Note: Descriptions are shown in the official language in which they were submitted.
20767~
WO91/13157 PCT/AU91/00064 :
'
This invention relates to DNA vectors, and is
particularly concerned with DNA shuttle vectors which are
capable of replication in mycobacteria and Eschrichia
coli cells. The invention also relates to bacterial ~-
20 hosts containing such DNA vectors, and further relates to `~
vaccines containing such bacter:ia. ~ :
Mvcobacterium bovis BCG (hereinafter BCG) has been ~:
used for many years as a ~accine against tuberculosis
tTB). Th~ vaccination programme has been extremely
25 effective in controlling human TB firstly because BCG ~ ~:
stimulates long term cell-mediated immunity and secondly -~;
because it has had an outstanding safety record. These ~:
: characteristics make BCG a good candidate to form the
basis of a live delivery system for recombinant vaccines. ~ :
A number of problems are associated with DNA
manipulations involving mycobacteria. These include very
few (one) plasmid vectors, poor growth rates of .~.
mycobacteria and low transformation rates when compared
to bacterium such as E. coli. Given these problems, it ..
is desirable to produce a shuttle vector which is capable
of replication in a standard bacterial work horse such as
E. coli for day to day genetic manipulations, and is ~ .
~'~
: , : ., . ~ . ,. . . . -:
~: -- - ~ ,. :.
WO91/1315, 2 ~ 7 ~ 7 ~ ~ PCT/AU91/000~ - ~
- 2 -
further capable of replication in Mycobacterium at or
near tAe final stage of genetic manipulation. In this
way, genes can be inserted into mycobacteria and ~time
delays associated with MYcobacterium growth and
transformation can be largely avoided. Gicquel Sanzey et
al. (Acta Lepologica 1989, 7, Suppl (l): 208-211) have
described a mycobacteria-E. coli plasmid shuttle vector
known as pAL8 which comprises two origins of replication,
the first for mycobacteria and the second for E. coli.
Multiple origins of replication have been necessary due
to the evolutionary distance betwsen mycobacteria and E.
coli such that a mycobacterial plasmid having a
mycobacterial origin is not capable of growth in E. coli
and vice versa. Such vectors suffer from the problem
that they are a considerable size due to the inclusion of
two origins of replication, this decreasing cloning
efficiency, the size of desired DNA fragments which may
be inserted into such plasmids, and also increasing the
number of unique restriction s~tes which may be
introduced into the plasmids.
The presen~ applicant has overcome problPms
associated with prior art shuttle vectors by providing a
DNA shuttle vector which carries a single origin of
replication which confers the ability of the vector to
25 replicate in mycobacteria and E. coli cells. In a ~-
particularly preferred aspect of this invention as will
be described hereinafter, the replication region
- corresponds to that of the corynebacterial plasmid pNG2
or fragments thereof.
In accordan~e witA a first aspect of this invention, ~-
there is provided a DNA shuttle vector carrying a
replication region comprising a single origin of
replication, said origin of replication allowing
replication of said vector in MYcobacteria and E. coli ~ -
cells; and a selectable marker. The shuttle vector may
addltionally comprise a nucleotide sequence containing
one or more restriction site~ for the insertion of a
., . . :: ~ . : -
- - ~
.
207~7~
WO9~/13ls7 PCT/~U91/00064
-- 3 --
desired nucleotide sequence.
Preferably, the repllcation reglon corresponds to
the replication region of the corynebacteria plasmid pNG2
or a fragment thereof which permits repllcation of said
vector in mycobacteria and E. coli.
Surprislngly, the replication region of plasmid pNG2
confers the ability of replication in various bacterial
species, apart from Corynebacteria.
The DNA shut~le vector may further comprise a
promoter with one or more restriction endonuclease sites
downstream of said promoter, such that when a nucleotide
sequence is inserted into one or more of these sites, the
promoter allows DNA transcription to proceed. The DNA
shuttle vector may contain multiple promotsrs and
downstream restriction endonuclease sites.
The term "shuttle vector" as used herein includes
plasmid DNA which may be double-stra~ded linear or
double-stranded circular. The shuttle vector may be
introduced into a bacterial cell by any number of ~;
techniques well known in the art, such as conjugation,
mobilisation, transformation, transfection, transduction
or electroporation.
