Note: Descriptions are shown in the official language in which they were submitted.
WO 91/18883 2 0 8 3 ~ ~ ~ Pcr/ussl/o394o
Amide Linked Pyridyl-Benzoic Acid Derivatives
For Treating Leukotriene-related I)iseases
Scope of the ~nvention
This invention relates to amide linked pyridyl-benzoic acid
5 derivatives which are useful for treating diseases associated wilh
leukotrienes. These compounds are particularly useful in treating
diseases attributable to hydroxyleukotrienes, especially I,TB4 and
LTB 4-agonist active substances.
Background of the Invention
The family of bioactive lipids }cnown as the leukotrienes exert
pharmacological effects on respiratory, cardiovascular and .
gastrointestinal systems. The leukotrienes are generally divided into
two sub-classes, the peptidoleukotritones (leukotrienes C4, D4 and E4)
and the hydroxyleukotrienes (leulcotriene B4). This invention is
primarily concerned with the hydroxyleukotrienes (LTB) but is no
limited to this specific group of leukotrienes.
The peptidoleukotrienes are implicated in the biological
response associated with the "Slow Reacting Substance of
Anaphylaxis" (SRS-A). This response is expressed in vivo as
2 0 prolonged bronchoconstriction, in cardiovascular effects such as
coronary artery vasoconstriction and numerous other biological
responses. The pharmacology of the peptidoleukotrienes include
smooth muscle contractions, myocardial depression, increased
vascular permeability and increased mucous production.
2 5 By comparison, LTB4 exerts its biological effects through
stimulation of leukocyte and Iyrnphocyte functions. It stimulates
chemotaxis, chemokinesis and aggregation of polymorphonuclear
leukocytes (PMNs).
They are critically involved in mediating many . types of
3 0 cardiovascular, pulmonary, dermatological, renal, allergic, and
inflammatory diseases including asthma, adult respiratory distress
syndrorne, cystic fibrosis, psoriasis, and inflammatory bowel disease.
Leukotriene B4 (LTB4) was first described by Borgeat and
Samuelsson in 1979, and later shown by Corey and co-workers to be
3 5 5(S),12~R)-dihydroxy-(Z,E,E,Z)-6,8,10,14-eicosatetraenoic acid.
~ Fig. I
2~3~6 2
wo 91/18883 PCr/US~1/03940
It is a product of the arachidonic acid cascade that results from the
enzymatic hydrolysis of LTA4. It has been found to be produced by
mast cells, polymorphonuclear leukocytes, monocytes and
macrophages. LTB4 has been shown to be a potent stimulus in ~iv~
5 for PMN leukocytes, causing increased chemotactic and chemokinetic
migra~ion, adherence, aggregation, degranulation, superoxide
production and cytotoxicity. The effecl~s of LTB4 are mediated
through distinct receptor sites on the leukocyte cell surface which
exhibit a high degree of stereospecificity. Pharmacological studies on
10 human blood PMN leukocytes indicate the presence of two classes of
LTB4-specific receptors that are separate from receptors specific for
the peptide chemotactic factors. Each of the sets of receptors appear
to be coupled to a separate set of PMN leukocyte functions. Calcium
mobilization is involved in both mechanisms.
l S LTB4 has been established as an inflammatory mediator in vi~o.
It has also been associated with airway hyper responsiveness in the
dog as well as being found in increased levels in lung lavages from
humans with severe pulmonary dysfunction.
By an~agonizing the effects of LTB4, or other pharmacologically
2 0 active mediators at the end organ, for example airway smooth muscle,
the compounds and pharmaceutical compositions of ~he instant
invention are valuable in the treatment of diseases in subjects,
including human or animals, in which leukotrienes are a factor. Sume
of these compounds may also inhibit the 5-lipoxygenase enzyme or
2 5 may be Ll`D4 antagonists.
