Note: Descriptions are shown in the official language in which they were submitted.
20~37 ~ ~
WO91/19711 PCT/SE91/~15
Substituted benzimidazoles, ~rocess for their PreParation
and their ~har~aceutical use
D~SCRIPTION
Field of the invention
The object of the present invention is to provide novel
compounds, which inhibit exogenously or endogenously
stimulated gastric acid secretion and thus can be used in
the prevention and treatment of peptic ulcer.
The present invention also relates to the use of the
compounds of the invention for inhibiting gastric acid
secretion n mammals including man. In a more general
sense, the compou~nds of the inYention may be used for
prevention and treabment o$ gastrointeætinal inflammatory
diseases, and gastric acid-related disease~ in mammals
including man, ~uch as gastritis, gastric ulcer, duodenal
ulcer, reflux esophagitis, and Zollinger-Ellison syndrome.
Furthermore, the compounds may be used for treatment of
other gastrointestinal disorders where gastric
antisecretory effect is desirable e.g. in patients with
gastrinomas, and in patients with acute upper
gastrointestinal bleeding. They may also be used in
patients in intensive care situations, and pre- and
postoperatively to prevent acid aspiration an~ stress
ulceration. The compounds of the invention may also be
used for treatment or prophylaxis of inflammatory
conditions in mammals, including man, especially those
involving lysozymal enzymes. Conditions that ~ay be
specifically mentioned are rheumatoid arthritis and gout.
The compounds may also be useful in the treatment of
diseases related to bone metabolism disorders as well as
'~t~71~
WO91/l9711 PCT/SE91/~15
the treatment of glaucoma. The invention also relates to
pharmaceutical compositions containing the ~ompounds of
the invention, as active ingredient. In a further aspect,
the invention relates to processe for preparation of such
S new compounds and to the use of the active comFounds for
the preparation of phar~aceutical compositions for the
medical use indicated above.
It is a specific primary object of the invention to
provide compounds with a high level of biovailability. The
compounds of the invention will also exhibit good
stability properties at neutral and acidic p~ and a good
potency in regard to inhi~ition of gastric acid
secretion. The compounds of the invention will not bloc~
the uptake of iodine into the thyroid gland. It has
ear}ier been disclosed in several }ectures from the
company, where the inventors are working that thyroid
toxicity depends on if the com~:ounds are lipophilic or
not. The inventors have now unexpecte~ly found that it is
not the lipophilicity that is the critical parameter. The
claimed compounds, which include rather hydrophilic
compounds, do not give any thyroid toxic effect and have
at the same time high acid secretion inhibitory effect,
good bioavailability and stability.
Prior art and backqround of the invention
Benzimidazole derivatives intended for inhibiting gastric
acid secretion are disclosed in numerous patent documents.
Among these can be mentioned GB 1 500 043, GB 1 525 958,
Us 4 182 766, US 4 255 431, Us 4 599 347, BE 898 880,
EP 124 495, EP 2~8 452, EP 221 041, EP 279 149, EP 176 308
and Der~ent abstract 87-294449/42. Benzimidazole
derivatives proposed for use in the treatment or
prevention of special gastrointestinal inflammatory
diseases are disclosed in US 4 359 465.
2~'~37~ ~
W091/19711 PCT/SE91/~15
The invention
The csmpounds of the formula I are effective as
inhibitors of gastric acid secretion in mammals including
man and in addition do not block the uptake of iodine
into the thyroid gland. It has also been found that the
compounds of the following formula I show high
bioavailability. Further, the compounds of the invention
exhibit a high chemical stability in solution at neutral
and acidic p~. The high chemical stability also at acidic
pH makes the compounds useful for non-enteric coated
peroral formulations.
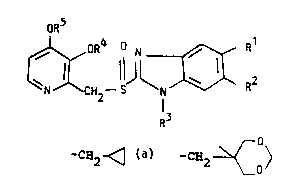
The compounds of the invention are of the following
formula I:
oR5 1 . I
~ R4 N ~ 2
wherein
Rl and R2, which are different, is each H, alkyl
containing 1-4 carbon atoms or -C(O)-R6, one of Rl or R2
is always selected from the group -C~O)-R6;
wherein
R6 is alkyl containing 1-4 carbon atoms or alkoxy
containing 1-4 carbon atoms
R3 is the group -CH20CooR7~ wherein R7 is alkyl containing
1-6 carbon atoms or benzyl;
R4 and R5 are the same or different and selected from
7 ~ i~
WO91/19711 PCT/SE91/~15
3 2 5~ H2 ~ , CH2 ~ 0~ , and -CH2C~20CH3,~or R4
and R5 form together with the adjacent oxygen atoms
attached t~ the pyridine ring and the carbon atoms in the
pyridine ring a ring, wherein the part constituted by R4
and R5 is -CH2CH2cH2-, -CH2CH2 2
It should be understood that the expressions "alkyl" and
"alkoxy" include straight and branched structures.
The structural isomers of the invention descxibed in
examples 1-6 may be used separately, or in egual or
unequal mixtures.
The compounds of the invention of the for~ula I have an
asymmetric centre in the sulfur atom, i.e. exists as two
optical isomers (enantiomers) or if they also contain one
or more asymmetric carbon atoms, the compounds have two or
more diastereomeric forms, each existiny in the two
enantiomeric forms. Both the pure enantiomers, racemic
mixtures (50% of each enan~iomer) and unequal mixtures of
the two are within the scope of the present invention. It
should also be understood that all the diastereomeric
forms possible (pùre enantiomers or racemic mixtures) are
within the scope of the invention.
