Note: Descriptions are shown in the official language in which they were submitted.
WO 91/06548 PCT/US90/05756
,.-...
0. F ~ W J. l=N
INHIBITORS OF PURINE NL1CI~EOSIDE PHOSPHORYLASE
The present invention relates to derivatives of 2-amino-
3H,5H_-pyrrolo[3,2-d_]pyrimidin-4-one and to derivatives of
4-oxo-3H_,5H_-pyrrolo[3,2-d]pyrimidine. It also relates to
4-oxo-3H,5H-pyrrolo[3,2-d]pyrimidine derivatives
substituted at the 7-position.
Purine nucleoside phosphorylase (PNP) catalyzes the
phosphorolysis of purine nucleosides in a reversible
reaction. Individuals who are deficient in PNP exhibit
impaired T-cell development, resulting in lowered cell-
mediated immunity, but normal B-cell development, resulting
in normal humoral immunity. Accordingly, specific
inhibitors of PNP that selectively inhibit T-cell
development without damaging humoral immunity could be
potentially effective against disorders in which activated
T-cells are pathogenic. '
As a PNP inhibitor, the present invention provides a 2-
amino-7-(R)-3H_,5H_-pyrrolo[3,2-d]gyrimidin-4-one wherein R
is optionally substituted cyclohexenyl, cyclohexyl, or
-CH2-R1, wherein R1 is an optionally substituted
heteroalicyclic, pyridinyl or alicyclic group.
The present invention is also directed to a compound of
the formula '
y)P-(CH2)m X
wherein R1 is H, NH2, or OCH3, R2 is an optionally
substituted cyclic group of 5-7 carbon atoms optionally
WO 91/06548 PCT/US90/05756
t ,
2
containing one or more heteroatoms, R3 and R4 are '
independently H or C1-4 alkyl, m is 0-4, n is 0-6, p is 0-
l, X is CN, CSNH2, PO(OH)2, COON, S02NH2, NH2, OH, CNHNH2, '
tetrazole, triazole or CORS where R5 is C1-4 alkyl, CF3,
NH2, or OCl-4 alkyl, and Y is 0 or NH. The compound of the
present invention is useful as a PNP inhibitor. Also
'contemplated according to the present invention is a
pharmaceutical composition for the selective suppression of
mammalian T-cell immunity comprising an pharmaceutically
effective amount of the compound of the present invention
and a pharmaceutically acceptable carrier or diluent and a
method for the selective suppression of mammalian T-cell
immunity without diminished effect on humoral immunity
comprising administering to a subject a pharmaceutically ,
effective amount of the compound of the present invention.
In one aspect of the invention there is provided a
compound 2-amino-7-(R)-3~,5F~-pyrrolo[3,2-d]-pyrimidin-4-one
(I) wherein R is
-CH2-R1
and R1 is an optionally substituted heteroalicyclic group.
Preferably, the heteroalicyclic group is a 5 or 6 membered
saturated ring having oxygen, nitrogen, or sulfur as the
heterocyclic atom. More preferably the heteroalicyclic
group is 2- or 3-tetrahydrothienyl, 2-, 3-, or 4-
piperidinyl, 2- or 3-tetrahydrofuranyl, 2-, or 3-
pyrrolidinyl, or 2-, 3-, or 4-tetrahydropyranyl. In a
preferred aspect, R1 is unsubstituted, e.g., the compound
(I) is 2-amino-7-(2-piperidinylmethyl)-3H,5H_-pyrrolo[-
3,2-d]pyrimidin-4-one (IB), 2-amino-7-(3-piperidinyl-
methyl)-3H,5H-pyrrolo[3,2-d]pyrimidin-4-one (IC), 2-amino-
7-(4-piperidinylmethyl)-3_H,5H_-pyrrolo[3,2-d]pyrimidin-4-one
(ID), 2-amino-7-(2-tetrahydrofuranylmethyl)-3N,5H_-pyr-
rolo[3,2-d]pyrimidin-4-one (IE), 2-amino-7-(3-tetrahy-
drofuranylmethyl)-3H,5H_-pyrrolo[3,2-d]pyrimidin-4-one (IF),
WO 91/06548 PCT/U590/05756
. . ~~'w~~.~~
~1Y ~ ~Tl J. ~~
3
2-amino-7-(2-tetrahydrothienylmethyl)-31i,5H-pyrrolo[-
3,2-d]pyrimidin-4-one (IG), 2-amino-7-(3-tetrahydrothieny-
lmethyl)-3H,5H-pyrrolo[3,2-d]pyrimidin-4-one (IH), 2-amino-
7-(2-pyrrolidinylmethyl)-3H,5H_-pyrrolo[3,2-d]pyrimidin-4-
one (II), 2-amino-7-(3-pyrrolidinylmethyl)-3H,5H-pyr-
rolo[3,2-d]pyrimidin-4-one (IJ), 2-amino-7-(2-tetrahydro-
pyranylmethyl)-3#~,SIB-pyrrolo[3,2-~,]pyrimidin-4-one (IL), 2-
amino-7-(3-tetrahydropyranylmethyl)-3~,5~-pyrrolo[3,2-~]py-
rimidin-4-one (IM), or 2-amino-7-(4-tetrahydropyranyl-
methyl)-3I~,5H_-pyrrolo[3,2-d_]pyrimidin-4-one (IN). In an
alternative preferred embodiment the R1 has one or two
substituents selected from the group consisting of halogen,
hydroxy, alkoxy, alkyl, or trifluoromethyl. As halogen is
preferably mentioned chloro or fluoro. As alkoxy is
preferably mentioned lower alkoxy, including methoxy,
ethoxy, propoxy and butoxy. As alkyl is preferably
mentioned lower alkyl, including methyl, ethyl, propyl and
butyl.
In another aspect of the invention there is provided
2o a compound 2-amino-7-(R)-3H_,5H_-pyrrolo[3,2-d]-pyrimidin-4-
one (II) wherein R is
-CH2-R1
and R1 is optionally substituted pyridinyl. In a preferred
aspect, R1 is unsubstituted, i.e., the compound (II) is 2-
amino-7-(3-pyridinylmethyl)-3H_,5H_-pyrrolo[3,2-d]pyrimidin-
4-one (IIA), 2-amino-7-(2-pyridinylmethyl)-3H,5H-
pyrrolo[3,2-d]-pyrimidin-4-one (IIB), or .2-amino-7-(4-
pyridinylmethyl)-3H,5H-pyrrolo[3,2-d]-pyrimidin-4-one
(IIC). In an alternative preferred embodiment the R1 group
has one or two substituents selected from the group
consisting of halogen, hydroxy, alkoxy, alkyl, or
trifluoromethyl. As halogen is preferably mentioned chloro
or fluoro. As alkoxy is preferably mentioned lower alkoxy,
including methoxy, ethoxy, propoxy and butoxy. As alkyl is
WO 91/06548 PCT/US90/05756
(:~;~
.. ''
4
preferably mentioned lower alkyl, including methyl, ethyl,
propyl and butyl.
In another aspect of the invention there is provided
a compound (III) 2-amino-7-(R)-3H,5H-pyrrolo[3,2-d]
pyrimidin-4-one wherein the R group is unsubstituted or
substituted 1-, 2-, or 3-cyclohexenyl or cyclohexyl. In a
preferred aspect, R is unsubstituted, i.e., the compound
(III) is 2-amino-7-(1-cyclohexenyl)-3H_,5H_-pyrrolo[3,2-d]
pyrimidin-4-one (IIIA), 2-amino-7-(2-cyclohexenyl)-3I~,5H_
pyrrolo[3,2-d]pyrimidin-4-one (IIIB), 2-amino-7-(3
cyclohexenyl}-3H_,5H_-pyrrolo[3,2-d]-pyrimidin-4-one (IIIC),
or 2-amino-7-(cyclohexyl)-3H,5H-pyrrolo[3,2-d]-pyrimidin-4
one (IIID). In an alternative preferred embodiment the R
has at least one substituent selected from the group
~cnsisting of halogen, hydroxy, alkoxy, alkyl, or
.=ifluoromethyl. As halogen is preferably mentioned chloro
or fluoro. As alkoxy is preferably lower alkoxy, including
mathoxy, ethoxy, propoxy and butoxy. As alkyl is
preferably mentioned lower alkyl, including methyl, ethyl,
propyl and butyl.
In another aspect of the invention there is provided a
compound 2-amino-7-(R)-3~,5F~-pyrrolo[3,2-d]-pyrimidin-4-one
(IV) wherein R is
-CH2-R1
and R1 is an optionally substituted alicyclic group.
Preferable alicyclic groups include, e.g., single-ring
cycloparafins such as cyclopentyl, cyclohexyl, and
cycloheptyl, multi-ring cycloparafins such as 1- and 2-
adamantyl, 1-norbornanyl, 2-exo-norbornanyl, 2-endo-
norbornanyl, 1- and 2-bicyclo[2.2.2]-octanyl, 1-, 2-, 3-,
6-, and 8-bicyclo[3.2.1]octanyl, and 1-, 2-, and 3-
bicyclo[3.3.1]nonanyl and cycloolefins such as 1- and 2-
norbornenyl. Examples of the preferred compound (IV) are
2-amino-7-(2-adamantylmethyl)-3H,5H-pyrrolo[3,2-d]-
WO 91/06548 PCT/US90/05756
., y.
~~ A'~.r9'~
~w.~.d~ r.,~~
pyrimidin-4-one (IVA), 2-amino-7-(1-adamantylmethyl)-
3H,5H-pyrrolo[3,2-d]pyrimidin-4-one (IVB), 2-amino-7-
(cyclopentylmethyl)-3H_,5H-pyrrolo[3,2-d]-pyrimidin-4-one
(IVC), 2-amino-7-(cyclohexylmethyl)-3H,5H-pyrrolo[3,2-d]-
5 pyrimidin-4-one (IVD), 2-amino-7-(cycloheptylmethyl)-3_H,SH-
pyrrolo[3,2-d]pyrimidin-4-one (IVE), 2-amino-7-(1-
norbornanylmethyl)-3I~,,5~,-pyrrolo[3,2-d]pyrimidin-4-one
(IVF), 2-amino-7-(2-exo-norbornanylmethyl)-3H,5~i-
pyrrolo[3,2-d]pyrimidin-4-one (IVG), 2-amino-7-(2-endo-
norbornanylmethyl)-3H_,5I~,-pyrrolo[3,2-d]pyrimidin-4-one
(IVH), 2-amino-7-(1-norbornenylmethyl)-3H,5H-
pyrrolo[3,2-d]-pyrimidin-4-one (IVI), 2-amino-7-(2-
norbornenylmethyl)-3H,5H-pyrrolo[3,2-d]pyrimidin-4-one
(IVJ), 2-amino-7-(1-bicyclo[2.2.2]-octanylmethyl)-3H,5H_-
pyrrolo[3,2-d]-pyrimidin-4-one (IVK), 2-amino-7-(1-bicyclo-
[3.2.1]octanyl- methyl)-3H_,5H__-pyrrolo[3,2-d]pyrimidin-4-one
(IVL), and 2-amino-7-(1-bicyclo[3.3.1]nonanylmethyl)-3H,5H-
pyrrolo[3,2-d]-pyrimidin-4-one (IVM), and 2-amino-7-(1-
noradamantyl-methyl)-3H_,5H-pyrrolo[3,2-d]-pyrimidin-4-one
(IVN). In an alternative preferred embodiment the R1 group
has one or two substituents selected from the group
consisting of halogen, hydroxy, alkoxy, alkyl, or
trifluoromethyl. As halogen is preferably mentioned chloro
or fluoro. As alkoxy is preferably mentioned lower alkoxy,
including methoxy, ethoxy, propoxy and butoxy. As alkyl is
preferably mentioned lower alkyl, including methyl, ethyl,
propyl and butyl.
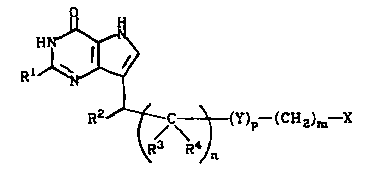
The present invention is also directed to a compound of
the formula
Ri
Y)P (CHZ)m X
WO 91/06548 PCT/US90/05756
t'_:':)
~~?'~ ~~ n~o
6
wherein R1 is H, NH2, or OCH3, R2 is an optionally '
substituted cyclic group optionally containing one or more
heteroatoms, R3 and R4 are independently H or C1-4 alkyl, m
is 0-4, n is 0-6, p is 0-1, X is CN, CSNH2, PO(OH)2, COOH,
S02NH2, NH2, OH, CNHNH2, tetrazole, triazole or CORS where
R5 is C1-4 alkyl , CF3 , NH2 , or OCl_4 alkyl , and Y is O or
NH.
The optionally substituted cyclic group (hereinafter
referred to as cyclo) recited for the above formula
l0 includes aromatic, heteroaromatic, alicyclic, and
heteroalicyclic groups preferably containing five to nine
atoms. Preferred optional substituents include halogen,
hydroxy, alkoxy, alkyl, and trifluoromethyl. Exemplary
substituents include chloro, fluoro, methoxy, ethoxy,,
propoxy, butoxy, methyl, ethyl, propyl, and butyl.
Preferred heteroatoms include oxygen, nitrogen, and sulfur,
which can be present in combination in the same group. The
preferred aromatic and heteroaromatic groups are phenyl, 2-
or 3-thienyl, 2- or 3-furanyl, 2-, 3-, or 4-pyridinyl, 2
or 3-pyrrolyl, 2-, 4-, or 5-thiazolyl, 2-pyrazinyl, 3- or
4-pyridazinyl, and 3-, 4-, or 5-pyrazolyl. The preferred
alicyclic and heteroalicyclic groups are 1- or 2-adamantyl,
cyclohexyl, cycloheptyl, 2- or 3-tetrahydrofuranyl, 2- or
3-tetrahydrothienyl, 2- or 3-tetrahydropyranyl, 2-, 3-, or
4-piperidinyl, 3- or 4-pyrazolidinyl, 2-, 4-, or
5-thiazolidinyl, 2- or 3-piperazinyl, 2- or 3-morpholinyl,
or 3- or 4-hexahydropyridazinyl.
In another aspect of the invention there is provided a
method for the selective suppression of mammalian T-cell
function without diminished effect on humoral immunity
which comprises administering to a mammal the compound (I),
whereby said compound inhibits purine nucleoside
phosphorylase and thereby T-cell formation.
WO 91/06548 PCT/US90/05756
~''4Y YufJ'~ '
7
In a further aspect of the present invention there is
provided a pharmaceutical composition for the selective
suppression of mammalian T-cell function without diminished
effect on humoral immunity which comprises an effective
amount of the compound and a pharmaceutically acceptable
diluent therefor.
The invention further relates to pharmaceutical
compositions suitable for enteral, such as oral or rectal,
transdermal and parenteral administration to mammals
including man, which are useful to inhibit purine
nucleoside phosphorylase activity and for the treatment of -
disorders responsive thereto, comprising an effective
amount of a pharmacologically active compound of the
invention, alone or in combination, with one or more
pharmaceutically acceptable carriers.
Preferred pharmaceutical compositions are tablets and
gelatin capsules comprising the active ingredient together
with a) diluents, e.g., lactose, dextrose, sucrose,
mannitol, sorbitol, cellulose and/or glycine; b)
lubricants, e.g., silica, talcum, stearic acid, its
magnesium or calcium salt and/or polyethyleneglycol; for
tablets also c) binders, e.g., magnesium aluminum silicate,
starch paste, gelatin, tragacanth, methylcellulose, sodium
carboxymethylcellulose and/or polyvinylpyrrolidone; if
desired d) disintegrants, e.g., starches, agar, alginic
acid or its sodium salt, or effervescent mixtures; and/or
e) absorbents, colorants, flavors and sweeteners.
Injectable compositions are preferably aqueous isotonic
solutions or suspensions, and suppositories are
advantageously prepared from fatty emulsions or
suspensions. Said compositions may be sterilized and/or
contain adjuvants, such as preserving, stabilizing, wetting
or emulsifying agents, solution promoters, salts for
regulating the osmotic pressure and/or buffers. In
WO 91/0648 PC1'/U590/Oa756
(~y;i
... ,.:~:,,.~ .
~~;'~! :,.s~..:.d
8
addition, they may also contain other therapeutically
valuable substances. Said compositions are prepared
according to conventional mixing, granulating or coating
methods, respectively, and contain about 0.1 to 75%,
preferably about 1 to 50%, of the active ingredient.
Suitable formulations for transdermal application include
an effective amount of a compound of the invention with a
carrier. Advantageous carriers include absorbable
pharmacologically acceptable solvents to assist passage
ZO through the skin of the host. Characteristically,
transdermal devices are in the fona of a bandage comprising
a backing member, a reservoir containing the compound
optionally with carriers, optionally a rate controlling
barrier to deliver the compound to the skin of the host at
a controlled and predetermined rate over a prolonged period.
of time, and means to secure the device to the skin.
