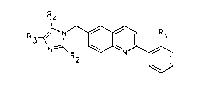Note: Descriptions are shown in the official language in which they were submitted.
OUINOLINE DERIV~TIVE8, PRC)CE:SE~ FOR THEIR PRl~PARZ~TION~,
AND ~HEIR T~E~PEUTIC APPLICATION~3
The present invention relates to quinoline
derivatives, their preparation and their therapeutic
applications.
The compounds of the invention are of the general
formula (I)
R4
~ ~N~ ~ ~ (I)
in which
Rl represents either ~ lH-tetrazol-5-yl group, or a CO2H group,
R2 represents either a (C17)alkyl group or a (C26)alkenyl
group,
R3 and R4 represent, independently of each other, either a
hydrogen atom or a halogen atom or a cyano group or a
(C17)alkyl group or a (C37~cycloalkyl(C14)alkyl group or an
aryl group or an aryl(C14)alkyl group or an aryl(C26)alkenyl
group or a (CH2)m-COR5 group in which m = 0 to 4 and ~
represents a hydrogen atom, an OH group, a (Cl6)alkoxy group or
an NR7R8 group, R7 and R8 representing, independently of each
other, a hydrogen atom or a (Cl4)alkyl group or a (CH2)n-R6
group in which n = 1 to 4 and R6 represents an OH group, a
(C16)alkoxy group, a (Cl4)alkoxy(Cl4)alkoxy group or a
(C37)cycloalkyl(Cl4)alkoxy group.
The preferred compounds of the invention are those
for which R1 represents either a lH-tetrazol-5-yl group or a
CO,H group,
R2 represents a (C17~alkyl g~oup,
R3 represents either a halogen atom or a ~C17)alkyl group or an
aryl(C14)alkyl group,
R4 represents either a (CH2)m-CO~ group in which m = 0 to 4 and
R~ represents a hydrogen atom, an OH group, a (C16)alkoxy group
or an NR7R~ group, R7 and R~
representing, independently of each other, a hydrogen atom or a
(C~4)alkyl group,
or a (CH2)n-R6 group in which n = l to 4 and R6 represents an OH
group or a (C16)alkoxy group.
Finally, the preferred compounds are those for which R
represents a lH-tetrazol-5-yl group,
R2 represents a butyl group,
R3 represents either a chlorine atom or an ethyl group or a
phenethyl group,
R4 represents a CH2OH, CHO, CO2H, CO2CH3, CO2C2H5 or CH2OC~3 group-
The compounds of the invention may be in free form orin the form of pharmaceutically acceptable organic or inorganic
salts.
The compounds of the invention for which R1
represents a lH-tetrazol~5-yl group may be prepared according
to the scheme in Appendix I.
In a first stage, 4-methylbenzenamine(para-toluidine)
is reacted at the reflux temperature with a benzaldehyde of
formula (II), in which X represents a bromine or iodine atom,
in the presence of a catalyst such as 4-methylbenzenesulphonic
acid (para-toluenesulphonic acid or PTSA), in solution in
benzene. After cooling, propiolic acid is added and the mixture
3 ~ f '~
is heated at the reflux temperature in order to obtain a
compound of formula (III).
In a ~econd stage, a mixture of the compound (III)
and of copper(I) cyanide in a solvent such as pyridine is
heated in order to obtain 2-(6-methylquinolin-2-yl)benzonitrile
(IV).
In a third stage, 2-(6-methylquinolin-2-yl)benzo-
nitrile is reacted with an organometallic azide such as
trimethyltin azide or a metallic azide such as sodium azide in
order to obtain a compound over which a stream of gaseous
hydrochloric acid is passed in order to obtain 6-methyl-
2-[2-(lH-tetrazol-5-yl)phenyl]quinoline (V). The first reaction
is carried out in a solvent such as xylene at the reflux
temperature; the second reaction is carried out in a solvent
such as toluene/tetrahydrofuran mixture at room temperature.
