Note: Descriptions are shown in the official language in which they were submitted.
WO91~15579 2 ~ 7 ~ ~ ~ 2 PCT/US91/023~
MUTAGENESIS TESTING USING TR~NSGENIC NON-HUMAN
ANIMALS CARRYING TEST DNA SEQUENCES
Cross Reference to Related A~plications
5This application is a continuation-in-part
application of U.S. Patent Application Serial No.
505,676, field April 5, l990 which is a continuation-
in-part of U.S. Patent Application Seriai No. 045,037,
filed May l, 1987 for "Mutagenesis Testina Using
Transgenic Non-Human Animals Carrying Test DNA
Seauences," the disclosures of which are specifically
incorporated herein by reference.
Technical Field
15This invention relates to transgenic animals
and to tests for monitoring mutagenic agents in live
animals. More specifically, this invention relates to
the creation of transgenic non-human animals carrying
test DNA se~uences and to methods for monitoring and
assessing the mutagenic potential of agents by
exposing the transgenic animal to one or more
suspected mutagens, and optionally recovering the test
DNA seauence, and examining the test DNA seauence for
mutations. Novel methods for increasing the
efficiency of test DNA seauence recovery and rapid
analysis of specific test DNA mutations are also
described.
Backaround
Various agents, such as radiation, ultraviolet
light, synthetic chemicals, natural substances, and
aberrations in genetic replication and repair can
produce mutations in DNA. The results of a
representative study indicate that as many as 60% of
the cancers that develop in women and as many as 40%
- - ; ~ - , : .
- ,. . .
- .. . , : -:
. : ,: ; ~ - . . - . .
:- : .: :
~: .
.
~ () 7 ~ 7 ~ ~ - 2 - PCT/US9l/023
of those that develop in men result from avoidable
exposure to mutagens from dietary intake. Vuoto et
al., Environ. Mutaaen, 7:577-598 (1985). Exposure to
environmental mutagens such as nitroaromatic compounds
found in automobile exhaust, chlorination by-products
used in drinking water, and acrylamide and
formaldehyde used extensively in industrial
laboratories is also of major concern. Quantitative
measurement of the effect of suspected mutagens is --
essential to control exposure to harmful agents.
Additionally, whenever a new chemical, drug, or food
additive, for example, is to be taken from the
laboratory to the marketplace, it must be tested for
its toxicity and cancer-causing potential. As a
result, significant effort has gone into the
development of assays that detect the ~utagenic
potential of various compounds.
Existing tests that assess the mutagenic --
potential of substances focus either on alterations of
DNA in cultured cells or bacteria or alterations in
the health of test animals. However, few tests that
monitor alterations in DNA actually expose live
animals to the agent to be tested. This is because it
is very difficult to rapidly monitor alterations in
the genetic code simultaneously in many different
organs. Tests to detect these mutations must be very
sensitive. They must be able to detect a single
mutation amongst millions of normal genetic units.
The difficulty of this task currently makes this
approach for live animal studies prohibitively
expensive as well as time intensive. Therefore, most
current live animal genotoxicity tests use disease
~ormation or large scale chromosomal alterations as an -
assay for gene alteration.
- .. , . . -
.
WO9t/t5579 2~`7 9 7 ~ 2
The problem of readily detecting small scale
DNA alterations that are caused by potential mutagenic
agents has generally been approached by performing
studies on procaryotic or eukaryotic cells in culture
(in vitro tests). The well-known Ames' test uses a
special strain of bacteria to detect these mutations.
Ames, et al., An Improved Bacterial Test System for
the Detection and Classification of Mutagens and
Carcinogens, Proc. Nat. Acad. Sci., 70:782-86 (1973).
This test and many analogues that use other types of
bacterial or animal cells permit the rapid screening
of very large numbers of cells for the appearance of
an altered phenotype. The appearance of this altered
phenotypic trait reflects the occurrence of a mutation
within the test gene. These tests are, however,
inse~sitive to or nonspecific for many mutagens that
result from metabolic activation of the agent being
screened. Although attempts have been made to
increase their sensitivity and specificity by
activation of such metabolites with liver and other
extracts it is noted that, for instance, the --
metabolites produced by these extracts are often not
present at the same concentrations as in the live
tissues of an animal. Metabolites that are only
produced in other organs are not detected at all.
Eukaryotic cell lines have also been used to
detect mutations. E.g., Glazer et al., Detection and
Analysis of W -induced Mutations in Mammalian ~ell DNA
using Lambda Phage Shuttle Vector, Proc. Natl. Acad.
Sci. USA, 83:1041-1044 (1986). In this test a target
test gene, the amber suppressor tyrosine tRNA gene of
E. coli in a bacteriophage shuttle vector, was
integrated into a genomic host mammalian cell line by
DNA transfection of cultured cells in vitro. After
exposing the host cell line to putative mutagenic
-:- . - : - . . -
2 ~ 7 ~ 7 9 ~ PcT/US9l/023~
agents, test genes were reisolated, propagated in
bacteria, and analyzed for mutations. Because the
host is only a mammalian cell line and not a live
animal, the test is incapable of accurately monitoring
mutagenic metabolites of the agent being tested that
are only produced at the appropriate concentrations by
differentiated cells or the tissue of live animals.
A two year study by the NIH concluded that
data obtained from four different prokaryotic and
eukaryotic in vitro assays had only a 60% concordance
with whole animal carcinogenicity studies. Tennant et
al., Science, 236:933-941 (1987). The study suggests
that the high rate of error may result from potential
variation in genetic susceptibility between in vitro
systems and whole animals. For example, m~tabolites,
frequently involved in activation of promutagens, are
not present in in vitro systems, allowing mutagenic
potential to go undetected. In addition, differences
in DNA repair mechanisms between prokaryotes and
eukaryotes may account for some discrepancies in -
results.
Test genes and large scale screening assays
used for in vitro assays are not available for live
animal studies. Short of relying on long term animal
studies that detect phenotypic changes that require a
long time to be identifiable, such as tumors, organ - -
failure, coat color, etc., current tests do not
provide a means for monitoring organ-specific
mutations o~ DNA. Hence, there exists a need for a
system that places a test DNA sequence within an
animal and is subsequently assayed on a large scale
for mutations. There also exists a need for methods --
that detect mutations caused by chemical metabolites
of the agent being tested. To be most effective the
system needs to be capable of monitoring genetic
, .. .... .. ... .. , . . . ~ ... . . . . .................. - . -
- - . -~
,
' :~ , . ~ ~ ' .' ,. '
WO91/l5579 2 ~ 7 9 7 ~ 2 PCT/US9l/023~
changes in as many tissues of an animal and as easily,
rapidly, and inexpensively as possible.
The present invention, providing novel
transgenic non-human mammals and methods utilizing
such mammals for mutagenesis testing, satisfies these
needs. More specifically, the present invention
provides a sensitive screen for the mutagenicity of
suspected agents and permits the monitoring of the
mutagenic effects of such agents and the mutagenic
effects of the metabolites of such agents.
Additionally, the invention can permit the
identification of ~he nature of the mutation, e.g.,
DNA transition, transversion, deletion, or a point or
frameshift mutation. Further, the methods of the
invention offer the significant advantage of being
rapid to perform, thus permitting the identification
of potential mutagens appreciably before other tests
can be completed, and is inexpensive relative to other
whole animal tests. And, the present invention
substantially reduces the number of animals which must
be used for mutagenesis testing.
Summary of the Invention
The present invention contemplates a method
for assaying the mutagenic potential of an agent. The
method comprises administering a predetermined amount
of the agent to an animal containing cells having a
genome characterized by the presence of an excisably-
integrated lambda phage. The phage contains a target
gene operatively linked to prokaryotic expression
signals. The exposed animal is typically maintained
ali~e for a predetermined period of time, usually from
hours to months, and preferably from days to weeks,
such as about 4 days to two weeks. A predetermined
amount of the excisably-integrated lambda phage is
wO91/15~79 2 ~ ~ 9 7 ~ 2 PCT/US91/023~
- 6 -
then rescued from cells harvested from the maintained
animal. The rescued lambda phage is then introduced
into and expressed in a restriction system deficient
microorganism.
In a preferred embodiment, the animal is a
transgenic animal, such as a transgenic rodent,
preferably a transgenic rat or mouse.
In another preferred embodiment, the cells are
human cells present in a SCID mouse.
As described below, a preferred method has
been developed which uses a target gene system
comprised of a set of two genes, a test gene and a
reporter gene, incorporated into animals or animal
cells to screen for compounds having mutagenic,
carcinogenic or teratogenic activity. Exposure of the
animals or animal cells to compounds having any of -
these activities causes mutations resulting in
alterations in expression of the reporter gene. Both
the target gene and the reporter gene constructs are
operatively linked to a prokaryotic promoter.
This method has several advantages over the
prior art methods of screening for compounds having ~ -
mutagenic or teratogenic activity. The most
significant advantage is the ease in detection and
decrease in number of ~alse positives. Although the
mutation of genes encoding reporter proteins has
previously been used to assay for mutagenic activity,
the mutational event resulted in the protein not being
expressed. Detecting a single cell, or even a few
cells, not expressing a protein, while surrounded by
cells which express the protein, is difficult,
tedious, and subject to a high percentage of error.
In contrast, in the present method, the mutational
event ultimately results in the expression of a
reporter molecule which would otherwise not be
- . '
WO91/15579 PCTIUS91/023~
~ ~ 7 -2~7~2
expressed, and which is readily detected.
AS used herein, unless specifically stated
otherwise, "animal cells" will be used to include
cells in cell cultures, embryos, and differentiated
animals. As also used herein, "mutagen" will be used
to include toxins, carcinogens, teratogens, and other
agents which alter DNA or RNA sequence or expression,
unless stated otherwise.
The selection of reporter genes is based on
the following criteria: (i) the reporter gene product
cannot be detrimental or lethal to the transformed
cells, (ii) the gene product should provide a simple
and sensitive detection system for its quantitation,
and (ii) non-transformed cells should have a low
constitutive background of gene products or activities
that will be assayed. Reporter genes which encode
enzymes; antigens or other biologically active
proteins which can be monitored easily by biochemical
techniques are preferred. These include beta-
galactosidase (Norton, P. A. and Coffin, J. M., Mol.
C~L~ }ol, 5:281-290 (1985), peroxidase and
luciferase (de Wet, J. R. et al., Mol. Cell. Biol,
7:725-737 (1987).
The test gene is selected from the group of
sequences which encode regulatory molecules that bind -
to a sequence controlling reporter gene expression.
These can be repressors or other regulatory molecules,
including anti-sense RNA. In the most preferred
embodiment, the lacI repressor gene is used as a
mutagenssis target. The inactivation of the repressor
gene by a mutagenic even causes the transcription and
translation of a defective repressor protein that is
no longer able to repress expression of the reporter
gene the lacz gene encoding beta-galactosidase.
Alteration of the operator region for the reporter
- , , .
.
. - . . : . :
WO91/15579 - 8 - PCT/US91/023
gene in a manner that prevents binding of the
repressor protein produces the same effect.
Derepression of the reporter gene can then be
monitored by assaying for defined functions of the
gene product.
The bacterial lac operator-repressor system is
preferred because it is one of the most basic and
thoroughly studies examples of a protein-nucleic acid
` interaction that regulates transcription of a gene, as
described by Coulondre and Miller, Mol. Biol., 117:577
(1977) and Niller, Ann. Rev. Genet., 17:215 (1983).
This bacterial regulatory system has been transfected
into mammalian cells and expression detected by
addition of an inducer, isopropyl beta-D
thiogalactoside tIPTG), as reported by Hu and
Davidson, Cell, 48:555 (1987), and Brown, et al.,
Cell, 4~:603 (1987). An important difference between
previous uses of the lac operator-repressor system and
the present method is that mutation rather than
induction is used to derepress the reporter genes to
express protein whose function is solely to serve as
an indicator. Another difference is that, in the
preferred embodiment, the target gene system is - -
excisable as an infectious lambda phage.
For the repressor protein to control the
expression of a reporter gene, the operator se~uence
has to be built into the reporter gene at the location
between the transcription initiation site and the
initiation codon ATG. Either an original lac operator
sequence (5'-GGAATTGTGAGCGGATAACAATCC-3'), or a mutant
lac operator (lacIq), for example, a sequence which
binds repressor eight time tighter (5'-
ATTGTGAGCGCTCACAAT-3'~, can be used in vector
construction. -~
.
. .. . ., . .. ~ . . . . . .. . .. . . ... . . . .
WO91/15579 ~ ~ 7 ~ ~ ~ 2 PCT/US9l/023~
g
Detailed DescriPtion of the Invention
The present invention contemplates engineered
animal cells, animal embryos and differentiated
animals having a genome characterized by the presence
of an excisably-integrated lambda phage containing a
target gene system. The target gene system comprises
a test gene (transcribable, and preferably
translatable DNA sequence). Preferably, the test gene
is operatively linked to prokaryotic expression
signals, such as a promoter, ribosome binding site,
stop codon, and the like.
An rDNA of this invention (subject rDNA)
contains a target gene system operatively link for
prokaryotic expression to a lambda phage. A target
gene system typically is operatively linked to a
lambda phage and can be comprised of any of a variety -
of test genes whose transcription results in a
detectable phenotype or genotype where a mutation in
the nucleotide sequence of a test gene measurably
alters the detectable phenotype or genotype. Typical
test genes include genes that confer drug resistance
or other selective advantage, or genes whose
expression alters the expression of a second reporter
gene. Exemplary drug resistance genes confer
resistance to ampicillin, kanamycin, chloramphenicol
and the like.
A preferred test gene is a repressor or
activator gene product whose expression directly
alters the expression of a reporter gene. Exemplary
is the repressor protein encoded by the lacI gene, and
genetic variants of the lacI gene that function to
block transcription of the beta-galatosidase gene
(lacZ) by binding to the operator region of the lacZ
gene's expression signals. In this system, lacZ is a
reporter gene. A preferred lacI gene is the lacIq
.. . ........... , . . - . . . . .
: ~... .-: . , . ~ . , : -
WO91/15~79 PCT/US91/023~
2a~979'~ -lo- ~ -
variant that expresses eight- to ten-fold elevated
levels of repressor protein and more tightly represses
expression of the lac~ gene.
In preferred embodiments, the test gene of the
target gene system is operatively linked to a reporter
gene.
A reporter gene provides a means for detecting
mutations in the test gene. Typical reporter genes
are expressed when the test gene is not mutated, and
are not expressed when the test gene is expressed.
Thus a reporter gene is a gene that encodes a
detectable phenotype or genotype and whose expression
is under the control of the test gene.