The present invention provides a method of inhibiting
purine nucleoside phosphorylase activity in mammals and
treating diseases and conditions responsive thereto, e.g.,
autoimmune disorders, rejection of transplantation, or
psoriasis, which comprises administering to a mammal in
need thereof an effective amount of a compound of the
invention or of a pharmaceutical composition comprising a
said compound in combination with one or more
pharmaceutically acceptable carriers.
A further aspect of the invention relates to a method of
inhibiting the phosphorolysis and metabolic breakdown of
antiviral or antitumor purine nucleosides in mammals which
comprises administering in conjunction therewith to a
mammal in need thereof, either separately or in combination
therewith, an effective purine nucleoside phosphorylase
inhibiting amount of a compound of the invention or of a
said compound in combination with one or more
pharmaceutically acceptable carriers. More particularly,
WO 91/06548 PCT/US90/05756
_ ~~ w~~~~
j~ ~ L.b i~Y
.Yi. ;
. ' 9' nn. '~, ~ v
9
such relates to a method of inhibiting the phosphorolysis
and metabolic breakdown of purine nucleosides known in the
art, e.g., of 2'-deoxyguanosine, 2',3'-dideoxyinosine,
2',3'-dideoxyguanosine or 2',3'-dideoxyadenosine.
Furthermore, the invention thus relates to a method of
potentiating the antiviral or antitumor effect of 2' or 3'-
monodeoxypurine nucleosides or of 2',3'-dideoxypurine
nucleosides in mammals which comprises administering in
conjunction therewith to a mammal in need thereof, either
separately or in combination with a said nucleoside, an
effective purine nucleoside phosphorylase inhibiting amount
of a compound of the invention preferably in combination
with one or more pharmaceutically acceptable carriers.
More particularly, such relates to a method of enhancing or
potentiating the effect of 2',3'-dideoxypurine nucleosides
known in the art, e.g., of 2',3'-dideoxyinosine, 2',3'-
dideoxyguanosine or 2'-3'-dideoxyadenosine for the
treatment of retrovirus infections, e.g., HIV-retrovirus
infections (acquired immunodeficiency syndrome, AIDS).
2o 2',3'-Dideoxypurine nucleosides are known in the art as
inhibitors of HIV retrovirus infectivity and to be
metabolically degraded by PNP, e.g., as described in
Biochemical Pharmacolocrv 22, 3797 (1987). Such are
administered at a pharmaceutically acceptable dose which
is effective in inhibiting HIV-retrovirus infections.
Preferably the lowest possible effective dose is used.
The pharmaceutically acceptable effective dosage of
active compound of the invention to be administered is
dependent on the species of warm-blooded animal (mammal),
the body weight, age and individual condition, and on the
form of administration.
The pharmaceutical composition may be oral, parenteral,
suppository or other form which delivers the compound into
the bloodstream of a mammal to be treated. An oral form
WO 91/06548 PCT/US90/05756
~:..
~: "~..:1
has from about 1 to about 150 mg of the compound for an
adult (5o to 70 kg) which is mixed together with
pharmaceutically acceptable diluents such as lactose. In a
typical capsule, 25 mg of the compound are mixed together
5 with 192 mg lactose, 80 mg modified starch and 3 mg
magnesium stearate. Injectable forms of the compound are
also contemplated for administration.
The present invention is also useful with other
therapeutic agents. A daily dosage for a human weighing 50
10 to 70 kg of 1-50 mg/kg inhibits metabolic destruction of
certain anticancer agents such as beta-2'-deoxy-6-
thioguanosine and antiviral agents such as 2',3'-
dideoxyinosine, an anti-AIDS drug. These types of agents
are known to be susceptible to. cleavage. Upon cleavage,,
the agents lose effectiveness. The compounds of the
present invention are capable of reducing such cleavage.
This protection, therefore, enhances the efficacy of other
chemotherapeutic agents.
One method of making the compound (I) of the present
invention uses 3-substituted propionitriles as starting
materials. Such starting materials can be obtained by a
variety of methods that are well documented in the
literature. The compound (I) is then prepared from the
starting material by an adaptation of the synthetic
methodology disclosed in M. I. Lim, R. S. Klein, and J. J.
Fox, J. Ora. Chem., 44, 3826 (1979); M.I. Lim, R.S. Klein,
and J.J. Fox, Tetrahedron Lett.,. 21, 1013 (1980); M.I. Lim
and R.S. Klein, Tetrahedron Lett., 22, 25 (1981): M. I.
Lim, W. Y. Ren, B.A. Otter, and R.S. Klein, J. Orcr. Chem.,
48, 780 (1983).
A method of making the compound (II) of the present
invention uses a 3-(pyridinyl)propionitrile as the starting
material to make the compound (II). The appropriate 3-
(pyridinyl)propionitrile can be produced by converting the
WO 91/06548 PCT/US90/05756
t..::-: ~;
~.a,'~'a.
' ~Oh.Y f.b.~_. _ .
lI
corresponding 3-(pyridinyl)propionyl chloride to the
corresponding amide by ammonolysis. with, e.g., ammonium
hydroxide, which is then dehydrated to the desired nitrile
by distillation with a dehydrating agent, such as POC13 or ,
SOC12. Alternatively, the starting material is produced by
condensation of the 3-aldehyde with cyanoacetic acid
followed by decarboxylation to give the corresponding
substituted acrylonitrile, which is hydrogenated to give
the corresponding 3-(pyridinyl)propionitrile by either
catalytic hydrogenation or magnesium metal dissolving in
methanol at 0 'C, such as disclosed in Profitt, J., et al.,
J. Org. Chem., 40, 127 (1975). The compound (II) is then
prepared from the starting material by an adaptation of the
synthetic methodology disclosed in M. I. Lim, R. S. Klein,
and J. J. Fox, J. Org. Chem., 44, 3826 (1979); M.I. Lim,,
R.S. Klein, and J.J. Fox, Tetrahedron Lett.; 21, 1013
(1980); M.I. Lim and R.S. Klein, Tetrahedron Lett., 22, 25
(1981) ; M. I. Lim, W. Y. Ren, B.A. Otter, and R.S. Klein,
J. Orq. Chem., 48, 780 (1983).
Another method of making the compound of the present
invention uses a known compound, i.e., cyclohexenyl-
acetonitrile, as the starting material. The compound (III)
of the present invention is made by reacting the starting
material in an adaptation of the synthetic methodology
disclosed in M. I. Lim, R. S. Klein, and J. J. Fox, J. Orcx.
Chem., 44, 3826 (1979); M.I. Lim, R.S. Klein, and J.J. Fox,
Tetrahedron Lett., 21, 1013 (1980); M.I. Lim and R.S.
Klein, Tetrahedron Lett., 22, 25 (1981); M. I. Lim, W. Y.
Ren, B.A. Otter, and R.S. Klein, J. Orcr. Chem., 48, 780
(1983). Catalytic hydrogenation of either (IIIA), (IIIB),
or (IIIC) yields the compound (IIID).
A method of making the compound (IV) of the present
invention uses 3-substituted propionitriles as starting
materials. Such starting materials can be obtained by a
WO 91/06548 PCT/US90/05756
t ;5e-;,
((/~~ ~,..a.:~;~ .;~ ..
~~ ~'lY ~ ~~1~~~~Y'n.~
12
variety of methods that are well documented in the
literature. The compound (IV) is then prepared from the
starting material by an adaptation of the synthetic
methodology disclosed in M. I. Lim, R. S. Klein, and J. J.
Fox, J. Orcr. Chem., 44, 3826 (19?9); M.I. Lim, R.S. Klein,
and J.J. Fox, Tetrahedron Lett., 21, 1013 (1980); M.I. Lim
and R.S. Klein, Tetrahedron Lett., ~, 25 (1981)t M. I.
Lim, W. Y. Ren, B.A. Otter, and R.S. Klein, J. org. Chem.,
48, 780 (1983).
Another aspect of the present invention concerning a
compound of the formula
R1
tY)p-(CH2)m-X
R'~
n
provides a method of making a 2-amino compound (R1 = NH2)
and intermediates thereof. The first step of the method
involves reacting an optionally substituted cyclic aldehyde
with cyanoacetic acid at a molar ratio of about 1/1 to 1/5
in the presence of ammonium acetate at about reflux
temperature for about 10 hours to 8 days to make a 3-cyclo-
substituted pentanedinitrile as an intermediate. In the
second step, the 3-cyclo-pentanedinitrile is reacted with
an alkyl formate such as ethyl formate and a strong base
such as the metal-containing bases sodium hydride or sodium
alkoxide, e.g., sodium methoxide, at a molar ratio of about
1-2/3-6/1-3 and at a temperature of about 20-f5°C for about
10 hours to 8 days to make a 3-cyclo-2-formylpentanedini-
WO 91/06548 PCT/US90/05756
fry °.',
13
trile as a further intermediate. The next step involves
reacting the 3-cyclo-2-formylpentanedinitrile with a
glycine alkyl ester hydrochloride and sodium or ammonium
acetate at a molar ratio of about 1-2/1.5-4/1.5-4 and at a
temperature of about 20-60°C for about 10-48 hours to make
methyl N-((3-cyclo-2,4-dicyano)-2-butenyl]glycine as an
intermediate. In the subsequent step, the methyl N-[(3-
cyclo-2,4-dicyano)-2-butenyl]glycine is reacted with an
alkyl chloroformate such as ethyl chloroformate and 1,5-
diazabicyclo(4.3.0]non-5-ene (DBN) or 1,8-diazabicyclo
[5.4.0]undec-7-ene (DSU) at a molar ratio of about 1-2/1.5-
5/1.5-4 and at a temperature of about 0-50°C for about 10
hours to 10 days to make methyl 3-amino-4-(2-cyano-1-cyclo-
ethyl)-1-ethyl-1H-pyrrole-1,2-dicarboxylate as an,
intermediate. The next step involves reacting the methyl
3-amino-4-(2-cyano-1-cyclo-ethyl)-1-ethyl-1H-pyrrole-1,2-
dicarboxylate with a base such as sodium carbonate at a
molar ratio of about 2/1 to 1/5 and at about room
temperature for about 10-48 hours to make methyl 3-amino-4-
2~ (2-cyano-1-cyclo-ethyl)-1H-pyrrole-2-carboxylate as an
intermediate. In the next step, the methyl 3-amino-4-(2-
cyano-1-cyclo-ethyl)-1H-pyrrole-2-carboxylate is reacted
with benzoylisothiocyanate at a molar ratio of about 2/1 to
1/2 and at about room temperature for about 30 minutes to 3
2S hours to make N-benzoyl-N--[4-(2-cyano-1-cyclo-ethyl)-2-
methoxycarbonyl-1H-pyrrol-3-yl]thiourea as an intermediate.
The next step reacts the N-benzoyl-N--[4-(2-cyano-1-cyclo-
ethyl)-2-methoxycarbonyl-1H-pyrrol-4-3-yl]thiourea with an
alkyl halide such as methyl iodide at a molar ratio of
30 about 1/1 to 1/6 and at a temperature of about 0-30°C for
about 10 minutes to 10 hours to make N-benzoyl-N--[4-(2-
cyano-1-cyclo-ethyl)-2-methoxycarbonyl-1H-pyrrol-3-yl]S-
methylthiourea as an intermediate. In the following step,
the N-benzoyl-N'-[4-(2-cyano-1-cyclo-ethyl)-2-
WO 91/06548 PCTlUS90l05756
~.,~y IJJ.v:~'.Y~~:- .,
14
methoxycarbonyl-1H-pyrrol-3-yl]-S=methylthiourea (about 1-
2 mol) is reacted with methanolic or ethanolic ammonia at a
ratio of about 1/1 to 1/20 and at a temperature of about
20-130'C for about 16-60 hours to make a mixture of a 2-
amino compound of the present invention 3-cyclo-3-[2-amino-
4-oxo-3H-5H-pyrrolo[3,2-d]pyrimidin-7-yl]propanenitrile and
a 3-cyclo-3-[2-methylmercapto-4-oxo-3H,5H-pyrrolo[3,2-
d]pyrimidin-7-yl]propanenitrile as an intermediate in
making another compound of the present invention.
In a further aspect concerning the compound of the
formula
R1
)p (CH2)m_!C
there is provided a method of making a 2-methoxy compound
(R1 = OCH3) and intermediates thereof. The intermediate 3-
cyclo-3-j2-methylmercapto-4-oxo-3H,5H-pyrrolo[3,2-
d]pyrimidin-7-yl]propanenitrile is reacted with an
oxidizing agent such as permanganate or hydrogen peroxide
at a molar ratio of about 1/1 to 1/10 and at a temperature
of about 25-120'C for about 3-48 hours to make 3-cyclo-3-
[2-methylsulfonyl-4-oxo-3H,5H-pyrrolo[3,2-d]pyrimidin-7-
yl]propanenitrile as an intermediate. In the next step,
the 3-cyclo-3-[2-methylsulfonyl-4-oxo-3H_,5H-pyrrolo[3,2-
d_]pyrimidin-7-yl]propanenitrile is reacted with a sodium
alkoxide such as sodium methoxide at a molar ratio of about
1/1 to 1/10 and at_a temperature of about 25-100°C for
WO 91/06548 ~ PCT/US90/05756
. ~~"~ M'~_ ~,3 ; '
about 1-48 hours to make a 2-methoxy compound of the
present invention 3-cyclo-3-[2-methoxy-4-oxo-3H,5H-
pyrrolo[3,2-d]pyrimidin-7-yl]propanenitrile.
In a further aspect, there is provided a method of making
5 a compound wherein R1 is hydrogen. The methyl 3-amino-4
(2-cyano-1-cyclo-ethyl)-1H-pyrrole-2-carboxylate
intermediate described supra is reacted with
dimethylformamide dimethyl acetal at a molar ratio of about
1/1 to 1/4 and at a temperature of about 25-100'C for about
10 1-10 days to make methyl 4-(2-cyano-1-cyclo-ethyl)-3-[N-
(dimethylaminomethylene)amino]-1H-pyrrole-2-carboxylate as
an intermediate. The next step involves reacting the
methyl 4-(2-cyano-1-cyclo-ethyl)-3-[N-(dimethylamino-
methylene)amino]-1H-pyrrole-2-carboxylate with methanolic
15 or ethanolic ammonia at a molar ratio of about 1/1 to 1/20.
and at a temperature of about 20-130'C for about 10-68
hours to make the compound of the present invention 3-
cyclo-3-[4-oxo-3H,5H-pyrrolo[3,2-d]pyrimidin-7-
yl]propanenitrile.
As will be apparent to the skilled artisan, variations of
the aforesaid procedures are useful in making the variety
of compounds of the present invention without departing
from the spirit thereof. For example, compounds having
different values for "n" and "m" in accordance with the
formula of the present invention are obtained by either
stepping up or stepping down the series by the necessary
number of carbon atoms in accordance with known procedures.
Also, reactions involving some intermediates require
protection of nitrogen or oxygen atoms on the intermediates
30, using known procedures.
In order to more fully describe the present invention the
following non-limiting examples are provided. In the
examples all parts and percentages are by weight unless
WO 91/06548 PC'T/US90/05756
': )
16
indicated otherwise. Proportions of solvent mixtures used
as chromatographic eluents are by volume.
EXAMPLE 1
3-(3-Pyridinyl)propionitrile is prepared in this example.
A three-neck flask carrying a magnetic stir bar is fitted
with, a thermometer, pressure equalizing addition funnel,
and a reflux condenser carrying an argon inlet. Freshly
powdered potassium hydroxide (6.6 g, 0.1 mol) and anhydrous
acetonitrile (150 ml) are charged into the flask and heated
at reflux while 3-pyridinecarboxaldehyde (10.7 g, 0.1 mol)
in anhydrous acetonitrile (50 ml) is added dropwise over a
period of about five minutes and refluxing continued for
about another three minutes. The resulting hot reaction
mixture is poured into an ice/water mixture (100 g), and ,
the resulting solution is extracted with CH2C12 (3 x
100 ml), dried with Na2S04, and evaporated to give crude 3-
(3-pyridinyl)acrylonitrile, which is purified by column
chromatography over silica gel using CHC13 as the eluent;
yield 3.3 g (25.6%).
Under an argon atmosphere, a stirred solution of the
acrylonitrile (2.662 g, 0.02 mol) in 99% ethanol (100 ml)
is treated with a drop of 4% aqueous sodium hydroxide
followed by sodium borohydride (0.378 g, 0.01 mol).
Additional sodium borohydride (0.378 g) is added twice more
at four-hour intervals. The mixture is stirred at room
temperature overnight, diluted with water, extracted with
EtOAc and dried with Na2S04. The solvent is removed under
reduced pressure and the crude product obtained is
chromatographed over a column of silica using
chloroform/methanol (40/1) as the eluent to give 2.2 g
(84.6%) of the product as a colorless oil.