The term "selectable marke!r" as used herein refers
to any selectable characteristic provided by or encoded
for, by a nucleotide sequence. Suitable detectable
markers include resistance to antibiotics or enzymes or
immunologically detectable proteins or chemicals capable
of causing a detectable reaction when provid~d with a
suitable substrate. Examples include resistance to
ampicillin, streptomycin, penicillin, hygromycin,
kanamycin, and the like, ~-galactosidase, urease,
alkaline phosphatase and the like. ;~
The term "promoter" is used in its broadest sense
and refers to any nucleotide sequence which binds to RNA
polymerase and which directs the transcription of
nucleotide sequences downstream (3' "or operably linked")
to the promoter. Suitable promoters include prokaryotic
.
.
,
WO91/131~7 2 0 7 6 7 ~ ~ PCT/AU91/000~
_ a, .
promoters such as the Pl promoter of bacterlophage
lambda, trp promoter, lac promoter, kanamycin resistance
promoters of transposon Tn903 and transposon Tn5,
mycobacterial promoters such as that of the promoter of
the com~on mycobacterial 65Kd antigen, rtbosomal RNA
promoter of mycobacteria, promoters of M.bovis antigens
MPB70, MPB59 and MPB64 and the like, hybrids betwPen
eukaryotic and prokaryotic promoters, and eukaryotic
promoters such as the metallothionine promoter, growth
hormone promoter, and the like.
Restriction endonuclease sites provided on the
vector may correspond to the cleavage of one or more
known restriction endonucleases, such as EcoRl, BamHl,
Pstl, Clal, Kpnl, HindIII, HincI and HincII, and the
like. Restriction endonuclease sites may be provided in
the form of one or more polylinkers which contain a
number of closely grouped restriction endonuclease sites.
Nucleotide ~equences of interest may be inserted
into the endonuclease cleava~e sites provided on the
vector by ligation of DNA fragments having complementary
"sticky ends" to allow annealing thereof, or by ligation
of nucleotide sequence having "flush" ends (ends having
no unpaired nucleotides) by methods well known in the
art, and described for example, in Sambroo~ et al.
(Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Laboratory, 2nd Edition, 1989). A nucleotide
sequence fDr insertion into the vector of this invention
may include a promoter to direct transcription of
downstream (3') sequences. Promoters may be the natural
promoter of the gene to be transcribed or may be a
different promoter. As previously mentioned, the shuttle
vector of this invention may itself contain one or more
promoters upstream (5') from nucleotide sequences
encoding one or more restriction endonuclease cleavage
sites. In such an embodiment, a desired nucleotide
sequence lacking a promoter may be inserted into the
vector with transcription of the desired nucleotide
::.
.: .,. , : , - . -
.: :. .: ,., .. , ., :-.. , ... - . :.
' .:, . . ' :.""'~"' ' : , :
... . . : . :: . .
, . ,
WO91/13157 2 0 7 6 7 ~ ~ PCT/AU91/00064
-- 5 --
sequence belng driven by the promoter resident on the
vector.
Nucleotide sequences for insertion into the vector
of this invention may, for example, encode one or more
antigens from disease causing bacteria, viruses or
- parasites, such as Taenia ovis, rotavirus, Babesia bovis,
Mycobacterium bovis, MYcobacterium tuberculosis,
MYcobacterium leprae, MYbacterium paratuberculosis,
Bacteriodes nodosus or Bordetella. Desired nucleotide
sequences may also encode hormones such as LHRH, growth
hormone or epitopes or analogues thereof; or DNA
sequences capable of recombination with nucleotide
sequences within a bacterial host cell; or nucleotide
sequences capable of mutagenising DNA sequences within a
host cell. Where the shuttle vector of this invention
functions as an expression vector, nucleotide sequences
encoding desired products may include a signal or leader
sequence to allow insertion in~o membranes of a suitable
host cell or secretion from a host cell. Absence of a
secretory leader will cause th,e accumulation of antigen
or other protein product within the cytoplasm of a
bacterial host cell, where it may be recovered by well
known methods.