Genes that encode detectable phenotypes
include drug resistance markers, enzymes whose
activity produces a detectable reaction product and
the like. A preferred reporter gene is the beta-
galacto~idase gene (lacZ).
A preferred lacZ gene is one that utilizes
alpha complementation, as described herein, whereby
functional lacZ activity requires the association of
the alpha portion of the lacZ gene product with the
complementary portion of the lacZ referred to as the
lacZ Ml5 gene product.
The test gene is operatively linked to
prokaryotic expression signals to facilitate the rapid
detection of mutations by the present inv0r.tion. Upon
excision rescue of a target gene system, the test gene
system is introduced into a prokaryotic expression
system, such as a bacterial cell lawn, so that
dilutions of the test genes can be expressed and
thereby observed (reported) to quantify the extent of
test gene mutation.
Insofar as the expression of a test gene is
measured in a prokaryotic expression system such as a
, - . ,, ~ . : .. :. . , : . - - . . .
WO91/tS579 PCT/US91/023~
~ 2~7~2
bacterial cell, it is understood that the mutations
can occur either in the structural portion~ of a test
gene or in the expression control signals such as the
prokaryotic promoter for expressing the test gene.
Thus in preferred embodiments the test gene comprises
a lacI or lacIq gene and includes a lacI promoter
region.
A lambda phage of this invention comprises a
target gene system that is excisably-integrated into
the genome of an animal cell or embryo. By excisably-
integrated is meant that the lambda phage comprises
excision elements operatively linked to the genome
that provide a means to conveniently remove the test
gene system from the animal, cell or embryo genome
subjected to mutagenesis conditions for the purpose of
assessing the possible occurrence of mutation.
Exemplary excision elements are nucleotide
sequences flanking the target gene, and if present the
reporter gene, and other elements of the target gene
system, that allow site-specific excision out of the
genome to which the target gene system is operatively
linked (integrated). Preferred nucleotide sequences
are the cos sites, flp recombinase recognition sites,
or loxP sites recognized by the Cre protein, all of
whom are described more fully herein.
Preferred are cos site excision elements
because of the convenience and the efficiency of
excision of the genes contained between the two cos
site nucleotide sequences when utilizing lambda
bacteriophage in vitro packaging extracts as described
herein.
The excision elements of a target gene system
confer the ability to readily recover the target gene
system from the mutagen exposure conditions to the
prokaryotic expression medium in which the reporter
.: - .-. : ~ .: ~ , , ,, -
WO9l/15579 PCT/US91/023
gene2i~s me~sured. - 12 -
An exemplary and preferred test gene system is
the lambda LIZ alpha vector described herein in which
a lacIq test gene is operatively linked to the alpha-
complementation-based lacZ alpha gene, where both test
and reporter genes are under the control of
prokaryotic expression signals, namely, lacI promoter
and lacZ promoter/operator sequences.
This preferred system further contains
nucleotide sequenc~s operatively linked to the test
gene that define a prokaryotic origin of replication,
a selectable marker (ampR) and a filamentous phage
origin of replication such that the test gene can
readily be transformed into a "fl-type" nucleic acid
sequencing vector for rapid determination of the
nature of the mutation in the test gene. This latter
feature is provided according to the teachings of
Short et al., Nucl. Acids Res., 16:7583-7600 (1988),
where the terminator and initiator domains of the fl
intergenic region are separated and flank the test -
gene sequences of this invention to be recovered and
sequenced.
A promoter is a sequence of nucleotides that
forms an element of a structural gene transcriptional ~
unit which controls the gene's expression by providing ~-
a site for RNA polymerase binding resulting in the
initiation of the process of transcription whereby a
gene is transcribed to ~orm a messenger ribonucleic
acid (mRNA3 molecule.
An operator is a sequence of nucleotides that
forms a site for specific repressor binding. Thus,
Gperators are specific for a particular repressor.
A repressor binding site is considered
specific if the equilibrium binding constant for
repressor binding to the operator is greater than 10-9
- : .~...... ~ . : : ,
WO91/15579 - 13 2 ~ ~ 9 ~ ~ 2 PCT/US91/023~
molar (M), preferably greater than 101 M, and more
preferably greater than 10 1l M. The equilibrium
binding constant for a repressor binding to an
operator can readily be measured by well known
equilibrium dialysis methods, or in a nitrocellulose
filter binding assay where repressor is immobilized on
nitrocellulose and 32P-labeled operator-containing DNA
segment is presented in solution for ~inding to the
immobilized repressor. See, Miller "Experiments in
Molecular Genetics", p367-370, Cold Spring Harbor
Laboratory, New York, 1972.
The operator for the lac repressor has been
well characterized and is used as exemplary herein.
See Miller et al., in "The Operon", Cold Spring Harbor
Laboratory, New York (1980), for a detailed study.
Alternative nucleotide sequences have been described
for a lac repressor operator that specifically binds
to repressor. See, for example, the description of
numerous lac operator variants and the methods for
characterizing their repressor-binding activity
reported by Sartorius et al., EMBO J., 8:1265-1270
(198q); and Sadler et al., Proc. Natl. Acad. Sci. USA,
80:6785-6789 (1983). Any nucleotide sequence that
binds lac repressor specifically can be used in the
present invention, although wild type and optimized
operators are preferred and used as exemplary herein.
The two optimized operators derived from the lac
operon include the nucleotide sequences shown in SEQ.
ID. NO. 1 and NO. 2 as follows:
3~ (SEQ. ID NO. 1) 5'-TGT GGA ATT GTG AGC GCT
CAC AAT TCC ACA-3'
(SEQ. ID NO. 2) 5'-ATT G'G AGC GCT CAC AAT-3'
Operators function to control the promoter for
a structural gene by a variety of mechanisms. The
operator can be positioned within a promoter such that
, . ,: ,,
: : .
~ WO91/15579 PCT/US9t/023~
2~3rl979'~ - 14 - ~
the binding of the repressor covers the promoter's
binding site for RNA polymerase, thereby precluding
access of the RNA polymerase to the promoter binding
site. Alternatively, the operator can be positioned
downstream from the promoter binding site, thereby
blocking the movement of RNA polymerase down through
the transcriptional unit.
Multiple operators can be positioned on a rDNA
molecule to bind more than one repressor. The
advantage of multiple operators is several fold.
First, tighter blockage of RNA polymerase binding or
translocation down the gene can be effected. Second, ` -
when spaced apart by at least about 70 nucleotides and
typically no more than about 1000 nucleotides, and
preferably spaced by about 200 to 500 nucleotides, a
loop can be formed in the nucleic acid by the
interaction between a repressor protein bound to the
two operator sites. The loop structure formed
provides strong inhibition of RNA polymerase
interaction with the promoter, if the promoter is -
present in the loop, and provides inhibition of
translocation of RNA polymerase down the
transcriptional unit if the loop is located downstream
fxom the promoter.
In preferred embodiments, a vector
contemplated by the present invention includes a
procaryotic replicon, i.e., a DNA sequence having the
ability to direct autonomous replication and
maintenance of the reco~binant DNA molecule
extrachromosomally in a procaryotic host cell, such as
a bacterial host cell, transformed therewith. Such
replicons are well known in the art and include OriC
as described herein. In addition, those embodiments
that include a procaryotic replicon may also include a
gene whose expression confers a selective advantage
:
:, -
, , ' ': ~ : '. `, '' .- . : .
WO91/15579 PCT/US91/023
- 15 _ 2'~79 ~9 2
such as amino acid nutrient dependency or drug
resistance to a bacterial host transformed therewith
as is well known, in order to allow selection of
transformed clones. Typical bacterial drug resistance
genes are those that confer resistance to ampicillin
as used herein, tetracycline, kanamycin, and the like.
Those vectors that include a procaryotic
replicon may also include a procaryotic promoter
capable of directing the expression (transcription and
translation) of the gene transformed therewith. A
promoter is an expression control element formed by a
DNA sequence that permits binding of RN~ polymerase
and transcription to occur. Promoter seguences
compatible with bacterial hosts are typically provided
in plasmid vectors containing convenient restriction
sites for insertion of a DNA segment of the present
invention. Bacterial expression systems, and choice
and use of vectors in those systems is described in
detail in "Gene Expression Technology", Meth.
Enzvmol., Vol 185, Goeddel, Ed., Academic Press, NY
(1990). Typical of such vector plasmids are pUC8,
pUC9, pBR322 and pBR329 available from Bio-Rad
Laboratories, (Richmond, CA) and pPL and pKK233-2,
available from Pharmacia, (Piscataway, NJ), or Clone
Tech (Palo Alto, Ca). ~ -
Transqenic Animals and Their Use
In one embodiment, the present invention
provides novel transgenic non-human animals and
methods for monitoring the mutagenic effects of
potential mutagenic agents. In accordance with this
invention, at least one copy of at least one target
test DNA sequence is introduced into cells of a non-
- human mammal thereafter bred to produce test animals.
Preferably, substantially all of the cells will
. :
WO91/15579 ~ PCT/US91/023~
2~ 16 - ~
contain the test DNA sequence. The test transgenic
animal is then exposed to an agent suspected to be
mutagenic and the test DNA sequence may be
subse~uently recovered from individual tissues of the
transgenic animal. The test DNA sequence may be ~--
transferred into a microorganism, although such
recovery and transfer is not requisite, and assayed
for mutations, allowing rapid examination of multiple
tissue specific genetic mutations. Other methods to ~-
monitor mutations in the test DNA need not rely on
rescue and involve either direct examination of the
test DNA in situ, PCR amplification of the test DNA,
examination of RNA transcription products of the test
DNA or protein translation products of said RNA, or
effects of said proteins or substrates for said
proteins. `-
Theoretically, any animal suitable for
mutagenic testing may be used as the starting
organism. In order to allow for ubiquitous insertion
of the novel test sequence, single cell animal embryos :
are harvested, although there may be other cells -~
facilitating the uptake and ultimate ubiquitous
presence of the marker DNA in cells of a
differentiated animal. ~ `
In accordance with the invention, any number
or variety of sequences coding for a phenotype or
genotype that is detectable upon mutation may be used
for introduction into the transgenic non-human mammals
of the invention. Vectors capable of facilitating the
recovery of the test DNA sequence from the host mammal
cells, and capable of allowing replication and
expression of the sequence in a bacterial host, are
preferably used as carriers for the target test DNA
sequence. Accordingly, the construct for such a
vector and insert preferably should contain regions
. . , ,,~
., :. : - ~ - -:
. , ~ ,
:
WO9l/15579 PCT/US91/02364
~ - 17 - 2$797Q2
for excision from the mammal host genome, and regions
that allow replication in a bacterial host cell, as
well as regions that permit expression and assay of
the test DNA sequence. If integration into the host
genome is not required, desired regions that allow for
replication of the test DNA sequence in the animal
host cells should be present. Elbrecht et al.,
Episomal Maintenance of a Bovine Papilloma Virus
Vector in Transgenic Mice, Mol. Cell. Biol., 7:1276-
1279 (1987~.
Further, in accordance with the invention, thetest DNA sequence is introduced into the host mammal,
preferably (but not necessarily) at the single-cell
embryo stage, so as to provide the stable presence of
the test sequence throughout cells of the
differentiated animal. The use of chimeric animals is
also contemplated herein. Typically, this involves
the integration of the test DNA sequence into the
mammal host genome, although methods that allow the
test sequence to be stably and heritably present
through the use of autonomously replicating vectors
will also be useful. Elbrecht et al., Episomal
Maintenance of a Bovine Papilloma Virus Vector in
Transgenic Mice. Mol. Cell. Biol., 7:1276-1279
(1987). At the c~llular level, this may be
accomplished using the techniques of microinjection,
electroporation, dielectrophoresis or various
chemically mediated transformation techniques, all of
which are well known in the art. At the
differentiated tissue level, other techniques may be
necessary.
Following the introduction of the test DNA
sequence and integration into the genome or cell, the
transgenic cell or cells must be allowed to
differentiate into a whole organism. This may be
'' . '
, . -
, . , ,~,
.:. . : - , . . . ~ ,
.
W091/15579 ~ 7 ~ 7 9 ~ - 18 - PCT/US91/023
accomplished, for example, by embryo implantation into
pseudopregnant females, or by other techniques
allowing maturation of trans~enic embryos. Once such
maturation and differentiation has occurred, the
animal is assayed for the presence of the test DNA
sequence. Typically this involves removing small
portions of tissue from the animal and using standard
DNA hybridization assay techniques to detect the
presence of the test DNA sequence.
Transgenic animals carrying the test DNA
sequence are thereafter bred and offspring carrying
the test DNA sequence my be selected for mutagenesis
testing. In accordance with the invention, the :
selected transgenic mammals are exposed to agents or
substances in question under appropriate conditions.
Such conditions will depend, for example, on the
nature of the agent or substance, the purpose of the
mutagenesis study and the type of data desired.
After exposure of test transgenic animals to
the agent to be tested under the desired conditions,
desired tissue may be removed from the test animal.
Because in the preferred embodiment the test DNA
se~uence is present in essentially all tissues, the
tissue type tested is not limited by the process of
insertion of the test DNA sequence. Any desired
tissue may be removed and assayed at the DNA, RNA,
protein or substrate/product level, by various methods
including, but not limited to, in situ hybridization
to the DNA or RNA, PCR, protein or enzymatic assays
(PCR Protocols,_A Guide to Methods and APplications,
eds. Innis, M. et al., Academic Press, Inc., l990;
Maniatis et al., Molecular Cloninq, A Laboratory
Manual, Cold Spring Harbor, New York 1982).
Alternatively, genomic DNA may be purified
from the tissue. The target test DNA sequence which
, ~ .
: ~ .. . ,, - .: , . . .
. - : :~ :
: ~ :~ , -
;'
~O~1/15579 PCT/US91/023~
19 - 2a79~
is integrated may then be rescued from the total
genomic DNA of the host. This may be accomplished by
excising it from the host genome or by suitable
procedures allowing separation by size, weight or
charge density. The method of rescue is dependent
upon whether test DNA sequence is inserted into the
genome, and whether flanking regions allow for
excision, or whether the test DNA sequence is part of
a replicating element allowing for separation
techniques. -
The rescued test DNA sequences may then be
transferred into and expressed by microorganisms
suitable for large scale screening techniques. In a
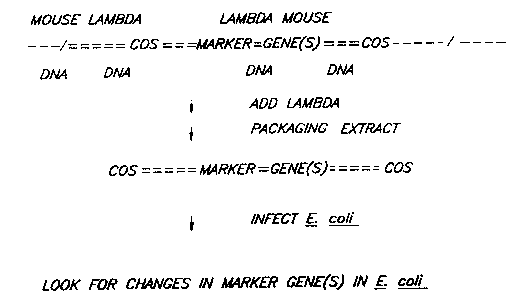
preferred embodiment, this involves excising the test
DNA sequence vector from the genomic DNA by packaging
the test DNA sequence with bacteriophage packaging
techniques. This may require ligating the test DNA
sequence into an appropriate vector or merely involve
direct transformation into a microorganism.