EXAMPLE 2
3-(3-Pyridinyl)propionitrile of. Example 1 is further
treated in the synthesis of the present invention. Under
WO 91/06548 PCT/US9Q/05756
s r~
a
17 ..
an atmosphere of dry N2, a mixture of 3-(3-
pyridinyl)propionitrile (0.661 g, 5.0 mmole), sodium
hydride (0.240 g, 10.0 mmole), and ethyl formats (1.11 g,
15.0 mmole) in anhydrous tetrahydrofuran (20 ml) is stirred
for 48 hours with protection from air and moisture.
Volatile matter is evaporated, and a solution of the solid
residue in 15 ml of sold water is adjusted at 0 ° C to a pH
of 6 with cold 6N HCl. The resulting oily mixture is
extracted with CHC13, and the extract is washed with water,
dried using Na2S04, and evaporated to give a dark oil,
which is a mixture of 2-formyl-3-(3-pyridinyl)propionitrile
and the nitrile starting material. This crude product is
;sed in the next reaction without further purification.
EXAMPLE 3
i5 Glycine methyl ester hydrochloride (0.942 g, 7.5 mmole)
and anhydrous sodium acetate (0.615-g, 7.5 mmole) are added
to a solution of the crude formyl compound (0.89 g) in
MeOH/H20 (4:1, 50 ml). After 24 hours, the MeOH is
evaporated in vacuo, and the mixture of water and oil is
extracted with CHC13. The CHC13 layer is dried (Na2S04)
and evaporated to give an amber oil which is applied to a
silica gel column. Elution with CHC13 gave two major
bands: (1) 3-(3-pyridinyl)propionitrile (used as starting
material in the previous step), and (2) the desired
enamine.
EXAMPLE 4
Under a nitrogen atmosphere, ethyl chloroformate
(0.521 g, 4.8 mmole) is added dropwise to a solution of the
enamine of Example 3 (0.513 g, 3.2 mmole) and 1,5-
diazabicyclo[4.3.0)non-5-ene (nBN, 1.37 g, 11.1 mmole) in
dry CH2C12 (15 ml) with external cooling in an ice bath.
After stirring at 0 °C for one hour, the solution is
allowed to a stand at room temperature overnight. After
checking progress by TLC, additional ClCO2Et (0.1 ml) and
WO 91/06548 PCT/US90/OS756
('~":''.;
18
DBN (1.o ml) are added to complete the conversion, and the
solution is allowed to stand for 24 hours. Volatile matter
is evaporated in vacuo and the viscous residue is purified
on a short silica gel column (whose main purpose is to
remove the less-mobile DBN) to give an N-blocked pyrrole;
which is used for the next step without further
purification.
EXAMPLE 5
To a solution of the N-blocked pyrrole of Example 4
(0.635. g, 2.0 mmole) in MeOH (50 ml) is added solid Na2C03
(0.212 g, 2.0 mmole), and the reaction mixture is stirred
at room temperature for 48 hr with separation of the
resultant deblocked pyrrole. The mixture is evaporated to
dryness, and the residue is triturated thoroughly with H20 .
(25 ml) to dissolve inorganics and extracted with CHC13 (3
x 100 ml). The extract is dried (Na2S04) and evaporated to
give a viscous gum that crystallized upon drying in vacuo
for use as an intermediate without further purification.
More extensive purification can, however, be effected by
using either column chromatography employing silica
gel/CHC13 or recrystallization from toluene/cyclohexane
(1:3).
EXAMPLE 6
Benzoyl isothiocyanate (0.232 g, 1.42 mmole) is added
dropwise to a solution of the unblocked pyrrole of Example
5 (0.290 g, 1.18 mmole) in dry CH2C12 (100 ml). After 1 h
at room temperature, solution is evaporated, and the gummy
residue is stirred in Et20/cyclohexane (1:1, 20 ml). The
resulting suspension of yellow solid is filtered under N2
pressure, and the thioureido product is dried in vacuo over
P205.
EXAMPLE 7
Methyl iodide (0.228 g, 1.61 mmole) is added to a
solution of the thioureido product of Example 6 (0.383 g,
WO 91/06548 PCT/US90/05756
r.,
~~a~~~~i~C ..
19
0.94 mmole) and DBN (0.140 g, 1.12 mmole) in dry CH2C12 (10
ml) at 0 'C. The solution is stirred at 0 °C for 15 min.,
at ambient temperature for 1 h, and then evaporated in
vacuo. A solution of the residue in CHC13 is
chromatographed on a silica gel column with CHC13/methanol
(97:3) as eluent to give homogeneous fractions of the
methylthio intermediate compound.
EXAMPLE 8
A solution of the methylthio compound of Example ?
(0.358 g, 0.85 mmole) in 100 ml of MeOH that has been
saturated with NH3 at 0 'C is heated at 90-95 °C for 24
hours in a glass-lined stainless steel bomb. The contents
of the chilled bomb are evaporated in vacuo to give a
mixture of the desired 2-amino intermediate compound,
benzamide, and a by-product that is a 2-methylthio .
derivative, as opposed to the 2-amino compound. The
mixture is dissolved in methanol and the solution is
evaporated with silica gel (about 5 g). The mixture is
then carefully layered onto the top of a silica-gel
chromatography column, which is then eluted with CHC13/MeOH
(9:1) to give the methylthio by-product and the desired 2
amino-7-(3-pyridinylmethyl)-3#~,5i~-pyrrolo(3,2-d_]pyrimidin
4-one intermediate. Further purification is obtained by
recrystallization from boiling isopropyl acetate in a
Soxhlet apparatus.
A solution of the pyridinylmethyl intermediate in O.1N
HCl is hydrogenated with a platinum catalyst at 60 lb/in2
H2 pressure. The catalyst is generated by brief .
hydrogenation of Pt02 in O.1N HC1. When the reaction is
complete, the catalyst is removed by filtration under N2
pressure, and the filtrate is evaporated. A solution of
the residue in the minimum amount of ethanol is diluted
slowly with a large amount of Eto2, and the hydrochloride
WO 91/06548 PCT/US90/05756
~~:~'~ ~~.~3
salt is obtained as a white hygroscopic solid of the
compound (IC).
EXAMPLE 9
The compound (IC) of Example 8 is tested foz enzyme
5 inhibition activity. A purine nucleoside phosphorylase
(PNP) enzyme assay is performed in which PNP activity
(IC50) for the compound is observed, which is determined
radiochemically by measuring the formation of [14C]
hypoxanthine from (14CJ-inosine (see Biomedicine, 33, 39
10 (1980)) using calf spleen as the enzyme source.
EXAMPLES 10-14
The following compounds of the present invention are
prepared . that are 2-amino-7-(R)-3H_,5H-pyrrolo[3,2-d_]-
pyrimidin-4-ones wherein R is -CH2R1 in which the R1 group ,
15 is as follows:
Example 10 R1 = 2-methyl-3-piperidinyl
Example 11 R1 = 2-chloro-3-piperidinyl
Example 12 R1 = 2-trifluoromethyl-3-piperidinyl
Example 13 R1 = 2-methoxy-3-piperidinyl
20 Example 14 R1 = 2-fluoro-3-piperidinyl
The compounds are prepared following the procedures set
forth in Examples 1-8 using the appropriate 3-(substituted
3-pyridinyl)-propionitriles as starting materials.
EXAMPLE 15
3-(2-Pyridinyl)propionitrile is prepared in this example
using the procedure of V. Boekelheide, et al., J. Am. Chem.
Soc., 75, 3243 (1953). A solution of potassium cyanide
(83.74 g) in water (160 ml) is added to a solution of
freshly distilled 2-vinylpyridine (67.59 g) in acetic
anhydride (131.30 g) with the rate of addition adjusted to
maintain gentle refluxing. When the addition is complete,
the resulting dark red mixture is heated for about 17 hours
at 105°C in an oil bath with vigorous stirring. The cooled
reaction mixture is adjusted to pH 8 with a saturated
WO 91/06548 PCT/US90/05756
21 ~o~~ ~ ~~ r-'~'N-~
Na2C03 solution. The mixture is extracted with CHC13 (4 x
150 ml), washed with water, dried with Na2S04, and
evaporated to a dark viscous oil. The oil is fractionally
distilled in vacuo through a vigreaux column. After a
small forerun containing 2-vinylpyridine (0.35 g), the
desired 3-(2-pyridinyl)propionitrile is collected as a
clear, fluorescent yellow-green, viscous oil: yield 59.8 g;
70.4%; by 86°C at 1.0 mm Hg.
EXAMPLES 16-20
The following compounds of the present invention are
prepared that are 2-amino-7-(R)-3H,5H-pyrrolo[3,2-d]
pyrimidin-4-ones wherein R is -CH2R1 in which the R1 group
is as follows:
Example 16 R1 = 2-piperidinyl
Example 17 R1 = 4-piperidinyl
Example 18 R1 = 3-trifluoromethyl-4-piperidinyl
Example 19 R1 = 3-methoxy-2-piperidinyl
Example 20 R1 = 3-fluoro-4-piperidinyl
The compounds are prepared following the procedures set
2o forth in Examples 1-8 and 15 using the appropriate 3-(2- or
4-pyridinyl)-propionitriles as starting materials.
EXAMPLE 21
A pharmaceutical composition for intraperitoneal
injection is prepared for testing the compound (IC). An
intraperitoneal injection solution containing the compound
of Example 8 is dissolved in an aqueous carrier that
contains ten percent DMSO.
EXAMPLE 22
The compound (IC) is intraperitoneally injected into
Lewis Rats via the test composition of Example 21 to
provide 30 mg of the compound (IC), with an injection given
twice per day. Controls are used, which receive only the
vehicle. At specific times after administration, the
animals are sacrificed and plasma samples are prepared.
WO 91/06548 PCT/US90/05756
l~fe~'r~ :e~ y4J ~~ i
22
The plasma is extracted with cold 0.5 N HC104 and
neutralized with solid NH4HC03. After removal of
perchlorate salts, the extract is subjected to HPLC on a
reversed phase column (Spherisorb ODSI). A significant
increase in plasma inosine,is observed in the plasma taken
from animals receiving the compound (IC).
EXAMPLES 23-33
Compounds prepared as in Examples 10-20 are each made
into a pharmaceutical formulation in accordance with the
preparation of Example 21 and the resultant injectable
solutions are tested in accordance with the procedure of
Example 22. A significant increase in plasma inosine is
observed in the plasma taken from animals receiving the
compounds of the present invention.
EXAMPLE 34
3-(2-Furanyl)propionitrile is prepared in this example.
In a three-neck flask fitted with a condenser and drying
tube, magnesium turnings (20 g) are added to a solution of
3-(2-furanyl)acrylonitrile (67.2 g) in dry methanol (2 1).
Additional magnesium is added in parts as the reaction rate
would allow until a total of 145 g has been added. After
about five hours, the solvent is evaporated until the
contents set to a solid paste, which is adjusted to a pH of
about 6.5 With 6N HC1 with cooling. The mixture is
extracted with several portions of CHC13, dried with
Na2S04, and concentrated to a dark oil. Distillation in
vacuo through a short Vigreaux column gives the
propionitrile as a colorless oil; yield 49.5 g (72%); by
46.0-46.5°C at 0.5 mm.
EXAMPLE 35
3-(2-Furanyl)propionitrile of Example 34 is further
treated in the synthesis of the present invention. Under
an atmosphere of dry N2, the propionitrile (48.7 g), sodium
hydride (10.28 g), ethyl formate (32.74 g), and anhydrous
WO 91/06548 PCT/US90/05756
23
tetrahydrofuran (200 ml) are stirred at room temperature
with protection from moisture for about 18 hours. The
~_'
volatile matter i~'Hthen evaporated, the resulting yellow
solid is dissolved in about 20 ml of cold water with ice
s bath cooling, and the solution is adjusted to a pH of 6.0
with cold 6N HCl. The resulting heavy oil precipitate is
extracted into CHC13, and the extract is washed with water,
dried with Na2S04, and evaporated to give a thin oil which
contains the crude formyl compound; yield 53.06 g.
EXAMPLE 36
Glycine methyl ester hydrochloride (67.05 g) and
anhydrous sodium acetate (43.79 g) are added to a solution
of the crude formyl compound of the previous example
(53.05 g) in MeOH/H20 (4:1, 1500 ml). After 24 hours, the.
MeOH is evaporated in vacuo, and the mixture of water and
oil is extracted with CHC13. The CHC13 layer is dried
(Na2S04) and evaporated to give an amber oil (59.1 g).
EXAMPLE 37
Under a nitrogen atmosphere, ethyl chloroformate
(43.68 g) is added dropwise to a solution of the amber oil
of Example 36 (59.1 g) and DBN (133 g) in dry CH2C12 (400
ml) with external cooling in an ice bath. After stirring
at 0 ° C for one hour, the solution is allowed to stand at
room temperature overnight. After checking progress by
TLC, additional C1C02Et and DBN are added to complete the
conversion, and the solution is allowed to stand for 24
hours. Volatile matter is evaporated in vacuo, the viscous
residue purified on a silica gel column (whose main purpose
is to remove the less-mobile DBN) to give an N-blocked
pyrrole, a corresponding pyrrole without the ethoxycarbonyl
blocking group on the pyrrole nitrogen, and the starting
propionitrile.
x
WO 91/06548 PCT/US9U/05756
~.~,,,~.:1
~~? r m~~~ 2~~
EXAMPLE 38
To a solution of the N-blocked pyrrole of Example 37
(11.66 g) in MeOH (200 ml) is added solid Na2CO3 (4.23 g),
and the reaction mixture is stirred at room temperature for
24 hr with separation of the resultant deblocked pyrrole.
The mixture is evaporated to dryness, and the residue is
triturated thoroughly With H20 (200 ml) to dissolve
inorganics and extracted with CHC13 (3 x 200 ml). The
extract is dried (Na2S04) and evaporated to give a viscous
gum that crystallized upon drying in vacuo for use as an
intermediate without further purification. More extensive
purification can, however, be effected by using either
column chromatography employing silica gel/CHC13 or
recrystallization from toluene/cyclohexane (1:3).
EXAMPLE 39
Benzoyl isothiocyanate (4.58 g) is added dropwise to a
solution of the unblocked pyrrole of Example 38 (5.15 g) in
dry CH2C12 (75 ml). After 1 h at room temperature,
solution is evaporated, and the gummy residue is dissolved
in Et20 (100 ml) with almost immediate separation of the
crystalline solid, which is collected by filtration. The
Et20 filtrate is heated to boiling and diluted with an
equal volume of warm cyclohexane. On cooling slowly the
solution gives additional thioureido product.
EXAMPLE 40
Methyl iodide (8.70 g) is added to a solution of the
thioureido product of Example 39 (10.68 g) and DBN (4.15 g)
in dry CH2C12 (250 ml) at 0 °C. The solution is stirred at
0 °C for 15 min., at ambient temperature .for 1 h, and then
evaporated in vacuo. A solution of the residue in CHC13 is
chromatographed on a silica gel column with CHC13 as eluent
to give homogeneous fractions of the methylthio
intermediate compound.
WO 91/06548 PCT/US90/05756
~, rah
,: ~~: ti.~:~.r~'~.~
. ~.a
EXAMPLE 41
A solution of the methylthio compound of Example 40
(0.80 g, 2.0 mmole) in 25 ml of MeOH that has been
saturated with NH3 at 0 °C is heated at 90-95 °C for 24
5 hours in a glass-lined stainless steel bomb. The contents
of the chilled bomb axe evaporated in vacuo to give a
mixture of the 2-amino compound, benzamide and a by-product
that is a 2-methylthio derivative, as opposed to the 2-
amino compound. The mixture is stirred vigorously for
10 several minutes with 30 ml of 2:1 Et20/cyclohexane, and the
insoluble white solid is filtered off and washed with Et20.
The filtrate contained most of the benzamide and 2-
methylthio components. A solution of the Et20/cyclohexane-
insoluble solid (0.425 g) in MeOH is evaporated with appr.
15 10 g of silica gel. The powdered residue is layered evenly.
onto the top of a silica gel column, which is then eluted
with CHC13/MeOH/HOAc (95:5:1) to give the 2-methylthio by-
product and the desired furanyl intermediate. The
intermediate is recrystallized by extraction into boiling
20 isopropyl acetate in a Soxhlet apparatus. The white
crystals are collected in three crops and dried in vacuo
over P205 at 110°C for 7h.
A solution of the intermediate (116 g, 0.5 mmole) in
methanol (50 ml) is hydrogenated with 30% palladium-on
25 charcoal (40 mg) at 62 lb/in2 H2 pressure for about 36
hours. The catalyst is removed by filtration under N2
pressure, and the filtrate is evaporated and reevaporated
with toluene. The solid residue is recrystallized by
extraction into boiling isopropyl acetate in a soxhlet
apparatus. The 2-tetrahydrofuranyl compound (IE) is
obtained as a white crystalline solid, which is dried in
vacuo over P205 at 110°C for about six hours; yield 73 mg
(61.8%): m.p. 284-286°C dec.
