Note: Descriptions are shown in the official language in which they were submitted.
2~
N-substituted heterocyclic derivatives, their
preparation and the pharmaceutical compositions in
which they are present
S
The present invention relates to N-substituted
heterocyclic derivatives, to their preparation and to
the pharmaceutical compositions in which they are
present.
The compounds according to the invention
antagonize the action of angiotensin II, which is a
peptide hormone of the formula
H-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-OH
Angiotensin II is a potent vasopressor and the
hiologically active product of the renin-angioterlsin
system: renin acts on the angiotensinogen of the plasma
to produce angiotensin I, which is converted to
angiotensin II by the action of the angiotensin I
converting enzyme.
The compounds of the present invention are non-
peptide compounds which antagonize angio~ensin II. By
inhibiting the action of angiotensin II on its
receptors, the compounds according to the invention
prevent especially the increase in blood pressure
produced by the hormone-receptor interaction; they also
have other physiological actions on the central nervous
system and on the kidneys, for example.
Thus the compounds according to the invention
are useful in the treatment of cardiovascular
complaints such as hypertension and heart failure, as
well as in the treatment of complaints of the central
nervous system and in the treatment of glaucoma and
diabetic retinopathy and renal insufficiency.
2~
Non-peptide compounds which antagonize -th~
action of angiotensin II are already describe in many
patents or patent applications. Particularly, reference
could be made to CA application 2 050 769 published on
S March 11, 1992. Said CA application generally relates
to compounds of formula :
N
in which notably :
~ X represents a group of formula :
-c(RIIIRIv-cc~Rv)RvI]p-[c(RvII )RVIII]q~
- RI represents an alkyl or an alkenyl group, said
groups being unsubstituted or substituted by an
halogen,
- RII represen~s a carboxy or a tetrazolyl,
- RIII and RIV represent each independently an alkyl
unsubstituted or substituted by or more halogen
atoms, a cycloalkyl or an aromatic radical.
Said CA application 2 050 769 does not
specifically disclose compounds of formula 1 in which
RIII is an alkyl and RIV is a cycloalkyl.
2~
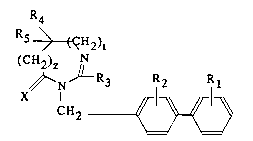
The present invention relates to compounds of
the formula
R4
R5 ~ H2)
(CH2)z
N ~ R3 R~ R1 (I)
CH2 ~\
S in which:
- R1 and R2 ar similar or di~f~rent and are aach
independently hydrogen or a group selected ~rom a Cl-
C6 alkyl, a C1-C4 alkoxy, an amino, an aminomethyl, a
carboxyl, an alkoxycarbonyl in which the alkoxy is
C1-C4, a cyano, a tetrazolyl, a methyltetrazolyl, a
methylsulfonylamino, a trifluoromethylsulfonylamino,
a trifluoromethylsulfonylaminomethyl, an N-cyano-
acetamide, an N-hydroxyacetamide, an N-(4-carboxy-
1,3 thiazol-2-yl)acetamide, a ureido, a 2-cyano-
guanidinocarbonyl, a 2-cyano-guanidinomethyl, an
imidazol-1-ylcarbonyl and a 3-cyano-2-methyliso-
thioureidomethyl, with the proviso that at least one
of the substituents Rl or R2 is other than hydrogen;
- R3 is a hydrogen, a C1-C6 alkyl which is
unsubstituted or substituted by on~ or more halogen
atoms ; a Cz-C6 alkenyl, a C3-C7 cycloalkyl, a
phenyl, a phenylalkyl in which the alkyl is Cl-C3, or
a phenylalkenyl in which the alkenyl is C2-C3, said
phenyl groups being unsubstituted or monosubstituted
or polysubstituted by a halogen atom, a Cl-C4 alkyl,
a Cl-C4 halogenoalkyl, a Cl-C~ polyhalogenoalkyl, a
hydroxyl or a Cl-C4 alkoxy ;
- - ~
, ,: . '
,
z~
- R4 is a Cl-C6 alkyl which is unsubstituted or
substituted by one or more halogen atoms ;
- Rs is a cycloalkyl or a cycloalkylmethyl, said
cycloalkyl being C3-C7, which is unsubstitutecl or
S substituted by one or more halogen atoms ;
- or R4 and R5 are each a cyclopropyl ~
- X is an oxygen atom or sulfur atom; and
- z and t are zero or one is zero and the other is
one ;
and their salts.
If a compound according to the invention has an
asymmetric carbon, the invention includes the 2 optical
isomers of this compound and their racemic mi~ture.
The salts of the compounds of formula (I)
lS according to the present inven~ion include those with
mineral or organic acids which permit separation or
suitable crystallizatlon of the compounds of formula
(I), such as picric acid, oxalic acid or an optically
active acid, for example a mandelic acid or
camphosulfonic acid, and acids which form
pharmaceutically acceptable salts such as the
hydrochloride, the hydrobromide, the sulfate, the
hydrogensulfate, the dihydrogenphosphate, the
methanesulfonate, the methylsulfata, the maleate, the
fumarate and the naphthalene-2-sulfonate.
The salts of the compounds of formula (I) also
include the salts with organic or mineral bases, for
example the salts of alkali or alkaline earth metals,
such as the sodium, potassium and calcium salts, the
sodium and potassium salts being preferred, or with a
tertiary amine such as trometamol, or else the salts of
arginine, lysine or any physiologically acceptable
~t~?~
amine.
According to the present description and in the
claims which follow, halogen atom is understood as
meaning a bromine, chlorine or fluorine atom; N-
S protecting group (also designated by Pr) is understoodas meaning a group conventionally used in peptide
chemistry for affording temporary protection of the
amine group, for example a Boc, z or Fmoc group or a
benzyl group; esterified carboxyl group is understood
as meaning an ester which is labile under appropriate
conditions, such as, for example, a methyl, ethyl,
benzyl or tert-butyl ester. "Alkyl" denotes linear or
branched saturated aliphatic hydrocarbon radicals.
The compounds of formula (I) in which R1 is in
lS the ortAo position and is a carboxyl or tetrazolyl
group and R2 is hydrogen are preferred compounds.
The compounds of formula (I) in which R4 is
methyl and R5 is cyclohexyl are also preferred
compounds.
Likewise, the compounds of formula (I) in which
R3 is a linear C1-C6 alkyl group are preferred
compounds.
The compounds of formula (I) in which X is an
oxygen atom are also preferred compounds.
Finally, the compounds of formula (I) in which
z = t = 0 are preferred compounds.
2-n-Butyl-4-methyl-4-cyclohexyl-1-[(2'-(tetrazol
-5-yl)biphenyl-4-yl) methyl]-2-imidazolin-5-one or one
of its salts with acids or bases are particularly
preferred.