Microorganisms containing the test DNA
sequence vector are thereafter grown on indicator
plates or in selective media. Those organisms have a
phenot~pe indacating mutation of the test DNA sequence
are considered to contain a mutated test DNA sequence.
The ratio of those organisms expressing mutated
phenotype of test sequences to the total number of
organisms containing the test DNA sequence is a
measure of the mutagenicity of the agent and
metabolites of it present in the tested tissue.
Bacteriophaqe packaging techniques involve the
use of bacteriophage-infected host cell extracts to
supply the mixture of proteins and precursors required
for encapsidating the bacteriophage DNA from exogenous
sources. We have recently discovered that the rescue
efficiency of the test DNA sequence can be
-, , - , ~ , , , . - . -
-: : -
-. . .
WO91/15579 ~ PCT/US91/023~
2~7~ 20 - ~
significantly increased by eliminating the restriction
systems in the strain of host microorganism used both
for preparing the packaging extracts as well as those
microorganisms used for plating to detect mutagenesis.
Additionally, other recovery systems, e.g., DNA
transformation of isolated genomic DNA, would be -:
improved by removal of such restriction systems or
activities.
By removing these restriction systems which
recognize and deactivate foreign DNA, rescue
efficiencies may be increased up to at least 12,000
pfu~g genomic DNA. These rescue efficiencies enable
several million target genes from each tissue be
analyzed, generating a large number of data points and
resulting in a significant reduction in the numbers of ~ -
animals required for mutagenesis testing with greater
statistical significance.
Accordingly, the integrated target test DNA
sequence is, preferably, rescued from the total
genomic DNA of the host mammal using a lambda
packaging extract deficient in restriction systems
which recognize and deactivate foreign DNA. The
rescued test DNA sequences may then be transferred
into and expressed by restriction system deficient ;
microorganisms.
- Alternatively, a shuttle vector system can be ~ -
`constructed which provides rapid analysis of test DNA
sequence. The test DNA sequence may be contained
within a system which allows excision and
recircularization of the test DNA sequence, which
system is contained by a bacteriophage genome.
Following rescue of the bacteriophage genome
containing test DNA sequence using packaging extracts,
t~e test DNA may be further excised from the
bacteriophage genome and recircularized to provide for
: , : . ............. i ~ - - .
.: . . . , :
:
WO91/15579 2 ~ 7 ~ 7 ~ 2 PCT/US9t/023~
21
rapid mutation analysis.
Further, the present invention ~ontemplates
the performance of mutagenesis testing by examining
the phenotypes of cells containing the test DNA
sequence without recovery of the test DNA sequence
from the cell. This may be accomplished by the
sectioning of tissues of the transgenic mammal of the
invention, after exposure to a potential mutagenic
agent, and assaying the genotype or phenotype of the
test DNA sequence by in situ hybridization or, e.g.,
by staining of the tissue sections.
The present invention has application in the
genetic transformation of multicellular eukaryotic
organisms which undergo syngamy, i.e., sexual
1~ reproduction by union of gamate cells. Preferred
organisms include non-human mammals, bixds, fish,
gymnosperms and angiosperms.
Thus, the present invention contemplates a
non-human mammal containing a modified lambda
bacteriophage (rDNA) of the present invention
excisably-integrated in the genome of the mammal's
somatic and germ cells, i.e., a transgenic mammal.
Mammals containing a rDNA of the present invention are
typically prepared using the standard transgenic
technology described in Hogan et al., Manipulatina the
Mouse Embrvo: A LaboratorY Manual, Cold Sprin~ Harbor,
NY (1987); and Palmiter et al., Ann. Rev. Genet.,
20:465-499 (1986); which methods are described further
herein. Production of transgenic mammals is also
possible using the homologous recombination transgenic
systems described by Capecchi, Science, 244:Z88-292
(1989). Preparation of transgenic mammals has also
been described in U.S. Patent No. 4,736,866, No.
4,870,009, No. 4,873,191 and No. 4,873,316.
... .. -. . - . . . : . .
- . -, . . .~ , . , . ~ .-. .
WO91/15579 PCT/US91/023
- 22 -
2 ~ 7~n~e technique for transgenically altering a
mammal is to microinject a rDNA into the male -
pronucleus of the fertilized mammalian egg to cause
one or more copies of the rDNA to be retained in the
cells of the developing mammal. Usually up to 40
percent of the mammals developing from the injected
eggs contain at least 1 copy of the rDNA in their
tissues. These transgenic mammals usually transmit
the gene through the germ line to the next generation.
The progeny of the transgenically manipulated embryos
may be tested for the presence of the construct by
Southern blot analysis of a segment of tissue.
Typically, a small part of the tail is used for this
purpose. The stable integration of the rDNA into the
genome of the transgenic embryos allows permanent
transgenic mammal lines carrying the rDNA to be
established.
Alternative methods for producing a non-human
mammal containing a rDNA of the present invention-
include infection of fertilized eggs, embryo-derived
stem cells, totipotent embryonal carcinoma (Ec) cells,
or early cleavage embryos with viral expression
vectors containing the rDNA. See fox example,
Palmiter et al., Ann. Rev. Genet., 20:465-499 (1986)
and Capecchi, Science, 244:12B8-1292 (1989).
A transgenic mammal can be any species of
mammal, including agriculturally significant species,
such as sheep, cow, lamb, horse and the like.
Preferred are animals significant for scientific
purposes, including but not limited to rabbits,
primates and rodents, such as mice, rats and the like.
A transgenic mammal is not human.
Methods of Genetically Proarammina a Cell Within an
oraanism With A Target Gene System
... , , . - : ,
- , -
.. .. : .
WO9t/15579 PCT/US91/023~
~ - 23 2~7~2
The present invention also contemplates a
method of introducing a target gene system into a
cell, i.e., genetically programming a cell within an
organism by introducing a modified lambda genome
containing a target gene system of the present
invention into the genome of a zygote to produce a
genetically altered zygote, or into the genome of
individual somatic cells in the organism. The
genetically altered zygote is then maintained under
appropriate biological conditions for a time period
equal to a gestation period or a substantial portion
of a gestation period that is sufficient for the
genetically altered zygote to develop into a
transgenic organism containing at least l copy of the
rDNA.
The term "genetically programming" as used
herein means to permanently alter the DNA content of a
cell within an organism such as a mammal so that a
prokaryotic target gene system has been introduced
into the genome of the cells of the organism.
Any multicellular eukaryotic organism which
undergoes sexual reproduction by the union of gamete
cells may be genetically programmed using an rDNA of
the present in~ention. Examples of such multicellular
eukaryotic organisms include amphibians, reptiles,
birds, mammals, bony fishes, cartilaginous fishes,
cyclostomes, arthropods, insects, mollusks,
thallaphytes, embryophytes including gymnosperms and
angiosperms. In preferred embodiments, the
multicellular eukaryotic organism is a mammal, bird,
fish, gymnosperm or an angiosperm.
A transgenic organism is an organism that has
been transformed by the introduction of a recombinant
nucleic acid molecule into its genome. Typically, the
recombinant nucleic acid molecule will be present in
-
-- . . .
.~ . . .. . ... . . . .
W091/15~79 PCT/US91/023~
2~7~ 24 - ~
all of the germ cells and somatic cells of the
transgenic organism. Examples of transgenic organisms
include transgenic mammals, transgenic fish,
transgenic mice, transgenic rats and transgenic plants
including monocots and dicots. See for example,
Gasser et al., Science, 244:1293-1299 (1989); European
Patent Application No. 0257472 filed August 13, 1987
by De La Pena et al.; PCT Pu~. No. WO 88/02405 filed
October 1, 1987 by Trulson et al.; PCT Pub. No. WO
87/00551 filed July 16, 1986 by Verma, and PCT Pub.
No. WO 88/09374 filed May 20, 1988 by Topfer et al.
Methods for producing transgenic organisms
containing a rDNA of the present invention include
standard transgenic technology; infection of the
zygote or organism by viruses including retroviruses;
infection of a tissue with viruses and then
reintroducing the tissue into an animal; and
introduction of a rDNA into an embryonic stem cell of
a mammal followed by appropriate manipulation of the
embryonic stem cell to produce a transgenic animal.
See for example, Wagner, et al., U.S. Patent No.
4,873,191 ~Oct. 10 l989!; Rogers, et al., Meth. in
Enzymol., 153:253-277 (1987); Verma et al., PCT
Publication No. WO87/00551; Cocking et al., Science,
236:1259-1262 (1987); and Luskin et al., Neuron 1:635- -
647 (1988).
Transgenic mammals having at least 1 cell -
containing the rDNA's of a prokaryotic gene regulation
system of the present invention can be produced using
methods well known in the art. See for example,
Wagner et al., U.S. Patent No. 4,873,191 (Oct. 10,
1989); Hogan et al., Manipulating the Mouse Embryo: A
Laboratory Manual, Cold Springs Harbor, New York
(1987); Capecchi, Science, 244:288-292 (1989); and
Luskin et al., Neuron 1:635-647 (1988).
; . ~ , :: . . ;
., : . . : , , :. ~ :
. '-'' ' . ~
, :, . . . .
, , - . ..
-, ~ . . . :
WO91/15579 2 ~ 7 ~ 7 ~ 2 PCT/US9-,023~
In preferred embodiments the transgenic mammal
of the present invention is produced by:
1) microinjecting a subject rDNA into a
fertilized mammalian egg to produce a
genetically altered mammalian egg;
2) implanting the genetically altered
mammalian egg into a host female mammal;
3) maintaining the host female mammal for a
time period equal to a substantial
portion of a gestation period of said
mammal;
4) harvesting a transgenic mammal having at
least one cell containing a rDNA that has
developed from the genetically altered
mammalian egg.
A fertilized mammalian egg may be obtained
from a suitable female mammal by inducing
superovulation with gonadotropins. Typically,
pregnant mare's serum is used to mimic the follicle-
stimulating hormone (FSH) in combination with human
chorionic gonadotropin (hCG~ to mimic luteinizing
hormone (LH). The efficient induction of
superovulation in mice depends as is well known on
several variables including the age and weight of the
females, the dosP and timing of the gonadotropin
administration, and the partieular strain of mice
used. In addition, the number of superovulated eggs
that become fertilized depends on the reproductive
performance of the stud males. See, for example,
Mani~ulatina the Embryo: A Laboratorv Manual, Hogan
et al., eds., Cold Spring Harbor, NY (1986).
The rDNA may be microinjected into the
mam~alian egg to produce a genetically altered
mammalian egg using well known techniques. Typically,
the rDNA is microinjected directly into the pronuclei
-
I WO91/15579 PCT/US91/023~
~ 79~ - 26 - ~
of the fertilized mouse eggs as has be~n described by
Gordon et al., Proc. Natl. Acad. Sci.. USA, 77:7380-
7384 (1980). This leads to the stable chromosomal
integration of the rDNA in approximately 10 to 40
percent of the surviving embryos. See for example,
Brinster et al., Proc. Natl. Acad. Sci. USA, 82:4438-
4442 (1985). In most cases, the integration appears
to occur at the 1 cell stage, as a result the rDNA is
present in every cell of the transgenic animal,
including all of the primordial germ cells. The
number of copies of the foreign rDNA that are retained
in each cell can range from 1 to several hundred and
does not appear to depend on the number of rDNA
injected into the egg as is well known.
An alternative method for introducing genes
into the mouse germ line is the infection of embryos
with virus vectors. The embryos can be infected by
either wild-type or recombinant viruses leading to the
stable of integration of viral genomes into the host
chromosomes. See, for example, Jaenisch et al., Cell,
24:519-529 (1981). One particularly useful class of
viral vectors are virus vector derived from retro-
viruses. Retroviral integration occurs through a
precise mechanism, leading to the insertion of single
copies of the virus on the host chromosome. The
frequency of obtaining transgenic animals by
ret~oviral infection of embryos can be as high as that
obtained by microinjection of the rDNA and appears to
depend greatly on the titre of virus used. See, for
example, van der Putten et al., Proc. Natl. Acad.
Sci.. USA, 82:6148-6152 (1985).
Another method of transferring new genetic
information into the mouse embryo involves the
introduction of the rDNA into embryonic stem cells and
then introducing the embryonic stem cells into the
- . : , , - , :
WO91/15579 2 `~ ~ ~ 7 ~ 2
embryo. The embryonic stem cells can be derived from
normal blastocysts and these cells have been shown to
colonize the germ line regularly and the somatic
tissues when introduced into the embryo. See, for
example, Bradley et al., Nature, 309:255-256 (1984).
Typically, the embryo-derived stem cells are
transfected with the rDNA and the embryo-derived stem
cells further cultured for a time period sufficient to
allow the rDNA to integrate into the genome of the
cell. In some situations this integration may occur
by homologous recombination with a gene that is
present in the genome of the embryo-derived stem cell.
See, for example, Capecchi, Science, 244:1288-1292
(1989). The embryo stem cells that have incorporated
the rDNA into their genome may be selected and used to
produce a purified genetically altered embryo derived
stem cell population. See, for example, Mansour et
al., Nature, 336:348 (1988). The embryo derived stem
cell is then injected into the blastocoel cavity of a
preimplantation mouse embryo and the blastocyst is
surgically transferred to the uterus of a foster
mother where development is allowed to progress to
term. The resulting animal is chimeric in that it is
composed from cells derived of both the donor embryo
derived stem cells and the host blastocyst.
Heterozygous siblings are interbred to produce animals ~ -
that are homozygous for the rDNA. See for example,
Capecchi, Science, 244:1288-1292 (1989).
The genetically altered mammalian egg is
implanted into host female mammals. Methods for
implanting genetically altered mammalian eggs into
host females are well known. See, for example, Hogan
et al., Manipulatin~ the Mouse Embryo: A LaboratorY
Manual, Cold Spring Harbor Laboratory, Cold Spring
Harbor, New York (1986). Pseudopregnant recipient
- ' , . , ' . - -, , ' .. - ::'
.-
i
'
WO91/1S~79 PCT/US91/023
~37 ~r~ 28 -
females may be produced by mating females in natural
estrus with vasectomized or genetically sterile males.