In ~ fourth stage, the tetrazole group of 6-methyl-
2-[2-(lH-tetrazol-5-yl)phenyl]quinoline (V) is protected with a
protecting group of formula CR13R14R15, in which R13, R14 ~nd R1s
each represent independently of each other a (C12)alkyl group
or an aryl group; in this stage, the compound (V) is reacted
with a protecting agent such as for example trityl chloride, at
room temperature in a solvent such as dichloromethane in the
presence of a base such as N-methylmorpholine or triethylamine
and a compound of formula (VI) is obtained in which R13, R14 and
R15 are as previously defined. The protection of the tetrazole
group preferably occurs in position 2.
In a fifth stage, the methyl group in position 6 of
the quinoline of formula (VI) is functionalised by introducing
therein a departing group. If the departing group is a bromo
J
-- 4
radical, then a compound of formula (VI) is reacted with
N-bromosuccinimide in order to obtain a compound of formula
(VII) in which CRl3Rl4R15 is as previously defined; the reaction
is carried out at the reflux temperature in a solvent ~uch as
carbon tetrachloride in the presence of an initiator such as
benzoyl peroxide or ~,~' or azobisisobutyronitrile.
In a sixth staye, a compound of formula (VII) is
reacted with an imidazole of formula (VIII) in which ~2~ R3 and
R4 are as previously defined, in order to give a derivatîve of
formula (IX). The reaction is carried out in a solvent such as
dimethylformamide at a temperature of O-C to 50DC in the
presence of a base such as potassium carbonate.
In a seventh s'age, deprotection of the tetrazolyl
group is carried out in order to obtain a compound of formula
(I a).
The compounds of the invention for which R1
represents a CO2H group may be prepared according to the scheme
in Appendix II.
In a first stage, 4-methylbenzenamine (para-
toluidine) is reacted at the reflux temperature with abenzaldehyde of formula (II) in which X represents a bromine or
an iodine atom in the presence of a catalyst such as
4-methylbenzenesulphon.ic acid tpara-toluenesulphonic acid or
PTSA) in solution in benzene. After cooling, propiolic acid is
added and the mixture is heated at the reflux temperature in
order to obtain the compound of formula (III).
In a second stage, a mixture of the compound (III)
and copper(I) cyanide in a solvent such as pyridine is heated
in order to obtain 2-(6-methylquinolin-2-yl) benzonitrile (IV).
S~.,t~ Cj'j!
In a third sta~e, 2-(6 methylquinolin-2-yl)-
benzonitrile is rPacted with an alcohol R-OH (V'), where R is a
branched or unbranched (C14)alkyl radical, in the presence of
an acid, for example sulphuric acid, in order to obtain a
quinoline of formula (VI'~ in which R is as previously defined.
In a fourth stage, the methyl group in position 6 of
the quinoline is functionalised by introducing therein a
departing group~ If the departing group is a bromo radical,
then the quinoline of formula ~VI') is reacted with
N-bromosuccinimide in order to obtain the quinoline of formula
(VII') in which R is as previously defined; the reaction is
carried out in a solvent such as carbon tetrachloride in the
presence of an initiator such as benzoyl peroxide or
~,~'-azobisisobutyronitrile at the reflux temperature.
In a fifth stage, the quinoline of formula (VII') is
reacted with an imidazole of formula (VIII), in which R2, R3
and R4 are as previously defined, in order to give a derivative
of formula (IX'~. The reaction is carried out in
dimethylformamide at a temperature of 0~C to 50~C in the
presence of a base such as potassium hydroxide or potassium
carbonate.
In a sixth stage, the ester functional group of the
quinoline of formula (IX') is hydrolysed in order to obtain a
compound of formula (I b).
The intermediate compounds are novel and are part of
the invention. They are of the formula (X)
- 6 ~ d~". ~ ?~
W~ (X)
in which
W represents either a m~thyl group or a -CH2R1l group in which
R11 represents a chlorine atom, a bromine atom or a departing
group ORl2, R12 being a group such as a tosyl group or a mesyl
group,
Z represents either a halogen atom or a cyano group or a
lH-tetrazol-5-yl group or a lH-tetrazol-5-yl group protected by
a protecting group of formula CR13Rl4R1s~ R13, R,4 and R15
representing independently of each other a (C12)alkyl group or
an aryl group, or a COOR group, R being a branched or
unbranched (C14)alkyl group.