The following abbreviations are used in the
description and in the Examples:
`s~
alcohol : ethyl alcohol
Et : ethyl
nBu, tBu n-butyl, tert-butyl
DMF : dimethylformamide
THF : tetrahydrofuran
DCM : dichloromethane
NBS : N-bromosuccinimide
DCC : dicyclohexylcarbodiimide
DIPEA : diisopropylethylamine
ether : ethyl ether
TFA : trifluoroacetic acid
Z : benzyloxycarbonyl
Boc : tert-butoxycarbonyl
BOP : benzotriazolyloxytrisdimethylamino-
phosphonium hexafluorophosphate
Fmoc : fluorenylmethoxycarbonyl
Lawesson's r~agent : [2,4-bis(4-methoxyphenyl)-
1,3-dithia 2,4-diphosphetane 2,4-disulfide].
The present invention further relates to the0 method of preparing the compounds (I). In said method :
al) a heterocyclic derivative of the formula
R4
S ~ (C~2)t
(CH2)~ N
//~ N ~LL R3
O I ,
H
in which z, t, R3, R4 and R5 are as defined above for
~I), is reacted with a (biphenyl-4 yl)methyl. derivative
of the formula
2~ 3~7
R'2 R 1
Hal-cH2 ~ ~ 3
in which Hal i5 a halogen atom and R'1 and R'2 are
respectively either Rl and ~2 or a precursor group of
Rl and/or R2;
bl) if appropriate, the resulting compound of
the formula
R4
(CH2)t
~CH2)Z N
/~N~R3 R'~ R'
1~ _ r~" ~
is treated wi~h Lawesson's reagent [2,4-bis(4-methoxy-
phenyl)-1,3-dithia-2,4-diphosphetane 2,4-disulfideJ,
and
cl) the compound obtained in al) or bl), of the
formula
Z~
R4
~ )t
(CH2)~ ~ 5
/~ N ~ R3 R'2 R l
CH 2 ' ~--`\
in which X is an o~ygen atom or a sulfur atom, is
treated to give the compound (I) by conversion of the
groups R'l and/or R'2 to the groups R1 and/or R2
respectively ;
dl) the compound so obtained is converted if
approprlate, into one of its salts, according to known
method~ well known by the skilled in the art.
Among the compounds 2, some of them are prior
art compounds, the others are new and belong to the
scope o the invention.
EP patent application 144 748 discloses
herbicides of formula :
lS
X
Y~\ 1~
H
in which one of X and Y may represent an alkyl and the
other a cycloalkyl ; furthermore the phenyl group may
be substituted.
Thus the present invention further relates to
the compounds (II) of the formula
- .
2'~
S " ~ H2)t
~(CH~ N fl
X ~ N 3
H
in which:
- z and t are zero or one is zero and the other is one;
R3 is a hydrogen, a C1-C6 alkyl which is
unsubstituted or substituted by one or more halogen
atoms ; a C2-C6 alkenyl, a C3-C7 cycloalkyl, a
phenyl, a phenylalkyl in which the alkyl is C1-C3, or
a phenylalkenyl in which the alkenyl is C2-C3, said
phenyl groups being unsubstltuted or monosubstituted
or polysubs~ituted by a halogen atom, a C~-C4 alkyl,
a Cl-C~ halogenoalkyl, a Cl-C4 polyhalogenoalkyl, a
hydroxyl or a Cl-C~ alkoxy;
- R4 is a Cl-C6 alkyl which is unsubstituted or
lS substituted by one or more halogen atoms ; and
- R5 is a cycloalkyl or a cycloalkylmethyl, the
cycloalkyl being C3-C7, which is unsubstituted or
substituted by one or more halogen atoms ;
- or else R4 and R5 are each a cyclopropyl ;
- X represents an oxygen atom,
with the proviso that R3 is other than a phenyl or a
substituted phenyl when z = t = 0, R5 represents a
cycloalkyl.
Among the derivatives (II), the compounds in
which z = t = 0 of the formula :
R4
~5 N
X ~ \ ~ R3
H
in which X, R3, R4 and R5 are as defined above for
(II~, with the proviso that R3 is other than a phenyl
S or a substituted phenyl when R5 represents a cycloalkyl
are preferred compounds.
The compounds (II) in which z = 0 and t = 1, of
the formula
R\4
R5- "^'N
xi ~ ~ -R3
in which R3, R4, R5 and X are as defined above for
(II), are preferred compounds.
Finally, the compounds (II) in which z - 1 and
t = 0, of the formula
R ~ R5
N
H (II"')
in which R3, R4, Rs and X are as defined above for
(II), are preferred compounds.
The derivatives 2 are prepared by known
.
2~
methods. For example, it is possible to use the method
described by Jacquier et al. (Bull. Soc. Chim. France,
1971, ~, 1040-1051) and by Brunken and Bach (Chem.
Ber., 1956, ~9, 1363-1373) and to react an alkyl
imidate with an amino acid or its ester in accordance
with the following reaction scheme: -
R4 (CH2)z~co2R ~NH
C ~ \
R5 (CH2)t-NH2 OR
_' _
R4
R5 - ~ ~I2~t
(CH2)z N 2
/~N~R3
O
H
in which R is a Cl-C4 alkyl, R' is hydrogen or a Cl-C4
alkyl and R3, R4, R5, z and t are as defined above for
I).
This reaction is carried out in an acid medium
by heating in an inert solvent such as xylene or
toluene.
The compound 5' are known compounds or are
prepared by known methods. ThP compounds ~' can be
obtained optically pure using methods of asymetric
synthesis or methods of resolving the racemic mixture,
such as those described in "Synthesis of Optically
Active Alpha-aminoacids, R.M. Williams, Pergamon Press,
1989".
12
2~ t~
According to another procedure, the compound 2
can be prepared by reacting an aminoalkylamide (5'')
with an alkyl ortho-ester ~10) in an acid medium in
accordance with the following react~on scheme:
R4 (CH~ CONH2
\ /
C ~ +R3~C(0R)3 -- ~ 2
~5 (CH2)t~H2
in which R is a C1-C4 alkyl.
Using a procedure described by H. Takenaka et
al. (Heterocycles, 1989, ~9(6), 1185 89), it is also
possible to prepare the compound 2 by reacting an acid
halide of the formula
R3-CO-Hal 12
in which Hal is a halogen, preferably chlorine, with
lS the deriv~tive 5'' ; ~he cyclization of the diamine is
then carried out in a basic medium.
If z = t = O, a derivati~e 5" can be prepared
from a ketone by a procedure described in US patent
4 017 510 or in Swiss patent 540 271 :
cyanure H+ ou OH
R ~ CO ~ R4-c-~N _ ~ 5
R5 NH40H ~5 NH~
NH4CI
",
. -~
.
`
21~? ~
The ketone R4R5CO is treated with cyanide
(hydrocyanic acid, sodium cyanide or potassium cyanide)
and ammonium ions (aqueous ammonia and ammonium
chloride) ; the aminonitrile compound 5"' i5 then
S hydroly2ed to 5"', either in a strongly acidic medium
or in a basic medium. If appropriate, the aminonitrile
5"' can be resolved with an optically active acid by
the procedure in European patent 158 O00, in which case
it is possible to prepare the compound 5" and then the
compound 2 in optically pure form.
