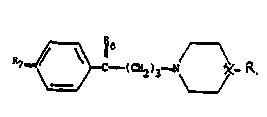Note: Descriptions are shown in the official language in which they were submitted.
W O 92/06977 PCT/FR91/0081~
~7 ~
.
NCUVE~UX DERIVES p-SUBSllTUES DE P9ENYL 4--CXYBUnANE ArINE
LEURS PROCEDES DE pREpARAlIoN ET r.F~ ooMposIIloN~
La présente invention a pour objet de nouveaux dérivés de phényl
oxybutanes, substitués en 1 par un groupe aminé, leurs procédés de
.préparation et les compositions pharmaceutiques en renfermant.
L'invention a plus particulièrement pour objet de nouveaux dérivés du
phényl butane portant en position l de la chaîne butanique, un radical
aminé cyclique.
L'invention concerne spécifiquement de nouveaux dérivés du
4-phényl 4-oxyl l-aminobutane de formule générale I
7 ~ (C~2)3-N ~ - R (I)
dans laquelle R6 est le groupe HOH, ~0, ou ~
O -tétrahydropyranyle
X est un substituant choisi dans le groupe constitué par >N- et
>CH-NH - FH3
représente COOH, terbutyle ou - C - COOH
et R represente un substituant cyclique choisi dans lc groupe con8titu~
P~
~) un substituant Xanthiquo do formule
O~
R~ R~
W O 92/06977 PCT/FR91/00811
z~ 7iL~? ~t ~
où R~ et R2, identiques ou différents, représentent un atome
d'hydrogène ou un radical alcoyle renfermant de l à 5 atomes de carbone
en chaine droite ou ramifiée, et R3 représente un radical
halogénobenzyle de la forme
CH2 ~
ou Z est un atome d'halogène
ou bien un radical -CH2, CH2 OC2 H5 (ethoxyethyle)
et b) un substituant Benzimidazoligue de formule générale
R4 ~ ~
ou R4 représente un at e d'hydrogène ou un halogène
et R5 représente un radical halogénobenzyle
CH2 - ~
dans laqualle Z est un ato~e d'halog~ne
ou ethoxy othyle
on distlngue donc parmi les composés de formule générale I deux
sous-groupes, à savoir :
-. . .: .
. . ' ' ' .:' ' -
..
. . ~.
W O 92/06977 PCT/FR91/0081I
- ~ 7 ~
~ ~, . s . ~
1. les composés de formLle générale IA pour lesquels R est un radical
Xanthine, choi6is dans le groupe constitué par les
piperazinylxanthines de formLle générale IA,
R7~ 1CH2l3 N ~ N-R (I~,
dans laquelle R est un substituant xanthique
et les plpéridylaminoxanthines de form~le générale IA"
R7 ~ (CH2)3--N~w-R ~ ")
dan~ laqyelle R e~t un substituant xanthique
2. 1-6 compo66s de formLle ,* n~ral~ ~ dan5 laquelle R ~st un radical
Benzimidazollque, choi~i6 tans le groupe constltué par les
piperazinyl benzimidazoles de ~ormule gén~rale IB,
.
W O 92/06977 PCT/FR91/00811
~7~o~
R7~ C -~ 2)3 - N ~ N ~
R~,
dans laquelle R4, R5, R6 et ~ sont définis comme précéde Y nt
et les pipéridinylamino benzimidazoles de formLle IB"
R7~ ~ C~213 - N 3 N~R~
R ~I~")
~n~ l~quelle R4, R5, R6 et ~ sont définis comme précédemment
Panmi les composés de formule * nérale I on distinguera tout
particulièrement les composés pour lesquels ~ représente un radical
terbutyle et plus sp~cifiquement les composés de formLle générale IB
dans laquelle R est un radical Benzimidazolique et notamment les
plpérazinyl benzimidazoles de fonmlle générale IB,
1 3 ~ ~
CE13 ~ t~H2) 3--N~ N--~ t~Bl)
R5
W o 92/06977 PCT/FR91/00811
2G~
dans laguelle R4 et R5 sont définis comme précédemmemt
et les pipéridinylamino benzimidazoles de formule IB"
3 ~ (C~2)3--N~h ~H~ J R~
Rs ~I6 '
dans laquelle R4 et R5 sont définis c 3 e précédemment.
D'une manière générale, les composés actuellement préférés sont ceux
pour lesquels R5 est un radical p-fluorobenzyle et R6 est un hydroxyle.
L'invention a encore pour objet les sels de composés de formLle
générale I avec un acide minéral ou organique, de préférence
thérapeutiguement-compatible. Ceux qui ne peuvent pas être utilisés en
thérapeutique comme les iodates, les périodates, les reineckates ou les
picrates servent de yen d~isolement, de purification ou de
caractérisation.
Par aillours, les lécules de formule générale I comportent au ins
un atome de Carbone asymétrique et peuvent de ce fait être dédoublées
en leurs i~ooères optiques, notamment par salification par un acide
chiral comme l'acide d-camphosulfonique, l'acide d-dibenzoyltartrique,
l'acide NNLdiethyltartramique, ou l'aclde d-glucose l-phosphorique.
Les cocpos~s de formule g ff rale ~ peuvent également être dédoublés en
leurs is d res optiquement-actifs par esterification à l'aide d'un
acide optiquement actif comme l'acide l-menthoxyacétique puis
saponification ménagée. Il est possible encore de dedoubler ces esters
W O 92/06977 PCT/FR91/00811
~r~
par hydrolyse enzymatique ou par chromatographie sur une colonne
chargée avec un absorbant chiral.
La présente invention a également pour objet un procédé de préparation
des dérivés de formLle I caractérisé en ce que :
- pcur les dérivés Xanthiques de formLle IA on fait réagir un dérive
halogéné de Xanthine ayant la formLle générale (II)
.Q ,.
~ al (II)
P~
dans laquelle ~ et R2 ont les significations précédemment définies
et Hal représente un atome de Chlore ou de Brome
avec un dérivé halogéné choisi dans le groupe formé t'un dérivé
benzylique de formule gén~rale (III)
,~3CH2,~
tIII)
ou un dérivé ethoxyethyle III'
C2~5 0CH2 CH2 Hal
cu Hal r-pr~ente un atomo t~ Chloro ou de ~romo et Z ropr~onte un
halog~ne
et obtient le dérivé Xanthique de formule *nérale rv
: . :
.. . .
. ~
- ,
W O 92/06977 PCT/FR91/00811
2 ~ 7 ~ r ~
-- 7 --
~i '
R5
dans laquelle Rl, R2 et R5 sont définis comme précédemment
puis fait réagir ce dernier co~posé avec un 4-(4-phényl~ 4-oxy
l-aminobutane de formule générale v
R7 _~c~ R
~ (v)
dans laquelle X, R6 et ~ possèdent les significations antérieures.
et R représente un substituant Xanthique
pour obtenir un ccmposé de formule IA"
Lorsque X est egal à >N- et R à xanthyle, on peut préparer les composés
de formule IA, en condensant le dérivé 4-halogéno l-~R7-phényl)butyle
de formule VI
~ C - lCH213- ~al (V11
.
:
;
.
