l/0~725 PCT/VS90/03630
. '
s5
:... .. ...
The term "substituted alkenylene" as used herein
refers to an alkenylene group substituted with one or two
substituents independently selected from loweralkyl,
haloalkyl, halo and cyano.
The term "cycloalkylalkylene" as used herein refers
to a cycloalkyl group appended to an alkylene radical.
The term "substituted amino" as used herein includes
-N(Rl)(R2) wherein R1 and R2 are independently selected
from
(1) hydrogen,
(2~ loweralkyl,
(3) haloalkyl,
(4) alkoxyalkyl,
(5) alkenyl,
(6) aryl,
(7) arylalkyl,
(8) cycloalkyl,
(9) cycloalkylalkyl,
(10) cyanoalkyl,
(11) loweralkyl substituted by -CO2R3 wherein R3
(i) hydrogen,
(ii) loweralkyl,
(iii) cycloalkyl,
(iv) aryl or
(v) arylalkyl,
(12) loweralkyl substituted by -C(O)N(Rs)~R6)
wherein R5 and R6 are independently selected from
(i) hydrogen,
(ii) loweralkyl,
.. . , . . , . .. . ,- . . . .
WO9l/00725 PCT/US9D/03630
.. ..
Z~ . 55
-12-
(iii) cycloalkyl,
(iv) alkoxyalkyl,
(v) alkenyl,
(vi) aryl and
(vii) arylalkyl,
(13) -W-CO2R3 wherein R3 is defined as above and
W is
(i) cycloalkylalkylene,
(ii) arylalkylene or
(iii) heteroarylalkylene,
(14) adamantyl,
(15) indanyl and
(16) -CH(aryl)-X wherein X is arylalkyl;
with the proviso that R1 and R2 are not both hydrogen.
Substituted amino also includes
~ H~R8
--N /J
(CH2)r
wherein n is 1 to 3, r is 1 to 3 and J is
(1) --S--,
(2) -S(O)-,
(3) -S(O)2-~
(4) -O-,
(5) -CH2-r
~6) -N~Rs)- wherein RS is defined as above or
(7) -N (C (O) R4) wherein R4 is defined as above and
R8 represents one, two or three substituents independently
WO91/00725 PCT/US90/03630
.
2~
selected from
(1) hydrogen,
~2) loweralkyl,
(3) haloalkyl,
~4) aryl,
(5) -C~O)R4 wherein R4 is independently defined as
above,
(6) -C(O)N(RS)(R6) wherein Rs and R6 are
independently defined as above,
(7) -0Rl6 wherein R16 is
(i) hydrogen or
(ii) hydroxyl protecting group,
(8) hydroxyalkyl,
(9) alkoxyalkyl,
(10) -NH~R1s) wherein Rls is
(i) hydrogen or
(ii) an N-protecting group,
(11) cyano and
(12) halo.
The term "alkylamino" as used herein refers to -NHR40
wherein R40 is a loweralkyl group.
The term "dialkylamino" as used herein refers to
-NR4lR42 wherein R4l and R42 are independently selected
from loweralkyl.
The term "aminocarbonyl" as used herein refers to
--C (O) NH2 -
The term "alkylaminocarbonyl" as used herein refers
to -C(0)Rso wherein Rso is an alkylamino group.
The term "dialkylaminocarbonyl" as used herein refers
to -C(O)Rs1 wherein Rs1 is a dialkylami.o group.
.
, . ,, . . . . . . . -
. : , ; -
WO91/00725 PCT/US90/03630
S5
-14-
The term "alkenylaminocarbonyl" as used herein refers
to -C(O)NHRs2 wherein Rs2 is an alkenyl group.
The term "halogen" or "halo" as used herein refers to
F, Cl, Br, I.
The terms "alkoxy" and "thioalkoxy" as used herein
refer to R13O- and R13S- respectively, wherein R13 is a
loweralkyl group.
The term "alkoxycarbonyl" as used herein refers to
-C(O)OR43 wherein R43 is loweralkyl.
The term "aryl" or "aryl group" as used herein refers
to a monocyclic, bicyclic or tricyclic carbocyclic ring
system containing one or more aromatic carbocyclic rings
including, but not limited to, phenyl, naphthyl, indanyl,
fluorenyl, ~1,2,3,4)-tetrahydronaphthyl, indenyl,
isoindenyl and the like. Aryl groups can be unsubstituted
or substituted with one, two, or three substituents
independently selected from loweralkyl, alkoxy,
thioalkoxy, carboxy, alkoxycarbonyl, arylcarbonyloxy,
arylalkylcarbonyloxy, heterocycliccarbonyloxy,
heterocyclicalkylcarbonyloxy, arylalkoxy,
heterocylicalkoxy, -OSO3H, cyano, nitro, haloalkyl,
hydroxy, amino, aminocarbonyl, alkylaminocarbonyl,
dialkylaminocarbonyl, alkenylaminocarbonyl, alkylamino and
dialkylamino.
The term "arylalkyl" as used herein refers to an aryl
group appended to a loweralkyl radical.
The term "arylalkylene" as used herein refers to an
aryl group appended to an alkylene radical.
The term "arylcarbonyloxy" as used herein refers to
Rs4C(O)O- wherein Rs4 is an aryl group.
.
W091/00725 PCT/US90/03630
''''
2~ - ?q55 .
-15-
The term "arylalkylcarbonyloxy" as used herein refers
to RssC~O)O- wherein Rss is an arylalkyl group.
The term "arylalkoxy" as used herein refers to R56O-
wherein Rs6 is an arylalkyl group.
The term "heteroaryl" as used herein refers to a
monocyclic or bicyclic aromatic ring system, each ring
having 5 or 6 atoms, one to four of which are
independently selected from oxygen, sulfur and nitrogen.
Heteroaryl groups also include a heteroaryl ring as
defined above fused to a benzene ring. Heteroaryl groups
can be unsubstituted or substituted with one, two or three
substituents independently selected from loweralkyl, halo,
hydroxy, cyano, nitro, haloalkyl, alkoxy, thioalkoxy,
amino, aminocarbonyl, alkylaminocarbonyl,
dialkylaminocarbonyl, alkenylaminocarbonyl, alkylamino,
dialkylamino, N-protected amino, protected hydroxyl,
carboxylic acid, carboxamide, arylcarbonyloxy,
arylalkylcarbonyloxy, heterocycliccarbonyloxy,
heterocyclicalkylcarbonyloxy, arylalkoxy,
heterocylicalkoxy, -OSO3H, carbamyl and aryl.
The term "heteroarylalkyl" as used herein refers to a
heteroaryl group appended to a loweralkyl radical.
The term 'Iheteroarylalkylene'' as used herein refers
to a heteroaryl group appended to an alkylene radical.
The term "heterocyclic ring" or "heterocyclic" as
used herein refers to any 3- or 4-membered ring containing
a heteroatom selected from oxygen, nitrogen and sulfur; or
a 5- or 6-membered ring containing one, two or three
nitrogen atoms; one nitrogen and one sulfur atom; or one
nitrogen and one oxygen atom. The 5-membered ring has 0-2
~. ; ' -
,
W091t0072~ P~T/US90/03630
~.
z~755
-16-
double bonds and the 6-me~bered ring has 0-3 double bonds.
The nitrogen and sulfur heteroatoms can be optionally
oxidized. The nitrogen heteroatoms can be optionally
quaternized. The term "heterocyclic" includes any
bicyclic or tricyclic group wherein the heterocyclic ring
is fused to one or two benzene rings or one or two
heterocyclic groups independently defined as above.
Heterocyclics include thienyl, furanyl, pyrrolyl,
imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl,
pyrrolidinyl, pyrrolinyl, imidazolidinyl, imidazolinyl,
pyrazolidinyl, tetrahydrofuranyl, pyranyl, pyronyl,
pyridyl, pyrazinyl pyridazinyl, piperidyl, piperazinyl,
morpholinyl, thionaphthyl, benzofuranyl, isobenzofuryl,
indolyl, oxyindolyl, isoindolyl, indazolyl, indolinyl, 7-
azaindolyl, isoindazolyl, benzopyranyl, coumarinyl,
isocoumarinyl, quinolyl, isoquinolyl, naphthridinyl,
cinnolinyl, quinazolinyl, pyridopyridyl, benzoxazinyl,
quinoxadinyl, chromenyl, chromanyl, isochromanyl,
carbolinyl, and the like. Heterocyclic groups can be
unsubstituted or substituted with one, two or three
substituents independently selected from loweralkyl,
haloalkyl, oxo, hydroxy, protected hydroxyl, alkoxy,
thioalkoxy, amino, aminocarbonyl, alkylaminocarbonyl,
dialkylaminocarbonyl, alkenylaminocarbonyl, alkylamino,
dialkylamino, N-protected amino, cyano, nitro, carboxylic
acid, carboxamide, arylcarbonyloxy, arylalkylcarbonyloxy,
heterocycliccarbonyloxy, heterocyclicalkylcarbonyloxy,
arylalkoxy, heterocylicalkoxy, -OSO3H, carbamyl and aryl.
The term "heterocyclicalkyl" as used herein refers to
a heterocyclic group appended to a loweralkyl group.
: . , .: ~ -
,~ . .
:: .- , . ~ :. -
W09t~00725 PCT/US9Q/03630
"
2~ ~55
-17-
The term "heterocycliccarbonyloxy" as used herein
refers to Rs7C(O)O- wherein R57 is a heterocyclic group.
The term "heterocyclicalkylcarbonyloxy" as used
herein refers to RsgC~O)O- wherein R5g is a
heterocyclicalkyl group.
The term "heterocyclicalkylene" as used herein refers
to a heterocyclic group appended to an alkylene radical.
The term "heterocyclicalkoxy" as used herein refers
to RsgO- wherein Rsg is a heterocyclicalkyl group.
The term "N-protecting group" or "N-protected" as
used herein refers to those groups intended to protect the
N-terminus of an amino acid or peptide or to protect an
amino group against undesirable reactions during synthetic
procedures or to prevent the attack of exopeptidases on
the compounds or to increase the solubility of the
compounds and includes, but is not limited to, sulfonyl,
acyl, acetyl, pivaloyl, t-butyloxycarbonyl (Boc),
carbobenzyloxy (Cbz), benzoyl or an a-aminoacyl residue,
which may itself be N-protected similarly.
The term "hydroxyl protecting group" as used herein
refers to a substituent which protects hydroxyl groups
against undesirable reactions during synthetic procedures
and includes, but is not limited to, substituted methyl
ethers, for example methoxymethyl, benzyloxymethyl, 2-
methoxyethoxymethyl, 2-(trimethylsilyl)ethoxymethyl,
benzyl, and triphenylmethyl; terahydropyranyl ethers;
substituted ethyl ethers, for example, 2,2,2-
trichloroethyl and t-butyl; silyl ethers, for example,
trimethylsilyl, t-butyldimethylsilyl and
-:
;
. .
WO91/00725 PCT/VS90/03630
2~?5~755
t-butyldiphenylsilyl; cyclic acetals and ketals, for
example, methylene acetal, acetonide and benzylidene
acetal; cyclic ortho esters, for example,
methoxymethylene; cyclic carbonates; cyclic boronates; and
esters, for example acetates or benzoates.
Exemplary compounds of the present invention include:
N-(3'-Quinolylcarbonyl)-R-Valine-di-n-pentylamide;
N-(2'-Indolylcarbonyl)-R-Valine-di-n-pentylamide;
N-(4',8'-Dihydroxy-2'-quinolylcarbonyl)-R-Valine-di-n-
pentylamide;
N-(2'-Naphthoyl)-R-Valine-di-n-pentylamide;
N-~3'-Quinolylcarbonyl)-R-Norleucine-di-n-pentylamide;
N-(2'-Indolylcarbonyl)-R-Norleucine-di-n-pentylamide;
N-(3'-Quinolylcarbonyl-R-(O-benzyl)Serine-di-n-
pentylamide;
N-(3'-Quinolylcarbonyl)-(2R,3S)-(O-benzyl)Threonine-di-n-
pentylamide;
N-(3'-Quinolylcarbonyl)-(2R,3S)-Threonine-di-n-
pentylamide;
N-(3'-Quinolylcarbonyl)-(2R,3S)-(O-acetyl)Threonine-di-n-
pentylamide;
N-(3'-Quinolylcarbonyl)-(2R,3S)-(O-methyl)Threonine-di-n-
pentylamide;
N-(3'-Quinolylcarbonyl)-3-(2'-thienyl)-R-Alanine-di-n-
pentylamide;
N-(2l-Indolylcarbonyl)-R-Histidine-di-n-pentylamide;
N-(3'-Quinolylcarbonyl)-R-Histidine-di-n-pentylamide;
N -(3'-Quinolylcarbonyl)-N -(benzyloxycarbonyl)-R-Lysine-
di-n-pen~ylamide;
:,,' '- ,
:. , .: ,: . : .
, , , - , : :
. . ,
WO91/00725 PCT/US90/03630
, :.
29~ 55
--1 9--
N-~3'-Quinolylcarbonyl)-R-Phenylalanine-di-n-pentylamide;
Na-(3'-Quinolylcarbonyl)-N-(2'-chlorobenzyloxycarbonyl)-
R-Lysine-di-n-pentylamide;
N-(3'-Quinolylcarbonyl)-R-(4'-hydroxyphenyl)glycine-di-n-
pentylamide;
Na-(3'-Quinolylcarbonyl)-NE-(acetyl)-R-Lysine-di-n-
pentylamide;
N-(2'-Indolylcarbonyl)-R-Tyrosine-di-n-pentylamide;
N-(3',4'-Dichlorobenzoyl)-R-Tyrosine-di-n-pentylamide;
N-(2'-Naphthoyl)-R-Tyrosine-di-n-pentylamide;
N-(3'-Quinolylcarbonyl)-R-Tyrosine-di-n-pentylamide;
Methyl N-(3'-Quinolylcarbonyl)-R-Tyrosyl-S-
phenylglycinate;
N-(2'-Indolylcarbonyl)-R,S-Homoserine-di-n-pentylamidei
N-~3'-Quinolylcarbonyl)-R,S-Homoserine-di-n-pentylamide;
N-(2'-Indolylcarbonyl)-R-MethioninesulfGxide-di-n-
pentylamide;
N-(3'-Quinolylcarbonyl)-R-Methionine-di-n-pentylamide;
N-(3'-Quinolylcarbonyl)-R-Methioninesulfoxide-di-n-
pentylamide;
- Na-(3'-Quinolylcarbonyl)-N~-phenylthiolcarbonyl-R-Lysine-
di-n-pentylamide;
N-(3'-Quinolylcarbonyl)-R-Tyrosine-di-n-pentylamide
hydrochloride;
N-~3'-Quinolylcarbonyl)-R-Histidine-di-n-pentylamide
dihydrochloride;
N-(2'-Indolylcarbonyl)-glycine-di-n-pentylamide;
N-(3'-Quinolylcarbonyl)glycine-di-n-pentylamide;
N-(3'Quinolylcarbonyl)-R-phenylglycine-di-n-pentylamide;
' ~
-, ,- , ~' -
i WO91/00725 PCT/US9OtO3630
.,
21~
-20-
N-(4',8'-Dihydroxy-2'-quinolylcarbonyl)-R-Phenylglycine-
di-n-pentylamide;
N-~5'-Fluoroindolylcarbonyl)-R-phenylglycine-di-n-
pentylamide;
N-(4',8'-Dihydroxy-2'-quinolylcarbonyl)glycine-di-n-
pentylamide;
N-(2'-Naphthoyl)glycine-di-n-pentylamide;
N-(3'-Methylphenylaminocarbonyl)glycine-di-n-pentylamide;
N-(4',8'-Dihydroxy-2'-quinolylcarbonyl)-R-(4'-
hydroxyphenyl)-glycine-di-n-pentylamide;
N-(q',8'-Dihydroxy-2'-quinolylcarbonyl)-(2R,3S)-(O-
benzyl)-Threonine-di-n-pentylamide;
Methyl N-(3'-Quinolylcarbonyl)-R-Methionine-S-(p-
hydroxy)-phenylglycinate;
N-(3'-Quinolylcarbonyl)-R-Serine-di-n-pentylamide;
N-~8'-Hydroxy-2-quinolylcarbonyl)-glycine-di-n-
pentylamide;
N-Methyl-N-~3'Quinolylcarbonyl)-glycine-di-n-
pentylamide;
N-(3'-Iodo-2'-indolylcarbonyl)-glycine-di-n-
pentylamide; and
N-(2'-Indolylcarbonyl)-R-Alanine-di-n-pentylamide.
The compounds of the invention may be made as shown
in the following scheme(s). The compounds of the
invention having one asymmetric center can exist as
separate enantiomers or as mixtures or enantiomers. The
compounds of the invention which contain two or more
asymmetric carbon atoms can exist as pure diastereomers,
mixtures of distereomers, dias~ereomeric racemates or
mixtures of diastereomeric racemates. The present
., ~ , , . ~ - ~ . - . .. . .. . ..
: : :. - . . : . .. ... .
.:
..
- ... . . :
WO9l/00725 PCT/US90~03630
2(~
.`:` . ~`.
invention includes within its scope all of the isomeric
forms
A number of synthetic pathways exist for the
production of a-amino acids and their derivatives. The
invention is not limited to those methods discussed here -
for the synthesis of a-amino acids but is meant to include
those variations and methods encompassed by ~he prior art
as discussed in the chemical literature in its entirety.
a-Amino acids ~refer to Scheme 1) can be produced directly
by the displacement of a-halogenated esters (1, x is
halo) and the like or other a-situated leaving sroups by
ammonia and or other substituted amines (Rg is hydrogen,
loweralkyl, carboxyester-substituted alkyl) and/or their
analogs ~e.g., carbamates, hydrazines, azides) (e.g.,
Marvel nL~ ~YUL;h ~Q, 81, 1940; 106, 1940; ~1, 60, 1941;
79, l9ql; Birnbaum, ~ h8m, 333, 1953). The amino
group is then unmasked, for example by reduction, and the
ester group (amide, etc.) is saponified to the acid in a
standard fashion.
A second method involves the condensation of an a-
ketoester (amide, etc) with an amine or amine equivalent
(e.g., hydroxylamine, hydrazine, carbamate, etc.) and the
subsequent reduction of this product (2) to the a-
aminoester (amide, acid, etc. (e.g., Can ~ Chem, 29, 427,
1951; J Ora hem, 38, 822, 1973; J Q~ ~h~m, ~, 878,
1941~). Alternatively, an organometalic reagent can be
added to the oxime 2 ~imine, etc.) to provide as final
products either monosubstituted a-amino acids in the case
where D is hydrogen, or disubstituted amino acids in the
.
,
: .
WO91/00725 PCTIUS90/03630
Z~755
-22-
case where D is other than hydrogen (e.g., Tetrahedron
L~S~ 2), 4973, 1987).
A third method is the alkylation of a carbanion
resulting from compound (3) with an electrophilic nitrogen
source ~eg. diethylazodicarboxylate). The intermediate
product can subsequently be unmasked to provide the
desired a-amino acid. A similar method involves
alkylation of the carbanion derived from compound (4) with
an appropriate alkylating agent. This method also allows
for the possibility of disubstitution of the a center.
A fifth route involves the Strecker reaction and its
modifications. Reaction of cyanide and ammonium on
aldehydes and ketones (S) provides the amino acid.
A last method involves the direct reduction of
unsaturated heterocyclic carboxylic acids (6) to directly
provide the cyclic amino acids (7), (wherein D and R9 are
encompassed in a ring).
With suitably available a - amino acids (8) (Scheme 2)
the amino group is protected with an N protecting group
(most frequently Boc or Cbz) and, if the carboxylic acid
has not been unmasked, it is saponified with base to
provide the parent carboxylic acid (9). The N-protected
intermediate is then coupled with the amine HNR1R2 using
any of a number of standard coupling techniques
(carbodiimide, BOPCl, chloroformates, oxalylchloride,
etc.). Preferred secondary amines are of the type where R
and R2 are alkyl, arylalkyl, aryl, or represent another
amino acid. The resulting product (10) is then N-
deprotected using HCl or trifluoroacetic acid to remove a
Boc group and hydrogenolysis or HBr to remove a Cbz group.
.. .
: ~ ' ' , , . ~ , . . '' , . .
WO91/00~25 PCT/US90/03630
2 ~ 5 5
-23-
The resultant amine ~11) is then coupled with aromatic
carboxylic acids, aromatic acid halides, heteroaromatic
carboxylic acids, aromatic isocyanates, aromatic sulfonic
acids, aromatic sulfonyl chlorides, and the like using
standard coupling techniques to provide the desired
products ~12), (13), (14), and (15). Preferred acyl
coupling partners groups include: quinoline carboxylic
acids, indole carboxylic acids, substituted benzoic acids
and benzoyl chlorides, arylisocyanates and
arylisothiocyanates, naphthoic acids, benzothiofuranyl
carboxylic acids and the like.
WO 9l/00725 PCI`/USgO/03630
21?~755
Scheme 1
O H2~R9 1
D I D
o H2/cDtDlyst (Rlo=H) H O
R g _ N~l~ o ~ R r R ~ ~ o R
D 2 Rlo~ R10 D
0 1. bDse IH O
~O R R ~N ~I~O ~R
D 3 2. ":~H2 ' D
1. bDse/DX ,H O
`1/~ ~o.R , R ~N~I~ R
2. H D
o Strecker l O
RgJ~D Rlo D
C~ U,
... : '
.
~ . . ~ . . ..
WO gl/00~25 PCI'/US90/03630
Z~755
: i . . .
, ,. . . .. ~
Scheme 2
~N3~ `H D~o
Rg Rg 0 9
8 -
coupling agent
HNRIR2
D R10 R1 D R10 ~R1
R2 deprotect P N~ R2
11 10
\\ \
~rCOz~ I \ \ArSO3H/coupllng agent or
\ ArS02CI
coupllng¦ \ \ArNCO
agent ~
0~ N ~ N `R \ Ar r3~ R2
Rg O O D R10 ,R1 14
12 ~N~N.R D R10 ,
Ar Rg O NH ~N~ 'R2
1~ Alr R g O
.. ~ . . . . ,, , . -. , , ~ . .
. . .
WO91/00725 PCT/US90/03630
.
2~ S5
-26-
Intermediates for the preparation of the compounds of
formula I include compounds of the formula:
D R1o
P~- N
Rg O
whereln G is
(l) NH2 or
~2) substituted amino;
Rg is
~l) hydrogen,
~2) loweralkyl,
~3) carboxy-substituted alkyl or
~4) carboxyester-substituted alkyl;
Rlo is
(l) hydrogen,
(2) loweralkyl,
(3) functionalized alkyl or
(4) cycloalkyl;
D is
(l) hydrogen,
(2) loweralkyl,
(3) functionalized alkyl,
(4) cycloalkyl,
(5) aryl,
(6) functionalized oxyalkyl or
~ 7) heterocyclic;
or Rlo t~ken together with D is
~l) C4 to C6 alkylene,
, :;: . ''
WO 91/00725 PCT/US90/03630
: '~
ZC?5-77~55 . `. .: `
--2 7--
(2) - (CH2) q~V~ (CH2) r~ wherein q is 1 to 3, r is 1
to 3 and
V is
(i) --o--,
( i i ) --S--,
(iii) -CH2- or
~iv) -N(R25)- wherein R2s is hydrogen,
loweralkyl, haloalkyl, alkoxyalkyl, arylalkyl,
aryl or an N-protecting group;
or Rg taken together with D is
(1) C3 to C5 alkylene or
(2) -(CH2)p-V-(CH2)t- wherein p is 1 to 3, t is 1
to 3 and V is defined as above; and
P1 is hydrogen or an N-protecting group.
Other intermediates for the preparation of compounds
of the formula I include compounds of ~he formula:
~ B ~ Z
wherein Z is
(1) --C (O)--,
(2) -C(S)- or
(3) -S(0)2-;
B is
(1) absent,
(2) alkylene,
(3) alkenylene,
(4) substituted alkenylene,
~5~ -R2Ç-R27- wherein R2ç is absent, or -C~2- and
R27 is -O-, -S-, -NH- or -N(lo~-eralkyl)- or
', . .- !' . .
., ` . . . .
' ' . '".,. '' ' ', ' ,
WO9l/0072~ PCT/US90/03630
,~
2~ ? ~ S 5
-28-
~6) -R27-CH2- wherein R27 is defined as above;
Ar is
(1) aryl or
~2) a heterocyclic group; and
Z' is an activating group; or B-Z-Z' taken together
represent -N=C=O, -N=C=S, -CH2-N=C=O or -CH2-N=C=S.
Activating groups are those functional groups which
activate a carboxylic acid or sulfonic acid group toward
coupling with an amine to form an amide or sulfonamide
bond. Activating groups Z' include, but are not limited
to, -OH, -SH, alkoxy, thioalkoxy, halogen, formic and
acetic acid derived anhydrides, anhydrides derived from
alkoxycarbonyl halides such as isobutyloxycarbonylchloride
and the like, N-hydroxysuccinimide derived esters, N-
hydroxyphthalimide derived esters, N-hydroxybenzotriazole
derived esters, N-hydroxy-5-norbornene-2,3-dicarboxamide
derived esters, 4-nitrophenol derived esters, 2,4,5-
trichlorophenol derived esters and the like.
The following examples will serve to further
illustrate preparation of the novel compounds of this
invention.
Example 1
N-(t-Butvlo~ycarbonyl)-R-Valine-di-n-pentylamide
N-t-Butyloxycarbonyl-R-Valine (2.5 g, ll.S mmol) was
stirred at 0 C in 30 mL of methylene chloride ~CH2Cl2)
with bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BOPC1,
3.5 g, 13.8 mmol) and 1.5 mL (11.5 mmol) of triethylamine
(TEA). To this reaction mixture was added
dl-n-pentylamine (11.6 mL, 58 mmol). The mi:cture was
stirred overnight and allowed to warm to room temperature.
. . . .
~'' ` . .
WO9l/00725 PCT/US90/03630
2~ 55
r
An additional equivalent of BOPCl was added after 18 hrs
and the reaction mixture was stirred an additional day at
ambient temperature. The solvents were evaporated in
vacuo and the residue taken up in ethylacetate ~EtOAc) and
washed with water, 1 N hydrochloric acid (HCl) solution,
saturated sodium bicarbonate solution (NaHCO3), water.
The organic solution was dried over magnesium sulfate
(MgSO4). After filtration and concentration of the
filtrate in vacuo, the residue was chromatographed using
ethyl acetate-hexane as the solvent system in the ratio
(1:4). The product was isolated as an oil 79% yield (3.25
g). ~a]D= +21.2 (c=1.5, MeOH). MS~CI) m/e 357(m+H) .
1H NMR(CDCl3,300MHz) ~ 0.85-1.0(m,12H), 1.32(m,8H),
1.4-1 5(m,4H), 1.5(s,9H), 1.84(m,lH), 3.05(m,lH),
3.2(m,lH), 3.35(m,lH), 3.55(m,lH), 4.42(m,lH),
5.25(d,J-7Hz,lH).
R-Valine-di-n-pentylamide hydrochloride
The product of example 1 (0.2 g, 0.6 mmol) was
dissolved in 4 N HCl in dioxane (10 mL) and stirred under
inert atmosphere (N2) for an hour. When the reaction was
complete by tlc the solvents were evaporated in vacuo and
hexane and diethylether were added. The residue was
triturated with these two solvents and the solvents again
evaporated in vacuo. This procedure was repeated several
times until product was obtained as a glass in
quantitative yield. MS(CI) m/e 293(m+H) .
-- . ~: . , : :
.: .,, , .,
:: ~,- ~ : . . .
WO 91/00725 PCI`/US90/03~30
.. ;: .
z~ 755
--30--
Example 3
N-(3'-Ouinolylcarbonyl)-R-Valine-di-n-~entvlamide
The hydrochloride of example 2 (150 mg, 0.5 mmol), 1-
ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI, 100
mg), HOst (135 mg) and quinoline-3-carboxylic acid (88 mg)
were stirred at 0C under nitrogen in 5 mL of anhydrous
CH2C12. To this mixture was added 120 IlL of N-
methylmorpholine (NMM) and the mixture was stirred
overnight (warming to ambient temperature). The reaction
mixture was poured into ethyl acetate and water and the
organic extract was washed successively with water, 10g~
citric acid solution, and saturated aqueous NaHCO3. The
solution was dried over MgSO4, filtered and concentrated.
The residue was chromatographed using ethylacetate ~EtOAc)
and hexane as the elutant mixture to provide 110 mg of an
oil (54~ yield) after removal of the volatiles. [a]D= -
14.8 ~cz0.5, MeOH). MS(CI) m/e 412(m+H) . lH
NMR(CDC13,300MHz) ~ O.92(m,6H), 1.05(m,6H), 1.35 (m,8H),
1.5-1.7(m,4H), 2.15(m,lH), 3.05(m,lH), 3.3-3.4(m,lH),
3.5(m,lH), 3.65(m,lH), 5.08(dd,J=3, 9Hz,lH),
7.25(d,J=9Hz,lH), 7.62 (t,J=7Hz,lH), 7.8(t,J=7Hz,lH),
7.91(d,J=lOHz,lH), 8.16(d,J=lOHz,lH), 8.6(d,J=3Hz,lH),
9.35(d,J=3Hz,lH). Analysis calculated for C25H37N3O2: C
72.95, H 9.06, N 10.21; found: C 72.61, H 9.21, N 9.97.
Example 4
N-(2'-Indolylcarbonyl)-~ aline-~ -pentylamide
The hydrochloride of example 2 ~130 mg, 0.45 mmol),
EDCI (90 mg), HOBt (120 mg) and indole-2-carboxylic ac d
,
- . :
W09lt00725 PCT/US90tO3630
2~ SS
~,,.. ...:,...
(75 mg) were stirred at 0C under nitrogen in 5 mL of
anhydrous CH2Cl2. To this mixture was added 100 ~L of NMM
and the mixture was stirred overnight (warming to ambient
temperature). The reaction mixture was poured into
ethylacetate and water and the organic extract was washed
successively with water, 10% citric acid solution, and
saturated aqueous NaHCO3. The solution was dried over
MgSO4, filtered and concentrated. The residue was
chromatographed using ethylacetate and hexane as the
elutant mixture to provide 36 mg of product (75% yield)
after evaporation of the volatiles. mp= 132-4C. [a~D= -
9.2 (c=0.5, MeOH). MS(CI) m/e 400(m+H) . lH
NMR~CDCl3,300MHz) ~ 0.9(t,J=7Hz,6H), 1.0(m,6H), 1.2-
1.4(m,8H), 1.5-1.6(m,4H), 2.12(m,lH), 3.05(m,lH),
3 3(m,lH), 3.42(m,lH), 3.63(m,lH), 5 0(q,J=3,6Hz,lH),
7.0(m,1H), 7.1(d,J=9Hz,lH), 7.25~t,J=7.5Hz,lH),
7.3(t,J-7.5Hz,lH), 7.41(d,J=7Hz,lH), 7.65(d,J=7Hz,lH),
9.3(bs,1H). C,H,N analysis calculated for C2qH37N3O2 C
72.14, H 9.34, N 10.52; found: C 72.52, H 9.25, N 10.49.