More particularly, according to another object
of the present invention, the compound 2 is prepared by
a method which comprises reacting a compound of the
formula
lS R3-B 14
in which B is
- a group C~OR)3
NH~
- a group C or
20 OR/
- a group COHal
R being a Cl-C4 alkyl and Hal denoting a halogen atom,
preferably chlorine, with a compound of the formula
R4 \ (CH2)zCOA l3
R5 (CH~)tNH2
in which A is an OH group, an NH2 group or a group OR',
R' being hydrogen or a Cl-C4 alkyl.
The (biphenyl-4-yl)methyl derivative (~) is
prepared by a method described ~n European patent
30 application 324 377.
14
The conversion of a group R'1 and/or R'2 to a
group R1 and/or R2 is effected by methods well known to
those skilled in the art. Thus, i~ the compound (I) to
be prepared possesses a group Rl and/or R2 = carboxyl,
S R'l and/or R'2 are an esterified carboxyl group. If
the compound (I) to be prepared possesses a group Rl
and/or R2 - tetrazolyl, R'l and/or R'2 can be either a
tetrazolyl protected for example by a trityl group, or
a cyano group which will subsequently be replaced with
a tetrazolyl group protected if necessary by a trityl.
The conversion of the cyano group to a tetrazolyl can
be effected with an azide, for example tributyltin
azide or sodium azide.
It is also possible to use groups R'l and/or
R'2 such as nitro, carboxyl, cyano or acid chloride
groups and ~hen to convert them by reactions well known
to those skilled in the art to give groups R1 and/or R2
as defined ~or the compound ~I).
Thus, if R'1 and/or R'~ are a carboxyl, they
can be converted to R1 and/or R2 in the form of an
imidazol-l-ylcarhonyl or elsa an N-(4-carboxy-1,3-
thiazol-2-yl)acetamide.
The group R'1 and/or R'2 in the form of an acid
chloride can be converted to Rl and/or R2 in the form
of N-hydroxyacetamide, N-cyanoacetamide, ureido or 2-
cyanoguanidinocarbonyl.
The group R'l and/or R'2 in the form of a nitro
can be converted to amino, from which Rl and/or R2 is
prepared in the form of methylsulfonylamino, trifluoro-
methylsulfonylamino or trifluoromethylsulfonylamino-
methyl.
The group R'1 and/or R'2 in the form of a cyano
. - '
2~?~
can be converted to aminomethyl, from which a 3-cyano-
2-methylisothioureidomethyl is prepared (according to
C. Gordon et al., J. Org. Chem.~ 1~70, ~(6~, 2067-
2069) or a 2-cyanoguanidinomethyl is prepared
S (according to R.W. Turner, Synthesis, 1975, 332).
Step al) is carried out in an inert solvent
such as DMF, DMSO or THF, in a basic medium, for
example in the presence of potassium hydroxide, a m~tal
alcoholate, a metal hydride, calcium carbonat~ or
triethylamine.
Step bl) is carried out by heating under
nitrogen in a solvent such as toluene, according to the
method described by M.P. Cava et al., Tetrahedron,
1985, 41, 22, 5061.
In the description below, the method camprising
steps al, bl and cl is referred to as method 1.
Alternatively, the compounds (I) can be
prepared by another method, which is also a sub~ect of
the present invention. In this method:
a2) an amino acid of the formula
4 \ / (CH2)t NHPr
/ 1 7
S (CH2)z
COOH
in which z, t, R4 and R5 are as defined above for (I),
and of which the amine group is protected by the Pr
group, is reacted with a (biphenyl-4-yl)methylamine
derivative of the formula
.
- , :
16
R'2 R'
9 9 8
in which R'l and R'2 are respectively either Rl and R2
or a precursor group of Rl and R2;
S b2) a~ter deprotection of the amina, the
resulting compound of the formula
R'~ R'
R5 / ¦ Z CH2 ~ ~\ ~ 2
is then treated wlth an alkyl ortho-ester of the
formula R3C(OR)3 (lO), in which R3 is as defined above
for (I) and R is a C1-C4 alkyl;
c2~ if appropriate, the resulting compound of
the ~ormula
: 15
: R4
: R5 ~ (CH )
(C~I2)z N 4
/~ N ~ R3 ~ 2 R'1
CH2--~\
is treated with Lawesson's reagent [2,4-~is(4-
methoxyphenyl)-1,3-dithia-2,4-diphosphetane 2,4-disul-
fide];
~ . ' ' ' ': . ,
.
s~
and
d2) the compound thus obtained in b2 or c2, of
the formula
R~
S--~ ~CH2)t
(CH2)z N S
/~ N ~ R3 R 2 R'1
X CH2 ~ ~ \>
is then treated under suitable conditions for preparing
the compound (I) by conversion of the groups R'2 and/or
R'l to the groups R2 and/or Rl respectively ;
e2) the compound so obtained is converted if
appropriate, into one of its salts according to known
methods well known by the skilled in the art.
The compounds 7 are known or are prepared by
known methods (Chemistry of the Amino Acids, Greenstein
lS and Winitz, published by ~ohn Wiley, 1961, vol. I, p.
697). If appropriate, these compounds can be obtained
: optically pure using methods of asymetric synthasi~ or
methods of resolving the racemic mixture, such as those
: : dsscribed in 'ISynthesis of Optically Active Alpha-
aminoacids, R.M. Williams, Pergamon Press, 1989".
The compounds 8 are prepared according to
European patent application 324 377. Step a2~ is
carried out under the usual conditions for the coupling
of an acid with an amine, for example in the presence
of BOP and DIPEA.
Step b2), which is the cyclization of the
18
~,~ 3 i~
compound 9 in the presence of 10, is carried out
according to Jacquier et al. (Bull. Soc. Chim. France,
1971, (3), 1040-1051) and according to srunken and Bach
(Chem. Ber., 1956, ~9, 1363-1373).
S In the description below, the method comprising
steps a2 to d2 is referred to as method 2.
In one variant of method 2, in step b2, it is
possible, if appropriate, to isolate an intermediate 9
of the formula
-(CH2)z ~0 - NH- CH2 ~ \
(CH2)t- NH-C0-R3
and then to prepare the compound 4 by cyclization in an
acid or alkaline medium.
The affinity of the products according to the
invention for angiotensin II receptors was studied in a
test for the binding of angiotensin II, labeled with
iodine 125, to rat liver mambrane receptors. The
method used is the one described by S. KEPPENS et al.
20 in Bio~hem. J., 1982, 20~, 80~-817.
The IC50, namely the concentration which gives
a 50~ displacement of the labeled angiotensin II bound
speaifically to the receptor, is measured. The IC50 f
the compounds according to the invention is less than
25 10-6 M.
Also, the effect of the products according to
the invention as angiotensin II antagonists was
observed on different animal species in which the
renin-angiotensin system had been activated befor~hand
19
~,t~ 3~t~
(C. LACOUR et al., J. Hypertension, 1989, 7 (suppl~ 2),
S33-S35).
The compounds according to the invention are
active after administration by diffarent routes,
especially after oral administration.
No signs of toxicity are observed with these
compounds at the pharmacologically active doses.