W O 92/06977 PCr/FRs1/00811
2~ 8 -
dans laquelle R6 et R7 sont définis comme précédemment
Hal est un atome de chlore ou de brome
avec une pipérazine bloguée par un radical Xanthique de formLle VII
O
~ a, (VII)
dans laguelle Rl, R2 et R3 ont les significations antérieures~a.condensation s'effectue de préférence dans un solvant choisi parmi
les éthers cycliques et/ou un alcool renfermant jusgu'à 3 atomes de
carbone en chaine linéaire ou ramifiée, à la température de reflux de
l'alcool ou de l'éther choisi, pendant 2 à 12 heures. On obtient ainsi,
le dérivé de formule IA,
7 ~ ~ - lCNz)3 ~ 1 ~ N ~ 0 ~IA')
R3 R2 '
qu lle Rl, R2, R3, R6 t R7 sont d~finls comme pc~céde _ nt
Lorsque X est égal à >CH-NR-, on pr~pare le composé de focmule V
correspondant en condensant un dér1vé de formLle g~nérale VI d~fini
- .
~ ~ , ........ .
W O 92/06977 PCT/FR91/00811
_ g _
ccmme précédemment, avec une 4-oximino pipéridine de formule VIII
~N - N - O~
/ (VIII )
~a condensation s'effectue de préférence dans un solvant choisi parml
les alcools renfermant jusqu'à 3 atomes de carbone en chaine linéaire
ou ramifiée, en présence d'un agent alcalin comme les carbonates
alcalins à la t~mr~rature du reflux de l'alcool choisis pendant 8 à
12 heures. On obtient ainsi le dérive ~IX)
H3C--C ~--(CH~I 3~3 N-OH
CE13
dans lequel R6 est un hydroxyle et R7 est défini comme précédemment
~e dérivé oximino ~IX) est ensuite réduit en amine primaire dans un
alcool renfermant de 1 ~ 3 at es de carbone en chaine linéa~re ou
ramifi~e, par un m~tal alcalin, à une température choisie entre 20 et
le reflux de l'alcool, pendant 2 à 12 heures, selon la température
choisie. On obtient ainsi le dérive aminé de fonmule X
7~ ~CH~ ~lH~
`:
W O 92/06977 PCT/FR91/00811
.
~~ o-
dans laquelle ~ a les significations antérieures
que l'on condense avec un composé de formule générale IV, pour former
un composé de formLle IA".
~ ~(CH2~3--N~Ri
RS R2
dans laquelle Rl, R2, ~ et R5 ont les significations antérieures.
Cette condensation s'effectue de préférence dans un solvant choisi
parmi les alcools renfermant de l à S atomes de carbone en chaine
linéaire ou ramifiée, ou d'un solvant polaire en présence d'un agent
alcalin, un carbonate de métal alcalin ou alcalino-terreux et d'un
catalyseur comme un iodure de métal alcalin. Les carbonates peuvent
être ceux de calcium, de sodium, de potassium. La condensation so fait
au reflux du solvant choi~i, pendant un temps qui peut varier de 4 à 30
heures selon le~ conditions.
an obtient ainsi le~ d~rivés do formule * nérale IA en série (pipéridyl
amino) ou pip~razinyl Xanthique. Ceux-ci peuvent ensuite être oxydés en
cétanes (R6 - 0) par action d'un agent métallique oxydant.
Les nouveaux dériv~s Xanthiqye~ de formul~ g~n~rale IA, et ~" ainsi
obtenu- peuvont atre tran~formfs en 8el~ d'atdltlon avec des ac~des
min~raux ou organigues, de pr~f~eence thérapeutiquement compatible6.
Ces sel6 d'addition font partie de l'invention.
, .
.
W O 92/06977 PCT/FR91/00811
-- 11 --
Comme principaux acides utilisables pour la focmation de sels, on
pourra citer à titre d'exemple les acides chlorhydrique, bromhydrique,
sulfurique, phosphorique, acétique, propionique maléique, fumarique,
citrique, benzo~que ou méthar.e sulfonique.
Les nouveaux dérivés de formule générale IA sont purifiés lorsque cela
est nécessaire, par les méthodes physiques ou chimiques comme par
exemple par recristallisation ou par chrcmatographie.
Ces dérivés ont été identifiés et dosés en we d'essais
pharmacologiques par les méthodes classiques d'analyse comme l~analyse
élémentaire, la spectrophotométrie infra-rouge, ultra-violet, la
résonnance magnétique nucléaire, la chromatogcaphie liquide haute
performance.
Pour produire les composés de formule générale I dans laquelle R e6t un
radical Benzimidazole et X est un groupe )N-, on condense le
benzimidazole halogéné de formule générale XI
4 ~ ~ H~-
dans laquelle R4 a les signlflcat~ons d~flnies pr~céde _ nt
et H~l repcésente un atome de chloro ou te brome
avec un derivé halogéné choisl dans le groupe formé des dérivés
bcnzyliques de fonmule * nérale III
.
~, .
W o 92/06977 PCT/FR31/00811
- 12 -
~ CH2 ~
Z (III)
et des halogénures d'éthoxyethyle de formule générale III~
CH3-CH2-O-CH2 CH2 ~al
dans laguelle Hal est du chlore ou du brome et z un halogène
pour obtenir le dérivé benzimidazoligue halogéné substitué, de formLle
générale XII
N ~ ~XII)
R5
dans laquelle R4 a les valeurs fournies précédemment et en particulier
le chlore ou l'hydrogène. On condense ce composé XII avec la pipérazine
dans un solvant cho~si de préférence parmi les hydrocarbure~
aromatiques, non ~ubstitues ou substitués, comme le toluène, les xylènes
ou Ie benzène. Il est avanta~eux d'opérer à une température comprifie
entre 40 et le point d'~bullition du solvant pendant 4 à 24 heures
selon la tcmpé~ature choisie. On obtient a~nsi un d~rlv~
~enzimidazol~que d formul- * néral~ X~I~
' ' : .
~ ., .
W O 92/06977 PCT/FR91/0081l
~7:~..L I.
- 13 -
t~ / (XIII)
I
dans laguelle R4 est de l'hydrogène ou du chlore et R5 est défini comme
précédemment
que l'on condense avec un 4-halogéno 1-~4-~ -phényl) butane de
formule Vl
R7 ~ C _' (CNzl3 _ Nal (Vll
dans laguel~e Hal est un at e de chlore ou de brome et R6 et ~ ont
les significations fournies précédemment pour former un composé de
fonmule g;né~ale IB,
~ (C~2l3 - N ~ ~ ~ R~ I!3~t
RS
.
.. .
, ~ :- - ,
. ' '.
WO 92/O~i977 PCI/FR91/0081 1
;~7~ 14-
SCE~. DE SYN~SE DES DERIVES BE21ZIIIIDAZOLIQOES I n
B
(X ~ >CH-NH- et R = Benzimidazolyl)
lère ETAPE:
. Les composés de fonmule A sont convertis en o.intro aniline
I Hal ~_ _ ~ ~ 5
(A) 2 20CH2CH3 ~
2`e~e ET~PE: ~éduction de la fonction nitro o.phenylène diamine en
correspondante XVI
IIH (XV~)
3~me EIAPE : A partir du dérivé de formule X, on prépare le dérivé
tétrahydropyranique de fonmule XIV
,)3_ tH~R,
(XIV)
où la fonction alcool est bloquée sous forme d~éther
tétr~hydropyranique (~THP).