Preferred groups of compounds of the formula I are:
l. Compounds, wherein R3 is -CH2OCOOCH2CH3.
2. Compounds, wherein Rl and R are selected from H,
methyl or -C(O)-R6, wherein R6 is alkyl containing l-
4 carbon atoms or alkoxy containing 1-4 carbon
atoms.
3. Especially preferred benzimidazole structures are:
WO91/19711 2 ~, 3 ~ PCT/SEgl/~lS
o o
N~COCH3 ,~ ;~CH3
J~N ~ H J~N I~OCH3 N CH3
N~3~1CIH33 ~10CH~CH~
J~
N COCH2CH~
4. Especially preferred are compounds, wherein R4 and R5
are methyl.
5. Especially preferred specific compounds of the
invention are the compounds listed in the following
tabulation
X~ R3 R~ . R5
CH3 C(O)OCH3 CH2Ocoo 2 3 CH3 CH3
30 C(O)OCH3 CH3 CH2OCOOCH2CH3 CH3 CH3
CH3 C(O)CH3 CH2OCOOCH2CH3 CH3 CH3
C(O)C~3 CH3 CH2ocoocH2cH3 CH3 CH3
It is believed that compounds of formula I are metabolized
W091/19711 PCT/SE91/~15
into the corresponding co~pounds, wherein R3 i8 ~ before
exerting their effect.
Pre~aration
The compounds of the invention may be prepared according to
the following methods:
a) Reacting a compound of the formula II
1 0 oR5
1 ~ H2 50 ~ ~ 2 II
wherein Rl, R2, R4 and R5 are as defi~ed under for~ula I,
and 2, is either a metal cation such as Na+, R+, Li+ or Ag+
or a quaternary ammonium ion, such as tetrabutyla~onium
with alkyl chloromethyl carbonate or benzyl chloromethyl
carbonate.
b) Reacting a compound of the formula II, wherein Rl , R2,
R4 and R5 are as defined under formula I and Z is
hydroxymethyl with a compound of the formula III,
X-C(o)-o-R7 III
wherein R7 is as defined above and X is Cl or imidazole or
p-nitrophenoxy or a functionally equivalent group, in the
presence of a suitable base such as triethylamine.
The reactions according to a) and b) are suitably carried
out under protective gas in absence of water. Suitable
solvents are hydrocarbons such as toluene or benzene or
halogenated hydrocarbons such as methylene chloride or
2 ~ " 3 ~
~091/19711 PCT/SE91/~415
chloroform, or acetone, acetonitrile or d~methylformæmide.
The reactions may be carried out at a temperature between
the ambient temperature and the boiling temperature of the
reaction mixture.
c) Oxilizing a compound of the formula IV
oR5
~ ; ~ ~ R2 IV
wherein Rl, R2, R3, R4 and R5 are as defined under formula
I.
This oxidation may be carried out by u~ing an oxidizing
agent such as nitric acid, hydrogen pero~ide, ~optionally in
the presence of vanadium compounds), peracids, peres~ers,
ozone, dinitrogentetraoxide, iodosobenzene, N-
halosuccinimide, l-chlorobenzotriazole, t-
butylhypochlorite, diazabicyclo-t2,2,2]-octane bromine
complex, sodium metaperiodate, selenium dioxide, manganese
dioxide, chromic acid, cericammonium nitrate, bromine,
chlorine, and sulfuryl chloride. The oxidation usually takes
place in a solvent such as halogenated hydrocarbons,
alcohols, ethers, ketones.
The oxidation may also be carried out enzymatically by
using an oxidizing enzyme or microbiotically by using a
suitable microorganism. The structural isomers obtained,
may be separated by means of crystallization or
chromatography.
Racemates obtained can be separated according to known
methods, e.g. recrystallization from an optically active
WO91/19711 ' ~ ; PCT/SE91/~15
solvent. In the case of racemic diastereomeric mixtures
these may be separated into diastereomeric pure enantiomers
by means of chromatography or ~ractional crystall~zation.
The starting materials utilized in the methods a)-c) are in
some cases unknown. These unknown starting materials may,
be obtained according to processes known E~
Alkyl chloromethyl carbonate and benzyl chloromethyl
carbonate may be obtained from the pertinent alcohol by
treatment with chloromethyl chloroformate in the presence
of pyridine.
Intermediates of the formula II, wherein Z is hydroxymethyl
are obtained by reaction of the corresponding benzimidazole
compound carrying H in the N-l position with formaldehyde.
Starting materials of the fonmula III ~ay be obtained by
known methods, e.g. from an alcoho~ HoR7 by treatment with
phosgene or l,ll-carbonyldiimidazole or p-nitrophenyl
chloroformate.
For clinical use a compound of the invention is formulated
into pharmaceutical formulations for oral, rectal, or other
modes of administration. The pharmaceutical formulation
contains a compound of the invention normally in combination
with a pharmaceutically acceptable carrier. The carrier may
be in the form of a solid, semi-solid or liquid diluent, or
a capsule. These pharmaceutical preparations are a further
object of the invention. Usually the amount of active
compound is between 0.1-95% by weight of the preparation,
and between 1-50% by weight in preparations for oral
administration.