Example ~
N-(2'-Ouinolylcarbonvl)-R-Valine-di-n-pentylamide
The reaction was performed in a similar manner to
that in example 3 utilizing 0.2 g of the hydrochloride
salt of example 2, quinoline-2-carboxylic acid (0.12 g),
EDCI (0.15 g), HOBt (0.1 g), and NMM (0.18 mL). The
product was isolated in 80% yield (0.225 g). mp= 78-79C.
[a)Dc -13.1 (c=l.l, MeOH). MS(CI) m/e 412(m+H) . lH
NMR(CDC13,300MHz) ~ 0.9(m,6H), 1.05(m,6H), 1.2-1.4(m,8H),
1.55(m,4H), 2.22(m,lH), 3.08(m,lH), 3.4(m,2H), 3.64(m,lH),
.
'
W09l/00725 PCT~US90/~3630
21?~55
-32-
5.0~dd,J=3,7Hz,lH), 7.62~t,J=7Hz,lH), 7.78~t,J=7Hz,lH),
7.85~d,J=9Hz,lH), 8.15(d,J=9Hz,lH), 8.35(m,2H),
8.85(d,J=10Hz,lH). C,H,N analysis calculated for
C25H37N3O2, H2O: C 72.17, H 8.96, N 10.10; found: C
72.36, H 8.93, N 10.03.
Example 6
N-~E-2'-Cyano-3'-~4''-hydro~yphenyl)prop-2'-enoyll-R-
Valine-di-n-pentylamide
The hydrochloride of example 2 (300 mg, 1.03 mmol)~
EDCI (200 mg), HOBt (280 mg) and a-cyano-q-hydroxycinnamic
acid (195 mg) were stirred at 0C under nitrogen in 15 mL
of anhydrous CH2Cl2. To this mixture was added 250 ~L of
NMM and the mixture was stirred overnight (warming to
ambient temperature). The reaction mixture was poured
into ethylacetate and water and the organic extract was
washed successively with water, 10% citric acid solution,
and saturated aqueous NaHCO3. The solution was dried over
magnesium sulfate, filtered and concentrated. The residue
was chromatographed using ethylacetate and hexane as the
elutant mixture to provide 225 mg of an oily product (57
yield) after evaporation of the volatiles. [~]D= -9.8
(c=1.15, MeOH) MS(CI) m/e 428(m+H) . H
NMR(CDCl3,300MHz) ~ O.92(m,6H), 1.08(m,6H), 1.35(m,8H),
1.56-1.75(m,9H), 2.15(m,lH), 3.1(m,lH), 3.3-3.5(m,2H),
3.7(m,lH), 4.65(m,lH), 6.73(d,J=9Hz,lH), 6.85(d,J=9Hz,2H),
7.65(d,J=9Hz,2H), 7.72(s,1H), 9.28(s,1H). C,H,N analysis
calculated for C25H37N3O3: C 70.22, H 8.72, N 9.83, found:
C 69.88, H 8.39, N 9.60.
- - . . - . - . - - - . - -
: : , ~
W091t00725 PCT/US90/03630
Z~ SS
-33-
Example 7
N-(2'-Benzothiofuranylcarbonyl)-R-Valine-di-n-p~ntylamide
The reaction was performed in a similar manner to
that in example 3 utilizing 0.3 g of the hydrochloride
salt of example 2, benzothiofuran-2-carboxylic acid (0.205
g), EDCI (0.22 g) HOBt (0.28 g), and NMM (0.22 mL). The
oily product was isolated in 58% yield, 0.28 g [a]D= ~
5.85 (c=2.0, MeOH). MS(CI) m/e 417(m+H) , 158. H
NMR(CDCl3,300MHz) ~ 0.g-l.l(m,12H), 1.2-1.3(m,8H), 1.5-
1.6(m,4H), 2.15(m,lH), 3.05(m,lH), 3.3(m,lH), 3.42(m,lH),
3.65(m,lH), 5.0(q,J=3,6Hz,lH), 7.00(d,J=9Hz,lH),
7.41(m,2H), 7.80(s,lH), 7.86(m,2H). C,H,N analysis
calculated for C24H36N22S' 0-25 H2O
6.65; found: C 68.73, H 8.48, N 6.71.
~m~l~ 8
N- (4 ' . 8 ' -Dihydroxy-2'-quinolylcarbonyl)-~-Valine-
di-n-pentylamide
The hydrochloride salt of example 2 (0.95 g, 3.22
mmol) was stirred in 25 mL of CH2C12 with NMM (0.7 mL)
under nitrogen at 0 C. EDCI (0.7 g) and HOBt (0.11 g)
were added followed by the addition of 4,8-
dihydroxyquinoline-2-carboxylic acid (0.66 g, 3.22 mmol).
The reaction mixture was stirred overnight (warming to
ambient temperature). The solvents were evaporated in
vacuo and the residue taken up in ethylacetate and washed
successively with water, 0.1 N solution of HCl, water and
brine. The organic solution was dried over MgSO4 and then
filtered. Solvents were evaporated in vacuo and the crude
product subjected to flash chromatography using
, . -
- ~:: , . , . : .
- ::
WO91/00725 PCT/US90/03630
~`
. , , ' . ' ';
Zc`~-~'55
-39-
ethylacetate, hexane and methanol as the elutant mixture.
The product was crystallized from methanol-water to
provide 0.82 g ~56%). mp= 233-235C. ~a]D = -15.6
(c=0.5, MeOH). MS(CI) m/e 444(m+H) . lH .
NMR(DMSOd6,300MHz) ~ 0.84(m,6H), 0.92(m,6H), 1.1-
1.35(m,8H), 1.4-1.6(m,4H), 2.33(m,lH), 3.1-3.45(m,2H),
3.55(m,2H), 4.67(m,1H), 7.1(d,J=9Hz,lH), 7.42(t,J=7Hz,lH),
7.55(m,2H), 9.62(d,J=9Hz,lH), 10.3(s,lH), 11.75(s,lH).
C,H,N calculated for C25H37N3O4: C 67.69 H 8.41, N 9.47;
found: C 67.47 H 8.45, N 9.39.
Examp~e ~
N-(2l-Benzofuranvlcarbonvl)-R-Valine-di-n-~entvlamide
The reaction was performed in a similar manner to
that in example 8 utilizing 0.3 g of the hydrochloride
salt of example 2, benzofuran-2-carboxylic acid (0.19 g),
EDCI (0.22 g~, HOBt (0.28 g), and NMM (0.22 mL). Product
was isolated in 56% yield (0.225 g). [a]D= -29.2 (c=l.l,
MeOH). MS(CI) m/e 401(m+H) . H NMR(CDC13,300MHz) ~
0.9-1.0(m,6H), 1.05(m,6H), 1.25-1.4(m,8H), 1.5-1.68(m,4H),
2.15(m,lH), 3.1(m,lH), 3.28-3.5(m,2H), 3.62(m,lH),
5.0(dd,J=3,6Hz,lH), 7.28(t,J=8Hz,lH), 7.4(t,J=8Hz,2H),
7.45(s,lH), 7.52(d,J=9Hz,lH), 7.65(d,J=9Hz,lH). C,H,N
analysis calculated for C24H36N2O3: C 71.96, H 9.06, N
6.99; found: C 72.09, H 9.08, N 6.99.
: ~ . . :
W09t/00725 PCT/US90/03630
.
Z ~ ~ ? 7 5 5
..
-35-
~xam~le lQ
~-r4'-Hydroxy-2'-phenyl-3'-quinolylcarbonyll-R-Valine- ~
~ '.
The reaction was performed in a similar manner to
that in example 8 utilizing 0.2 g of the hydrochloride
salt of example 2, 4-hydroxy-2-phenyl-quinoline-3-
carboxylic acid (0.18 g), EDCI (0.16 g), HOBt (0.19 g),
and NMM (0.16 mL). Product was isolated in 64% yield
(0.22 g). mp= 154-155C. [a]D = -30.0 (c=0.4, MeOH).
MS(CI) m/e 504(m+H) . H NMR(DMSOd6~300MHz)
0.82(m,14H), 1.2(m,8H), 1.38(m,4H), 1.94(m,lH),
3.02tm,2H), 3.2~m,lH), 3.4(m,lH), 4.55(m,lH), 7.43(m,5H),
7.7(m,2H), 8 2 ~d,J=7Hz,lH), 12.02(s,lH). C,H,N analysis
calculated for C31H41N3O3: , ,
found: C 73.73, H 8.18, N 8.34.
Example 11
~-(4'-Hydroxy-7'-~-ifluoro-3'-quinolylcarbonyl~
Valine-di-n-pentvlamide
The reaction was performed in a similar manner to
that in example 8 utilizing 0.21 g of the hydrochloride
salt of example 2, 4-hydroxy-7-trifluoro-quinoline-3-
carboxylic acid (0.185 g), EDCI (0.15 g), HOBt (0.2 g),
and NMM (0.16 mL). Product was isolated in 37~ yield,
0.16 g. mp= 194-195C. [a]D = -79.2 (c=0.5, MeOH).
MS~CI) m/e 497(m+H) . lH NMR(DMSOd6,300MHz) ~
0.88(m,12H), 1.35(m,8H), 1.45(m,2H), 1.6(m,2H), 2.05(m,lH)
3.0(m,2H), 3.25-3.4(m,2H), 3.48(m,lH),
4.85(dd,J-3,9Hz,lH), 7.8(d,J=7Hz,lH), 8.1(s,lH),
8.45(d,J=7Hz,lH), 8.9(s,1HJ, 10.2(d,J=7Hz,lH), 12.9(bs,1H)
. . .:, ~ : . .
:-: . . : . - . : .
. , .; :
.
WO91/00725 PCT/US90/0363
Z ~ ~ ? 7 5 5
-36-
C,H,N analysis calculated for C26H26F3N3O3, 0.2 H2O: C
62.56, H 7.35, N 8.41; found: C 62.57, H 7.17, N 8.38.
Example 1~-(7'-Chloro-4'-~ydroxy-3'-auinolylcarbonyl~-R-Valine-
di-n-pentylamide
The reaction was performed in a similar manner to
that in example 8 utilizinq 5.0 g of the hydrochloride
salt of example 2, 4-hydroxy-7-chloro-quinoline-3-
carboxylic acid (3.8 g), EDCI (3.5 g), HOBt (4.6 g), and
NMM (3.8 mL) and 10 mL DMF. Product was isolated in 54%
yield, 4.25 g. mp= 205-206 C. ~a]D = -93.8 (c=0.5,
MeOH). MS~CI) m/e 463~m+H) . H NMR~DMSOd6,300MHz)
0.95(m,6H), 1.15(d,J=8Hz,3H), 1.26(d,J-8Hz,3H),
1.38(m,8H), 1 65(m,2H), 1.8(m,lH), 2.0(m,lH), 2.23(m,lH),
3.15(m,lH), 3.35(m,lH), 3.48(m,lH), 3.72(m,lH),
4.6(t,J=6Hz,lH), 7.2(dd,J=3,9Hz,lH), 7.6(d,J=9Hz,lH)
7.68(d,J=2Hz,lH), 8.26(d,J=7Hz,lH), 10.25(d,J=6Hz,lH),
12.25(d,J=9Hz,lH). C,H,N analysis calculated for
C25H36ClN3O3: C 64.98, H 7.85, N 9.09, Cl 7.67; found: C
65.16, H 8.04, N 8.94, Cl 7.91.
Example 13
N-(9'-Hydroxy-2'-quinolylcarbonyl)-~-Valine-di-n-
pentylamide
The reaction was performed in a similar manner to
that in example 8 utilizing 0.2 g of the hydrochloride
salt of example 2, 4-hydroxyquinoline-2-carboxylic acid
~0.13 g) EDCI (0.14 g), HOBt ~0.19 g), and NMM /0.15 mL).
Product was isolated in 71~ yield (0.207 g). mp= 70-
,
..
' ~ " ' '
' '
WO9l/00725 PCT/US90/03630
2~ 7S5
~'.' '`.''':,;
-37-
71C [~]D = -13.3 (c=0.6, MeOH). MS(CI~ m/e 428(m+H) .
H NMR~DMSOd6,300MHz) ~ 0.85-l.l(m,12H), 1.2-1.4(m,8H),
1.5-1.7(m,4H~, 2.15(m,lH), 3.02(m,1H), 3.25(m,lH),
3.45(m,lH), 3.64(m,lH), 4.95(dd,J=3,6Hz,lH), 6.7(bs,lH),
7.35-7.5(m,2H), 7.6~(t,J=7Hz,2H), 8.35(d,J=8Hz,lH),
10.4(bs,1H). C,H,N analysis calculated for C25H37N3O3: C
70.22, H 8.72, N 9.83; found: C 69.91, H 8.71, N 9.68.
Example 1N-r5'-(N-Allylcarbamyl)pyridyl-3'-carbonyll-R-Valine-
di-n-pentylamide
The hydrochloride salt of example 2 (0.20 g, 0.69
mmol) was stirred in 15 mL of CH2C12 with NMM, (0.15 mL,
1.4 mmol) under nitrogen at 0C. EDCI (0.135 g, 0.69
mmol) and HOBt (0.19 g, 0.14 mmol) were added followed by
the addition of 5-allylcarbamylnicotinic acid (0.142 g,
0.69 mmol). The reaction mixture was stirred overnight
(warming to ambient temperature). The solvents were
evaporated in vacuo and the residue taken up in
ethylacetate and washed successively with water, saturated
NaHCO3, a saturated solution of citric acid, water, and
brine. The organic solution was dried over MgSO4 and then
filtered. Solvents were evaporated in vacuo and the crude
product subjected to flash chromatography using
ethylacetate and hexane as the elutant mixture. The oily
product was isolated in 56% yield (0.17 g). MS(CI) m/e
445 (m+H) . H NMR(CDC13,300MHz) ~ 0.85-l.l(m,12H), 1.2-
1.4(m,8H), 1.5-1.6(m,4H), 2.1(m,lH), 3.05(m,lH),
3.3(m,lH), 3.48(m,lH), 3.65(m,lH), 4.15(m,2H),
5.0(dd,J=3,6Hz,lH), 5.25(m,2H), 5.95(m,lH), 6.95(m,lH),
' ,' ," . : ':, "~ ', "' ' : ' , ,' . ' ', ''. ., ' ' " , ~ . '
WOgl/0072~ PCT/US90/03630
.
~' . . ;''
Zc~ 5S
-38-
7.15~d,J=9Hz,lH), 8.98(s,1H), 9.15(s,2H). C,H,N analysis
calculated for C25HgoN403 C 67.53, H 9.07, N 12.60;
found: C 67.27, H 8.97, N 12.53.
~
N-(l'-Ethyl-7'-methvl-4'-oxo-1',8'-na~hthyridinyl-3'-
carbonyl)-R-Valine-di-n-pentvlam~de
The hydrochloride salt of example 2 (0.2 g, 0.69
mmol) was stirred in 15 m~ of CH2Cl2 with NMM (0.15 mL,
1.4 mmol) under nitrogen at 0 C. EDCI (0.135 g, 0.69
mmol) and HOBt (0.190 g, 1.38 mmol) were added followed by
the addition of nalidixic acid (0.160 g, 0.69 mmol). The
reaction mixture was stirred overnight (warming to ambient
temperature). The solvents were evaporated in vacuo and
the residue was taken up in ethylacetate and washed
successively with water, saturated NaHCO3, a
saturated solution of citric acid, water and brine. The
organic solution was dried over MgSO4 and then filtered.
Solvents were evaporated in vacuo and the crude product
subjected to flash chromatography using ethylacetate and
hexane as the elutant mixture. The purification provided
0.19 g (59%) of an oil. MS(CI) m/e 471(m+H) . lH
NMR(CDC13,300MHz) ~ 0.9(m,6H) 1.05(m,3H), 1.20-1.4(m,10H),
1.48-1.8(m,8H), 2.1(m,lH), 2.65(s,3H), 3.05(m,lH),
3.4(m,2H), 3.6(m,lH), 4.5(dd,J=3,9Hz,lH), 4.6(m,lH),
4.95(dd,J=3,6Hz,lH), 7.25(m,2H), 8.68(m,lH), 8.85(m,lH).
C,H,N analysis calculated for C27H42O3N4, 0.25 H2O: C
68.34, H 9.03, N 11.82; found: C 68.12, H 8.83, N 12.07.
~. . .
.
WO91/0072~ PCT/US90/03630
~?~ 55``-
-39-
~ ~ .
N-~Z-2'-Fluoro-3'-Dhenyl~ro~-2'-enoyll-R-Valine-di-n-
~ntylamide
The reaction was performed in a similar manner to
that in example 3 utilizing 0.27 g of the hydrochloride
salt of example 2, a-fluorocinnamic acid (0.16 g~, EDCI
~0.19 g), HOBt (0.25 g), and NMM (0.21 mL). The oily
product was isolated in an 68% yield, 0.25 g [a] D= +7.1
(c=l.1, MeOH). MS(CI) m/e 405(m+H) . H
NMR(CDCl3,300MHz) ~ O.82-1.0(m,12H), 1.2-1.5(m,8H), 1.5-
1.7(m,4H), 2.1(m,lH), 3.05(m,lH), 3.25(m,lH), 3.4(m,lH),
3.6(m,lH), 4.85(m,lH), 7.05~d,J=42Hz,lH),
7.1(d,J=laHz,lH), 7.3-7.45~m,3H), 7.62(d,J=9Hz,2H). C,H,N
analysis calculated for C24H37FO2N2: C 71.25, H 9.22, N
6.93; found: 70.99, H 9.14, N 6.95.
E~mRle 17
N-(2' Naphthoyl)-R-Valine-di-n-~entvlamide
The reaction was performed in a similar manner to
that in example 3 utilizing 0.2 g of the hydrochloride
salt of example 2, 2-naphthoic acid (0.12 g), EDCI (0.13
g), HOBt (0.18 g), and NMM ~0.16 mL). The product was
isolated as an oil in 72% yield, 0.2 g. [a]D= -13.0
~c=1.0, MeOH). MS~CI) m/e 411(m+H) . H
NMR(CDCl3,300MHz) ~ 0.8-0.9(m,6H), l.l(m,6H), 1.2-
1.4(m,8H), 1.55-1.67(m,4H), 2.13(m,lH), 3.0-3.1(m,lH),
3.25-3.3(m,lH), 3.5lm,lH), 3.65(m,lH),
5.08(dd,J=3,6Hz,lH), 7.11(d,J=9Hz,lH), 7.52(m,2H),
7.9(m,4H), 8.33(s,lH). C,H,N analysis calculated for
~::.. : . .- . . . , : . ~:
WO9l/00725 PCTtUS90/03630
755
-40-
C26H38N2O2: C 76.05, H 9.33, N 6.82; found: C 76.20, H
9.32, N 6.98.
~xample 1~
N-r3'-(3''-Pyridyl)prop-2'- noyll-R-Valine-di-n-
pentyl~mide
The reaction was performed in a similar manner to
that in example 3 utilizing 0.3 g of the hydrochloride
salt of example 2, 3-(3'-pyridyl)acrylic acid (0.17 g),
EDCI (0.22 g), HOBt ~0.28 g), and NMM (0.22 mL). An oil
was isolated in 76~ yield, 0-3 g [a~D = +10.0 (c=0.85,
MeOH). MS(CI) m/e 388(m+H) . lH NMR(CDCl3,300MHz)
0.8-1.05(m,12H), 1.2-1.4(m,8H), 1.45-1.72(m,4H),
2.06(m,lH), 3.1(m,lH), 3.2-3.5(m,2H), 3.5-3.65(m,lH),
4.92(dd,J=2,6Hz,lH), 6.6(d,~z15Hz,lH), 7.28(d,J=9Hz,lH),
7.3(m,lH), 7.6(d,J=15Hz,lH), 7.8(d,J-9Hz,lH),
8.58(d,J=6Hz,lH), 8.74(d,J=2Hz,lH). C,H,N analysis
calculated for C23H37N3O2, 0.75 H2O Ç
10.48; found: C 68.74, H 9.31, N 10.21.
Example 1~
N-(1',2'.(3'S) 4'-Tetrahvdrocarbolinyl-3'-carbonvl)-R-
Valine-di-n-~entylamide
The reaction was performed in a similar manner to
that in example 3 utilizing 250 mg of the hydrochloride
salt of example 2, N-L-1,2,3,4-tetrahydroharman-3-
carboxylic acid ~270 mg), EDCI (160 mg), HOBt (235 mg),
and NI~M (190 mL). The oily product was isolated in 38%
yield (148 mg). [a]D= -5.5 (c=0.2, MeOH). MS(CI) m/e
455(m+H) . H NMR(CDC13,300MHz) ~ 0.8-1.0(m,12H), 1.2-
., .
. ~
~ ' ' ,~ ' ' - ,
WO91/~0725 PCT/US90/03630
~ ~ 5 ? ~ 5 S
-41-
1.35~m,8H), 1.5(m,4H), 1.6(m,lH), 2.05(m,lH), 2.55-
2.82(m,lH), 3.1-3.4(m,4H), 3.55(m,2H), 4.1(m,lH),
4.75(m,lH), 7.0-7.15(m,2H), 7.25(d,J=9Hz,lH),
7.45(d,J=9Hz,lH), 7.8(bs,1H), 7.85(bs,1H), 8.26(s,1H).
C,H,N analysis calculated for C27H42N4O2, 0.75 H2O: C
69.27, H 9.36, N 11.97; found: C 69.58, H 9.16, N 11.91.
Example 20
N-(l'-Hydroxy-2'-naphthoyl)-R-Valine-di-n-pentylamide
The reaction was performed in a similar manner to
that in example 3 utilizing 250 mg of the hydrochloride
salt of example 2, 1-hydroxy-2-naphthoic acid (160 mg),
EDCI (180 mg), ~OBt ~240 mg), and NMM ~200 ~L). Product
was isolated in 85~ yield ~310 mg). mp= 85-86C. ta]D=
+90.5 ~c=0.6, MeOH). MS(CI) m/e 427(m~H) . H
NMR(CDCl3,300MHz) ~ O.9(m,6H), 1.05(m,6H), 1.25-1.4(m,8H),
1.5-1.7(m,4H), 2.15(m,lH), 3.05(m,lH), 3.25(m,lH),
3.5(m,lH), 3.65(m,lH), 5.06(dd,J=3,9Hz,lH),
7.2(d,J=9Hz,lH), 7.35~d,J=lOHz,lH), 7.45~d,J=lOHz,lH),
7.5(dd,J=3,6Hz,lH), 7.6(dd,J=3,6Hz,lH), 7.75(d,J=7Hz,lH),
8.4(d,J=9Hz,lH), 10.6(bs,lH). C,H,N analysis calculated
for C26H38N2O3: C 73.20, H 8.98, N 6.57; found: C 73.24,
H 9.02, N 6.55.
Example 21
~ -(t-Butyloxycarbonyl)-R-Norleucine-di-n-pentylamide
N-(t-Butyloxycarbonyl)-R-Norleucine (1.2 g, 5.2 mmol)
was sti.red at 0 C in 40 mL of CH2Cl2 with BOPCl (1.5 g,
5.9 mmol?, and TEA (0.7 mL, 5.2 mmol). To this reaction
mixture was added di-n-pentylamine (2.5 mL, 10.5 mmol).
.
WO ~l/00725 PCT/US90/03630
7SS
-42-
The mixture was stirred overnight and allowed to warm to
room temperature. An additional equivalent of BOPCl was
added after 18 hrs and the reaction stirred an additional
day at ambient temperature. The solvents were evaporated
in vacuo and the residue taken up in ethylacetate and
washed with water, 1 N HCl, saturated NaHCO3 solution,
water and then the organic solution was dried over MgSO4.
After filtra~ion and concentration of the filtrate in
vacuo, the residue was chromatographed using ethylacetate-
hexane as the solvent system in the ratio (1:4). The
product was isolated as an oil in 75% yield (1.45 g).
MS(CI) m/e 371(m+H) , lH NMR(CDC13,300MHz) ~ 0.9-
1 2~m,9H), 1.24-1 35~m,12H), 1.5~s,9H), 1.55-1.6~m,4H),
1.88~m,2H), 3.1~m,lH), 3.32~m,lH), 3.92~m,lH), 3.6~m,lH),
5.15(m,lH), 6.9~d,J=lOHz,lH).
ExampLe ~
R-Norleucine-di-n-pentylamide hydrochloride
The product of example 21 (1.4g, 3.8 mmol) was
dissolved in 4 N HCl in dioxane (25 mL) and stirred at
room temperature for an hour. When the reaction was
complete by tlc the solvents were evaporated in vacuo and
hexane and diethylether were added. The residue was
triturated with these solvents and the solid product was
filtered away in quantitative yield. [a]D= -1.4 ~c=0.6,
MeOH). MS(CI) m/e 271(m+H) . H NMR(DMSOd6,300MHz)
0.87(m,9H), 1.2-1.4(m,12H), 1.42-1.6(m,4H), 1.7(m,2H),
3.0(m,lH~, 3.1-3.3(m,2H), 3.53(m,lH), 4.14(bs,lH),
8.25(bs, 2r;, .
:
:, . . . . - , ~:
. " .. . ..
W09l/00725 PCTtUS90/03630
.:
2~ 55`
-43-
Example 23
N-(3'-Ouinolylcarbonyl~-R-Norleucine-di-n-pentylamide
The hydrochloride of example 22 (240 mg, 0.87 mmol),
EDCI (170 mg), HOBt (240 mg) and quinoline-3-carboxylic
acid (150 mg) were stirred at 0C under nitrogen in 20 mL
anhydrous CH2C12. To this mixture was added 200 ~L of NMM
and the mixture was stirred overnight (warming to ambient
temperature). The reaction mixture was poured into
ethylacetate and water and the organic extract was washed
successively with water, 10% citric acid solution, and
saturated aqueous NaHCO3. The solution was dried
over MgSO4, filtered and concentrated. The residue was
purified by chromatography using ethylacetate and hexane
as the elutant mixture to provide 200 mg of the glassy
product (54% yield) after evaporation of the volatiles.
[a]Ds -10.5 (c=1.0, MeOH). MS(CI) m/e 426(m+H) . 1H
NMR(CDCl3,300MHz) ~ 0.9(m,9H), 1.35(m,12H), 1.55(m,2H),
1.65-1.80(m,4H), 3.10(m,lH), 3.25-3.35(m,lH), 3.4(m,lH),
3.55-3.6(m,lH), 5.15(m,lH), 7.4(d,J=9Hz,lH), -
7.6(dd,J=3,7Hz,lH), 7.8(dd,J=3,7Hz,lH), 7.9(d,J=9Hz,lH),
8.15(d,J=9Hz,lH), 8.6(d,J=2Hz,lH), 9.35(d,J=3Hz,lH).
C,H,N analysis calculated for C26H39N3O2, 0.3 EtOAc: C
72.27, H 9.23, N 9.27; found: C 72.26, H 9.01, N 9.54.
Example 24
N-(2'-Indolylcarbonyl)-R-Norleucine-di-n-pentylamide
The hydrochloride salt of example 22 (0.30 g, 1.0
mmol) was stirred in 10 mL of CH2C12 with NMM (0.2 mL, 2.0
mmol) under nitrogen at 0 C. EDCI (0.2 g, 1.1 mmol) and
HOBt (0.27 g, 2.0 mmol) were added followed by the
.. . . , .. . ( ,,
- . . - .
.
:. , -
: : . : . --
;: :. -' .: :
W091/00725 PCT/US90/03630
5~
addition of indole-2-carboxylic acid (0.162 g, 1.0 mmol).
The reaction mixture was stirred overnight (warming to
ambient temperature). The solvents were evaporated in
vacuo and the residue taken up in ethylacetate and washed
successively with water, saturated NaHCO3, a saturated
solution of citric acid, water and brine. The organic
solution was dried over MgSO4 and then filtered. Solvents
were evaporated in vacuo and the crude product subjected
to flash chromatography using ethylacetate and hexane as
the elutant mixture. The product was crystallized from
ethylacetate and hexane to provide a glass 0.285 g (69~).
[a]D= -10.6 (c=0.8, MeOH). MS(CI) m/e 414(m+H) . lH
NMR(CDC13,300MHz) ~ 0.9(m,9H), 1.2-1.4(m,10H), 1.5-
1.7~m,6H), 1.86(m~2H), 3.15(m,1H), 3.3-3.4(m,2H),
3.58(m,lH), 5.1(m,lH), 7.0(d,J=2Hz,lH),
7.15(dd,J=3,7Hz,lH), 7.3(m,2H), 7.4(d,J=9Hz,lH),
7.67(d,J=9Hz,lH), 9.4(s,1H). C,H,N analysis calculated
for C25H39N3O2, 0.75 H2O: C 70.30, H 9.55, N 9.84; found:
C 70.38, H 9.20, N 9.85.
Example 25
N-(t-Butyloxycarbonyll-R-(O-benzvl)Serine-di-n-
pentylamide
N-(t-Buiyloxycarbonyl)-R-(O-benzyl)serine (3.0 g,
10.15 mmol) was stirred at 0C in 50 mL of CH2C12 with
BOPCl (2.8 g, 11 mmol) and 2.0 mL (1.5 mmol) of TEA. To
this reaction mixture was added di-n-pentylamine (7 mL, 35
mmol). The mixture was stirred overnight and allowed to
warm to room temperature. An additional equivalent of
BOPCl was added after 18 hrs and the reaction stirred an
, - :
.. .
W091/00725 PCT/US90/03630
'
, 55
~ . ..
-45-
additional day at ambient temperature. The solvents were
evaporated in vacuo and the residue taken up in
ethylacetate and washed with water, 1 N HCl solution,
saturated NaHCO3, water and then the organic solution was
dried over MgSO4. After filtration and concentration of
the filtrate in vacuo, the residue was purified by
chromatography using ethylacetate-hexane as the elutant
system in the ratio (1:9). The product was isolated as an
oil in 44% yield (1.9 g). MS(CI) m/e 435(m+H) . H
NMR(CDCl3,300MHz) ~ 0.89(m,6H), 1.28(m,8H), 1.4(s,9H),
1.55(m,4H), 3.05-3.2(m,2H), 3.4-3.65(m,4H), 4.5(m,2H),
4.85(m,lH), 5.35(d,J=7Hz,lH), 7.31(m,5H).