W O 92/06977 PCT/FR91/00811
2~7~
4ème ET~PE : Formation du Thiocyanate Xv par action du sulfure de
carbone en milieu sodique sur le dérivé tétrahydropyranique XIV
S-C~ (CH2,)3-CH~3~7
5`eme EIAPE : Condensation de la benzylamine XVI avec le thiocyanate.XV
pour former une thiourée XVII
S ~R5 l ( m
8~PNO~-(C4I3- IH~I17
t~,
6`eme EI~PE : Cyclisation dans un hydrocarbure aromatique en présence
d'un agent de désulfurisation pour former un composé benzimidazolique
XVIII
R4 ~ ~H. ~ -~CH~L13--CH ~ R7
~5 (XV~II)
O 92/06977 PCT/FR91/00811
7 ~ ~ 1.
-- 16 --
7è~e El~pe Hytrolyse acide de l'éther tétrahydropyranigue XVIII
Cette reaction s'effectue dans un alcool dans des conditions très
douces et on obtient un dérivé benzimidazoligue de fonmule ~enérale I~"
dans laquelle x - >C-NH-
~S ~--H~ cH~l5cH
L (IB"~
Les composés de formule générale I porteurs d~un substituantbenzimidazolique, de fonmules générales (IB) et (IB~) ainsi obtenus
peuvent être
- oxydés en cétone correspondante par action d'un agent oxydant
métallique
- transformé6 en sels d'addition avec un acide minéral ou organique
- pur~fiés par les m&mes ~éthodes que celles décrites pour les dérivés
Xanthiq,ues
- identifiés et do és en vue d'essais parmacologiques par lés m~mes
technique~ cell-- décrites pr~c~domment
Les cc~y~,sés de formLle * nérale I se caractérisent par des propriétés
p~armacologique6 ~nt~res6ante6, anti-histaminiques et anti-allergiques
Les derivés t formule générale IB pour laquelle R est un ratical
benzimidazol- t leurs ~ ls physiolog~quement tolérables possadent
notaooent des propri~t~s ~nti-hl-~a~niqye- p~r action sur l~s
r~c-pt urs Nl d m~me tyF~ qy celles r-ncontr~-~ avec l'Ast~mlzole
~es composés de formule gén~rale T trouvent de ce fait un emplo~ on
thérapeutique notam~ent dan~ les désordres allergigyes et d~ns le
': . - . .
W O 92/06977 PCT/FR9l/0081]
- 17 - 2~
traitement de l'asthme.
La présente invention a également pour objet les compositions
pharmaceutiques contenant comme principe actif un dérivé de formule
générale I, ou un de ses sels physiologiquement toléra~le, mélangé ou
associé à un excipient pharmaceutique approprié.
Les compositions pharmaceutiques ainsi obtenues sont présentées
avantageusement sous les formes appropriées pour la voie orale, telles
que par exemple des comprimés, des dragées, des gélules, ou des
préparations appropriées pour des administrations par voie sublinguale,
nasale, injectable, buvable, ophtalmique ainsi que par aérosols ou par
voie rectale.
La posologie unitaire pourra s'échelonner entre 1 mg et 20 mg. La
posologie journalière varie entre 1 et 100 mg chez l'adulte.
Les exemples suivants sont donnés à titre non limitatif et illustrent
l'invention. Les points de fusion, sauf mention contraire, ont été
d~terminés à l'appareil Mettler. Les spectres infra-rouge sont
effectués à l'aide d'un spectrophot ètre Perkin Elmer 1600 serie FIIR.
Les spectres U.V à l'aide du spectrophotomètre Kontron, Uvikon 860. Les
dosages par HPLC sont pratiqués avec l'appareil Waters 600E équipé de
l'intégrateur APC4.
E~E~PLF I :
1,3-di~éthyl-7-(4-fluorobenzyl)-8~ (4-~4terbutyl-pb~ny1)-4-hydrosytu-
tyl)4-p~péridyl)asino]-s~nthine (~A~)
STADC A : Byd~ooyi-ino pip~r~dino
On chauffe au reflux sou8 agitation 46,2 g de chlorhydrate de
pipéridine -4-one avec 3~,4 g de chlorhydrate d'hydroxyhmine
dans 250 ml d'éthanol en présence de 100 9 de carbonate de
soude. Après 3 h ures de reflux, on filtre la suspension
bouillante et lave 4 fois le solide avec 100 ml d'éthanol
bouillant. On concentre la solution alcoolique sous vide. On
reprend le solide dans du chlorure de methylène, filtre sur
Célite et concentre ~ nouveau à sec. On obtient 34,1 g de
produit blanc PF - 117,2, notache en ca dans un système
méthanol 100, am~oniaque 2, sur plaque de silice MERCR et
W O 92/06977 PCT/FR91/00811
- 18 -
révélation par CuCl2 Ninhydrine.
ST~DE B : 1- [4-~4-terbutyl)- p~ényl-4-hydroxy 1-butyl]-4-hydroxy-
iminopipéridine.
On chauffe au reflux pendant 8 heures sous agitation un
mélange de 11,4 g de 4-oximinopipéridine et 24 g de
4-chloro-4-(4-terbutyl-phényl)-1-butanol et 100 g de
carbonate de soude dans 100 ml d'éthanol. Après ce temps, on
concentre le solvant sous vide et reprend après
refroidissement, avec de l'eau pour éliminer les sels
minéraux. On extrait le produit au chlorure de méthylène et
après séchage sur sulfate de sodium et élimination du
solvant, on recristallise le produit dans l'éther
isopropylique. On obtient 27 g de cristaux blancs.
PF: 128,5 - 131C.
ST~DE C : 1,3 DLméthvl-7(4-fluorobenzyl-)8-chloroxanthine.
on porte à 70-80C une solution de 43 g de 1,3 diméthyl
8-chloroxanthine dans 200 ml d'eau et 20 ml de soude 10 N et
introduit à cette température en 4 heures 27,6 ml de
chlorure de 4-fluorobenzyle. Pendant cette introduction on
ajoute par petites portions 20 ml de soude 10 N pour
maintenir le pH alcalin. Lorsque l'introduction est terminée,
on chauffe encore 2 heures à 70-80C puis laisse refroidir
sous agitation. On essore, lave à l'eau jusqu~à neutralité et
après séchage on recristallise le produit dans l'acétone et
on obtient 19 g de produit pur, PF 183-184C.
SIWDE D : 1-4-[~4t~rbutyl-phényl)-4-hydroxy]-1-butyl-4~oino-
plp~
On r~duit une solutlon de 25,5 g d'oxime du stade A dans
l'~thanol ~séch~ une nuit sur tamis léculaire 4 W) avec du
sodium i ébullition; lorsque tout le sodium a disparu
~23,5 g) on poursuit l';bulition pendant 1 heure puis
refroidit sous azote. On ajoute une solution de 57 g de
ClH N4 dans 200 ml d'eau, on élimine l'éthanol au maximum par
distillation sous vide. on extrait l~mine formée avec du
n-butanol et après lavage à l'eau, on concentre à sec sous
vide. On reprend dans du chlorure de méthylène séché sur
.