In the preparation of pharmaceutical formulations
containing a compound of the present invention in the form
Wosl/19711 PCT/SE91/00415
of dosage units for oral admini~tration a co~pound selected
may be mixed with a solid, powdered carrier, such as
lactose, saccharose, sorbitol, mannitol, ~tar~h,
amylopectin, cellulose derivatives, gelatin, or another
suitable carrier, stabilizing substances such as alkaline
compounds e.g. carbonates, hydroxides and oxides of sodium,
potassium, calcium, magnesium and the like, as well as with
lubricating agents such as magnesium stearate, calcium
stearate, sodium stearyl fumarate and polyethylenglycol
waxes. The mixture is then processed into granules or
pressed into tablets. Granules and tablets may be coated
with an enteric coating which protects the active compound
from acid catalyzed degradation as long as the dosage form
remains in the stomach. The enteric coating is chosen among
pharmaceutically acceptable enteric-coating materials e.g.
beeswax, shellac or anionic film-formi~g polymers such as
celluloce acetate phthalate, hydroxypropyl-methylcellulose
phthalate, partly methyl esterified methacrylic acid
polymers and the like, if preferred in combination with a
suitable plasticizer. To the coating various dyes may be
added in order to distin~uish amon~ tablets or granules
with different active compounds or with different amounts
of the active compound present.
Soft gelatine capsules may be prepared with capsules
containing a mixture of an active compound of the
invention, vegetable oil, fat, or other suitable vehicle
for soft gelatine capsules. Soft gelatine capsules may also
be enteric-coated as described above. Hard gelatine
capsules may contain granules or enteric-coated granules of
the active compound. Hard gelatine capsules may also
contain the active compound in combination with a solid
powdered carrier such as lactose, saccharose, sorbitol,
mannitol, potato starch, amylopection, cellulose
deriva~ives or gelatine. The hard gelatine capsules may be
enteric-coated as described above.
WO91/19711 2 ~ ~ 3 ~ ~ ~ ` PCT~SE91/~415
Dosa~e units for rectal administration may be prep~red in
the form of suppositories which co~tain an active substance
mixed with a neutral fat base, or they may be prepared in
the form of a gelatine rectal capsule which contains the
active substance in a mixture with a vegetable oil, paraffin
oil or other suitable vehi~le for gelatine rectal capsules,
or they may be prepared in the form of a ready-made micro
enema, or they may be prepared in the form of a dry micro
enema formulation to be reconstituted in a suitable solvent
just prior to administration.
Liquid preparation for oral administration may be prepared
in the form of syrups or suspensions, e.g. solutions or
suspensions containing from 0.2% to 20% by weight of the
active ingredient and the remainder consisting of sugar or
sugar alcohols and a mixture of ethanol, water, glycerol,
propylene qlycol and~or polyethylene glycol. If de~ired,
such liquid preparations may contain colouring agents,
flavouring agents, saccharine and carboxymethyl cellulose
or other thickening agents. Liguid preparations for oral
administration may also be prepared in the form of a dry
p~wder to be reconstituted with a suitable solvent prior to
use.
The typical daily dose of the active substance will depend
on various factors such as for example the individual
requirement of each patient, the route of administration and
the disease. In general, oral dosages will be in the range
of 5 to 500 mg per day of active substance.
The invention is illustrated by the following examples.
~xam~le l. Preparation of 5-carbomethoxy-6-methyl-2-tt(3,4-
dimethoxy-2-pyridinyl)methyl3sulfinyl]-lH-benzimidazole-l-
ylmethyl ethyl carbonate and 6-carbomethoxy-5-methyl-2-
7 1~
WO91/19711 PCT/SE91/~15
[[~3,4-dimethoxy-2-pyridinyl)methyl3~ulfinyl]~
benzimidazole-l-ylmethyl ethyl carbonate, as an isomeric
mixture.
To a suspension of 0.45 g (1.1 mmol) of 5-carbomethoxy-6-
methyl-2-~(3,4-dimethoxy-2-pyridinyl)methyl~-sulfinyl]-
lH-benzimidazole and 0.25 g (1.8 mmol) of potassiu~
carbonate anhydrous in 45 ml of dry aoetonitrile, 0.21 g
(1.5 mmol) of chloromethyl ethyl carbonate dissolved in 5 ml
of acetonitrile was added. The reaction mixture was stirred
at room temperature over night. The solvent was then
removed in vacuo and the residue was diluted with methylene
chloride and water. The organic solvent was dried over
anhydrous sodium sulfate. Removal of the solvent in Yacuo
gave the crude product, which was chromatographed with
silica gel and eluted with ethyl acetate to provide 0.94 g
of a yellow oil which slowly crystallized. Recrystallization
with ethanol yielded 0.25 g (44 %) of the title compounds a~
an isomeric mixture.
NMR data for the products are given below.
Exam~le 2. Preparation of 6-carbomethoxy-5-methyl-2-~[(3,4-
dimethoxy-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole-1-
ylmethyl ethyl ~arbonate.
The title compound was obtained by crystallizing the
isomeric mixture given in example 1 from ethanol.
NMR data are given below.
Exam~le 3. Preparation of 5-acetyl-6-methyl-2-t[(3,4-
dimethoxy-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole-1-
ylmethyl ethyl carbonate and 6-acetyl-5-methyl-2-lt(3,4-
dimethoxy-2-pyridinyl)methyl]sulfinyl~-lH-benzimidazole-1-
yl~ethyl ethyl carbonate, as an isomeric mLxture.