Thus the compounds accordin~ to the invention
can be used in the treatment of various cardiovascular
complaints, especially hypertension, heart failure and
venous insufficiency, as well as in the treatment of
~laucoma, diabetic retinopathy and various complaints
of the central nervous system, for example anxiety,
depression, memory deficiencies or Alzheimer's disease~
lS The present invention further relates to
pharmaceutical compositions containing an effective
dose of a compound according to the inven~ion, or of a
pharmaceutically acceptable salt, and suitable
excipients. Said excipients are chosen according to
the pharmaceutical form and the desired mode of
administration.
In the pharmaceutical compositions of the
present invention for oral, sublingual, subcutaneous,
intramuscular, intravenous, topical, intratracheal,
intranasal, transdermal or rectal administration, the
activa principles of formula I above, or their salts if
appropriate, can be administered to animals and humans
in unit forms of administration, mixed with
conventional pharmaceutical carriers, for the
prophylaxis or treatment of the above disorders or
diseases. The appropriate unit forms of administration
include forms for oral administration, such as tablets,
2~, ,j~
gelatin capsules, powders, granules and solutions or
suspensions to be taken orally, forms for sublingual,
buccal, intratracheal or intranasal administration,
forms for subcutaneous, intramuscular or intravenous
S administration and forms for recta:L administration.
For topical application, the compounds according to the
invention can be used in creams, ointments or lotions.
To achieve the desired prophylactic or
therapeutic effect, the dose of active principle can
vary between 0.01 and 50 mg per kg o~ body weight per
day.
Each unit dose can contain from 0.1 to 1000 mg,
preferably 1 to 500 mg, of active ingredients in
combination with a pharmaceutical carrier. This unit
lS dose can be administered 1 to 5 times a day so as to
administer a daily dosage of 0~5 to 5000 mg, preferably
1 to 2500 mg.
When a solid composition in the form of tablets
is prepared, the main active ingredient is mixed with a
pharmaceu~ical vehicle such as gelatin, starch,
lactose, magnesium stearate, talc, gum arabic or the
like. The tablets can be coated with sucrose, a
cellulose derivative or other appropriate substances,
or else they can be treated so as to have a ~rolonged
or delayed activity and so as to release a
predetermined amount of active principle continuously.
A preparation in th~ form of gelatin capsules
is obtained by mixing the active ingredient with a
diluent and pouring the resulting mixture into soft or
hard gelatin capsules.
I A preparation in the form of a syrup or elixir
or for administration in the form of drops can contain
3,~
the active ingredient in conjunction with a sweetener,
which is preferably calorie-free, methylparaben and
propylparaben as antiseptics, as well as a flavoring
and an appropriate color.
S The water-dispersible granules or powders can
contain the active ingredient mixed with dispersants or
wetting agents, or suspending agents such as
polyvinylpyrrolidone, as well as with sweeteners or
taste correctors.
Rectal administration is effected using
suppositories prepared with binders which melt at the
rectal temperature, for example cacao butter or
polyethylene glycols.
Parenteral administration is effected using
aqueous suspensions, isotonic saline solutions or
sterile and in~ectable solutions which contain
pharmacologically compatible dispersants and/or wetting
agents, for example propylene glycol or butylene
glycol.
The active principle can also be formulated as
microcapsules, with one or more carriers or additives
if appropriate.
In addition to the products of formula I above
or one of their pharmaceutically acceptable salts, the
compositions of the present invention can contain other
active principIes such as, for example, tranquilizers
or other drugs which can be useful in the treatment of
the disorders or diseases indicated above.
Thus the present invention relates to
pharmaceutical compositions containing several active
principles in association, one being a compound
according to the invention and it being possible for
the other or others to be a beta-blocking compound, a
calcium antagonist, a diuretic, a non-steroidal
antiinflammatory or a tranguilizer.
The following Examples illustrate the invention
without however implying a limitation. The following
abbreviations are used in these Examples: d denotes
density, RT denotes room temperature, KHS04-K2S04
denotes an aque3us solution containing 16.6 g of
potassium bisulfate and 33.3 g of potassium sulfate per
liter.
The melting points (m.p.) are given in degrees
Celsius; unless indicated otherwise, they were measured
without recrystallization of the product.
The purity of the products is checked by thin
layer chromatography (TLC) or HPLC. The products are
characterized by their NMR spectra run at 200 MHz in
deuterated DMS0 with tetramethylsilans as the internal
reference.
The specific optical rotation ~]D are measured
at 22'C ; path length : lO cm, concentration l g per
100 ml.
The following abbreviations are used in the
lnterpretation of the NMR spectra:
s : singlet
sb : broad singlet
d : doublet
t : triplet
q : quadruplet
quint : quintuplet
sext : sextuplet
m ; unresolved signals or multiplet
In addition, im denotes imidazole.
EXAMPLE 1
(R,S)-2-n-Butyl~ (2'-carboxybiphenyl-4-yl)-
methyl]-4-cyclohexyl-4-methyl-2-imidazolin-5-one tri-
fluoroacetate
S A) 5-Cyclohexyl-5-methylhydantoin
This compound is prepared accordin~ to J. Org.
Chem., lg60, 2S, 1920-1924.
A solution of 50 g of cyclohexyl methyl ketone
in 4~0 ml of 95- alcohol is added over 30 minutes to
29.4 g of sodium cyanide and 192 g of ammonium
carbonate in 400 ml of water. The mixture is heated
at 55-60- C for 4 hours and then evaporated to half
its volume under vacuum and left to stand overni~ht at
+4~ C. The precipitate formed is filtered off, washed
with wa~er and then dried under vacuum over phosphorus
pentoxide to give 65.5 g of the expected hydantoin,
wh~ch is identified by its IR and NMR spectra.
M.p. = 220- C.
B) tR,S)-2-Amino-2-cyclohexylpropionic acid
This compound is prepared according to J. Org.
Chem., 1960, 2~, 1920-1924.
A mixture containing 7 g of the hydantoin
prepared in the previous step and 28 g of barium
hydroxide octahydrate in 150 ml of water is heated at
160- C for 5 hours in a steel tube. The reaction
medium is saturated with dry ice, the insoluble.
material formed is filtered off and the -filtrate is
then concentrated under vacuum. The solid residue is
taken up in acetone, filtered off and dried to give
5.25 g of the expected acid, which is identified by
its IR and NMR spectra. The product melts at 350- C
with decomposition.
24
C) Ethyl ester of (R,S)-2-amino-2-cyclohexylpropionic
acid
3 g of the acid prepared in the previous step
are added to 40 ml of absolute alcohol saturated with
S gaseous hydrochloric acid and the mixture is then
refluxed for 20 hours, with stirring. The reaction
medium is evaporated under vacuum and the residue is
taken up in an ether/water mixture, which is brought
to pH 9 by the addition of a saturated solution of
sodium bicarbonate. The organic phase is decanted,
washed with a saturated solution of sodium chloride
and then evaporated under vacuum to give 2.1 g of the
expected ester in the form of an oil. Identification
by IR and NMR spectra.
lS D) Ethyl valerimidate
This compound is prepared in the form of the
hydrochloride according to Mac Elvain (J. Amer. Chem.