. .
'' ` '~ ' ,.
W O 92/0697~ PCT/FR91/0081l
Z~
-- 19 -- .
SO4Na2 filtré et concentré à sec. on recristallise dans un
mélange hexane ~100 parties)-méthanol(1 partie); on obtient
13,3 g de produit, PF : 99,2C.
sr~DE E : 1,3-diméthyl-7-(4-fluorQbenzyl) 8 [1-(4terbutyl-phényl-
4-hydroxy)-butyl)4-pipéridyl)-a~ino]-xanthine (IA").
On chauffe au reflux sous azote pendant 24 heures un mélange
de 32 g de 8-chloro-7-(4-fluorobenzyl)- théophyline, 16 g
d'iodure de sodium, 30 g de 1-l4-~4terbutyl-phényl)
-4-hydroxy]-butyl-4-amino pipéridine, 12 g de carbonate de
soude et 200 ml de diméthylformamlde. Après refroidissement,
on verse dans 800 ml d'eau glacée, on essore et lave à
neutralité, On purifie le produit sur colonne d'alumine avec
de l'hexane, de l'éther isopropylique puis de l'acétone.
Après concentration de la phase acétonique on obtient 17,6 g
de solide que l'on recristallise dans un mélange de 3
parties d'hexane et 1 partie d~acétone.
(Point de fusion : 95C )
Anal~se : C33 H43 N6 3
Calc. % : C 67,09 H 7,3 N 14,22F 3,22
Tr. : 67,10 7,27 14,03 3,18
W (MeOH) : ~ max en nm : 296 et 216,2
IR (R~r) : cm : 1224, 1625, 1640, 1695, 3330, 3400
RMN 1H ~CDC13, r~f.interne TMS, ~ en ppm) : 1,3 (S, 9H,
C (CH3)3), 3,38 (S, 3H, N'CH3), 3,52 (S, 3H, N-CH3),
4,60 (td, lH, - CH - Ol, 7 à 7,40 ~m, 8 H arom).
Chlorhydrate : PF 167.169
Fumarate : PF 179
E~EMP$E II
1,3-DLméthy1,7-(4-fluorobenzyl),8-[4-(4-terbutylph&ny1)-4-hydroxybutyl)
-pip~razinyl)-xanthine (IA,)
SIUDE A : 1-14-terbutylph~nyl)-4-hydroxyl-butylpipérazine
Rn disscut à 20C 6,36 g de pipérazine base anhydre dans
19 ml d'éthanol. Lorsgue toute la pipérazine est passée en
solution on ajoute d'un seul coup 8,90 g de 4-chloro-1-
l4-(4-terbutyl pényl)]-l-butanol.
W O 92/06977 PCT/FR91/0081~
2 ;'~
- 20 -
Le mélange est porté à 80C pendant 6 heures, on laisse
revenir à 20C et extrait le milieu réactionnel avec de
l'éther isopropylique. Le produit est recristallisé dans
l'acétate d~éthyle. On obtient 6 g de produit.
Point de fusion : 114-115C
S~ADE B : 1,3-Diméthyl 7-(4-fluorobenzyl)8-[4(4-terbutylphényl)
4-hydroxy)-butyl)-pipérazinyl]-xanthine
on mélange dans 100 ml d'éthanol absolu 16,53 g de
7-(4-fluorobenzyl)-8-chlorothéophylline et de 14,86 g de
1-[4-(4-terbutylphényl)4-hydroxy]-butylpipérazine. Le mélange
est porté à 80C pendant 9 heures. Lorsque la réaction est
terminée, l'éthanol est concentré sous vide et le produit est
extrait au chlorure de méthylène. Après cristallisation dans
1 vol w d'éthanol et 2 volumes d~éther isopropylique, on
obtient 6 g de produit.
Point de fusion : 145C
Analy---se : C32 H41 N6 3
Calc % : C 66,63 H 7,11 N 14,57F 3,29
Tr. : 66,24 7,08 14,53 3,65
W (EtOH abs.) : ~ max nm : 291,6 et 205,8
IR (XBr) : ~ cm 1 : 1222, 1440, 1510, 1660, 1700, 3180
RMN llH] (CDC13, Ref.Interne TMS, ~ en ppm) : 1,3 (S, 9H, C(CH3)3, 3,35
(S, 3H, NLCH3), 3,52 (S, 3H, -N-CH3), 4,65 (Ex, lH, -CH-O),
6,9 a 7,40 (m, 8H arom)
CH~ORD RATE : PF. 202-204C
FUMARAIE : PF a8-89Oc
E~EMPLE III
1-t4-fluorobonzyl)-2-[4-(4-terbuty1ph&nyl)-4-hydroxy-butyl)-pipérazino]
-benzi~idazole (IB,)
ST~DE A : 2-chloro bcnzi ~dazole
Une solution de 2-hydroxy benzimidazole (40,2 g) dans 93 ml
d'oxychlorure de phosphore est portée à reflux sous agitation
pendant 6 heures. En fin de réaction, on ramène à -10C et
hydrolyse par 100 g de glace pilee et 100 ml d'eau glacée. On
~;,
- ~,
W O 92/06977 PCT/FR91/00811
~ 7 ~
rajoute lentement 249 ml de soude 10N pour avoir un pH
neutre.
Le produit est filtré, lavé avec un minimum d'eau. Les
cristaux sont repris dans de l~éthanol à chaud. Après
refroidissement, on filtre et concentre les jus alcooliques à
sec.
ST~DE B: 2-chloro 1-(4-fluorQ~enz~yl)-benzimidazole
On met 11 g de 2-chlorobenzimidazole en solution dans 74 ml
d'eau et 17,35 ml de soude à 30%. Le mélange est porté à 82C
et l'on ajoute lentement 23,76 g de chlorure de
4-fluorobenzyle en 5 heures. Lorsque la réaction est
terminée, on refroidit à 20C et extrait au chlorure de
méthylène. Le produit est recristallisé dans l'acétate
d'éthyle.
STADE C : 1-(4-fluorobenzyl)-2-pipérazinylbenzimidazole
Dans 168 ml de xylène, on introduit 40,8 g de pipérazine
anhydre et 60 g de 2-chloro-(4-fluorobenzyl)-benzimidazole.
Le mélange est porté à 80C pendant 10 heures.
Lorsque la réaction n'évolue plus, on ramène la température à
20C et porte le pH à 2 avec une solution aqueuse d~acide
chlorhydrique à 30%. On lave à l'eau puis remonte à pH 11 par
du carbonate de sodium. on extrait le produit par de l'éther
isopropylique. Finalement, on obtient 46 g de produit.