The starting compounds are commercially available or
are described in the literature or can be prepared using
methods which are described therein or which are known to a
person skilled in the art.
The following examples illustrate the invention.
Microanalyses and IR and NMR spectra confirm the
structure of the compounds obtained.
Example 1
2-butyl-4 chloro-l [[2-[2-(lH-tetrazol-5-yl)phenyl]-
quinolin-6 yl]methyl]~lH-imidazole-5-carboxaldehyde,
hydrochloride.
1.1 2-(2 bromophenyl)-6-methylquirloline
50 g (270 mmol~ of 2-bromobenzaldehyde are heated to
the reflux temperature, in a round-bottomed flask fitted with a
Dean-Stark, with 29.5 g (~76 mmol) of para-toluidine and 0.5 g
of para-toluenesulphonic acid in solution in one liter of
benzene. When the removal of water has been completed (about 5
ml), 8.3 ml (135 mmol) of propiolic acid are added to the
reaction medium previously cooled to around 50C. A substantial
release of CO2 is observed and the mixture i5 refluxed for 3
hours. The reaction is monitored by thin-layer chromatography
in a dichloromethane and hexane mixture (70/30). Under our
experimental conditions, it was necessary to add a 20 % excess
of propiolic acid followed by refluxing for 1 hour in order to
bring the reaction to completion. The solvent is evaporated
under reduced pressure and th~ residue is purified by
chromatography on a silica column, eluting with a
dichloromethane and hexane mixture (70/30).
22 g of the expected derivative are recovered in the
form of a crystallised compound.
wt. = 22 g m.p. = 92C Yield = 27 %
lH NMR ~200 MHz, CDC13): ~ 2.55 (sl 3H), 7.25-7.70 (m, 7H),
8.02-8.15 (m, 2H).
Similarly, 2-(2-iodophenyl)-6-methylquinoline is
prepared from 2-iodobenzaldehyde.
m.p. -- 77-77.5C
1.2 2-(6-methylquinolin-2-yl)benzonitrile
A mixture containing 15 g (50 mmol) of the compound
previously obtained in 1.1 and 5 g (56 mmol) of
copper(I)cyanide in 60 ml of pyridine is heated at 160DC for 12
hours under argon. The reaction is monitored by thin-layer
8 ~ r
chromatography (TLC) in dichloromethane. The pyridine is
evaporated under reduced pressure and the residue is taken up
in dichloromethane. The organic phase is washed several times
with an aqueous solution of ammonium hydroxide until the
aqueous phase i8 colorless. After a last wash with water, the
organic phase is dried over magnesium sulphate and the solvent
is evaporated. The residue is taken up in petroleum ether.
wt. = 9.6 g m.p. = 157C Yield = 78 %
1.3 6-methyl-2-[2-(lH-tetrazol-5-yl)phenyl]quinoline
hydrochloride
9.6 g (39.29 mmol) of the nitrile previously obtained
in 1.2 and 14.96 g (72.7 mmol) of trimethyltin azide are
introduced into 110 ml of xylene. This mixture is refluxed for
15 hours. After cooling, the solid is ~iltered and suspended in
115 ml of toluene and 7 ml of tetrahydrofuran. The mixture,
which is cooled using an ice bath, is subjected to bubbling of
hydrochloric gas for 2 hours. The insoluble fraction is
recovered by filtration, then washed with toluene and with
water.
wt. = 13 g
1.4 6-methyl-2-[2-[2-(triphenylmethyl)-1ll-tetrazol-
5-yl]phenyl]quinoline
80.5 g (0.219 mol) of the compound previously
obtained in 1.3, 60 ml (0.547 mol) of N-methylmorpholine and
73.1 g (0.262 mol) of trityl chloride are added to one litre of
dichloromethane at room temperature. I'he solution is stirred
overnight, taken up in water, and the organic phase is washed
twice with water and then dried. The solvent is evaporated and
the residue is crystallised from a minimum amount of ether.