After mating with a sterile male, the female
reproduction tract becomes receptive for transferred
embryos even though her own unfertilized eggs
degenerate. The genetically altered mammalian eggs
are then transferred to the ampullae or the uterine
horns of the pseudopregnant recipient. If the
genetically altered mammalian egg is transferred into
the ampullae it must be enclosed in a zona pellucida
membrane. If it is transferred into the uterine horns
the genetically altered mammalian egg does not require
a zona pellucida membrane.
The host female mammals containing the
implanted genetically altered mammalian eggs are
maintained for a sufficient time period to give birth
to a transgenic mammal having at least l cell
containing a rDNA of the present invention that has
developed from the genetically altered mammalian egg.
Typically this gestation period is between 19 to 20 -
days depending on the particular mouse strain. The
breeding and care of mice is well known. See for
example, Manipulating the Mouse Embryo-. A Laboratory
Manual, Hogan et al., eds., Cold Spring Harbor, New
York, (1986).
The infection of cells within an animal using
a replication incompetent retroviral vector has been
described by Luskin et al., Neuron, 1:635-647 (1988).
In one embodiment, an animal that contains a
target gene system in specific tissues or cells is
used to test the effect of a material, composition, or
compound suspected of being a carcinogen on the
specific tissue. The animal is exposed to the
particular material or compound and the mutagenic
effect on the animal is determined by the derepression
.
wo gl/t5579 2 ~ 7 9 7 ~ 2 PCTtUS91tO23~
- 29 -
of the operator-regulated reporter gene segment as an
indication of the carcinogenicity of the compound or
material.
The composition suspected of having
carcinogenic activity is introduced into the animal by
any suitable method including injection, or ingestion
or topical administration.
The animal is then maintained for a
predetermined time period that is sufficient to allow
the composition to produce a mutagenic effect on the
genes of the target gene system. Typically, this time
period ranges from several minutes to several days
depending on the time the composition requires to
mutagenize the genes.
The physiological process or parameter assayed
as an indication of mutagenesis depends upon the
particular physiological alteration produced by the -
expression of the reporter gene.
A change in a physiologic parameter is
determined by measuring that parameter before
introduction of the composition into the animal and
comparing that measured value to a measured value
determined in identical manner after introduction of
the composition into the animal.
The copy number of the test gene in the
transgenic animals alters the sensitivity of
transgenic animals to the effects of the suspected
carcinogen. Therefore, selection of transgenic
animals with varying transgene copy numbers of the
test gene will alter the sensitivity of the transgenic
mice to the suspected carcinogen.
Examples
The following examples are intended to
illustrate, but not limit, the present invention.
W091J15~9 PCT/US91/023~
2~ 30 _ ~
Accordingly, variations and equivalents, now known or
later developed, that would be within the purview of
one skilled in this art are to be considered to fall
within the scope of this in~ention, which is limited
only as set forth by the appended claims.
1. Construction of E. coli RecA- Lysoaen Strains
Strains B~B2688R and ~HB2690R are
constructed using recA+ transformants of E. coli
strains BHB2688 and BHB2690, respectively, as the
recipients and any E. coll K-12 strain that carries a
TnlO (tetracycline resistant) in (or near) the mcrA
gene (relevant genotype = mcrA:TnlO(tetR)) as the
donor. BHB2688, which is E , and BHB2690, which is
D , are available from the American Type Culture
Collection (ATCC), Rockville, MD under the accession
numbers 35131 and 35132, respectively. RecA+
transformation is accomplished by standard methods,
typically using a recA expressing plasmid. Step_l: A
Pl lysate is made from the E. coli K-12 strain
described above. Step ?: B~B2688 and BHB2690 are
transduced with the Pl lysate (Miller, J., Experiments
in Molecular Genetics, Cold Spring Harbor Lab., Cold
Spring Harbor, New York (1972)). Step 3:
Tetracycline (tet~) resistant colonies are selected
and purified. Ste~ 4: ~oss of tetracycline
resistance is selected for on Bochner plates (Bochner,
B. R., et al., J. Bacteriol., 143:926-933 (1980)), and
colonies are purified. Step 5: Lack of mcrA
restriction activity is tested by comparing
transformation efficiency of unmethylated pBR322
versus pBR322 that has been in vitro methylated by
HpaII methylase (Raleigh, supra). A mcrA+ strain will
show a greatly reduced efficiency with the methylated
plasmid. If mcrA activity is absent, this strain is
,
WO91/15579 PCT/US91/023~
`` - 31 - 2~79792
then called BHB2688mcrA and BHB2690mcrA .
To delete the mcrB locus in the above mcrA
strains, a donor E. coli K-12 strain with the relevant
genotypes mcrB::TnlO(tetR), mrr::Tn5(kanR) was used.
Make a Pl lysate from an E. coli K-12 strain that
carries a TnlO(tetR) in the mcrB gene. The strain
should also have a Tn5(kanR) in the mrr gene. Stem 7:
Transduce the BHB2688 mcrA and BHB2690 mcrA
recA+(tets) strain. Ste 8: Select for tetR
colonies. Purify one colony that is also kanR. Step
2: Select for loss of tetR on Bochner plates
(Bochner, suDra). Step 10: Purify several colonies
and test for sensitivity to tetracycline and
kanamycin. Select colonies that are both tetS and
kan . SteD 11: Test for lack of mcrB restriction
activity as done for the mcrA test, however in this
case, the pBR322 should be in vitro methylated by AluI
methylase (Raleigh, supra; Ross, sumra). A mcrB+
strain will show a greatly reduced efficiency with the
methylated plasmid. Test for mrr restriction activity
by comparing plating efficiency of lambda versus
lambda which has been in vivo methylated by Pst I
methylase (Heitman, supra). An mrr+ strain will show
reduced efficiency with the methylated lambda. Test
for hsdR restriction activity by comparing plating
efficiency of lambda versus lambda which has been in
vito methylated by hsdM methylase (Wood, W., J. Mol.
Biol., 16:118-133, (1966); Adams, Bacteriophages, New
York: Interscience 1959; Bickle, supra, at pp. 95-
100). An hsdR+ strain will show reduced efficiencywith unmethylated lambda. If a strain (purifi~d
colony) lacks all restriction activity mcrA, mcrB,
mrr, hsdR and was constructed by this mathod, it
should then contain a deletion throughout the mcrB
region (-mcrB). These strains are BHB2688R and
. , , . . . . . . . . . .. ~ - , . .. . .
WO91/15579 PCT/US91/023~
2~ 7~ 32 - ~-
BHB269OR .
A. E. coli BHB2690R-
(1) Transduction to Obtain BHB2690mcrA-
a) Preparation of a P1 Lysate
A bacteriophage P1 lysate,
hereinafter referred to as Pl, was made from any E.
coli K12 strain that carries a tetracyline resistant
Transposon 10 (TnlO) in or near the mcrA gene
(TnlO::mcrA). Briefly, one drop from an overnight
culture of K12 was admixed into 5 ml of LB broth
(Luria-Bertani broth was prepared by admixing and
dissolving 10 grams (g) bacto-tryptone, 5 g bacto-
yeast extract and 10 g NaCl into 1 liter deionized
water) containing 5 X 10 3 molar (M) CaCl2. The
admixture was aerated by swirling until the cells were
in exponential log phase growth and had reached a
density of 2 X 108 cells/ml. Pl was preadsorbed by
admixing 107 phage to 1 ml of the above admixture
followed by maintenance at 20 minutes in a 30 degrees
Celsius (30 C) waterbath to form a phage-cell
admixture. LB-top agar, 2.5 ml, maintained at 45C,
was then admixed with the phage-cell admixture. The
resultant agar-containing cell suspension was plated
onto a freshly made LB plate which was maintained at
30 C for 8 hours. At the end of the maintenance
period, the soft agar layers was scraped into a small
centrifuge tube. The scraped surface of the plate was
then washed with 1 ml broth and the wash was collected
for admixture with the scraped soft agar. Five drops
of chloroform were added to the centrifuge tube
followed by centrifugation to pellet cell debris. The
resultant supernatant, containing the Pl lysate, was
collected.
b) Transduction with Pl Lysates
q
- ,: . .. . . . : .- -
WO 91/15579 2 ~ ~ ~ 7 ~3~ PCT/US91/023~
~ ~ S7 b V ~ '
33 -
In this invention, E. coli lysogen
BHB2690 (ATCC # 35132) was used as the specific strain
for transduction. E. coli BRB2690, which was RecA-,
was first transformed with pJC859 to introduce a
functional RecA protein into the lysogen. pJC859 was
a plasmid in which the nucleotide sequence encoding
RecA had been inserted at the Bam HI site of the
plasmid E. coli vector, pBR322 (ATCC # 31344).
Maniatis et al., Molecular Clonina: A Laboratory
Manual, Cold Spring Harbor Laboratory, 2nd ed.,
Sections 1.12 and 5.88, 1989; and US Patent 4843006
(in which the RecA promoter/operator and 150 amino
acid residues of RecA coupled to a heterologous
polypeptide sequence is described). For the
transformation, E. coli BHB2690 competent cells were
prepared following standard procedures familiar to one
skilled in the art. Maniatis et al., supra, Section
1.76~ Alternatively, competent cells can be obtained
commercially.
Five ml of a fresh overnight culture of -
BHB2690 recA+ to be transduced was resuspended in 5 ml
of buffer consisting of 0.1 M MgSO4 and .5 mM CaCl2
according to the procedure by Miller. Miller,
Experiments in Molecular Genetics., Cold Spring Harbor
Laboratory, 1972. The BHB2690 recA~ cell suspension
was then aerated by swirling at 30C for 15 minutes.
To each of 5 small test tubes, 0.1 ml of the aerated
suspended cells was added. One hundred microliters
(ul) of Pl lysate, prepared above, was added to the
first tube. The Pl lysate was serially diluted 10-
fold for addition to the remaining tubes except for -
the last tube which did not receive Pl and, thus,
served as a control. A tube without cells containing
P1 was also used as an additional control. The P1
lysate was preabsorbed by maintaining the tubes at
- , . - ...... . . -, . -, ' - ~ : ' . - ~:
WO91/15~79 PCT/US91/023~
2~7~ 34 _ ~
30OC in a water bath for 20 minutes. Two hundred ul
of 1 M sodium citrate was then added to each of the
prepared tubes and the contents of each tube was then
plated on tetracycline-containing plates to select for
tetracycline resistant (tetR) colonies.
After maintaining the plates at 30C to allow
for growth of colonies, tetR colonies were selected
and purified following procedures well known to one
skilled in the art. The tetR colonies were then
replated on Bochner plates to select for the loss of
tetR as described by Maloy. Maloy et al., J.
Bacteriol., 145:1110-1112 (1981). Briefly, tet-
sensitive, tetS, colonies were selected on a medium
consisting of the following: 15 grams/liter (g/l)
agar; 5 g/l tryptone broth; 5 g/l yeast extract; 4
milliliters/l (ml/l) chlortetracycline hydrochloride
(12.5 milligram (mg)/ml); 10 g/l NaCl; 10 g/l NaH2PO4-
; 6 ml/l fusaric acid (2 mg/ml); and 5 g/l ZnCl2
~20 millimolar (mM)). Chemicals were obtained from
Sigma. (Sigma, St. Louis, MO).
Selected tetS colonies were then purified and
tested for the lack of mcrA restriction activity. The
determination of mcrA- strains was accomplished by
comparing transformation efficiency of unmethylated
pBR322 versus pBR322 that had been in yitro methylated
by Hpa II methylase. A mcrA+ strain showed a greatly
reduced efficiency with the methylated plasmid.
BHB2690RecA+mcrA-: hereinafter designated
BHB2690mcrA-, strains were, thus, determined and used
to make BHB2690mcrB- transductions as described below.
(2) Tr~nsdu~tion to Obtain BHB2690mcrB-
The BHB2690mcrA- strains prepared above
were used in similar transductions to select for
BHB2690mcrB- strains. For this procedure, a Pl lysate
WO9l/15579 2 g 7 ~ ~ ~ 2 PCT/US9l/023~
- 35 -
was prepared as described above from any E. coli K12
strain that carried a TnlO (tetR) in the mcrB gene
(mcrB::TnlO (tetR). The strain selected also carried
the Tn5 with kanamycin (antibiotic) resistant gene
(kanR) in the mrr gene (mrr::Tn5 (kanR).
The E. coli BHB2690mcrA- (tetS) strains were ~ -
then transduced with P1 lysate [(mcrB::TnlO (tetR) and
(mrr::Tn5 (kanR) as described in Example la above.
TetR colonies that were also kanR were selected and
purified. The loss of tetR on Bochner plates was
measured as described above. Colonies that were both
tetS and kanS after selection on Bochner plates were
purified. -
The lack of mcrB restriction activity was
performed as described for determining the lack of
mcrA activity with the exception that pBR322 was in
vitro methylated by Alu I methylase. A mcrB+ strain
showed a greatly reduced efficiency with the
methylated plasmid. The test for mrr restriction
activity was accomplished by comparing plating
efficiency of lambda versus in vivo methylated lambda
(by Pst I methylase). A mrr+ strain showed reduced
efficiency with the methylated lambda. A separate
test for hsdR restriction activity was also performed
as the lack of activity confirmed the deletion of the
entire mcrB region. The hsdR restriction activity
test was performed by comparing plating efficiencies
of lambda versus lambda which had been in vivo
methylated by hsdM methylase. A hsdR+ strain showed
reduced efficiency with the unmethylated lambda. With
these tests, a selected colony which lacks all
restriction activity, mcrA, mcrB, mrr, and hsdR, and
constructed using this transduction approach was shown
to contain a deletion throughout the mcrB region. This
strain was designated BHB2690R- and used in the
: . .... .
-
,. . .. ; . ., :, ... :
- . ::. . , , ~ . , .: . ::; -: . , -
I, WO91/1557~ PCTtUS91/023~
~ 7'~ 36 - ~
preparation of extract for prehead as described in
Example 2a.
B. E. coli BHB2688R-
E. coli BHB2688 strains containing RecA+
but lacking mcrA, mcrB, mrr and hsdR were prepared
using the approach described above for preparing E.
coli BHB2690R-. For the transductions, E. coli
lysogen BHB2688 (ATCC # 35131) was used. The
resultant strain, designated BHB2688R-, was used in
the preparation of extract for protein donor a
described in Example 2 below.