ST~DE D : 1-(4-flw robenzyl)2-[4-(4-(terbutylpényl)4-hydroxy-butyl)
-pip~razino]-benzim~dbzolo
Dans 80 ml de n-but~nol, on mélange 28,45 g de
2-chloro-1~4-fluorobenzyl)bonzimidazole, 9,7 g de carbonate
de sodium, 13,66 g d'iodure de sodium et 30 g de 4-chloro
1-~4-terbutyll-phényl-1-tétrahydropyranyloxy butane. La
solution est portée à 100C pendant 16 heures. ~orsque la
réaction est terminée, le produit est extrait à l~éther
isopropylique, la phase organique est lavée à neutralité puis
concentrée à sec. La fonction alcool est libérée par
hydrolyse avec une solution d'acide chlorhydrique à 0,6%
pendant 45 minutes. On extrait à nouveau le produit à
1'éther, neutralise par du carbonate de sodium et on
. -
W O 92/06977 PCT/FR9l/00811
~ ~ ~.~ ~ 3~.
- 22 -
recristallise dans l~éther isopropylique avec un mlnimum
d~éthanol. on obtient finalement 19.90 g de produit pur,
ayant un point de fusion : 179,2C.
Analyse : C32 H39 N4 OF
Calc % : C 74,70 H 7,58 N 10,89 F 3,69
Tr. : 73,57 7,73 10,71 3,66
W (MeoH) : A max nm : 285,6- 248,8 et 213
IR (R3r) : cm : 954, 1080, 1129, 1609, 3222
RMN [1H] (CDC13, Ref.Interne TMS, ~ en ppm) : 1,3 ~S, 9H, C(CH3)3),
4,65 (td, lH, -CH-O), 5,15 (5, 2H, -CH2 - benzylique),
6,90-7,65 (m, 12 arom)
CHLORYDRATE : PF. 173-175,5C
FUMARATE : PF 196-197C
E~PLE IV '
- 1-(4-fluorobenzyl)-2-[4-terbutyl)phényl-4-hydroxy-butyl)-4-pipéridyl)- a~inol -benzimidazole (IB~)
STWDE A : l-chloro-[4-terbutyl-ph~nyl)]-butanol
On dissout 200 g de 1-chloro-(4-terbutyl-phényl) 3-butanone
dans 1680 ml de méthanol. Puis on introduit en 30 minutes une
solution de 16,78 g de borohydrure de sodium dans 151 ml
d'eau t 1,31 g de soude. La température du milieu
réactionnel est maintenue entre 10 et 20C pendant
l'introduction du borohydrure de sodium. On laisse 60US
agitation pendant S heures puis le m~thanol ~st concentré
wus vid . Le mllieu est extrait ~ ther i~opropylique et
le prodult e~t recristalli6é dans 2 volumes d'hexane. On
obtient 183 g de produit.
Point de fw ion : 50,9-51,3C
SIADE ~ chloro-1-t4-terbutyl-phe'nyll-4-tetrahydropyranyloxy butane
Dans 8,7 ml de d$hydropyran on introduit 10 mg d'acide
p.toluène sulfonique (APTS). A ce mélange on ajoute en 30
minutes et à 60C le 1-chloro-t(4-terbutyl-phényl- butanol-4
(20g). Après addition on chauffe encore entre 60 et 65
pendant 30 minutes. On laisse sous agitation pendant 90
W O 92/06977 PCT/FR91/00811
2~,Y7
- 23 -
mlnutes et l~on ajoute 0,5 g de bicarbonate de sodium. Onagite encore 1 heure. Le dérivé tétrahydro-pyranylé est remis
directement en réaction sans autre purification.
SI~DE C : N(4-fluorobenzyl) pénylène-diamine
On réduit 56 g de N (4-fluorobenzyl) 2-nitro aniline en
présence de 34 g de charbon à 5% de Pd dans le méthanol (5~
ml), par un courant d~hydrogène à température et pression
ordinaire. Après fin de réaction, on purge à l'azote, filtre
sur Célite( ) et concentre à sec, on recristallise dans
l'éther isopropylique. On obtient ainsi la phénylène diamlne
deslree.
Poids obtenu : 38,5 g.
On prépare }a N-(4 fluorobenzyl) 2-nitro aniline nécessaire
par alcoylation de la 2-nitro aniline au yen du chlorure de
4-fluorobenzyle dans la méthyl éthyl cétone.
SI~DE D : 1-[(4-terbutylphényl)-4-Tetrahydropyranyloxy butyl] 4-hydko- xy~ino pipéridine.
on chauffe au reflux sous agitation 58,4 g de 1 chloro
-l4-terbutYl-phényl] 1-(tétrahydropyranyloxy) butane avec 20
g de 4-hydroxymino pipéridine et 20 g de carbonate de soude
dans 200 ml d'éthanol. Après 20 heures de reflux on concentre
à sec et reprend avec de l'eau, on extrait au chlorure de
méthylène séché et concentre à sec sous vide. On a obtenu
70,5 g de produit.
On recristallise dans le méthanol et on obtient 40g de solide
de PF: 150 - 151C .
SI~De E : 1-[(4-to~butyl-ph~nyl)-4-Tbtr~kydropyr~nyloxy~butyl¦-4-a lno pip~ridln~
Dans un réacteur de 1 litre on réduit par 25,5 g de sodiu~
métal au reflux une solution de l'oxime correspondante ~35 g
dans 235 ml d'éthanol) en lheure et apres disparition
c~Lylèto du sodium, on chauffe au reflux 1/2 houre puis
refroidit sous azote. On introdu~t dans le milieu w us
agitation une solution de 60 g de chlorure d'ammonium dans
250 ml d'eau. on extrait à l'éther isopropylique, lave à
l'eau, sèche sur sulfate de soude puis, concentre à sec. On
. - . . ,:
. ' ' :
W O 92/06977 PCT/FR91/00811
2~7~
- 24 -
purifie par recristallisation dans le méthanol afin
d'éliminer l~oxime non réduite eventuellement présente.
L'amine brute après concentration du méthanol est purifiée
par passage sur colonne de silice montée avec de i~hexane
puis élution au méthanol et concentrée à sec. On obtient 25 g
de produit sous forme d~une huile épaisse.
ST~DE F: 1-[4-terbutyl-pheny1-4-tetrahydrcpyranyloxy-butyl] isothio
cyanato pipéridine.
Dans un réacteur de 250 ml on place 10 ml de NaOH 10 N et 50
ml d'eau puis après refroidissement à 5 on ajoute 6 ml de
sulfure de carbone. On introduit en 1 heure entre 0-10 . une
solution de 19,5g d~amine du stade E. précédent dans 100 ml
d~eau et de l'acide acétique pour obtenir un pH de 7-8. On
agite encore 2 heures entre 0 et 10C. On suit la disparition
de l'amine par CCM sur silice dans un mélange CHCl3 -éthanol
7-3. On porte la température à 20C et introduit
goutte-à-goutte, sans dépasser 30, 10 ml de chlorocarbonate
d'éthyle en 3/4 heure. On chauffe ensuite pendant 2 à 3
heures entre 50-60 sur bain-marie jusqu'à fin de coloration
en brun du papier à l'acétate de plomb.
(CCM sur Si 02 avec cyclohexane : 3-acétate d'éthyle 7).
Après refroidissement, on extrait au chlorure de méthylène,
lave, s~che et concentre à sec. Dn purifie le produit sur
colonne de silice ntée à l'hexane et élution par un mélange
cyclohexane-5 acétate d'éthyle-S. On obtient 15.3 g d'un
produit visqueux.