~L~ ~ '.
R-(O-Ben ~ drochloride
The product of example 25 (0.43 g, 1.0 mmol) was
dissolved in 4 N HCl in dioxane (10 mL) and stirred under
inert atmosphere (N2) for a~ hour. When the reaction was
complete by tlc the solvents were evaporated in vacuo and
hexane and diethylether were added. The residue was
triturated with these two solvents and the solvents again
removed in vacuo. This procedure was repeated several
times until the product was obtained as a glassy solid in
93% yield (0 35 g). [a]D= +1.6 (c=0.5, MeOH). MS(CI)
m/e 335(m+H) .
::
-
. . ,
WO 91/00725 PCT/US90/03630
~(~ ? ~ 5 5
-46-
~mL1~ 27
N-(3'~Ouinolylcarbonyl-R-(O-benzyl)Serine-di-~-
pentylamide
The hydrochloride salt of example 26 (0.35 g, 0.95
mmol) was stirred in 25 mL of CH2C12 with NMM, (0.22 mL, 2
mmol) under N2 at 0C. EDCI (0.19 g, 1.0 mmol) and HOBt
(0.27, 2 mmol) were added followed by the addition of
quinoline-3-carboxylic acid (0.165 g, 0.95 mmol). The
reaction mixture was stirred overnight (warming to ambient
temperature). The solvents were evaporated in vacuo and
the residue taken up in ethylacetate and washed
successively with water, saturated NaHCO3, a saturated
solution of citric acid, water and brine. The organic
solution was dried over MgSO4 and then filtered. Solvents
were evaporated in vacuo and the crude product subjected
to flash chromatography using ethylacetate and hexane as
the elutant mixture. The product was crystallized from
ethylacetate and hexane to provide a semisolid, 0.44 g
(94%). [a]D= -4.0 (c=0.45, MeOH). MS(CI) mte
490~m+H) . H NMR~CDC13,300MHz) ~ 0.9(m,6H), 1.2-
1.4~m,8H), 1.5-1.6~m,4H), 3.05-3.28~m,2H), 3.5-3.7~m,2H),
3.8(m,2H), 4.57(m,2H), 5.4(m,lH), 7.3(m,5H),
7.4(d,J=9Hz,lH), 7.62(dd,J=2,7Hz,lH), 7.81(dd,J=2,7Hz,lH),
7.9(d,J=8Hz,lHj, 8.15(d,J=9Hz,lH), 8.58(d,J=3Hz,lH),
9.3(d,J=3Hz,lH). C,H,N analysis calculated for
C30H39N3O3, 0.75 H2O: C 71.61, H 8.11, N 8.35; found: C
71.73, H 8.01, N 8.21.
.
.: .
WO91/00725 PCT/US90/03630
2 ~ ~ ? ~ 5 5; ;
-47-
Exam~le 28
N-(t-Butyloxycarbonyl)-R-Phenvlalanine-di-n-
pentylamide
The reaction was performed in a similar manner to
that in example 2 utilizing N-(t-Butyloxycarbonyl)-R-
Phenylalanine (0.8 g, 3.1 mmol), BOPCl (1.2, 4.06 mmol),
dipentylamine (3.1 mL, 15 mmol), and TEA (0.4 mL, 3.1
mmol). The oily product was isolated in 65.5% yield (0.87
g). [a]D= +7.0 (c=1.0, MeOH). MS(CI) m/e 405(m+H) .
H NMR(CDC13,300MHz) ~ 0.85(m,6H), 1.15-1.45(m,8H),
1.5(s,9H), 1.55-1.6(m,4H), 2.9-3.1(m,5H), 3.5(m,lH),
4.25(m,lH), 5.3~d,J=9Hz,lH), 7.25~m,SH).
Exam~le 22
N-(t-Butyloxycarbonyl)-(2R 3S)-(O-benzyl)Threonine-
di-n-Dentylamide
The reaction was performed in a similar manner to
that in example 1 utilizing N-~t-Butyloxycarbonyl)-D-(O-
benzyl)- threonine ~5 g, 16.2 mmol), BOPCl ~8.2 g, 16.2
mmol), dipentylamine (16 mL, 78.5 mmol), and TEA (2.1 mL,
16.2 mmol). The product was isolated in 58% yield (4.15
g). MS(CI) 449(m+H) . H NMR(CDCl3,300MHz) ~
0.85(t,J=6Hz,6H), 1.18(d,J=6Hz,3H), 1.2-1.35(m,8H),
1.45(s,9H), 1.5-1.6(m,4H), 3.0-3.18(m,2H), 3.41-
3.63(m,2H), 3.75~m,lH), 4.57(dd,J=12,18Hz,2H), 4.65(m,lH),
5.5(d,J=9Hz,lH), 7.30(m,SH).
.. . ..... ... . ~. ,
. ': .. :. ' : .
. .- ,
....
WO91/00725 PCT/US9OtO3~30
r~_,
,'755
-48-
Example 30
(2R 3S~-(O-Benzyl)Threonine-di-n-~entylamide
hydrochloride
The product of example 29 (1 g, 2.22 mmol) was
deprotected and isolated in a similar manner to that in
example 2. The product was isolated as an oil. [a] D =
+13.3 (c=l.l, MeOH). MS(CI) m/e 359(m+H) . H
NMR(DMSOd6,300MHz) ~ 0.86(m,6H), 1.08-1.32(m,11H),
1.48(m,4H), 3.03(m,2H), 3.42~m,2H), 3.88(m,lH),
g.2(d,J-6Hz,lH), 4.56(m,2H), 7.35(m,5H), 8.35(bs,2H).
~ ~ .
N-~3'-Ouinolvlcarbonyl)-(2R.3S)-lO-ben~yl)Threonine-
di-n-pentylamide
The hydrochloride salt of example 30 (0.25 g, 0.65
mmol) was stirred in 15 mL of CH2C12 with NMM (0.175 mL,
1.3 mmol) under nitrogen at 0C. EDCI (0.15 g, 0.8 mmol)
and HOBt (0.18 g, 1.3 mmol) were added followed by the
addition of quinoline-3-carboxylic acid (0.115 g, 0.65
mmol). The reaction mixture was stirred overnight
(warming to ambient temperature). The solvents were
evaporated in vacuo and the residue taken up in
ethylacetate and washed successively with water, saturated
NaHC03, a saturated solution of citric acid, water and
brine. The organic solution was dried over MgS04 and then
filtered. Solvents were evaporated in vacuo and the crude
product subjected to flash chromatography using
ethylacetate and hexane as the elutant mixture. The oily
product was isolated in 62% yield (0.2g). [a~D= -4.1
- . - , . , :
.
: , . ~ . ,
WO9l/~0725 PCT/US90tO363~
, . .
2~ 55 .- ,,
-49-
lc-1.0, MeOH). MS(CI) m/e 504(m+H) . H
NMR(CDCl3,300MHz) ~ 0.9(m,6H), 1.2-1.45~m,11H), 1.5-
1.7~m,4H), 3.0-3.25~m,2H), 3.56-3.7~m,2H), 3.9~m,lH),
4.5~m,2H), 5.3~apparent q,J=4.5Hz,lH), 7.2-7.3~m,5H),
7.56~d,J=6Hz,lH), 7.65~t,J=7Hz,lH), 7.8~t,J=7Hz,lH),
7.92~d,J=9Hz,lH) 8.15~d,J=9Hz,lH), 8.63~d,J=2Hz,lH),
9.35~d,J=3Hz,lH). C,H,N analysis calculated for
C31H41N3O3, 1.6 H2O: C 69.92, H 7.89, N 8.37; found: C
69.81, H 7.78, N 8.08.
E~m~l~ 32
N-(3'-Ouinolvlcarbonyl)-(2R 3S)-Threonine-di-n-
pentylam;de
The product of example 31 (1 g, 2 mmol) was stirred
in 20 mL of CH2C12 and 7 mL of borontristrifluoroacetate
~1.0 M solution in trifluoroacetic acid) was added at 0C.
The mixture was stirred approximately 1 hour The tlc
revealed some starting material therefore ano~her 5 mL of
borontristrifluoroacetate and 5 mL trifluoroacetic acid
were added. The reaction proceeded overnight to completion
by tlc analysis. The reaction mixture was diluted with
MeOH and then concentrated in vacuo. The residue was
purified by chromatography using ethylacetate and hexane
as the elutant mixture. The pure fractions were pooled
together and the desired product characterized as the di-
trifluoroacetic acid salt. mp= 84-6 C. [~]D= -11.6
~c=0.55, MeOH). MS~CI) m/e 414~m+H) . 1H
NMR~CDC13,300MHz) ~ 0.85~m,6H), 1.13~d,J=7Hz,3H), 1.15-
1.38~m,8H), 1.48~m,2H), 1.6~m,2H), 3.1~m,1H), 3.32-
3.53(m,4H), 4.05~m,1H), 4.9~t,J=6Hz,lH), 7.7~t,J=6Hz,lH),
! . . , , ~
'
, ~, ,
` ' ' ` : ' ' ' :
WO91/0072~ PCT/US90/03630
~5 ~75~5
-50-
7 88~t,J-7Hz,lH), 8.1~d,J=9Hz,lH), 8.8~d,J=9Hz,lH),
8.93~bs,lH), 9.31~bs,lH), 10.02~bs,lH). C,H,N analysis
calculated for C24H35N33~ 2 CF3C2
6.55; found: C 52.31, H 5.62, N 6.66.
Example ~
N-(3'-Ouinolylcarbonyl)-(2R 3S)-(O-acet-~l)Threonine-
di-n-pentylamide
Pyridine ~20 ~L) and acetic anhydride ~60 ~L) were
added to the product of example 32 ~51 mg, 0.125 mmol)
which was dissolved in acetonitrile ~2 mL). The reaction
mixture w~s stirred overnight at room temperature.
Ethylacetate was added and this solution was washed
~uccessively with water and brine. The organic solution
was dried over MgSO4. After filtration and concentration
of the filtrate in vacuo, the residue was purified by
chromatography using ethylacetate and hexane as the
elutant system in the ratio ~9:1). The product was
isolated as a glass in 44% yield (25 mg). MS~CI) m/e
456(m+H) . H NMR~CDC13,300MHz) ~ 0.9~m,6H), 1.25-
1.45~m,11H), 1.52~m,2H), 1.7~m,2H), 2.05~s,3H), 3.1(m,2H),
3.3-3.6 (m,3H), 5.28(m,lH), 5.44(m,lH), 7.35~d,J=9Hz,lH),
7.65~t,J=7Hz,lH), 7.82~t,J=7Hz,lH), 7.95~d,J=7Hz,lH),
8.18~d,J=9Hz,lH), 8.6~d,J=3Hz,lH), 9.35~d,J=3Hz,lH).
C,H,N analysis calculated for C26H37N3O4, 0.4 H2O: C
67.48, H 8.23, N 9.08; found: C 67.69, H 8.20, N 8.60.
.. . , . . - . : .~
: .- ~ . : ., - .
. : '', . . .
WO91/00725 PCT/US90/0363~
. .
2a~?~ss~ ~;;, ~--'
~xamDle N-(3'-Ouinolvlcarbonyl)-~2R.3S)-~O-methyl)Threonine-
di-n-pentylamide
Lithium bis(trimethylsilyl)amide in THF ~0.15 mL of
1.0 M solution in THF) was added to a cooled ~-10C)
solution of the product of example 32 ~55 mg, 0.14 mmol)
in 2 mL tetrahydrofuran (THF) and then methyl iodide
~0.015 mL) was added. The reaction mixture was stirred
approximately 1 hour and slowly wa-med to room
temperature. Tlc revealed some starting material
therefore another equivalent of methyl iodide t0.01 mL)
was added. The reaction then proceeded to completion by
tlc. The reaction mixture was concentrated in vacuo.
Ethylacetate was added to the residue, which was then
washed with water and brine. The ethylacetate extract was
dried over MgSO4. Filtration and concentration of the
filtra~e in vacuo, provided a residue which was purified
by chromatography using ethylacetate and hexane as the
elutant mixture. An oil was isolated in 47% yield (28 mg).
MS(CI) m/e 428(m+H) . H NMR(CDCl3,~00MHz) ~ 0.92(m,6H),
1.25(d,J=6Hz,3H), 1.25-1.4(m,8H)j 1.55-1.6(m,4H),
3.05(m,lH), 3.2-3.3(m,2H), 3.35(s,3H), 3.58-3.82(m,2H),
5.25(m,lH), 7.45(d,J=9Hz,lH), 7.65(t,J=6Hz,lH),
7.8(t,J=6Hz,lH), 7.9(d,J=9Hz,lH), 8.18~d,J=9Hz,lH),
8.6(d,J=3Hz,lH), 9.35(d,J=3Hz,lH).
. :., ' ~ : .
; : -
WO9l/00725 PCT/US90/0363
~, :
2~ 5~
-52-
N-(t-Butyloxycarbonyl)-3-(2'-thienyl)-R-Alanine-di-n-
~ ,.
N-~t-Butyloxycarbonyl)-R-3-~2'-thienyl)-Alanine ~0.78
g, 3.25 mmol) was stirred at 0C in 25 mL of CH2Cl2 with
BOPCl ~0.44 g, 3.25 mmol) and 0.5 mL, (3.25 mmol) of TEA.
To this reaction mixture was added di-n-pentylamine ~2 mL,
10 mmol). The mixture was stirred overnight and allowed
to warm to room temperature. An additional equivalent of
BOPCl was added after 18 hrs and the reactions stirred an
additional day at ambient temperature. The solvents were
evaporated in vacuo and the residue taken up in
ethylacetate and washed with water, 1 N HCl solution,
saturated NaHCO3 solution, water and then the organic
soluti~n was drièd over magnesium sulfate. After
filtration and concentration of the filtrate in vacuo, the
residue was purified by chromatography using ethylacetate-
hexane as the solvent system in the ratio (1:4). The
product was isolated as an oil in 57~ yield ~0.76 g).
[a~D= -2.27 (c=0.66, MeOH). MS(CI) m/e 411(m+H) , 355,
311. lH NMR(CDCl3,300MHz) ~ 0.85(m,6H), 1.15-
1.38(m,10H), 1.45(s,9H), 1.51(m,2H), 3.1(m,4H),
3.22(m,lH), 3.4(m,lH), 4.75(apparent q,J=lOHz,lH),
5.45(d,J=9Hz,lH), 6.83(d,J=6Hz,lH), 6.9(t,J=4Hz,lH),
7.15(d,J=6Hz,lH).
- : .
: - ~
. ~ ',, - .:
..
.
WO 91~00725 PCT/US90/0363
:'
2~755
-, .,~
-53-
~xample
R-3-(2'-Thienyl)-Alanine-di-n-pentylamide hydrochlorid~
The product of example 35 (0.22 g, 0.59 mmol) was
deprotected and isolated in the same manner as that in
example 2 in quantitative yield. MS(CI) m/e 327~M+H) .
Example 37
N-(3'-Ouinolylcarbonyl)-3-(2'-thienyl)-R-Alanine-
di-n-pentylamide
The reaction was performed in a similar manner to
that in example 3 utilizing ~80 mg, 0.23 mmol) of the
hydrochloride salt of example 36, quinoline-3-carboxylic
acid ~40 mg), EDCI ~50 mg), HOBt (62 mg), and NMM (51 ~L).
An oil was isolated in 45% yield, ~48 mg). MS~CI) m/e
466~m+H) . H NMR~CDC13,300MHz) ~ 0.9~m,6H), 1.2-
1.4(m,8H), 1.45-1.65~m,4H), 3.05-3.4(m,4X), 3.45-
3.6~m,2H), 5.35~dd,J=6,7Hz,lH), 6.87~d,J=3Hz,lH),
6.99~m,lH), 7.18~d,J=6Hz,lH), 7.4~d,J=9Hz,lH),
7.63~dd,J=3,7Hz,lH), 7.8(dd,J=3,7Hz,lH), 7.9(d,J=8Hz,lH),
8.15(d,J=8Hz,lH), 8.6~d,J=3Hz,lH), 9.32~d,J=3Hz,lH).
C,H,N analysis calculated for C27H35N3O2S, 0.9 H2O: C
67.29, H 7.70, N 8.72; found: C 67.60, H 7.47, N 8.98.
E~ample 38
N-(t-Butyloxycarbonyl)-S-Valir.~-di-n-pentylamide
The reaction and product isolation were performed in
a similar manner to that in example 1 utilizing N-~t-
Butyloxycarbonyl)- S-Valine (2.5 g, 11.5 mmol), BOPCl (3.5
g, 13.8 mmol) and dipentylamine (11.6 mL, 58 mmol), and
'
-
WO91/0072~ PCT/US90/~3630
~': '' ` ' ,' ,
;~?~755
TEA (1.6 mL, 12 mmol). The oily product was isolated in
55% yield (2.25 g). [a]D= -21.1 (c=1.0, MeOH). MS(CI)
m/e 357~m+H) . H NMR(CDC13,300MHz) ~ O.9(m,6H),
1.05(m,6H), 1.25-1.35(m,8H), 1.95(s,9H), 1.5-1.55(m,4H),
1.95(m,lH), 3.0(m,lH), 3.2(m,lH), 3.36(m,lH), 3.6(m,lH),
4.4(dt,J=3,7Hz,lH), 5.24(d,J=9Hz,lH).
Example 39
S-Valine-di-n-pentylamide hydrochloride
The product of example 38 (0.2 g, 0.57 mmol) was
deprotected and the product isolated as in example 2 in
quantitative yield. MS(CI) m/e 257(m+H) .
~xample ~Q
N-(3'-Ouinolylcarbonyl)-S-Valine-di-n-pent~lamide
The reaction sequence was performed in a similar
manner to that in example 3 utilizing 175 mg of the
hydrochloride salt of example 39, quinoline-3-carboxylic
acid (110 mg), EDCI (125 mg), HO~t (165 mg), and NMM (75
~L). The glassy product was isolated in 80% yield, (198
mg)- [a]D= +12.95 (c=0.8, MeOH). MS(CI) m/e 412(m+H) .
H NMR(CDCl3,300MHz) ~ 0.8-1.05(m,12H), 1.2-1.44(m,8H),
1.55(m,4H), 2.15(m,lH), 3.1(m,lH), 3.3(m,lH), 3.5(m,lH),
3.65(m,1H), 5.1(dd,J=3,6Hz,lH), 7.25(d,J=7Hz,lH),
7.62(t,J=7Hz,lH), 7.8(t,J=7Hz,lH), 7.9(d,J=8Hz,lH),
8.15(d,J=9Hz,lH), 8.61(d,J=3Hz,lH), 9.35(d,J=3Hz,lH).
C,H,N analysis calculated for C25H37N3O2, 0.25 H2O: C
72.16, H 9.09, N 10.10; found: C 72.41, H 9.21, N 9.97.
,
: , , : -. . .. .: ,
WO91/00725 PCT/US9OtO3630
2C?5~5S
.. ...
. ....
-55-
N-tt-Butyloxycarbonyl)-~im-tosyl)-R-Histidine-di-n-
pentylamide
N-(t-Butyloxycarbonyl)-R-(Nim-tosyl)-histidine, (4.95
g, 12.6 mmol) was stirred at 0C in 50 mL of CH2C12 with
BOPCl (3.2 g, 12.6 mmol) and 1.65 mL (12.6 mmol) TEA. To
this reaction mixture was added di-n-pentylamine (7.7 mL,
38 rnmol). The mixture was stirred overnight and allowed
to warm to room temperature. An additional equivalent of
BOPCl was added after 18 hrs and the reaction stirred an
additional day at ambient temperature. The solvents were
evaporated in vacuo and the residue was taken up in
ethylacetate and washed with water, 1 N HCl solution,
saturation NaHC03, water. The organic solution was dried
over MgS04. After filtration and concentration of the
filtrate in vacuo, the residue was purified by
chromatography using ethylacetate-hexane as the solvent
system in the ratio (1:4). The product was isolated as an
oil in 75% yield (5.1 g). [a]D= +8.8 (c=1.0, MeOH).
MS(CI) m/e 549(m+H) . H NMR(DMSOd6,300MHz) ~ 0.85(m,6H),
1.05-1.46(m,21H), 2.42(s,3H), 2.67(m,2H), 3.03-3.15(m,4H),
4.52(m,1H), 7.0(s,1H), 7.28(d,J=7Hz,lH), 7.49(d,J=7Hz,2H),
7.9(d,J=7Hz,2H), 8.28(s,lH). C,H,N analysis calculated
for C28H44N405S: C 61.28, H 8.08, N 10.21; found: C 61.04,
H 8.05, N 10.10.
~ '' ' .
.... . . .
WO91/0072~ PCT/US90/0363~
~'
755
-56-
E~ample~ 42
(Nim-Tosyl)-R-Histidine-di-n-pentylamide
To a solution of the product of example 41 (6.7 g,
12.21 mmol) in CH2C12 (100 mL) was added trifluoroacetic
acid (TFA, 40-50 mL). The reaction mixture was stirred at
room temperature 60 minutes. When reaction was complete
by tlc, the solvents were evaporated several times in
vacuo and CH2C12 was added with a saturated solution of
NaHCO3. The reaction mixture was stirred vigorously
another 1 hr and after separation of layers, the organic
layer was washed several times with water and brine. The - -
CH2C12 layers and washings were dried over magnesium
sulfate. The product was then concentrated in vacuo. The
semisolid product was isolated and dried in a vacuum oven
over P2O5 at room temperature, 5.1 g (93% yield).
[a]D= -9.4 (c=1.0, MeOH). MS~CI) m/e 449(m+H) , 264,
295. lH NMR(CDCl3,300MHz) ~ O.85(m,6H), 1.1-1.35(m,8H),
1.47-1.6(m,4H), 2.45(s,3H), 2.9-3.2(m,6H), 3.4-3.55(m,2H),
4.5(m,lH), 7.18(s,lH), 7.35(d,J=8Hz,2H), 7.82(d,J=8Hz,2H),
7.95(s,lH).
Example 43
N-(2l-Indolylcarbonyl)-R-~istidine-di-n-pentylamide
The compound of example 42 (170 mg, 0.5 mmol), EDCI
(105 mg), HOBt (135 mg) and indole-2-carboxylic acid (85
mg) were stirred at 0 C under nitrogen in 10 mL of
anhydrous CH2C12. To this mi:cture was added 110 ~L of NMM
and the mlxture was stirred overnight (warming to ambient
temperature). The reaction mixtu e was poured into
' .. : ' .'.. . . -- :- .. , , . '
:. , . ~: .. . . .. . . -
: ~
:-, . - . : ,. :- . - : -
- : .. : . . . .
WO 9l/00725 PCTtUS90/03630
-57-
ethylacetate and water and the organic extract was washed
successively with water, 10% citric acid solution, and
saturated aqueous NaHCO3. The soiution was dried over
MgSO4, filtered and concentrated. The residue was
purified by chromatography using chloroform/methanol/
ammonia as the elutant mixture to provide 98 mg of the
semisolid product (45% yield) after evaporation of the
~olatiles. [a]D= +9.8 (c=0.46, MeOH). MS(CI) m/e
438(m+H) , 253, 281. H NMR(CDCl3,300MHz) ~ O.75-
0.95(m,6H), 1.2(m,8H), 1.5(m,4H), 3.13(m,4H), 3.3(m,lH),
3.4(m,lH), 3.5(m,2H), S.32(m,lH), 6.8(s,lH), 6.9(s,lH),
7.1(t,J=7Hz,2H), 7.2(t,J=7Hz,2H), 7.35(d,J=9Hz,lH),
7.59(d~J=9Hz,lH)~ 9.8(s,lH). C,H,N analysis calculated
for C25H35N5O2, 0.5 H2O: C 67.23, H 8.13, N 15.68; found:
C 67.24 H 8.06, N 15.24.
~am~le 44
-(t-Butyloxycar~on~ N -(benzyloxycarbonyl)-R-
Lysine-di-n-pentylamide
The reaction was performed in a similar manner to
that in example 1 utilizing N -t-Butyloxycarbonyl-R-(N -
benzyloxycarbonyl)Lysine (S g, 13.1S mmol), BOPCl (6.7 g,
26.3 mmol), di-n-pentylamine (26 mL, 131 mmol) and TEA
(1.8 mL, 13.S mmol) in CH2Cl2 (25 mL). The oily product
was isolated in 64.5% yield (4.4 g). [a]~= +65.3
(c=0.15, MeOH). MS(CI) m/e 520(m+H) . H
NMR(CDCl3,300MHz) ~ 0.9(m,6H), 1.2-1.35(m,12H),
1.41(s,9H), l.S-1.66(m,4H), 3.05-3.25(m,9H), 3.3(m,2H),
3.5(m,2H), 9.53(m,lH), 4.9~m,lH), 5.1(s,2H),
5.38(d,J=9Hz,lH), 7.3(m,SH).
:: . , . -
: : - -
-: "
::
W091/00725 PCT/US90/03630
z~755
-58-
~ample q5
N-(t-Butyloxycarbonyl)-3-(1'-naphthvl)~R-~lanine-
di-n-pentylamine
N-(t-Butyloxycarbonyl)-3-~1'-naphthyl)-R-Alanine
(0.35 g, 1.1 mmol) was stirred at 0C in 25 mL of CH2C12
with BOPCl, (0.3 g, 1.2 mmol), and 0.15 mL of TEA (1.2
mmol). To this reaction mixture was added di-n-
pentylamine (0.8 mL, 4 mmol). The mixture was stirred
overnight and allowed to warm to room temperature. An
additional equivalent of BOPC1 was added after 18 hrs and
the reaction stirred an additional day at ambient
temperature. The solvents were evaporated in vacuo and
the residue taken up in ethylacetate and washed with
water, 1 N HC1 solution, saturated NaHCO3, water and then
the organic solution was dried over MgSO4. After
filtration and concentration of the filtrate in vacuo, the
residue was purified by chromatography using ethylacetate-
hexane as the solvent system in the ratio (1:4). The
product was isolated as an oil in 65% yield (0.25 g).
MS(CI) m/e 455(m+H) . H NMR(CDC13,300MHz) ~ 0.7-
0.8(m,6H), 0.9(m,8H), 1.2-1.3(s,4H), 1.35(s,9H),
3.0(m,2H), 3.35(m,2H), 3.5-3.6(m,2H), 4.3(m,lH),
7.4(m,lH), 7.45-7.55(m,2H), 7.6(m,lH), 7.8 (d,J-9Hz,lH),
7.85(d,J=9Hz,lH), 8.35(d,J=9Hz,lH), 8.9(bs,lH).
Ex~mple 46
3-(l~-Naphthy~ lanine-di-n-pent~lamide hydrochloride
The product of example 45 (0.32 g, 0.72 mmol) was
dissolved in 4 N HCl in dioxane ~10 mL) and stirred under
. .
. - : . : , , : ::: -
: : .
W09l/00725 PCr/US90/03630
.
-59-
inert atmosphere (N2) for an hour. When the reaction was
complete by tlc the solvents were evaporated in vacuo and
hexane and diethylether added. The residue was triturated
with these two solvents until the product was obtained as
a glassy solid in quantitative yield.
MS(CI) m/e 391(m+H) . lH NMR(CDC13,300MHzl: ~ 0.63(m,3H),
0.85(m,3H), l.OS-1.45(m,10H), 1.5-1.72(m,2H), 2.62(m,lH),
2.85(m,lH), 3.6-3.92(m,4H), 4.85(m,lH), 9.73(m,2H),
7.36(m,lH), 7.5(m,lH), 7.7(d,J=6Hz,lH), 7.75(d,J=6Hz,lH),
8.35(d,Jz8Hz,lH), 8.92(bs,2H), 9.9(s,lH).
E~mple 47
N-~3'-Ouinolylcarbonyl)-3-(l'-Naphthyl)-R-Alanin~-
di-n-pentylamide
The hydrochloride of example 96 (200 mg, 0.52 mmol),
EDCI, HO~t (70 mg) and quinoline-3-carboxylic acid (90 mg)
were stirred at 0C under N2 in 5 mL of anhydrous CH2Cl2.
To this mixture was added lO~L of NMM and the mixture was
stirred overnight (warming to ambient temperature). The
reaction mixture was poured into ethylacetate and water
and then the separated organic extract was washed
successively with water, 10% citric acid solution, and
saturated aqueous NaHC03. The solution was dried over
MgS04, filtered and concentrated. The residue was
purified by chromatography using ethylacetate and hexane
as the elutant mixture to provide 180 mg of the oily
product (68% yield) after removal of the volatiles.
MS(CI) m/e 510(m+H) , 280. lH NMR(CDC13,300MHz) ~
0.72(m,3H), O.9(m,3H), 1.1-1.45(m,10H), 1.5.1-6(m,2H),
2.38-2.6(m,2H), 2.85(m,lH), 3.47(m,2H), 3.9(m,lH),
.. ~.... ...
.. : . , ,.. ;., - ' ~ ~ . . :
: .
WO91/00725 PCT/US90/03630
Z~ ,, S5
-60-
5.6(m,lH), 7.35(d,J=6Hz,2H), 7.52(t,J=7Hz,2H), 7.6-
7.7(m,3H), 7.72-7.93(m,3H), 8.15(d,J=9Hz,lH)
8.55(d,J=9Hz,lH), 8.6(d,J=3Hz,lH), 9.4(d,J=3Hz,lH).
ExamDle ~
N-(t-Butyloxycarbonyl)-3-~2'-naphthyl)-R-Alanine-
di-n-pentylamide
N-(t-Butyloxycarbonyl)-3-(2'-naphthyl)-R-Alanine
(0.31 g, 1.0 mmol) was stirred at 0C in 25 mL of CH2Cl2
with BOPCl, (0.38 g, 1.5 mmol) and 0.2 mL of TEA (1.5
mmol). To this reaction mixture was added di-n-
pentylamine (0.7 mL, 3.5 mmol). The mixture was stirred
overnight and allowed to warm to room
temperature. An additional equivalent of BOPCl was added
after 18 hrs and the reaction stirred an additional day at
ambient temperature. The solvents were evaporated in
vacuo and the residue taken up in ethylacetate and washed
with water, 1 N HCl solution, saturated NaHCO3, and water.