In accordance with a specific embodiment of this
invention ~here is provided a DNA shuttle vector pEP2,
said vector having a size of about 3.l kh, as detsrmined
by agarose ~el electrophoresis, a replication region of
about l.85 kb comprising single origin of replication
derived from the Corynebacterium replicon pNG2, an
antibiotic r sistance gene to kanamycin, and a nucleotide
sequence containing a number of restriction endonuclease
cleavage sites for the insertion o a DNA sequence of
interest. The 4.5 kbr plasmid pEP3 contains the same
sequences of replication as pEP2 plus a marker encoding
hygromycin resistance effective in both E.coli and
mycobacteria.
Shuttle vectors of this invention are capable of
.
,. . . ~ ,
,, : -. : .. :
.. ..
:: . : . : . i " : :
: ~ . .: . . : , :
".
.
WO91/13157 ~ 7 6 7 .S C) PCT/AU91/00064 --
- 6 -
replication in species of both gram nega~ive and gram
positive hosts, such as Mvcobacterium, E. ccli,
Corynebacterlum, and Ac-tinomvcetes, as defined in
Bergey's Manual of Determinative Bacteriology, 8th
Edition, pp. 599-861. ~he vectors of this invention do
not appear to replicate in Bacillus species. Any
bacterial strain may be readily tested according to
methods well known in the art, to ascertain whether or
no~ the shuttle vector of this invention is capable of
replication therein~ Advantageously, the shuttle vectors
of this invention are capable of replication to high copy
numbers in bacterial strains such as in the attenuated
strain of Mycobacterium bovis BCG, which as previously
stated has been extensively used in vaccination
programmes throughout the world, and is a potent
adjuvant, which stimulates long-term cell mediated
immunity.
Shuttle vectors incorporating the origin of
replication of pNG2 are particularly efficient in
organisms of the genus CorYnebacterium.
In accordance with a further aspect of this
invention, there is provided a bacterial host which
contains a shu~tle vector as herein defined. The shuttle
vector may be present as a single copy, or ~ore
pre~erably as multiple copies thereof within the
bacterial host. Preferably, but in no way limiting the
invention, the bacterial host is a Mvcobacteria,
Corynebacteria or E. coli strain, such as C.
~seudotuberculosis, M. sme~matis, and M. bovis BCG. ~he
vector may be used to deliver antigen or other protein
genes into the bacterial host for expression thereof, as
an excreted product from the host cell as previously .
described. Proteins may be expressed while residing on
the plasmid vector or after recombination or insertion .
into the chromosome. Alternatively, expressed products
may be inserted into or associated with the host cell
membrane or cell wall or reside~t within the host cell
:
'~ . ' `'. ',' , '' ~ `' ' ',' ' , ' ' ' , ''' :'' . ' ' ' '
:: ' ' , ' ' ' ' ' ~ ' '
W0~1/13157 20767r~ Pcl/~u9l/ono64
itself. Bacterlal hosts expressing deslred an~igens may
be provided as vaccines, for example, as M. bovis BCG,
expressing a desired antigen. On immunisation, an immune
response would be mounted to the host, such as M. bovis
BCG as well as the desired antigen of the disease causing
bacterium, virus or parasite.
In a further aspect of this invention there is
provided a polypeptide when expre~sed by a bact~rial host
cell containing a shuttle vector as herein defined.
This invention contemplates deletions or insertions j~A
of nucleotide sequences to or from the replication region
of pNG2 as long as such modifications do not prevent the
ability of such sequences to confer replication in
Mycobacterium and Eo coli. Techniques for lr.sertion or
deletion of nucleotide sequences are well known in the
art. Mutants could be readily tested for the ability to
; confar r~plication in gram negative and gram positive
host cells, such as E. coli and corynebacteria.
This invention also extends to replication region of
20 pNG2 itself or fragments thereof, which, on inser~ion ~-
intc suitable vector are capable of permitting
replication of said vector mycobacteria and E. coli.
A culture of Eschrichia coli containing plasmid p~P2
was deposited under the terms and conditions of the
Budapest Treaty at the Australian Government Analytical
Laboratory (AGAL), Pymble, New 'outh Wales, Australia on
23rd February, l990 and accorded Accession No.