- SUMMARY OFTHE ~VENl`ION
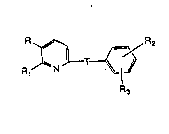
The compounds of this invention are represented by formula (I)
R~
R3 (I)
or a pharmaceutically acceptable salt or N-oxide thereof where
T is the amide linking group
Oq/
HN
3~ ,
wo gl/18883 2 ~ ~ 3 ~ 5 ~ pcr/ussl/o394o
where the carbonyl carbon is bonded to the pyridyl ring;
R is Cl to C20-aliphatic, unsubstituted or substituted phenyl C
to Cl o-aliphatic where substituted phenyl has one or more radicals
selected from the group consisting of lower alkoxy, ]ower alkyl,
trihalomethyl, or halo, or R is Cl to C20-aliphatic-O-, or R is
unsubstituted or substituted phenyl Cl to Clo-aliphatic-O- where
substituted phenyl has one or more radicals which are lower allcox~
Iower alkyl, trihalomethyl, or halo;
R1 is R4,-(Cl to Cs aliphatic)R4, -(Cl to C5 aliphatic)CHO, -(Cl to
10 C5 aliphatic)CH2ORg, -CH2OH or -CHO;
R2 is hydrogen, -CORs where R~ is -OH, a pharmaceutically
acceptable ester-forming group -OR6, or -OX where X is a
pharmaceutically acceptable cation, or Rs is -N(R7)2 where R7 is H, or
an aliphatic group of 1 to 10 carbon atoms, a cycloalkyl-(CH2)n- group
of 4 to 10 carbons where n is 0-3 or both R7 groups combine to form
a ring having 4 to 6 carbons, or R2 is NHSO2Rg where Rg is -CF3, Cl to
C6 alkyl or phenyl;
R3 is hydrogen, lower alkoxy, halo, -CN, CORs, or OH;
R4 is -CORs where Rs is -OH, a pharmaceutically ac~eptable
ester-forming group -ORs, or -OX where X is a pharmaceutically
acceptable cation, or Rs is -N(R7)2 where R7 is H, or an aliphatic group
of 1 to 10 carbon atorns, a cyc~oalkyl-(CH2)n- group of 4 to 10 carbons
where n is 0-3 or both R7 groups combine to form a ring having 4 to 6
carbons;
~8 is hydrogen, Cl to C6 alkyl, or C] to C6-acyl.
In another aspect, this invention covers pharmaceutical
compositions containing the instant compounds and a
pharmaceutically acceptable excipient.
Treatment of diseases related to or caused by leukotrienes,
3 0 particularly LTB4, or related pharmacologically active mediators at
the end organ, are within the scope of this invention. This treatment
can be effec~ed by administering one or more of the compounds of
formula I alone or in combination with a pharmaceutically acceptable
excipient.
3 5 In yet another aspect, this invention relates to a method for
making a compound of formula I, which method is illustrated in the
Reaction Schemes given below and in the Examples set forth below.
WO91/18883 ~ 4 PCI/US91/03940
DETAII,ED DESCRIPTION OF THE ~NVENT~ON
The following definitions are used in describing this invention
and setting out what the inventors believe to be their invention
herein .
"Aliphatic" is intended to include saturated and unsaturated
radicals. This includes norma] and branched chains~ saturated or
mono or poly unsaturated chains where both double and triple bonds
may be present in any combination. The phrase "lower alkyl" means
an alkyl group of 1 to 6 carbon atoms in any isomeric form, but
particularly the normal or linear form. "Lower alkoxy" means the
group lower alkyl-O-. "Acyl" means the radical having a terminal
carbonyl carbon. "Halo" refers to and means fluoro, chloro, bromo or
iodo. The phenyl ring may be substituted with one or more of these
radicals. Multiple substituents may be the same or different, such as
where there are three chloro groups, or a combination of chloro and
alkyl groups and further where this latter combination may have
different alkyl radicals in the chloro/alkyl pattern.