The organic solution was dried over MgSO9. After
filtration and concentration of the filtrate in vacuo, the
residue was purified by chromatography using ethylacetate-
hexane as the solvent system in the r~tio (1:4). The
product was isolated as an oil in 62% yield (0.28 g).
MS(CI) m/e 455(m+H) , 355.
Ex~-m-pl~ 49
3-(2'-Naphthvl~-R-Alanine-di-n-~entylamide h~drochloride
The product of example 48 (0.28 g, 0.6 mmol) was
dissolved in 4 N HCl in dioxane ~10 mL) and stirred under
N2 for an hour. When the reaction was complete by tlc the
,,. , . ~ -
~: ., .. - ....................... . .
,: . : : . ~ . . .
WO91/00725 PCT/US90/03630
, .
2~ 5S
-61-
solvents were evaporated in vacuo and then hexane and
diethylether were added. The residue was triturated with
these two solvents until the product was obtained as a
glassy solid in 93% yield. MS(CI) m/e 355(m+H) .
Example 50
N-(3'-Ouinolylcarbonyl)-R-Histidine-di-~-pentylamide
The free base of example 42 ~3.7 g, 9.26 mmol), EDCI,
(1.7 g, 9 mmol), HOBt (3.65 g) and 1.5 g quinoline-3-
carboxylic acid were stirred at 0C in 50 mL of anhydrous
~imethylformamide (DMF) and CH2Cl2 in 1:1 ratio. After
reaction was complete by tlc, solvents were evaporated
under vacuum and the residue dissolved in large excess of
ethylacetate (300 mL). Water was added and the organic
extract was washed with 10% citric acid solution, and
saturated NaHCO3. The solution was dried over MgSO4,
filtered and concentrated. The residue was purified by
chromatography using chloroform-methanol and ammonium
hydroxide as the elutant mixture to provide 1.98 g (68.3~)
product. [~]D= -6.9 (c=0.25, MeOH). MS(CI) m/e
450(m+H) , 156. H NMR(CDC13,300MHz) ~ 0.9(m,6H),
1.29(m,8H), 1.45-1.6(m,4H), 3.08-3.2(m,3H), 3.23-
3.4(m,2H), 3.5-3.6(m,lH), 5.3(apparent q,J=9Hz,lH),
6.85(s,lH), 7.6(m,3H), 7.(t,J=6H,lH), 7.88(d,J=8Hz,lH),
7.97(d,J=8Hz,lH), 8.15(d,J=8Hz,lH), 8.6(d,J=3Hz,l~),
9.3(d,J=3Hz,lH). N-(3'-Quinolylcarbonyl)-(Nim-tosyl)-R-
histidine-di-n-pentylamide ~0.2 g) also was isolated refer
to example 51.
. . -: . - - .:
~, , ' , . '
.
' . ', . .
WO91~00725 PCT/US90/03630 ~
',~
i
~1?~ 55
-62-
Exam~le ~1
N-3'-Ouinolylcarbonyl-(Nlm-tosyl)-R-Histidine-di-n-
pentylamide
The title compound of example 51 was isolated as a
side product in the procedure in example 50. la]D= +13.3
(c=1.05, MeOH). MS(CI) m/e 604~m+H) , 450. H
NMR(CDC13,300MHz) ~ 0.9~m,6H), 1.3(m,8H), 1.45-1.7(m,4H),
2.25~s,3H), 3.0-3.13(m,3H), 3.25(m,1H), 3.35(m,1H),
3.5(m,1H), 5.36(apparent q,J=6Hz,lH), 7.15(m,3H),
7.6(t,J=7Hz,2H), 7.7(d,J=9Hz,2H), 7.8-7.9(m,2H),
7.95(d,Jz2Hz,lH), 8.13(d,J=7Hz,lH), 8.45(d,J=3Hz" H),
9.18(d,J=3Hz,lH). C,H,N analysis calculated for
C33H41N5O9S: C 65.64, H 6.85, N 11.60; found: C 65.58, H
6.84, N 11.50. ?
Example 52
~_(Benzyloxycarbony~ -R-Lvsin~-di-n-oentylamide
hydroc~1o~;~e
The compound was prepared in similar manner to
example 2 via deprotection of the product of example 44
using 4 N HCl in dioxane. The product was isolated in
quantitative yield. MS(CI) m/e 420(m+H) .
~xample 53
Na-(3'-Ouinolylcarbonyl)-N -(benzyloxycarbonyl)-~-
Lysine di-n-oentyl2mide
The reaction was performed in the similar manner to
that in example 3 utilizing 1.0 g of hydrochloride salt of
example 52 quinoline-3-carboxylic acid (0.38 g), EDCI
(0.95 g), HOBT (0.6 g), and NMM (0.48 mL). The oily
: ,:: ' -. .: -, ,
.. . . . . .
WO91/00725 PCT/US90/03630
r -
2(~ ~jr?~s5
,
-63-
product was isolated in 72~ yield. [~D= +2.7 (c=0.7,
MeOH). MS(CI) m/e 575(m+H) . lH NMR(C~Cl3,300MHz)
0.9(m,6H), 1.3-1.62(m,8H), 1.53(m,6H), 1.65(m,2H),
1.85(m,2H), 3.05-3.55(m,lH), 5.05(m,lH), 5.15(m,2H),
7.28(m,SH), 7.55(t,J=8Hz,lH), 7.8(m,3H), 8.18(d,J=9Hz,lH),
8.58~d,J=2Hz,lH), 9.32~d,J=2Hz,lH). C,H,N calculated for
C34H96N4O4: C 71.05, H 8.07, N 9.75; found: C 71.00, H
8.18, N 9.68.
Ex~mRle 54
N-(3'-Ouinolylcarbonyl~-R-~ysine-di-n-pentylamide
To a suspension of 0.5 g 10~ Pd/C in methanol ~MeOH,
25 mL) and cyclohexadiene (3 mL) under N2 was added a
solution of the product of example 53 (0.51 g, 0.89 mmol)
in methanol via cannula. The reaction mixture was stirred
overnight at ambient temperature. Cyclohexadiene (2 mL)
was added and the reaction was continued overnight. The
mixture was filtered through celite and washed several
times with methanol. The filtrate and washings were
combined and concentrated in vacuo. The residue was
subjected to flash chromatography using chloroform-
methanol and ammonium hydroxide 90:10:1 as the elutant
mixture. Lyophilization provided product in 64% yield
(0.25 g). MS(CI) m/e 441(m+H) . H NMR(DMSOd6,300MHz)
0.85(m,6H), 1.15-1.35(m,8H), 1.4-1.65(m,4H), 1.7(m,2H),
1.75(m,2H), 2.7(m,2H), 3.1-3.5(m,8H), 4.9(m,lH),
7.7~t,J=6Hz,lH), 7.88(t,J=6Hz,lH), 8.1(d,J=8Hz,2H),
8.9(d,J=3Hz,lH), 9.0(d,J=3Hz,lH), 9.3(d,J=3Hz,lH). C,H,N
analysis calculated for C26H40N402, H2O: C 69.45, H 8.97,
N 12.46; found: C 69.98, H 8.76, N 12.03.
: - ,, . :: . ,
,: . ~' . ' ~ ' '
WO~1/00725 PCT/US90/03630
~'
2~5~755
-64-
ExampleN-(t-Butyloxycarbonyl)-R-(4'-Hydroxy~henyl)~lycine-
di-n-pentylamide
The reaction was performed in a similar manner to
that in example 1 utilizing N-~t-Butyloxycarbonyl)-R-4'-
hydroxy phenylglycine (5 g, 18.7 mmol), BOPCl (5.1 g, 20
mmol), dipentylamine (8 mL, 37 mmol), and TEA (2.6 mL).
The product was isolated in 78% yield (5.9 g). MS(CI) m/e
407(m+H) . H NMR(CDC13,300MHz) ~ 0.85(m,6H), 1.1-
1.35~m,8H), 1.3(s,9H), 1.45-1.58(m,9H), 3.0(m,lH~,
3.15(m,2H), 3.45~m,lH), 5.42~d,J=9Hz,lH),
6.02~d,J=9Hz,lH), 6.5~s,lH), 6.75~d,J=9Hz,2H),
7 18~d,J-gHz,2H).
FxampleN-(8'-~ydroxy-2'-~uino~lcz hony~ al;~-d;-n-
pe~tylamide
The title compound was prepared in a similar fashion
to that in example 3. mp= 143-4 C. MS~CI) m/e 428~m+H) ,
293, 158. lH NMR~CDC13,300MHz) ~ 8.58~d,J=lOHz,lH),
8.31(s,2H), 8.09(s,lH), 7.54(m,lH), 7.39(dd,J=1,8Hz,lH),
7.24(m,lH), 5.01(dd,J=7,10Hz,lH), 3.65(dt,J=7,16Hz,lH),
3.28-3.55(m,2H), 3.06(dt,J=7,14Hz,lH),
2.22(septet,J=7Hz,lH), 1.50-1.75(m,9H), 1.25-1.42(m,8H),
1.06(d,J=7Hz,3H), 1.03(d,J=7Hz,3H), 0.92(t,J=7Hz,3H),
0.89~t,J=7Hz,3H). C,H,N analysis calculated for
C25H37N3O3, 0.1 H2O: C 69 93, H 8.73, N 9.79; found: C
69.78, H-8.51, N 9.61.
' ' ~ . ' ! : ~ ,
WO91/0072~ PCT~US90/03630
2~'5~5S
., ;, ,
-65-
E~a~le 57
R-Phenylalanine-di-n-pentylamide hydrochloride
The compound was prepared in similar manner to
example 2 via deprotection of N-t-Butyloxycarbonyl-R-
Phenylalanine- di-n-pentylamide, the product of example
28, using 4 N HCl in dioxane. The product was isolated in
quantitative yield. MS(CI) m/e 305(m+H) .
Example ~
N-(3'-Ouinolylcarbonyl)-R-Phenylalanine-di-n-
pentylamide
The hydrochloride of example 57 (870 mg, 2.46 mmol),
EDCI (550 mg), HOBt ~300 mg), and quinoline-3-carboxylic
acid (430 mg) were stirred at 0C under N2 in 25 mL of
anhydrous CH2CL2. To this mixture was added 550 ~L of NMM
and the mixture was stirred overnight (warming to ambient
temperature). The reaction mixture was poured into
ethylacetate and water and the organic solution was
separated. The organic extract was washed sùccessively
with water, 10% citric acid solution, and saturated
aqueous NaHCO3. The solution was dried over MgSO4,
filtered and concentrated. The residue was purified by
chromatography using ethylacetate and hexane as the
elutant mixture to yield 870 mg of product (77%) after
removal of the volatiles. [a]D= +12.9 (c=1.05, MeOH).
MS(CI) m/e 460~m+H) . lH NMR(CDC13,300MHz) ~ 0.9(m,6H),
1.15-1.4(m,8H), 1.5-1.55~m,4H), 2.9-3.12tm,3H), 3.2(m,2H),
3.48-3.6(m,lH), 5.35(m,lH), 7.27~m,5H), 7.48(d,J=10Hz,lH),
7.62(t,J=8Hz,lH), 7.8(t,J=8Hz,lH), 7.9(d,J=9Hz,lH),
8.15(d,J=9Hz,lH), 8.55(d,J=3Hz,lH), 9.38(d,J~3Hz,lH).
- .
WO91/00725 PCT/US90/03630
~,:
.
~ .
SS
-66-
C,H,N analysis calculated for C29H37N3O2, 0.5 H2O: C
74.32, H 8.39, N 8.97; found: C 73.92, H 8.05, N 8.83.
~L~ ~
N-(2'-MethylDhenylaminocarbonyl)~R-Valine-di-n-
~entylamide
A solution of hydrochloride of example 2 (0.15 g,
0.52 mmol), 2-methyl-phenylisocyanate ~0.1 g) and
triethylamine (0.1 mL) was allowed to react at ambient
temperature. The solvent was removed in vacuo and the
residue dissolved in ethylacetate. Water was added and
the mixture extracted several times with EtOAc. The
combined ethylasetate extracts were washed with brine and
dried over MgS04. The volatiles were removed in vacuo and
the residue purified by chromatography. The oily product
was isolated in 80% yield. [a~D= +1.5 (c=0.4, MeOH).
MS(CI) m/e 390~m+H) . H NMR~CDCl3,300MHz) ~ 0.8-
1.0(m,12H), 1.12-1.41(m,8H), 1.42-1.78~m,4H), 2.01~m,lH),
2.22~s,3H), 3.25~m,lH), 3.35~m,2H), 3.51(m,lH), 4.7~m,lH),
6.5~m,lH), 6.7~s,lH), 7.04~t,J=6Hz,lH), 7.16~m,2H),
7.53~d,J=9Hz,lH). C,H,N analysis calculated for
C23H39N3O2: C 70.91, H 10.09, N 10.79; found: C 70.57, H
9.46, N 10.57.
Example 60
Na-(t-Butyloxycarbor.yl)-N (2'-chlorobenzyloxycarbonyl)-
R-L~sine-di-n-pentylamide
N -~t-Butyloxycarbonyl)-N -~2'-
chlorobenzyloxyCarbonyl)-R-Lysine ~1.0 g, 2.4 mmol) was
stirred at 0 C in 25 mL of CH2Cl2 with BOPCl, ~0.65 g, 2.6
~ ., . . ,, , .. - - . , . . , : .
. . , ~ , , ,, . : , .~ : -
,. , ,: . ~ .; , . - :
.
wo 9l/00725 PCrtVS90/03630
f:- ;
2~ 55
~ . ! ', ' '. .
-67-
mmol), and TEA (0.35 mL, 2.4 mmol). To this reaction
mixture was added di-n-pentylamine (2.5 mL, 12 mmol). The
mixture was stirred overnight and allowed to warm to room
temperature. An additional equivalent of BOPCl was added
after 18 hrs and the reaction stirred an additional day at
ambient temperature. The solvents were evaporated in
vacuo and the residue taken up in ethylacetate and washed
with water, 1 N HCl solutionj saturated NaHCO3, and water.
The organic solution was dried over MgSO4. After
filtration and concentration of the filtrate in vacuo, the
residue was purified by chromatography using ethylacetate-
hexane as the solvent system in the ratio ~1:4). The
product was isolated as an oil in 53~ yield ~0.7 g).
MS~CI) m/e 554~m+H) , 326. 1H NM~CDC13,300MHz) ~
0.g(m,6H), 1.2-1.38~m,12H), 1.42~s,9H), 1.5-1.7~m,4H),
3.02-3.45~m,4H), 3.48~m,4H), 4.5~m,lH), S.01~m,lH),
5.2~s,2H), 5.4~d,J=9Hz,lH), 7.25~m,2H), 7.3-7.45~m,2H).
Example 61
_(2'-Chlorobenzyloxycarbonyl)-R-Lysine-di-n-
pentylamide hydrochloride
The compound was prepared in similar manner to
example 2 via deprotection of the product of example 60,
using 4 N HCl in dioxane. The product was isolated in
quantitative yield. MS(CI) m/e 454(m+H) , free base.
.. . .. . .
. . .
WO91/00725 PCT/US90/U3630
~ '55 -68-
Exam~le ~2
~ -(3'-Ouinalylcarbonyl~-N -(2'-
chlorobenzyloxycarbonyl)-R-Lysine-di-n-pentylamide
The hydrochloride salt of example 61 (0.5 g, 1.02
mmol) was stirred in 15 mL of CH2C12 with NMM (0.29 mL,
2.2 mmol) under N2 at 0C. EDCI (0.25 g, 1.3 mmol) and
HOBt (0.3 g, 2.2 mmol) were added followed by the addition
of quinoline-3-carboxylic acid (0.1 g, 1.1 mmol). The
reaction mixture was stirred overnight and allowed to
slowly warm to ambient temperature. The solvents were
evaporated in vacuo and the residue taken up in
ethylacetate and washed successively with water, saturated
NaHCO~, a saturated solution of citric acid, water and
brine. The organic solution was dried over MgSO4 and then
filtered. Solvents were evaporated in vacuo and the crude
product subjected to flash chromatography using
ethylacetate and hexane as the elutant mixture. The
product was isolated as an oil, 0.46 g (74~). MS(CI) m/e
609(m+H) . H NMR(CDCl3,300MHz) ~ 0.8-0.96(m,6H) 1.16-
1.42(m,12H), 1.45-1.6(m,2H), 1.8-2.0(m,2H), 2.7(m,2H),
3.07-3.45(m,4H), 3.5-3.65(m,2H), 5.15(m,3H),
6.85(d,J=12Hz,lH), 7.2(d,J=9Hz,2H), 7.4(d,J=9Hz,2H),
7.6(m,2H), 7.8(t,J=7Hz,lH), 7.9(t,J=7Hz,lH),
8.15(d,J=9Hz,lH), 8.6(s,1H), 9.35(d,J=3:-.7,1H). C,H,N
analysis calculated for C34H45ClN404, 0.6 H2O: C 65.86, H
7.41, N 9.04; found: C 65.63, H 7.29, N 9.92.
... .... ~ . . ,
-. . .: . - , . . .
. . , ~, . . .
.~ . . .
WO 9l/00725 PCT/US90/03630
!
S~
-69-
~1m~l~ 63
N-(3'-Ouinolylcarbonyl)-3-(2'-Naph~hyl)-R-Alanine-
di-n-dipentylamide
The reaction was performed in a similar manner to
that in example 3 utilizing 75 mg of hydrochloride salt of
example 49, quinoline-3-carboxylic acid (34 mg), EDCI (q0
mg), HOBt (50 mg), and NMM (22 ~L). The oily product was
isolated in 31% yield, (32 mg). MS(CI) m/e 510(m+H) .
lH NMR(CDC13,300MHz) ~ 0.85(m,6H), 1.06-1.35(m,12H),
2.85(m,lH), 3.0(m,2H), 3.35(m,2H), 3.55(m,lH),
5.45(apparent q,J=7Hz,lH), 7.32-7.5(m,4H),
7.62(t,J=6Hz,lH), 7.68-7.82(m,5H), 7.88(d,J=7Hz,lH),
8.15(d,J=7z,lH), 8.52~d,J=2Hz,lH).
E~ample 64
R-~4'-Hydroxyphenyl)-glycine-di-n-pe~ L~mide
hydrochloride
The compound was prepared in similar manner to
example 2 via deprotection of the product of example 55,
using 4 N HCl in dioxane. The oily product was isolated
in 90% yield. []D= -87.0 (c=0.2, MeOH). MS(CI) m/e
307(m+H) H NMR(DMSOd6,300MHz) ~ 0.82(m,6H), 1.02-
1.2(m,8H), 1.3-l.S(m,4H), 3.05-3.3(m,2H), 3.32-3.4(m,2H),
5.22(bs,lH), 6.83(d,J=9Hz,2H), 7.25(d,J=9Hz,2H),
8.4(bs,3H).
"' ' ;. .
-, . .
WO91/0072~ PCT/US90/03630
~'
Z~?si~755
-70-
Fxample 65
N-~3 -Ouinolylcarbonyl)-R-(4 -hydroxyphenyl~alycine-
di-n-pentylamide
The reaction was performed in a similar manner to
that in example 3 utilizing (300 mg, 2.6 mmol) of
hydrochloride salt of example 64, quinoline-3-carboxylic
acid ~450 mg), EDCI (550 mg), HOBt (380 mg), and NMM (0.62
mL). Product was isolated in 53% yield (0.78 g). mp= 79-
80 C. (a]D= -99.6 (c=1.0, MeOH). MS(CI) m/e 462(m+H) .
lH NMR(CDC13,300MHz) ~ 0.85(t,J=7Hz,6H), 1.1-1.3(m,10H),
1.4-1.5(m,2H), 3.1-3.2(m,2H), 3.25-3.5(m,2H),
5 9(d,J=9Hz,lH), 6.6(d,J=9Hz,2H), 7.25(d,J=9Hz,2H),
7 7(t,J=7Hz,lH), 7.85(t,J-7Hz,lH), 8.08(d,J=9Hz,2H),
3.9(d,J=3Hz,lH), 9.1(d,J=6Hz,lH), 9.25(d,J=3Hz,lH),
9.53(s,1H). C,H,N analysis calculated ~or C28H35N3O3: C
72.85, H 7.64, N 9.10; found: C 72.65, H 7.65, N 9.08.
Example 66
N -~3 -Ouinolylcarbonyl~-N -(acetyl)-R-~ysine-di-n-
pentylamide
The reaction was performed in a si~ilar manner to
that in example 33 utilizing 60 mg of t:~e product of
example 54 and pyridine with acetic anhydride. The oily
product was purified by standard chroma=ography and
isolated in 33% yield (22 mg). [a]D= -1.3 (c=0.5, MeOH).
MS(CI) m/e 483(m~H) . H NMR (CDC13,30~MHz) ~ 0.92(m,6H),
1.23-1.4(m,8H), 1.45-1.7(m,8H), 1.8~m,2:-), 1.98~s,3H),
3.1~m,lH), 3.25(m,2H), 3.32(m,lH), 3.6~.~,2H), 5.15(m,lH),
5.85(bs,lH), 7.5(d,J=8Hz,lH), 7.65(t,J=-.Hz,lH),
.
~, .. . . . . . . . .
-, ., ~ . - : -
' - ~
.
,
.. .. .
WO9l/00725 PCT/US90/03630
z~'?7S5
7.82~t,J=6Hz,lH), 7.94~d,J=8Hz,lH), 8.18~d,J=8Hz,lH),
8.62~d,J=2Hz,lH), 9.36~d,2Hz,lH).
~
N-(5'-Hydroxyindolyl-2'-carbonyl)-R-Valine-di-n-
pentylamide
The 5-hydroxyindole-2-carboxylic acid ~95 mg),
hydrochloride of example 2 (150 mg), NMM (0.12 mL), HOBt
(70 mg), and EDCI (105 mg) reacted under similar
conditions to those described in example 3. The product
was isolated in 74~ yield. MS(CI) m/e 916(m+H) . 1H
NMR~CDCl3,300MHz) ~ 0.9~m,6H), 1.0(apparent q,J=7Hz,6H),
1.32~m,8H), 1.62(m,4H), 2.11(m,lH), 3.15(m,lH), 3.2(m,lH),
3.43~m,lH), 3.62~m,lH), 4.95~m, lH), 5 . 6 ~ s, lH), 6 . 78~m,lH),
6.88~dd,J=2,9Hz,lH), 6.98~d,J=9Hz,lH), 7.02~d,J=2Hz,lH),
7.25~d,J-9Hz,lH), 9.3~s,lH).
Example 68
N-(4'-Chlorobenzenesulfonyl)-~-Valine-di-n-
pentylamide
The hydrochloride of example 2 (60 mg, 0.22 mmol),
NMM (25 ~L), was dissolved in 10 mL of CH2C12 and 4-
chlorophenylsulfonyl chloride (46 mg) was added to this
reaction mixture and stirred overnight (warming to ambient
temperature). The solvent was evaporated in vacuo and
ethylacetate and water both in large ~xcess were added to
the residue. The organic extrac~s were successively
washed with saturated aqueous NaHCO3, 0.1 HC1 solution and
brine. The combined extracts were dried over MgSO9,
.. ~ :.' ' ' ':,
WO91/00725 PCT/US90/03630
Zl?~75~
-72-
filtered and concentrated. The product was purified by
chromatography using ethylacetate and hexane as elutants.
The pure product was isolated in 75% yield (59 mg). mp=
89-90 C. [~]D= -61.8 (c=0.5, MeOH). MS(CI) m/e
431(m+H) . H NMR(CDC13,300MHz): ~ 0.9(m,12H),
1.15(m,8H), 1.3~m,4H), 1.85(m,lH), 2.9(m,2H), 3.02(m,lH),
3.22(m,lH), 3.8(m,lH), 5.75(d,J=9Hz,lH), 7.43(m,2H),
7.75(m,2H). C,H,N analysis calculated for C21H35ClN2O3S:
C 58.52, H 8.18, N 6.50; found: C 58.56, H 8.22, N 6.48.
Exam~le 69
4-Chlorocinnamic acid N-hydroxysuccinimide ester
To a solution of 4-chlorocinnamic acid (0.8g, 4.38
mmol) in CH2C12 was added N-hydroxysuccinimide (0.55 g,
4.8 mmol) and EDCI and the reaction mixture was stirred at
ambient temperature overnight. The solvents were removed
in vacuo and the residue dissolved in ethylacetate and
water. Combined EtOAc extracts were dried over MgSO4 and
the solution concentrated in vacuo. The residue was
crystallized from a mixture of ethylace-ate and hexane.
The product was isolated in 72% yield (0.88g). mp= 192-
193 C. MS(CI) m/e 297(m+NH4 ). H NMR(DMSOd6,300MHz)
2.87(s,4H), 7.05(d,J=17Hz,lH), 7.56(d,J=9Hz,2H),
7.92(d,J=9Hz,2H), 7.99(d,J=17Hz,lH).
~.,.. ,......... ~: :
. .
.~
. . ' ' .
~' , . ' '
WO 91/00725 PCT~US90/03630
. .
2(?~55.
. :., .
Exam~le 70
-(3'-Oui~lylcarbonyJIL-N-rE-3'-(4''-chlorophenyl)prop-
2'-enoyll-R-Lysine-di-n-pentylamide
To a solution of example 54 (60 mg, 0.14 mmol) in
dimethylformamide (8 mL) cooled to 0C were added NMM (35
~L) and the active ester of example 69 (40 mg,0.14 mmol).
The mixture was stirred overnight with warming to ambient
temperature. The DMF was removed in vacuo and the residue
was chromatographed on silica using ethylacetate-hexane as
the elutant mixture. The oily product was isolated in 40%
yield (35 mg). MS(CI) m/e 605(m+H) . H NMR(CDCl3,300MHz)
~ 0.92(m,~H), 1.3(m,8H), 1.62(m,8H), 1.83(m,2H),
3.14~m,lH), 3.35(m,4H), 3.58(m,lH), 5.15(m,lH),
6.18~m,lH), 6.35(d,J-17Hz,lH), 7.25~m,6H),
7.48(d,~=17Hz,lH), 7.62(t,J=8Hz,lH), 7.83(t,J=8Hz,lH),
8.15(d,J=9Hz,lH), 8.62(d,J=2Hz,lH), 9.37(d,J=2Hz,lH).
Example 71
N-(t-Butyloxycarbonyl)-R-Tyrosine-di-n-pentylami~
N-t-Butyloxycarbonyl-R-Tyrosine (4.5 g, 15.4 mmol)
was stirred with BOPCl (3.92 g, 15.4 mmol) and
dipentylamine (7.9 mL, 39 mmol) in 100 mL of
tetrahydrofuran (THF) at 4C and allowed to warm to room
temperature overnight. After one day, additional BOPCl
(800 mg) was added and, after two days, the volatiles were
evaporated. The residue, dissolved in EtOAc, was
extracted with 0.1 M citric acid solution, 0.1 M sodium
carbonate (Na2CO3) solution, and water; then dried over
magnesium sulfate (MgSO4), filtered and concentrated in
,: . . .
:; ~ ' ' . , : '
,: , ' . . . .
: . . -
WO91/00725 PCT/US90103630
~.;
755
-74-
vacuo to yield an oil, 5.67 g, 13.4 mmol ~87.4%). Rf=
0.45 (2:1 hexanes-EtOAc). la]D = +2.8 (c=0.76, MeOH).
MS(CI) m/e 421(m+H) . lH NMR(CDC13,300MHz) ~
0.88(apparent q,J=7Hz,6H), 1.15-1.32(m,10H), 1.36-
1.47(m,11H), 2.80-3.07(m,5H), 3.38-3.48(m,1H),
4.72(apparent q,J=6Hz,lH), 5.41(d,J=8Hz,lH),
6.70(d,J=8Hz,2H), 7.02(d,J=8Hz,2H).
Example 72
R-Tyrosine-di-n-pentylamide hydrochloride
The product of example 71 (2.0 g, 4.75 mmol) was
dissolved in 4 N HCl in dioxane (20 mL, 80 mmol) that was
precooled to 4C. After 3 hours, the excess reagent was
evaporated and the oily residue was placed under high
vacuum overnight to yield a glass, 1.5 g, 4.2 mmol (87%).
[a~D = -42.8 (c=1.2, MeOH). MS(CI) m/e 321(m+H) . lH
NMR(DMSOd6,300MHz) 8 0.82-0.89(m,6H), 1.1-1.4(m,12H),
2.70-3.04(m,5H), 3.37-3.50(m,lH), 4.22(dd,J=5,7Hz,lH),
6.70(d,J=8Hz,2H), 6.99~d,J=8Hz,2H), 8.37(bs,3H),
9.48(s,lH).
E~m~L~ 73
N~O-Di-(3'-Ouinolylcarbonyl)-R-Tyrosine-di-n-pentylamide
The product of example 72 (357 mg, 1 mmol),
quinoline-3-carboxylic acid (173 mg, 1 mmol), HOBt (13 mg,
0.1 mmol), and TEA (279 ~L, 2 mmol) were dissolved in 10
mL methylene chloride and EDCI (191 mg, 1 mmol) was then
added in one portion. After 3 days, the volatiles were
evaporated and the residue, in EtOAc, was e:~tracted as in
example 71. The residue was then purifie~ by
. ~, . . . .
:'~ : ; ', ' '' ',, : ' ,
, " : ~
: . -
WO91/00725 PCT/US90/03630
/
-75-
chromatography on silica gel eluted with 1% ethanol in
chloroform to provide first the mono-acylated material (19
mg, see example 80) followed by an oily product, (108 mg,
0.17 mmol, 17% yield). Rf= 0.36 (18:1 chloroform-
ethanol). []D = +5.8 (c=0.5, CHC13). [a]D = +53.2
(c=0.73, MeOH). MS(CI) m/e 631(m+H) , 518, 458, 446, 368.
lH NMR(CDCl3,300MHz) ~ 0.88-0.94(m,6H), 1.22-1.91(m,10H),
1.50-1.59(m,2H), 2.96-3.30(m,5H), 3.52-3.62(m,lH), 5.33-
5.92(m,lH), 7.22(d,J=8Hz,lH), 7.30(d,J=8Hz,lH),
7.37(d,J=8Hz,2H), 7.63(dt,J=1,7Hz,lH),
7.68(dt,J=1,7Hz,lH), 7.79-7.93(m,3H), 8.0(dd,J=1,8Hz,lH),
8.16(d,J=3Hz,lH), 8.22(d,J=8Hz,lH), 8.56~d,J=2Hz,lH),
9.02(d,J=2Hz,lH), 9.32(d,J=2Hz,lH), 9.54(d,J=2Hz,lH).