S5~DE G : N-l1-~4-~terbutyl-ph~nyl)-4-t~tr~hydropyranyl]- pip~ridinyl]
-oxybutyll N~t2-~4- nuorobenzyl)-amincph~nyl~- thiour~e.
on mélange dans 150 ml de méthanol, 25,4 g de 1-~4-(terbutyl
-phényl)4-Tétrahydropyranyloxy butyl]4-isocyanato-pipéridine
et 18,3 g de N-(4-fluorobenzyl) 1,2-phénylène-diamine et
laisse au repos pendant 24 heures. on concentre à sec sous
vide et passe sur coloMe de silice montée avec du
cyclohexane pour éliminer l'excès d'amine aromatique.
On extrait la thiourée avec de l'acétate d'éthyle. On obtient
33 g de substance huileuse.
W O 92/06977 PCT/FR91/00811
: ~'`' J.. '.. '.,L._
- 25 -
ST~DE ~ (4-fluorobenzyl)2-[4-(terbutyl- pényl)-4-hydroxybutyl)
4-pipéridyl-amino]-benzimidazole.
On chauffe au reflux dans 200 ml de benzène, 33 g de thiourée
obtenue au stade G avec 16 g de Dicyclohexyl-carbodiimide
pendant 5 heures. La réaction terminée, on re~roidit, lave à
l'eau puis avec une solution aqueuse de car~onàte de sodium
et encore à l'eau. On sèche et concentre à sec sous vide, on
reprend le résidu dans 100 ml de méthanol et 20 ml d'eau puis
ajuste le pH à 1 avec de l'acide chlorhydrique concentré on
contrôle la fin de la réaction par CCM sur silice avec
chloroforme 7. Ethanol 3 (révélateur W et iodoplatinate). on
neutralise après dilution par l'eau avec du carbonate de
soude puis de la soude pour avoir un pH - 12. On extrait au
chlorure de méthylène, on lave à l'eau, sèche, filtre et
concentre à sec. On obtient 37 g de produit brut que l'on
la~e à l'éther isopropylique et on obtient 2~ g de produit
pur. Cn recristallise le produit dans de l'acétate d'éthyle
contenant un peu de méthanol.
PF: 164,8-165,2
Analyse C33 H41 N4 OF
Cal % : C 74,96 H 7,82N 10,60 F 3,59
Trouvé : 73,80 7,80 10,30 3,50
W ( MeOH ) : ~ max nm : 286,2 249,4 et 218,4
IR ( KBr ) : cm~1 : 1087, 1227, 1570, 1616, 3068, 3227
RMN [lH] ~CDC13, réf.interne TMS, ~ en ppm) : 1,3 (S, 9H, C~CH3~3),
4,02 (td, lH, -CH-0), 5,03 (S, 2H, -CH2 - bQnzylique), 6,90 -
7,55 (m, 12 arom).
CH W RYD~ATE : 204-206C
FUMARATE : 177C
V
E~EMoeLES De ReA~ISAI5oN De ~M~S PHDRriCEUnIQUES
. . ..:
W O 92/06977 PCT/FR91/00811
- 26 -
A) Gélules
Principe actif 20 g
Lactose 100 g
Cellulose microcristaline23 g
Stéarate de magnésium 1 g
Silice colloidale 1 g
qs. pour 1.000 gélules
B) Comprimés
Principe actif 50 g
Lactose pour compression1360 g
Avicel PH 101 200 g
Précirol 20 g
Silice colloidale 20 g
qs. pour 10.000 comprimés
C) Soluté buvable
Principe actif 150 mg
Conservateurs 1 mg
Sorbitol sol 70% 50 g
Alcool 95 20 g
Eau purifiée gsp 150 g
D) Injectable
Principe actif ~S/forme de sel) en base 1 mg
Chlorure de sodium 5 mg
Phosphate no~odique qs Ph 5,5/6
Eau PPI qsp S ml
~) suPpositoir~6
Principe actif 2,5 mg
Witepsol H35/H37/ 50/50 qsp 3 g
E) Aerosols
Principe actif 3 %
Sorbitol trioléate 2 à 2,5
Agent propulsant qsp 100
. ~ , . . ..
. , .
W O 92/0~977 PCT/FR91/00811
- 2~ ~ ~? ~ ~ .
- 27 -
F) Collyre
Principe actif sous forme de
chlorhydrate 1 mg
Chlorure de Benzalkonium 0,0012; mg
eau purifiée qsq 5 ml
ETUDE P9ARMPOOLOGIQUE DES oo~pDsEs sELoN L' rNVENlIoN
Les études pharmacologiques des composés selon l'invention ont comporté
deux aspects : la caractérisation de llactivité principale de type
anti-histaminique H1 et la recherche d~éventuels effets secondaires
indésirables sur le systè~e nerveux central (SNC). Chacun de ces deux
volets a pu mettre en oeuvre aussi bien des techniques in vitro de
mesure d'affinité pour des récepteurs spécifiques ou d'études sur
organe isolé, que des modèles d~activités pharmacologiques in vivo.
I--CAFUC19PlSAlIoN DE L'ACIIVITE ANnq-~l
1.1 Me~ure de l~affinite pour les récepteur~ Hl
Des concentrations croissantes des composés à étudier sont incubés avec
des préparations membranaires d'homogénats de cortex de rat ou de
poumon de cobaye en présence d'un ligand spécifique des récepteurs H1 :
la mépyramine, marquée au tritium, à la concentration fixe de 2 nM.
Après 30 mn à 25C, à pH 7,4 dans un tampon 50 mM Tris-HCl pour les
membranes de cortex, ou 0.5 M Na2HP04/RH2P04 pour les membranes de
poumon, la fra--ion liée résiduelle de ligand tritié est comptee par
scintillation en milieu liquide apras filtration sur appareil de type
8randel et plusieurs lavages. ~a relation entre la concentration du
composé à étudler et l'inhlbition de la liaison sp~cifique de la
mépyramine tritiée aux récepteurs H1 permet de calculer la
concentration inhibitrice à 50 % tC150).
Dans ces conditions exp~rimentales, le composé de l'exe~ple III
présente une C15g de 1,1 x 10 6M sur~les récepteurs Hl de cortex de rat
et de 2,7 x 10 M sur les récepteurs H1 de poumon de cobaye, contre
respectivement 1 x 10 6M et 3,2 x 10 6M pour la terfénadine, substance
anti-Hl non sédative de référence tWocdward and Munro, 1982). Les deux
,~ ;
.
.. . ..
W O g2/06977 - PCT/FR91/00811
- 28 -
produits possèdent donc in vitro une affinité très semblable pour les
récepteurs H1 dans deux espèces différentes.
1.2 Etudes sur l'iléon isolé de cobaye
Des concentrations successives des composés à étudier sont testées
contre une même gamme de concentrations croissantes d~histamine, qui
agit pa ses récepteurs de type H1 sur les fibres musculaires lisses de
la paroi de l'ileon mis dans un bain de survie selon la technique
décrite en détail par PERRY et coll. (1970). Le déplacement de la
relation entre effet contracturant et concentration d'histamine par le
composé à étudier, signe une activité anti-Hl.