To a magnetically stirred suspension of potassium carbonate
20~7~ 1
WO91/19711 PCT/SE91/~15
anhydrous (O.48 g, 3.47 mmol) in 80 ml of dry acetonitr~le
0.80 g (2.14 mmol) of 5-acetyl-6-methyl-2-~ E (3,4-dimethoxy-
2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole and 0.39 g
(2.8 mmol) of chloromethyl ethyl carbonate dissolved in 10
ml of acetonitrile was added dropwise. Stirring was
continued at room temperature for 20 hours. The solvent was
removed in vacuo, the residue diluted with methylene
chloride, the methylene chloride solution washed with water
and dried over anhydrous sodium sulfate. Removal of the
solvent in vacuo gave the crude product which was
chromatographed with silica gel and eluted with ethyl
acetate to yield 0.63 g of an almost white crystalline
solide. The product was recrystallized frGm ethyl acetate to
give 0.50 g (49 %) of the title compounds as an isomeric
mixture.
NMR data for the products are given below.
kxamPle 4. Preparation of 5-acetyl-6-methyl-2-[[(3,4-
dimethoxy-2-pyridinyl~methyl]sulfinyl~_lH_benzimidazole-1-
ylmethyl ethyl carbonate.
The title compound was isolated from the isomeric mixturegiven in example 3 by chromatography on a silica column with
methylene chloride - acetonitrile ~ratio 6:4) as eluent. The
title compound was crystallized from ethanol.
NMR data are given below.
ExamPle 5. Preparation of 6-acetyl-5-methyl-2-[[(3,4-
dimethoxy-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole-1-
ylmethyl ethyl carbonate.
The title compound was isolated from the isomeric mixturegiven in example 3 by chromatography on a silica column with
methylene chloride-acetonitrile (ratio 6:4) as eluent. The
title compound was crystallized from ethanol.
NMR data are given below.
2Q837~ l~
~O91/19711 PCT/SE91/~15
ExamDle 6 Preparation of 5-carbethoxy-2-[[(3,~-dimetho~y-2-
pyridinyl)methyl]sulfinyl]-lH-benzimidazole-1-yl~ethyl ethyl
carbonate and 6-carbethozy-2-t[(3,4-dimethoxy-2-
pyridinyl)methyl]s~lfinyl~-lH-benzimidazole-l-ylmethyl ethyl
carbonate, as an isomeric mtxture.
To a suspension of 0.28 g (0.72 mmol) 5-carbethoxy-2-~t(3,4-
dimethoxy-2-pyridinyl)methyl]sulfinyl~-l_ benzimida~ole and
0.16 g tl.2 mmol) anhydrous potassium carbonate in 20 ~1 of
dry acetonitrile 0.16 g (1.2 mmol) chloromethyl ethyl
carbonate dissolved in 2 ml dry acetonitri~e was added. The
mixture was stirred at ambient temperature over night. The
solvent was evaporated off and the crude product was
chromatographed on a silica column using ethyl acetate as
eluent. Crystallizing fr~m ethanol gave the title compounds
as an isomeric mixture, (0.13 g, 37%).
NMR data for the products are given below.
Table 1
Ex. Solvent NMR data ~ P~m
1 CDC13 1.20-1.30 (m, 3H), 2.70 (s,1.8H),
(300 MHz) 2.75 (s, 1.2H), 3.85-3.95 (m,9H),
4.15-4.25 (m,2H), 4.85-5.05 (m,2H),
6.40-6.55 (m,2H), 6.75 (d,lH), 7.45
(s, 0.6H), 7.65 (s, 0.4 H), 8.10
(d, lH), 8.20 (s, 0.4 H), 8.40 (s,
0.6 H).
2 CDCl3 1.30 (t, 3H), 2.70 (s, 3H)
(300 MHz) 3.90 (s,3H), 3.90 (s, 3H), 3.95 (s,
3H), 4.25 (g, 2H), 4.95 (d, lH), 5.05
(d, lH), 6.50 (m, 2H), 6.75 (d, lH),
7.65 (s, lH), 8.10 (d, lH), 8.20 (s,
2 Q ~ 3 ~
WO91~19711 PCT/SE91/~15
lH)
3 CDC13 1.30 (t, 3H) 2.60-2.70 (m, 6~),
(300 MHz) 3.85-3.90 (m, 6H), 4.25 (g,2H),
4.85-5.05 (m, 2H), 6.75 (d,lH), 7.45
(s, 0.7 H), 7.60 (s, 0.3H), 8.05 (s,
0.3H), 8.10 (d, lK), 8~20 (s, a.7H)
4 CDC13 1.30 (t, 3H), 2.60 (s, 3~), 2.70
(300 MHz) (s, 3H), 3.90 (8, 3H), 3.90 (s, 3H),
4.20 (g, 2H), 4.90 ~d, lH), 5.05 (d,
lH), 6.50 (m, 2H), 6.80 (d, lH),
7.50 (s, lH), 8.15 (d, lH), 8.20 (s,
lH)
CDCl3 1.30 (t, 3H), 2.60 (~, 3H), 2.70
~300 MHz) (s, 3H), 3.90 (s, 3H), 3.90 (s, 3
- H), 4.25 (q, 2~), 4.90 (d, 1~),
5.05 (d, lH), 6.55 Im, 2H), 6.80
(d, lH), 7.60 (s, lH), 8.05 (s, 1
H~, 8.15 (d, lH)
6 CDC13 1.30 (m, 3H), 1.45 (m, 3H), 3.90
(300 MHz) (s, 3H), 3.90 (s, 3~), 4.25 (m,
2H), 4.45 (m, 2H), 5.00 (m, 2H),
6.55 (m, 2H), 6.80 (d, lH), 7.70
(d, 0.55H), 7.80 (d, 0.45H), 8.10
(m, 2H), 8.35 (s, 0,45H), 8.50 (d,
0.55H).