2. Preparation of Packaginq Extracts from Two
Lysogens
A. Preparation of Sonic,ated Extract from
Induced E.coli Strain BHB2690R- Cells
For preparing a sonicated extract of the ' '
E. coli lysogen, strain BHB2690R ~prehead donor)
prepared in Example la, the genotype of the strain is
first verified before large-scale culturing. The
presence of the mutation that renders the
bacteriophage cI gene product temperature-sensitive is
determined by streaking from the master stocks of E.
~gli BHB2690L- onto two LB agar plates. One of the
plates is maintained at 32-C while the other is
maintained at 45C. Bacteria with intact cI only grow
on the plates maintained at 32-C. A single small
colony of E. coli BHB2690R- is picked and maintained
overnight at 32-C and 45-C. The bacteria with the
mutation only grow at 32-C and grow slowly due to the ,
recA- mutation present in the BHB strains. A 100 ml
subculture of the verified master stock of E. coli
strain BHB2690R- is then prepared and maintained
overnight at 32-C.
.. . .. . . . .. ..
. - . - . . -~ - . ~ : .
WO91/15579 3 2 ~ 7 ~ 7 ~ 2 PCT/US9lt023~
After maintaining the E. coli BHB2690R-
culture overnight, the optical density (OD) is
measured at a wavelength of 600 nm. An aliquot of the
overnight culture is admixed into 500 ml of NZM broth
(NZM broth is prepared by admixing lO g NZ amine, 5 g
NaCl, and 2 g MgSO4-7H2O to 950 ml of deionized water;
the pH of the solution containing dissolved solutes is
adjusted to pH 7.Q with 5 N NaOH), prewarmed to 32C,
in a 2-liter flask, to result in a starting OD~o f
approximately 0.1. The bacterial admixture is then
maintained at 32C with vigorous agitation (300
cycles/minute in a rotary shaker) until an OD~o of
approximately 0.3 is reached. The OD~ of 0.3 is
generally attained within 2 to three hours of
maintaining the culture. The cultures must be in the
mid-log phase of growth prior to induction as
described below.
The lysogen is induced by placing the flask in
a water bath preheated to 45C. The flask is swirled
continuously for 15 minutes. An alternative approach
for inducing lysogen is to immerse the flask in a
shaking water bath set at 65C. The temperature of ~ - -
the fluid contents of the flask is monitored. When
the fluid reaches 45 C, the flask is then transferred
to a water bath set at 45-C and maintained for 15
minutes. The induced cells are then maintained for 2
to 3 hours at 38 to 39-C with vigorous agitation as
described above. A successful induction is verified
by the visual clearance of an added drop of chlorofor~
to the culture.
Following the 2 to 3 hour maintenance period,
the cells are recovered from the admixture by
centrifugation at 4000g for 10 minutes at 4 C. The
resultant supernatant is decanted and any remaining
liquid is removed wi~h a pasteur pipette and a cotton
:, . - . - . - .
,_., . !.-- :, ,~. .;
~, '- ` .,. -: ' ~ ":""............... - .
- . '~ ' ' :: . '
WO91/15579 PCT/US91/023~
2 ~ 7 9 7 ~ ~ 38 !r
swab. The walls of the centrifuge bottle are wiped
dry with towels. To the pelleted induced bacterial
cells, 3.6 ml of freshly prepared sonication buffer is
admixed. Sonication buffer consists of 20 mM Tris-
HCl, pH 8.0, (Tris[hydroxymethyl]-aminomethane
hydrochloride), 1 mM EDTA, pH 8.0,
(ethylenediaminetetraacetic acid) and 5 mM beta-
mercaptoethanol. The bacterial cell pellet is
resuspended in the sonication buffer by mixing to form
a homogenized cell suspension.
The resultant suspension is transferred to a
small, clear plastic tube (Falcon 2054 or 2057,
Falcon, Oxnard, California) for subsequent sonication.
The cells are disrupted by sonication with l0 second
bursts at maximum power using a microtip probe. For
sonication, the tube containing the suspension is
immersed in ice water and the temperature of the
sonication buffer should not be allowed to exceed 4OC.
Th~ sample is cooled for 30 seconds in between each
sonication burst. The sonication procedure is
continued until the solution in the tube clears and
its viscosity decreases. The sonicated bacterial
sample is transferred to a centrifuge tube and debris
is pelleted by centrifugation at 12,000g for 10
minutes at 4-C forming a clear supernatant.
The resultant supernatant containing preheads
is removed and admixed with an equal volume of cold
sonication buffer and one-sixth volume of freshly
prepared packaging buffer to form a diluted prehead
admixture. Packaging buffer consists of the
following: 6 mM Tris-HCl, pH 8.0; 50 mM spermidine;
50 mM putrescine; 20 mM MgCl2; 30 mM ATP, pH 7.0; and
30 mM beta-mercaptoethanol. The admixture is then
dispensed into pre-cooled to 4-C 1.5-ml microfuge tube
in 15 ul aliquots. The caps of the microfuge tubes
: . .
-- . . : . . : - :
W091/15579 2 9 7 9 7 ~ ~ US91/023~
- 39 -
are then closed and the tubes are immersed briefly in
liquid nitrogen for freezing. The frozen preheads in
packaging buffer are then stored at -70C for long- -
term storage.
B. PreParation of Frozen-~hawed Lysate of
Induced E. coli BHB2688R- Cells
A subculture of E. coli BHB2688R- ,
prepared in Example lb above, for obtaining an extract
of packaging protein donor is verified for the
genotype and is prepared as described above for
preparing E. coli BHB2690R-. Overnight cultures are
maintained and lysogen is induced also as described
above.
The induced E. coli BHB2688R- cells are
pelleted by centrifugation at 4000g for 10 minutes at
4C. The resultant supernatant is removed and any
excess liquid is removed. The pelleted cells are
resuspended in a total of 3 ml of ice-cold sucrose
solution (10~ sucrose in 50 mM Tris-HCl, pH 8.0) to
form a cell suspension. The resultant cell suspension
is dispensed in 0.5 ml aliquots into each of six
precooled to 4C microfuge tubes. Twenty-five ul of
fresh, ice-cold lysozyme solution ~2 mg/ml lysozyme in ;~
10 mM Tris-HCl, pH 8.0) is admixed to each tube
containing the cell suspension. The cell-lysozyme
admixture is gently mixed to form an E. coli extract
an-d then immersed in liquid nitrogen for freezing.
The frozen tubes are removed from the liquid
nitrogen and the extracts are thawed on ice. Twenty-
five ul of packaging buffer, as prepared above, is
admixed to each tube containing thawed extract to form
a packaging buffer-extract admixtuxe. The separately
prepared admixtures are then combined in a centrifuge
tube and centrifuged at 45,000g for 1 hour at 4C to
:. ` ~ ` ~ : .
WO 91/15579 r~ ~ PC~/US91/02364
~ 40 -
form an supernatant containing packaging protein
donor.
The resultant supernatant is removed and
dispensed in 10 ul aliquots into precooled at 4C
microfuge tubes. The caps of the tubes are closed and
the tubes are then immersed in liquid nitrogen. The
tubes are then removed from the liquid nitrogen and
stored at -70C for long term storage.
3. Pre~aration of In Vitro Packaaing_Using Two
Extracts
The frozen tubes containing prehead and
packaging donor extracts prepared in Example 2a and
2b, respectively, are removed from storage at -70C
and allowed to thaw on ice. The frozen-thawed lysate
containing the protein donor thaws first and is
admixed to the still-frozen sonicated prehead extract
to form a prehead-protein donor admixture. The ~-
resultant admixture is mixed gently until almost
totally thawed. The DNA to be packaged (up to 1 ug
dissolved in 5 ul of 10 mM Tris-HCl, pH 8.0, 10 mM -
MgCl2) is admixed with the thawed combined extracts
and mixed with a fine glass stirring rod to form a
DNA-extract admixture. The admixture is then
maintained for 1 hour at room temperature. To the
admixture, 0.5 to 1 ml of SM (SM buffer is prepared
by admixing 5.8 g NaCl, 2g MgS04-7H20, 50 ml Tris-HCl,
pH 7.5, and 5 ml 2% gelatin (w/v) to 1 liter of
deionized water and adjusting the pH to 7.5) and a
drop of chloroform is added and gently mixed. Debris
is removed by centrifugation at 12,000g for 30 seconds
at room temperature in a microfuge. The resultant
supernatant is removed and contains packaged
bacteriophage DNA particles.
. . .
.. , ., . .,- . , . .~ .-
.. ~ .. . , :, ~ . . .. : ; . . .
WO91~15579 - 4l 2 ~ 7 ~ 2 PCT/US91/02
The titer of the viable bacteriophage
particles is measured by plating on the appropriate
indicator strains. Recombinant DNAs that are 90% or
80~ of wild-type bacteriophage lambda in length are
packaged with efficiencies that are 20-fold to 50-fold
lower, respectively, than those obtained with unit-
length bacteriophage lambda. The same packaging
extracts may be used for the packaging of both
bacteriophage lambda and cosmids.
4. Preparation of_Transgenic Mice and Their Use
The following studies provide details of the
manner in which the present invention may be made and
used in order to achieve the rapid recovery and -
examination of test DNA sequences from transgenic - -
animals.
A. DNA Test Sequence
The test sequence DNA can, theoretically,
contain any number or variety of genes or other
identifiable test DNA sequences. In the prototype
described herein, an E. coli bacteriophage lambda `
genome has been engineered to carry lacZ, a beta-
galactosidase test DNA sequence. Lam~da shuttle
vectors L2B (46.5kb) or C2B (48.0kb) may be used. The
genotype of the modified lambda genome L2B is Lac5
delta (shind III lambda 2-3-) srI lambda 3-5 cI857
sXhL lambda l sScII lambda 4-. Before injecting it
into mouse embryos as described below, this lam~da DNA
was diluted to a concentration of lO micrograms per
milliliter and the cos ends were annealed and ligated
under conditions predominantly forming circular lambda
phage monomers. Maniatis et al., Molecular Cloninq. A -
Laboratory Manual, pp. lO9-llO, 383-389 (Cold Spring
Harbor, New York 1982). `
.: . , . ........ - , . , .~
WO91/15579 PCT/US91/023
42 -
In addition, a variation of L2B may be
constructed that contains a plasmid sequence that can
be readily excised from the lambda phage and contains
the lacI repressor gene. This variation has several
advantages. First, as discussed below, physical
identification of phage carrying mutations will be
facilitated since they will grow as blue plaques on a
white background in the presence of X-gal (5-bromo-4-
chloro-3-indolyl-~-D-galactopyranoside) without IPTG
(isopropyl~-D-thiogalacto-pyranoside). This advantage
will also simplify and reduce the cost of the assay
since it will permit an increase in the density of
phage per plate. Additionally, the lacI genetic
systems of E. coli are the first systems that
conveniently permitted the study of large numbers of
mutations within procaryotes at the DNA level (Miller
et al., J. Mol. Biol., 109:275-302 (1977), Coulondre
and Miller, J. Mol. Biol., 117:275-302 (1977),
Schaaper, J. Mol. Biol., 189:273-284, (1986)), and the
use of lacI will provide a test gene with significant
historical mutational data for comparison between
mutagenesis assays. ~-
B. Creation of a Transgenic Animal
Mice were used as the test animal.
(Hogan et al., Manipulat n~ the Mouse Embrvo: A
Laboratory Manual, ~old Spring Harbor Laboratory,
1986). Single cell mouse embryos were harvested from
female mice that were impregnated the evening before.
The embryos were treated with hyaluronidase and
briefly cultured in M16 medium. The embryos were
transferred to M2 medium on a microscope glass
depression slide. The embryos were observed with a
40X objective and a lOX eyepiece using a Nikon Diaphot
microscope equipped with ~offman optics. The embryos
- . - . . . . ... .. . . . . ... ........ . . ... . . .
:-: :- ,, . . : . , -
. .
. : . : .. .. - - -,: . ~ , , , ~: : ..
- . - . -
- . - . - -
WO91/15579 PCT/US91/023~
~ _ 43 _ 2 ~ 7 ~ ~ 9 2
were held in place with a holding pipet that had been
rounded with a microforge. The positions of both the
holding pipets and the injection pipets were
controlled with micromanipulators. DNA as described
above was loaded in the injection pipet at a
concentration of l to l0 micrograms per milliliter. ~ :
Approximately one picoliter, as judged by a refractile
change (Hogan et al., supra) of the pronucleus, of DNA
solution was injected into the male pronucleus. ~ -
After DNA injection, four hundred embryos were
transferred to Ml6 medium and incubated at 37-C in a 5
% CO2 atmosphere for one to two hours. One hundred
fifty embryos survived microinjection. Lysed embryos
were discarded and 30 embryos that appeared normal
were transferred to one of the fallopian tubes of each
of 5 pseudopregnant foster mothers. The transfers
were performed under a dissecting microscope using
general anesthesia (avertin).
Seven pups were born. After birth, newborn
mice were kept with their foster mothers for 2 weeks,
at which point they were then weaned and screened for
DNA integration. A 2 cm portion of the tail was
removed and homogenized in 2 ml of a solution of 0.l M
NaCl, 50 mM Tris-HCl, pH 7.5, lmM EDTA for short
duration, but long enough to disrupt cell and nuclear
membranes. The homogenized tissue was treated with 50
U/ml RNaseA and 0.l~ SDS for 15 minutes at 37C. The
mixture was exposed to Proteinase K digestion for 3
hours at 55 C followed by three extractions with
phenol/chloroform. DNA was then precipitated by the
addition of ethanol. After resuspending the
precipitated DNA in l0 mM Tris pH 8.0, 0.5 mM EDTA,
some of it was digested with BamHI endonuclease and
electrophoresed through an 0.8% agarose gel. The DNA
was denatured by soaking the gel in l.5 M NaCl, 0.5 M
.. . . . . .. . . .
- ~ . -
- - . - . ~ .
,
:~
.
W091/15579 PCT/US91/023~
2~ 2 44 ~
NaOH for one hour and then neutralizing the DNA by
soaking it in 1.5 M NaCl, 0.5 M Tris, pH 7.4 for 30
minutes. The gel was then soaked in 10X SSC for one
hour. The DNA was then transferred from the gel into
a nitrocellulose filter by the method of Southern, as
described in Maniatis, supra.
The filter with transferred DNA was hybridized
overnight with 32p labeled lambda DNA prepared
according to standard procedures by the method of nick
translation. Maniatis, supra. Following this
overnight hybridization, the filter was washed in 0.1
x SSC, 0.1% SDS at 50C and Kodak XAR film was exposed
to it in order to identify lambda DNA present within
the mouse genome. Lambda DNA, used as standards, that
had been electrophoresed alongside the mouse genomic
DNA were compared in intensity to the transgenic mouse
DNA hybridized to the 32p labeled lambda DNA to
estimate the number of copies of test DNA per mouse
cell. Three transgenic animals have been produced and
identified by this technique.