The phrase "a pharmaceutically acceptable ester-forming group"
in R2 and R3 covers all esters which can be made from the acid
2 0 function(s) which may be present in these compounds. l`he resultant
esters will be ones which are acceptable in their application to a
pharmaceutical use. By that it is meant that the mono or diesters will
retain the biological activity of the parent compound and will not
- have an untoward or deleterious effect in their application and use in
2 5 treating diseases. Such esters are, for example, those formed with
one of the following radicals representing -OR6 where R6 is: Cl to Cl O
alkyl, phenyl-CI-C6 alkyl, cycloalkyl, aryl, arylalkyl, alkylaryl,
alkylarylalkyl, aminoalkyl, indanyl, pivaloyloxymethyl,
acetoxymethyl, propionyloxymethyl, glycyloxymethyl,
3 0 phenylglycyloxynlethyl, or thienylglycyloxymethyl. Aryl includes
phenyl and naphthyl, or heteroaromatic radicals like furyl, thienyl,
imidazolyl, triazolyl or tetrazolyl. Most preferred ester-forming
radicals are those where R6 is alkyl, particularly alkyl of 1 to 10
carbons, [i.e. CH3-(CH2)n- where n is 0-9], or phenyl-(CH2)n- where n
3~ is 0-4.
Pharmaceutically acceptable salts of the instant compounds are
also intended to be covered by this invention. These salts will be
ones which are acceptable in their application to a pharmaceutical
use. By that it is meant that the salt will retain the biological activity
wo g~ 883 ~ ~ ~ 3 ~ ~ 6 P~/U~91/03940
of the parent compound and the salt will not have untoward or
deleterious effects in its application and use in treating diseases.
Pharmaceutically acceptable salts are prepared in a standard
manner. The parent compound in a suitable solvent is reacted with
5 an excess of an organic or inorganic acid, in the case of acid addition
salts of a base moiety, or an excess of organic or inorganic base where
R4 is OH. Representative acids are hydrochloric acid, hydrobromic
acid, sulfuric acid, phosphoric acid, acetic acid, maleic acid, succinic
acid or methanesulfonic acid. Cationic salts are readily prepared from
l 0 alkali metal bases such as sodium, potassium, calcium, magnesium,
zinc, copper or the like and ammonia. Organic bases include. the mono
or disubstituted amines, ethylene diamine, amino acids,
caffiene, tromethamine, tris compounds, triethyl amine, piperazine
and the like.
l 5 Oxides of the pyridyl ring nitrogen may be prepared by means
known in the art and as illustrated herein. These are to be
considered part of the invention.
If by some combination of substituents, a chiral center is
created or another form of an isomeric center is created in a
2 0 compound of this invention, all forms of such isomer(s~ are intended
to be covered herein. Compounds with a chiral center may be
administered as a racemic mixture or the racemates may be
separated and the individual enantiomer used alone.
As leukotriene antagonists, these compounds can be used in
2 S treating a variety of diseases associated with or attributing their
origin or affect to leukotrienes, particularly LTB4. Thus it is expected
that these compounds can be used to treat allergic diseases including
~hose of a pulmonary and non-pulmonary nature. For example these
compounds will be useful in antigen-induced anaphylaxis. They are
3 0 useful in treating asthma and allergic rhinitis. Ocular diseases such as
uveitis, and allergic conjunctivitis can also be treated by these
compounds .
The preferred compounds of this invention are those where R is
alkoxy, particularly alkoxy of 8 tv l 5 carbon atoms or substituted or
35 unsubstituted phenyl-Cl to Clo-aliphatic-O-; Rl is -(Cl to C~
aliphatic)R4 or -(Cl to C5 aliphatic)CH20Rg; and R2 is COOH or an alkali
metal salt thereof or NHS02Rg where Rg is -CF3, Cl to C6 alkyl or
phenyl. The more preferred compounds of this invention are those
where R is alkoxy of 8 to l 5 carbon atoms or alkoxy-substitu~ed
wo 9l/18883 2 ~ 8 3 9 5 ~ PCr/US91/03~40
phenyl-CI to Cg-alkenoxy or -Cl to c8-alkoxy; Rl is -CORs,
-CH2CH2CORS or -CH=CH-CORs R2 is -~COOH or -NHSO2Rg, par~icularly
where Rg is -CF3; and R3 is hydrogen or chloro.