Soc., 1942, 64, 1825-1827)o It is freed from its
hydrochloride by reaction with potassium carbonate and
then extracted with DCM.
E) (R,S)-2-n-Butyl-4-cyclohexyl-4-methyl-2-imidazolin-
5-one
2 g of the ester prepared in step C and 2.35 g
of ethyl valerimidate are mixed in 6 ml of xylen~, to
which 6 drops of acetic acid are added; the reaction
medium is re1uxed for 6 hours. It is then concen-
trated under vacuum and the residue is chromatographed
on fine silica gel using a chloroform/methanol/acetic
acid mixture (95/9/3; v/v/v) as the eluent. The
~ractions containing the desired product are com~ined
and then evaporated under vacuum; the residue is taken
up in an ethyl acetate/water mixture and the pH is
brought to 9 by the addition of a solution of sodium
hydroxide. The organic phase is decanted, washed with
water and then with a saturated solution of sodium
chloride, dried over sodium sulfate and then
S evaporated to dryness. The expected product is
obtained in the form of a thick oil, which solidifies
to give an amorphous solid.
m = 1.56 mg.
- IR (chloroform):
1720 cm~1 : C=0
1640 cm~1 : C=N
- NMR consis~ent.
F) 4-Bromomethyl-2'-tert-butoxycarbonylbiphenyl
This compound is prepared by the method
described in European patent application 324 377.
G) (R,S)-2-n-Butyl-4-cyclohexyl-4~methyl-1-C(2'-tert-
butoxycarbonylbiphenyl-4-yl)methyl~-2-imidazolin-5-
one
1.5 g of the imidazolinone prepared in the
previous step are dissolved in 20 ml of DMF. 250 mg
of sodium hydride as an 80% dispersion in oil are
added. After stirring for 20 minutes, 2.48 g of the
compound prepared in step F) are added and the
reaction medium is left to stand for 2 hours at RT.
It is taken up in an ethyl acetate/water mixture; tha
organic phase is decan~ed washed with water and with
a saturated solution of sodium chloride, dried over
sodium sulfate and then concentrated under vacuum.
The residue is chromatographed on silica using an
ethyl acetate/toluene mixture ~1/4; v/v~ as the eluent
to give 1.8 g of the expected product in the ~orm of a
white wax.
26
- IR (chloroform):
1710 1730 cm~l : ester and imidazolinone C=0
1630 cm~1 : C=N
H) (R,S)-2-n-Butyl-1-[(2'-carboxybiphenyl-4-yl)methyl]-
4-cyclohexyl-4-methyl-2-imidazolin-5-one trifluoro-
acetate
1.5 g of the compound obtained in the previous
step are stirred for 40 minutes in 7 ml of TFA and 7
ml of ~CM. The reaction medium is concentrated under
vacuum and taken up in ether to give a white solid,
which is filtered off, washed with ether and dried
under vacuum.
m = 1.40 g.
M.p. = 171- C.
MH+ : 447.
- NMR:
7.10-7.70 ppm : m : 8 aromatic H
4.45 ppm : s : 2 H : N-C~2-C6H4-
1.25 ppm : s : CH3 in the 4 position
EXAMPLE 2
(R,S)-2-n-Butyl-1-[(2'-carboxybiphenyl-4-yl)-
methyl]-4-cyclohexyl-methyl-4-m~thyl-2-imidazolin-5-one
trifluoroacetate
The ~ollowing compounds are prepared from
cyclohexyl methyl ketone according to the procedures
described in Example 1:
A) 5-Cyclohexylmethyl-5-methylhydantoin
M.p. = 205-206- C.
B~ (R,S)-2-Amino-3-cyclohexyl-2-methylpropionic acid
Characterized by its IR and NMR spectra.
2~
C) Ethyl ester of (R,S)-2-amino-3-cyclohexyl-2-methyl-
propionic acid
Characterized by its IR and NMR spectra.
D) (R,S)-2-n-Butyl-4-cyclohexylmethyl-4-methyl-2-
S imidazolin-5-one
This product is obtained in the form of an
oil, which solidifies. Identification by I~ and NMR.
- IR (chloroform):
1720 cm~1 : imidazolinone C=0
1630 cm~1 : C=N
E) (R,S)-2-n-Butyl-4-cyclohexylmethyl-4-methyl-1-[(2'-
tert-butoxycarbonylbi-phenyl-4-yl)methyl]-2-imicla-
zolin-5-one
This product is obtained by treatiny the
compound of step D with 4-bromomethyl-2'-tert-
butoxycarbonylbiphenyl in the presenca of sodium
hydride. After purification by ahromatography, the
product is in the form of an oil, which crystallizes
in the refrigerator.
Yield : 51%.
M.p. = 73-75- C.
- IR tKBr):
1700-1730 cm~1 : imidazolinone and ester C=0
1630 cm~1 : C=N
F) (R,S)-2-n-Butyl-1-~(2'-carboxybiphenyl-4-yl)methyl]-
4-cyclohexylmethyl-4-methyl-2-imidazolin-5-one
trifluoroacetate
Yield : 90~.
M.p. = 143-146- C.
- NMR:
7.20-7.80 ppm : 8 H : aromatic protons
4.85 ppm : m : 2 H : N-CH2-C6H4-
28
2 ~ ~3'~ 7
2-70 ppm : t : 3 H : -cH2-cH2-cH2-cH3
1.80-0.85 ppm : m : 20 H : 4-methyl
4-cyclohexylmethyl
-CH2-CH2-CH2-CH3
0.80 ppm : t : 3 H : CH2-CH2-CH2-CH3
EXAMPLE 3
(R,S)-2-n-Butyl-1-[(2' carboxybiphenyl-4-yl)-methyl]
-4-cyclohexyl-4-ethyl-2-imidazolin-5-one trifluoro-
acetate
A) (R,S)-2-Amino-2-cyclohexylbutyronitrile hemioxalate
1.18 g of ammonium chloride and 1.5 ml of a
32~ aqueous solution of ammonia are added successively
to a solution of 1.03 ~ of sodium cyanide in 6 ml of
lS water, followed, over 15 minutes, by 2.8 g of
cyclohexyl ethyl ketone in 5 ml of methanol, and tha
reaction medium is heated at 60- C for 6 hours. It is
cooled and extracted 4 times with DCM and the extracts
are then evaporated under vacuum. The residue is
treated again with the same amounts of cyanide,
ammonium chloride, aqueous ammonia, water and methanol
at 60- C for 6 hours. The reaction medium i5 then
cooled and extracted 4 times with DCM and the extracts
are dried and then evaporated under vacuum. The
residue is taken up in 30 ml of acetone, and a
solution of 1.1 g of o~alic acid dihydrate in 10 ml of
acetone is added dropwise. After 15 minutes, the
precipitate formed is filtered off, washed with
acetone and then with ether and dried under vacuum to
give 2.87 g of product, which becomes pasty at 120- C.
Identification by IR and NMR.
- IR of the free base: -
29
J.t
2220 cm~l : C-N
B) (R,S)-2-Amino-2-cyclohexylbutyramide
2.84 g of the nitrile ob~ained in step A are
added over 30 minutes to 6 ml of pure sulfuric acid.