Newborn mice tested for the presence of the
test DNA sequence by the tail-blotting procedure
(Hogan et al., Manipulating the Mouse Embryo: A
Laboratory Manual, pp. 174-183, Cold Spring Harbor
Laboratory, 1986) were found to carry the test DNA
sequence in DN~ isolated from their tails. Eight
weeks after birth these transgenic mice were mated and
their progeny were examined for the test DNA sequence.
Approximately 50% of the resulting offspring carried
the test DNA sequence, demonstrating that the original
transgenic mice carried the test DNA sequence in their
germ line and that this sequence was inherited
normally. While transgenic lines having approximately
one copy of the test DNA sequence per cell can be
obtained, it will be understood by one skilled in the
-- - . .. i . ~ . , -, . . . .
.: - . . -
,: ' ' ~.
,. . ... ... .
' ' ~ ' ' . . .
WO91/1~579 PCT/US91/023~
~ _ 45 _ 2 ~ 7~ J
art that multiple copy numbers per cell are obtainable
and may be useful for many different applications.
C. Mutaqen Treatments -
Six to eight week old transgenic male
mice were treated on day 1 and day 4 by
intraperitoneal injection of either 125 or 250 mg N-
ethyl-N-nitrosourea (EtNu), per kg body weight.
Control animals were injected with lOOmM phosphate
buffer at 10 ml/kg body weight. Tissues were
collected two hours after final injection.
D. Recoverv of the Test DNA Sequence and
Mutaaenesis Testing -
1. Recovery
In the embodiment described here,
rescue of the marker DNA sequence was accomplished by
containing it within a lambda bacteriophage genome.
The entire lambda bacteriophage genome is excised from
the mouse chromosome by the in vitro packaging
extract. The packaging extract recognizes the cos
sites of the integrated lambda DNA and packages the
sequences between the cos sites into lambda phage
particles, as shown in Figure 1.
The test DNA sequence may be found within the
genomic DNA purified from any tissue of the transgenic
mouse. Since the test DNA sequence is contained
within a lambda phage genome, it can be excised away
from the remainder of genomic DNA by using a lambda
phage packaging extract. Packaged lambda phage such
as L2B or C2B, may then be plated on E. coli cells for
further evaluation.
Bacteriophage lambda DNA can be packaged in
vitro using protein extracts prepared from bacteria
infected with lambda phage lacking one or more genes
,. . , . ~ . . . . . . ~ , ,
..
', , . ': : . .
: .
: - : :: -, , :
WO9l/l5s79 PCT/US91/023
~ rl~3r~ 46 - -
~for producing the proteins required for assembly of
infectious phage particles. Typical in vitro
packaging reactions are routinely capable o~ achieving
efficiencies of 108 plaque from units (pfu) per ~g of
intact bacteriophage lambda DNA. About 0.05 - 0.5
percent of the DNA molecules present in the reaction
can be package into infectious virions.
Various genetic mutations affect different
stages of bacteriophage lambda DNA packaging. For
instance, the E protein is the major component of the
bacteriophage head and is required for the assembly of
the earliest identi~iable precursor. Bacteriophages
mutant in the E gene (E ) accumulate all of the
components of the viral capsid. The D protein is
localized on the outside of the bacteriophage head and
is involved in the coupled process of insertion of
bacteriophage lambda DNA into the "prehead" precursor
and the subsequent maturation of the head.
Bacteriophages mutant in the D gene (D) accumulate
the immature prehead but do not allow insertion of
bacteriophage lambda DNA into the head. The A protein
is involved in the insertion of bacteriophage lambda
DNA into the bacteriophage prehead and cleavage of the
concatenated precursor DNA at the cos sites.
Bacteriophages mutant in the A gene (A) also
accumulate empty preheads. Complementing extracts
have been prepared from cells infected ~ith A and E
or D and E strains; alternatively, extracts prepared
from cells infected with A mutants can be
complemented by the addition of purified wild-type A
protein.
A bacteriophage lambda DNA packaging extract
is a proteinaceous composition that is capable of
packaging bacteriophage lambda DNA into infectious
virus particles. Preferably, the lambda DNA packaging
. -
- .
- : ~ : . - ~ . . . .
,, .. . .. , . -., - ~ .
- . :. . :.: ~ ., .. , . . : ,. ~ .- :
W091/15579 PCT/US91/023
47
extracts useful in this invention have a packaging
efficiency of at least lO8, and more preferably at
least 109, pfu/~g of intact lambda DNA.
The packaging extracts of this invention are
usually prepared from cells containing bacteriophage
lambda lysogens of the appropriate genotype, e.g.,
amber mutations in genes A, D, E and the like. In
addition to lacking a functional lacZ gene, useful
lysogens preferably have one or more of the following
mutations:
cIts857 - specifies a temperature-sensitive
bacteriophage lambda repressor molecule.
This mutation causes lambda DNA to be
maintained in the lysogenic state when
the host bacteria are grown at 32C;
bacteriophage growth is induced by
transiently raising the temperature to
42-45C to inactivate the repressor
specified by the cI gene.
Sam7 - an amber mutation in the bacteriophage
S gene that is required for cell lysis.
This mutation causes capsid components to
accumulate within SuIII bacterial cells
for 2-3 hours following induction of the
cIts857 lysogen.
b-ragion deletion (b2 or blO07) - a deletion
in the bacteriophage genome that
effectively removes the lambda DNA
attachment site (att). This mutation
reduces, but does not entirely eliminate,
the packaging of endogenous lambda DNA
molecules in extracts made from the
induced cells.
red3 (in lambda) and recA (in E. coli) -
mutations that inactivate the generalized
.. ... . . .
. . . ,
- , . - .:
. . : : - . - . . . .
WO91/15579 PCT/US91tO23~
2~'~37(~ - 48 - ~
recombination systems of bacteriophage
lambda and the host, thereby minimizing
recombination between the endogenous
lambda DNA in the extract and the
exogenously added recombinant genomes.
Thus, in preferred embodiments, a lambda lysogen
useful for producing a packaging extract is one
deficient in one or more of the mcrA, mcrB, hsd and
mrr restriction systems. The genes comprising these
10 systems can be removed or inactivated by well known
methods, such as by transduction, transposon (Tn)
mutagenesis, and the like.
Packaging extracts are usually prepared from a
lysogenic bacteria having one or more of the following
15 mutations: mcrA , mcrB , mrr , hsdR , and preferably
from K-12 ^mcrB, BHB2690R , or BHB2668R , by growing
the appropriate lysogenic bacteria to mid-log phase at
3Z C, inducing lytic functions by inactivating the cI
repressor protein by raising the temperature to 45C
20 for 15 minutes, and then growing the cultures for an
additional 2-3 hours at 38-39-C to allow packaging
components to accumulate. Cell extracts are then
prepared as described further herein.
2. Testinq for Mutagenesis
For plating bacteria, ~-galactosidase -
deficient E. coli, are grown in lX TB (5g/L NaCL,
lOg/L tryptone) supplemented with 0.2% maltose and
lOmM MgSO4 overnight at 30 C. Cells are harvested by
centrifugation and resuspended in lOmM MgSO4 in
30 preparation for plating (Maniatis, supra).
In a typical experiment, 1-5 ~g of genomic DNA
are exposed to in vitro lambda phage packaging extract
and incubated for 2 hours at room temperature. The
packaging reaction is then diluted in 500 ~1 SM buffer
35 (lOOmM NaCL, 8mM MgSO4, 50mM Tris, pH 7.5, and 0.01%
: .,: , , : . - . : . -
WO91/1~579 2 ~ W ~ ~ ~ 2 PCT/US9t/023~
gelatin) and incubated with the above described
bacteria (2.0 mL of OD~o = 0.5), and then plated onto
NZY/agar Nunc Bioassay Dishes (245mm x 245mm x 20mm)
with molten top agar containing l.25 mg/mL X-gal and
2.5 mM IPTG at a density of less than 20,000 pfu per
plate. The plates are incubated overni~ht at 37C.
For the lambda genomes containing the ~-gal
(not the lacI) gene, in the presence of X-gal and
IPTG, the phage plaques turn blue if the beta-
galactosidase sequence within the lambda genome has
not mutated. However, a white plaque or faint blue
plaque on the petri dish is evidence that a mutation
in the beta-galactosidase sequence has, for example,
altered the reading frame, altered essential codons,
or has created stop codons in the sequence. These
white or faint blue plaques will be scored as positive
for mutations and they can be plaque purified and
saved for further analysis. The ratio of white or
faint blue to blue plaques minus background (mutation
rate from non-mutagenesis potency of the agent being
tested when compared with DNA extracted from mice that
have not been treated with potentially mutagenic
agents. -
E. Methods for Increasinq Efficiency of Test
DNA Seouence Rescue
l. pemethylation
It is anticipated that test DNA sequence
rescue efficiency can be influenced by the state of
CpG methylation in the mouse chromosome. Highly
methylated DNA may not be efficiently excised by
lambda packaging extract, presumably because of
inhibition of cleavage at the cos sites, inhibition of
expression of lambda genes encoded on lambda phage, or
restriction by E. coli restriction systems. This may
~ WO91/15579 PCT/US91/023~
` 2~97~2 50 _ ~
be alleviated by placing transcriptional enhancers,
promoters and/or other regions of the DNA which
inhibit methylation near critical sites such as the
cos site to reduce CpG methylationO The drug 5'-
azacytidine can also be used to reduce the level ofDNA methylation in the target cells prior to DNA
purification and rescue. Jaenisch, R., et al., Proc.
Natl. Acad. Sci. USA, 82:1451-1455 (1985). In such a
procedure, fibroblast cell lines are obtained from
organisms containing the test DNA sequence of
interest. Adams, R.L.P., Cell Culture for
Biochemists, pages 68-83 (1980) Elselvier/North Hollan
Biomedical Press). The cells are exposed in vitro at
37 C, within 50 ~M 5'azacytidine supplementing the -
culture medium. Upon DNA replication, the daughter
DNA loses its CpG methylation, which eliminates the
methylation of sites in the target vector, where the -
target vector is a lambda phage. The DNA from these
fibroblasts is then exposed to in vitro packaging
extract, as previously described. -
Alternatively, organisms containing the test
DNA sequence ~an be directly injected with a 1 mg/ml
solution of 5'-azacytidine in 0.15 M NaCl. This is
done over a period of at least about 4 days, with a
total of 400 ~g administered. Jaenisch, supra. After
this treatment, DNA can be extracted from various ;
tissues and packaged as before.
2. Removal of Packaaina Extract and
Plating Strain Restriction Svstems
We have determined that the efficiency of
test DNA sequence recovery is dependent on the
genotype of both the bacterial strain used to generate
the packaging extract as well as the plating strains
used ~or mutagenesis t0sting. This is due to host-
controlled restriction systems that enable a bacterial
: . . ,, ~ . - : , ~ : - .
''
WO91/15579 PCT/US91/023~
~ - 51 _ ~ ~7~7~2
cell to identify and inactivate foreign DNA by
end~nuclease cleavage. DNA is susceptible to
restriction by the endonucleic activity of the host
unless it is protected by modifications, such as
methylation of specific nucleotides. While
methylation of specific nucleotides usually serves to
protect DNA from restriction by the endonucleolytic
activity of the host, methylation at some DNA -
sequences actually confers sensitivity to restriction.
One example, the mcrB restriction system of E. coli K-
12, is responsible for the biologic~l inactivation of
foreign DNA that contains 5-methylcytosine residues.
Ross et al., Journal of Bacteriolooy, 171:1974-1981
(1989).
There are a number of restriction/methylation
systems endogenous to E. coli which are capable of
inactivating foreign DNa by endonuclease cleavage.
The most widely known systems are hsd (Bickle, T.
Nucleases, p. 85, Cold Spring Harbor Lab., Cold Spring
Harbor, N.Y. 1982), mrr (Heitman, J. et al., J.
Bacteriol., 169:3243-3250 (1987)), mcrA (Raleigh et
al., PNAS, 83:9070-9074 (1986)) and mcrB (Raleigh,
supra). The hsd system works by selectively
restricting DNA that is not protected by adenine
methylation at the N-6 position in the sequence,
A~#ACNNNNNNGTGC or GC~X-ACNNNNNNGTT. The mrr system
also involves adenine methylation, however, in this
case the methylation does not serve to protect the
DN~, but serves to make the DNA vulnerable to the
restriction system. The systems mcrA and mcrB are
similar to mrr in that they recognize and restrict
methylated DNA. However, these two systems differ
from mrr in that they recognize methylated cytosine.
Further, the mcrB function is provided by the products
of at least two genes, mcrB and mcrC (Ross et al., J.
, : , . . :. ~ . .
'
- ' , '
.
- . - :
WQ91/15579 PCT/US91/023~
~ ~ 7 ~ 52 - ~
Bacteriol., 171:1974-1981 (1989)). The recognition
se~uences for mcr and mrr are contemplated in the
literature, but precise sequences are as yet unknown.
We found that efficiency of recovery of the
lacZ construct from the transgenic animal genome was
increased without the use of 5-azacytidine, by using
lambda packaging extracts and E. coli plating strains
lacking restriction systems that cleave eukaryotic
DNA. By remoYing these restriction systems, rescue
efficiencies have been increased up to at least 12,000
pfu/~g genomic DNA. Of course, one skilled in the art
will recognize that "removal" of these restriction
systems may be effected by deleting or inhibiting the
activity of these restriction systems, and the term
"restriction system deficient" includes, ~ut is not
limited to, removal of the restriction systems by
either method. In addition, naturally occurring
strains of E. coli that are deficient in these
restriction systems may be isolated and used.
Identification of the genes responsible for
the E. coli restriction systems was achieved by
examination of the inhibitory effect of certain E.
coli strains on the ability to recover lambda phage.
Isolation of the responsible genes was achieved
through the use of interrup~ed matings and Pl
transduction. An approximately 200 kb region of DNA
in E. coli K-12 was found to pr~duce an inhibitory
effect on the plating efficiency of the rescued
vector. Further, the region responsible for
decreasing rescue efficiency was found to be near 98
minutes in the E. coli K-12 genetic map (Bachmann, B.
E. coli and S. Typhimurium: Cellular and MOlecular
Biolo~y, eds. Neidhart et al., ASM, WA, DC, 1987) in -
the approximately 2.6 kb mcrB region containing mcrB
and mcrC.
- - . , - ., . .. - .