The most preferred compounds are set out in Figure Il.
Figure .lI
RX~-- R2
_ R__ R]__ R2
HN * H2] Clo HOOC~CH2~2- m-~OOH
7C8 HOOC CH~ ** ,.
CIo ,.
sCI2 ..
" .H29C14 ll ..
. p-MeO-Ph-(CH2)8- .l . ..
* The carbonyl carbon is substitut :d on the pyridyl
l 0 ** Trans configuration.
Synthesis
These compounds may be made by the intermediates and
reagents as in the following reaction schemes. This specific set of
l 5 intermediates is used to illustrate the general method. Scheme l
illustrates a method for making compounds useful in making the R
group. The other schemes use the ma~erials whose preparation is
described in scheme l, or intermediates from commercial sources, to
form the R group, ~hen illustrate a method for making the compounds
2 0 of formula I.
The R groups in formula I are available from chemical supply
houses or can be made by one of the two methods outlined in
Reaction Scheme I.
Scheme I(a) illustrates a method for making an unsaturated
2 5- phenyl-alphatic R group.
Scheme l~a)
/==\ (Ph)3P=cH(cH2)3co2- CH o ~=~ H
H3CO~CHO ~ ~ 3 ~
(a)
WO 9~/18X~3 ~! 0 8 3 ~ 5 ~ PCr/US91/03940
TsCI
LiAlH4 ~~~,OH py r.
(b)
.
CH30 {~ -- OT'
(C)
While the methoxyphenyl compound is illustrated here, this
series of steps and reagents may be used to malce other substituted
phenyl~ aliphatic groups denoted by R. The starting material, the
ben~aldehydes, are commercially available or can be readily made by
10 known methods.
To make the acid (a), first an.alkylsilazide is added to an inert
solvent under an inert atmosphere. Then the phosphonium salt
added. This addi~ion can be done at room temperature or
thereabouts. After a brief period of mixing, this mixture is usually ;
15 suspension, the benzaldehyde is added slowly at about room
temperature. A slight molar excess of the phosphonium salt is
employed. After an additional brief period of stirring at :about roorr
temperature, the reaction is quenched with water. The solution is
acidified and the acid extracted with a suitable organic solvent.
2 0 Further standard separatory and purification procedures may be
employed as desired.
The alcohol is made by reducing the acid using a reducing agent.
Lithium aluminum hydride or similar reducing agents may be
employed and conditions may be varied as needed to effect the
2 5 reduction.
'rhe tosylate is prepared in an inert solvent employing p-
toluene sulfonyl chloride and a base such as pyridine. Suitable
conditions include carrying out the reaction at room temperature or
thereabouts for a period of 1 to ~ hours. Other suitable leaving
3 0 groups similar in function to the tosylate may be prepared and will
be useful as a means for adding this R moiety to the pyridyl ring.
Reaction Scheme I(b) outlines one method ~or making an
alkoxyphenylalkyl R group. This method could be used to mal~e other
R groups where phenyl is the c,) group on the aliphatic chain, including
3 5 substituted phenyl-containing groups.
-
wo 9l/l8883 5~ 8 PCI/US91/039q0
Scheme ( I b!