N90/007030.
~his invention will now be illustrated with
30 reference to the following non-limiting Figures and ~-
Examples.
FIGURES
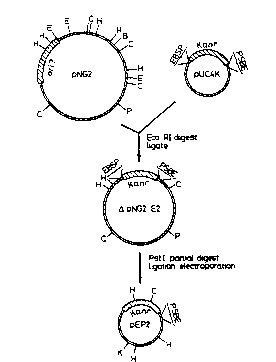
Figure 1 shows the construction of plasmid pEP2.
Plasmid pN~2 (14.5) was digested with EcoRI and the
largest fragment ligated to the Kanr cartridge of pUC4K.
Following electroporation into E.coli the resulting
plasmid DNA was extracted and partially digested with
:. : , :, - , ., . : :, , :: :- .
.: ., ., . ... , . : : : .
.. . ,., ., . . . .. . ~ , : . .
; : : : ., :, : . ~:: - :
: .
: .: . ,
h13~ b ~
WO91/1315/ PCT/AU91/00064
-- 8
; Pstl, and then religated and again electroporated wlth
E.coli. One of the resulting Kan' colonies contained
plasmid pEP2. Restriction siles: BtBamHl)~ E( EcoRI),
~(HindIII), Hc(HlncII), P(PstI), S(SalI). Kan refers to
kanamycin resistance gene from pUC4K.
Figure 2A is a circular map of the Mycobacterium-E.
coli shuttle vector pEP2. The restriction map is given
only for relevant cleavage sites: B (BamHI), E. (EcoRI),
H (HindIII), Hc (HincII), K (X~nI), p (PstI), S (SalI).
Kanamycin reslstance gene \\\\\ from pUC4K. -
B Sequencing strategy. Arrows indicate the extent and
direction of sequence generated from restriction
fragments cloned in Ml3 vectors. X denotes sequence ;~
'~ derived using oligonucleotide primers.
Figure 3 is a complete nucleotide sequence of the -
replication region of pEP2. Relevant restriction sites
are marked for comparison with Figure l (E (EcoRI), H
(HindIII), Hc (HincII), K (K~nI). IR and DR, inverted
and direct repeat sequences respectively: i~BS, putative
ribosome binding site; ------> <~ dyad symmetry
associated with putative rho-dependent transcriptional
terminator, T. ORFA, major open reading frame.
` Figure 4 shows an agrose gel (right hand plate) and
a Southern blot (left hand plate) of that gel probed with
, 25 plasmid pEP2 extracted from E.coli. Tracks contain (A)
Undigested pEP2 DNA from E.coli 500ng, (B) Pst l diyested
pEP2 DNA from E.coli 200n~, ~C) PstI digested whole DNA
extract of M. bovis BCG pEP2, 2.5ug, (D) PstI digested M.
bovis BCG DNA, 2.5 ug (E) Undigested DNA extract of M.
bovis ~CG pEP2, 2.5ug, (F) Undigested DNA extract of M.
- bovis BCG, 2.5ug, (G) HindIII digested lambda DNA
markers.
' EXAMPLE 1
Recombinant DNA Procedures:
~ 35 Unless otherwise specified herein, manipulation of
; recombinant molecules and the preparation of solutions
, are by standard known techniques. Such techniques are
.', ~
,
. .
:. : . . . - .
" .:, . . . . ~ :
.: . , . ~ ::
.. :.. : .. ~ : . :~ . :.
. . , .,. , ~ , ,:
~767~3
WO9l/l31~7 PCT/AU91/000~
g
described in Sambrook et al. Molecular Cloning, A
Laboratory Manual, Cold Spring Harbor Laboratory, Cold
Spring Harbor [1989], pp. 1-500.