WO gl/1~579 ~ 7 ~ 7 ~' 2 PCT/US91/023~
The comparison of the rescue efficiency using -
E. coli strains with different restriction genotypes
is shown in Table 1. The bacterial strains listed in
table 1 are available from the following source or
reference: ED8767 (Ishiura, M. et al., Anal.
Biochem., 176:117-127 (1988); ER1451 (New England
BioLabs, ~everly, MA); LCK8 ~B. Bachman, Yale E. coli
Center); NM621 (N. Murray, Univ. of Edinburgh); K802,
IT392, NM554, PLK-A, PLK-17, Y1088, E. coli C, Sure
(Stratagene, La Jolla, CA)).
Table 1
Strain Restriction GenotyPe Platinq EfficiencY
hsdR mcrA mcrB mrr
ED8767 - - - +
ER1451 - - - +
K802 - - - +
LCK8 - - - +
LE392 - - + +
NM554 - - - +
NM621 - - - +
PLK-A - - - +
PLK-17 - - - +
Y1088 - - + (+)
E. coli C - - ~ - +
RR1-A - - - - +
K-12 mcrB - +
sureTm ~ +
Strain RRl-A and K-12~mcrB are constructed as
described below.
Strain RR1-A is constructed with strain RR1
(Maniatis, suDra) (relevant genotype = mcrA+, (tetS))
as the recipient and any E. coli K-12 strain that
carries a TnlO (tetracycline resistant3 in (or near)
.
: '
.- -
~W091/l5~7~ r PCTtUS9~/023~
~9 ~ - 54 - ~ '
the mcrA gene (relevant genotype = mcrA:TnlO(tetR)) as
the donor~ Step_I: A Pl lysate is ~ade from the E.
coli K-12 strain described above. Step 2: RRl is
transduced (Miller, J., Expariments in Molecular
Genetics, Cold Spring Harbor Lab., Cold Spring Harbor,
New York (1972)). Step 3: Tetracycline resistant
colonies are selected and purified. Step 4: Loss of
tetracycline resistance is selected for on Bochner
plates (Bochner, 8.R., et al., J. Bacteriol., 143:926-
933 (1~80)), and colonies are purified. step 5: Lack
of mcrA restriction activity is tested by comparing
transformation efficiency of unmethylated pBR322
versus pBR322 that has been in v tro methylated by
HpaII methylase (Raleigh, supra). A mcrA+ strain will -
show a greatly reduced efficiency with the methylated
plasmid. If mcrA activity is absent, this strain is
then called RR1-A.
Strain K-12-mcrB is constructed using two
donor E. coli K-12 strains with the relevant genotypes
mcrB::TnlO(tetR), mrr::Tn5(KanR) and mcrA::TnlO(tetR)
and a recipient E. cQli K-12 with the relevant
genotype recA+, tetS. Steps 1-5: Perform steps 1-5
as described for construction of RRl-A. In step 2,
transduce any E. coli K-12 recA+ strain. Step_6:
Make a Pl lysate from an E._coli K-12 strain that
carries a Tnlo(tet~ in the ~ 9~ T d
recA+(tets) strain. Step_8: Select the tetR colonies.
Purify one colony that is also kanR. Ste~ 9: Select
for loss of tetR on Bochner plates ~Bochner, supra).
Step 10: Purify several colonies and test for
sensitivity to tetracycline and kanamycin. Select
colonies that are both tetS and kanS. Step ll: Test
for lack of mcrB restriction activity as done for the
mcr~ tet, however in this case, the pBR322 should be
in vitro methylated by AluI methylase (Raleigh, supra;
WO91/lS579 PCT/US91/023~
~ _ 55 _ ~ 9~2
Ross, suPra). A mcrB+ strain will show a greatly
reduced efficiency with the methylated plasmid. Test
for mrr restriction activity by comparing plating
efficiency of lambda versus lambda which has been in
vivo methylated by Pst I methylase (Heitman, su~ra).
An mrr+ strain will show reduced efficiency with the
methylated lambda. Test for hsdR restriction activity
by comparing plating efficiency of lambda versus
lambda which has been in vitro methylated by hsd
Mmethylase (Wood, W., J. Mol. Biol., 16:118-133
~1966); Adams, Bacteriophages, New York: Interscience
1959; Bickle, supra, at pp. 95-100). An hsdR+ strain
will show reduced efficiency with unmethylated lambda.
If a strain (purified colony) lacks all restriction
activities, namely, mcrA, mcrB, mrr, hsdR and was
constructed by this method, it should then contain a
deletion throughout the mcrB region (~mcrB). It will
then also very efficiently plate la~bda that has been
rescued from the mouse. This strain is called K-
12-mcrB.
The "-" symbol in Table 1 indicates that the
strain contains a large deletion in the mcrB region.
All other mcrB- strains listed in TAble 1 are K-12
derivatives believed to contain a small mutation in
the mcrB region, with the exception of E. coli C which
does not contain the K-12 mcrB region and RR1-A which
carries the wild type mcrB locus of E oli B. It is
known that all of these strains plate control L2B
phage (amplified in hsdM+ E. coli K-12 rather than
rescued from the mouse) with equal efficiency (within
1-4 fold). Rescued L2B phase were recovered from the
mouse genome using mcr~ E. coli K-12 lambda packaging
extracts (Gigapack II - Stratagene, La Jolla, CA) and
plated onto the indicated bacterial strains. A "+"
plating efficiency of phage indicates that
~ -: - . ~ - - , . - . , - :
- . ~
. ,- . .- ~
.
. :: :... i : . , .:
- . .: , . ~ .
- . - ,. - -
WO9l/15579 PCT/US91/023
r~ ~ 5 6 ~
approximately 500 pfu/0.05 ~g of transgenic mouse
genom~ DNA was observed, while a "-" plating
efficiency indicates that less than 5 pfu/0.05 ~g of
transgenic mouse genome DNA was observed. Note also
that (+) indicates that the mrr activity has not been
confirmed in Y1088.
In order to determine more precisely the
region of DNA responsible for the inhibition of dC-
methylated lambda phase genome, the 98 minute region
of E. coli K-12 LCK8 was cloned. A partial LCK8
genomic library was made in pOU61cos. (Knott, V. et
al., Nucleic Acid Res., 16:2601-2612 (1988)), packaged
with Gigapack~ II XL (Stratagene, La Jolla, Calif.),
and plated on E. coli C. Clones containing the 98
minute region were identified by colony hybridization
using an oligonucleotide (ATGAGTGCGGGGAAATTG) probe
specific to the hsd region (Gough, J. A., et al., J.
MOl. Biol., 166:1-19 (1983)). All clones were
propagated in the host RRl-A when tested for plating
efficiency of phage. As shown in Figure 3, in panels
3A and 3B, the activity was isolated to a 2.6 kb
fragment containing the mcrB gene. The mcrB region
including open reading frames (Ross et al., supra) is
shown in Figure 3A. The subclones corresponding to
these groups are shown directly below. The table on
the far left gives information pertaining to th~ DNA
fragment shown on the right. (The restriction map
depicted in Figure 3A and 3B, showing the location of
the hsdS gene and adjacent McrB region of the E. coli
K12 chromosome, is from Ross, T., et al., J. Bact.,
171:1974-1981 (1989).)
.
The results in Table 1 support the observation
that the restriction activities of the minute 98
region have a negative effect on rescue efficiency.
To obtain high plating efficiencies, a complete
SUBSTITUTE SHEET
,. . . ~ . .: . . . . ......................... `~.:
, ` . . . ' . :. -. :. ; . . ~ .
~ WO91/15579 PCT/US91/023~
- 57 _ 2 ~ 7 ~
deletion of the minute 98 mcrB region (mcrB through
mrr) is preferred, as opposed to a small mutation of
mcrB present in most commonly used mcrB- lab strains.
This is because despite the mcrB- phenotype exhibited
by these mcrB- strains (using ALuI methylase modified
pBR322 transformation as the assay (Ross, su~ra,))
some inhibitory activity of the mcrB region remains.
Complete deletion resulted in optimal efficiency,
accounting for a greater than lO00-fold improvement in
rescue efficiency using eukaryotic modified DNA.
Preferred E. coli strains for rescue of the
lacZ construct from the transgenic animal genome are
SCS-8 (Catalog Number 200,288) and VCS257 (Catalog
Number 200,256) which are commercially available from
Stratagene Cloning Systems and are also contained in a
kit (Big BlueTM Mouse Mutagenesis System, Catalog
Number 720,000). SCS-8 has the following genotype:
recAl, endAl, mcrA, (mcrBC-hsdRMS-mrr), (argF-
lac)Ul69, phi80~lacZ-Ml5, TnlO(tetr). SCS-8 provides
the lacZ-Ml5 gene which allows for alpha-
complementation when SCS-8 is infected by the packaged
bacteriophage. Additional commercially available E.
coli strains which contain the lacZ~Ml5 genotype for
use in this invention include the following: XLl-Blue
(Stratagene, Catalog Number 200,236); SureTM
(Stratagene, Catalog Number 200,238); PLK-F'
(Stratagene, Catalog Number 200,237); JMlOl
(Stratagene, Catalog Number 200,234); JMlO9
(Stratagene, Catalog Number 200,235); and NM522
(Stratagene, Catalog Number 200,233).
While the use of mcrB deletion strains is
described herein for use in mutagenesis testing and
recovery of lambda phage DNA from mammalian cells, it
is apparent that restriction system deficient strains
~ay be used for other eukaryotic DNA cloning projects.
- ' ::
,' ~ ,
'~ WO91/15579 PCT/US91/023
2 ~7 ~ ~2 - 58 -
0~ course, any number or variety of test DNA
sequences or genes can be inserted between lambda cos
sites. The in vitro packaging extract would still
excise the DNA between the cos sites and package it
into a lambda phage particle. Thus, a variety of
recombinant lambda genomes or cosmids may be used for
this excision event.
F. Construction of Shuttle Vector Systems
for Rapid DNA Sequence Identification of
Mutations in Test DNA
Mutations evidenced by the production of -
white plaques resulting from disruption of the ~-
galactosidase (~-gal) gene are useful for determining
the mutation rate of a mutagen, but give little -~
information regarding the specific mutation within the
DNA. In addition, analysis of the specific mutation
is hampered somewhat by the size of the test ~-gal
gene (i.e., about 3200 b.p.).
To help increase the effectiveness of the
procedure, the target lambda phage can be made to
provide a target gene with reduced size ~e.g., the
lacI gene having about l000 b.p.), and a rapid means
with which the target gene is transferred from the ;-
lambda phage into plasmid vectors for se~uence
analysis.
9Oth the lacI and ~-gal genes are inserted
within a l2~bda vector, such that if the mutation
occurs within the lacI gene, the repressor activity is
lost allowing the ~-gal gene to be expressed giving
rise to blue plaques in the absence of IPTG. In the
described embodiment, the lacI gene is positioned
upstream of the alpha 2ortion of the lacZ gene in the
vector (~iller, J.H. and Reznikoff, W.S., The Operon,
2nd Ed. Cold Spring Harbor Laboratory, 1980, pp. 104- -
- , ~ '' ~ - . ..
,
,
' ' ' ', ' ' ' ' . -' .,, ` ',
~ ' . ~ ' . '
WO91/1~S79 _ 59 _ 2~797~w~
105). When the host E. coli (which is infected by the
bacteriophage vector) provides the complementary
portion of the lacZ gene (referred to as lacZ-M15)
(Miller, J.H. and Reznikoff, W.S. sura), the gene
products synthesized by these two partial genes
combine to form a functional ~-galactosidase protein
(referred to as alpha-complementation) giving rise to
blue plaques in the presence of Xgal when a mutation
has occurred in the lacI gene or in the presence Xgal
and IPTG when the lacI gene is not mutated. The ~M15
portion of the lacZ gene provided by the host is
provided either episomally (via a low copy number
plasmid or F-factor) or stably integrated into the
bacterial chromosome. The alpha portion of lacZ is
used because 1) the ~-gal protein formed by alpha-
complementation is known to be weaker in activity than
the contiguous protein, minimizing the possibility of
background blue plaques due to inefficient repression
by lacI, and 2) to provide a smaller and thus more
easily characterized lacZ target should this gene be
used in mutagenesis studies. The re~uirements of the
host E. coli in this system are the following:
lacI(-), laeZ-M15, restriction(-). All cloning steps
are outlined in the figures 4 through 8 and are done
using standard procedures (Sambrook, J. et al.,
Molecular Clon na. A Laborator~ Manual. 2nd. Ed. Cold
Spring ~arbor Laboratory 1989).
The embodiment described utilizes the alpha
portion of lacZ with lacI. The complete lacZ can also
be used by providing a means to maintain complete
repression by lacI until induction is desired. This
can be done in a variety of ways including control of
M15 laZ expression by a lambda specific promoter
(PR') which prevents lacZ expression in the host E.
coli until several minutes following infection by the
- .:
. - , ~ : . ~- ;
:: . : ~, ~ ~
~ ~ 7 ~ PCT/US91/023~
- 60 -
bacteriophage, allowing lacI levels to build up to
suitable levels to enable complete repression.
Additionally, low lavels of lac repressor can be
maintained in the host to assist in repression by lacI
until induction occurs, either by a mutation in lacI
or by addition of IPTG to the system. A third
alternative is to use an altered lacI gene which gives
rise to a repressor protein with higher specific
- activity, thereby allowing stronger repression of ~- -
galactosidase production.
The source of starting materials for the
cloning procedures are as follows: the pBluescript II
SK+ and SK-, pBS~+), lambda gtll, and lambda L2B are
available from Stratagene Cloning Systems, La Jolla, -
CA. Lambda L47.1 and pPreB: Short, J.M., et al.,
Nucleic Acids Res., 16:7583-7600. pMJR1560 is
available from Amersham Corp., Arlington Heights,
Illinois.
Rapid sequencing of the mutagenized lacI gene
within the làmbda vector is facilitated by
incorporating "lambda ZAP" excision sequence within
the lambda vector. (Short, J.M. et al., Nucleic Acids
Res., 16:758~-7600 (1988). Lambda ZAP is a lambda
phage vector which permits in vivo excision of inserts
from the lambda vector to a plasmid. This is possible
because the lambda phage contains the two halves of an
fl bacteriophage origin of replication. In the
presence of proteins supplied by fl helper phage, all
~NA present between the two partial fl origins is
automatically excised from the lambda phage. The two
halves come together to form an intact fl origin. The
resulting phagemid contains a Col El origin of
replication and an ampicillin resistance gene, thus
the insert is effectively subcloned into a plasmid
vector. All sequences between the two partial fl
. .