S The mixture is heated for 1 hour at 85- C and then 30
minutes at 100- C. After cooling, the reaction medium
is added dropwise to 20 ml of iced 32% aqueous
ammonia. The mixture is extracted with chloroform and
the extract is then dried over sodium sulfate and
evaporated under vacuum to give 2.5 g of the expected
product in the form of a wax. Identification by IR
and NMR.
C) (R,S)-2-n-Butyl-4-cyclohexyl-4-ethyl-2-imidazolin-
5-one
2.45 g of the product obtained in the previous
step are dissolved in 30 ml of THF; 1.84 ml of
triethylamine are added, followed, over 25 minutes, by
a solution of 1.73 ml of valeroyl chloride in 10 ml of
THF. After stirring for 2 hours, 3.57 g of potassium
hydroxide pellets, then 4 ml of water and then 10 ml
of methanol are added and the mixture is refluxed for
3 hours. After cooling, 6 g of ammonium chloride are
added and the reaction medium is concentrated to half
its volu.ne and then extracted with ethyl acetate. The
organic phase is washed with a saturated solution of
sodium chloride, dried over sodium sulfate and then
concentrated under vacuum. The residue is taken up in
10 ml of hexane and left to stand for 4 hours at 0- C.
The solid formed is filtered off and dried.
m = 2.62 g.
M.p. = 80-85- C.
Identification by NMR and IR (chloroform and KBr).
2 ~'3~
D) (R,S)-2-n-Butyl-1-[(2'- tert -butoxycarbonylbiphenyl-
4-yl)methyl]-4-cyclohexyl-4-ethyl-2-imidazolin-5-one
The procedure described in Example 1, step G),
is followed; 1.50 g of the expected product are
obtained, after chromatography, by treating 1 g of the
product of step C) with 4-bromomethyl-2'-tert-
butoxycarbonylbiptlenyl.
- IR (chloroform):
17G0-1720 cm~l : imidazolinone and ester C=0
1635 cm~1 : C=N
E) (R,S)-2-n-Butyl-1-[(2'-carboxybiphenyl-4-yl)methyl]- -
4-cyclohexyl-4-ethyl-2-imidazolin-5-one
tri~luoroacetate
The expected product is obtained by treating
lS the compound obtained in the previous step by the
mathod described in Example 1, step H).
Yield : 85%.
M.p. = 159-161- C.
- NMR:
7.10-7.7Q ppm : m : 8 H : aromatic protons
4.75 ppm : s : 2 H : -N-CH2-C6H4-
2.60 ppm : t : 2 H : -CH2-~H2-CH2-CH3
0.90-1.90 ppm : m : 17 H : cyclohexyl + -CH2-CH3 +
-CH2-cH2-cH2-cH3
0.80 ppm : t : 3 H : -C~3
0.60 ppm : t : 3 H : -CH3
EXAMPLE 4
(R,S)-2-n-Butyl-4-cyclohexyl-4-methyl-1-[(2'-(tetra-
zol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5~one
A) 4-Methyl-2'-(tetrazol-5-yl)biphenyl
2'-Cyano-4-methylbiphenyl is prepared according
31
to European patent application 324 377.
2 g of this compound are placed in a round-
bottomed flask in the presence of 4 g of tributyltin
azide and 20 ml of xylene and the mixture is reflu~ed
S for 110 hours. After it has returned to RT, the
reaction medium is diluted with toluene and the
organic phase is then extracted 3 times with 50 ml of
1 N sodium hydroxide solution. The aqueous phases are
combined, washed with ether and then cooled in a bath
of iced water and acidified to pH 1-2 by the addition
of concentrated hydrochloric acid. The precipitate
formed is filtered off, washed with water and dried
under vacuum over phosphorus pentoxide to give 2.18 g
of the expected product.
M.p. = 146-148- C after recrystallization from
ethyl acetate.
B) 4-Methyl-2'-(triphenylmethyltetra~ol-5-yl)biphenyl
5.46 g of the compound obtained in step A,
6.9 g of trityl chloride, 100 ml of DCM and 4 ml o
triethylamine are mixed in a round-bottomed flask.
The medium is refluxed for 4 hours and then
evaporated. The residue is taken up in ethyl acetate
and washed with water, a 3% solution of potassium
hydrogensulfate, 1 N sodium hydroxide solution, water
and then a saturated solution of sodium chloride. It
is dried over sodium sulfate, filtered and avaporated
to give 11 g of the expected product.
M.p. = 161-164- C.
C) 4-Bromomethyl-2'-(triphenylmethyltetrazol-5-yl)
biphenyl
A mixture containing 11 g of the compound
prepared in step B, 140 ml of carbon tetrachloride,
32
4.12 g of NBS and 0.4 g of benzoyl peroxide is
refluxed for 3 hours. After it has returned to RT, it
is filtered and the filtrate is then evaporated. The
residue is taken up in 30 ml of isopropyl ether. The
precipitate ormed is filtered off and then dried
under vacuum,
The product obtained is used as such in the
next step.
D) ~R,S)-2-n-Butyl-4-cyclohexyl-4-msthyl-1-[(2'-~tri-
phenyl-methyltetrazol-5-yl)biphenyl-4-yl)me~hyl]-2-
imidazolin-5-one
394 mg of sodium hydride as an 80% dispersion
in oil are suspended in 100 ml of anhydrous DMF under
nitrogen, 1.71 g of 2-n-butyl-4-cyclohexyl-4-methyl-2-
imidazolin-5-one in 10 ml of anhydrous DMF, prepared
in Example 1, step E), are added gradually, with
stirring, and the mixture is stirred for 30 minutes.
4.86 g of the compound prepared in step C) are added
and the mixture is stirred for 3 hours at RT. It is
evaporated to dryness, the residue is then taken up in
60 ml of ethyl acetate and the medium is filtered and
evaporated to dryness. The oil obtained is chromato-
graphed on silica using an ethyl acetate/hexane
mixture (1/3; v/v) as the eluent. ~fter evaporation
of the solvents, 3.45 g of the expected product .~re
obtained in the form of a solid foam.
- NMR (CDC13):
0.9 ppm m : 3 H : -CH2-CH2-CH2-cH3
1-1.8 ppm : m : 18 H : 4-methyl + 4-cyclohexyl
CH2-CH2-CH2-CH3
2.35 ppm : distorted t : 2 H : CH2-CH2-CH2-CH3
4.5 pprn : s : 2 H : N-CH2-C6H4-
.
33
2~ 3'7
6.70-8 ppm : m : 23 H : aromatic protons
E) (R,S)-2-n-Butyl-4-cyclohexyl-4-methyl-1-[(2'-(tetra-
zol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one
3.38 g of the compound prepared in step D) are
S dissolved in a mixture of 40 ml of methanol and 20 ml
of THF. 3.5 ml of 4 N hydrochloric acid are added and
the mixture is stirred for 3 hours at RT. After
evaporation ~o dryness, the residue is taken up in lO
ml of 2 N sodium hydroxide solution and 10 ml of ether
and the mixture is then stirred until a solution is
formed. The aqueous phase is extracted twice with
ether. The a~ueous phase is acidified to pH 6 with
dilute hydrochloric acid and then extracted 3 times
with ethyl acetate and the extracts are dried over
sodium sulfate and evaporated to dryness to give 1.72
g of the expected product in the form of a white solid
~oam.