C,H,N analysis calculated for C39H42N4O4, H2O: C 72-20, H
6.84, N 8.64; found: C 72.38, H 6.62, N 8.50.
Exam~le 74
N-(2'-In~olyl~carbonyl)-R-Tyrosine-d~-n-pentylamide
The product of example 72 (200 mg, 0.56 mmol),
indole-2-carboxylic acid (97 mg, 0.6 mmol) and TEA (84 ~L,
0.6 mmol) were dissolved in 5 mL methylene chloride and
treated with EDCI (115 mg, 0.6 mmol) at room temperature.
After 3 days, the solvent was evaporated and the residue
was extracted as in example 71. Column chromatography on
silica gel eluted with 1% ethanol in methylene chloride
provided product. Rf= 0.38 (18:1 methylene chloride-
ethanol). mp= 124-7 C.l [~]D = +21.4 (c=1.17, MeOH).
MS(CI) m/e 964(m+H) . H NMR(CDCl3,300MHz) ~
0.88(apparent q,J=8Hz,6H), 1.15-1.56(m,12H), 2.46-
3.22~m,5H~, 3.48-3.54(m,lH), 5.23-5.32(m,lH), 6.12(s,lH),
: :,
-
.
WO 91tO0725 PCTtUS90/03630
SS
-76-
6.70(d,J=8Hz,2H), 6.95(d,J=lHz,lH), 7.05(d,J=8Hz,2H),
7.13(dt,J=1,7Hz,lH), 7.18(d,J=8Hz,lH),
7.27(dt,J=1,7Hz,lH), 7.90(d,J=8Hz,lH), 7.64(d,J=8Hz,lH),
9~22(s,lH). C,H,N analysis calculated for C28H37N3O3: C
72.54, H 8.05, N 9.06; found: C 72.37, H 8.10, N 8.80.
Example 75
N-(3',4'-Dichlorobenzoyl)-R-Tyrosine-di-n-pentylamide
The product of example 72 (103 mg, 0.29 mmol) was
dissolved in 5 mL methylene chloride and treated with 3,4-
dichlorobenzoylchloride (126 mg, 0.6 mmol) and TEA (84 ~L,
0.6 mmol) at room temperature. After 2 hours, additional
acid chloride (13 mg) and TEA (8 ~L) were added and the
reaction was stirred overnight. The volatiles were
evaporated and the residue, in EtOAc, was extracted with
0.1% citric acid, H2O; then dried over MgSO4, filtered and
concentrated in vacuo. The resulting diacylated product
residue was dissolved in 10 mL of 1:1 THF-methanol and
treated with 1 N NaOH (290 mL, 0.29 mmol). After 1 hour,
tlc revealed complete reaction and the solvent was
evaporated in vacuo. The residue was dissolved in EtOAc
and acidified with 0.1 M citric acid. The EtOAc layer was
then washed until neutral, dried over MgSO4, filtered and
concentrated in vacuo. The residue was warmed with 80%
aqueous ethanol and cooled overnight to provide a solid,
64 mg, 0.13 mmol (45% yield). mp= 148-52 C. [a]D =
+15.6 (c=1.0, MeOH). MS(CI) m/e 493(m+H) . H
NMRtCDCl3,300MHZ) ~ 0.88-0.92~m,6H), 1.2-1.6(m,12H), 2.93-
3.22(m,5H), 3.50-3.60(m,lH), 5.21-5.28(m,lH), 6.29(s,lH),
6.68(d,J=8Hz,2H), 7.02(d,J=8Hz,2H), 7.15(d,J=8Hz,lH),
- .
- , . .
'' ~ .,
- -, .
W091/00725 PCT~US90/03630
2~ 55
-77-
7.47(d,J=8Hz,lH), 7.59(dd,J=2,8Hz,lH), 7.91(d,J=2Hz,lH).
C,H,N analysis calculated ~or C26H34Cl2N2O3: C 63.28, H
6. 94, N 5.68; found: C 63.39, H 7.00, N 5.54.
Example 76
N-(2'-Naphthoyl)-R-Tyrosine-di-n-pentylamide
The product of example 72 (100 mg, 0.28 mmol) was
acylated with 2-naphthoic acid (52 mg, 0.30 mmol) in the
presence of TEA (39 ~L, 0.28 mmol) and EDCI ~57 mg, 0.30
mmol) in 5 mL methylene chloride. The reaction and
extractive workup were performed as in example 71 to yield
120 mg, 0.25 mmol (89%). mp= 128-133C. [a]D = +11.8
(c=0.68, MeOH). MS(CI) m/e 975(m+H) , 303, 290. H
NMR(CD30D,300MHz) ~ 0.88-0.93(m,6H), 1.19-1.38(m,9H),
1.44-1 62(m,3H), 2.99(dd,J=7,13Hz,lH), 3.08-3.29(m,4H),
3.37-3.47(m,lH), 5.22(dd,J=7,9Hz,lH), 6.72(d,J=8Hz,2H),
7.13(d,J=8Hz,2H), 7.53-7.62(m,2H), 7.84(dd,J=2,9Hz,lH),
7.90-7.99(m,3H), 8.37(s,lH). C,H,N analysis calculated
for C30H38N2O3: C 75.91, H 8.07, N 5.90; ound: C 75.57,
H 7.97, N 5.83.
Example N-t-Butyloxycarbonyl-(O-benzyl)-R-Tyrosine-di-n-
pentylamide
N-t-Butyloxycarbonyl-(O-benzyl)-R-Tyrosine (3.71 g,
10 mmol) was stirred with di-n-pentylamine (5.1 mL, 25
mmol), HOBt (1.4 g, 10 mmol) and TEA (1.4 mL, 10 mmol) in
150 mL methylene chloride at 4C and then BOPCl (2.6 g, 10
mmol) was added. The reaction was allowed to reach room
temperature overnight. After one day, additional BOPCl
,~
~: : `: . : - -
WO91~00725 PCT/US90/03630
_~,
1' ~` .
;~?~ S5
-78-
(260 mg) and TEA (140 ~L) were added. After 2 days, the
volatiles were evaporated and the residue (in EtOAc) was
extracted with 0.1 M H3PO4, 0.1 M Na2CO3, H2O; then dried
over MgSO4, filtered and concentrated in vacuo. The
residue was chromatographed on silica gel eluted with 2:1
hexanes-EtOAc to yield an oil, 1.3 g, 2.55 mmol (25%).
[a]D = +5.8 (c=1.5, MeOH). MS(CI) m/e 511(m+H) , 456,
393. H NMR~CDC13,300MHz) ~ 0.84-0.93(m,6H), 1.1-
1.35(m,12H), 1.41(s,9H), 2.81-3.04(m,5H), 3.36-3.46(m,1H),
4.15-4.23(m,1H), 5.03(s,2H), 5.32(d,J=8Hz,lH),
6.87(d,J=8Hz,2H), 7.11(d,J=8Hz,2H), 7.32-7.43(m,5H).
~xam~ 78
(O-Benzyl)-R-Tyr~sine-di-n-pentylamide hydrochloride
The product of example 77 (1.3 g, 2.55 mmol) was
treated with 5 mL of 4 N HCl in dioxane, precooled to 4C.
The reaction mixture was then allowed to reach room
temperature. After 1 hour tlc revealed complete reaction
and the excess reagent was evaporate. The residue was
placed under high vacuum overnight to yield an oil, 1.2 g.
Rf= 0.59 (80:20:1 chloroform-methanol-ammonium hydroxide).
[~]D = -32.5 (c=2.2, MeOH). MS(CI) m/e 411(m+H) . H
NMR(DMSOd6,300MHz) ~ 0.85(apparent q,J=7Hz,6H), 1.07-
1.38(m,12H), 2.68-2.97(m,4H), 3.05(dd,J=5,13Hz,lH), 3.32-
3.92(m,2H), 4.27(dd,J=5,8Hz,lH), 5.09(s,2H),
6.93(d,J=8Hz,2H), 7.12(d,J=8Hz,2H), 7.32-7.43(m,5H),
8.37~s,3H).
... . .
WO91/00725 PCT/US90/0363~
,
21?~755
-79-
E~m~l~ 79-(3'-Ouinolylcarbonyl;-(O-benzyl)-R-Tyrosine-di-n-
pentylamide
EDCI (290 mg, 1.5 mmol) was added to a cooled (4C)
solution of quinoline-3-carboxylic acid (260 mg, 1.5
mmol), the product of example 78 (650 mg, 1.35 mmol), and
TEA (418 ~L, 3.0 mmol) in 5 mL methylene chloride. The
stirred reaction mixture was allowed to warm to room
temperature overnight. After evaporation of the
volatiles, the residue was dissolved in EtOAc and
extracted with 0.1 M H3PO9 (3x), 0.1 M Na2CO3 (3x), brine
(3x); then dried over MgSO4, filtered and concentrated in
vacuo to yield an oil, 650 mg, 1 15 mmol ~85%). Rf- 0.77
~18:1 chloroform-ethanol), 0.40 ~1:1 hexanes-EtOAc). [~]D
~0.21 ~c=0.47, CHC13). MS~FAB) m/e 566(m+H) , 393,
381. H NMR(CDC13,300MHz) ~ 0.91(apparent q,J=7Hz,6H),
1.17-1.38(m,10H), 1.43-1.6(m,2H), 2.86-3.17(m,SH), 3.49-
3.59(m,lH), 5.03(s,2H), 5.26-5.33~m,lH), 6.90(d,J-8Hz,2H),
7.16~d,J=8Hz,2H), 7.28-7.43(m,6H), 7.62(dt,J=1,7Hz,lH),
7.82(dt,J=1,8Hz,lH), 7.90(d,J=8Hz,lH), 8.18(d,J=8Hz,lH),
8.54(d,J=2Hz,lH), 9.32(d,J=2Hz,lH). C,H,N analysis
calculated for C36H43N3O3: C 76.55, H 7.88, N 7.29;
found: C 76.43, H 7.66, N 7.43.
Example 80
N-(3~-ouinolylcarbonyl)-p-T~rosin~-c~-n-~entylamide
The product of example 79 (614 mg, .09 mmol) was
dissolved in 30 mL methanol and treated ~Jith 10% Pd/C (200
mg, pre-wetted with solvent under nitroa~n) under 1
: : , - ~ 1, ' . . ..
: . ~
: . - , , . :
:, , .' ' ~ :,. ~ ... . .
,. . . ... .. . . .. . . . .. .. . . . .. .
WO91/00725 PCT/US90/03630
755
-80-
atmosphere hydrogen gas. Another 200 mg of catalyst was
added after 4 hours and the reaction mixture was stirred
overnight. The mixture was then filtered and the filtrate
concentrated in vacuo. Silica gel column chromatography
of the residue (eluted with a 2:1 to 1:1 hexane-EtOAc step
gradient) provided 270 mg, 0.57 mmol (52% yield). mp=
135-37 C. [~]D = +12.6 (c=0.5, MeOH). MS(CI) m/e
476(m+H) , 347, 321, 291. H NMR(CDC13,300MHz) ~
0.91(t,J-7Hz,6H), 1.29-1.38(m,8H), 1.48-1.62(m,9H), 3.0-
3.28(m,5H), 3.51-3.61(m,lH), 5.30-5.38(m,lH),
6.72(d,J=8Hz,2H), 6.78(s,lH), 7.06(d,J=8Hz,2H),
7.38(d,Jz8Hz,lH), 7.60(t,J=7Hz,lH), 7 80(dt,J=1,7Hz,lH),
7.88(d,J=8Hz,lH), 8 15~d,J-9Hz,lH), 8.58(d,J=2Hz,lH),
9.27(d,J=2Hz,lH). C,H,N analysis calculated for
C29H37N3O3: C 73.23, H 7.84, N 8.83; found: C 73.23, H
7.89, N 8.76.
Example ~1
N-(3'-Ouinolylcarbonyl)-(O-bisulfat~l)-R-Tyrosine
di-n-pentylamide ammonium salt
The product of example 80 (59 mg, 0.12 mmol) was
dissolved in 2 mL DMF and treated with rreshly prepared
pyridine-sulfur trioxide complex (H.C.Reitz et al J. Amer.
Chem. ~QÇ~ 68, 1031-5, 1946) overnight at room
temperature. The pyridine was evaporated in vacuo and the
DMF solution was poured into water and t:~e pH adjusted to
7 with 1 N NaOH. The homogeneous solution was then frozen
and lyophilized. Preparative C-18 chrom~tography of the
residue eluted with a gradient from 100~ aqueous buffer
(0.05 M ammonium acetate, pH 6.2) to 50~
.
W09t/00725 PCT/US90/03630
I
2~ SS
.', , ~ "5
-81-
acetonitrile/aqueous buffer over 10 minutes provided
product fractions which were pooled, frozen and
lyophilized to yield 48 mg, 0.08 mmol (67%). mp= 113-6C.
[a]D = +12.2 ~c=0.88, MeOH). MS(FAB) m/e 559~m-H) , 368,
302, 298. H NMR(D20,300MHz) ~ 0.68-0.75(m,6H), 0.98-
1.43(m,12H), 2.98-3.28(m,6H), 5.22(t,J=7Hz,lH),
7.24~d,J=8Hz,2H), 7.30(d,J=8Hz,2H), 7.44(t,J=8Hz,lH),
7.62(d,J=8Hz,lH), 7.69(t,J=8Hz,lH), 7.82(d,J=8Hz,lH),
8.36(s,1H), 8.78(s,lH). C,H,N analysis calculated for
C29H40N4O6S, 0.50 H2O: C 59.88, H 7.10, N 9.63; found:
C 59.77, H 6.82, N 9.11.
~xample
~L
3~5-Di-iodo-N-(3'-quinolylcarbonyl)-R-Tvr-di-n-pentylamide
(b)
3-Iodo-N-(3'-quinolylcarbonyl)-R-T~r-di-n-pentylamide
Iodine (27 mg, 0.11 mmol) was mixed with morpholine
(40 ~L, 0.46 mmol) in 5 mL methanol and added to the
product of example 80 (50 mg, 0.11 mmol) in 15 mL methanol
at room temperature. The reaction was stirred until tlc
indicated complete reaction. After evaporation of the
solvent, chromatography of the residue on silica gel
eluted with a step gradient of chloroform to 1~ ethanol in
chloroform provided first the diiodo product followed by
the monoiodo compound. Diiodo product (a): [a~ D = +18
(c=0.11, MeOH). MS~CI) m/e 728(m+H) , 602. H
NMR(CDCl3,300MHz) ~ 0.92(apparent q,J=7Hz,6H), 1.2-
1.45(m,12H), 2.92-3.13~m,SH), 3.53-3.67(m,lH), 5.22-
.
:: .: -: ,
.. . . .. . .. ....
.
WO91/00725 PCT/US90~3630
~ ....
,
Z~
-82-
5.28(m,1H), 5.72(s,1H), 7.27(d,J=7Hz,lH), 7.56(s,2H),
7.63(dt,J=1,8Hz,lH), 7.83(dt,J=1,8Hz,lH),
7.93(d,J=8Hz,lH), 8.18(d,J=8Hz,lH), 8.55(d,J=2Hz,lH),
9.33(d,J=2Hz,lH). C,H,N analysis calculated for
C29H35I2N3O3, 0.4 EtOAc: C 48.19, H 5.05, N 5.51; found:
C 48.43, H 5.03, N 5.79. Monoiodo product (b): mp= 75-
85 C. MS(CI) m/e 602(m~H) . H NMR(CDCl3,500MHz)
0.84(apparent q,J=7Hz,6H), 1.13-1.35(m,9H), 1.37-
l.S3(m,3H), 2.90-2.98(m,3H), 3.02-3.08(m,2H), 3.48-
3.5S(m,lH), 5.18-S.23(m,lH), 6.83(d,J=8Hz,lH),
7.05(dd,J=1,8Hz,lH), 7.22(d,J=8Hz,lH), 7.46(d,J=2Hz,lH),
7.57(dt,J=1,8Hz,lH), 7.76(dt,J=1,8Hz,lH),
7 84(d,J=8Hz,lH), 8.10~d,J=8Hz,lH), 8.98~d,J=2Hz,lH),
9.24~d,J~2Hz,lH). C,H,N analysis calculated for
C29H36IN3O3, 1.5 H20: C 55.42, H 6.25, N 6.69; found: C
55.19, H 5.95, N ~.17.
Example 83
N-(3'-Ouinolylcarbonyl)-(O-methyll-R-T~roslne-di-n-
pentylamide
The product of example 80 (25 mg, 0.053 mmol) was
dissolved in 1 mL acetone and K2CO3 ~8 mg, 0.058 mmol) and
methyl iodide (5 ~L, 0.08 mmol) were added. After 3 hours
at reflux, additional methyl iodide (5 mL) and acetone (2
mL) were added. After 2 days, the volatiles were
evaporated and the residue, in EtOAc, was extracted with
0.1% aqueous citric acid, water; then dried over MgSO4,
filtered and concentrated in vacuo. MS~CI) m/e 490~m+H) ,
476, 361, 347, 317. H NMR~CDC13,300MHz) ~ 0.86-
0.93(m,6H), 1.2-1.56~m,12H), 2.42-3.15(m,5H), 3.49-
, .. .. .
,
- : . ~: . . .
.. - . . - :
WO91/00725 PCT/USgO/03~30
;~'5~7S5
.. V
-83-
3.59(m,1H), 3.78(s,3H), 5.27-5.39(m,1H), 6.77(d,J=8Hz,lH),
6.82(d,J=8Hz,lH), 7.08(d,J=8Hz,lH), 7.16(d,J=8Hz,lH),
7.41-7.46(m,1H), 7.56-7.63(m,1H), 7.76-7.82(m,1H), 7.83-
7.88(m,lH), 8.14(d,J=8Hz,lH), 8.53(d,J=2Y.z,lH),
9.29(t,J=2Hz,lH).
Example 8Methyl N-t-Butyloxycarbonyl-(O-benzyl)-R-Tyrosyl-S-
phenylglycinate
N-t-Butyloxycarbonyl-(O-benzyl)-R-Tyrosine (1.0 g,
2.7 mmol), methyl S-phenylglycinate hydrochloride (540 mg,
2.7 mmol), HOBt (362 mg, 2.7 mmol) and TEA (374 ~L, 2.7
mmol) were dissolved in 20 mL THF and treated with BOPCl
(682 mg, 2.7 mmol). The reaction was followed by tlc
(18:1 chloroform-ethanol) and additional BOPCl ~200 mg)
and TEA (374 ~L) were added after 1,2 and 4 days.
Methylene chloride (20 mL) also was added after 2 days.
After 1 week, the volatiles were evaporated in vacuo and
the residue, in EtOAc, was extracted as in example 71.
Chromatography of the residue on silica gel eluted with a
step gradient from 9:1 to 2:1 hexanes-EtOAc yielded 485
mg, 1.13 mmol (42%). mp= 138-39C. [a]D = +48.7 (c=1.0,
MeOH). MS(CI) m/e 519(m+H) , 463, 419. lH
NMR~CDC13,300MHz) ~ 1.41(s,9H), 2.92-3.04~m,2H),
3.71(s,3H), 4.35~bs,lH), 5.01(s,3H), 5.43-5.46(m,lH),
6.78(d,J=7Hz,lH), 6.82(d,J=8Hz,2H), 7.02(d,J=8Hz,2H),
7.19-7.23(m,lH), 7.30-7.45(m,10H).
:
- .: - ~ .. .~.. ..
.~
W091~00725 PCT/US9OJ03630
~,
;~(?~i~755
-84-
ExamDle ~ethyl (O-Benzyl)-R-Tyrosyl-S-phenylglycinate
hydrochloride
The product of example 84 (450 mg, 1.05 mmol) was
dissolved in 4 N HCl in dioxane (5 mL, 20 mmol) precooled
to 4C. After 1 hour, the excess reagent was evaporated
in vacuo and the product used directly in the next step.
mp= 153-6C. [a]D = +43-7 (c=0.76, MeOH). MS(FAB) m/e
419~m+H) , 403, 226. H NMR(DMSOd6,300MHZ) ~ 2-86-
3.00(m,2H), 3.67(s,3H), 4.13(bt,J=5Hz,lH), 5.03(s,2H),
5.45(d,J=7Hz,lH), 6.88(d,J=8Hz,2H), 7.05(d,J=8Hz,2H),
7.22-7.25(m,2H), 7.33-7.46(m,8H), 8.28~s,3H),
9.35(d,J=7Hz,lH).
Example 86
Methyl N-t3'-Ouinolvl~arbonyl~-(O-benzyl)R-
Tyrosyl-S-phenylalycinate
Quinoline-3-carboxylic acid (182 mg, 1.05 mmol), TEA
(146 ~L, 1.05 mmol) and the product of example 85 (1.05
mmol) were dissolved in 20 mL methylene chloride and EDCI
(201 mg, 1.05 mmol) was added at ambient temperature.
After 4 days, the volatiles were evaporated and the
residue was extracted as in example 71. The solvents were
evaporated in vacuo to provide 407 mg, 0.71 mmol (68%
yield). mp= 153-8 C. [a]D = +73.0 (c=1.2, CHC13-
MeOH/l:l). MS(FAB) m/e 574(m+H) , 419, 381. H
NMR(CDC13,300MHz) ~ 3.06(dd,J=8,14Hz,lH),
3.20(dd,J-5,14Hz,lH), 3.70(s,3H), 4.94-5.02(m,3H),
5.53(d,J=7Hz,lH), 6.78(d,J=8Hz,2H), 6.83(d,J=7Hz,lH),
-' -: :
: .. .': '. : : ~ :
W O 91/00725 PC~rtUS90/03630.`
2(?5i~55 ~;
-85-
7.01(d,J=8Hz,2~), 7.14~d,J=7Hz,lH), 7.20-7.23(m,2H), 7.33-
7.36~m,4H), 7.39-7.49(m,4H), 7.62(dt,J=1,7Hz,lH),
7.82(dt,J=1,7Hz,lH), 7.88(d,J=8Hz,lH), 8.15(d,J=8Hz,lH),
8.54(d,J=2Hz,lH), 9.28(d,J=2Hz,lH). C,H,N analysis
calculated for C35H3lN3Os~ 0-5 H2O C 72.15~ H 5-54~ N
7.21; found: C 72.05, H 5.63, N 6.88.
Exa~L~ 87
Methyl N-(3'-OuinolylcarbQnyl)-R-Tyrosyl-S-phenylglycinate
The product of example 86 (200 mg, 0.35 mmol) was
dissolved in 10 mL methylene chloride and treated with
trimethylsilyliodide (TMSI, 198 ~L, 1.39 mmol) at room
temperature. Additional TMSI (198 ~L) was added after 1
day After 3 days, the reaction was quenched with
methanol for 5 minutes and then poured into 0.1 M citric
acid and extracted with ethylacetate (3x). The combined
ethylacetate solution was washed with water; then dried
over MgSO4, filtered and concentrated in vacuo. The crude
solid was purified by chromatography on silica gel eluted
with a s~ep gradient of 1 to 5% ethanol in methylene
chloride and then crystallized from EtOAc and hexane to
yield 51 mg (30%). mp= 238-40 C. [a~D = +72.6 (c=0.23,
MeOH). MS(CI) m/e 484(m+H) , 319. H NMR(CDCl3-
CD3OD,300MXz) ~ 3.0-3.16~m,2H), 3.72(s,3H), 4.92-
5.01(m,lH), 5.50(d,J=7Hz,lH), 6.67(d,J=8Hz,2H),
6.99(d,J=8Hz,2H), 7.21-7.24(m,2H), 7.35-7.38(m,3H),
7.40(s,1H), 7.68(dt,J=1,7Hz,lH), 7.86(dt,J=1,7Hz,lH),
7.98(d,J=8Hz,lH), 8.12(d,J=8Hz,lH), 8.14(d,J=6Hz,lH),
8.22~d,J=8Hz,lH), 8.68(d,J=2Hz,lH), 9.21(d,J=2Hz,lH).
:. : ..
, , , , -
.: . . . . .
.- , . - . . ~ ., .
: . . .: : .. . , ~ ~ ,
~ : . , . . ~ , ~ , . , . -
W091/0072~ PCT/US90/03630
2~ ' . 55
-86-
C,H,N analysis calculated for C28H25N3O5: C 69.55, H 5.21,
N 8.69; found: C 69.20, H 5.29, N 8.60.
.Example 88
N'-Benzyloxycarbonyl-(2 R)-aminobutyrolactone
N-Benzyloxycarbonyl-R-methionine (283 mg, 1.0 mmol)
and a-iodo acetamide (555 mg, 3.0 mmol) were dissolved in
6 mL of 50% aqueous ethanol and warmed to 4C for 4 days.
Citric acid was added (3 mL of a 0.1 M solution) and the
mixture was refluxed for 4 hours. After evaporation of
the volatiles, the residue was poured into water and
extracted with ethyl acetate (3x). The combined
ethylacetate solution was extracted with 0.5 N HCl, water;
then dried and concentrated in vacuo. The resulting
residue was chromatographed on silica gel eluted with 1:1
hexanes-ethylacetate to yield 106 mg, 0.52 mmol (52%).
(cf: Ozinskas, A.J., Rosenthal, G.A., ~. Oraanic Chem. 51,
5047, 1986). mp= 124-5 C. [a]D z +31.3 (c=1.2, MeOH).
H NMR(CDC13,300MHz) ~ 2.16-2.28(m,lH), 2.76~2.86(m,lH),
4.2-9.31(m,1H), 4.37-4.50(m,2H), 5.13(s,2H), 5.32(bs,1H),
7.32-7.38(m,5H).
.
Example 89
N-Benzyloxycarbonyl-Homoserine-di-n-pentylamide
The product of example 88 (620 mg, 2.8 mmol) and
dipentylamine (1.4 mL, 7 mmol) were dissolved in 60 mL
acetonitrile and then heated to reflux overnight. After
evaporation of the volatiles, the residue was
chromato~raphed on silica gel eluted with a step gradient
from chloroform to 1% ethanol in chloroform to yield an
, . . . - .
:
. . . . ... . . .
: :
WO91/00725 PCT/US90/03630
, .
755
-87-
oil, 580 mg, 1.6 mmol (56~). [a]D = +0.31 (c=0-96,
MeOH). MS(CI) m/e 393(m+H) , 253, 236, 192. lH
NMR(CDC13,300MHz) ~ 0.87-0.93(m,6H), 1.22-1.38(m,8H),
1.47-1.63(m,4H), 1.86-1.97(m,lH), 3.01-3.20(m,2H), 3.34-
3.43(m,2H), 3.52-3.72(m,4H), 4.76(dt,J=3,11Hz,lH),
5.1(d,J=12Hz,lH), 5.13(d,J=12Hz,lH), 5.93(d,J=8Hz,lH),
7.31-7.38(m,5H).
Example 90
N'-(2'-Indolylcarbonyl)-(2,RS)-aminobutyrolactone
EDCI (191 mg, 1.0 mmol) was added to a solution of
indole-2-carboxylic acid (161 mg, 1.0 mmol), a-
aminobutyrolactone hydrobromide (182 mg, 1.0 mmol), HOBt
~135 mg, 1.0 mmol), and TEA (279 ~L, 2.0 mmol) irl 15 mL
methylene chloride at room temperature. Additional EDCI
(120 mg) and TEA (56 ~L) were added after 1 day. After 5
days, the volatiles were evaporated and the residue, in
EtOAc, was extracted with 1 M H3PO4, 0.1 M Na2CO3, and
brine. The solution was dried over MgSO4, filtered and
concentrated in vacuo. The product was crystallized from
EtOAc to yield 147 mg, 0.6 mmol, 60%. Rf= 0.17 (1:1
hexanes-EtOAc). mp= 235-6C. MS(CI) m/e 245(m+H) , 144.
H NMR(CDC13-CD3OD,300MHZ) ~ 1.86-2.51(m,lH), 2-19-
2.79(m,lH), 4.32-4.42(m,lH), 4.56(dt,J=2,11Hz,lH),
4.82(dd,J=8,11Hz,lH), 7.1-7.15(m,2H), 7.28(dt,J=1,8Hz,lH),
7.40(s,0.5 H), 7.46(d,J=8Hz,lH), 7.66(d,J=8Hz,lH).
.
. . :- . . . ~ :
- . . ; ., . : , ' ~ ' , ., :: .
:.
--
.
WO91/0072~ PCT/US90103630
. .
Z ~
-88-
E~ample ~1
~-~2'-Indolylcarbonyl)-R,S-Homoserine-di-n-pentylamide
The product of example 90 (25 mg, 0.1 mmol) and
dipentylamine ~50 ~L, 0.25 mmol) were dissolved in 2 mL
THF and warmed to 50C. Additional dipentylamine (250 ~L)
was added after several hours. After 4 days, the
volatiles were evaporated and the residue was
chromatographed on silica eluted with 2:1 hexanes-EtOAc.
Yield: 26 mg, 0.06 mmol, 60%. mp= 128-139 C. MS(CI) m/e
402(m+H) , 158. H NMR(CDCl3,300MHz) ~ 0.92(t,J=7Hz,6H),
1.26-1.42(m,10H), 1.52-1.72(m,3H), 1.98-2.11(m,lH),
2.69~t,J=8Hz,lH), 3.06-3.26(m,2H), 3.42-3.52(m,lH), 3.60-
3.77(m,3H), 5.12-5.20(m,lH)~ 7.03~d,J=lHz,lH),
7.16(dt,J-1,8Hz,lH), 7.31~dt,J=1,7Hz,lH),
7.42(dd,J=1,8Hz,lH), 7.48~d,J=8Hz,lH), 7.67(d,J=8Hz,lH),
9.13(s,1H). C,H,N analysis calculated for C23H35N3O3, 0.5
H2O: C 67.28, H 8.84, N 10.24; found: C 67.42, H 8.64, N
10 . 10 .
E~mple 92
N'-(3'-Ouinolylcarbonyl)-(2,RS)-aminobutyrolactone
Quinoline-3-carboxylic acid (5.2 g, 30 mmol) was
coupled to a-aminobutyrolactone (5.5 g, 30 mmol) in a
manner similar to that in example 90 to provide 2.62 g,
10.2 mmol (34~ yield). Additional extraction of the
aqueous layer with EtOAc yielded another 820 mg, 3.2 mmol
(10.7~). R~= 0.26 (18:1 chloroform-ethanol). mp= 160-
63 C. MS(CI) m/e 257(m+H) . H ~MR(CDC13,300MHz) ~ 2.32-
2.46(m,lH~, 2.91-3.01(m,lH), 4.35-4.43(m,lH),
,,
:: :: : . : ~. :
.. . . .
.