... . . .
- : - , . :: . . . : : :
WO9l/15S79 2 ~ ~ ~ 7 ~ ~ PCT/US91/023~
~` - 61 -
origins are excised as a plasmid within hours.
In the mutation analysis vector, these fl
origins are positioned so that the lacI gene can be
automatically excised from the lambda phage from the
mouse genomic DNA. Following this conversion from
phage to plasmid, the insert may be rapidly sequenced
or characterized by other known methods.
Characterization of a large number of mutations within
the lacI gene can be completed within 3 days following
isolation of mouse genomic DNA, as opposed to several
months using standard techniques.
In the example described herein, a lambda ZAP
is used to convert the test DNA inserts from
integration in the lambda vector to a plasmid. Other
systems may also be used which allow excision and
recircularization of a linear sequence of DNA thereby
providing a rapid means with which the test DNA
sequence may be transferred from the phage to a form
suitable for analysis. Such other systems include,
but are not limited to, the use of FLP-mediated
(Senecoff, J. et al., Proc. Natl. Acad. Sci. USA, -
82:7270-7274 (1985); Jayaram, M., Proc. Natl. Acad. --~
Sci USA, 82:5875-5879 (1985); McLeod, M., Mol. Cell.
Biol., 6:3357-3367 (1986); Lebreton, B. et al.,
Genetics, 118:393-400 (1988)) or Cre-lox site specific
recombination techniques (Hoess, R. et al., J. Mol.
Biol., 181:351-362 (1985); Hoess, R. et al., Proc.
Natl. Acad. Sci. USA, 81:1026-1029 ~1984)).
The embodiments described above utilize the E.
coli beta-galactosidase gene as a test DNA sequence,
which allows phenotypeæ that are positive and negative
for mutation to be observed. Other potential test DNA
sequences include (but are not limited to): the lac I
repressor, the cl repressor, any antibiotic resistance
gene sequence (ampicillin, kanamycin, tetracycline,
- . .: . : "
- . . ~ ~ : :
- . , , . . ~ . . ~ . .
~ WO91/15579 PCT/~S91/023~
2~7'~ 62 - ~
neomycin, chloramphenicol, etc.), the lambda red and
gam gene sequences, a thymidine kinase gene sequence,
a xanthine-guanine phosphoribosyl transferase gene
sequence, sequences that code for restriction enzymes
or methylation enzyme~, a gene sequence that codes for
luciferase, and/or a tRNA stop codon or frameshift
suppresscr gene sequence.
Even more general models can be made that
eliminate the cos sites, although the excision
mechanism now becomes different. By bracketing the ~ -
test DNA sequence(s) with convenient restriction
sites, as shown in Figure 2, the test sequence(s) can
be separated away from the mouse DNA with restriction
enzymes and subsequently ligated with lambda or cosmid
vectors which contain cos sites or if the test
sequence is linked to a replication origin it can be
transformed directly. Background can be reduced in
such a system by including with the test DNA sequences
a sequence that is necessary for lambda phage
replication, which is then cloned with the test DNA
sequence into a lambda genome deficient or defective
in that sequ~nce.
5. Preparation of a Modified Lambda Genome
2S Containinq a Target Gene System
The modified Lambda genome, designated Lambda
LIZ alpha, is prepared through a series of molecular
gene manipulations as diagrammed in Figures 4 through
8.
Figure 4 depicts the construction of pBlue
MI-. p81uescript SK- (Stratagene, La Jolla,
California) is modified using site directed
~utagenesis to introduce an Ava III restriction site
at a position 5' to the open reading frame for the
LacI gene, but downstream from the ampicillin
.. :- . - .: .: .. . . . - .: . .: .. . .: .. .~ , .: , . : - .. . . .
WO91/15579 63 2 ~ ~ 9 7 q 2 PCT/US91/023~
resistance gene and the ColEl origin of replication
present on pBluescript to form pBlue MI-.
Figure 5 depicts the construction of pLacIq.
pBluescript II SK+ (Stratagene) is digested with the
restriction enzymes PstI and EcoRI, both which cleave
in the polylinker region to form linearized
pBluescript SK+ lacking the small fragment derived
from the polylinker. pMJRl560 (Amersham Corporation,
Arlington Heights, Illinois) is digested with the
restriction enzymes PstI and EcoRI to release a LacIq-
containing fragment that is separated by agarose gel
electrophoresis and eluted from the gel. The lacIq-
containing fragment is then ligated into the
linearized pBluescript SK~ to form pLacIq.
Figure 6 depicts the construction of pInt.l.
A double stranded DNA segment defining multiple
cloning sites (a polylinker) is produced by synthetic
olignucleotide synthesis and annealing. The polylinker
contains multiple restriction endonuclease recognition
sequences including two Ava III sites flanking XbaI,
KpnI and PvuI sites. The polylinker is digested with
Ava III to form Ava III cohesive termini on the
polylinker. pBlueMI- is digested with Ava III and the
polylinker is ligated into pBlueMI-to introduce the
PvuI site into the Ava III site and form pInt.l
Figure 7 depicts the construction of
pPreL2cIqZ (pPRIAZ~. To that end, pPre B is first
prepared as described by Short et al, Nucl. Acids
Res., 16:7583-7600 (l988).
To prepare pPre B, plasmid pUC l9 (ATCC
#37254) described by Yanisch-Perron et al, Gene,
33:103-ll9 (1985), was digested with EcoRI,
dephosphorylated and ligated to complementary
oligonucleotides, each having compatible EcoRI ends
and defining a T7 RNA polymerase promoter as described
: . . - . . . . . .. : . -: ~ :
WO91/15579 ~ 7~ 64 - PCT/US91/023
by McAllister et al, Nucl. Acids Res., 8:4821-4837
(1980)to form pJF3 having the T7 promoter oriented to
direct RNA synthesis towards the multiple cloning site
of pUC 19. pJF3 was digested with HindIII,
dephosphorylated and ligated to complementary
oligonucleotides having HindIII-compatible ends and
defining a T3 RNA polymerase promoter as described by
Morris et al, Gene, 41:193-2000 (1986). A resulting
plasmid, designated pBluescribe (pBS) was isolated
that contained the T3 promoter oriented to direct RNA
synthesis towards the multiple cloning site of pUC 19.
pBS was then digested with AatII and NarI, treated
with mung bean nuclease and alkaline phosphatase, and
ligated to a 456 base pair (bp) RsaI/DraII blunt-end -
fragment isolated from the pEMBL8 plasmid described by --
Dente et al, Nucl.Acids Res., 11:1645-1655 (1983).
The 456 bp fragment contains the intergenic region of
fl phage, but does not contain the fl gene II promoter
sequence. Phagemid clones were isolated from the
resulting ligation mixture and clones were isolated
containing both orientations (+ or -) of the
intergenic region and are designated pBS(+) or pBS(-),
where "+" indicates that the intergenic region is in
the same orientation as the lacZ gene. pBluescript
SK(-) and SK(+) were produced from pBS(-) and pBS(+),
respectively, by digestion of each with EcoRI and
HindIII, followed by blunt ending with Klenow fragment
of DNA polymerase I. The blunt-ended molecules were
ligated to a blunt-ended synthetic polylinker
containing 21 unique restriction sites as described by
Short et al, Nucl.A~cids Res., 16~7583-7600 (1988), to
form pBluescript SK(-) or SK(~), respectively. A
majority of the terminator portion of the fl
intergenic region is contained on the RsaI (position
5587) to HinfI (position 5767) restriction fragment
.. ,. . . , , ,,, , -
, . . - . - .~ . , - , - ~ - - : . .
WO~l/15~79 2 ~ PCT/US9l/023
65 -
isolated from pEMBL8. The remaining terminator
sequences were provided by preparing synthetic
oligonucleotides as described by Short et al, suDra,
to provide a complete terminator, a gene II cleavage
signal and unique restriction sites for EcoRV and
NdeI. The synthetic oligonucleotide and the RsaI/HinfI
fragment were ligated with a 3009 bp DraI/NdeI
fragment obtained from pBS to form the plasmid pBST-B.
The initiator domain of the fl intergenic region was
separately cloned by digesting pEMBL8 with Sau961 and
DraI to form a 217 bp fragment that was then blunt-
ended with Klenow and then subcloned into the NarI
site of pBST-B to form pBSIT0#12. pBluescript SK(-)
was digested with NaeI and partially digested to cut
only at the PvuI site located adjacent to the fl
origin, and the resulting fragment lacking the fl
origin was isolated. The isolated fragment was
ligated to the NaeI/PvuI fragment of pBSIT0#12 that
contains the terminator and initiator regions of the
fl intergenic region to form plasmid pPre B.
Lambda gtll (ATCC #37194) was digested with
KpnI and XbaI to produce a 6.3 kilobase (Xb) fragment
containing the LacZ gene, which was agarose gel
purified. pBS(+) prepared above and available from
Stratagene was digested with RpnI and XbaI, and the
resulting LacZ fragment was ligated into pBS(+) to
form pBS(LacZ). pLacIq from above was digested with
NarI and Sal I and the resulting small fragment
containing the LacIq gene was isolated. pBS(LacZ) was
digested with NarI and SalI and the resulting large
fragment containing the LacZ gene was isolated and
ligated to the small LacIq-containing fragment to form
pLacIgZ. pInt.1 prepared above was digested with
KpnI, and the resulting linear molecule was ligated to
the LacI-LacZ fragment, produced by digesting pLacIqZ
.. . . . . . ..
,,, ~ ~ . . . ~ :
WO91/l5~79 PCT/US91/023
2,a~7~ 66 -
prepared above with KpnI, to form pIntLacIqZ.
pIntLacIqZ was then digested with XbaI, blunt-ended
with Klenow, digested with ScaI, and the resulting
large fragment containing LacZ-LacIq and most of the
ampicillin resistance gene was isolated. pPre B
prepared above was digested with PvuI, blunt ended
with mung bean nuclease, digested with ScaI and the
resulting fragment containing the terminator and
initiator fl intergenic region components was isolated
and ligated to the pIfntLacIqZ-derived large fragment
to form plasmid pPreLacIqZ (pPRIAZ)
Figure 8, shown in two panels 8A and 8B,
depicts the construction of Lambda LIZ alpha. To that
end, Lambda L47.1, described by Loenen et al, Gene,
10:249-259 (1980), by Maniatis et al, in "Molecular
Cloning: a Laboratory Manual" , at page 41, Cold
Spring Harbor, New York (1982), and by Short et al,
supra, and having the genetic markers (srIlambdal- -~
2)delta, imm434 cI-, NIN5, and chi A131, was digested
with EcoRI, HindIII and SmaI to form a Lambda L47.1 ~ -
digestion mixture. Lambda L2B, available from
Stratagene, was first digested with XbaI, then treated -
with Rlenow to fill-in the 5' XbaI overhang, then
digested with MluI to form a L2B digestion mixture.
The L47.1 and L2b digestion mixtures were ethanol-
precipitated to prepare the DNA for ligation, and were
then ligated to pPRIAZ prepared above that had been
linearized with NdeI to form Lambda LIZ alpha.
The final construction, Lambda LIZ alpha, is a
preferred modified Lambda bacteriophage for use in the
present invention because it combines the elements of
1) a reporter gene in the form of the alpha component
of LacZ, 2) the lambda bacteriophage excision
capability provided by the presence of the cos sites
at the termini, 3) an indicator gene system in the
~-
:
.
SUBSTITIJTE SHEET
~ . . ... - . . . . ~ . . . . . . . - . ..... . .. . .. . . .
! '
.' ~. ', ' ' :
', . . . ',, ' ~.. . .
WO9l/15579 PCT/US91/023
- 67 - 2 ~ 7~.~Cl2
form of the LacIq target gene, including a lacI
promoter and the repressor structural gene sequences,
and ~) an fl origin of replication arranged according
to the in vivo excision system of Short et al, su~ra,
that allows quick isolation of the mutated test gene
from positive colonies containing the mutated test
gene for sequencing.
6. Production of Chimeric SCID/hu Mice
A. Preparation of Human Lymphocytes
Peripheral blood lymphocytes (PBLs) are
isolated from venous blood drawn from volunteer
donors. First, one hundred milliliters (ml) of blood
are anticoagulated with a mixture of 0.14 M citric
acid, 0.2 M trisodium citrate, and 0.22 M dextrose.
The treated blood is layered on Histopaque-1077
(Sigma, St. Louis, Missouri) to form isolated PBLS
which are recovered by centrifugation at 400 X g for
30 minutes at 25C. Isolated PBLs are then washed
twice with phosphate buffered saline (PBS) (150 mM
sodium chloride and 150 mM sodium phosphate, pH 7.2 at
25C). The resulting PBL cell pellet is resuspended
in PBS to a concentration of 1 X Io8 cells/ml.
Lymphocytes are also isolated from tonsils
obtained from therapeutic tonsillectomies from
consenting patients. The tonsils are first
homogenized and then lymphocytes are isolated over
Histopaque as described above.
Subject rDNA are then inserted into the
isolated lymphocytes using techniques known to one
skilled in the art. Preferred techniques are
electroporation of lymphocytes and calcium chloride
permeabilization of the lymphocytes.
B. Preparation of_SCID Mice
- ~ . .: .
:
:. ~ : . : : . , : . , . : .- .
i 091~1557~ 7 ~ 68 - PCTtUS~1/023
SCID mice having the autosomal recessive
mouse mutations scid are obtained from Imdyme (San
Diego, California). Alternatively, SCID mice are
derived from an inbred strain of mice, C.B-17 (Balb/c-
C57BL/Ka-Igh~lb/ICR (N17F34) as described by ~osma et
al., Nature, 301: 527-530 (1983). Analysis of the
pedigree of mice lacking IgM, IgGl or IgG2a determined
that the defect was inheritable and under the control
of the recessive scid gene. Bosma et al., supra. A
colony of mice can be established which are homozygous
for the defective gene. The SCID mice are maintained
in microisolator cages (Lab Products, Maywood, New
Jersey) containing sterilized food and water.
C. Preparation of SCID~hu Chimeras
SCID mice obtained in Example 6b are
reconstituted by intraperitoneal injection with at
least 5 X 107 human PBLs or tonsil lymphocytes
prepared in Example 6a. The recipient SCID mice are
designated SCID/hu chimeras which contain the subject
rDNA. The human PBL reconstituted SCID mouse model is
then used for a~saying the effects of mutagens on
human cells as described in this invention.
The foregoing is intended as illustrative of
the present invention but not limiting. Numerous
variations and modifications can be effected without
departing from the true spirit and scope of the
invention.
.
. .