WO 91/06548 PCT/US90/05756
t,'T~:.'
.....
26
EXAMPLE 42
The product of Example 41 is tested for enzyme inhibition
activity according to the procedure of Example 9, and PNP
activity (IC50) for the compound is observed.
EXAMPLE 43
A pharmaceutical composition for intraperitoneal
injection is prepared for testing the compound (IE). An
intraperitoneal injection solution containing the compound
of Example 41 is dissolved in an aqueous carrier that
l0 contains ten percent DMSO.
EXAMPLE 44
Using the procedure of Example 16, the compound (IE) is
intraperitoneally injected into Lewis Rats via the test
composition of Example 43 to provide 30 mg of the compound,
(IE) and the results are analyzed compared to controls. A
significant increase in plasma inosine is observed in the
plasma taken from animals receiving the compound (IE).
EXAMPLES 45-49
The following compounds of the present invention are
prepared that are 2-amino-7-(R)-3F~,5H_-pyrrolo[3,2-d_]
pyrimidin-4-ones wherein R is -CH2-R1 in which the R1 group
is as follows:
Example 45 R1 = 3-tetrahydrofuranyl
Example 46 R1 = 3-chloro-2-tetrahydrofuranyl
Example 47 R1 =~ 3-trifluoromethyl-2-tetrahydrofuranyl
Example 48 R1 = 3-methoxy-3-tetrahydrofuranyl
Example 49 R1 = 3-fluoro-2-tetrahydrofuranyl
The compounds are prepared following the procedures set
forth in Examples 34-41 using the appropriate 3-(furanyl)
acrylonitriles as starting materials.
EXAMPLE 50
Under a nitrogen atmosphere, a solution of the
tetrahydrofuranyl compound (IE) (50o mg) obtained from
Example 41 and a crystal of phenol in 2N HBr (20 ml) is
WO 91/06548 PCT/US90l05756
~, r.~" j .~y , , .
~..' .f :.., ~.: .,~
27
stirred for 18 hours at 40'C. The solvent is evaporated
vacuo at low temperature, and the residue is washed with a
few ml of Et20 by decantation to remove any tribromophenol.
A suspension of the residue in H2o is adjusted to pH 7 with
1N NaOH to give the bromoalcohol as its free base. The
solid is collected, washed with cold water, and dried at
room temperature.
A solution of the bromoalcohol in anhydrous dimethyl-
acetamide is chilled in an ice bath and treated with an
equimolar portion of PBr3. The solution is allowed to stir
at room temperature for one hour, then the
dimethylacetamide is evaporated in vacuo at low temperature
using an air pump and dry-ice trap. A suspension of the
residue in ice/water is adjusted to pH 7 using 1N NaOH to
give the crude reaction product containing the 1,4-dibromo
compound. After filtration, washing with cold water, and
drying at room temperature, the resulting product is
refluxed with sodium sulfide in 50% ethanol/water, or
alternatively warmed with Na2S in N,N-dimethylacetamide
solution; the solvent removed under vacuum, the residue
washed with water to remove NaBr, and the pH adjusted to
about 7 to precipitate the 2-tetrahydrothienyl compound
(IG) .
EXAMPLE 51
The compound of Example 50 is tested for enzyme
inhibition activity as in Example 9, and PNP activity
(IC50) for the compound is observed.
EXAMPLE 52
A pharmaceutical composition for intraperitoneal
injection is prepared for testing the compound (IG). An
intraperitoneal injection solution is made containing the
compound of Example 50 is dissolved in an aqueous carrier
that contains ten percent DMSO.
WO 91/06548 PCT/US90/05756
28
EXAMPLE 53
Using the procedure of Example 16, the compound (IG) is
intraperitoneally injected into Lewis Rats via the test
composition of Example 52 to provide 30 mg of the compound
(IG) and the results are analyzed compared to controls. A
significant increase in plasma inosine is observed in the
plasma taken from animals receiving the compound (IG).
EXAMPLES 54-58
The following compounds of the present invention are
prepared that are 2-amino-7-(R)-3$,5H-pyrrolo[3,2-d_]
pyrimidin-4-ones wherein R is -CH2-R1 in which the R1 group
is as follows:
Example 54 R1 = 3-tetrahydrothienyl
Example 55 R1 = 3-chloro-2-tetrahydrothienyl
Example 56 R1 = 3-trifluoromethyl-2-tetrahydrothienyl
Example 57 R1 = 3-methoxy-2-tetrahydrothienyl
Example 58 R1 = 3-fluoro-2-tetrahydrothienyl
The compounds are prepared following the procedures set
forth in Examples 34-41 and 50 using the appropriate 3
(furanyl)-acrylonitriles as starting materials.
EXAMPLE 59
Using the procedure of Example 1, 3-(2-pyrrolyl)-
propionitrile is prepared using pyrrole-2-carboxaldehyde as
the starting material. Following Examples 2-8, the
intermediate 2-amino-7-(2-pyrrolylmethyl)-3H,5Fi-
pyrrolo[3,2-d]pyrimidin-4-one is prepared from the
propionitrile, from which the 2-pyrrolidinyl compound (II)
is obtained by reduction of the intermediate.
EXAMPLE 60
Using the procedure of Example 1, 3-(3-pyrrolyl)-
propionitrile is prepared using pyrrole-3-carboxaldehyde as
the starting material. Following Examples 2-8, the
intermediate 2-amino-7-(3-pyrrolylmethyl)-3H,5H-pyrrolo-
[3,2-d]pyrimidin-4-one is prepared from the propionitrile,
WO 91/0648 PCTlUS90l05756
.~_~.
29 ~~~ ~s~M.J:.i~:Y~'~
from which the 3-pyrrolidinyl compound (IJ) is obtained by
reduction of the intermediate.
EXAMPLE 61
Using the procedure of Example 1, 3-(2-pyranyl)
propionitrile is prepared using 2-pyrancarboxaldehyde as
the starting material. Following Examples 2-8, the
intermediate 2-amino-7-(2-pyranylmethyl)-3I~,5~i-pyrrolo-
[3,2-_d]pyrimidin-4-one is prepared from the propionitrile,
from which the 2-tetrahydro-pyranyl compound (IL) is
obtained by reduction of the intermediate.
EXAMPLE 62
Using the procedure of Example 1, 3-(3-pyranyl)
propionitrile is prepared using 3-pyrancarboxaldehyde as
the starting material. Following Examples 2-8, the
intermediate 2-amino-7-(3-pyranylmethyl)-3H,5H-
pyrrolo[3,2-dJpyrimidin-4-one is prepared' from the
propionitrile, from which the 3-tetrahydro-pyranyl compound
(IM) is obtained by reduction of the intermediate.
EXAMPLE 63
Using the procedure of Example 1, 3-(4-pyranyl)
propionitrile is prepared using 4-pyrancarboxaldehyde as
the starting material. Following Examples 2-8, the
intermediate 2-amino-7-(4-pyranylmethyl)-3H,5H-pyrrolo-
[3,2-d)pyrimidin-4-one is prepared from the propionitrile,
from which the 4-tetrahydro-pyranyl compound (IN) is
obtained by reduction of the intermediate.
EXAMPLE 64
A solution of the methylthio compound of Example 7
(0.358 g, 0.85 mmole) in 100 ml of MeOH that has been
saturated with NH3 at 0 °C is heated at 90-95 °C for 24
hours in a glass-lined stainless steel bomb. The contents
of the chilled bomb are evaporated in vacuo to give a
mixture of the desired 2-amino intermediate compound,
benzamide, and a by-product that is a 2-methylthio
WO 91/06548 PCTlUS90/05756
~~,.i
~o~ti.i~ :aas,.~~y , ,
derivative, as opposed to the 2-amino compound. The
mixture is dissolved in methanol and the solution is
evaporated with silica gel (about 5 g). The mixture is
then carefully layered onto the top of a silica-gel
5 chromatography column, which is then eluted with CHC13/MeOH
(9:1) to give the methylthio by-product and the desired 2
amino-7-(3-pyridinylmethyl)-3I~,5H_-pyrrolo[3,2-d_]pyrimidin
4-one, compound (IIA). Further purification is obtained by
recrystallization from boiling isopropyl acetate in a
10 Soxhlet apparatus.
EXAMPLE 65
The compound (IIA) of Example 64 is tested for enzyme
inhibition activity. A purine nucleoside phosphorylase
(PNP) enzyme assay is performed in which PNP activity
15 (IC50) for the compound is observed, which is determined
radiochemically by measuring the formation of [14C]-
hypoxanthine from [14C]-inosine (see Biomedicine, 33, 39
(1980) using calf spleen as the enzyme source.
EXAMPLES 66-68
2~ The following compounds of the present invention are
prepared that are 2-amino-7-(R)-3H_,5H_-pyrrolo[3,2-d]
pyrimidin-4-ones wherein R is -CH2-R1 in which the R1 group
is as follows:
Example 66 R1 = 2-pyridinyl
25 Example 67 R1 = 4-pyridinyl
Example 68 R1 = 3-chloro-2-pyridinyl
The compounds are prepared following the procedures set
forth in Examples 1-7 and 64 using the appropriate 3-
(pyridinyl)-propionitriles as starting materials.
30 EXAMPLE 69
A pharmaceutical composition for intraperitoneal
injection is prepared for testing the compound (IIA). An
intraperitoneal injection solution containing the compound
WO 91/06548 PCT/US90/05756
r~.
f...
~~',, s~,~~~''3
31
of Example 64 is dissolved in an aqueous carrier that
contains ten percent DMSO.
EXAMPLE 70
The compound (IIA) is intraperitoneally injected into
Lewis Rats via the test composition of Example 69 to
provide 30 mg of the compound (IIA), with an injection
given twice per day. Controls are used, which receive only
the vehicle. At specific times after administration, the
animals are sacrificed and plasma samples are prepared.
The plasma is extracted with cold 0.5 N HC104 and
neutralized With solid NH4HCOg. After removal of
perchlorate salts, the extract is subjected to HPLC on a
reversed phase column (Spherisorb ODSI). A significant
increase in plasma inosine is observed in the plasma taken .
from animals receiving the compound (TIA).
EXAMPLES 71-73
Compounds prepared as in Examples 66-68 are each made
into a pharmaceutical formulation in accordance with the
preparation of Example 69 and the resultant injectable
solutions are tested in accordance with the procedure of
Example 70. A significant increase in plasma inasine is
observed in the plasma taken from animals receiving the
compounds of the present invention.
EXAMPLE 74
1-Cyclohexenylacetonitrile is treated in the synthesis of
the present invention. Under an atmosphere of dry N2, a
solution of 1-cyclohexenylacetonitrile (9.2 g: 75.92 mmole)
in anhydrous tetrahydrofuran (THF, l0 ml) is added to a
stirred mixture of sodium hydride (3.18 g; 132.86 mmole)
and ethylformate (30.14 g; 406.93 mmole) in 50 ml THF, and
the resulting mixture is stirred at room temperature for
about 18 hours. Volatile matter is evaporated in vacuo at
room temperature. Water (30 ml) is added to the residue at
0'C, and the solution adjusted to a pH of 6 by dropwise
WO 91/06548 PCT/US90/05756
,r..~,,
iv.';
~~; t,.'~""'~ r' ~~
32
addition of 6N HC1. The resulting precipitate of heavy oil
is extracted into CHC13. The extract is washed with water
and dried with Na2SO4, and the resulting organic layer
evaporated to give a crude formyl compound as a red-brown
oil (9.6 g).
EXAMPLE 75
Glycine methyl ester hydrochloride (16.68 g, 132.85
mmole) and anhydrous sodium acetate (10.89 g, 132.85 mmole)
are added to a solution of the crude formyl compound (9.6
g) of Example 74 without further purification in MeOH/H2o
(4:1, 150 ml). After 24 hours, the MeOH is evaporated in
vacuo, and the mixture of water and oil is extracted with
CHC13. The CHC13 layer is dried (Na2S04) and evaporated to
give an amber oil which is applied to a silica gel column.
_.. ~lution with CHC13 gave the desired enamine: yield 4.5 g.
EXAMPLE 76
Under a nitrogen atmosphere, ethyl chloroformate (3.04 g;
28.06 mmole) is added dropwise to a solution of the enamine
of Example 75 (4.12 g, 18.70 mmole) and 1,5-di-
azabicyclo[4.3.0]-non-5-ene (DBN, 6.96 g, 56.11 mmole) in
dry CH2C12 (100 ml) with external cooling in an ice bath.
After stirring at 0 'C for one hour, the solution is
allowed to stand at room temperature overnight. After
checking progress by TLC, additional C1C02Et (0.5 ml) and
DBN (3.0 ml) are added to complete the conversion, and the
solution is allowed to stand for 24 hours. Volatile matter
is evaporated in vacuo, the viscous residue purified on a
short silica gel column (whose main purpose is to remove
the less-mobile DBN) to give an N-blocked pyrrole, which is
used for the next step without further purification.
EXAMPLE 77
To a solution of the N-blocked pyrrole of Example 76
(3.0 g, 10.26 mmole) in MeOH (100 ml) is added solid Na2C03
(2.71 g, 25.65 mmole), and the reaction mixture is stirred
WO 91/06548 PCF/US90/05756
...,.
.:,
33
at room temperature for 48 hr with separation of the
resultant deblocked pyrrole. The mixture is evaporated to
dryness, and the residue is triturated thoroughly with H2o
(50 ml) to dissolve inorganics and extracted with CHC13 (3
x 50 ml). The extract is dried (Na2so4) and evaporated to
give a viscous gum, which is purified on a silica gel
column using CHC13 as the eluent; yield 2.04 g; m.p. 125°C.
EXAMPLE 78
Benzoyl isothiocyanate (0.76g, 4.65 mmole) is added
dropwise to a solution of the unblocked pyrrole of Example
77 (0.91 g, 4.13 mmole) in dry CH2C12 (30 ml). After 1 h
at room temperature, the solution is evaporated, and the
gummy residue is triturated with methanol to give a
thioureido product: yield 0.70 g~ m.p. 170°C.
EXAMPLE 79
Methyl iodide (0.678 g, 4.78 mmole) is added to a
solution of the thioureido product of Example 78 (0.630 g,
1.64 mmole) and DBN (0.230 g, 1.85 mmole) in dry CH2C12 (50
ml) at 0 °C. The solution .is stirred at 0 °C for 15 min.,
at ambient temperature for 1 h, and then evaporated in
vacuo. A solution of the residue in CHC13 is
chromatographed on a silica gel column with CHC13 as eluent
to give homogeneous fractions of the methylthio
intermediate; yield 0.7 g.
EXAMPLE 80
A solution of the methylthio compound of Example 79
(0.70 g, 1.76 mmole) in 50 ml of MeOH that has been
saturated with NH3 at 0 °C is heated at 90-95 °C for 24
hours in a glass-lined stainless steel bomb. The contents
of the chilled bomb are evaporated in vacuo to give a
mixture of the compound (IIIA), benzamide and a by-product
that is a 2-methylthio derivative, as opposed to the 2-
amino compound (IIIA). The mixture is stirred vigorously
for several minutes with appr. 50 ml of Et20, and the
WO 91/06548 PCT/US90f05756
~'~. ;1
34
insoluble white solid is filtered off and washed with Et20.
The filtrate contained most of the benzamide and 2-
methylthio components. A solution of the Et20-insoluble
solid (0.342 g) in MeOH is evaporated with appr. to g of
silica gel. The powdered residue is layered evenly onto
the top of a silica gel column, which is then eluted with
CHC13/MeOH/HOAc (95:5:1) to give the 2-methylthio by-
product and the desired 2-amino product (IIIA). (IIIA) is
recrystallized by extraction into boiling isopropyl acetate
in a Soxhlet apparatus. The white crystals are collected
in three crops and dried in vacuo over P2o5 at 110 ' C for
7h; yield 44%: mp 280 'C dec., anal. calcd. for
C12H14N40'0.6H20: C, 59.78; H. 6.35; N, 23.23. Found: C,
59.98; H, 6.46; N, 23.15.
EXAMPLE 81
The compound of Example 80 is tested for enzyme-
inhibition activity. A purine nucleoside phosphorylase
(PNP) enzyme assay is performed in which PNP activity for
the compound is determined radiachemically by measuring the
formation of [14C]-hypoxanthine from [14C]-inosine (see
Biomedicine, 33, 39 (1980) using calf spleen as the enzyme
source. At 1 mM phosphate the IC50 is 1.9 ~.M, and at 50 mM
phosphate the IC50 is 19 ~M.
EXAMPLE 82
Following the procedures set forth in Examples 74-80,
compounds (IIIB) and (IIIC) are made using 2- and 3-
cyclohexenyl-acetonitrile, respectively, as starting
materials. The compounds are tested as in Example 81 and
significant enzyme-inhibition activity is observed.