Dans ces conditions expérimentales, le coL4usé de 1'exemple III et la
terfénadine se comportent de façon très voisine : aucun effet à 10 7M,
inhibition de type compétitive (c~est-à-dire permettant de retrouver
l'intégralité de l'action de l'histamine à une concentration plus
élevée) à 10 6M, et inhibition partiellement irréversible (c'est-à-dire
antagonisant une fraction de la contraction induite par l~histamine
quelle gue soit sa concentration) à 10 5M. Si le compoé de l'exemple
III apparait lé * rement plus actif que la terfénadine à 10-6M,
l'inverse semble se produire à 10 SM, sans gu'aucune de ces nuances ne
soit significative entre ces deux produits.
1.3 Btudes sur le brcnch~sp~ l'histamins chez le cobaye
L'inhalation d'un aérosol à 1 p. mille d'histamine (solution dans l'eau
distillé~e, nébulisée par appareil Hospltak) provoque chez le cobaye un
bronchospasme gui s'accompagne de l'immobilisation de l'anlmal sur le
flanc. ~e temps guo mettent les animaux ~ tomber sur le ~lanc est
mesuré pendant 5 minutes maximum. Cette durée est appelée "temps de
riésistance" et s'alIonge lorsque les animaux ont recu préalablement une
substance douée de propriétés anti-H1. Au-delà des 300 secondes
d'observation, les animaux sont considérés comme "protégés" de l'action
bronchoconstrictrice de l'histamine (Olsson, 1971).
Par voie intrapéritonéale, une protection presgue complète de tous les
animaux soumis au test est obtenue 30 minutes après administration de
10 mg/kg de terfénadine ou du composé de l~exemple III, leur activité
,
W O 92/06977 pcr/FR9l/oogll
~ ~
- 29 -
se trouvant encore très semblable. A 1 et 3 mg/kg, terfénadine et lecomposé de l'exemple III demeurent peu efficaces dans ces conditions
expérimentales. Dans cette plage de doses, les courbes de régression
permettent le calcul de la dose efficace à 50 ~ (DE;o) pour chacun des
produits : environ 6,7 mg/kg pour le composé de l~exemple III, et
eniron 7 mg/kg pour la terfénadine.
Par voie orale, la dose unique de 10 mg/kg a été étudiée en fonction du
temps. Les cobayes ayant recu cette dose de terfénadine ou du composé
de l'exemple III ont passé le test de l'aérosol à l'histamine 30 mn, 1,
2, 4, 6, 8, 12 et 24 heures après gavage (8 animaux par produit pour
chaque temps). La terfénadine n~a protégé tous les animaux traités que
pendant 4 heures, tandis que tous les animaux ayant recu le composé de
l'exemple III ont totalement résisté aux effets de l'histamine inhalée
jusqu'à 12 heures après ingestion du composé anti-H1. Au temp~
24 heures, la protection totale (>5 mlnutes) se manifeste encore chez
50% des animaux traités par le composé de l'exe~ple III, qui présente
donc une durée d'action plus longue que celle de la terfénadine.
En conclusion, le composé de l'exemple III se distingue parmi les
- composés selon l'invention comme étant porteur d'une activité
principale anti-H1 de même puissance intrinsèque gue la terfénadine
(Cl50 sur les récepteurs Hl, inhibition de la contraction à l'histamine
de l'iléon isolé de cobaye, DE50 par voie intrapéritonéale contre le
bronchospasme à l'histamine chez le cobaye), mais de durée d'action
supérieure après administration par voie orale (bronchospasme à
l'histamine chez le cobaye).
oecEoear~E D~ENENn~ElB EFPEIS SECCNCPIRES SUR LE SNC
2.1 Mesures d'~ffinit~ p~ur diver8 r~cepteurs neuromédiateurs
Le composé de l'exemple III, sélectioMé sur la base de son activité
principale anti-Hl a été comparé à deux autres molécules anti-Hl
représentatives des deux générations de cette classe d'agents
phanmacologiques. La mépyramine appartient à la première génération gui
présente des propriétés résiduelles sédatives sur le SNC. La
terfénadine est le premier représentant de la seconde génération
d'anti-Hl, gui est désormais dénuée d~effets secondaires sur le $NC
(Garrison, 1990).
.. .. .
-
W O 92/06977 PCTtFR91/00811
- 30 -
Une batterie de récepteurs membranaires de divers organes de rat a été
testée selon le même principe que le récepteur Hl dans les ~onditions
résumées suivantes :
. Adénosine ~ : cortex, 90 mn à 25C dans 50 mM Tris-HC1 à pH 7,7
contre 1 nM de 3H-cyclohexyladénosine,
. Adénosine A2 : striatum, 60 ~n à 25C dans 50 mM Tris-HC1 à ph 7,7
additionné de 50 nM de cyclopentyladénosine, de 10 mM de MgC12 et
d~adénosine-désa~inase (0.1 unité/ml), contre 4 nM de 3H-NECA
(5'-N-éthylcarboxamidoadénosine).
. Muscarinique Ml : pool de cortex, striatum et hippocampe, 60 mn à
25C dans 10 mM Na2HP04/KH2P04 à pH 7,4 contre 2 nM de H-pirenzépine
. Muscarinique M2 : pool de coeur, d~iléon et de cervelet, 60 mn à 22C
dans 50 mM Tris-HCl à pH 7,5 contre 0,5 nM de H-aNB
(quinuclidinyl-benzylate)
. Sites centraux aux benzodiazépines : cerveau entier, 90 mn à 0C dans
50 mM Tris-HCl à pH 7,1 contre 0,3 nM de 3H-flunitrazépam
. Sites périphériques aux benzodiazépines : cortex, 90 mn à 25C dans
90 mM Na2HPOq/RH2P04~ 81 mM NaCl et 9,5 mM RC1 à pH 7,4 contre 1 nM
de 3H-PR11195
. ~ : cerveau entier, 20 mn à 4C, dans 50 mM Tris-HCl à pH 7,2
additionné de 0,5 p.mille de Triton X-100, contre 5 nM de 3H-muscil
. ~ : cervelet, 10 mn à 22C, dans 50 mM Tris-HCl à pH 7,4
additionné de 1,2 mM MgS04 et 2,5 mM CaCl2, contre 20 nM de
3H-baclofen
. Récepteur Sigma : cerveau entier, 2 heures à 22C, dans 50 mM
Tris-HCl à pH 8, contre 2 nM de 3H-~+)3PPP t3-(3-hydroxyphémyl)-
-N-~l-propyl)pipéridine].
Comme dans le cas du récepteur Hl, la relation entre les pourcentages
d'inhibition de la liaison spécifique du ligand tritié en fonction des
concentrations croissantes de mépyramine, de terfénadine ou du composé
de l'exemple III permet de calculer la Cl50 de chacun des produits pour
chaque récepteur. Plus la Cl50 est faible, plus l'affinité est forte.
Lorsque l'inhibition n'est que partielle et ne permet le calcul que
d'une C150 théorique supérieure à 10 5M, on peut considérer que
l'affinité est très faible, voire nulle.