wt. = 119 g m.p. = 176-177C Yield = 87 %
1.5 6-bromomethyl-2-[2-[2-(triphenylmethyl)-lH--tetrazol-
5-yl]phenyl]quinoline
10 g (0.189 mol) of the compound previously obtained
in 1.4 is added to 300 ml of carbon tetrachloride and the
mixture is heated to around 60C until complete dissolution has
taken place. 3.7 g (0.208 mol) of N-bromosuccinimide and 60 mg
(0.0037 mol) of ~,~'-azobisisobutyronitrile are added all at
once at this temperature. The mixture is refluxed for 2 to 3
hours until the N-bromos~ccinimide disappears. 100 ml of water
and 300 ml of dichloromethane are added to the cooled mixture.
The organic phase is washed several times with water and then
dried~ The solvent is evaporated and the residue is triturated
in diisopropyl ether. A 90 % pure product is obtained which
will be used as it is.
wt. - 10.3 g
1.6 2-butyl-4-chloro-1-[[2-[2-t2-(triphenylmethyl)-
lH-tetrazol-5-yl]phenyl~quinolin-6-yl]methyl]-lH-imidazole-
5-carboxaldehyde
a) 2-butyl-5-chloroimidazole-4-methanol
9.52 g (0.071 mol) of N-chlorosuccinimide are
introduced, at a temperature of between 0 and 5C, into 10 g
(0.065 mol) of 2-butylimidazole-4-methanol in
suspension in 200 ml of ethyl acetate. The mixture is stirred
overnight while maintaining this temperature. The solution is
filtered and the solid is washed with 20 ml of ice-cold ethyl
acetate, drained and washed with water in order to remove
traces of succinimide and starting product, and then dried.
wt. = 7.3 g m.p. = 143~C Yield = 73 %
b) 2-butyl-4-chloroimidazole-5-carboxaldehyde
A solution containing 17.3 g (0.092 mol) of the
compound previously obtained, dissolved in 52 ml of acetic
acid, is added dropwise, so that the temperature of the medium
is kept between 22 and 28~C, to 133 g (0.243 mol) of ammonium
cerium nitrate solubilised in 200 ml of water. The reaction
medium is allowed to stand for 3 to 5 hours until the solution
becomes colourless. The medium is cooled and the pH is adjusted
to 5-6 by adding 10 N sodium hydroxide. The product formed is
extracted using 3 portions of ether and the organic phase is
washed with sodium bicarbonate, dried and the solvent is
evaporated. 16.4 g of compound are obtained which are
recrystallised from cyclohexane.
wt. = 13.9 g m.p. = 92-93~C Yield = 81 %
c) 2-butyl-4-chloro-l-[[2-[2-[2-(triphenylmethyl)~lH-tetrazol-
5-yl]phenyl]quinolin-6-yl]methyl]-lH-imidazole-5-carboxaldehyde
5.11 g (0.037 molj of potassium carbonate and l9.1 g
(0.028 mol), in portions, of the compound previously obtained
in 1.5 are added, while cooling on an ice bath, to 5.07 g
(0.027 mol) of 2-butyl-4-chloroimidazole-5-carboxaldehyde in
solution in 40 ml of dimethylformamide. The mixture is stirred
under argon overnight, allowing the temperature to return to
room temperature. The reaction medium is poured into water, and
the solid formed is recovered and dried. The compound obtained
is purified by passing through a silica column, eluting with a
t) ~ 6
toluene/ethyl acetate mixture (90/10).
wt. = 11.9 g m.p. = 165C Yield = 57 %
1.7 2-butyl-4-chloro-1-[[2-[2-(lH-tetrazol-
5-yl)phenyl~quinolin-6-yl]methyl]-lH-imidazole-5-car~oxaldehyde
10 ml (0.040 mol) of 4 N hydrochloric acid are added
to 11 g (0.015 mol) of the compound previously obtained in 1.6,
in solution in 130 ml of tetrahydrofuran. The mixture is
stirred under an argon atmosphere overnight at room
temperature.