2~3~ ~
091/19711 PCT/SE91/~15
Preparation of intRr~edi~tes
~xamDle I l
S Preparation of 5-carbomethosy-6-Eethyl-2-[[(3,~-dimethosy-
2-~.idinyl)~ethyl]thio]-l~-benz~oidazole
5-carbomethoxy-6-methyl-2-mercapto-lH-benzimida~ole ~0.67
g, 0.003 mol) and NaOH (0.12 g, 0.003 mol) in H2O (0.6 ml)
were dissolved in CH30H (l5 ml). 3,4-dimethoxy-2-
chloromethylpyIidine hydrochloride, (=0.0036 mol) as a
crude material in CH3OH (l0 ml) and NaOH (0.144 g, 0.0036
mol) in H2O ~0.72 ml) were added. The mixture was heated to
reflux and the reflux was continued for 1 hour. CH30H was
lS evaporated off and the crude material was purified by
chromatography on a silica column using C~2Cl2-CH3OH (98-2)
as eluent, giving 1l.03 g, 92%) of the pure title compound.
NMR data are given below.
ExamDle I 2
Preparation of 5-carbomethQxy-6-~ethyl-2-tt(3,~-dimetko~y-
2-pyridinyl)methyl]~lf ~ l1-lH-benzi~ida~ole
5-carbomethoxy-6-methyl-2-[[(3,4-dimethoxy-2-
pyridinyl)methyl]thio]-lH-benzimidazole (1.03 g, 0.00276
mol) was dissolved in CH2Cl2 (30 ml). NaHCO3 (0.46 g,
0.0055 mol) in H2O (l0 ml) was added and the mixture was
cooled ~o 12C. m-chloroperbenzoic acid 69.5% (0.62 g,
0.0025 mol) dissolved in CH2Cl2 (5 ml) was added dropwise
under stirring. Stirrin~ was continued at +2C for 15 min.
After separation the organic layer was extracted with an
aqueous 0.2 M NaOH solution (3x15 ml, 0.009 mol). After
separation the aqueous solutions were combined and
neutralized wi~h methyl formate (0.56 ml, 0.009 mol) in the
S ~
WO9~/19711 PCT/SE91/~415
presence of CH2C12 (25 ml). After separation the organic
layer was dried over ~a2S04 and evaporated under reduced
pressure. The residue was crystallized from CH3CN (l0 ml)
giving the title compound (0.68 g, 70 %).
NMR data are given below.
Exam~le I 3
Preparation of 5-acetvl-6-~sthyl-2- U(3,4-di~etho ~-2-
Rqridinyl)methYl]thio]-l~-beD~idaz~le
5-acetyl-6-~ethyl-2-mercapto-lH-benzimidazo}e (4.2 g, 20
mmol) and NaOH (0.8 g, 20 mmol) in H20 (l ml) were dissolved
in 60 ml ethanol. 3,4-dimethoxy-2-chloromethylpyridine
hydrochloride (z17 mmol) as a crude material was added and
the mixture was heated to boiling. NaOH (0.7 g, 17 mmol) in
H20 (1 m}) was added and the reflux was continued for 6
hours. The solvent was evaporated off and the residue was
diluted with methylene chloride and water. The organic phase
was dried over Na2SO4 and the solvent was removed under
reduced pressure. Crystallizing from acetonitrile gave the
title compound, ~3.75 g, 62%).
NMR data are given below.
Example I 4
Pre~aration of 5-acetYl-6-~ethyl-2-[1(3,4-dimethQ~y-2-
Pqridi~yllmetbYl1sulfinvll-lff-ben~IEida~ole
5-acetyl-6-methyl-2-[[(3,4-dimethoxy-2-
pyridinyl)methyl]thio3-lH-benzimidazole (3.75 g, l0 mmol)
was dissolved in CH2Cl2 (70 ml). NaHCO3 (1.76 g, 21 mmol) in
H2O (25 ml) was added and the mixture was cooled to -+3C.
m-Chloroperbenzoic acid 69.5~ (2.43 5, 9.8 mmol) dissolved
2~37'~
WO91/19711 PCT/SE91/~15
in CH2C12 (20 ~l) was added dropwi~e under stirring.
Stirring was continued for l0 min. The phases were separat~d
and the organic ~hase was dried over Na2SO4 and evaporated
under reduced pressure. The residue was crystallized from
CH3CN giving the title compound (2.25 g, 60%).
NMR data are given below.
Example I 5
Preparation of 5-carbetho~y-2-lt(3~4-di~etho~y-2
pyridinyl)meth~l]thiol- y-benzieida~ole
5-carbethoxy-2-mercapto-l~-benzimidazole (2.0 9, 9 mmol) and
NaOH (0.36 g, 9 mmol) in H2O (l ml) were dissolved in
ethanol (30 ml). 3,4-dimethoxy-2-chloromethylpyridine
hydrochloride ( 6.6 mmol) as a crude material were added and
the mixture was heated to boilin~. ~aO~ (0.26 g, 6.6 ~mol)
in H20 (l ml) was added and the reflux was continued for 6
hours. The solvent was evaporated off and the residue was
diluted with methylene chloride and water. The organic phase
was dried over Na2SO4 and the solvent removed under reduced
pressure. Crystallizing from CH3CN gave the desired product
~1.75 ~, 71 %).
NMR data are given below.