- NMR (CDCl3):
0.9 ppm : t : 3 ~ : C~2-CH2-CH2-CH3
0.95-2.7 ppm : m o 18 H : 4-cyclohaxyl ~ 4-methyl +
-CH2-CH2-C~2-cH3
2.1 ppm : t : 2 H : -CH2-CH2-CH2-CH3
4.4-4.7 ppm : AB system : 2 H : N-CH2-C6H4-
6.95-7.1 ppm : q : 4 H : aromatic protons
7.3-7.6 ppm : m : 3 H : aromatic protons
7.8 : d : 1 H : aromatic protons
EXAMPLE 5
2-n-Butyl-4-cyclohexyl-4-methyl-1-[(2'-(tetra-
zol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one,
levorotatory
A) 5-Methyl-5-phenylhydantoin
34
2~ 3~
30 g of acetophenone diluted in 250 ml of 95-
alcohol are added over 30 minutes to a mixture of
18.35 g of sodium cyanide and 125 g of ammonium
bicarbonate in 250 ml of water and the reaction medium
S is heated at 60-65- C for 22 hours, with stirring. It
is concentrated to half its volume under vacuum and
the solld which has precipitated is filtered off,
washed with watsr and ether and then dried under
vacuum to give 38 g of a white solid, which is
identified by IR.
M.p. = 190-192- C.
B) ~R,S)-2-Amino-2 phenylpropionic acid
20 g of the compound prepared in the previous
step are added to a mixture of 75 g of barium
hydroxide octahydrate and S00 ml of water and the
reaction madium is then heated at 160- C for S hours
in a steel tube. It is saturated with dry ice and the
precipitate is then filtered off. The filtrate is
concentrated under vacuum and the white solid formed
is taken up in acetone, filtered off, washed with
acetone and ether and then dried to give 15.3 g of the
expected acid.
M.p. = 260-265~ C (with decomposition).
C) EthyI ester of (R,S)-2-amino-2-phenylpropionic acid
Using a spatula, 24 g of the acid prepared in
the previous step are added to a solution of 80 g of
gaseous hydrochloric acid in 210 ml of absoluta
ethanol, with stirring, and the mixture is refluxed
for 6 snd a half hours. It is concentrated under
vacuum, the residue is taken up in 600 ml of ethyl
acetate and 100 ml of water, and 2 N sodium hydroxide
solution is added until the pH is 9. The organic
phase is decanted, washed with water and a saturated
solution of sodium chloride, dried over sodium sulfate
and then concentrated under vacuum to give 25.5 g of
the expected product in the form of an oil.
Identification by IR.
D) Dextrorotatory ethyl ester of 2-amino-2-phenylpro-
pionic acid
The enantiomers of the ester prepared in the
previous step are separated by the method described by
Y. Sugi and S. Mitsui in Bull. Chem. Soc. Japan, 1969,
42, 2984-2988. 19.8 g of (L)(+)-tartaric acid are
added to th~ 25.5 g of ester obtained in step C,
diluted in 210 ml of absolute ethanol. The mi~ture is
heated to 60- C to give a total solution, ~hich is
then left to stand at RT for 4 hours. The precipitate
formed is filtered off, then rinsed with twice 70 ml
of absolute alcohol and then redissolved in ~00 m:L of
alcohol at the boil and the solution is then left to
stand at RT for 72 hours. The acicular crystals
formed are filtered off, rinsed twice with 30 ml of
alcohol and then dried under vacuum to give 11.9 g of
the tartaric acid salt of the expected dextrorotatory
ester.
M.p. = 172-173- C.
[~]D = ~44-5 (C = 1, water).
The remaining alcoholic solu~ion is enriched
in the tartaric acid salt of the levorotatory ester.
6.1 g of the tartaric acid salt of the dextro-
rotatory ester are taken up in 30 ml of water and then
200 ml of ethyl acetate, and 5 N sodium hydroxide
solution is added until the pH is 9. The organic
phase is decanted, washed with water and a saturated
36
f
solution of sodium chloride, dried over sodium sulfate
and than concentrated under vacuum to give 3.33 g of
the expected product in the form of an oil.
[~]D = +24- (C = 2, ethanol). Identification
S by NMR.
E) Dextrorotatory ethyl ester of 2-amino-2-cyclohexyl-
propionic acid
3.30 g of the dextrorotatory ester obtain~d in
the previous step are diluted in 120 ml of acetic
acid; 1.5 g of platinum oxide are added and the
mixture is then hydrogenated at atmospheric pressure.
After hydrogenation for 40 hours, the reaction medium
is filtered and then concentrated under vacuum. The
residue is taken up in an ether/water mixture and 6 N
lS hydrochloric acid is added until the pH is 2. The
organic phase is separated from the agueous phase.
Ethyl acetate is added, followed by 5 N sodium
hydroxide solution until the pH is 9.5. The organic
phase is decanted, washed with water and a saturated
solution of sodium chloride, dried over sodium sulfate
and then concentrated under vacuum to give 3.10 g of
the expected product.
[~]D = +18- (C = 2, ethanol). Literature :
W~A. Bonner et al., J. Amer. Chem. Soc., 1956, 7~,
3218-3221.
Identification by NMR.
F) 2-n-Butyl-4-cyclohexyl-4-methyl-2-imidazolin-5-one,
levorotatory
A mixture containing 3 g of the dextrorotatory
ester obtained in the previous step, 4.7 g of ethyl
valerimidate and 8 drops of acetic acid in 15 ml of
xylene is brought to the reflux point, with stirring.
3~
After refluxing for 7 hours, the reaction
medium is concentrated under vacuum. The residue is
chromatographed on silica using a chloroform/methanol/
acetic acid mixture (95/9/3) as the eluen-t; the
S fractions containing the product are combined and
concentrated under vacuum. The residue is taken up in
an ethyl acetate/water mixture and the pH is brought
to 9 by the addition of 5 N sodium hydroxide solution.
The organic phase is decanted, washed wi-th water and a
saturated solution of sodium chloride, dried over
sodium sulfate and concentrated under vacuum to give
an oil, which changes to an amorphous solid.
m = 2.36 g.
~]D = -57-2- (G = 1, chloroform).
lS ~ IR (chloroform):
1720 cm~l : C=0
1640 cm~1 : C=N
The IR spectrum confirms the 5-one form of the
imidazolinone in solution.
G~ 2-n-Butyl-4-cyclohexyl-4-methyl-1-[(2l-(triphenyl-
methyl-tetrazol-5-yl)biphenyl-4-yl) methyl]-2-imida-
zoline-5-one, levorotatory
This compound is prepared from the product
prepared in step F by following the procedure
2S described in Example 4, step D.
Yield : 73%.
[~]D = -22.8- (C = 1, chloroform).