The hydrochloride salt of the expected product
crystallises from the reaction medium. It is recovered by
filtration and washed with 10 ml of ice- cold tetrahydrofuran.
wt. = 7.4 g m.p. = 185~C (dec) Yield = 94 %
lH NMR (200 MHz, DMS0): ~ 0.8 (t, 3H), 1.2-1.4 (m, 2H),
1.5-1.65 (m, 2H), 5.8 (s, 2H), 7.6-8.0 t~, 8H), 8.6 (d, lH),
9.7 (s, 1~)
Example 2
2-butyl-4-chloro-1-[[2-[2-(lH-tetrazol-
5-yl)phenyl]quinolin-6-yl]methyl]-lH-imidazole-5-carboxylic
acid
5 ml of methanol, 241 mg (3.7 mmol) of potassium
cyanide, 64 ~1 of acetic acid and 1.5 g of manganese dicxide
are added to 344 mg (0.73 mmol) of the aldehyde derivative
previously obtained in 1.6. The mixture is stirred for 48 hours
at room temperature. The methyl ester formed is recovered after
filtration and evaporation. 2.55 ml (2.55 mmol) of lN sodium
~ Y~ 3
- 12 -
hydroxide arP immediately added and the solution is left for 3
hours at room temperature. The pH of the solution is adjusted
to 3.5 and the insolubles are filtered, washed and driedn
wt. = 230 mg m.p.=170~C (dec) Yield = 70 %
1H NMR (200 MHz, DMS0): ~ 0.8 (t, 3H), 1.2-1.4 (m, 2H),
1.5-1.65 (m, 2H), 2.65 (t, 2H), 5.8 (s, 2H), 7.45-8 (m, 8H),
8.6 (d, lH)
Example 3
2-butyl-4-chloro-1-[~2-[2-(lH-tetrazol-
5-yl)phenyl]quinolin-6-yl]methyl]-lH-imidazole-5-methanol
3.1 2-butyl-4-chloro-1-[[2-[2-[2-(triphenylmethyl)-
lH-tetrazol-5 yl]phenyl]quinolin-6-yl]methyl]-lH-imidazole-
5-methanol
330 mg (6.9 mmol) of sodium borohydride are added in
small portions to 1.65 g (2.3 mmol) of the compound obtained in
1.6, in solution in 150 ml of methanol. After reacting for 30
minutes, the mixture is concentrated and poured in a 2N
solution of sodium hydroxide. The compound obtained is
extracted with dichloromethane. The crude product thus obtained
is purified by chromatography on a silica column with a
chloroform/ethyl acetate (80/20) mixture.
wt. = 1.12 g m.p. = 175C Yield = 68 %
3.2 2-butyl-4-chloro-1-[[2-[2-(lH-tetrazol-
5-yl)phenyl]quinolin-6-yl]methyl]-lH-imidazole-5-methanol
1.1 g of the compound obtained previously in 3.1 is
J ~"
-- 13 --
dissolved in a mixture containing 31 ml of tetrahydrofuran, 31
ml of methanol and 5 ml of acetic acid. The solution is
refluxed for 24 hours. The solvents are evaporated, the residue
is triturated in ether and the insolubles are recovered by
filtration. The product is recrystallised in 2-butanoneO
wt. = 450 mg m.p. = 150-152-C Yield = 62 %
H NMR (400MHz, DMS0): ~ 0.75 (t, 3H), 1.25 (m, 2H), 1.5
~m, 2H), 2.53 (m, 2H), 4.36 (s, 2H), 5.2 (s, lH), 5.46 (s, 2H),
7.53, (m, 3H), 7.7-7.79 (m, 4H), 7.93 (d, lH), 8.3 (d, lH).