EXAMPLES 83-87
The following compounds of the present invention are
prepared that are 2-amino-7-(R)-3H,5H-pyrrolo[3,2-d]
pyrimidin-4-ones in which the R group is as follows:
Example 83 R = 3-methyl-2-cyclohexenyl
WO 91/06548 PCT/US90/05756
r'r' ;.
.. w "~" p:'
Example 84 R = 2-chloro-3-cyclohexenyl
Example 85 R = 3-trifluoromethyl-1-cyclohexenyl
Example 86 R = 3-methoxy-1-cyclohexenyl
Example 87 R = 2-fluoro-3-cyclohexenyl
5 The compounds are prepared following the procedures set
forth in Examples 74-81 using the appropriate substituted
cyclohexenyl acetonitriles as starting materials.
EXAMPLE 88
A pharmaceutical composition for intraperitoneal
10 injection is prepared for testing the compound (IIIA). An
intraperitoneal injection solution containing the compound
(IIIA) is dissolved in an aqueous carrier that contains ten
percent DMSO.
EXAMPLE 89
15 The compound (IIIA) is intraperitoneally injected into
Lewis Rats via the test composition of Example 88 to
provide 30 mg of the compound (IIIA), with an injection
given twice per day. Controls are used, which receive only _
the vehicle. At specific times after administration, the
20 animals are sacrificed and plasma samples are prepared.
The plasma is extracted with cold 0.5 N HC104 and
neutralized with solid NH4HC03. After removal of
perchlorate salts, the extract is subjected to HPLC on a
reversed, phase column (Spherisorb. ODSI). A significant
25 increase in plasma inosine is observed in the plasma taken
from animals receiving the compound (IIIA).
EXAMPLES 90-94
Compounds grepared as in Examples 83-87 are each made
into a pharmaceutical formulation in accordance with the
30 preparation of Example 88 and the resultant injectable
solutions are tested in accordance with the procedure of
Example 89. A significant increase in plasma inosine is
observed in the plasma taken from animals receiving the
compounds of the present invention.
WO 91/06548 PCT/US90/05756
~~ 1~,~ ~~
36
EXAMPLE 95
The compound (IIID) is prepared using 2-amino-7-(1-
cyclohexenyl)-3H,5H-pyrrolo[3,2-d]pyrimidin-4-one as an
intermediate. A solution of the intermediate (0.2 g; 0.86
mmole) in ethanol (50 ml) is hydrogenated with 10% Pd-C (50
mg) at 45 lb/in2 for 16 h and filtered hot through Celite.
The filtrate is evaporated to dryness, and the residue is
crystallized from hot ethanol to give the compound (IIID);
yield 157 mg (78%), mp >300 'C dec. anal. calcd. for
C12H16N40'0. EtOH: C, 61.80; H, 7.10; N, 23.51. Found:
C, 61.95; H, 7.43: N, 23.56.
EXAMPLE 96
The compound (IIID) prepared in Example 95 is tested for
enzyme-inhibition activity as in Example 81. At 1 mM.
phosphate the IC50 is 1.3 uM, and at 50 mM phosphate the
IC50 is 145 ACM.
EXAMPLE 97
3-(2-Adamantyl)propionitrile is prepared in this example
using a modification of the procedure of M. Ohno, et al. ,
J. Org. Chem. 53, 1285 (1988). A Solution of 2-
bromoadmantane (20 g;,92.96 mmole); Bu3SnH (32.46 g; 111.5
mmole), acrylonitrile (9,86 g; 185.92 mmole), and AIBN (740
mg) in toluene (280 ml) is stirred at reflux temperature
for 3 h. The reaction mixture is washed with ammonia water
(0.4 M, 500 ml), the organic layer is washed with H20 and
dried aver MgS04 and evaporated. The residue is distilled
between 110-118'C (and about 0.2 mmHg); fractions are
combined to give a crude sample of contaminated 3-(2-
adamantyl)propionitrile with tin complexes, which is
purified on silica gel column with hexanes; followed by
hexanes/ethylacetate 97:3 and hexanes/ethylacetate 95:5,
yield 9.4 g (53.4%); mp semi-solid.
WO 91/06548 PCT/US90/05756
37 ~~,~;~~~,,~~3
EXAMPLE 98
3-(2-Adamantyl)propionitrile of Example 97 is further
treated in the synthesis of the present invention. Under
an atmosphere of dry N2, a mixture of 3-(2-adamantyl)
propionitrile (7.0 g, 36.99 mmole), sodium hydride (1.7 g,
73.95 mmole), and anhydrous tetrahydrofuran (?5 ml) is
heated at 52'C in a water bath for 15 min., and a solution
of ethyl formate (13.69 g, 184.89 mmole) in THF (100 ml) is
added over a period of 45 min. After two hours at 50 - 55
°C, a second portion of NaH.(0.8 g) and HC02Et (7.5 ml) are
added, and the reaction mixture is stirred for about two
days. A third portion of HC02Et (7.5 ml) and NaH (0.8 g)
are added and left at room temperature for about 24 hours
(unreacted nitrile is inert in the next step and can be
recovered at the first purification step). The thick paste
is stirred overnight and allowed to cool to room
temperature. Volatile matter is evaporated under reduced
pressure, and the residual pale yellow crust is dissolved
in the minimum volume of cold water (appr. 150 ml) at 0 °C.
The solution is adjusted to pH 6.0 by addition of 6N HC1
and extracted with CHC13 (3 x 100 ml). The extract is
washed with H20, dried over Na2SO4, and evaporated in vacuo
to a thick amber oil. This crude product is used in the
next reaction without further purification.
EXAMPLE 99
Glycine methyl ester hydrochloride (6.96 g, 55.46 mmole)
and anhydrous sodium acetate (4.55 g, 55.46 mmole) are
added to a solution of the crude formyl compound (8.0 g) in
MeOH/H20 (4:1, 500 ml). After 24 hours, the MeOH is
evaporated in vacuo, and the mixture of water and oil is
extracted with CHC13. The CHC13 layer is dried (Na2S04)
and evaporated to give an amber oil which is applied to a
silica gel column. Elution with CHC13 gave two major
bands: (1) 3-(2-adamantyl)-propionitrile (used as star'cing
WO 91/06548 PCT/US90/05756
~,~;~~?~ ~~=3
38
material in the previous step), and (2) the desired
enamine: yield 6 g.
EXAMPLE 100
Under a nitrogen atmosphere, ethyl chloroformate (2.82 g,
26.0 mmole) is added dropwise to a solution of the enamine
of Example 99 (5.0 g, 17.34 mmole) and 1,5-diazabicyclo
[4.3.0]non-5-ene ("DBN,~~ 6.46 g, 52.0 mmole) in dry CH2C12
(50 ml) with external cooling in an ice bath. After
stirring at 0 'C for one hour, the solution is allowed to a
stand at room temperature overnight. After checking
progress by TLC, additional C1C02Et (1.81 ml) and DBN
(3.23 g) are added to complete the conversion, and the
solution is allowed to stand for 24 hours. Volatile matter
is evaporated in vacuo, the viscous residue purified on a
short silica gel column (whose main purpose is to remove.
the less-mobile DBN) to give an N-blocked pyrrole, which is
used for the next step without further purification.
EXAMPLE 101
To a solution of the crude N-blocked pyrrole of Example
100 (6.0 g, 16.59 mmole) in MeOH (100 ml) is added solid
Na2C03 (4.39 g, 41.49 mmole), and the reaction mixture is
stirred at room temperature for 48 hr with separation of
the resultant deblocked pyrrole. The mixture is evaporated
to dryness, and the residue is triturated thoroughly with
H20 (50 ml) to dissolve inorganics and extracted with CHC13
(3 x 100 ml). The extract is dried (Na2S04) and evaporated
to give a viscous gum, which crystallized by triturating
with ether: yield 4 g; m.p. 162-163°C.
EXAMPLE 102
Benzoyl isothiocyanate (1.22 g, 7.47 mmole) is added
dropwise to a solution of the unblocked pyrrole of Example
101 (1.91 g, 6.62 mmole) in dry CH2C12 (50 m1). After 1 h
at room temperature, solution is evaporated, and the gummy
residue is dissolved in Et20 (100 ml) with almost immediate
WO 91/06548 PCT/US90/05756
,~'aF~"a'~ '?~
..'~~.sa. r-.r~
39
separation of the crystalline solid. The Et20 filtrate is
heated to boiling and diluted with an equal volume of warm
cyclohexane. On cooling slowly the solution gives
additional thioureido product: yiled 2.81 g (95%); m.p.
193-194°C.
EXAMPLE 103
Methyl iodide (2.46 g, 17.39 mmole) is added to a
solution of the thioureido product of Example 102 (2.7 g,
5.98 mmole) and DBN (0.82 g, 6.57 mmole) in dry CH2C12 (50
ml) at 0 °C. The solution is stirred at 0 °C for 15 min.,
at ambient temperature for 1 h, and then evaporated in
vacuo to give a crude sample of the methythio intermediate
compound; yield 2.78 g (crude).
EXAMPLE 104
A solution of the methylthio compound of Example 103
(2.78 g, 5.18 mmole) in 150 ml of MeOH that has been
saturated with NH3 at 0 °C is heated at 90-95 °C for 24
hours in a glass-lined stainless steel bomb. The contents
of the chilled bomb are evaporated in vacuo to give a
mixture of the compound (IVA), benzamide and a by-product
that is a 2-methylthio derivative, as opposed to the 2-
amino compound (IVA). The mixture is stirred vigorously
for several minutes with appr. 75 ml of Et20, and the
insoluble white solid is filtered off and washed with Et20.
The filtrate contained most of the benzamide and 2-
methylthio components. A solution of the Et20-insoluble
solid (1.38 g) in MeOH is evaporated with appr. 25 g of
silica gel. The powdered residue is layered evenly onto
the top of a silica gel column, which is then eluted with
CHC13/MeOH/HOAc (95:5:1) to give the 2-methylthio by-
product and the desired 2-amino product (IVA). (IVA) is
recrystallized by extraction into boiling isopropyl acetate
in a Soxhlet apparatus. The white crystals are collected
in three crops and dried in vacuo over P205 at 110 °C for
WO 91/06548 PCT/US90l05756
~r,,V>
~D'~
.J I IV.f.~i~i~
7h; yield 51.6%; mp >350 'C dec.; anal. calcd. for
C17H22N40'0.21MeOH~0.22H20: C, 66.88; H, 7.59; N, 18.12.
Found: C, 66.86; H, 7.59; N, 18.12.
EXAMPLE 105
5 The compound of Example 104 is tested for enzyme
inhibition activity. A purine nucleoside phosphorylase
(PNP) enzyme assay is performed in which the PNP activity
(IC50) for the compound is found, which is determined
radiochemically by measuring the formation of [14C]-
10 hypoxanthine from [14C]-inosine (see Biomedicine, 33, 39
(1980)) using calf spleen as the enzyme source. At 1 mM
phosphate the IC50 is 0.090 uM, and at 50 mM phosphate the
IC50 is 2.5 ~M.
EXAMPLES 106-110
15 The following compounds of the present invention are r
prepared that are 2-amino-7-(R)-3H,5H-pyrrolo[3,2-d]-
pyrimidin-4-ones wherein R is -CH2-R1 in which the R1 group
is a 2-adamantyl group as follows:
Example 106 R1 = 2-(1-methyl)-adamantyl
20 Example 107 R1 = 2-(1-chloro)-adamantyl
Example 108 R1 = 2-(1-trifluoromethyl)-adamantyl
Example 109 R1 = 2-(1-methoxy)-adamantyl
Example 110 R1 = 2-(1-fluoro)-adamantyl
The compounds are prepared following the procedures set
25 forth in Examples 98-104 using the appropriate 3-(2
adamantyl)-propio-nitriles as starting materials.
EXAMPLES 111-116
The following compounds of the present invention are
prepared that are 2-amino-7-(R)-3H,5H-pyrrolo[3,2-d]
30 pyrimidin-4-ones wherein R is -CH2-R1 in which the R1 group
is a 1-adamantyl group as follows:
Example 111 R1 = 1-(2-methyl)-adamantyl
Example 112 R1 = 1-(2-chloro)-adamantyl
Example 113 R1 = 1-(2-trifluoromethyl)-adamantyl
WO 91!06548 PCT/US90/05756
. . ~d?! °a'~
:"r.....;.,.
41
Example 114 R1 = 1-(2-methoxy)-adamantyl
Example 115 R1 = 1-(2-fluoro)-adamantyl
Example 116 R1 = 1-adamantyl
The compounds are prepared following the procedures set
forth in Examples 98-104 using the appropriate 3-(1
adamantyl)-propio-nitrites as starting materials.
EXAMPLE 117
A pharmaceutical composition for intraperitoneal
injection is prepared for testing the compound (IVA). An
intraperitoneal injection solution containing the compound
(IVA) is dissolved in an aqueous carrier that contains ten
percent DMSO.
EXAMPLE 118
The compound (IVA) is intraperitoneally injected into
Lewis Rats via the test composition of Example 117 to .
provide 30 mg of the compound (IVA), with an injection
given twice per day. Controls are used, which receive only
the vehicle. At specific times after administration, the
animals are sacrificed and plasma samples are prepared.
The plasma is extracted with cold 0.5 N HC104 and
neutralized with solid NH4HC03. After removal of
perchlorate salts, the extract is subjected to HPLC on a
reversed phase column (Spherisorb ODSI). A significant
increase in plasma inosine is observed in the plasma taken
from animals receiving the compound (IVA).
EXAMPLES 119-129
Compounds prepared as in Examples 106-116 are each made
into a pharmaceutical formulation in accordance with the
preparation of Example 117 and the resultant injectable
solutions are tested in accordance with the procedure of
Example 118. A significant increase in plasma inosine is
observed in the plasma taken from animals receiving the
compounds of the present invention.
WO 91/06548 PCI'/US90/05756
42
EXAMPLE 130
3-Cyclopentylpropionitrile is prepared in this example.
3-Cyclopentylpropionyl chloride (57.7 g, 0.36 mole) is
added dropwise to a large excess of concentrated ammonium
hydroxide (400 ml) cooled in an ice/salt bath. The heavy
suspension of white solid is stirred overnight, collected
by filtration, washed with cold water, and recrystallized
fram about 2 liters of boiling water. The lustrous white
plates of the amide are dried in vacuo over P205; yield
31.6 g (62.3%); mp 122 °C.
With protection from atmospheric moisture, a solution of
the amide (23.5 g, 0.166 mole) in POC13 (150 ml) is heated
at 120 °C for 1 h. The oil bath is cooled to about 70 °C,
and excess POC13 is distilled off under vacuum, and the
cooled residue is poured onto ice (about 300 g). The
mixture is neutralized by cautious addition of solid Na2C03
and extracted with several portions of Et2O. The dried
(Na2S04) extract is evaporated to give a clear, pale yellow
oil which is distilled in vacuo to give the desired
nitrile; yield, 16.86 g (82%) by 88.0 - 88.5 °C/8.7 mm).
MS (EI): m/z 122 (M-H)+; IR (cap. film), 2245cm-1(CN); 1H
NMR, d 1.67 (q, 2, -CH2CH2CN), 2.36 (t, 2, -CH2CN), complex
multiplets centered about 1.11, 1.63, 1.86 (cyclopentyl
protons).
EXAMPLE 131
3-Cyclopentylpropionitrile of the previous example is
further treated in the synthesis of the present invention.
Under an atmosphere of dry N2, a mixture of 3-cyclopentyl-
propionitrile (14.8 g, 0.12 mole), sodium hydride (5.8,
0.24 mole), and anhydrous tetrahydrofuran (30o ml) is
heated at 52 °C in a water bath for 15 min., and a solution
of ethyl formate (13.3 g, 0.18 mole) in THF (100 ml) is
added over a period of 45 min. After two houxs at 50 - 55
°C, a second portion of NaH (1.9 g) and HC02Et (5.0 ml) are
WO 91/06548 PCf/US90/05756
43
~~ ~f.M./:..,
iG~_.h-(n~r~~l ~ ;,~
added, followed in 30 min. by a third portion of HC02Et
(unreacted nitrile is inert in the next step and can be
recovered at the first purification step). The thick paste
is stirred overnight and allowed to cool to room
temperature. Volatile matter is evaporated under reduced
pressure, and the residual pale yellow crust is dissolved
in the minimum volume of cold water (about 150 ml) at 0'C.
The solution is adjusted to pH 6.0 by addition of 6N HCl
and extracted with CHC13 (3 x 100 ml). The extract is
washed with H20, dried over Na2S04; and evaporated in vacuo
to a thick amber oil. This crude product (15.6 g) is used
in the next reaction without further purification.