Bacterial S~rains and Plasmids:
S Mycobacterium bovis BCG variant CSL was obtained as
lyophylised human vaccine from the Commonwealth Serum
Laboratories (CSL) Australia. M. sme~matis and M. Dheli
were obtained from the Fairfield Hospital, Melbourne,
Australia and Escherichia JM109 was obtained from Promega
(Madison, Wisconnsin, U.S.A.~. Plasmids pNG2 (Serwold-
Davis et al. 1987, Proc. Natl. Acad. Sci. USA B4: 4964-
4968) and pPB3 was obtained from Dr. Philip Bird (Monash
University Faculty of Medicine, Alfred ~ospi~al,
Melbourne). Plasmid pAL8 (Gicquel-Sanzey et al. 1989
15 Acta Leprol. 7: 207-211) was obtained from Dr. Brigette
Gicquel-Sanzey (Pasteur Institute, Paris) and plasmid
pUGC4~ was purchased from Pharmacia LKB (Uppsala,
Sweden).
Media:
E. coli strains were grown Luria broth, (LB:10 grams
- tryptone, 5 grams yeast extract, 10 grams NaCl per
litre). Coryneform bacteria were cultured in LB media
(Oxoyd~ and mycobacterium species were grown in Dubos or
7H11 media (Oxoid, Australia).
Electroporation of Bacteria:
Mvcobacter~um speci~s and CorYnebacterium ~seudo-
tuberculosis were electroporated according to Lugosi et
al. (~uber~ule 70. 159-170 tl98g]) using a Gene Pulser -~
commercially available from Bio-Rad Laboratories Inc.
Transformants were selected on 7Hll or nutrient media
containing 100 ~g kanamycin per ml or 200~g hygromyan B
per ml.
DNA Isolation and Hybridization AnalYsis:
DNA was extracted from MYcobacterium using a
modification of a method used to isolate DNA from yeast
(Mann and Jeffrey, Anal. ~iochem. 1981, 178: 82-87).
Mycobacterial cells were harvested from 400 mls of Dubos
" . -: : : .: , : . - .: .: . ~ : :
: ' '''' ' ' ' : .. ' ' ' ,, . :
. .
WO91/131~7 2 0 7 S 7 ~ ~ PCT/AU91/000~
-- 10 -
broth by centrifugatlon, resuspended in 5 ml of TE buffer
and heat-killed by treatment at 70C for l hour. Glass
beads (3-6 mm) were added and the mix vortexed vigorously
for a minimum of 30 seconds to disperse the bacteria.
Bacterial suspensions were then transferred to liquld
nitrogen in a mortar pre-cooled in a bath of liquid
nitrogen and crushed into a fine powder uslng a pre-
cooled pestle. The crushing of cells was performed in a
biohazard hood. Frozen crushed cells were added in small
portions (spatula loads) to 15 ml of lysis bufer (6.6 mM
Tris, 30 mM EDTA, 1.2% w/v sodium lauroylsarkosinate).
Five mg of protease K was added and the mixture incubated
for 90 minutes at 37~C. The mix was then extracted with
an equal volume of phenol-chloroform and the aqueous
phase precipitated with isopropanol at 4C for l0
minutes. The pellet was dissolved in l.0 ml of TE buffer
and extracted with phenol-chloroform and then water-
` saturated ether prior to ethanol precipitation and
resuspension in TE.
Genomic DNA was isolated from C. Dseudotuberculosis
c as previously described (Hodgson et al., l990).
pNG2RI DNA was digested with various restriction
enzymes and the fragments Southern blotted (Reid et al.) ~;
to Hybond N (Amersham) nylon filters. ~ilters were
25 hybridised overnight at 37C with pEP2 labelled with 32P
using random hexamer primers, washed at increasing
stringency as necessary (up to 65C) and exposed to X-ray
film (Fuji RX). Restriction digested total genomic DNA
isolated from M. bovis BCG and that transformed with pEP2
was Southern blotted to nylon and probed with labelled
pEP2 as described above. Other transformed MYcobacterium
, species were analysed with the pEP2 probe using DNA do~
`~ blot hybridisation. C~ pseudotuberculosis transformants
were analysed in the same fa~hion.
Total cell DNA was used to isolate and transform
E.coli to kanamycin or hygromycin resistance.
, ' ,
.
; .
. . . . .