Example 4
2-butyl 4 -phenethyl-1-[[2-[2-(lH-tetrazol-
5-yl)phenyl]quinolin-6-yl]methyl]-lH-imidazole-5-carboxaldehyde
4.1 2-butyl-4-phenylethenylimidazole-5-carboxaldehyde
10.2 g of (E)-~-tri-n-butylstannylstyrene and 1 g of
tetrakis(triphenylphosphine)palladium(0) are added to 6 g of
2-n-butyl-4-iodoimidazole-5-carboxaldehyde under an argon
atmosphere in 80 ml of dry toluene. The mixture is refluxed for
6 hours. The solution i5 clarified by filtration after adding
animal charcoal and the solvent is evaporated. The residue is
taken up in hexane in order to remove the tin derivatives. The
compound obtained is purified in hydrochloride or oxalate form.
hydrochloride m.p. = 225~C (decomposition)
oxalate m.p. = 217C
wt. = 5.1 g Yield = 99 %
4.2. 2-butyl-4-phenethylimidazole-5-carboxaldehyde
- 14 - 2 ~ 3 ~
A solution of 2.5 g of the compound obtained
previously in 4.1 and dissolved in 50 ml of ethanol is
subjected to catalytic hydrogenation at room temperature and at
atmospheric pressure in the pre~ence of palladium on carbon as
catalyst. After 30 minutes, the catalyst is removed by
filtration and the solvent is evaporated. 2.5 g of the expected
compound are obtained in the form of a gum. The compound
obtained is purified in hydrochloride form.
Hydrochloride m.p. = 179.5~C
4.3 2-butyl-4-phenethyl-
1-[[2-[2-[2-(triphenylmethyl)-lH-tetrazol-5-yl]phenyl]quinolin-
6-yl~methyl]-lH-imidazole-5-carboxaldehyde
This compound is obtained by reaction between
2-butyl-4-phenethylimidazole-5-carboxaldehyde and the
bromine-containing derivative described in Example 1.5
according to the process described in Example 1.6.
m.p. = 142.5C
4.4 2-butyl-4-phenethyl-1-[[2-[2-(lH-tetrazol-
5 yl)phenyl~quinolin-6-yl]methyl]lH-imidazole-5-carboxaldehyde
This compound is obtained by detritylation of the
compound obtained previously in 4. by heating for 24 hours in
methanol at the reflux temperature.
lH NMR (200MHz, CDCl3): [sic] 0.8 (t, 3H), 1.25 (m, 2H), 1.6
(m, 2H), 2.5 (t, 2H), 3.1 (m, 4H), 5.75 (s, 2H), 7-7.5 (m,
llH), 7.6 (d, lH), 7.75 (d, lH), 7.9 (d, lH), 9.55 (s, lH).
Example 5
2-butyl-4-phenethyl-1-C[2-[2-~lH-tetrazol-
5-yl)phenyl]quinolin-6-yl~methyl]-lH-imidazole-5-methanol.
5.1 2-butyl-4-phenethyl-1-[[2-[2-[1-(triphenylmethyl)-
lH-tetrazol-5-yl]phenyl~quinolin-6-yl]methy~]-lH-imidazole-
5-methanol
This compound is obtained from the compound obtained
previously in 4.3, according to the process described in
Example 3.1.
m.p. = 150-C
5.2 2-butyl-4-phenethyl-1-[[2-[2-(lH-tetrazol
5 yl)phenyl]quinolin-6-yl]methyl]-lH-imidazole-5-methanol
This compound is obtained by detritylation of the
compcund obtained previously in 5.1 by heating for 24 hours in
methanol at the reflux temperature.
H NMR (200MHz, CDCl3-DMSO): a 0.8 (t, 3H), 1.3 (m, 2H), 1.6
(m, 2H), 2.65 (t, 2H), 2.9 (m, 4H), 4.15 (s, 2H), 5.4 (s, 2H),
20 7.15-7.9 (m, 13H), 8.1 (d, lH).
~m~
6-L[2-butyl-4-chloro-5-(methoxymethyl)-lH-imidazol-
1-yl]methyl]-2~[2-(lH-tetrazol-5-yl)phenyl]quinoline
1.5 g of the compound described in Example 3 are
introduced into 36 ml of methanol and 0.18 ml of concentrated
sulphuric acid are added. The mixture is refluxed for 15 hours,
the methanol is evaporated and the residue is taken up in a
s.,~ J~
mixture of 1 N svdium hydroxide and toluene. The aqueous phase
is recovered and acidified to pH 3 with hydrochloric acid. The
precipitate formed is filtered and it is purified by
chromatography on a silica gel column, eluting with a
dichloromethane/methanol/acetic acid mixture (95/5/0.1).
wt. = 0.4 g m.p. = 10~C (dec) Yield = 40 %
The following table illustrates the structures and
the physical properties of some compounds according to the
invention.