EXAMPLE 132
Glycine methyl ester hydrochloride (19.31 g, 0.154 mole)
and anhydrous sodium acetate (12.61 g, 0.154 mole) are
added to a solution of the crude formyl compound of the
previous example (15.6 g) in MeOH/H20 (4:1, 500 ml). After
24 hours, the MeOH is evaporated in vacuo, and the mixture
of water and oil is extracted with CHC13. The CHC13 layer
2o is dried (Na2S04) and evaporated to give an amber oil which
is applied to a silica gel column. Elution with CHCl3 gave
two major bands: (1) 3-cyclopentylpropionitrile (8.22 g,
66.: mmole or 55.6% of the nitrile used as starting
material in the previous step), and (2) the desired enamine
(3.45 g; 29.1% based on theoretical yield corrected for
amount of nitrite present in starting material; MS (FAB):
223 (M + H)+) .
EXAMPLE 133
Under a nitrogen atmosphere, ethyl chloroformate (2.53 g,
23.3 mmole) is added dropwise to a solution of the enamine
of the previous example (3.45 g, 15.5 mmole) and DBN (5.78
g, 46.6 mmole) in dry CH2C12 (50 ml) with external cooling
in an ice bath. After stirring at 0 °C for one hour, the
solution is allowed to a stand at room temperature
WO 91/06548 PCT/US90/05756
~~~.r-w,r~,..~ .~
tGo.~~~ r:w.,.::.::~
44
overnight. After checking progress by TLC, additional
C1C02Et (0.5 ml) and DBN (3.0 ml) are added to complete the
conversion, and the solution is allowed to stand for 24
hours. Volatile matter is evaporated in vacuo, the viscous
residue purified on a short silica gel column (whose main
purpose is to remove the less-mobile DBN) to give an N-
blocked pyrrole (4.50 g; 98%), which is used for the next
step without further purification.
EXAMPLE 134
l0 To a solution of the N-blocked pyrrole of the previous
example (4.50 g, 15.3 mmole) in MeOH (100 ml) is added
solid Na2C03 (1.62 g, 15.3 mmole), and the reaction mixture
is stirred at room temperature for 48 hr with separation of
the resultant deblocked pyrrole. The mixture is evaporated
to dryness, and the residue is triturated thoroughly with
H2o (50 ml) to dissolve the inorganics and extracted with
CHC13 (3 x 100 ml). The extract is dried (Na2S04) and
evaporated to give a viscous gum that crystallized upon
drying in vacuo; yield 2.97 g (87.4%) of material suitable
for use as an intermediate without further purification.
More extensive purification can, however, be effected by
using either column chromatography employing silica
gel/CHC13 or recrystallization from toluene/cyclohexane
(1:3) .
EXAMPLE 135
Benzoyl isothiocyanate (2.62 g, 16.03 mmole) is added
dropwise to a solution of the unblocked pyrrole of Example
134 (2.97 g, 13.36 mmole) in dry CH2C12 (100 ml). After 1
h at room temperature, solution is evaporated, and the
gummy residue is dissolved in Et20 (100 ml) with almost
immediate separation of crystalline solid; yield 1.75 g.
The Et20 filtrate is heated to bailing and diluted with an
equal volume of warm cyclohexane. On cooling slowly the
solution gave an additional 1.58 g of thioureido product;
WO 91/06548 PCf/US90/05756
~.;~-,~~"~
~~... r;.",.,:_;~~
total yield 3.33 g (64.6%). A small amount of the
thioureido product is recrystallized from warm
Et20/cyclohexane (15 ml each); mp 123 - 125 °C. MS (FAB):
386 (M + H)+. Anal. Calcd, for C20H23N303S~0.45C6H12: C.
5 64.40; H, 6.76; N, 9.93. Found: C, 64.51. H, 7.10 N,
9.93.
EXAMPLE lab
Methyl iodide (2.60 g, 18.32 mmole) is added to a
solution of the thioureido product of Example 135 (3.21 g,
10 8.33 mmole) and DBN (1.24 g, 9.99 mmole) in dry CH2C12 (80
ml) at 0 °C. The solution is stirred at 0 °C for 15 min.,
at ambient temperature for 1 h, and then evaporated in
vacuo. A solution of the residue in CHC13 is
chromatographed on a silica gel column with CHC13 as eluent
15 to give homogeneous fractions of the methylthio
intermediate compound; yield, 2.46 g (74%).
EXAMPLE 137
A solution of the methylthio compound of Example 136
(2.07 g, 5.18 mmole) in 150 ml --of MeOH that has been
20 saturated With NH3 at 0 'C is heated at 90-95 °C for 24
hours in a glass-lined stainless steel bomb. The contents
of the chilled bomb are evaporated in vacuo to give a
mixture of the compound (IVC), benzamide and a by-product
that is a 2-methy?thio derivative, as opposed to the 2-
25 amino compound (IVC). The mixture is stirred vigorously
for several minutes with appr. 75 ml of Et2o, and the
insoluble white solid is filtered off and washed with Et20.
The filtrate contained most of the benzamide and 2-
methylthio components. A solution of the Et20-insoluble
30 solid (1.13 g) in MeOH is evaporated with appr. 10 g of
silica gel. The powdered residue is layered evenly onto
the top of a silica gel column, which is then eluted with
CHC13/MePH/HOAc (95:5:1) to give the 2-methylthio by-
product (252 mg; MS (FAB) : 264 (M + H)+) and the desired
WO 91/06548 PCT/US90/05756
..>
46
2-amino product (IVC) (679 mg, 56.4%). (IVC) is
recrystallized by extraction into boiling isopropyl acetate
in a Soxhlet apparatus. The white crystals are collected
in three crops and dried in vacuo over P205 at 110 °C for
7h; yield 5.40 mg (44.9%) ; mp 324 - 326 ° C dec. ; MS (FAB)
233 ( M + H)+: anal. calcd. for C12H16N40~ C, 62.05; H,
6.94; N, 24.12. Found: C, 62.04: H, ?.11; N, 24.48.
EXAMPLE 138
The compound of Example 137 is tested for enzyme°
inhibition activity in accordance with the procedure of
Example 9. At 1 mM phosphate the IC50 is 0.029 ~,M, and at
50 mM phosphate the IC50 is 1.8 ~,M.
EXAMPLES 139-142
The following compounds of the present invention are
prepared that are 2-amino-7-(R)-31_i,SH_-pyrrolo[3,2-d]-
pyrimidin-4-ones wherein R is -CH2-R1 in which the R1 group
is as follows:
Example 139 R1 = 3-methylcyclopentyl
Example 140 R1 = 2-chlorocyclopentyl
Example 141 R1 = 3-triflouromethylcyclopentyl
Example 142 R1 = 3-methoxycyclopentyl
The compounds are prepared following the procedures set
forth in Examples 130-137 using the appropriate 3
(substituted cyclopentyl)-propionitriles as starting
materials.
EXAMPLE 143
A pharmaceutical composition for intraperitoneal
injection is prepared for testing the compound (IVC). An
intraperitoneal injection solution is prepared containing
the compound (IVC) is dissolved in an aqueous carrier that
contains ten percent DMSO.
WO 91/06548 PCT/US90/05756
f %~M.L..i~RY
47
EXAMPLE 144
Using the procedure of Example 112, the compound (IVC) is
intraperitoneally injected into Lewis Rats via the test
composition of Example 143 and the results compared with
controls. A significant increase in plasma inosine is
observed in the plasma taken from animals receiving the
compound (IVC).
EXAMPLE 145
3-Cyclohexylpropionitrile is prepared in this example. A
solution of cyclohexanepropionic acid (50 g; 0.32 mole) and
thionyl chloride (152 g; 1.28 mole) in 100 ml benzene is
allowed to stand overnight and is then evaporated to an
oily residue. The residue is added in portions to 28%
aqueous ammonia (270 ml) at 25°C and the mixture stirred
for about two hours. The resulting product is collected by
filtration, washed with cold water, and recrystallized from
about 2 liters of boiling water. The lustrous white plates
of the amide are dried in vacuo over P205; yield 45.5 g.
With protection from atmospheric moisture, a solution of
the amide (45.5 g, 0.293 mole) in SOC12 (200.3 g; 1.68
mole) refluxed for about six hours. The oil bath is cooled
to about 70 °C, and excess SOC12 is distilled off under
vacuum, and the cooled residue is poured onto ice (about
300 g). The mixture is neutralized by cautious addition of
solid Na2C03 and extracted with several portions of Et20.
The dried (Na2S04) extract is evaporated to give a clear,
pale yellow oil Which is distilled in vacuo to give the
desired nitrile; yield 42.0 g.
EXAMPLE 146
3-Cyclohexylpropionitrile of Example 145 is further
treated in the synthesis of the present invention. Under
an atmosphere of dry N2, a mixture of 3-cyclohexyl-
propionitrile (22.3 g, 0.16 mole), sodium hydride (5.38,
0.224 mole), and anhydrous tetrahydrofuran (120 ml) is
WO 91/06548 PCT/U590/05756
48
heated at 52 °C in a water bath for 15 min., and a solution
of ethyl formate (55.4 g, 0.75 mole) in THF (50 ml) is
added over a period of 45 min. After two hours at 50 - 55
°C, a second portion of NaH (2.0 g) and HC02Et (5.0 ml) are
added (unreacted nitrile is inert in the next step and ca
be recovered at the first purification step), and the
reaction mixture is stirred for about three days at 55'C
and then allowed to cool to roam temperature. Volatile
matter is evaporated under reduced pressure, and the
residual pale yellow crust is dissolved in the minimum
volume of cold water (about 75 ml) at 0 °C. The solution
is adjusted to pH 6.0 by addition of 6N HC1 and extracted
with CHC13 ( 3 x 100 ml ) . The extract is washed with H20,
dried over Na2S04, and evaporated in vacuo to a thick amber
oil. This crude product is used in the next reaction
without further purification.
EXAMPLE 147
Glycine methyl ester hydrochloride (30.60 g, 0.24 mole)
and anhydrous sodium acetate (19.99 g, 0.24 mole) are added
to a solution of the crude formyl compound of the previous
example (25.22 g) in MeOH/H20 (4:1, 500 ml). After 24
hours, the MeOH is evaporated in vacuo, and the mixture of
water and oil is extracted with CHC13. The CHC13 layer is
dried (Na2S04) and evaporated to give an amber oil which is
applied to a silica gel column. Elution with CHC13 gave
two major bands: (1) 3-cyclohexylpropionitrile (used as
starting material in the previous step), and (2) the
desired enamine: yield 16 g.
EXAMPLE 148
Under a nitrogen atmosphere, ethyl chloroformate (1.38 g,
12.7 mmole) is added dropwise to a solution of the enamine
of Example 147 (2.0 g, 8.46 mmole) and DBN (2.1 g, 16.9
morale) in dry CH2C12 (50 ml) with external cooling in an
ice bath. After stirring at 0 °C for one hour, the
WO 91/Ob~48 PGT/US90/0575b
I~ ~"tI .'r
~;s~ f IM.i:v~'t11
49
solution is allowed to a stand at room temperature
overnight. After checking progress by TLC, additional
C1C02Et (0.5 ml) and DBN (1.5 ml) are added to complete the
conversion, and the solution is allowed to stand for 24
hours. Volatile matter is evaporated in vacuo, the viscous
residue purified on a short silica gel column (whose main
purpose is to remove the less-mobile DBN) to give an N-
blocked pyrrole, which is used for the next step without
further purification.
EXAMPLE 149
To a solution of the N-blocked pyrrole of Example 148
(2.6 g, 8.43 mmole) in MeOH (100 ml) is added solid Na2C03
(2.23 g, 21.07 mmole), and the reaction mixture is stirred
at room temperature for 48 hr with separation of the
resultant deblocked pyrrole. The mixture is evaporated to
dryness, and the residue is triturated thoroughly with H2o
(50 ml) to dissolve inorganics and extracted with CHC13 (3
x 100 ml). The extract is dried (Na2S04) and evaporated to
give a viscous gum, which is purified on a silica gel
column using CHC13 as the eluent; yield 1.67 g (84%); m.p.
73-74'C.
EXAMPLE 150
Benzoyl isothiocyanate (0.74 g, 4.02 mmole) is added
dropwise to a solution of the unblocked pyrrole of Example
149 (0.95 g) in dry CH2C12 (20 ml). After one hour at room
temperature, the solution is evaporated, and the gummy
residue is dissolved in Et20 (100 ml) with almost immediate
separation of crystalline solid. The Et2o filtrate is
heated to boiling and diluted with an equal volume of warm
cyclohexane. On cooling slowly the solution gives
additional thioureido product; total yield 1.41 g (88%):
m.p. 156-157°C.
WO 91/06548 PCT/US90/05756
~~,lM.~~ ~ W1
FF~~ldd01 J s'~e.~l..:~n.;J~
EXAMPLE 151
Methyl iodide (1.1 g, 7.6 mmole) is added to a solution
of the thioureido product of Example 150 (0.96 g, 2.61
mmole) and 1,5-diazabicyclo[4.3.0]non-5-ene (0.38 g, 3.0
5 mmole) in dry CH2C12 (20 ml) at 0°C. The solution is
stirred at 0°C for 15 min., at ambient temperature for 1 h,
and then evaporated in vacuo. A solution of the residue in
CHC13 is chromatographed on a silica gel column with CHC13
as eluent to give homogeneous fractions of the methylthio
10 intermediate compound; yield 0.92 g.
EXAMPLE 152
A solution of the methylthio compound of Example 151
(0.8 g, 1.93 mmole) in 50 ml of MeOH that has been -
saturated with NH3 at 0 °C is heated at 90-95 °C for 24
15 hours in a glass-lined stainless steel bomb. The contents
of the chilled bomb are evaporated in vacuo to give a
mixture of the compound (IVD), benzamide and a by-product
that is a 2-methylthio derivative, as opposed to the 2-
amino compound (IVD). The mixture is stirred vigorously
20 for several minutes with appr. 75 ml of Et20, and the
insoluble white solid is filtered off and washed with Et2o.
The filtrate contained most of the benzamide and 2-
methylthio components. A solution of the Et2o-insoluble
solid (0.390 g) in MeOH is evaporated with appr. 10 g of
25 silica gel. The 'powdered residue is layered evenly onto
the top of a silica gel column, which is then eluted with
CHC13/MeOH/HOAc (95:5:1) to give the 2-methylthio by-
product and the desired 2-amino product (IVD). (IVD) is
recrystallized by extractian into boiling isopropyl acetate
30 in a Soxhlet apparatus. The white crystals are collected
in three crops and dried in vacuo over P205 at 110 °C for
7h; yield 49%, mp >300 °C; anal. calcd. for C13H18N40: C,
63.39, H, 7.36: N, 22.74. Found: C, 63.50; H, 7.74: N,
22.67.
WO 91/065x8 PCT/US90/05756
t.:N.a..' v
51
EXAMPLE 153
The compound of Example 152 is tested for enzyme
inhibition activity as in Example 105. At 1 mM phosphate
the IC50 is 0.037 uM, and at 50 mM phosphate the IC50 is
2.2 uM.
EXAMPLE 154
The compound of Example 152 is tested to determine its
effectiveness in potentiation of the toxicity of 2'-
deoxyguanosine (d-Guo) (see D. A. Schewach et al., Cancer
-ss. , 46, 519 (1986) , and J. C. Sircar et al. , Accents and
~tions, 21, 253 (1987)). CCRF-CEM cells are grown in
RPMI-1640 medium. To a suspension cultures of these cells,
d-Guo at a fixed concentration (5.62 ~,M) and the compound
at varied concentrations are added and the number of cells
are determined in a Coulter counter 24, 48, and 72 hours
thereafter. From these data, the IC50 is calculated to be
2.0 ~,M as the concentration of the compound required to
reduce the increase in cell number between 0 and 72 hours
to 50% of that of control cultures.
EXAMPLE 155
The compound 2-amino-7-(3-methylcyclohexylmethyl)-3~i,5H_-
pyrrolo[3,2-d]pyrimidin-~!-one is prepared. First, using
the procedures set forth in Examples 146-152 above, but
with 3-(3-methylbenzyl)-propionitrile as the starting
material, the aryl derivative 2-amino-7-(3-methylbenzyl)-
3H,5H-pyrrolo[3,2-d]-pyrimidin-4-one is made. A solution
of the aryl derivative (0.2 g, 0.78 mmole) in
trifluoroacetic acid (TFA) (20 ml) is hydrogenated with
Pto2 at 60 lb/in2 for 24 h. Catalyst is filtered off
through a Celite bed, and the filtrate is evaporated. The
residue is triturated with methanol and left in the
refrigerator overnight. The resulting crystallized
trifluoroacetate salt precipitates from the solution and is
collected by filtration. The TFA salt is suspended in 8 ml
WO 91/06548 PGT/US90/05756
. f~,~ s ~A ~y~..j1
n.>
52
of H20, adjusted to pH8 by cone. NH40H and sonicated. The
pure product is collected, washed with H20 and dried: yield
165 mg (81%); mp 282 °C. Anal: Calcd. for C14H20N40= . C,
64.60; H, 7.74; N, 21.52. Found: C, 64.24; H, 7.96; N,
21.51%.