- NMR: superimposable on that of the compound of
Example 4, step D.
H) 2-n-Butyl-4-cyclohexyl-4-methyl-l-[(2'-(tetrazol-5-
yl) biphenyl-4-yl)methyl]-2-imidazolin-5-one, levo-
rotatory
This compound is prepared from the product
prepared in step G by following the procedure
described in Example 4, step E.
Yield : 85%.
S [~D = -25.9- (C - 1, methanol).
- NMR: superimposable on that of the compound of
Example 4, step E.
EXAMPLE 6
2-n-Butyl-4-cyclohexyl-4-methyl-1~[(2'-(tetra-
zol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one,
dextrorotatory
A) Levorotatory ethyl ester of 2-amino-2-phenylpropio-
nic acid
The alcoholic solution obtained in Example 5,
step D), i5 concentrated after separation of the
crystals of the tartaric acid salt of the dextrorota-
tory ethyl ester of 2.-amino-2-phenylpropionic aaid.
The solid residue is ~aken up in 150 ml of water and
600 ml of ethyl aceta~e and the pH is brought to g by
the addition of 5 N sodium hydroxide solution. The
organic phase is decanted, washed with water and a
saturated solution of sodium chloride, dried over
sodium sulfate and then concentrated under vacuum to
give 20.6 g of the ester enriched in the levorotatory
form.
15.9 g of (D)(-~-tartaric acid are added to
20.5 g of this ester diluted in 200 ml of absolute
ethanol, and a solution is formed at the boiling point
of the alcohol. After 5 hours at RT, the acicular
crystals formed are filtered off, washed twice with 50
ml of absolute alcohol and then dried under vacuum to
39
give 16.3 g or the tartaric acid salt of the expected
product.
M.p. = 172-173- C.
[a]~ = -45.2- (C = 1, water).
S 6 g of- the salt obtained are taken up in 50 ml
of water and 200 ml of ethyl acetate and the pH is
brought to 9.5 by the addition of 5 N sodium hydroxide
solution. The organic phase is decanted, washed with
water and a saturated solution of sodium chloride and
then dried over sodium sulfate and concentrated under
vacuum to give 3.31 g of the expected product in the
form of an oil, which is identified by NMR.
[a]D = -25.5- (C = 2, ethanol).
B~ Levorotatory ethyl ester o~ 2-amino-2-cyclohexyl-
propionic acid
The procedure of Example 5, step E), is
followed to give 3.20 g of the expected product from
3.30 g of the compound of step A.
[a]D = -19.2- (C = 1, ethanol).
20 C) 2-n-Butyl-4-cyclohexyl-4-methyl-2-imidazolin-5-one,
dextrorotatory
The procedure of Example 5, step F), is
followed.
t~]D = +56.9- (C = 1, chloroform).
The NMR and IR spectra are superimposable on
those of the levorotatory isomer prepared in Example
5, step F).
D) 2-n-Butyl-4-cyclohexyl-4-methyl-1-~(2'-(triphenyl-
methyl-tetrazol-5-yl)biphenyl-4-yl)methyl]-2-imida-
zolin-5-one, dextrorotatory
The procedure described in Example 5, step G),
is followed to give 2.3 g of the expected product in
the form of a white solid from 1.1 g of the. compound
prepared in step C.
[~]D = -23.8- (C = 1, methanol) and NMR
spectrum superimposable on that of the compound
S prepared in Example 4, step D.
E) 2-n-Butyl-4-cyclohexyl-4-methyl-1-[(2'-~tetrazol-5-
yl~ biphenyl-4-yl)methyl]-2-imidazolin-5-one, dextro-
rotatory
The procedure described in Example 5, step H,
is followed to give 1.1 g of the expected produc~ in
the form of a white solid from 2.15 g of the compound
prepared in step D.
t~]D = +27.1' (C = 1, methanol~.
The NMR spectrum is superimposable on that of
lS the compound prepared in Example 4.
~ ikewise, the following compounds according to
the invention were prepared according to the procedure
described in Example 3:
EXAMPLE 7
(R,S)-2-n-Butyl-1-[(2'-carboxybiphenyl-4-yl~-
methyll 4~cyclopropyl-4-methyl-2-imidazolin-5-one tri-
fluoroacetate
M.p. = 149-150J C.
- NMR:
7.05-7.80 ppm : m : 8 H : aromatic proton.s
4.70 ppm : s : 2 H N-CH2-C6H4-
2.45 ppm : t : 2 H : CH2-CH2-CH2-CH3
1.05-1.45 ppm : m + s : 8 H : -CH2-CH2-CH2-CH3 + CH3
in the 4 position +
cyclopropane CH
0.70 ppm : t : 3 H : CH3-(C~2)3-
41
~:?~3' 3 ~3
0.05-0.45 ppm : m : 4 H : 2 cyclopropane CH2
EXAMPLE 8
(R,S)-2-n-Butyl-1-[(2'-carboxybiphenyl-4-yl)-
S methyl]-4,4-dicyclopropyl-2-imidazolin-5-one trifluorQ-
acetate
M.p. = 132-134- C.
- Mass spectrum:
MH+ : 431
- NMR:
7.15-7.80 ppm : m : 8 H : aromatic protons
4.75 ppm : s : 2 ~ : N-CH2-C6H4-
2.50 ppm : t : 2 H : CH2-CH2-CH2-CH3
1.1-1.60 ppm : m : 6 H : CH2-CH2-CH2-CH3 ~ 2 cyc
propane CH
0.80 ppm : t : 3 H : (CH2)~-CH3
0.10-0.80 ppm : m : 8 H : 4 cyclopropane CH2
EXAMPL~ 9
(R,S)-2-n-Butyl-1-[(2'-carboxybiphenyl-4-yl)-
methyl]-4-cyclopentyl-4-methyl-2-imidazolin-5-one
trifluoroacetate
M.p. = 104-107- C.
- NMR:
7.20-7.80 ppm : m : 8 H : aromatic protons
4.85 ppm : AB system : 2 H : N-CH2-C6H4-
2.75 ppm : distorted t : 2 H : CH2-CH2-CH2-CH3
2.20-1.00 ppm : m + s : 16 H : cyclopentane + CH3 in
the 4 position + -C~2-
CH2-CH3
0.80 ppm : t : 3 H : CH3-(CH2)3-
.
42
EXAMPLE 10
(R,S)-2-n-Butyl-4-cyclopentyl-4-methyl-1-[(2'-
t~trazol-5-yl)-biphenyl-4-yl) methyl]-2-imidazolin-5-
one.
S This compound is prepared by following the
procedure described in Example 4.
M.p. = 78-80-C
- NMR : (CDC13)
7.05-7.8 ppm : m : 8H : aromatic protons
4.70 ppm : s : 2 H : -N-CH2-C6H4-
2.40 ppm : t : 2 H : -CH2-CH2-CH2-CH3
2.15-1.20 ppm : m : 13 H : -CH2-CH2-CH2-CH3 +
4-cyclopentyl
1.20 ppm : s . 3 H : C~13-4
0.95 ppm : t : 3 H : -CH2-CH2-CH2-C~3