-- 17 --
o~J ~ .~ U , ~ ~ U .~ Lr
CL ~ ~ o~ _ co ~ r
__ _
~ ~ l .
_
~ O O O O 0~ ~ 0,~ O ~
_
Z~ ~ ~
\Z C:;~ O e~ ~ ~ S ~ V
~) .c:
___ __ _ __ _
:S ::: 3: ::C 5 ~_ :~ :1: ~s:
~ ~ N 3 N S N ~ N ~
_ ~~~ ~~) C~ ~~~ U ~~~ ~ ~J t
x~ ~ x~ ~r ~I~r x, ~:~ :~ x~
~- ~ O Z U C~ Z Z Z Z
. _ _
O rJ ~ ~r In ~ I_
1 8 ~ 3
U ~ U ~, ., ~ _~ ~ ~ ~ ~, ~=
E ~ ¦ ~ ~ o ~ ~ ~ I ~ ~ ~ ~ c,
~ ~ ~ ~ ~
~ ~ C~ C~ V C~ ~ ~ ~J ~ ~ V V V~ ~ O ~
~ S U~ U~ U~ U~ U U U~ U, U, U~ U, U~ U~ U~ U
- - - - ~ . -- -
~ ~c~
Z o ~ ~ ~ ~ ~ ~ r ~ ~ o ~ ~ ~ ~ n
$
_ ~ ~ a I
~ l o~ - l ~x~
r O O $ U O
_ _ _ D
:C' U ~ U U l~ ~ ~u
~:~ _ ~ ~ ~ o ~ ~ ~
:: ~: .a .. , ~ U
_ D ~ ~ ~ U ~ i U
Z (`~ `J ~ N 1~1
~,5,J~J~5~
- 20 -
The compounds of the invention have been the subject
of pharmacological studies which have demonstrated their
antagonistic properties to angiotensin II.
Test of binding of [3H]-anaiotensin II to rabbit adrenal
cortex
2 to 3-kg Fauves de Bourgogne male rabbits are used.
After sacrificing them by cervical dislocation, the adrenal
glands are excised and the cortex is dissected on a culture
plate cooled using ice. It is placed in 10 ml of an ice-cold
buffer solution at 10 mM of tris(hydroxymethyl)aminomethane
containing 0.33 M sucrose and 1 mM ethylenediaminetetraacetic
acid where the pH had been adjusted to 7.4 with hydrochloric
acid. The tissue is homogenised by means of an electric Potter
apparatus using 13 to and fro movements of the piston at a
speed of 1200 revolutions p~r minute. The volume of the
preparation is adjusted to 25 ml with tris-sucrose buffer
before centrifuging for 15 min at 1075 x g. The supernatant is
kept. The pellet is again homogenised after re-suspending in 10
ml of tris-sucrose buffer by passing through an electric Potter
and then centrifuged under the conditions previously described.
The supernatant obtained is added to the first supernatant and
they are centrifuged for 30 min at 47 800 x g. The pellets are
finally taken up in 150 volumes (that is to say 100 mg of
tissue in 15 ml of buffer) in a 50 mM solution of tris-HCl
buffer containing 150 mM of NaCl, 5 mM of
ethylenediaminetetraacetic acid, 1.25 ~g/ml of bacitracin,
100 ~M of phenylmethylsulphonyl fluoride and 0.2 % of bovine
serum albumin (pH = 7.4 at 25C).
2~
- 21 -
This suspension contains the adrenal cortex
microsomes and will be used as it is in the ctudies described
below.
Aliquot fractions of 100 ~1 of suspension are
incubated in the presence of [3H]-angiotensin II (New England
Nuclear, with a specific activity of 61 Ci/mmol) in a final
volume of 1 ml of tris-HCl buffer the composition of which has
previously been described. After incubating for 30 minutes at
25C, the microsomes are recovered by filtration on 0.45 ~m
Millipore HAWPTM cellulose nitrate filters previously
conditioned by soaking in a 1 % solution of bovine serum
albumin. The filters are washed three times with 5 ml of ice-
cold tris-HCl buffer. The amount of radioactivity bound to the
tissue and retained on the filters is measured by scintillation
spectrometry.