EXAMPLE 156
The procedure described in Example 155 is repeated to
prepare 2-amino-7-(3-trifluoromethylcyclohexyl-methyl)-
3~,5~i-pyrrolo-(3,2-d]pyrimidin-4-one using 3-(3-
trifluoromethylbenzyl)-propionitrile as the starting
compound: yield 69%; mp 165 'C. Anal. caled. for
C14H17N40F3'0.6H20: C, 51.72 H, 5.64; N, 17.23. Found:
C, 51.82; H, 5.71: N, 16.81%.
EXAMPLE 157
The compound prepared in Example 155 is tested for
enzyme-inhibition activity as in Example 105. At 1 mM
phosphate the IC50 is 0.025 ACM, and at-50 mM phosphate the
IC50 is 0Ø820 ~M.
EXAMPLE 158
The compound prepared in Example 156 is tested for
enzyme-inhibition activity as in Example 105. At 1 mM
phosphate the IC50 is 0.020 ~M, and at 50 mM phosphate the
IC50 is 0.740 ~M.
EXAMPLE 159
3-Cycloheptylpropionitrile is prepared in this example
according to the procedure of Example 97 using a solution
of 2-bromocycloheptane (25.57 g; 144.38 mmole); Bu3SnH
(50.42 g; 173.26 mmole), acrylonitrile (15.32 g; 288.77
mmole), and AIBN (1.13 g) in toluene (300 ml). Yield is
16 g; mp oil.
EXAMPLE 160
3-Cycloheptylpropionitrile of Example 159 is further
treated in the synthesis of the present invention. Under
an atmosphere of dry N2, a mixture of 3-cycloheptyl-
WO 91/06548 PCT/US90/05756
., ..d J'..:WI
n~F,f mp~ s~~,~
53
propionitrile (8.5 g, 56.19 mmole), sodium hydride (2.6 g,
112.39 mmole), and anhydrous tetrahydrofuran (100 ml) is
heated at 52 °C in a water bath for 15 min., and a solution
of ethyl formate (20.81 g, 280.99 mmole) in THF (100 ml) is
added over a period of 45 min. After two hours at 50-55'C,
a second portion of NaH (1.35 g) and HC02Et (10.4 g) are
added, followed in 30 min. by a third portion of HC02Et
(unreacted nitrile is inert in the next step and can be
recovered at the first purification step). The thick paste
is stirred overnight and allowed to cool to room
temperature. Volatile matter is evaporated under reduced
pressure, and the residual pale yellow crust is dissolved
in the minimum volume of cold water (about 150 ml) at 0'C.
The solution is adjusted to pH 6.0 by addition of 6N HC1
and extracted with CHC13 (3 x 100 ml). The extract is
Washed with H20, dried over Na2S04, and evaporated in vacuo
to a thick amber oil. This crude product is used in the
next reaction without further purification.
EXAMPLE 161
Glycine methyl ester hydrochloride (9.35 g, 74.47 mmole)
and anhydrous sodium acetate (6.10 g, 74.47 mmole) are
added to a solution of the crude formyl compound of Example
160 (8.9 gt 49.65 mmole) in MeOH/H20 (4:1, 250 ml). After
24 hours, the MeOH is evaporated in vacuo, and the mixture
of water and oil is extracted with CHC13. The CHC13 layer
is dried. (Na2S04) and evaporated to give an amber oil which
is applied to a silica gel column. Elution with CHC13 gave
two major bands: (1) 3-cycloheptylpropionitrile (used as
starting material in the previous step), and (2) the
desired enamine, which is recrystallized from a CHC13/Et20
mixture; yield 6.18 g; m.p. 57-58°C.
EXAMPLE 162
Under a nitrogen atmosphere, ethyl chloroformate (4.01 g,
37.03 mmole) is added dropwise to a solution of the enamine
WO 91/06548 PCT/US90/05756
~~ ;t ~, ~~~:.a
..
54
of Example 161 (6.18 g, 24.69 mmole) and DBN (9.19 g, 74.04
mmole) in dry CH2C12 (100 ml) with external cooling in an
ice bath. After stirring at 0 °C for one hour, the
solution is allowed to a stand at room temperature
overnight. After checking progress by TLC, additional
C1C02Et (0.5 ml) and DBN (3.0 ml) are added to complete the
conversion, and the solution is allowed to stand for 24
hours. Volatile matter is evaporated in vacuo, the viscous
residue purified on a short silica gel column (whose main
purpose is to remove the less-mobile DBN) to give an N-
blocked pyrrole, which is used for the next step without
further purification.
EXAMPLE 163
To a solution of the N-blocked pyrrole of Example 162
(7.8 g, 24.19 mmole) in MeOH (100 ml) is added solid Na2C03
(6.41 g, 60.48 mmole), and the reaction mixture is stirred
at room temperature for 48 hr with separation of the
resultant deblocked pyrrole. The mixture is evaporated to
dryness, and the residue is triturated thoroughly with H2o
(50 ml) to dissolve inorganics and extracted with CHC13 (3
x 100 ml). The extract is dried (Na2SO4) and evaporated to
give a viscous gum, Which was purified by column
chromatography employing silica gel/CHC13; yield 4 g; m.p.
88-X39 ° C.
EXAMPLE 164
Benzoyl isothiocyanate (1.5 g, 8.96 mmole) is added
dropwise to a solution of the unblocked pyrrole of Example
163 (1.99 g, 7.95 mmole) in dry CH2C12 (50 ml). After 1 h
at room temperature, solution is evaporated, and the gummy
residue is dissolved in Et2o (l00 ml) with almost immediate
separation of the crystalline solid. The Et2o filtrate is
heated to boiling and diluted with an equal volume of warm
cyclohexane. On cooling slowly the solution gives
WO 91/06548 PCT/US90/05756
3
additional thioureido product; yield 2.89 g (88%); m.p.
158-159'C.
EXAMPLE 165
Methyl iodide (1.7 g, 11.96 mmole) is added to a salution
5 of the thioureido product of Example 164 (1.7 g, 4.1 mmole)
and DBN (0.56 g, 4.52 mmole) in dry CH2C12 (80 ml) at 0 'C.
The solution is stirred at 0'C for 15 min., at ambient
temperature for 1 h, and then evaporated in vacuo. A
solution of the residue in CHC13 is chromatographed on a
10 silica gel column with CHC13 as eluent to give homogeneous
fractions of the methylthio intermediate compound.
EXAMPLE 166
A solution of the methylthio compound of Example 165
(1.72 g, 4.02 mmole) in 50 ml of MeOH that has been
'!5 saturated with NH3 at 0 'C is heated at 90-95 °C for 24
hours in a glass-lined stainless steel bomb. The contents
.of the chilled bomb are evaporated in vacuo to give a
mixture of the compound (IVE), benzamide and a by-product
that is a 2-methylthio derivative, as opposed to the 2-
20 amino compound (IVE). The mixture is stirred vigorously
for several minutes, with appr. 75 ml of Et20, and the
insoluble white solid is filtered off and Washed With Et2o.
The filtrate contained most of the benzamide and 2-
methylthio components. A solution of the Et20-insoluble
25 solid (0.850 g) in MeOH is evaporated with appr. 1o g of
silica gel. The powdered residue is layered evenly onto
the top of a silica gel column, which is then eluted with
CHC13/MeOH/HOAc (95:5:1) to give the 2-methylthio by-
product and the desired 2-amino product (IVE). (IVE) is.
30 recrystallized by extraction into boiling isopropyl acetate
in a Soxhlet apparatus. The white crystals are collected
in three crops and dried in vacuo over P205 at 110 °C far
7h: yield 54%, mp >300 °C dec.: anal. calcd. for C14H2pN40:
WO 91/06548 PCT/US90/05756
,..~-,,, ~ '° ;3
p ,.,, a._:.y ..
56
C, 64.60; H, 7.74; N, 21.52. Found: C, 64.78: H, 8.01; N,
21.61.
EXAMPLE 167
The compound of Example 166 is tested for enzyme
inhibition activity as in Example 105. At 1 mM phosphate
the IC50 is 0.030 ACM, and at 50 mM phosphate the IC50 is
0.840 uM.
EXAMPLE 168
Using the procedure of Example 97, 3-(1-norbornanyl)-
propionitrile is made from 1-bromonorbornane, and 3-(2-
norbornanyl)-propionitrile (mixture of 2-exo and 2-endo) is
made from 2-bromonorbornane. Following Examples 98-104,
the propionitriles are converted to the compounds (IVF),
IV(G), and (IVH).
EXAMPLE 169
Using the procedure of Example 97, 3-(1-bicyclo-
[3.2.1]octanyl)-propionitrile, 3-(2-bicyclo-
[3.2.1]octanyl)-propionitrile, 3-(3-bicyclo-
[3.2.1]octanyl)-propionitrile, and 3-(8-bicyclo-
[3.2.1]octanyl)-propionitrile are made respectively from 1-
bromo-bicyclo[3.2.1]octane, 2-bromo-bicyclo[3.2.1]octane,
3-bromo-bicyclo[3.2.1]octane, and 8-bromo-
bicyclo[3.2.1]octane. Following Examples 98-104, the
propionitriles are converted to the compound (IVL) and the
related, 2-bicyclo[3.2.1]octanyl, 3-bicyclo[3.2.1]octanyl,
and 8-bicyclo[3.2.1]octanyl derivatives.
EXAMPLE 170
Using a modification of the procedure disclosed in
D. Farcasiu, Synthesis, 615 (1972), 6-bicyclo[3.2.1]octane
carboxaldehyde is prepared by reacting bicyclo[3.2.1]octan
6-one with trimethylsulfoxonium iodide, giving an
intermediate epoxide, which is then converted to the
aldehyde by treatment with boron trifluoride etherate.
Following the procedure of Netherlands Pat. 6,610,204, the
WO 91/06548 PCT/LS90/05756
.~ ~-., r .'rA ~i ';Z "~
,C,s ~. ; ! /.., .i...'-~ a.
57
aldehyde is condensed with cyanoacetic acid by refluxing in
a pyridene/toluene solution with a catalytic quantity of
ammonium acetate for 48-72 hours to give the corresponding
acrylonitrile. The acrylonitrile is then hydrogenated
using a palladium-on-carbon catalyst in methanol as taught
in Profitt, et al, J. Orq. Chem., 40, 127 (1975) to give 3
(6-bicyclo-[3.2.1)octanyl)-propionitrile. Following
Examples 98-104, the propionitrile is converted to the 6
bicyclo[3.2.1]octanyl derivative related to the compound
( IVL) .
EXAMPLE 171
Using the procedure of Example 97, 3-(1-bicyclo[3.3:1]
nonanyl)-propionitrile and 3-(3-bicyclo-[3.3.1]nonanyl)-
propionitrile are respectively made from 1-bromo-
bicyclo[3.3.1]nonane and 3-bromo-bicyclo[3.3.1]nonane.
Following Examples 98-104, the propionitriles are converted
to the compound (IVM) and the related 3-bicyclo[3.3.1]-
nonanyl derivative.
EXAMPLE 172
Following the procedure of Example 171, bicyclo[3.3.1]
nonane-9-one is reacted to form the corresponding aldehyde,
from which is made the corresponding 3-substituted
propionitrile, which is then converted into the 9
bicyclo[3.3.1]nonanyl derivative related to the compound
(IVM).
EXAMPLE 173
Following the procedure of Example 170, 2-bicyclo[3.3.1]-
nonanecarboxaldehyde is reacted to form the corresponding
3-substituted propionitrile, which is then converted into
the 2-bicyclo[3.3.1]nonanyl derivative related to the
compound (IVM).
EXAMPLE 174
Using the procedure of Example 97, 3-(1-noradamantyl)-
propionitrile is made from 1-bromonoradamantane, and 3-(2-
WO 91/06548 PCT/US90/05756
.,..;,~ ~-, ~ ,
~'~' f : ~ :.
,, ..m.,.~. 58
noradamantyl)-propionitrile is made from 2-
bromonoradamantane. Following Examples 98-104, the
propionitrile is converted to the final compound (IVN).
EXAMPLE 175
Following the procedure of Example 170, 3-noradamantane-
carboxaldehyde is reacted to form the corresponding 3-
substituted propionitrile, which is then converted into the
3-noradamantyl derivative related to the compound (IVN).
EXAMPLE 176
l0 Following the procedure of Example 170, noradamantane-7-
one is reacted to form the corresponding aldehyde, from
which is made the corresponding 3-substituted
propionitrile, which is then converted into the 7-
noradamantyl derivative related to the compound (IVN).
EXAMPLE 177
Using the procedure of Example 9?, 3-(1-bicyclo[2.2.2]-
octanyl)propionitrile is made from 1-bromobicyclo(2.2.2]-
octane and 3-(2-bicyclo[2.2.2]octanyl)propionitrile is made
from 2-bromobicyclo[2.2.2]octane. Following Examples 98-
104, the propionitriles are converted to the compound (IVK)
and the related 2-bicyclo[2.2.2]octanyl derivative.
EXAMPLE 178
Using the procedure of Example 97, 3-(1-norbornenyl)
propionitrile is made from 1-bromonorbornene. Following
Examples 98-104, the propionitrile is converted to the
compound (IVI).
EXAMPLE 179
Following the procedure of Example 170, 5-norborene-2
.carboxaldehyde (a mixture of 2-endo and .2-exo) is reacted
to form the corresponding 3-substituted propionitrile,
which is then converted into the compound (IVJ).
WO 91/06548 PCT/US90/05756
' '1'~'~~~ r'
59 a~o~~' ~., .3' ~M.i...~'..'~
EXAMPLE 180
N
CI
~ C~~ N
C1
The above intermediate compound is prepared in this
Example by the modification of the procedure of Schiemenz,
G. P.; Engelhard, H. CChem. Ber:, 1962 , 95, 195).
A mixture of cyanoacetic acid (25.38 g, 298.38 mmol),
2,3,5-trichlorobenzaldehyde (25.0 g, 119.35 mmol), ammonium
acetate (500 mg), toluene (120 ml), and pyridine (65 ml) is
heated at reflux for l6 h in a flask fitted with Dean-Stark
trap and condenser. The solvents are evaporated in vacuo,
residue is extracted with CHC13, which is washed With H2O,
dried (Na2SO4), and evaporated to give the crude product,
which is purified by silica gel column chromatography using
hexane-EtOAc mixture as the eluent. Yield 23.69 g (73%)t
mp 90-91 °C.
EXAMPLE 181
N
C1 ~ CHO
(~-J) Cl CN
C1
The above intermediate compound is prepared in this
Example. To a stirred mixture of NaH (1.56 g, 65.05 mmol)
and ethyl formate (14.78 g, 199.51 mmol) in THF (100 ml) is
added substituted pentanedinitrile of Example 180 (10.17 g,
37.17 mmol) at room temperature under a nitrogen
WO 91/0654$ PCT/US90/05756
~~,~~.aa.~
r.
atmosphere, and the resulting reaction mixture is stirred
for 24 h. Volatile matter is evaporated in vacuo at room
temperature. Water (50 ml) is added to the residue at 0-5
°C, and the solution is adjusted to pH 5-6 by 20% conc. HC1
5 (v v). The heavy oil is extracted into ethyl acetate,
washed with H2o (1 x 100 ml) and dried (MgSO4). The ethyl
acetate layer is evaporated to give a red-brown oil (11.0
g) that is used in the next step without further
purification.
10 EXAMPLE 182
C1 H 0
'~~~OMe
C1 ~h.
15 C~ CN
The above intermediate compound is prepared in this
Example. Glycine methyl ester hydrochloride (8.17 g, 65.06
mmol) and sodium acetate (5.33 g, 65.06 mmol) are added to
20 a solution of the crude formyl compound of Example 181
(11.0 g) in a mixture of MeOH (80 ml) and H20 (20 ml), and
the resulting solution is stirred at room temperature for
22 h. After evaporation of solvent at room temperature,
the residue is extracted with ethyl acetate. The washed
25 (H20) and dried (MgS04) organic layer is evaporated to give
an oil. Flash column chromatography (silica gel) using
CHC13 as eluent gave the pure desired enamine as a mixture
of cis-trans isamers which is recrystallized from MeOH,
yield 10.41 g (75%), mp 142-143 °C.
WO 91/06548 PCt'/US90/05756
~-~,r . Ta.,~ '~.~
t t , .,. _: ° .
61
EXAMPLE 183
CI Et~O ( 0
N
~OMe
C1 \ / NHS
~cy
The above intermediate compound is prepared in this
Example. A solution of enamine of Example 182 (10.0 g,
26.84 mmol) in dry CH2C12 (100 ml) is cooled to 0 °C and
treated with 1,5-diazabicyclo[4.3.0]non-5-ene (10.53 g,
84.79 mmol) under a nitrogen atmosphere followed by ethyl
chloroformate (6.90 g, 63.57 mmol). The solution is
stirred at 0 °C for 1 h and then at room temperature for 48
h. Volatiles are evaporated in vacuo to give a viscous
dark gum which is purified by flash column chromatography
over silica gel using CHC13 as the eluent. All the
fractions containing the desired N-protected pyrrole are
pooled and evaporated to give a foamy light pale yellow
material which is stirred in MeOH (100 ml) to give the
crystalline material Which is recrystallized from CHC13-
MeOH, yield 8.92 g (74.7%), mp 160-161 °C.