, :. . ': ~ ~, '
WO91/0072S PCT/US90~03630
,
?5-'755
-89-
4.56~dt,J=2,10Hz,lH), 4.83-4.92(m,lH), 7.36(d,J=6Hz,lH),
7.60(dt,J=1,8Hz,lH), 7.81(dt,J=2,8Hz,lH),
7.86(d,Js8Hz,lH), 8.12(dd,J=1,8Hz,lH),
8.59(dd,J=1,2Hz,lH), 9.28(d,J=2Hz,lH). C,H,N analysis
14 12 2 3
found: C 65.42, H 4.82, N 10.82.
Example
N-(3'-Ouinolylcarbonyl)-R.S-Homoserlne-di-n-pentylamid~
The product of example 92 (500 mg, 2.0 mmol) was
treated with dipentylamine (1.5 mL, 7.4 mmol) in 25 mL of
toluene and refluxed. After 2 days, an additional 1 mL of
dipentylamine was added and the heating was continued.
After 1 week, the volatiles wer~ evaporated in vacuo and
the excess amine was removed by Kugelrohr distillation.
The residue was then chromatographed on silica gel eluted
with a step gradient of chloroform to 4% ethanol in
chloroform to yield an oil, 611 mg, 1.48 mmol (79%).
MS(CI) m/e 419(m+H) . 1H NMR(CDCl3,300MHz) ~ 0.88-
0.95(m,6H), 1.25-1.42(m,7H), 1.52-1.75(m,5H), 2.04-
2.15(m,lH), 3.06-3.28(m,2H), 3.46-3.57(m,2H), 3.62-
3.81(m,3H), 4.01(dd,J=5,9Hz,lH), 5.21-5.28(m,lH),
7.63(dt,J=1,8Hz,lH), 7.72(d,J=7Hz,lH),
7.83(dt,J=1,8Hz,lH), 7.93(dd,J=1,7Hz,lH),
8.18(d,J=8Hz,lH), 8.62(d,J=2Hz,lH), 9.37(d,J=3Hz,lH).
C,H,N analysis calculated for C24H35N3O3, 0.25 H2O: C
68.95, H 8.56, N 10.05; found: C 69.26, H 8.45, N 10.06.
:~ . , , . ...... ,.,:,,.. : : .: : ,. -
;, . ,. . , , :
WO91/0072~ PCT/US90/~3630
. ~ ~
2~C > ~ 55
--so--
Example 94
N-(3'-Ouinolvlcarbonyl)-R,S-Ho~oserine-n-pentylamide
The product of example 92 ~200 mg, 0.8 mmol) and n-
pentylamine (232 ~L, 2.0 mmol) were dissolved in 20 mL of
1:1 THF-acetonitrile and stirred at room temperature until
starting material was consumed (tlc: Rf= 0.15, 18:1
chloroform-ethanol). The volatiles were evaporated in
vacuo. The residue was mixed with hexanes and the product
filtered away to yield 273 mg, 0.79 mmol (99%). mp= 181-
3 C. MS(CI) m/e 344(m+H) . H NMR~CDC13,300MHz) ~
0.91(t,J-7Hz,3H), 1.30-1.38(m,4H), 1.51-1.58(m,2H), 1.95-
2.04(m,1H), 2.12-2.21(m,1H), 3.25-3.36(m,2H), 3.80(bs,2H),
4.26(bs,lH), ~.83-4.90(m,lH), 7.37(bt,J=3Hz,lH),
7.69(dt,J-1,5Hz,lH), 7.83(dt,J=1,6Hz,lH),
7.93(d,J=6Hz,lH), 8.10(d,J-6Hz,lH), 8.15(d,J-7Hz,lH),
8.68(d,J-2Hz,lH), 9.37(d,J-lHz,lH)~ C,H,N analysis
calculated for ClgH25N3O3, 0.25 CHC13: C 61.13, H 6.82, N
11.26; found: C 60.82, H 6.88, N 11.16.
Example 95
N-t-Butyloxycarbonyl-R-Methionine-di-n-~entylamide
BOPCl (5.1 g, 20 mmol) was added to a cooled solution
(4 C) of N-t-Butyloxycarbonyl-R-Methionine (5.0 g, 20
mmol), dipentylamine (8.0 mL, 40 mmol), in 60 mL of dry
THF and the stirred reaction was allowed to attain room
temperature overnight. The volatiles were evaporated in
vacuo. The residue was dissolved in EtOAc and extracted
successively with 1 M H3PO9 (3x), 1 M Na2CO3 (3x), brine
(3~.); then dried over MgSO4, filtered and concentrated in
~. . ,
: : .
~ , .
WO91/00725 PCT/US90/03630
,
xr~7ss
--91--
vacuo to yield an oil: 4.6 g, 11.7 mmol ~59%). Rf= 0.81
(1:1 hexanes-EtOAc). [a]D = +27.5 (c=2.7, MeOH). MS(CI)
m/e 389(m+H) , 333, 311, 258, 219, 202, 158. H
NMR(CDC13,300MHz) ~ O.86-0.93~m,6H), 1.21-1.37~m,9H),
1.42~s,9H), 1.43-1.66~m,3H), 1.76-1.96~m,2H), 2.11(s,3H),
2.54(t,J=7Hz,2H), 3.06-3.15~m,lH), 3.19-3.29~m,lH), 3.32-
3.42~m,lH), 3.46-3.56~m,lH), 4.68-4.75~m,lH),
5.37~d,J=9Hz,lH).
E~mple 96
N-(3'-Ouinolylcarbonyl)-tO-methyl)-R,S-Homoserine-di-
n-pentylamide
The product of example 93 was methylated in a similar
manner to that in example 34 to provide the title compound
after purification by chromatography.
Example 97
N-(3'-Ouinolylcarbonyl!-(O-benzyl)-R,S-Homoserine-di-
n-~entylamide
The product of example 93 was benzylated in a manner
similar to that in example 34 utilizing benzyl bromide as
the alkylating agent. The title compound was provided
after purification by chromatography.
Example 98
R-M~thionine-di-n-~entylamide trifluoroacetate sal~
The product of example 95 ~4 g, 10.3 mmol) was
dissolved in 30 mL trifluoroacetic acid precooled to 4C.
After 2 hours, the excess reagent was evaporated and the
residue was placed under high vacuum overnight. [a]D =
. . - : , . -. ,, :
.: . . . . :
' " - ~' '
W09l/00725 P~T/US90/036~0
-92-
+5.1 (c-1.4, MeOH). MS(CI) m/e 289(m+H) . H
NMR~DMSOd6,300MHz) ~ O.88(apparent q,J=8Hz,6H), 1.18-
1.35(m,8H), 1.42-1.58(m,4H), 1.89-1.96(bm,2H), 2.08(s,3H),
2.43-2.67(m,2H), 3.00-3.09(m,lH), 3.13-3.23(m,lH), 3.28-
3.38(m,1H), 3.q8-3.57(m,1H), 4.2-4.28(m,1H), 8.17~s,3H).
Exam~le 99
N-(3'-Ouinolylcarbonyl)-R-Methionine-di-n-pentylamide
Quinoline-3-carboxylic acid (0.43 g, 2.5 mmol), the
product of example 98 (1.0 g, 2.5 mmol), and TEA (697 ~L,
5 mmol) were dissolved in 15 mL of methylene chloride
cooled to 4 C and EDCI (0.48 mg, 2.5 mmol) was added. The
stirred reaction mixture was allowed to attain room
temperature overnight. The volatiles were evaporated and
the resi~ue in EtOAc was extracted with 0.1 M citric acid,
0.1 M Na2CO3, water; then dried over MgSO4, filtered and
concentrated in vacuo. Silica gel chromatography of the
residue eluted with a step gradient of chloroform to 0.5%
ethanol in chloroform yielded an oil, 572 mg, 1.29 mmol
(52%). Rf= 0.19 (1:1 hexanes-ethylacetate). [a]D = +8.0
(c=0.85, MeOH). MS(CI) m/e 444(m+H) . H
NMR(CDCl3,300MHz) ~ 0.91(t,J=7Hz,3H), 0.93(t,J=7Hz,3H),
1.23-1.42(m,8H), 1.52-1.62~m,2H), 1.63-1.75(m,2H), 2.02-
2.17~m,5H), 2.56-2.72(m,2H), 3.10(t,J=8Hz,0.5H),
3.14(t,J=8Hz,0.5H), 3.25-3.35(m,lH), 3.46-3.55(m,lH),
3.59(t,J=8Hz,0.SH), 3.63(t,J=8Hz,0.SH), 5.28-5.36(m,lH),
7.55(d,J=8Hz,lH), 7.12(dt,J=1,7Hz,lH),
7.81(dt,J=1,8Hz,lH), 7.88(dd,J=1,8Hz,lH),
8.15(d,J=8Hz,lH), 8.54(d,J=2Hz,lH), 9.33(d,J=2Hz,lH).
: ~ .
.
WO91t00725 PCT/US90/03630
f~
2~?755
--93--
C, H:, N analysis calculated for C25H37N3O2S, 0.5 H20: C
66.33, H 8.46, N 9.28; found: C 66.33, H 8.19, N 9.25.
Example 100
ouinolylc~~ ylL~-~e~ ioninesulfoxide-di-n
pentylamide
The product of example 99 (100 mg, 0.23 mmol) was
dissolved in 5 mL THF and m-chloroperbenzoic acid (47 mg,
0.23 mmol) was added at room temperature. The reaction
was stirred overnight. The volatiles were evaporated and
the residue, in EtOAc, was extracted with water until the
aqueous extract was neutral (pH=7); then the solution was
dried over MgSO4, filtered and concentrated. The residue
was purified by chromatography on silica gel eluted with
methylene chloride and ethanol to provide the product as
an oil- t~]D = 8i8 (c=0.73, MeOH). MS(CI) m/e
460(mlH) , 396. H NMR(CDC13,300MHz) ~ 0.92(apparent
q,J=7Hz,6H), 1.26-1.40(m,10H), 1.52-1.73(m,3H), 2.14-
2.26(m,lH), 2.39-2.52(m,lH), 2.71-3.02(m,3H), 3.08-
3 18(m,lH), 3.23-3.35(m,lH), 3.38-3.52(m,lH), 3.58-
3.68(m,1H), 5.20-5.34(m,1H), 7.62(tt,J=1,8Hz,2H),
7.72(d,J=7Hz,lH), 7.83(tt,J=1,8Hz,lH), 7.92(d,J=8Hz,lH),
8.17(d,J=8Hz,lH), 8.62~dd,J=2,5Hz,lH),
9.35(dd,J=2,3Hz,lH). C,H,N analysis calculated for
C25H37N3O3S, 0.1 EtOAc: C 65.13, H 8.13, N 8.97; found: C
65.31, H 8.30, N 8.73.
.......... . .
"::; ' :. . : . , :
-: -~, . .- ~:
.: .: , ............. , - .: :. . .:
. - ~ .
. . :: , . , , . :,
.: : , - . - . . -
.
WO91/00725 PCT/US90/03630
,,
. . .
~?~;~5
-94-
E~ample lQl
N-t-Butyloxycarbonyl-R-Proline-di-n-pentylamide
BOPCl (1.18 g, 9.69 mmol) was added to a cooled
solution (4C) of N-t-Butyloxycarbonyl-R-Proline (1.0 g,
9.64 mmol), dipentylamine (2.5 mL, 12.5 mmol), in 50 mL of
dry THF. The cooling bath was removed and the stirred
reaction mixture was allowed to warm to ambient
temperature gradually. After 5 hours, the volatiles were
evaporated in vacuo. The residue was dissolved in EtOAc
and extracted successively with 1 M H3PO4 (3x), 1 M Na2CO3
(3x), brine (3x); then dried over MgSOg, filtered and
concentrated in vacuo to yield an oil, 880 mg, 2.48 mmol
(54%). Rf= 0.28 (2:1 hexanes-EtOAc). [a]D = +28i7
(c=1.0, MeOH) MS(CI) m/e 355(m+H) , 299, 255. H
NMR(CDC13,300MHz) ~ 0.84-0 94(m,6H), 1 23-1.38(m,8H),
1.41(s,6H), 1.45(s,3H), 1.49-1.58(m,6H), 1.80-1.90(m,lH),
2.0-2.23(m,lH), 3.12-3.33(m,4H), 3.4-3.52(m,lH), 3.56-
3.67(m,lH), 4.44(dd,J=4,8Hz,0.6H), 4.58(dd,J=2,8Hz,0.4H).
Example 102
R-Proline-di-n-pentylamide hydrochloride
The product of example 101 (800 mg, 2.3 mmol) was
mixed with HCl-Dioxane (12.5 mL, 50 mmol, pre-cooled to
4C) under an N2 atmosphere at ambient temperature. After
1 hour, the volatiles were evaporated in vacuo and the
residue was mixed with toluene and concentrated (twice)
then placed under high vacuum overnight. The residue was
utilized directly.
` `' ,:~` . : .
.
...
: - , , . ,, ~ :
WO9l/00725 PCT~US90/03630
2~ 55
~; ,,~
~a~le 103
N-(2'-IndQlvlcarbonvl)-R-Proline-di-n-~entylamide
EDCI (440 mg, 2.3 mmol) was added to a cooled (9C)
solution of indole-2-carboxylic acid (371 mg, 2.3 mmol),
the product of example 102 (2.3 mmol assumed), HOBt (311
mg, 2.3 mmol), and TEA (321 ~L, 2.3 mmol) in 10 mL
methylene chloride. The stirred reaction was allowed to
attain ambient temperature overnight. The volatiles were
evaporated and the residue was dissolved in EtOAc and
extracted with 1 M H3PO4 (3x), 1 M Na2CO3 (3x), brine
(3x); then dried over MgSO4, filtered and concentrated to
an orange oil. The crude product was purified by
chromatography on silica eluted with 2:1 hexanes-EtOAc to
yield 0.92 g, 2.4 mmol (92%) as a slightly yellow glass
Rf- 0.22 (2:1 hexanes-EtOAc). The glass was dissolved in
hot hexanes-EtOAc, then cooled slowly to -20C. An oil
separated out and o~er 24 hours solidified. The solution
was decanted and the solid was collected using hexanes to
yield 769 mg (84%). mp= 63-7C. [a]D= -20.4 (c=1.0,
MeOH). MS(CI) m/e 398(m+H) , 241, 213. 1H
NMR(CDCl3,300MHz) ~ O.88(t,J=7Hz,3H), 0.93(t,J=6Hz,3H),
1.24-1.43(m,8H), 1.51-1.75(m,3H), 1.80-1.90(m,lH), 1.94-
2.28~m,3H), 2.32-2.45(m,lH), 3.16-3.37(m,2H), 3~43-
3.54(m,2H), 4.0-4.08(m,lH), 4,12-4.2(m,lH),
5.02~dd,J=4,8Hz,lH), 6.96(bs,1H), 7.12(dt,J=1,8Hz,lH),
7.28(dt,J=1,7Hz,lH), 7.48~dd,J=1,8Hz,lH),
7.67(d,J=8Hz,lH), 9.30(s,1H) C,H,N analysis calculated
for C24H35N3O2: C 72.50, H 8.87, N 10.57; found: C 72.55,
H 8.91, N 10.49.
,. ~ . . .. . . .. . . . . . .
- -
.: .. :- :: - .. -
.: . : - :: . . . - -
:: : : :--:. -: .: ., : .... , .: : . .. :.
WO 91tO0725 PCT/US90/03630
. . . ~
-96-
~m~ lQ~
Methyl 2-(3'-Ouinolylcarbonylamino)-2-methylpro~ionate
Quinoline-3-carboxylic acid (1.12g, 6.5 mmol~, methyl
a-aminoisobutyrate (l.Og, 6.5 mmol) and TEA (1.8 mL, 1.3
mmol) were dissolved in 50 mL methylene chloride and
treated with EDCI (1.2g, 6.5 mmol) overnight. The solvent
was evaporated and the residue was extracted as in example
71 to give a white solid, 660 mg, 2.58 mmol (40%). mp=
138-140 C. MS(CI) m/e 273(m+H) . H NMR(CDCl3,300MHz)
1.75~s,6H), 3.82(s,3H), 7.06(s,lH), 7.62(d,J=1,7Hz,lH),
7.81(dt,J=1,7Hz,lH), 7.91(dd,J=1,8Hz,lH),
8.15(d,J=8Hz,lH), 8.58(d,J=2Hz,lH), 9.28(d,J=2Hz,lH).
E~ 105
2-(3'-Ouinolvlcarbonvlamino)-2-methvlpropionic acid
The product of example 104 (620 mg, 2.42 mmol) was
dissolved in 50 mL methanol and treated with 1 N NaOH (2.5
mL, 2.5 mmol). An additional 2.5 mL was added after 1
day. After 2 days, the solvent was evaporated and the
residue was dissolved in water and extracted with
ethylacetate. The aqueous phase was then acidified and
re-extracted with ethylacetate. This second EtOAc layer
was dried over MgSO4, filtered and evaporated to yield 406
mg, 1.67 mmol (69%). Rf= 0.3 (80:20:~ CHCl3-CH30H-NH40H).
Example 1062-(3'-Ouinolylcarbo~ylamino)-2-m~thylpropion-
di-n-pent~ zmide
The product of example 105 (100 mg, 0.413 mmol),
dipentylamine (202 ~L, 1.0 mmol) and TEA (59 ~L, 0.42
' ' . ' ' . '
' ' ` ' ': '
WO 91~0072~ PCT/US90tO3630
2~ 55
.
- .
-97-
mmol) were dissolved in 15 mL methylene chloride, treated
with EDCI (80 mg, 0.42 mmol) and stirred at room
temperature overnight. The solvent was evaporated and the
residue was dissolved in ethylacetate and extracted as in
example 71. NMR indicated the presence of undesired
dehydrated product (oxazolone). MS(CI) m/e 241(m+H) . lH
NMR(CDCl3,300MHz) ~ 1.61(s,6H), 7.66(dt,J=l,'Hz,lH),
7.86(dt,J=1,7Hz,lH), 7.94(dd,J=1,8Hz,lH),
8.20(d,J=8Hz,lH), 8.74(d,J=2Hz,lH), 8.98(d,J=2Hz,lH). The
crude dehydrated product was redissolved in 25 mL THF and
treated with dipentylamine (202 ~L, 1.0 mmol). Another
400 mL of dipentylamine was added at 2 and 4 days. After
evaporation of the solvent, the residue was purified by
chromatography on silica gel eluted with a 4:1 to 1:1
hexane-ethylacetate step gradient to yield 51 mg, 0.13
mmol ~32~). mp= 139-5C. MS(CI) m/e 398(m+H) , 158. lH
NMR(CDC13,300MHz) ~ 0.92~t,J=7Hz,6H), 1.25-1.49(m,12H),
1.90(s,6H), 3.40(bs,4H), 7.61(dt,J=1,7Hz,lH),
7.80(dt,J=1,7Hz,lH), 7.91(dd,J=1,8Hz,lH) 8.15(d,J=8Hz,lH),
8.58(d,J=2Hz,lH), 8.69(s,lH), 9.37(d,J=2Hz,lH). C,H,N
analysis calculated for C24H35N3O2, 0.25 H2O: C 71.69, H
8.90, N 10.45; found: C 71.65, H 8.74, N 10.39.
~xample 107
N-(3'-Ouinolvlcarbonyl)-R-L~rsine-d--n-pentylamide
hydrobromide
The product of example 62 (1.61 g, 2.64 mmol) was
treated with 15 mL of HBr in HOAc (1.1 N, 16.5 mmol) for 2
hours under an inert atmosphere. The solvent was
evaporated and the residue was purified by chromatography
:: . : .- - - : .
-
:
, .: : .
':'.': ' ' ' '
WO9l/00725 PCT~US90/03630
. j,
.
2~ 755
-98-
on silica gel eluted with a methylene chloride to 1%
ethanol in CH2Cl2 step gradient to yield 1.25 g, 2.39 mmol
(91%) as a yellow glass. mp= 85-95 C. H
NMR~DMSOd6,300MHz) ~ 0.85(t,J=7Hz,6H), 1.23-1.83(m,18H),
2.78(t,J=7Hz,2H), 3.06-3.17(m,1H), 3.28-3.44(m,3H), 4.86-
4.93(m,lH), 7.57(bs,2H), 7.72(dt,J=1,7Hz,lH),
7.88(dt,J=1,7Hz,lH), 8.10(d,J=8Hz,2H), 8.92(a,J=2Hz,lH),
9.02(d,J=8Hz,lH), 9.32(d,J=2Hz,lH).
ExampLe 108
Na-t3'-Ouinolylcarbonyl)-~-~henylthiolcarbonyl-R-
Lysine dipentylamide
The product of example 107 (20 mg, 0.045 mmol) was
treated with carbonyldiimidazole (8.1 mg, 0.05 mmol) in 10
mL methylene chloride at room temperature overnight.
Thiophenol ~10.3 ~L, 0.10 mmol) and 10 mL THF were added
and the mixture was heated to 60C. After 1 day, the
reaction was eluted on silica gel with 1% ethanol in
methylene chloride to yield an oil. MS(CI) m/e 577(m+H) ,
467, 420. H NMR(CDCl3,300MHz) ~ 0.88-0.96(m,6H), 1.23-
1.86(m,18H), 3.12(dt,J=7,13Hz,lH), 3.22-3.44(m,4H),
3.59(dt,J=7,13Hz,lH), 5.0-5.17(m,lH), 5.70(t,J=SHz,lH),
7.32-7.37(m,3H), 7.47-7.51(m,3H), 7.62(dt,J=1,8Hz,lH),
7.82(dt,J=1,7Hz,lH), 7.91(dd,J=1,8Hz,lH),
8.16(d,J=8Hz,lH), 8.63(d,J=2Hz,lH), 9.37(s,J=2Hz,lH).
.
, . .
. ~ , ; - :
WO91/00725 PCT/US90/03630
,
i"755
.,, .~, . . .
_99_
~ample l
N-Benzyloxycarbonyl-R-Phenylalycine-(2'-
~ropylpiperidinyl)amide
N-Benzyloxycarbonyl-R-phenylglycine (1.0 g, 3.5
mmol), 2-propylpiperidine (1 mL, 6.64 mmol), HOBt (475 mg,
3.5 mmol) and TEA (490 ~L, 3.5 mmol) were dissolved in 25
mL of CH2C12 and treated with BOPCl (890 mg, 3.5 mmol).
Additional TEA (490 ~L) and BOPC1(890 mg) were added after
2 days. After 6 days, the solvent was evaporated and the
crude reaction was purified by chromatography on silica
gel eluted with a 9:1 to 4:1 hexane-ethylacetate step
gradient to yield 179 mg, 0.454 mmol (13%). mp= 100-
115C. ~a]D= -13.5 (c=1.0, MeOH). MS(CI) m/e 395(m+H) ,
261. H ~MR(CDC13,300MHz) ~ 0.52(t,J=7Hz,lH),
0.92(t,J=7Hz,2H), 1.18-1.70(m,10H), 2.56-2.67(m,0.33H),
3.01(dd,J=2,13Hz,0.67H), 3.57(bd,J-12Hz,0.67H),
3.80(bs,0.33H), 4.51(bd,J=13Hz,0.33H), 4.78(bs,0.67H),
4.98(d,J=llHz,lH), 5.12(d,J=llHz,lH), 5.54(d,J=7Hz,0.67H),
5.58(d,J=7Hz,0.33H), 6.46-6.55(m,1H), 7.28-7.43(m,10H).
~ 0
R-Phenylalycine-(2'-propylpiperidinyl)amide
The product of example 109 (150 mg, 0.38 mmol) was
treated with 25 mg of 10% Pd on carbon ln 5 mL of methanol
under one atmosphere of hydrogen for 24 hours. The
catalyst was filtered away and the filtrate was evaporated
to yield product.
,: . . . . .
:: . . : ' . . :
:: ,
'
W09l/0072~ PCT/US90/03630
5S
- 1 o o--
~L~ 1 1 1
N-(3'-Ouinolylcarhonyl)-R-Dhenylalyc~ne-
(2'-propylpi~eridinyl)amide
Quinoline-3-carboxylic acid (38.1 mg, 0.22 mmol), the
product of example 110 ~31 mg, 0.22 mmol) and TEA ~31 ~L,
0.22 mmol) were dissolved in 4 mL of 1:1 DMF-CH2Cl2 and
treated with EDCI ~42.1 mg, 0.22 mmol) with stirring at
room temperature overnight. The solvent was evaporated
and the residue was extracted as in example 71. Rf= 0.4
~1:1 hexane-ethylacetate). MS~CI) m/e 416~m+H) , 261,
154, 128. H NMR~CDCl3,300MHz) ~ 0.55~t,Jz7Hz,lH),
0.94~t,J=7Hz,2H), 1.23-1.72(m,10H),
2 71(dt,J=2,13Hz,0;33H), 3.08~dt,J=2,13Hz,0.67H),
3.68(bd,J=13Hz,0.67H), 3.93(bs,0.33H),
4.58(bd,J=13H~,0.33H), 4.85(bs,0.67H),
6.03(d,J-7Hz,0.67H), 6.07(d,J=7Hz,0.33H), 7.3-7.42(m,3H),
7.52-7.63(m,3H), 7.80(dt,J=1,7Hz,lH), 7.90(d,J=8Hz,lH),
8.14(d,J=8Hz,lH), 8.28(t,J=6Hz,lH), 8.59(d,J=2Hz,lH),
9.34(d,J=2Hz,lH). C,H,N analysis calculated for
C26H29N3O2, 0.5 H2O: C 73.56, H 7.12, N 9.90; found: C
73.60, H 7.10, N 9.61.
~xamPle 11~
N-(4',8'-Dihydroxy-2'-quinolylcarbonyl)-R-phenylglycine-
(2'-propylpiperidinyl)amide
4,8-Dihydroxyquinoline-2-Carboxylic acid (45 mg, 0.22
mmol), the product of example 110 (S2 mg, 0.20 mmol) and
TEA (31 ~L, 0.22 mmol) were dissolved in 4 mL of 1:1 DMF-
methylene chloride and treated with EDCI (42 mg, 0.22
mmol) with stirring overnight. The reaction was then
. ~
. , .. . : . .
~, .: :,
:- , .. : , ::
" ,,,
W091J0072~ PCT/US~0/03630
, . .
2~ 55
--101--
po~red into ethylacetate and extracted as in example 71.
The resulting residue was purified by chromatography on
silica gel eluted with a 1% to 9% ethanol in methylene
chloride step gradient. MStCI) m/e 448(m+H) , 293. H
NMR~DMSOd6,300MHz) ~ 0.71(t,J=7Hz,lH), 0.81-0.90(m,2H),
1.15-1.70(m,10H), 3.07(bt,J=13Hz,0.67H), 3.33(s,H2O),
3.68(bd,J=12Hz,0.67H), 4.02(bs,0.33H),
4.36(d,J=8Hz,0.33H), 4.68(bs,0.67H), 6.12-6.17(m,lH),
7.09(d,J=7Hz,lH), 7.32-7.56(m,8H), 9.89(d,J=8Hz,0.67H),
10.08(d,J=8Hz,0.33H), 10.23(s,0.67H), 10.24(s,0.33H),
11.73(bs,lH).
Example 113
-Benzyloxvcarbonyl-R-phenvlalvcine-(N-benzyl
~-2'-cyanoeth~l)am;de
N-Benzyloxycarbonyl-R-phenylglycine (285 mg, 1.0
mmol), 3-(benzylamino)propionitrile (391 ~L, 2.5 mmol) and
TEA (139 ~L, 1.0 mmol) were dissolved in 10 mL of CH2Cl2
and treated with BOPCl (256 mg, 1.0 mmol). After 1 day,
another 139 ~L of TEA was added. After 2 days, additional
BOPCl (256 mg), amine (391 ~L) and ~MF (5 mL) were added.
After 3 days, the solvents were evaporated and the residue
was extracted as in example 71. The crude residue was
recrystallized from hexanes-ethylacetate to yield 314 mg,
0.74 mmol (74~). Rf= 0.75 (1:1 hexanes-ethylacetate).
mp= 114-150 C. [a]D= -9. 4 (c=0.67, 1:1 DMF-MeOH).
MS(CI) m/e 428(m+H) , 445, 384, 375. H NMR(CDC13,300MHz)
2.45-2.66(m,2H), 3.33-3.42(m,lH), 3.46-3.52(m,0.5H),
3.66-3.75(m,lH), 4.38~d,J=16Hz,lH), 4.43-4.5(m,0.5H),
4.63(d,J=16Hz,lH), 4.69(s,0.5H), 5.01-5.2(m,3H),
... .. , - ~.:
.
~ ' ',
: ' .
WO91/00~2~ PCT/US90/03~30
-102-
5.59~d,J=7Hz,0.5H), 5.66(d,J-7Hz,lH), 6.88(s,0.5H), 6.18-
6.27(m,1.5H), 6.82(bs,0.5H), 6.95(t,J=4Hz,2H), 7.10-
7.18(m,2H), 7.28-7.39(m,15H). C,H,N calculated for
C26H25N3O3, 0.1 H2O: C 72.74, H 5.92, N 9.79; found: C
72.79, H 5.99, N 9.40.
Example 114
R-Phenvlglycine-(N-benzyl~N-2'-c~anoethyl)amide
The product of example 113 (225 mg, 0.53 mmol) was
dissolved in 25 mL of ethanol and treated with 100 mg of
10% Pd/C at room temperature. After 1.5 hours, the
catalyst was filtered and the filtrate was evaporated to
yield 158 mg, 0.54 mmol(quantitative). MS(CI) m/e
294(m+H) , 241.
~am~le 11~
N-(3'-Ouinolylcarbonyl)-R-phenylalycine
(N-benzyl.N-2'-cyanoethyl)amide
Quinoline-3~carboxylic acid (35 mg, 0.20 mmol) and
the product of example 114 (53 mg, 0.18 mmol) were
dissolved in 10 mL of methylene chloride and treated with
EDCI (38 mg, 0.20 mmol). After 1 day, the solvent was
evaporated and the residue was extracted as in example 71
to give 54 mg, 0.12 mmol (67%). [a]D= -0.42 (c-2.6,
CHCl3). mp= 57-63 C. MS(CI) m/e 449(m+H) . H
NMR(CDCl3,300MHz) ~ 1.90-2.02(m,0.25H), 2.27-
2.38(m,0.25H), 2.49-2.72(m,1.5H), 3.42(dt,J=7,13Hz,lH),
3.81(dt,J=7,13Hz,lH), 4.46(d,J=16Hz,lH),
4.73(d,J=16Hz,lH), 6.11(d,J=6Hz,0.25H),
6.16(d,J=7Hz,0.75H), 6.98-7.02(m,2H), 7.19-7.22(m,0.5H),
.