W O 92/06977 PCT/FRsl/00811
- 31 -
Le tableau ci-dessous résume les propriétés des trois
anti-histaminiques H1 étudiés :
Mepyrom~ne lerfén~dlneCom~os~ dc
Musc~rln1que Ml 3.8 x 10 6 ~ lO S ~ lO 5
Musc~rlnlquc M2 3~5 x lO 6 > 10'5 ~ 10 5
R~cepteur Slgma 5,9 x lO 7 ~ 10~5 ~ 10 5
Sites ~u Benzodlazéplnes :
- centr~ux > 10-5 > l~S ~ 1~.5
- periphériqJes ~ lO 5 ~ lO 5 ~ 10 5 .
Adenos1ne Al > 10-5 > 10-5 ' lO 5
Adenos1ne A2 > l 5 ~ l0~5 ~ 10-5
GABAA ~ ~ 5 ~ 10.5 ~ lO 5
GABAB > lO'S ~ 10-5 ~ lO 5
~a mepyramine possède une affinité modeste mais réelle pour les sites
muscariniques Ml et M2, environ 1000 fois plus faible que celle de
l'atropine, agent anticholinergique de référence. En outre, une
affinité non négligeable pour le récepteur Sisma a été mlse en
~vidence, de l'ordre de 20 fois plus faible que celle de la
pentazocine, ligand de référence pour ce récepteur. Certains des effets
secondaires de la mépyramine sur le SNC peuvont donc éventuellemont
trouver une biso fonct~oMelie dans ces a~finltos résiduelles non H1.
En revanche, l'absonco d'aff~nité décelable dans les m~mes conditions
oxp~rimentalos rapproche le composo de l'exemplo III de la terfénadine.
2.2 Etude de comportement chez la ~ouris
Le test d'lrwin, mLdifié selon la technique de Morpurgo (1971), a été
mis en oeuvre pour rechercher d~éventuelles modifications des
comportments spontané ou provogué sous l~effet du composé de l~exemple
III ou d~un antihistaminique Hl de première génération : la
dexchlorphéniramine, connue pour entrainer des ef~ets de somnolence
chez l'homme.
'' ' .
:
W O 92/06977 PCT/FR91~00811
2~?...~
- 32 -
Après administration par voie orale, les souris ont été observées
pendant 30 minutes, puis ont subi une série de tests simples de
screening comportemental aux temps 30 minutes, 3 heures et 24 heures.
La dexchlorphéniramine à la dose de 100 mg/Xg a entrainé une
hyperactivité et une irritabilité légère à 30 minutes, persistant à
3 heures mais disparaissant à 24 heures. D~autre part, une mydriase a
été notée à 3 heures. Le composé de l~exemple III n~a entrainé qu~une
très légère passivité sur la moitié des animaux ayant recu la dose
maximale testée de 400 mg/kg au temps 3 heures unique~ent. D~autre
part, dans une recherche de dose unique maximale non toxique par voie
orale dans la même espece, les souris ayant recu jusqu'à 1000 mqvkg
n~ont manifesté aucun signe clinique. Moins fine que le test d~Irwin au
cours duquel les animaux sont manipulés et interagissent avec
l'expérimentateur. Cette absence de symptôme à forte dose abonde dans
le sens d'une absence d'effets sur le $NC. L'hyperactivité due à la
dexchlorphéniramine ne correspond pas à l'effet sédatif rencontré chez
l'homme, mais traduit néanmoins une influence sur le SNC. La mydriase
résulte d'une action antichlolinergique au niveau oculaire,
caractéristique de l'atropine par exemple.
2.3 Te6t du Rota-rod chez la 60uris
Le principe du test consiste à chronométrer la durée pendant laquelle
des souris se maintiennent sur une tige tournant à vitesse constante.
Des substances douées d'activité sédative, myorelaxante ou 1nhlbitrlce
do la coordination trico vont entralncr lour chute pr~matur~e, avant
120 secondes qui est le temp5 maximum d'étude. Au 4 elà, les animaux
sont consitérés comme béneficiant de capacités psycho-motrices intactes
(Andrasi et Coll., 1982 ; Ongini et Coll., 1987). Le test a lieu 30 et
60 mn après administration. Par voie orale, le composé de l'exemple III
n'a pas altéré les performances des animaux testés jusqu'à la plus
forte dose testée de 100 mg/kg, c parées ~ celles enregistrées pour
des souris ne recevant que le véhicule d'administration. La
chlorpromnzine, neuroleptique majeur utilisé comme standard positif
dans le test du P~ota-rod, a entrainé la chute avant le délai imparti de
90 % des souris dès la dose de 4 mgvkg par voie intrapéritonéale. Dans
d~autres essais c~ylémentaires, le fumarate de kétotifène, molécule
W O 92/06977 PCT/FR9l/00811
2 ~ 7.~ ~, .~ ~.
anti-Hl à longue durée d'action, s'est montré capable de diminuer les
performances de 2~ à 35 % des souris traitées à 50 mg/kg par voie
orale.
En conclusïons, le composé de l'exemple III ne présente pas d'effet
notoire sur le SNC dans des situations expérimentales où d'autres
anti-histaminiques H1 entrainent des modifications neurologiques
diverses : hyperactivité, irritabilité et mydriase avec la
dexchlorphén,ramine, altérations des performances au Rota-rod avec le
fumarate de kétofifène. Enfin, les profils d'affinités aux récepteurs
spécifiques des principaux neurotransmetteurs permettent de distinguer
la mépyramine, anti-Hl de première génération d'une part, et la
terfénadine et le composé de l'exemple III d'autre part. Ces deux
derniers produits ne possèdent aucune affinité résiduelle sur ces
récepteurs, ce gui laisse présager d'un profil similaire de type non
sédatif.
III - CCNCLUSIoN DE L'ETUDE P9~RM~oOLOGIQUE
Les composés selon l'invention présentent à des degrés divers des
potentialités anti-histaminiques Hl. Parmi eux, le composé de l'exemple
III présente un profil pharmacologique voisin de celui de la
terfénadine, caractérisé par une puissance d'activité similaire in
vitro comme in vivo, et par une absence d'effets secondaires sur le
SNC. De plus, le composé de l'exemple III possède une durée d'action
supérieure à celle de la terfénadine.
~E~IaNCES BIBLICGRAE9ICpES
ANDFASI F, ~ORV~ , SrNEoER E, LERSENYL P, ~oRSY J, ~ENESSEY A,
T~RR ~, LANG T, ~OR06I J et EPMoFI T.
Arzneim Forsch/Drug Res. 1987, 37 : 1119-1124
G~RlSSoN J.C
in "The Pharmacological Basis of Therapeutics"
8ème édition par GILMAN A.G, RAEL T.W, NIES A.S et TAYLOR P
Pergamon Press Inc., New York l990, pp 575-588
.
: : .
; ~
W O 92/06977 PCT/FR91/00811
- 34 - -
~WRGO C.
Arzneim Forsch/Drug ~es. 1971, 11 : 1727-1734
OLSSoN O.A.T
Acta Allergologica, 1971, 26 : 438-453
PERRY et les membres du Département de Pharmacolsgie de l'Université
d'EDIMeOURG
in "Pharmacological experiments on isolated preparations"
édité par E & S LIVINGSTONE, Edimbourg et ~ondres, 1970 : pp 58-87
- W W OW~R3 J.~ et MUNCRO N.L
Arzneim Forsch/Drug Res. 1982, 32, 1154-1156
.
' ~
'-: ' .
'