. . , : ,.,
WO91/1315/ PCT/AU91/00064
-- 11 --
DNA Sequence Analysis:
Nucleotide sequence analysis was determined by the
dideoxy chain termination method (Sanyer et al., 1977,
Proc. Natl. Acad. Sci. USA 74: 5463-5467) using modified
T7 DNA polymerase (Pharmaoia LKB, Uppsula, Sweden). The
complete nucleotide sequence of pEP2 was derived on both
strands using universal or oligonucleotide primers
(Figure lB). The laLter was synthesized using a Gene
Assembler plus DNA Synthesizer (Pharmacia LKB, Supra).
DNA and amino acid sequence data were collated and an
analysed using the DNASIS and PROSIS software packages
(Pharmacia LKB).
EXAMPLE 2
Shuttle Vector Construction a~d Analysi~ Thereof:
The l.3 kb EcoRl fragment from pUC4K carrying the
~anamycin resistance gene was ligated to pNG2 DNA
digested wi~h EcoRl. The ligation mix was electroporated
into E.coli JMlO9 and transformant selected on L~ plates
supplemented with 50,ug kanamycin per ml. All
transformants contained plasmids with the 9.5 kb pNG2
EcoRl fragment. Plasmid DNA (2 ~g) from one of these
clones (pNG2~I) was partially digested with PstI and
blun~ed using T4DNA polymerase. DNA fra~ments smaller
than lO kb were purified from a l~ agarose gel and used
to transform E. coli JMl09 to kanamycin resistance. A
restriction map of a resulting plasmid tpEP2) isolated
from a transformant was derived using standard procedures
(Sambrook et al., Supra) and is shown in Figure lA.
Plasmid pEP2 has a molecular weight of 3.l kb as
determined by agarose gel electrophoresis. This plasmid
retains one of the pUC4K polylinkers and hence has unique
PstI, SalI, BamHl and EcoRI sites. Hybridization
analysis shows that plasmids pEP2 and pNG2RI (pNG2 after
digestion with EcoRI) both contain an 800 bp HindIII
fragment. Southern blotting of digests of sub-clones of
pNG2 with the pEP2 plasmid showed that the region of PNG2
marked in Figure l was that incorporated in the pEP2
- . -. , . :, , , . :.
: ~ . . :, . . , .. ~
: ~. . ` ' :
. , ', ' . ~ '''
;
, .. :
WO91/131~7 2 Q 7 6 7 A~ ~ PCT/AU91/00064 -
- 12 -
plasmid, and thus the area containing the origin of
replication.
To determine whether hygromycin resistance could be
used as a selectable marksr in Mycobacteria species and ~
5 C. pseudotuberculosis a hygromycin phosphotransferase ~.
gene (hph) was cloned into the MYcobacterium shuttle
vector pEP.
To clone the hygromycin phosphotransferase gene
(hph) from pPB3 into pEP2, pUC4K DNA was linearised.with
l0 XhoI and then blunted using the Klenow fragment of DNA - -
polymerase I. pPB3 DNA was digested for 30s on ice using
5 units each of AluI, HaeIII and RsaI. Fragments ~.Okb in
size were gel purified (Geneclean, Bresatech) and ligated
overnight with the linear, blunted pUC4K DNA. The
ligation mix was electroporated into JMlO9 and
recombinants were selected on LB plates supplemented wi~h
150ug hygromycin B (Sigma) per ml. A l.7kb SalI-ClaI~ ~
insert from a pUC4K chimera was ligated into the SalI- ~ .
ClaI site within the kanamycin resistance gene of pEP2.
20 JMl09 was transformed to hygromycin resistance with ~he ~.
ligation mix as described above and a res~riction map of :~
a hygromycin resistance plasmid (pEP3) was derived. The
plasmid pEP3 has a unique SalI site and $s approximately
4.5 kb in size (Fig. 2).
The nucleotide sequence of pEP2 excluding the
kanamycin resistant gene is presented in Figure 3. `~
Examination of the D~A sequence revealed a single open :
reading frame (ORF)~ A number of potential translational .:
start codons can be identified within this region but
only one is preceded by a putative ribosome binding site
(Figure 2). Although the sequence upstream of this ORF ~
(ORFA) does not possess an E. coli consensus promoter, a ~ .
putative rho-dependent transcriptional terminator was ~ .
found downstream of the stop codon (Figure 2). This
putative terminator has a 9O~ match to the TAATCAATAT
consensus sequence tRyder et al., Initiation of D~A
Replication [1981], Academic Press, New York) and as has
`'', ~:
WO91/13157 2 0 7 6 7 ~ ~ PCT/AU~1/OOn64
- 13 -
bePn descrlbed for other rho-dependent terminators
(Rosenberg et al., Nature 272, 414~428 [1978]), and is
preceded by a region of dyad symmetry (Figure 2). ORFA
is tharefore capable of coding for a 28kDa protein, a
size consistent with that reported for other Rep
proteins.