, .. , : . '
'' :~ -: ~,
- : .
WO9l/00725 PCT/US90/03630
21?5~55
-103-
7.30-7 33~m,2.5H), 7.38-7.46~m,3H), 7.53-7.64~m,3H),
7.82~dt,J=1,7H,lH), 7.85-7.94~m,2H), 8.15~d,lH,J=8Hz),
8.61~d,J=lHz,lH), 9.33~d,J=lHz,lH). C,H,N analysis
calculated for C28H24N42' 0-7 H2
12.15; found: C 72.86, H 5.58, N 11.77.
Exam~le 116
N-(4'.8'-Dihvd~xy-2'-quinolylcarbonyl)-R-phenylqlycine
(~-benzyl,N-2'-cyanoethyl)amide
4,8-Dihydroxyquinoline-2-carboxylic acid (41 mg, 0.20
mmol), the product of example 114 ~53 mg, 0.18 mmol), and
TEA ~28 ~L, 0.20 mmol) were dissolved in 5 mL of DMF and
treated with EDCI ~38 mg, 0.20 mmol). Additional TEA ~28
~L) and EDCI ~38 mg) were added after 2 hours and 1 day.
After 2 days HOBt (27 mg, 0.20 mmol) was added to the
reaction mixture. After 3 days, the solvent was
evaporated and the residue was extracted with 0.1 M citric
acid, and water and the organic solution was dried over
MgSO4 then filtered and concentrated. The crude product
was purified by silica gel chromatography eluted with 1:1
hexanes-ethylacetate to provide 22.6 mg, 0.05 mmol ~26~).
Rf= 0.4 ~1:1 hexane-ethylacetate). mp= 218-222 C. ~a]D=
-4.8 ~c=0.42, MeOH). MS~CI) m/e 481(m+H) , 428. lH
NMR~CD30D,300MHz) ~ 2.47-2.58~m,0.33H), 2.6-2.82(m,2H),
3.33-3.62~m,2.33H), 3.68-3.78~m,0.33H), 3.82-3.91~m,lH),
4.53~d,J=16Hz,lH), 4.62~d,J=14Hz,0.33H),
4.76(d,J=16Hz,lH), 4.87(s,H2O), 4.92~d,J=5H2,0.33H),
6.18(s,lH), 7.10~dd,J=1,7Hz,lH), 7.2-7.35~m,7H), 7.39-
7.46(m,3H?, 7.51-7.60~m,2H), 7.67(dd,J=1,8Hz,lH).
... , .. . , , . . - - -
: - ~, ,
WO91J00725 PCT/US90tO3630
7 5 5
~, . . .
-104-
Example 111
N-(3'-Ouinolvlcarbonyl)-R-Tyrosine-di-n-pentylamide
hydrochlor de
hvdrochloride
The product of example 80 (1.5 g, 3.0 mmol) was
treated with 1.4 N HCl in dioxane (11 mL, 15 mmol) for 10
minutes. The excess reagent was evaporated and the oily
residue was triturated with diethylether and filtered to
yield 1.3 g, 2.6 mmol (87%) of a pale yellow solid.
MS(CI) m/e 476(m+H) , 958. H NMR(DMSOd6,300MHz) ~
0.84(t,J=7Hz,6H), 1.15-1.62(m,12H), 2.87-3.22(m,3H), 3.29-
3.40(m,3H), 5.02(apparent q,J=7Hz,lH), 6.66(d,J=8Hz,2H),
7 11(d,Jz8Hz,2H), 7.78(dt,J=1,8Hz,lH),
7.96(dt,J=1,8Hz,lH), 8.17(t,J-7Hz,2H), 9.04~d,J-2Hz,lH),
9.22(d,J=8Hz,lH), 9.33(d,J-2Hz,lH). C,H,N analysis
calculated for C29H37N3O3, 1.3 HCl: C 66.60, H 7.38, N
8.03; found: C 66.43, H 7.38, N 7.99.
Example 118
N-(3'-Ouinolylcarbonvl)-R-Histidine-di-n-pentylamide
dihydrochloride
The product of example 50 (800mg, 1.78 mmol) was
dissolved in 13 mL of 1.4 N HCl in acetic acid for 10 mln
and then the volatiles were evaporated to remove excess
reagent. The oily residue was dissolved in a small amount
of CH2Cl2 and the product was precipitated with hexanes.
The solid was collected to yield 824 mg, 1.58 mmol (89~).
MS(CI) m;e 450(mtH) . H NMR(DMSOd6,300MHZ) ~
,
~, :' . '. , . . ?
, :. ' . . ~ ~ , . . . .. .
: ' :, : - . ,
. ~ . :-.:
WO 91/0072~ PCT/US90/0363~
S5
-105-
0.74(t~J=7Hz,3H), 0.85(t,J=7Hz,3H), 1.12-1.32(m,8H), 1.41-
1.52(m,4H), 3.08-3.43(m,6H), 5.24-5.31(m,lH), 7.45(s,lH),
7.77(dt,J=1,7Hz,lH), 7.94(dt,J=1,7Hz,lH),
8.15~dt,J=1,9Hz,2H), 9.02(s,2H), 9.31-9.33(m,2H),
14.18(s,lH), 14.57(s,1H). C,H,N analysis calculated for
C26H35N5O2, 2.6 HCl: C 57.36, H 6.96, N 12.87; found: C
57.30, H 6.96, N 12.86.
Example 119
N-(4',8'-Dihydroxy-2'-quinolylcarbonyl)-R-(4'-
hydro~yphenyl)-alycine-di-n-pentylamide
The reaction was performed in a similar manner to
that in example 8 utilizing 0.3 g of the compound of
example 64, 4',8'-dihydroxyquinoline-2-carboxylic acid
~0.2 g), EDCI ~0.21 g), HOBt ~0.13 g) and NMM ~0.22 mL).
The product was isolated in 75% yield ~0.37 g). MS~CI)
m/e 494~m+H) .
H NMR(DMSOd6,300MHz) ~ 0.85(m,6H), 1.1-1.35(m,10H), 1.38-
1.45(m,4H), 3.0-3.5(m,4H), 5.95(d,Jz9Hz,lH),
6.76(d,J=9Hz,2H), 7.08(d,J=9Hz,lH), 7.23(d,J=9Hz,2H), .
7.4(t,J=9Hz,lH), 7.55~m,2H), 9.5~bs,lH),
9.75(d,J=lOHz,lH). C,H,N calculated for C28H35N3O5, 0.5
H2O: C 66.91, H 7.22, N 8.36; found: C 66.76, H 7.20, N
8.18.
Example 120
N-Benzyloxycarbonyl-alycine-di-n-pentylamide
The compound was prepared in a manner similar to that
in example 1 utilizing N-t-butyloxycarbonylglycine.
MS~CI) m/e 349(m+1) , 305, 241, 215, 184. lH
WO91/00~2~ PCT/US90/03630
~,
2~ S5
-106-
NMR(CDC13,300MHz) ~ 7.30-740(m,5H), 5.86(bs,1H),
5.12(bs,2H), 4.0(bd,J=4.5Hz,2H), 3.32(t,J=7.5Hz,2H~,
3.15(t,J=7.5Hz,2H), 1.50-1.70(m,4H), 1.20-1.40(m,8H),
O.9(m,6H).
Example 1~1
N-(2'-Indolylcarbonyl)-~lycine-di-n-pentylamide
The product of example 120 was deprotected in a
manner similar to that in example 80. The free amine
product was then coupled with indole-2-carboxylic acid as
in example 4. mp= 98-100 C. MS(EI) m/e 3S7(m) , 287,
184. lH NMR(CDCl3,300MHz) ~ 9.27(s,lH), 7.67~d,J=6Hz,lH),
7.95(bd,J=7Hz,2H), 7.29(dt,J=1,6Hz,lH),
7.14(dt,J=1,6Hz,lH), 6.98(s,lH), 4.27(d,J=9Hz,2H),
3.~9(bt,J=7Hz,2H), 3.25(bt,Jz7Hz,2H), 1.55-1.70(m,4H),
1.25-1.40(m,8H), 0.93(t,J=6Hz,3H), O.91(t,J=6Hz,3H).
C,H,N analysis calculated for C21H31N3O2, 0.3 H2O: C
69.51, H 8.78, N 11.58; found: C 69.45, H 8.58, N 11.47.
Example 122
Ethvl N-(t-Blltyloxycarbonyl~alycinyl-lN-
benzyl)alycinate
N-t-Butyloxycarbonylglycine and ethyl N-
benzylglycinate were coupled in a manner similar to that
in example 1 to provide product.
. .
.. : : - '
: . .
.. , . ~ . . ..
,
WO91/00725 PCT/US90/03630
2~ 755
. . .
-107-
~xam~le 12~
Ethyl N-(3'0uinolvlcarbonvl)glycinyl (N-
benzyl)alvcinate
The product of example 122 was deprotected in a
manner similar to that in example 2 and then coupled in a
manner similar to that in example 3 to provide product.
MS(CI) m/e 406(m+H) , 334, 194. lH NMR(CDC13,300MHz)
9.37(d,J=2Hz,0.33H), 9.35(d,J=2Hz,0.67H), 8.65(bm,1H),
8.18(bd,J=7Hz,lH), 7.94~m,lH), 7.83~m,lH), 7.63(m,lH),
7.43-7.55(m,lH), 7.30-7.40(m,3H), 7.25(m,2H),
4 73(s,0.67H), 4.67(s,1.33H), 4.51(d,J=4Hz,1.33H),
4.33(d,J=4Hz,0.33H), 4.16-4.25(m,2H), 4.13(s,1.33H),
4.00(s,0.67H), 1.28(m,3H).
.,
E~melQ 1~4
N-(t-Butyloxvcarbonvl)-R-homo~henvlalanine-di-n-
pentylamide
The product was prepared in an analogous manner to
that in example 1 using t-Butyloxycarbonyl-R-
homophenylalanine. MS(CI) m/e 419(m+H) , 363, 345, 319.
H NMR(CDC13,300MHz) ~ 1.85(m,lH), 7.48(m,lH), 7.18-
7.32(m,5H), 5.39(bd,J=9Hz,lH), 4.56(m,lH),
3.48(dt,J=7,14Hz,lH), 3.39(t,J=7Hz,lH), 3.08(m,2H),
2.68(m,2H), 1.88(m,2H), 1.45(s,9H), 1.20-1.35(m,8HI,
1.13(m,2H), 0.88(m,6H).
:. ~. , : - .
. ., , . : . -. - , .
: ~ , : - :
:~ ,. , ",
W091/00725 PCT/VS90/03630
Z(.'~ ~ 5~
-108-
N-(3'-Ouinolylcarbonyl)-R-homophenylalanir.e-di-n-
pentylamide
The product was prepared in analogous manner to those
in examples 2 and 3 utilizing the product of example 124
as the starting material. MS~CI) m/e 97q(m+H) , 369, 319,
305, 289. H NMR~CDCl3,300MHz) S 9.32~d,J=2Hz,lH),
8.53(d,J=2Hz,lH), 8.16(bd,J=8Hz,lH), 7.90(dd,J=1,8Hz,lH),
7.82(m,lH), 7.62(m,1H), 7.40(bd,J=8Hz,lH), 7.30(m,4H),
7.20(m,lH), 5.19(m,lH), 3.55-3.70(m,lH), 3.05-3.20(m,3H),
2.78(bt,J=7.5Hz,2H), 2.15(m,2H), 1.50-1.65(m,4H), 1.15-
1.35(m,8H), 0.90(m,6H).
E~ ~
N-(3'-Q~innlylcarbonyl)al~cine
Quinoline-3-carboxylic acid and methyl glycinate
hydrochloride were coupled in a manner similar ~o that in
example 3. The resulting product was subjected to
saponification in methanol with 1 N NaOH. The desired
product was extracted with EtOAc from the acidified
solution or alternatively allowed to slowly precipitate
from the acidified solution. MS(CI) m/e 231(m+H) , 187.
H NMR(DMSOd6,300MHz) S 12.72~bs,1H), 9.32(d,J=9Hz,lH),
9.11(t,J=6Hz,lH), 8.87(d,J=3Hz,lH), 8.12(t,J=7H ,2H),
7.89~t,J=7Hz,lH), ?.71(t,J=7Hz,lH), 4.03(bs,2H).
.: . :: , , . ~
-:
W09lt00725 PCT/US90/03630
2(?~?755
--1 0 9--
Example 127
N- (3'-Ouinolylcarbonyl~alycine-di-n-pentylamide
The product of example 126 and di-n-pentylamine were
coupled in a manner similar to that in example 1. The
product was isolated by chromatography and solidifies upon
concentration. mp= 36-375. MS~CI) m/e 370~m+H) . lH
NMR~CDC13,300MHz) ~ 9.38~d,J=2Hz,lH), 8.65~d,J=1.8Hz,lH),
8.18~d,J=8.5Hz,lH), 7.93~dd,J=1,8Hz,lH), 7.83~m,lH),
7.69(m,2H), 4.32(d,J=3.7Hz,2H), 3.41(bt,J=8Hz,2H),
3.27(bt,J=8Hz,2H), 1.62(m,4H), 1.30-1.45(m,8H),
0.95~t,J=7Hz,3H), 0.92~t,J=7Hz,3H). C,H,N analysis
calculated for C32H31N32 C 71-49~ H 8-46~ N 11-37
found: C 71.28, H 8.42, N 11.36.
Example 1~
N-(3'-Ouinolylcarbonyl)alvcine-(~-
propyl)piperidinylamide
The acid from example 126 and 4-propylpiperdine were
coupled as in example 1. mp= 116-117C. MS~CI) m/e
340(m+H) , 279, 254, 201. lH NMR~CDCl3,300MHz) ~
9.36~d,J=2Hz,lH), 8.63~d,J=2Hz,lH), 8.16~d,J=8.5Hz,lH),
7.93~dd,J=1,8Hz,lH), 7.82(m,lH), 7.60(bs,lH), 7.63(m,lH),
4.61(dt,J=2,13Hz,lH), 4.31(m,2H), 3.79(bd,J=lOHz,lH),
3.07(dt,J=3,13Hz,lH), 2.70(dt,J=3,13Hz,lH), 1.81(bm,2H),
1.55(m,lH), 1.05-1.40(m,6H), 0.92(t,J=7Hz,3H). C,H,N
analysis calculated for C20H25N3O2, 0.1 H2O: C 70.40, H
7.44, N 12.31; found: C 70.19, H 7.44, N 12.15.
.. ~ , :
: - , . ::, ,. ~ : -
:, , - -:
,, "
WO91/00725 PCT/US90/0363~
2~ J
--110--
E~mel~ 129
N-Benzvlo~ycarbonyl-R-phenylalvcine-di-n-~entylamide
The product was obtained from the coupling of N-
Benzyloxycarbonyl-R-phenylglycine and di-n-pentylamine as
in example 1. MS(CI) m/e 425(m+H) , 333, 317, 291. lH
NMR(CDCl3,300MHz) ~ 7.27-7.45(m,10H), 6.48(bd,J=7.5Hz,lH),
5.53(d,J=7.5Hz,lH), 5.12(d,J=12Hz,lH), 5.01(d,J=12Hz,lH),
3.48(m,lH), 3.18(m,2H), 2.97(m,lH), 1.50(m,4H), 1.10-
1.35(m,8H), 0.87(t,J=7.5Hz,3H), 0.84(t,J=7.5Hz,3H).
Exam~le 130
R-Phenylalycine-dl-n-pentylamide
The product resulted from the hydrogenolysis of the
product of example 129. MS(CI) m/e 291(m+H) , 158. H
NMR(CDC13,300MHz) ~ 7.25-7.40(m,5H), 4.65~bs,lH),
3.52~m,lH), 3.08-3.22(m,2H), 2.92(m,lH)j 2.02~bs,2H),
1.50(m,3H), 1.10-1.35(m,9H), 0.88(t,J=7Hz,3H),
0.85(t,J=7Hz,3H).
N-(3~ouinolylcarbonyll-R-phenylalyclne-di-n-pentylamide
The product of example 130 was coupled in a similar
manner to that in example 3 to provide product. MS(CI)
m/e 446(m+H) . H NMR(CDCl3,300MHz) ~ 9.33(d,J=2Hz,lH),
8.58(d,J=2Hz,lH), 8.13(bt,J=8Hz,2H), 7.88(bd,J=8Hz,lH),
7.79(m,lH), 7.62(m,1H), 7.55(m,2H), 7.32-7.42(m,3H),
6.03(d,J=6Hz,lH), 3.55(m,3H), 1.15-1.40(m,9H),
0.90(t,J=7Hz,3H), 0.86(t,J=7Hz,3H).
.. ~ .......... . . .
,: .- . . . :~,
- ,- . . . :.:
WO91/On~25 PCT/US90/03630
`:
Z~. 5,_7155
--111--
Example 1~2
N-(4',8'-Dihydro~y-~'-~inQlylc~arbonyl)-R-
Phenylglycine-di-n-~entylamide
The product of example 130 was coupled in a similar
manner to that in example 8 to provide the title compound.
mp= 89-91C. MS(CI) m/e 478(m+H) , 293, 190, 177. lH
NMR(DMSOd6,300MHz) ~ 9.91(bd,J=8Hz,lH), 7.55(m,2H), 7.35-
7.45(m,7H), 7.08(dd,J=1,7.5Hz,lHj, 6.11(bd,J=8Hz,lH),
3.05-3.30(m,4H), 1.60(m,lH), 1.48(m,2H), 1.13-1.35(m,9H),
0.85(t,J=7Hz,3H), 0.78(t,J=7Hz,3H). C,H,N analysis
calculated for C28H35N34' 0-3 H2
8.70; found: C 69.61, H 7.40, N 8.65.
Example 133
N-(3'-Chlorophenylaminocarbonyl)-R-phenylglycine-di-
The product of example 13b was reacted with 3-
chlorophenylisocyanate to provide the title compound.
MS(CI) m/e 444(m+H) , 425, 317, 291, 259, 242. lH
NMR(CDC13,300MHz) ~ 7.95(bs,1H), 7.42(m,1H), 7.22-
7.34(m,5H), 7.13(d,J=7.5Hz,lH), 7.08(m,2H), 6.89(m,lH),
5.92(d,J=8Hz,lH), 3.50(m,lH), 3.00-3.30(m,4H), 1.43-
1.63(m,3H), 1.10-1.30(m,8H), 0.84(t,J=7Hz,3H),
0.78(t,J=7Hz,3H).
,. . i, , :- , .: , : : .
. . ,. : . -- . -:
, ,~ ...
- :
: . - . !
,'' ~ ~ ' " ',' '
W09l/00725 P~T/US90/03630
2~ , SS
-112-
~m~l~ 134
N-(3'-Methylphenylaminocarbonyl)-R-phenylqlYcine-di-
n-pentylamide
The product of example 130 was reacted with 3-
methylphenylisocyanate to provide the title compound.
MS(CI) m/e 424(m+H) , 374, 317, 291, 276, 239, 228. H
NMR(CDCl3,300MHz) ~ 7.27-7.48(m,5H), 7.18~m, L H),
7.12~d,J=8Hz,lH), 7.06~m,2H), 6.82~bd,J=8Hz,lH),
6.77(bd,J=8Hz,lH), 5.87(d,J=8Hz,lH), 3.51~m,lH),
3.20~m,2H), 3.04~m,1H), 2.28~s,3H), 1.50~bm,qH), 1.10-
1.30~m,8H), 0.84~t,J=7Hz,3H), 0.82~t,J=7Hz,3H).
Example 135
N-(5'-Fluoroindolylcarbonyl)-R-p:~enylalycine-di-n-
pentylamide
The product of example 130 was reacted with 5-
fluoroindole-2-carboxylic acid in a manner similar to that
in example 4 to provide the desired product. mp= 94-6C.
MS~CI) m/e 452~m+H) , 276, 267, 184. H NMR(CDCl3,300MHz)
9.36(bs,lH), 7.96(d,J=7Hz,lH), 7.50~m,2H), 7.30-
7.40~m,3H), 7.36~s,lH), 7.33(m,lH), 6.98(dt,J=2.5,9Hz,lH),
6.91(m,lH), 5.94(d,J=7Hz,lH), 3.53(m,lH), 3.13-3.30(m,2H),
3.04(m,lH), 1.45-1.65~m,4H), 1.10-1.40~m,8H),
0.89(t,J=7Hz,3H), 0.85(t,J=7Hz,3H). C,H,N analysis
calculated for C27H34FN3O2: C 71.81, H 7.59, N 9.31;
found: C 71.53, H 7.50, N 9.30.
.: . :
,: . . . : . . . -
.. . : . ~,: - : -
. . . ~, , : .
.
., :
.. , . . .. ~ . . . . .
WO91/00725 PCT/US90/0363~
2~5~isS
-113-
E~m~l~ 13~
N-(5'-Chloroindolylcarbonyl)-R-phenylalycine-di-n-
pentylamide
The product of example 130 was reacted with 5-
Chloroindole-2-carboxylic acid in a manner similar to that
in example 4 to provide the title compound. MS~CI) m/e
468(m+H) , 434, 302, 276, 212. H NMR~CDC13,300MHz)
9.36(bs,lH), 7.97~d,J=7Hz,lH), 7.59(m,lH), 7.50(m,2H),
7.35(m,3H), 7.22(m,2H), 6.89(m,lH), 5.94(d,J=7Hz,lH),
3.53(m,lH), 3.15-3.30(m,2H), 3.04~m,lH), 1.45-l.60(m,9H),
1.10-1.40~m,8H), 0,89~t,J=7Hz,3H), 0.85~t,J=7Hz,3H).
C,H,N analysis calculated for C27H34ClN3O2: C 69.29, H
7.32, N 8.98; found: C 69.49, H 7.36, N 8.95.
E~ ~
N-(2'-Ouinolylcarbonyl)-~-Phenylalycine-di-n-
~entylamide
The product of example 130 was coupled in a similar
manner to that in example 5 to provide the desired
compound. mpz 116-7 C. MS~CI) m/e 446~m+H) , 289, 277,
261, 246. H NMR~CDCl3,300MHz) ~ 9.62~d,J=8Hz,lH),
8.24(bs,2H), 8.17~d,J=8Hz,lH), 7.83~d,J=8Hz,lH),
7.74~m,lH), 7.59~m,3H), 7.30-7.40~m,3H), 6.06~d,J=8Hz,lH),
3.61~m,lH), 3.32~m,lH), 3.0-3.20~m,2H), 1.50-1.65~m,4H),
1.15-1.40~m,8H), 0.89~t,J=7Hz,3H), 0.87~t,J=7Hz,3H).
C,H,N analysis calculated for C28H35N3O2: C 75.47, H 7.92,
N 9.43; found: C 75.45, H 7.91, N 9.43.
- . .
~ ',, ', ' ':' `, ~- , ' '
- :, , ; .......... : . "
- ;, . . .
WO91/0072~PCTJUS90/0363~
~. :., .
21?~755
-114-
E~
N ' - (t-Butyloxycarbonyl)-l-amino-cvclch~x~ne-(di-n-
pentyl)carboxamide
The product was prepared as in example 1 from di-n-
pentylamine and N ' -t-Butyloxycarbonyl-l-aminocyclohexane
carboxylic acid. MS(CI) m/e 383(m+H) . H
NMR~CDCl3,300MHz) ~ 4.70(bs,1H), 3.20-3.50(m,4H), 1.85-
2.0(m,4H), 1.45-1.70(m,8H), 1.42(bs,9H), 1.20-1.90(m,10H),
0.92(bt,J=7Hz,6H).
~xample 139
N ' - (3'-Ouinolylcarbonyl~ amino-cyclohexane-(di-n-
pentyl)carboxamide
The desired product was prepared via deprotection of
the product of example 138 ~in a manner similar to that in
example 2) and coupling with quinoline-3-carboxylic acid
as in example 3. mp= 136-137C.
Example 140
N ' - (t-Butyloxycarbonyl)-l-am~n~=ÇYclohexane(N-
pentyl)carboxamide
The product was prepared via coupling of N'-t-
Butyloxycarbonyl-l-aminocyclohexane carboxylic acid and
pentylamine as in example 1. MS(CI) m/e 313~m+H) , 257,
239, 213, 198. H NMR(CDC13,300MHz) ~ 6.70(s,lH),
4.52(bs,lH), 3.23(m,2H), 1.80-2.05(m,4H), 1.65(m,4H),
1.99(s,9H), 1.25-1.38(m,8H), 0.88(t,J=7Hz,3H).
: ~: j ,. : . . . . . : . - .
i i . , .-; - ~ : : . . .
. : ~ ; ,
,: . :.
WO9l/0072~ PCT/US90/03630
. :
2(~ 7SS
. .
. . ..
--115--
E~ ~
N'-(3'-Ouinolylcarbonyl~-l-amino-cyclohexane-(N-
pentyl)carboxamide
The product was obtained in a similar manner to that
in example 139 using the product of example 140 as the
starting material. MS(CI) m/e 368~m+H) . H
NMR(CDCl3,300MHz) ~ 9.38(d,J=2Hz,lH), 8.58(d,J=2Hz,lH),
8.18(d,J=8Hz,lH), 7.94(bd,J=8Hz,lH), 7.83(m,lH),
7.65(m,lH), 7.12(bs,lH), 6.27(bs,lH), 3.38(m,2H),
2.34(m,2H), 2.03(m,2H), 1.65-1.80(m,4H), 1.50-1.60(m,4H),
1.25-1.40(m,9H), 0.88(t,J=7Hz,3H). C,H,N analysis
calculated for C22H29N3O2: C 71.91, H 7.95, N 11.43;
found: C 71.73, H 7.95, N 11.33.
N-(4',8'-Dihydroxy-2'-quinolylcarbonyl)alycine-di-n-
pentylamide
The product of example 120 was deprotected in a
manner similar to that in example 80 and the resulting
amine was then coupled in a manner similar to that in
example 8 to yield the title compound. mp= 158.5-159.5C.
MS~FAB) m/e 402(m+H) , 386, 245, 217. H
NMR(DMSOd6,300MHz) ~ 9.90~bs,lH), 9.80(bs,lH),
7.55(bt,J=8Hz,lH), 7.52(bs,lH), 7.42(m,lH),
7.11(bd,J=8Hz,lH), 4.20(bd,J=6Hz,2H), 3.36(bs,H20), 3.20 -
3.33(m,4H), 1.58(m,2H), 1.48(m,2H), 1.20-1.33(m,8H),
0.85(m,6H). C,H,N analysis calculated for C22H31N3O4,
H2O: C 62.99, H 7.93, N 10.02; found: C 53.12, H 8.02, N
10 . 01 .
.
.: . -
. . : .
. .
WO91/00725 PCT/US90/03630
. ~ .
Z~ . 55
-116-
Exam~le 143
N-(2'-NaDhthovl)~lycine-di-n-~entylamide
The product of example 120 was deprotected in a
manner similar to that in example 80 and the resulting
amine was then coupled in a manner similar to that in
example 17 to yield the title compound. MS~CI) m/e
369(m~H) , 200, 184, 172. H NMR(CDCl3,300MHz) S
8.38(s,lH), 7.85-7.95~m,4H), 7.50-7.60(m,3H),
9.30~d,J=4Hz,2H), 3.40(t,J=7.SHz,2H), 3.26(t,J=7.SHz,2H),
1.60(m,4H), 1.25-1.45(m,8H), 0.94(t,J=7Hz,3H),
0.92(t,J=7Hz,3H). C,H,N analysis calculated for
C23H32N2O2: C 74.96, H 8.75, N 7.68; found: C 74.44, H
8.75, N 7.55.
N-(6'-Hvdroxy-2'-nanhthoyl)alycine-di-n-pentylamide
The product of example 120 was deprot~cted in a
manner similar to that in example 80 and the resulting
amine was then coupled with 6-hydroxy-2-naphthoic acid in
a manner similar to that in example 17 to yield the title
compound. MS(CI~ m/e 385~m+H) , 228, 200, 184. 1H
NMR(DMSOd6,300MHz) S 8.58(bt,J=6Hz,lH), 8.36(bs,lH),
7.86(m,2H), 7.63(d,J=8Hz,lH), 7.15(m,2H),
4.14(d,J=5Hz,2H), 3.20-3.35(m,4H), 1.60(m,2H), 1.45(m,2H),
1.20-1.35(m,8H), 0.89(t,J=7Hz,3H), 0.86(t,J=7Hz,3H). C,H,N
analysis calculated for C23H32N2O3: C 71.84, H 8.39, N
7.29; found: C 71.73, H 8.36, N 7.21.
:: ., . .: :, ,, ,, , ". ::, .: : -
.: . . . .. . .~ -
.
WO91/00725 PCT~US90/03630
':
75S
-117-
~ample 145
N-~'-Methylphenylaminocarbonvl)alvcine-di-n-
E~nt~lamide
The product of example 120 was deprotected in a
manner similar to that in example 80 and the resulting
amine was then coupled with 3-methylphenylisocyanate to
yield the title compound. mp= 66-7C. MS(CI) m/e
348(m+H) , 241, 215, 200, 184. H NMR(CDCl3,300MHz)
7.08-7.20(m,3H), 7.03~bs,lH), 6.86(bd,J=7Hz,lH),
6.21(bs,lH), 4.13(bs,2H), 3.32(bt,J=7.SHz,2H),
3.21(bt,J=7.5Hz,2H), 2.30(s,3H), 1.45-1.65(m,4H), 1.20-
1.40(m,8H), 0.92(t,J=7Hz,3H), 0.86(t,J=7Hz,3H). C,H,N
analysis calculated for C20H33N3O2:
C 69.13, H 9.57, N 12.09; found: C 68.99, H 9.56, N 12.04.
~xample 196
N-(2'-Chloro~henylaminocarbonyl3-(2R.3S~-(O-
benzvl)Threonine-di-n-pentylamide
The reaction was performed in a similar manner as in
the example above utilizing 0.35 g of the hydrochloride
salt of example 30, 2-chlorophenylisocyanate (0.16 g), and
TEA (0.135 mL). The product was purified using chloroform
and methanol as the elutant mixture. The oily product was
isolated in 83% yield (0.42 g). [a]D= +21.8 (c=0.11,
MeOH). MS(CI) m/e 502(m+H) . H NMR(CDCl3,300MHz) ~
0.85(m,6H), 1.23(m,11H), 1.43-1.65(m,4H), 3.0-3.21(m,2H),
3.55(m,2H), 3.33(m,lH), 4.57(d,J=lSHz,lH),
4.63(d,J=15Hz,lH), 4.98(m,1H), 6.48(d,J=9Hz,lH),
6.95(t,J=7Hz,lH), 7.2(m,2H), 7.3(m,6H), 8.11(d,J=9Hz,lH).