The non-specifi~ binding of [3H]-angiotensin II is
measured by incubation in the presence of 1 ~M of non-
radioactive angiotensin II. This non-specific binding
represents S to 10 % of the total amount of radioactivity bound
on the filter. The specific binding is the difference between
the total radioactivity recovered on the filter and the non-
specific radioactivity. The binding of [3H]-angiotensin is
measured in the presence of various concentrations of the test
compounds and the ICso, the concentration of the test compound
which inhibits 50 % of the specific binding of [3H]-angiotensin
II, is graphically determined.
The IC50 values of the compounds of the invention are
between 5 nM and 10 ~M.
- 2~ 3
Inhibition of the response to angiotensin II on rat blood
pressure
Male rats (Sprague-Dawley, Charles River France)
weighing 250 to 280 g are used, they are anaesthetized with
sodium pentobarbital (55 mg/kg i.p.) and are maintained under
artificial respiration tHarvardT~ respirator; frequency of
respiration of 70 ml per minute, volume of air l ml per lO0 g
of body weight). The animals are "spinalised" by means of a
metal rod introduced through the orbit of the right eye and
taken down along the length of the vertebral column. The right
and left vagus nerves are sectioned (bivagotomy); the right
carotid artery is ligatured, the left carotid artery being
catheterised in order to measure the blood pressure using a
pressure cell (StathamTM P23Db type). A femoral vein i5
catheterised for the purpose of administering various
compounds.
The mean blood pressure variations induced by
angiotensin administered intravenously at the dose of 0.5 ~g/kg
before administering the compounds of the invention and those
induced by angiotensin administered under the same conditions 5
minutes after intravenous administration of the compounds of
the invention or 30 minutes after their oral administration are
measured. The compounds of the invention are administered at
doses ranging from O.Ol to lO0 mg/kg.
The percentage inhibition of the control response to
angiotensin II is used to evaluate the antagonistic potential
of the compounds of the invention to angiotensin II.
The compounds of the invention or their suitable
salts may be used as active therapeutic substances,
- 23 ~ r5t
particularly for the treatment of various forms of hypertensive
pathologies and of cardiac, renal or pulmonary insufficiencies
as well as for the treatment of glaucoma.
The invention further provides a pharmaceutical
composition which comprises a compound of the invention and a
pharmaceutically acceptable carrier or diluent. The compounds
of the invention or their suitable salts may also be used in
combination with other substances possessing cardiovascular
activity such as diuretics, ~-blockers, ~-blockers, calcium
antagonists or angiotensin I converting enzyme inhibitors.
The compounds of the invention or their suitable
salts may be provided in any pharmaceutical forms suitable for
treatment by means of or~l, parenteral, intramuscular or rectal
administration : tablets, capsules, hard gelatin capsules,
sterile solutions or suspensions, suppositories and the like~
For the treatment of glaucoma, the compounds of the
invention, may be provided in the form of tablets, hard gelatin
capsules, injectable solutions or topical eye formulations.
The compositions of the invention may be administered
to patients in an amount which may range from 1 tc 1000 mg per
day and per patient, in one or more doses.
- 2~
APPENDIX I
u~c~,~ x
~ !11!)
H~C ~ H (V)
~ CR~3R~RIS
H3C, ~ (Vl)
~C~ ~ 3R "R ~ ~
~3"~ (Vll)
(Vlll)
R2~ H H R~
. ~ CR~3RI~RH~
R3~ ~ (IX)
3 ~ ~ ( Ia )
-- 25 ~ 3 1
APPENDIX I I
S H~C~
X
N~ (!;I)
H'C~` CN
,~ (I~')
R--O}~
~c~ ~ 0~,, 0~ R
~`~, (`Il')
~~~3`R (VII~
2 N~ R,l ~ (t' 111 )
R`~5 (IX )
R ~ ~0~
(Ib)