EXAMPLE 184
C1 H 0
N
1 ~ 'OMe
C1 ~~ 'NHZ
C1 N
The above intermediate compound is prepared in this
3o Example. A suspension of N-protected pyrrole of Example
183 (8.6 g, 19.34 mmol) in MeOH (300 ml) is treated with
Na2C03 (5.12 g, 48.34 mmol) and the reaction mixture is
stirred at room temperature for 17 h with separation of the
deblocked pyrrole during the first hour. Solid sodium
WO 91/06548 PC1'/US90/05756
~~..'',..;..-~,~ ~r
f LJ .L..~'~D
62
carbonate is removed by filtration and washed well with
MeOH. The filtrate is reduced to a volume of 25 ml and
kept in a refrigerator for 1 h to give 5.23 g of
crystalline product. Further concentration of the mother
liquor gave an additional 0.14 g of pure product; total
yield 6.45 g (89.5 %), mp 7.78-181 °C.
EXAMPLE 185
CI H 0
~OMe
C1 ~NH-C-NHCOPh
C1 N S
The above intermediate compound is prepared in this
Example. To a suspension of pyrrole of Example 184 (5.83
g, 15.64 mmol) in dichloromethane (100 ml) is added
benzoylisothioeyanate (2.88 g, 17.64 mmol) at room
temperature under nitrogen. The reaction mixture is
stirred for 30 min with the separation of the desired
thioureido compound. Additional benzoyl isothiocyanate
(0.5 ml) is added to it and again stirred for 30 min. The
solvent is evaporated to dryness, and the light yellow
residue is triturated with methanol. The white crystalline
material is isolated by filtration and recrystallized from
a chloroform-ether mixture to give the required thioureido
compound as an analytically pure sample, yield 7.71 g
(92%), mp 210-211 °C.
EXAMPLE 186
H 0
~ Me
C1 ~N=j~NHCOPh
CI N SMe
WO 91/06548 PCT/US901Q5756
~fi ' f ;~'~ .'?'.'.3
'.,r.m..: a
63
The above intermediate compound is prepared in this
Example. A solution of thioureido compound of Example 185
(6.75 g, 12.6 mmol) and 1,5-diazabicyclo[4.3.0]non-5-ene
(1.76 g, 14.20 mmol) in dry CH2C12 (200 ml) is cooled to 0
°C and treated with methyl iodide (5.20 g, 36.65 mmol).
The reaction mixture is stirred at 0 °C for l0 min and then
for 1 h at room temperature. The solvent is evaporated at
room temperature, and the residue is extracted with CHC13,
washed with H20 (2 x 50 ml), dried (Na2S04) and evaporated
to give a glassy foam (6.95 g) which is used in the next
step without purification.
EXAMPLE 187
E
Hz,I hte
Cl H
C1
A
The above compounds A and B are prepared in this Example.
The compound A is a compound of the present invention and
the compound B is an intermediate. A solution of the
methylthio intermediate of Example 186 (6.90 g, 12.54 mmol)
in MeOH (200 ml) is saturated at 0 °C with ammonia and
heated at 100 °C for 20 h in a glass-lined stainless steel
bomb. The reaction mixture is brought to room temperature
and the solvent is evaporated at room temperature.
Purification of the crude mixture by flash column
chromatography over silica gel using CHC13 as eluent gave
8B (1.1 g, 21°s), mp 290-291 °C then CHC13-MeOH (95:5) gave
pure 8A (2.76 g, 57.5%), mp 284-285 °C.
WO 91/06548 PCT/US90105756
.~z~'r~~
~-; s~;,.,.r.'~''"
64
EXAMPLE 188
The compound of the present invention of Example 187 is
tested for enzyme inhibition activity. A purine nucleoside
phosphorylase (PNF) enzyme assay is performed in which the
PNP activity (IC50) for the compound (8A) is found, which
is determined radiochemically by measuring the formation of
(14C]-hypoxanthine from (14C]-inosine (see Biomedicine,
1980, 33, 39) using calf spleen as the enzyme source. At 1
mM phosphate the IC50 is 0.64 uM and at 50 mM phosphate the
IC50 is 10 ~,M.
EXAMPLE 189
H
C1
Following the procedure set forth in Examples 180-187, 3-
(3-chlorophenyl)-3-(2-amino-4-oxo-3H,5H-pyrrolo(3,2
~]pyrimidin-7-yl)propanenitrile is prepared using 3-(3
chlorophenyl)-pentanedinitrile as the starting material,
yield 54.5%, mp 157-158 °C.
EXAMPLE 190
Following the procedure set forth in Examples 180-187,
the following compounds are also prepared (1-9).
0
H
H
H~ N~N I ~
Ar CN
WO 91/06548 PCT/US90/05756
~;'! ~M ~_.'-. W
3-Aryl-3-(2-amino-4-oxo-3H,5H-pyrrolo[3,2-d]
pyrimidin-7-yl)propanenitrile
Where Ar is each of the following: (1) phenyl, 2,3
dichlorophenyl, 3-methylphenyl, and 3-methoxyphenyl, (2)
5 thienyl (2- and 3-), (3) furanyl (2- and 3-), (4) pyridinyl
(2-, 3-, and 4-), (5) pyrrolyl (2- and 3-), (6) thiazolyl
(2-, 4-, and 5-) , (7) 2-pyrazinyl, (8) pyridazinyl (3- and
4-), and (9) pyrazolyl.
EXAMPLE 191
10 Following the procedure set forth in Examples 180-187,
the following compounds 10-14 and 21 are prepared starting
from the appropriately substituted pentanedinitrile.
Compounds 15-20, and 22 are prepared from the corresponding
unsaturated Ar analogues in Example 190. In this
15 procedure, the nitrile group of the unsaturated analogue is
first converted to an amide group by acid- or base-
catalyzed hydrolysis, then the unsaturated Ar group is
converted to the saturated R2 group by known catalytic
hydrogenation, followed by reconverting the amide back to
20 the nitrile by known dehydration procedures.
0
H
H ( '
H2N ~ /
R N
3-(Substituted)-3-(2-amino-4-oxo-3H,5H-pyrrolo(3,2-d]-
pyrimidin-7-yl)propanenitrile
Where R2 is each of: 10) 1-adamantyl, 11) 2-adamantyl, 12)
cyclohexyl, 13) cycloheptyl, 14) cyclopentyl, 15)
tetrahydrofuranyl, 16) tetrahydrothienyl, 17)
tetrahydropyranyl, 18) pyrazolidinyl, 19) thiazolidinyl,
WO 91106548 PCT/US90/05756
PCe~r~~A a
f i..a.:,..
66
20) piperazinyl, 21) morpholinyl, and 22)
hexahydropyridazinyl.
EXAMPLE 192
H2
N
The above compound, 3-(2-amino-4-oxo-3H,5H_-pyrrolo[3,2-
d_]pyrimidin-7-yl)-3-phenylpropanenitrile, is prepared in
this Example. A solution of the compound A obtained in
Example 187 (2.0 g, 5.22 mmol) in warm ethanol (250 ml) and
dimethylformamide (DMF) (150 ml) is hydrogenated over 30%
Pd/C catalyst (1.0 g) in the presence of triethylamine
(2.64 g, 5.0 equivalent) at atmospheric pressure. After 5
h, the reaction is complete, and the catalyst is filtered
off under N2 pressure. The solid obtained by evaporation
of the filtrate is triturated and sonicated With H20 and
dried, yield 1.28 g (88%), mp 168-170 °C.
EXAMPLE 193
The compound prepared in Example 192 is tested for enzyme
inhibition activity as in Example 188. At 1 mM phosphate
the IC50 is 0.023 ACM and at 50 mM phosphate the IC50 is 4.7
~tM .
EXAMPLE 194
H2
a
WO 91/06548 PCT/US90/05756
~ Y~TT~ ~~
~~' ~:.,,.i...~ m.
67
The above compound, 3-(2-amino-4-oxo-3_H,5H-pyrrolo[3,2-
_d]pyrimidin-7-yl)-3-phenylpropanoic acid, is prepared in
this example. A solution of the compound obtained in
Example 192 (0.200 g, 0.72 mmol) in 6N HC1 (3.0 ml) is
heated at reflux for 18 h. The solvent is evaporated in
vacuo and the residue is triturated with H2o (6 ml),
adjusted to pH 10 by conc. ammonium hydroxide. Insoluble
material is collected by filtration and the filtrate is
readjusted to pH 6.8. White material which is
precipitated out is collected, washed with water and dried,
yield 0.19 g (89%), mp 290 °C dec.
EXAMPLE 195
The compound prepared in Example 194 is tested for enzyme
inhibition activity as in Example 188. At 1 mM phosphate
the IC50 is 0.012 ACM and at 50 mM phosphate the IC50 is
0.19 ~M.
EXAMPLE 196
Hz
The above compound, 3-(Z-amino-4-oxo-3H,5H-pyrrolo[3,2-
d_]pyrimidin-7-yl)-3-phenylpropanamide, is prepared in this
example. A solution of the compound obtained in Example
192 (0.200 g, 0.72 mmol) in conc. H2S04 (0.5 m1) is stirred
at room temperature for 20 h and then poured onto crushed
ice (5.0 g) and adjusted to pH 6.8 by conc. NH40H. The
precipitated solid is collected, washed with H2o and dried,
yield 0.180 g, mp 199-201 °C dec.
WO 91/06548 PCT/US90/05756
68
EXAMPLE 197
The compound prepared in Example 196 is tested for enzyme
inhibition activity as in Example 188. At 1 mM phosphate
the IC50 is 0.20 ACM and at 50 mM phosphate the IC50 is 6.6
~tM.
H2
a
The above compound, 3-(2-amino-4-oxo-3H,5H-pyrrolo[3,2-
d_]pyrimidin-7-yl)-3-phenylpropanoic acid, methyl ester, is
prepared in this example. Thionyl chloride (0..2 g, 0.17
mmol ) is added to stirred methanol ( 4 . 0 ml ) at 0 ° C. The
compound obtained in Example 194 (0.2 g, 0.67 mmol) is
added and the mixture is stirred at room temperature for 18
h. The solvent is removed on a water aspirator (30°C) and
vacuum pump (lyophilize) to give a semisolid mass which is
purified on a silica gel column using CHC13-MeOH as the
eluent, yield 0.1 g.
EXAMPLE 199
The compound prepared in Example 198 is tested for enzyme
inhibition activity. Significant activity (IC50) is found.
EXAMPLE 200
H.,
a
EXAMPLE 198
WO 91/06548 PCT/US90l05756
n~YH'~~ ~.
.~~, ~ f i~M.J...: 9
69
3-(2-Amino-4-oxo-3H,5H-pyrrolo[3,2-d]pyrimidin-7-yl)-3-
cyclohexylpropanoic acid is prepared in this example. A
solution of the compound obtained in Example 194 (83 mg,
0.28 mmol) in trifluoroacetic acid (TFA) (15 ml) is
hydrogenated with Pt02 (83 mg) at 60 lb/in2 for 24 h. The
catalyst is filtered off through a Celite bed, and the
filtrate is evaporated at 25 °C. The residue is suspended
in H20 (8 mI), and adjusted to pH 8.5 by cone. NH40H and
filtered through a Whatman filter paper to remove brown
colored impurities. The colorless filtrate is adjusted to
pH 6.8, and the precipitated compound is filtered, washed
with H20, and dried, yield 65 mg (77%), mp >300 °C.
EXAMPLE 201
The compound prepared in Example 200 is tested for enzyme
inhibition activity as in Example 188. At 1 mM phosphate
the IC50 is 0.097 ACM and at 50 mM phosphate the IC50 is 1.0
~1M .
EXAMPLE 202
A compound of the present invention is prepared wherein X
is PO(OH)2. The nitrile group of the compound of Example
192 is converted to the corresponding amide by treatment
with sulfuric acid. Using a Hoffman degradation reaction,
the amide is converted to the corresponding amine, which is
then converted to the corresponding pyridinium salt using a
pyrillium salt. Conversion of the salt to the
corresponding halide is accomplished using sodium bromide,
which is then converted to the phosphonic ester using
triethyl phosphate. Hydrolysis of the ester using
trimethylsilylbromide yields the corresponding phosphonic
acid wherein "n" is 1 and "m" is 0.
EXAMPLE 203
This Example makes a compound of the present invention by
stepping up the number of carbon atoms from "m" is 0 to "m"
is 1. The nitrile group of the compound of Example 192 is
.;
WO 91/06548 PCT/US90/05756
,~.~~-.r~ '.;.'.~
red~et~~f'°~'~o the corresponding aldehyde, which is then
converted to the corresponding alcohol. Using phosphorous
tribromide the alcohol is converted to the corresponding
alkyl bromide, which is then converted to the nitrile
5 compound of the present invention Wherein m is 1 using
potassium cyanide.
EXAMPLE 204
In this example a compound of the present invention is
prepared wherein "p'~ is 1 and '~Y" is oxygen. The alcohol
10 prepared as an intermediate in the previous example is
converted to the corresponding diethyl phosphonomethyl
ether using diethylchloromethyl phosphonate. Removal of
the ethyl groups of the ester is accomplished using
trimethylsilylbromide to give the phosphonic acid.
15 EXAMPLE 205
In this example a compound of the present invention is
made wherein "Y" is NH and "X" is S02NH2. The nitrile
group of the compound of Example 192 is reduced to the
amine using standard catalytic hydrogenation with palladium
20 in acidic media (usually 0.01 N to 1 N HC1), which is then
converted to the sulfamide using sulphamoyl chloride.
EXAMPLE 206
In this example a compound of the present invention is
prepared wherein "X" is COOH and "Y" is NH by reacting the
25 methyl amine intermediate prepared in the previous example
with chloroacetic acid.
EXAMPLE 207
In this example a compound of the present invention is
prepared wherein "X" is PO(OH)2 and "Y" is NH by reacting
30 the methyl amine intermediate prepared in Example 206 with
diethylchloromethyl phosphonate, and reacting the resulting
product with trimethylsilylbromide.
WO 91/06548 PCT/US90/05756
71
EXAMPLE 208
In this example a compound of the present inventionis
prepared wherein "X" is S02NH2 and "Y" is oxygen by
reacting the alcohol intermediate prepared in Example203
with sulphamoyl chloride.
EXAMPLE 209
In this example a compound of the present inventionis
prepared wherein R1 is H, R2 is phenyl, R3 and R4 are
hydrogen, m is 0, n is 1, p is 0, and X is CN. A
10modification of the procedure disclosed in Mu-I11 et
Lim,
al. , J. Orcr. Chem. , Vol. 44, No. 22, 3826 (1979)ed.
is us
A mixture of the compound of Example 184 and
dimethylformamide dimethyl acetal is reacted at oom
r
temperature for two days and then evaporated to in
dryness
15vacuo. The residue is crystallized to give the pureN-
(dimethylamino)methylene derivative, which is cyclizedith
w
saturated methanolic ammonia to give the desired end
product.
EXAMPLE 210
20In this example a compound of the present inventionis
prepared wherein R1 is OCH3, R2 is phenyl, R3 and are
R4
hydrogen, m is 0, n is 1, p is 0, and X is CN. Usingthe
compound B of Example 187, the S-methyl group is zed
oxidi
to methylsulfone, which then is converted to the
final
25methoxy compound by treatment with sodium methoxidein
methanol.
EXAMPLE 211
In this example a compound of the present invention is
prepared wherein X is tetrazole. The compound of Example
30 192 is treated with lithium azide in the presence of
ammonium chloride as a catalyst in dimethylformamide (DMF)
at 100 degrees C to give the desired tetrazole.
WO 91/06548 PCT/US90/05756
;~~'; "."'?~ '~'
r ... :~
72
EXAMPLE 212
In this example a compound of the present invention is
prepared wherein X is triazole. The compound of Example
198 is treated with hydrazine hydrate to give the
corresponding hydrazide, which is then treated with imino
ether to give the desired triazole.
EXAMPLE 213
The compound prepared in Example 189 is tested for enzyme
inhibition activity as in Example 188. At 1 mM phosphate
the IC50 is 0.012 ~,M and at 50 mM phosphate the IC50 is 2.0
f~M .
EXAMPLE 214
In this example an amidine compound of the present
invention is prepared, i.e., wherein X in the recited
generic formula is CNHNH2. The compound A from Example 187
is reacted with sodium methoxide in methanol at room
temperature for about 2 days to give a methyl-imidate
intermediate. The intermediate is. then reacted with
ammonia in methanol to give the amidine product.