Exam~le I 6
Prepæration of 5~ betbo~y-2-[t(3r~-di~etho~y-2-
pyridinyl)~ethyl]s~lfi~yl]-la-benzi~idazole
5-carbethoxy-2-[~(3,4-dLmethoxy-2-py~idinyl)methyl~thio]-
l~-benzimidazole (95.2% pure) (1.4 g, 0.0036 mol) was
dissolved in CH2C12 (30 ml). Na~C03 (0.6 g, 0.0072 mol in
H2O (l0 ml) was added and the mixture was cooled to +2C.
~0~7~
WO91/19711 PCT/SE91/~15
m-Chloroperbenzoic acid 69.5 % (0.87 g, 0.0035 mol)
dissolved in CH2C12 (5 ~1) was added dropwise under
stirring. Stirring was continued at +2C for 10 min. The
phases were separated and the organic phase was dried over
Na2S04 and evaporated under reduced pressure. The residue
was crystallized from CH3CN (15 ml ) giving the title
compound (0.76 g, 54 %).
NMR data are given below.
Table 2
Ex Solvent NMR data ~ pPm
15 I 1 CDC13 2.70 (s, 3H), 3.95 ~s, 3H),
~300 MHz) 3.95 (s, 3~), 4.00 (s, 3H),
4.40 (s, 2H~, 6.90 (d, 1~),
7.35 (s, lH), 8.23 (s, lH),
8.25 (d,lH).
I 2 CDCl3 2.70 (s, 3H), 3.85 (s, 3H),
(500 MHz) 3.90 (s, 3H), 3.95 (s, 3H),
4.70 (d, lH), 4.90 (d, lH),
6.8 (d, lH), 7.30 (b, lH), 8.20
- (d, lH), 8.35 (b, lH).
I 3 CDC13 2.60 (s, 3H), 2.65 (s, 3H), 3.90
(300 MHz) (s, 3H), 3.90 (s, 3H), 4.35 (s, 2H)
6.85 (d, lH), 7.25 (s,0.6H), 7.40
(s, 0.4H), 7.85 (s, 0.4H), 8.05
(s, 0.6H), 8.30 (m, lH)
I 4 CDC13 2.60 (s, 6H), 3.85 (s, 3H), 3.85
(300 MHz) (s, 3H), 4.70 (d, lH), 4.90
(d, lH), 6.80 (d, lH), 7.30
(b, lH), 8.15 (d, lH), 8.20 (b, lH)
~ V ~
WO91/19711 PCT/SE91/~415
19
I 5 CDCl3 1.40 (m, 3H), 3.90 (s, 3H), 3.90
(300 MHz) (s, 3H), 4.40 ~m, 4H), 6.90
(dd, lH), 7.45 (d, 0.4H), 7.60
(d, 0.6H), 7.90 (m, lH), 8.20
(s, 0.6H), 8.25 (m, lH), 8.25
(s, 0.4H)
lO I 6 CDCl3 1.45 (t, 3H), 3.8S (s, 3H),
(300 MHz) 3.90 (s, 3H), 4.40 (g, 2H),
4.65 (d, lH), 4.40 (d, lH),
6.80 (d, lH), 7.50 7.80 (b, lH)
8.05 (d, lH), 8.20 (d, lH),
8.25, 8.55 (b, lH)
The best mode of carrying o~t the invention known at present
is to use the compound mixture according to Bxan~le 3 and
the compound according to ~xample 4.
3 r ~ ~ `
WO 91/19711 PCI'/SE91/00415
Ul ~ ~ ~ h ~
l E ~ ~ e ~ --~ E _I E E JJ
o X o o o X O o o o o X
H E H ~ H E H rl H rl H E
~.i
J~
.~
o
_~
o
U~
~ .
~ ~r ~ ~ ~ ~ ~ ~
~ .
u~
0 ~ ~ $
,
E 8 8 8 8 8
~ ~ o o o o o o
~ N ~ 1 N
C~
~ C~
.,~ C~
U
~ ~ O O ~ O
_l ~ O O ~ O ~ O ~ O
U
o
~ ~ O ~ O
O O tr ~70 ~ O ~ O
C~
~1
O
Lq
_I ~1
0 ~ !i'
2 0 8 .~
WO91/19711 PCT/SE91/00415
21
Syru~
A syrup containing 1% (weight per volume) of active
substance was prepared from the following ingredients:
A compound according to Example 4 l.0 g
Sugar, powder 30.0 g
Saccharine 0.6 g
Glycerol 5.0 g
lO Tween l.0 g
Flavouring agent 0.05 g
Ethanol 96% 5.0 g
Distilled water g.s. to a final volume of lO0 ml
A solution of the compound mixture according to Example in
ethanol and Tween was prepared. Sugar and saccharine were
dissolved in 60 g of warm water. After cooling the
solution of the active compound was added to the sugar
solution and glycerol and a solution of flavouring agents
dissolved in ethanol were added. The mixture was diluted
with water to a final volume of lO0 ml.
Tablets
A tablet containing 50 mg of active compound was prepared
from the following ingredients:
I Compound mixture according to
Example 3 500 g
Lactose 700 g
Methyl cellulose 6 g
Polyvinylpyrrolidone cross-linked50 g
Magnesium stearate 15 g
Sodium carbonate 6 g
Distilled water q.s.
2~3'7 ~
WO91/19711 PCT/SE91/~15
22
II Hydroxypropyl methylcellulose 36 g
Polyethylene glyco l9 g
Colour Titanium dioxide 4 g
Purified water 313 g
I Compound mixture according to ~xample 3, powder, was
mixed with lactose and granulated with a water solution of
methyl cellulose and sodium carbonate. The wet ~RSS was
forced through a sieve and the granulate dried in an oven.