-t
. . , . , - . ,
, - . . . . . .
. : . ' :, . : :
,,
WO91/00725 PCT/US90/03630
,,~c~j
J~
-118-
C,H,N analysis calculated for C28H40ClN3O3, 0.3 CH~13: C
63.19, H 7.55, N 7.81; found: C 63.21, H 7.39, N 7.82.
~m~l~ 147
N-(4' 8'-Dihydroxy-2'-quinolylcar~onyl)-(2R 3S)-(O-
benzyl)-Threonine-di-n-pentylamide
The reaction was performed in a similar manner as in
example 8 utilizing 0.35 g of the hydrochloride salt of
example 30 4,8-dihydroxyquinoline-2-carboxylic acid (0.21
g), EDCI ~0.22 g), HOBt (0.14 g), and NMM (0.22 g). The
oily product was isolated in 60% yield (0.32 g)- [~]D=
+8.0 (c=0.125, MeOH). MS(CI) m/e 536(m+H) . 1H
NMR(DMSOd6,300MHz) ~ 0.82(m,6H), 1.15-1.3(m,11H), 1.4-
1.6(m,4H), 3.2-3.65(m,~H~, 4.08(m,lH), 4.52(d,J=12Hz,lH),
4.63(d,~-12Hz,lH), 4.98(t,J=9Hz,lH), 7.12(m,SH)
7.42(t,J=gHz,lH), 7.55(m,2H), 9.8(d,J=9Hz,lH),
10.4(bs,1H), 11 72(bs,2H) C,H,N analysis calculated for
C31H41N3O5, H2O: C 67.25, H 7.83, N 7.59; found: C 67.19,
H 7.60, N 7.38.
Example lq8
Methyl Boc-R-Methionine-S-(p-hydroxy)-phenylalycinate
Boc-R-methionine (250 mg, 1 mmol) , methyl p-
hydroxyphenylglycinate hydrochloride (217 mg, 1 mmol) and
triethylamine (139 ~L, 1 mmol) were combined in 10 mL of
dichloromethane at 0C and treated with BOPCl (254 mg,
lmmol). Additional BOPCl (254 mg) and TEA (134 ~L) were
added after one day. After two days, the reaction mixture
was poured into EtOAc and extracted successively with 0.1%
citric acid, 0 1 M NaHCO3 and water. The solution was
. . . . .
- - .. .
- . -: . , .,:: :.
W09l~00725 PCT/US90/03630
2~ ?~S
--119--
therl dried over MgSO4, filtered and evaporated to yield
288 mg, 0.7 mmol (70%). Rf = 0.56 ~1:1 hexanes - EtOAc)
mp = 158C ~dec). MS~CI) m/e 413(m+H)+, 357, 313.
1HNMR~CDCl3,300MHz) d 1.43(s,9H), 3.72(s,3H),
6.73(d,J=8Hz,2H), 7.17(d,J=8Hz,2H), 7.33~bs,1H).
Example 149
Methyl R-Methionine-S-(p-hydroxy)-phenylalycinate
hvdrochloride
The product of the example 148 ~250 mg, 0.6 mmol) was
treated with 5 mL of 4 N HCl in dioxane at room
temperature under a nitrogen atmosphere. After 30
minutes, the excess reagent was evaporated to yield
quantitatively the product.
~xampl~ 150
Methyl N-(3'-Ouinolylcarbonyl)-R-Methionine-S-(p-
hydrox.y)-phenylalycinate
The hydrochloride salt of example 149 ~50 mg, 0.14
mmol), 3-quinoline carboxylic acid ~26 mg, 0.15 mmol) and
TEA ~21 ~L, 0.15 mmol) were dissolved into 5 mL
methylenechloride and treated with EDCI ~29 mg, 0.15 mmol)
for 4 hours. The reaction was poured into EtOAc and
extracted with 0.1% citric acid and water followed by
drying over MgSO4. The resultant filtrate was
concentrated and chromatographed over silica gel eluting
with a 2:1 to 1:2 hexane - EtOAc gradient to yield 29 mg,
0.06 mmol (44%). MS~CI) m/e 468(m+H)+, 393, 287.
1HNMR(CDC13,300MHz) ~ 2.04~s,3H), 2.12-2.20~m,2H), 2.42-
.. "
:, :.
WO91/00725 PCT/VS90/03630
2~ _755
-120-
2.52(m,lH), 2.57-2.67(m,lH), 3.65(s,3H), 5.05(q,J=7Hz,lH),
5.41(d,J=6Hz,lH), 6.77(d,J=8Hz,2H), 7.16(d,J=8Hz,2H),
7.59(dt,J=1,7Hz,lH), 7.73-7.82(m,3H), 7.83(d,J=8Hz,lH),
8.12(d,J=8Hz,lH), 8.61(d,J=2Hz,lH), 9.30(d,J=2Hz,lH).
C,H,N analysis calculated for C2qH25N3O5S 0.5 H2O: C
60.99, H 5.60, N 8.81; found: C 60.69, H 5.63, N 8.35.
Example 151
N-(3'-Ouinolylcarbonyl)-R-Serine-di-n-pentylamide
BTFA (trifluoroacetoxyboronate) 0.154 g, 0.4 mmol was
added to the product of example 27 (71 mg, 0.145 mol)
dissolved in 2 mL of methylene chloride. Another mL of
methylene chloride was added and the reaction was
monitored by tlc. After 20 minutes of stirring at ambient
temperature, the starting material was consumed and the
solvents with methanul were evaporated under vacuum. This
evaporation sequence using methanol was repeated several
times . The residue was separated by chromatography using
EtoAc-hexane ~1:1) as the elutants. An oily product was
isolated in 69~ yield (40 mg). MS(CI) m/e 400 (m+H)+.
lHNMR(CD30D,300MHz) ~ 0.94 (m,6H), 1.26-1.44 (m,8H), 1.54-
1.64(m,2H), 1.68-1.86(m,3H), 3.25-3.35(m,lH), 3.43-
3.62(m,3H), 3.82-3.96(m,2H), 5.22(t,J=6Hz,lH),
7.73(t,J=6Hz,lH), 7.91(t,J=6Hz,lH), 8.07(d,J=9Hz,lH),
8.12(d,J=9Hz,lH), 8.9(s,lH), 9.28(s,lH).
E~ample 152
N-(8'-~ydro~.y-2-quinolylcarbonyl)-alycine-di-n-
pentylamide
:. . :. . . . . , ~
; ... . ,, - - : :
. .
- .
'. ' '~ :': '~ ' ' ' '' '
, ~ :
... . .
WO91/00725 PCT/US~0/03630
,~ .
7 5 5
-121-
Similar to example 121, the product of example 120
was deprotected and coupled to 8-hydroxy-2-quinolinic
carboxylic acid in a standard fashion utilizing EDCI etc.
to provide the product. MS(CI) m/e 386 (m+H)+.
lHNMR(CDC13,300MHz) ~ 8.96(bs,lH), 8.23(s,2H), 8.02(s,lH),
7.53(t,J=7.5Hz,lH), 7.36(dd,J=1,7.5Hz,lH),
7.23(dd,J=1,7.5Hz,lH), 4.34(d,J=5Hz,2H),
3.42(bt,J=8hz,2H), 3.28(bt,J=8Hz,2H), 1.55-1.70(m,4H),
1.25-1.40(m,8H), 0.93(apparent q,6H). C,H,N analysis
calculated for C22H31N3O3 0.2 H2O: C 67.91, H 8.13, N
10.80; found: C 67.90, H 8.14, N 10.69.
Example 153
N-Methyl-N-(3'Ouinolylcarbonyl)-alycine-di-n-
pentylamide
The product of example 127 was methylated using
bistrimethylsilylamide and methyl iodide in TH~ at -78C
warming to ambient temperature to provide product after
standard workup and purification. MS(DCI) m/e 384(m+H)+.
Example 154
N-(3'-Iodo-2'-indolylcarbonyl)-alycine-di-n-
pentylamide
The product of example 121- was iodinated with N-
iodosuccinimide to provide product after chromatographic
purification. MS(DCI) m/e 484(m+H)+. C,H,N analysis
calculated for C21H30IN3O2: C 52.18, H 6.25, N 8.69;
found: C 52.09, H 6.21, N 8.49.
~ ., .. ., , , .;, .
: ...... : . ::.:. . . :. . .
: ~ ,-: ; : . ~
: ' - : ' ~ , ~ -
WO 9l/00725 PCT/US90/03630
~''
, 5
-122-
~mDle 155
~-(2'-Indolvlcarbonvl)-R-Alan'ne-di-n-pentylamide
In a similar fashion to examples 57 and 58 the
product was prepared from the corresponding R-alanyl-di-n-
pentylamide hydrochloride and 3-quinoline carboxylic acid
to yield product. MS(CI) m/e 372~m+H)+. C,H,N analysis
calculated for titled product: C 71.1, H 8.95, N 11.31;
found: C 70.76, H 9.03, N 11.17.
The ability of the compounds of Formula I to interact
with CCK receptors and to antagonize CCK can be
demonstrated 1~ vitro using the following protocols.
~harmacoloaical Methods
CCK8 ~Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2] was
purchased from Peptide International (Louisville,XY) or
Cambridge Research Biochemicals (Atlantic Beach, NY)
EGTA, HEPES and BSA were purchased from Sigma Chemical Co.
(St. Louis, MO). [125I]BH-CCK8 tspecific activity, 2200
Ci/mmol) and Aquasol-2 scintillation cocktail were
obtained from New England Nuclear (Boston, MA). Bestatin
and phosphoramidon were purchased from Peptide
International. Male guinea pigs, 250 to 325 g, were
obtained from Scientific Small Animal Laboratory and Farm
(Arlington Heights, IL).
.. . ..
. - - . .:.
.. ~ ' '" " ~' .
~" ~ ' ' .
',,
. ~ ' ' . , ." ~ , ,, ,'.
. , .
WO91t00725 PCT/US90/03630
, . .
S5
-123-
Pr~tocol fQL B~si~aL~am~ ~i~di~a Experiments
1. Guinea Pig Cerebral Cortical and Pancreatic Membrane
~reparations
Cortical and pancreatic membranes were prepared as
described ~Lin and Miller; J. Pharmacol. E~. Ther. 232,
775-780, 1985). In brief, cortex and pancreas were
removed and rinsed with ice-cold saline. Visible fat and
connective tissues were removed from the pancreas.
Tissues were weighed and homogenized separately in
approximately 25 mL of ice-cold 50 mM Tris-HCl buffer, pH
7.4 at 4C, with a Brin~man Poloytron for 30 sec, setting
7 The homogenates were centrifuged for 10 min at 1075 x
g and pellets discarded. The supernatants were saved and
centrifuged at 38,730 x g for 20 min The resultant
pellets were rehomogenized in 25 mL of 50 mM Tris-HCl
buffer with Teflon-glass homogenizer, 5 up and down
strokes. The homogenates were centrifuged again at 38,730
x g for 20 min. Pellets were then resuspended in 20 mM
HEPES, containing 1 mM EGTA, 118 mM NaCl, 4.7 mM KCl, 5 mM
MgC12, 100 ~M bestatin, 3 ~M phosphoramidon, pH 7.4 at
22C, with a Teflon-glass homogenizer, 15 up and down
strokes. Resuspension volume was 15-18 mL per gram of
original wet weight for the cortex and 60 mL per gram for
the pancreas.
.. . .
, ,- ~ ., : :
,. ~ , . ,
W09l/00725 PCT~US90/03630
,~--
2~ ~ . 5 ~ -124-
2. Incubation Conditions
~ I~Bolton-Hunter CCX8 ([1 I]BH-CCK8), and the
test compounds were diluted with HEPES-EGTA-salt buffer
(see above) containing 0.5% bovine serum albumin (BSA).
To 1 mL Skatron polystyrene tubes were added 25 ~L of
[ I]BH-CCK8, and 200~L of membrane suspension. The
final BSA concentration was 0.1%. The cortical tissues
were incubated at 30C for 150 min and pancreatic tissues
were incubated at 37C for 30 min. Incubations were
terminated by filtration using Skatron Cell Harvester and
SS32 microfiber filter mats. The specific binding of
[125I]BH-CCK8, defined as the difference between binding
in the absence and presence of 1 ~M CCK8, was 85-90% of
total binding in cortex and 90-95% in pancreas. IC50's
were determined from the Hill analysis. The results of
these binding assays are shown in Table 1.
Protocol for Amylase Release
This assay was performed using the modified protocol
of Lin et al., J. pharmacol. E~- ~h~. 236, 729-739,
1986.
1. Guinea Pia a~ini Preparation
Guinea pig ocean were prepared by the method of
Bruzzone et al. (Biochem. J. 226, 621-624, 1985) as
follows. The pancreas was dissected and connective
tissues and blood vessels were removed. The pancreas was
cut into small pieces (2 mm) by a seizure and placed in a
" . ... . .
:
,
,
WO91/0~72~ PCT/US90/03fi30
2~?~55
-125-
15 mL conical plastic tube containing 2.5 mL of Krebs-
Ringer HEPES (KRH) buffer plus 400 units per mL of
collagenase. The composition of the KP~H buffer was:
HEPES, 12.5 mM; NaCl, 118 mM; KCl, 4.8 mM; CaCl2, 1 mM;
KH2PO4, 1.2 mM; MgS04, 1.2 mM; NaHC03, 5 mM; glucose, 10
mM at pH 7.4. The buffer was supplemented with 1% MEM
vitamins, 1% MEM amino acids and 0.001% aprotinin. The
tube was shaken by hand until the suspension appeared
homogeneous, usually 5-6 min. Five mL of the KRH, without
collagenase and with 0.1% BSA, was added and the tube was
centrifuged at 50 x g for 35 sec. The supernatant was
discarded and 6 mL of the KRH was added to the cell
pellet. Cells were triturated by a glass pipette and
centrifuged at 50 x g for 35 sec. This wash procedure was
repeated once. The cell pellet from the last
centrifugation step was then resuspended in 15 mL of KRH
containing 0.1% BSA. The contents were filtered through a
dual nylon mesh, size 275 and 75 ~M. The fil~rate,
containing the acini, was centrifuged at 50 x g for 3 min.
The acini were then resuspended in 5 mL of KRH-BSA buffer
for 30 min at 37C, under 100% oxygen atmosphere (2)'
with a change of fresh buffer at 15 min.
2. Amylase Assay
After the 30 min incubation time, the acini were
resuspended in 100 volumes of KRH-BSA buffer, containing 3
~M phosphoramidon and 100 ~M bestatin. While stirring,
400 ~L of acini were added to 1.5 mL microcentrifuge tubes
.. . ~ .
WO9l/007~5 PCT/US90/03630
Z~ , 55
-126-
containing 50 ~L of CCK8, buffer, or test compounds. The
final assay volume was 500 ~L. Tubes were vortexed and
placed in a 37C water bath, un-ler 100% 2~ for 30 min.
Afterward, tubes were centrifuged at 10,000 g for 1 min.
Amylase activity in the supernatant and the cell pellet
were separately determined after appropriate dilutions in
0.1% Triton X-100, 10 mM NaH2P04, pH 7.4 by Abbott Amylase
A-gent test using the Abbott Bichromatic Analyzer 200.
The reference concentration for CCK8 in determining the
IC50's of the compounds of Formula I was 3 x 10 OM. The
results of this assay are shown in Table 2.
In Vitro Results
The preferred compounds of Formula I are those which
inhibited specific ~125I]-BH-CCK8 binding in a
concentration dependent manner. Analysis of [125I]-BH-
CCK8 receptor binding in the absence and presence of the
compounds of formula I indicated the compounds of formula
I inhibited specific [125I]-BH-CCK8 receptor binding. The
IC50 values of the compounds of Formula I are presented in
Table 1.
.':' ', '' ' , ;' . ~ , :
, , - . : . -
;
- ~:
. , - , .
W09l/00725 PCT/us9O/03630
S5
-127-
TABLE 1
[ 25I]-BH-CCK8 Binding
Compound ofIC50 ~nM)
Exam~le Pancreas Cortex
3 40 17,000
4 100 ~10,000
S 27 >10,000
7 290 >10,000
8 12 <10,000
13 190 1-10,000
17 200 ~100,000
23 87 ~10,000
24 170 >10,000
27 140 7200
31 73 ~10,000
32 23 210,000
33 30 ~10,000
34 9 >10,000
37 210 ~10,000
43 48 1400
47 320 ~10,000
19 2qO0
53 24 ~10,000
56 530 >10,000
- 62 140 5200
41 <10,000
66 150 1-10,000
:~ . . ,~ . ,,- :.- ~ . . . : ,
WO 91/00725 PCr/US90~03630
~ .~
. SS
:,
--128--
260~10,000
73 180>10,000
74 70~10, 000
160>10, 000 .
76 92>10, 000
37~10,000
81 120 5300
82a 250>30, 000
87 29>10, 000
91 120 3000
93 195~10,000
99 56~10,000
100 63~10,000
117 7428, 000
118 42 3, 300
119 110 6,200
125 160~10, 000
131 9.3 1600
132 3.1 1700
133 210~10,000
135 69 6000
142 160>10, 000
193 130
- 145 100
147 86 2,900
150 980>10, 000
151 51
152 520>10,000
155 230<10,000
~,
.'~ ` - - ~........ -
: ' ., ~, ' , ' '` ' .
:
WO91/0072~ PCT/US90/03630
, .
. 55:
-129-
The results herein also indicate that compounds of
the invention possess selectivity for the pancreatic (type
A) CCK receptors.
Inhibition of
CCK8-induced
Compound of Amylase Release
E~m~le IC~Q ~nM)
3 290
4 <100,000
8 <100,000
17 <30,000
31 <100,000
32 <1000
34 <100,000
93 140
<100,000
59 -100,000
<100,000
74 <100,000
<10,000
81 <10,000
.. . ,. . - :
. ~ : , . ' . :?
.
: . . : :
. .
WO 91tO0725 PCr/US90/03~i30
Ç~:
Z~
-130-
91 <10,000
99 <10,000
131 <100,000
132 <100,000
141 <30,000
151 <10,000
These results indicate that compounds of the
invention are CCK antagonists.
In Vivo Results
The ability of the compounds of Formula I to interact
with CCK receptors and to antagonize CCK n vivo can be
demonstrated using the following protocols.
Ll~inn ~ Induced ,Gastric Emptyina
Three fasted mice were dosed (p.o.) with the test
compound. CCK8 (80 ~g/kg s.c.) was administered within 60
minutes and charcoal meal (0.1 mL of 10% suspension) was
given orally 5 minutes later. The animals were sacrificed
within an additional 5 minutes.
Gastric emptying, defined as the presence of charcoal
within the intestine beyond the pyloric sphincter, is
inhibited by CCK8. Gastric emptyinq observed in 2 or 3
mice (greater than 1) indicates antagonism of CCK8.
::-: , , ~ .
:
,, ~
.: : : . . .-.
:. : , , -. . , .. :
~' .
.
WO9l/0072~ PCT/US90/03630
; ''
2C~ 55 ; . ~ ;
-131-
Compound Number of mice
gf example Dose (p.o.) with Gastric Emptyina
118 100 mg/kg 2
Measurement of Plasma In~lin Level Followina Treatment
~l~h ~ and a Compound of Formula I
The ability of the compounds of Formula I to
antagonize CCK induced hyperinsulinemia can be
demonstrated in vivo using the following protocol.
Male mice, 20-30 g, were used in all experiments.
The animals were fed with laboratory lab chow and water ad
libitum. The compound of Formula I (1-100 mg/kg in 0.2 mL
of 0.9% saline) was administered i.p. Ten minutes later
CCK8 (0.2 to 200 nmole/kg in 0.2 mL of 0.9% saline) or
saline was injected into the tail vein. Two minutes later
the animals were sacrificed and blood was collected into
1.5 mL heparinized polypropylene tubes. The tubes were
centrifuged at 10,000 x g for 2 minutes. Insulin levels
were determined in the supernatant (plasma) by an RIA
method using kits from Radioassay Systems Laboratory
(Carson, CA.) or Novo Biolabs (MA.).
Antagonism of ~ Mediated Behavioral Effect n Mice ~i~h
Compounds of Formula I
Male Swiss CD-l mice (Charles River) (22-27 g) are
provided ample food (Purina Lab Chow~ and water until the
time of their injection with the test compounds.
: .- : .
,. , ~ :. , . : .
- - -: . ~
: ` :. ,i: . : '~, .~ :
WO91/00725 PCT/US90/03630
2~ 55
-132-
ICV injections were given by a free-hand method
similar to that previously described ~aley and McCormick,
~L~ J- Pharmacol- Chemother. 1~, 12-15 1957). The
animals were placed on a slightly elevated metal grid and
restrained by the thumb and forefinger at the level of the
shoulders, thus immobilizing their heads. Injections were
made with a 30 gauge needle with a "stop" cor.sisting of a
piece of tygon tubing to limit penetration of the needle
to about 4.5 mm below the surface of the skin. The needle
was inserted perpendicular to the skull at a midline point
equidistant from the eye and an equal distance posterior
from the level of the eyes such that the injection site
and the two eyes form an equilateral triangle. The
injection volume (5 ~L) was expelled smoothly over a
period of approximately 1 second.
Immediately after the injections the mice were placed
in their cages and allowed a 15 minute recovery period
~rior to the beginning of the behavioral observations.
For the behavioral observations, the mice were placed
in clear plastic cages. Each cage measured 19 x 26 x 15
centimeters and contained a 60-tube polypropylene test
tube rack (NALGENE #5970-0020) placed on end in the center
of the cage to enhance exploratory activity. Observations
were made every 30 seconds for a period of 30 minutes.
sehavior was compared between drug and CCK8 treated micei
CCK8 treated mice; and mice treated with an equal volume
of carrier (usually 0.9~ saline or 5% dimethylsulfoxide in
water). Locomotion as reported here consisted of either
floor locomotion or active climbing on the rack.
Differences among groups were analyzed by Newman-Kewels
-. ':
.. . . .
: . . . . . :
,' ' ~ ':, '
WO91/00725 PCT/US90/03630
2~'~ " 5~ ; ~
-133-
analysis and a probability level of p< 0.05 was accepted
as significant. Each group tested consisted of 10
animals. The results of this test indicate that compounds
of Formula I are antagonists of CCK in vivo. Minimally
effective doses ~MED) are defined as that dose at which a
statistically significant reversal of CCK-induced
inactivity was observed when the test compound of formula
I and CCK8 were coadministered.
Compound of Dose of
8 - MED
43 3 nmol 3 nmol
The compounds of Formula I antagonize CCK which makes
the compounds useful in the treatment and prevention of
disease states in mammals ~especially humans) wherein CCX
or gastrin may be involved, for example, gastrointestinal
disorders such as irritable bowel syndrome, ulcers, excess
pancreatic or gastric secretion, hyperinsulinemia, acute
pancreatitis, GI cancers ~especially cancers of the gall
bladder and pancreas), motility disorders, pain
~potentiation of opiate analgesia), central nervous system
disorders caused by CCK's interaction with dopamine such
as neuroleptic disorders, tardive dyskinesia, Parkinson's
disease, psychosis, including schizophrenia, or Gilles de
la Tourette Syndrome; disorders of the appetite regulatory
' ' ' ,., .' , '', '
. ', ; " , . .
-t:
.' ' '' ' , ' ',
WOgl/00725 PCT~US90/03630
t.._;
2~ J S S
-134-
systems, bulimia, Zollinger-Ellison syndrome, and central
G cell hyperplasia, and the treatment of substance abuse.
The compounds of the present invention can be used in
the form of salts derived from inorganic or organic acids.
These salts include but are not limited to the following:
acetate, adipate, alginate, citrate, aspartate, benzoate,
benzenesulfonate, bisulfate, butyrate, camphorate,
camphorsulfonate, digluconate, cyclopentanepropionate,
dodecylsulfate, ethanesulfonate, glucoheptonate,
glycerphosphate, hemisulfate, heptonate, hexanoate,
fumarate, hydrochloride, hydrobromide, hydroiodide, 2-
hydroxy-ethanesulfonate, lactate, maleate,
methanesulfonate, nicotinate, 2-naphthalenesulfonate,
oxalate, pamoate, pectinate, persulfate, 3-
phenylproplonate, picrate, pivalate, propionate,
succinate, tartrate, thiocyanate, tosylate, and
undecanoate Also, the basic nitrogen-containing groups
can be quaternized with such agents as loweralkyl halides,
such as methyl, ethyl, propyl, and butyl chloride,
bromides, and iodides; dialkyl sulfates, long chain
halides such as decyl, lauryl, myristyl, and stearyl
chlorides, bromides and iodides, arylalkyl halides like
benzyl and phenethyl bromides, and others. Water or oil-
soluble or dispersible products are thereby obtained.
The pharmaceutically acceptable salts of the present
invention can be synthesized from the compounds of Formula
I which contain a basic or acidic moiety Dy conventional
methods. Generally, the salts are prepared by reacting
the free base or acid with stoichiometric amounts or with
an excess of the desired salt forming inorganic or organic
- - .. : .
~: : ~: : - : - -: . -.. .. - . .. :.. .
: : -: .
:. : . .
:::: - : :. - ,. . .
:: . ... -:. :
WO9l/0072~ PCT/US90/03630
2~ 5S
-135-
acid or base in a suitable solvent or various combinations
of solvents.
Examples of acids which may be employed to form
pharmaceutically acceptable acid addition salts include
such inorganic acids as hydrochloric acid and phosphoric
acid and such organic acids such as oxalic acid, maleic
acid, succinic acid and citric acid. Other salts include
salts with alkali metals or alkaline earth metals, such as -
sodium, potassium, calcium, or magnesium or with organic
~ases.
The pharmaceutically acceptable salts of the acid of
Formula I are also readily prepared by conventional
procedures such as treating an acid of Formula I with an
appropriate amount of base, such as an alkali or alkaline
earth metal hydroxide e.g. sodium, potassium, lithium,
calcium, or magnesium, or an organic base such as an
amine, e.g., dibenzylethylenediamine, cyclohexylamine,
dicyclohexylamine, triethylamine, piperidine, pyrrolidine,
benzylamine, and the like, or a quaterary ammonium
hydroxide such as tetramethylammonium hydroxide and the
like.
When a compound of Formula I is used as an antagonist
of CCK or gastrin in a human subject, the total daily dose
administered in single or divided doses may be in amounts,
for example, from 0.001 to 1000 mg a day and more usually
1 to 1000 mg. Dosage unit compositions may contain such
amounts of submultiples thereof to make up the daily dose.
The amount of active ingredient thzt may be combined
with the carrier materials to produce a single dosage form
:.: . . .: ::
WO91/00725 PCT/US90~03630
Z~75~;
-136-
will vary depending upon the host treated, the particular
treatment and the particular mode of administration.
It will be understood, however, that the specific -
dose level for any particular patient will depend upon a
variety of factors including the activity of the specific
compound employed, the age, body weight, general health,
sex, diet, time of administration, rate of excretion, drug
combination, and the severity of the particular disease
undergoing therapy.
The compounds of the present invention may be
administered orally, parenterally, by inhalation spray,
rectally, or topically in dosage unit formulations
containing conventional nontoxic pharmaceutically
acceptable carriers, adjuvants, and vehicles as desired.
The term parenteral as used herein includes subcutaneous
injections, intravenous, intramuscular, intrasternal
injection, or infusion techniques.
Injectable preparations, for example, sterile
injectable aqueous or oleagenous suspensions may be
formulated according to the known art using suitable
dispersing or wetting agents and suspending agents. The
sterile injectable preparation may also be a sterile
injectable solution or suspension in a nontoxic
parenterally acceptable diluent or solvent, for example,
as a solution in 1,3-butandiol. Among the acceptable
vehicles and solvents that may be employed are water,
Ringer's solution, isotonic sodium chloride solution. In
addition, sterile, fixed oils are conventionally employed
as a solvent or suspending medium. ~or t~is purpose any
bland fixed oil may be employed includinc synthetic mono-
:: ,
.:. : : :- :.
,
, . .
" ':
WO91100725 PCT/US~0/03630
-
2(~?755 .
-137-
or diglycerides. In addition, fatty acids such as oleic
acid find use in the preparation of injectables.
Suppositories for rectal administration of the drug
can be prepared by mixing the drug with a suitable
nonirritating excipient such as cocoa butter and
polyethylene glycols which are solid at ordinary
temperatures but liquid at the rectal temperature and will
therefore melt in the rectum and release the drug.
Solid dosage forms for oral administration may
include capsules, tablets, pills, powders, and granules.
In such solid dosage forms, the active compound may be
admixed with at least one inert diluent such as sucrose,
lactose, or starch. Such dosage forms may also comprise,
as is normal practice, additional substances other than
inert diluents, e.g., lubricating agents such as magnesium
stearate. In the case of capsules, tablets, and pills,
the dosage forms may also comprise buffering agents.
Tablets and pills can additionally be prepared with
enteric coatings.
Liquid dosage forms for oral administration may
include pharmaceutically acceptable emulsion, solutions,
suspensions, syrups, and elixirs containing inert diluents
commonly used in the art, such as water. Such
compositions may also comprise adjuvants, such as wetting
agents, emulsifying and suspending agents, and sweetening,
flavoring, and perfuming agents.
The present agents can also be administered in the
form of liposomes. As is known in the art, liposomes are
generally derived from phospholipids or other lipid
substances. Liposomes are formed by mono- or multi-
. ~ . . . . .
- ; . ,~. : ~
.
WO91/00725 PCT/US90/03630
2~ J S S
-138-
lamellar hydrated liquid crystals that are dispersed in an
aqueous medium. Any non-toxic, physiologically acceptable
and metabolizable lipid capable of forming liposo~es can
be used. The present compositions in liposome form can
contain, in addition to the compounds of the present
invention, stabilizers, preservatives, excipients, and the
like. The preferred lipids are the phospholipids and the
phosphatidyl cholines (lecithins), both natural and
synthetic.
Methods to form liposomes are known in the art. See,
for example, Prescott, Ed., ~ethods 1~ Cell Bioloay Vol.
XIV, Academic Press, New York, N. Y. 1976, p.33 et seq.
The foregoing is merely illustrative of the invention
and is not intended to limit the invention to the
disclosed compounds. Variations and changes which are
obvious to one skilled in the art are intended to be
within the scope and nature of the invention which are
defined in the appended claims.
. .
.. . .
' ~
. .