KAPA OH t-Bu(Ph)2s1c
(a)
H3CO-Ph -I
=--(CH2)nosi(ph)2-t-Buph2 - ilD
(b) Pd ~(Ph~3P]2 Cl2
H3Co~(CH2)nos;(ph)2 l guph2 H2- Pd-C
(c)
H3co~}(cH2~n+2osi(ph)2-t-Bu BU4NF
(d)
1 0
H3CO~(CH2)n+2-OH TsCI ~ H3CO~(cH2)n+2-OTs
(~) (f)
In those instances where an ~-yn-1-ol is not commercially
available, it can be prepared from a corresponding 3-yn-l-ol by
l 5 treating the alcohol with a strong base. Here an alkali metal amide
was used. The alcohol is then protected in order to add the desired
phcnyl group at the ~erminal triple bond. A silyl ether was formed in
this insfance; it illustrates the general case. A halo-substituted-
phenyl adduct is used to add the phenyl group at the triple bond. At
2 0 this point, the triple bond can be reduced, most conveniently by
catalytic means, eg. palladium-on-carbon and hydrogen.
- Alternatively, the triple bond could be retained and the intermediate
carried on through as illustrated to the tosylate. The silyl group is
removed and the resulting alcohol is converted to the tosylate, or
2 5 another group which is sufficiently reactive so as to provide ready
formation of an ether later in the synthesis of these compound.
Using the compounds made in Scheme I and others purchased
or prepared by known methods, in the following reaction schemes,
one can prepare the compounds of formula 1 by following the
3 0 sequence of reactions outlined in the following Schemes. Again, these
schemes illustrate the general principle of how to make these
compounds using specific examples. These schemes can be used lo
W0 91/18883 2~ ~3,~ 5~ PCI/US91/03940
rnake the other compounds disclosed herein by varying or modifying
the chemistries illustrated here. Such variations or modifications will
be changes in the reaction conditions, eg. temperature, pressure,
length of reaction time, amount of reagents and the like. Reagents
5 may be substituted for their equivalent or for a similar reagent which
will effect the same or the equivalent product. Similarly, starting
materials and intermediates may be varied to accomodate the need
for making a particular compound.
One way of preparing compounds where the nitrogen of the
10 amide linking group is on the phenyl ring is set out in Reaction
Scheme 2.
Scheme 2
HO ~ MnO2 R-X
HO~ J Et3N H~ --
(a) (b) (c)
L~ ~ R-O,~ MnO2
1 5 (~)
(Ph)3PCHC02Me RO~, MCPBA RO~
_:~
MeO2C ~ N~ ~N+
(d) (e)
DMF R-O Tf20 ~
-MeO2C~O Py r. MeO2C N OTf
(f) (g)
~3 1 o PCr/US91/03940
H2N CO2Mc R-O~ q
1~ (h ) M~02C~ ~ 1. aq. LiOH, THF. McOH
~ ( i ) ~, C02MC
Pd(OAc)2, dppf I~J
CO, DMF
HO2C X r
HN ~,C02H
S The foregoing scheme illustrates one synthetic route for making
compounds of formula I where the carboxyl carbon is on the pyridyl
ring. I`he 3-hydroxy-2-(hydroxymethyl)pyridine is commercially
available or can be prepared by known, published means. This dio]
may be converted either to the aldehyde, then converted to the
3-alkoxy compound, or the 3-hydroxy group may be conYerted to the
ether first, then the 2-position hydroxymethyl is oxidized to the
aldehyde. Oxidizing the alcohol is readily accomplished using a mild
oxidizing agent; manganese dioxide is preferred but other oxidizing
agents could be successfully utilized in this step. Ethers are readi]y
prepared from the corresponding -halo-R group, or a compound such
as a tosylate, under basic conditions.
This 3-substituted-2-carboxyaldehyde (c) is then converted to
the 2-carbomethoxyethenyl form (d) by means of the appropriate
phosphoranylidene ester under conditions normally used for such a
2 0 reaction. The resulting ester is then treated with a peroxy acid to
make the N-oxide in preparation for rnaking the pyridone (e). This
step is illustrated by m-chloroperoxybenzoic acid7 but other similar
oxidizing agents could be used as well. Rearrangement of the N-oxide
- is then accomplished by means of trifluoroace~ic anhydride or a
2 5 similar reagent to produce the 2-pyridone (f).