After drying the granulate was mixed with
polyvinylpyrrolidone and magnesium stearate. The dry
mixture was pressed into tablet cores (lO 000 tablets),
each tablet containing 50 mg of active substance, in a
tabletting machine using 7 mm diameter punches.
II A solution of hydroxypropyl methylcellulose and
polyethylene glycol in puIified water was prepared. After
dispersion of titaniu~ dioxide the solution was sprayed
onto the tablets I in an Accela cotaR, Manesty coating
eguipment. A final tablet weight of 125 mg was obtained.
Ca~sules
Capsules containing 30 mg of active compound were prepared
from the following ingredients:
A compound according to Example 4 300 g
Lactose 700 g
30 Microcrystalline cellulose 40 g
Hydroxypropyl cellulose low-substituted 62 ~
Purified water q.s.
The active compound mixture was mixed with the dry
ingredients and granulated with a solution of disodium
2~3'~
WO91/19711 PCT/SE91/~15
23
hydrogen phoæphate. The wet mass was forced through an
extruder and spheronized and dried in a fluidi~ed bed
dryer.
500 g of the pellets above were first coated with a
solution of hydroxypropyl methylcellulose, 30 9, in water,
600 g, using a fluidized bed coater. After drying, the
pellets were coated with a second coating as given below:
Coating solution:
Hydroxypropyl methylcellulose phthalate 70 g
Cetyl alcohol 4 g
Acetone 600 g
Ethanol 200 g
The final coated pellets were filled into capsules.
Su~Dositories
Suppositories were prepared from the following ingredients
using a welding procedure. Each suppository contained
40 mg of active compound.
Compound ~ixture a~cording to Example 4 4 g
Witepsol ~-15 180 g
The active compound mixture was homogenously mixed with
Witepsol H-15 at a temperature of 41C. The molten mass
was volume filled into pre-fabricated suppository packages
to a net weight of 1.84 g. After cooling the packages were
heat sealed. Each suppository contained 40 mg of active
compound.
~ ~ 3 ~
WO91/19711 PCT/SE91/~M15
24
Bioloaical Effects
Biovailabilitv
Bioavailability, is assessed by calculating the quotient
between the areas under plasma concentration (AUC) curve
of a compound of the formula I wherein R3 is hydrogen
(herein defined as compound A), following l) intraduodenal
(id) or oral (po) administration of the corresponding
compound according to the invention and 2) intravenous
(iv) administration of compound A, from the rat and the
dog. Low, therapeutically relevant doses, were used. Data
are provided in Table 4.
Potency for inhibition of acid secretion
The potency for inhibition of acid secretion is measured
in the female rat orally and in the dog both
intraduodenally and orally.
Potency data are provided in Table 4.
Effects on the u~take of iodine into the thvroid qland.
The effect of a compound within the invention of the
formula I on the uptake of iodine into the thyroid gland
is measured as an effect on the accumulation of l25I in
the thyroid gland of the corresponding compound of the
formula I, wherein R3 is hydrogen, that is a metabolized
compound of the formula I.
Bioloqical Tests
Inhibition of Gastric Acid Secretion in the Conscious
Female Rat.
~837 ~ ~1
WO91/19711 PCT/SE91/~15
Female rats of the Sprague-Dawley strain are used. They
are equipped with cannulated fistulae in the ~tomach
(lumen), for collection of gastric secretions. A fourteen
days recovery period after surgery is allowed before
testin~ is commenced.
Before secretory tests, the animals are deprived of food
but not water for 20 h. The stomach is repeatedly washed
through the gastric cannula, and 6 ml of Ringer-Glucose
given s.c. Acid secretion is stLmulated with infusion
during 2.5 h (1.2 ml/h, s.c.) of pentagastrin and
carbachol (20 and 1~0 nmol~kg h, respectively), during
which time gastric secretions are collected in 30-min
fractions. Test substances or vehicle are given orally 120
min before starting the stimulation, in a volu~e of 5
ml/kg. Gastric jui~e samples are titrated to p~ 7.0 with
NaOH, 0.1 mol/L, and acid output is calculated as the
product of titrant volume and concentration. Further
calculations are based on group mean responses from 4-7
rats. Percentage inhibition is calculated from absolute
rates of acid output. ED50- values are obtained from
graphical interpolation on log dose-response curves, or
estimated from single-dose experiments ~ss-~;ng a si~;lar
slope for all dose-response curves. The results are based
on gastric acid secretion during the third hour after
drug/vehicle administration.
Bioavailability in the Male Rat.
Male adult rats of the Sprague-Dawley strain were used.
One day, prior to the experiments, all rats were prepared
by cannulation of the left carotid artery under
anaesthesia. The rats used for the intravenous
experiments, were also cannulated in the jugular vein.
2 ~
WO91/19711 PCT/SE91/~15
26
(~ef. V Popovic and P Popovic, J Appl Physiol 1960;15,727-
728). The rats used for the intraduodenal experi~ents,
were also cannulated in the upper part of the duodenum.
The cannulas were exteriorized at the nape of the neck.
The rats were housed individually after surgery and were
deprived of food, but not water, before administration of
the test substances. The same dose (4 ~mol/kg) were given
iv and id as a bolus for about one minute (2 ml/kg).
Blood samples (0.l-0.4 g) were drawn repeatedly from the
carotid artery at intervals up to 4 hours after given
dose. The samples were frozen as soon as po~sible until
analysis of the test compound.