In addition to the predicted size of the ORFA
protein, further evidence to suggest that we have
;~ identified the legitimate translated region arises from
an examination of the derived amino acid sequence.
Firstly, the codon bias for the putative ORFA product
(Phe, TTC; Asp, GAC; Arg, CGC; Ile, ATC; Val~ GTC; Ala,
Gcc; Thr, ACC) is the same as that for other
Corvnebacterium proteins. Secondly, the predicted
~; 15 protein encoded by ORFA is highly basic in nature (19
basic residues) which is a characteristic of Rep proteins
(and other DNA binding proteins) thought to be important
in the role they play in replication and inco~patibility.
Database searches were performed usin~ both the
complete 1.85kb nucleotide sequence and the predicted
amino acid sequence of ORFA, however no significant
homologies were found. In addition, more detailed
analyses revealed no similarities between the pEP2 Rep
region and either of the potentially related plasmids
from Corvnebacterium (pBL1, Martin et al., Biotechnol. 5:
137-146 [1987]) and MYcobacterium (pAL5000, Rauzier et
al., Gene II: 315-321 [19883).
;! .
Plasmid replication regions invariably have an
origin of replication. Most commonly, origins are
located in non-coding regions, possess clusters of direct
and inverted repeats and may be praceded by A+T rich
sequences (Kamio et al., J. Bacteriol. 258: 307-312
`~ [1984], Scott et al., Microbiol. Rev. 48: 1-23 [1984],
Rosen et al., Mol. Gen. Genet. 179: 527-537 C1980],
35 Rauzier et al., Gene 71: 315-321 [1988]). The region ~;
upstream of ORFA contains no ORFs and possesses a number
of direct and inverted repeat DNA sequences (Figure 2).
.,. ;~'~.
,,"
, . . .
, . . ~. . .
: ' ' ~' ', ' ~
WO91/13l~7 PC~/AU91/000~
2~767~3 14 -
In addition, the region between nt 86 and 171 ls 63.5
A~T compared with an average o 45~ for the entire Rep
region (Figure 2). We therefore believe this to be the
pEP2 origin of replication.
The replication region of pEP2 is capable of
encoding a single 28kDa protein and possesses a single
origin allowing plasmid replication in both Gram-positive
and negative bacteria. p~P2 is therefore a unique
shuttle vector and should be a useful tool, for example,
in the genetic analysis of MYcobacterium and in the
development of M. bovis BCG as a live recombinant
vaccine.
Electroporation of Mycobacteria with pEP2 and pEP3:
Electroporation of Mvcobacterium species with pEP2
DNA resulted in transformation to kanamycin resistance
respectively (Table 1). To confirm that the plasmids were -~
present in the mycobacteria, total cellular DNA
preparations were made from the kanamycin resistan~
transformants. Figure 4 shows the results of agarose gel
and Southern blot analysis of ~otal cell DNA extracted
from BCG CSL strains that had been transformed with pEP2.
The pEP2 plasmid is clearly present in the transformed
strains. Furthermore, the relat:Lve intensity of ~he gel
bands suggests that the pEP plasmids replicate to high
copy number in these bacteria. In additlon, when total
genomic DNA isolated from the drug resistant
transformants was used to electroporate E. coli,
approximately 1.0 X 105 kanamycin resistant clones were
obtained per ug DNA~ Taken together these data suggest
that the pEP2 replicon promotes stable plasmid
replication to high copy number in these bacteria.
Plasmid pEP3 was capable of transforming M.
smegmatis and M. bovis BCG CSL to resistance to at least
200ug hygromycin per ml (Table l). Total cellular DNA
isolated from pEP3 transformed M. s0eamatis was capable
of transforming E. coli to hygromycin resistance. This
shows that the php gene encoding hygromycin resistance is
::
;.