The area under the blood concentration vs time curve, AUC,
for the compound A, determined by the linear trapezoidal
rule and extrapolated to infinity ~y dividing the last
determined blood concentration by the eliminatio~ rate
constant in the terminal phaæe. The systemic
bioavailability (F%) of the compound A following
intraduodenal administration of compounds of the
invention of formula I was calculated as
AUC(ComPUnd A)id(~ompound of the
invention)
F(%) = x lO0
AUC(ComPUnd A)iv(compound A)
Inhibition of Gastric Acid Secretion and Bioavailability
in the Conscious Dog
Harrier-dogs of either sex were used. They were equipped
with a duodenal fistula for the administration of test
compounds or vehicle and a cannulated gastric fistula or a
Heidenhain-pouch for the collection of gastric secretions.
~ ~`35 ~
WO91/19711 PCT/SE91/~15
27
Before secretory tests the animals were fasted for about
18 h but water was freely allowed. Gastric acid secretion
was stimulated by a 4 h infusion of histamine
dihydrochloride (12 ml/h) at a dose producing about 80% of
the individual maximal secretory response, and gastric
juice collected in consecutive 30-min fractions. Test
substance or vehicle waæ given orally, id or iv 1 h after
starting the histamine in~usion, in a volume of 0.5 ml/kg
body weight. In the case of oral administration, it should
be pointed out that the test compound is administered to
the acid secreting main stomach of the Heidenhain-pouch
dog.
The acidity of the gastric juice samples were determined
by titration to pH 7.0, and the acid output calculated.
The acid ou~put in the collection periods after
administration of test substance or vehicle were
expressed as fractional responses, setting the acid output
in the fraction preceding administration to lØ
Per~entage inhibition was calculated from fractional
responses elicited by test com~ound and vehicle. EDSo~
values were obtained by graphical interpolation on log
dose - response curves, or estimated from single-dose
experiments under the assumption of the same slope of the
dose-response curve for all test compounds. All results
reported are based on acid output 2 h after dosing.
Blood samples for the analysis of test compound
concentration in plasma were taken at intervals up to 3 h
after dosing. Plasma was separated and frozen within 30
min after collection and later analyzed. AUC (area under
the plasma concentration - time curve) frsm time zero to 3
h after dose for compou~d A, was calculated by the linear
trapezoidal rule. The systemic bioavailability (F%) of the
f l l
WO91/19711 28 PCT/SE91/00415
compound A after oral or id admini~tr~tion of compound~ of
the invention was calculated as described above in the rat
model.
~ffect on t~e accumulation of 125I in the thyToid ~land
The accumulation of 125I in the thyroid gland was studied
in male, Sprague-Dawley rats which were deprived of food
for 24 hours before the test. The experimental protocol of
Searle, CE et al. ~Biochem J 1950; 47:77-81) was followed.
Test substances, suspended in 0.5% buffered tPH 9)
methocel, were administerd by oral gavage in a volumc of 5
ml/kg body weight.`After 1 hour, 125I (300kBq/ky, 3ml/kg)
was administered by intrapexitoneal injection. Four hours
after 125I-administration, the animals were killed by
- CO2-asphyxiation and bled. The thyroid gland together with
a piece of the trachea was dissected out and placed in a
small test tube for the assay of radioactivity in a gamma
counter (LgB-Wallac model 1282 Compugamma). Percentage
inhibition was calculated according to the formula 100 (1-
T/P), where T and P is the mean radioactivity of thyroid
glands from animals treated with test agent and placebo
(buffered methocel), respectively. ~he statistical
significanoe for a difference between test agent- and
placebo-treated animals was assessed with the Mann-Whitney
U-test (two-tailed). P~0.05 was accepted as significant.
Chemical Stabilitv
The chemical stability of the compounds of the invention
has been followed kinetically at low concentration at
37C in aqueous buffer solution at different pH values.
The results in Table 5 show the half life (t 1/2) at p~ 7,
that is the time period after which half the amount of the
~ ~ ~ 3 1 ~ 1
Wosl/19711 PCT/SE91/~
29
original compound romains unchanged, and ~10% at p~ 2,
that is the time period ~fter which 10% of t~e origtn~l
compound has decompoEed.
Results of biological and stabilitY tests
Table 4 and 5 give a summary of the te~t data available
for the compounds of the invention.
? ~
WO 91tl9711 PCI/SE91/00415
.,~ ~n ,
~ o~
.~ ,y ~ .,
~,,, O O o o r~
C
~o ~
oo ~s
J~
a~
a~ c
~ o~
.~ ~ ~ loD
,1 ~ ~ ,~
.,~ .,
,1 ~P 'O
~rl ~ ~ ~ ~D
~d ~
~ ~ _ _ _ _
m o ~ ~n In ~ In
o .~c
C ., ~,
o ~ o
o
C ~ 10 ~
~1 0 C _ _ _
W-r1 C~ ~ ~ CO
~,q Lq ,~ ~ _1 ~1 0
s-., ~
~ ~ ~ ~ cr o
_ ~ s~
Co rl C
~ 4~ O-rl ~ JJ O~
~o ~ ~ O ~s
I I~n q ~c
E~J' a~ c o .4 R R R ~ a~
.rl O ~ O Ul
C.e ~ ~n N ~ R
~a~
c
c U a~
I
E~ E~ X
WO 91/19711 2 9 ~ 3 'i' i ~i PCr/SE91/004lS
Table 5, Stability Data
Test compound Chemical
Example No. stability at
t 1/2 (h) t 10% (h)
879.S
2 506.5
3 517.5
4 82 1~
6 63 13