; : . .. . . . .
., : , : .. ~ :
:: '' ' ' '' ' , `" ` : ' ~ ,
: . ~ , . : ,, ; ;
. .
W091/13157 2 0 7 6 7 ~ ~ PCT/AU91/000~
- 15 -
effective in both mycobacteria and E, coll and thereby
constitutes a useful marker for these bacteria.
~ABI.~ 1
EL~CTROPORATIO~ 0~ MYCOBACT~RIA AND RE~AT~D SPECIES
_____________________________ _____ _ --
Plasmid electroporation efficiency:~anr CFU
per ~icrogram of plasm~d DNA
pEP2 pAL8 pEP3
Bacterl~l Specie~
Mvcobact@rium Dhlei --- 103
102
Mycobacterium smeqmatis lo2 lol lo2
01
~Y5353~3~3~9~ bovis 1o2 1o2 N/D
lO2 103
BCG v~r CSL lo2 104
Cor~nebacterium ovis 103 ___ lO2
t pseudot.uberculosis ) 104
________________________
N~D = Not done, ---= no transformation ~o resistant phenotype :~
Different f~gures for each electroporatlon represent the rosult
of ~epar~te expcrimente.
. .
.
- . .
: ; : . ; ~ ~ . ; ' .
' .
, :, . ~ . , ~ , , .:
WO9l/13t~7 ~ 7 6 7~ ~ PCT/AU91/00064
- 16 -
lectroporation of Other Bacterial Species with
pEP2 and ~EP3
~ . _
To determine the host range of the pEP replicon,
pEP2 and 3 plasmid DNA was electroporated into
Corn~bacterium ~eudotuberculosis. Transformation
occurred in C. pseudo tuberculosis tTable 1). These
results indicate that the pEP2 plasmids have a wide host ,~
range.
EXAMPLE 3
Generation and Expression of pEP2 Constructs:
A PCR derived DNA fragment carrying the promoter
from the Mycobacterial 65kDa heat shock protein (0~6Kb)
was cloned into the Pstl-BamHl sites of pEP2, generating
a plasmid referred to as pEP5. This promoter is known to
function in E. coli as well as Mycobacteria and is
therefore useful in this shuttle vector expression ~-
system.
A gene encoding for chloramphenicol-acetyl-
~ran~ferase (CAT) was cloned in~o the BamHl site of pEP5.
Expression of the CAT gene driven by the 65kDa promoter
was detected using the Pharmacia (Registered trademarX,
i Pharmacia, Pitcataway, N.J., U.';.A.) C~T detection kit. ~;
~ccording to this assay the 64kl)a promoter has a strength
comparable with the induced lac promoter in both E. coli
and C. pseudotuberculosis.
MPB70 is the major secreted protein of M. bovis and
a component of PPD. This gene and its signal sequence
was incorporated into the pEP2 expression system. A
truncated form of the MPB70 gene was generated by PCR
removing its promoter but leaving its ribosome binding -~
site (RBS) intact. This was cloned into pEP2 as a PStl~
BamHl fragmen~ (O.SKb). The 65kDa promoter was then
cloned upstream of the MPB70 BS on a Pstl-Scal fragment.
After electroporation into E. coli and C.
Dseudotuberculosis, expression was tested for by western
blots and Elisas. This construct did not express in E. ~;~
~.
', `,
" . : : : . ~ .
.
~ u ~
WO91/131~7 PCT/AU91/00064
- 17 -
coli. It did however express ln C. pseudotube.rculosis
with product detected in both the solicate and culture
filtrate. Ths indicated that the MPB70 RBS was inactive
in E. coli yet was recognised and functional in C
pseudotuberculosls.
Those skilled in the art will appreciate that the
invention described herein is susceptible to variations
and modifications other than those specifically
described. It is to be understood that ~he invention
includes all such variations and modifications which fall
within its spirit and scope. The invention also includes
all of the steps, features, compositions and compounds
referred to or indicated in this specification,
individually or collectively, and any and all
combinations of any two or more of said steps or
features.
,:
,