Converting the 2-pyridone to the amide is accomplished by
acylating the 2-pyridone (g) and ~hen reacting this ester with the t
desired aminobenzoate (h) in the presence of certain catalysts and
carbon monoxide. Trifluoromethanesulfonic anhydride illustrates the
3 0 acylation step. The amidation reaction is effected by bubbling carbon
wo gl/l8883 2 0 ~ 3 9 .~ ~ pcr/us91/o394o
monoxide through a solu~ion of the triflate in the presence of
Pd(OAc)2, l,l'-bis(diphenylphosphino)ferrocene. The resulting
diester (i) is then saponified using a mineral base to hydrolyze the
ester groups. The resulting salt may be neutralized in order to
recover the free acid. A free acid can be converted to another ester
or made into the co~Tesponding amide by known methods.
The saturated 3-position substituents are readily prepared from
the alkene analog by catalytic hydrogenation. ~eaction Scheme 3
illustrates this methodology.
1 0
Reaction ~cheme 3
RO~ H2, Pd C
MeO2C~ MeO2C~
~2H) ~CO2Me (3a) ~CO2Me
Aq. LiOH RO~
solvent HOOC~O
HN ~/=\~COOH
(3b) ~
The diester is catalytically Teduced (3a) by means of a heavy
metal catalyst and hydrogen in a classic catalytic reduction reaction.
Once the reduction is complete, a base can be used to hydrolyze the
diester if the diacid (3b) is desired. Either compound can be
converted to other compounds of this invention by the appropriate
2 0 oxidation, reduction, esterification, amidation reaction, or by other
means .
Carbon analogs of these compounds, that is those where the
atom linking the R group to the 3-position is methylel~e, may be
prepared by ~he sequence of steps set out in the fourth flow chart.
Scheme 4
HO~ 1.HCI / MeOH TfO~ R-B ~,
HO2C N 2. Tf2O/pyridine MeO2C N b
Pd~OAc)2 / dppf / DMF
(4a)
wo g / ~ 1 2 Pcr/ussl/o394o
MeO2C~ 2 (C6Hs)3FcHco2Me M O C~
(4b) (4c)
R~
HO2C~ N ~p
H N ~CO2H
3-Hydroxypicolinic acid is converted to the alkyl ester by
5 means of the corresponding alkanol and a acid catalyst. The hydroxyl
group is converted to the trifluoromethanesulfonate (4a) using
trifluoromethanesulfonic anhydride and pyridine. The lipid tail is
then attached (4b) using the appropriate alkyl catechol boronate,
prepared from 1-tridecene and catechol borane, using palladium
l 0 coupling conditions ([Pd(OAc)2]. Then the alkyl ester is transformed
into the corresponding aldehyde using an appropriate hydride, for
example diisobtylalurninum hydride. This aldehyde is then subjected
to a Wittig olefination, using for example, methyl(triphenyl-
phosphoranylidene)acetate. The resulting pyridyl acrylate is then
1~ converted to the target compound via the same set of steps outlined
in Scheme 2 above.
Reverse amides can be made by the sequence of steps given in
Scheme 5.
Scheme 5
HO~ 1. H' MeOH H25C120~ Tf20, pyridine
HO2C N 3 CMlC2Hp2B51, KC2HCOcl, DMF MeO2C~ N lo~- 3
4. TFM, DMF a)
H25C12~ 1 H2, Pd/C H2sC120
MeO2C N N3 3 (ph)3pcHco2Me MeO C'~l_NH2
(5c)
wo 91/18883 1 3 2 ~ ~ 3 9 a ~ Pcr/us91/0394o
Cl J~CO2Me H2sC,~
2. LiOH, THF, MeOH HO2C N NH
3. H+ O~CO2H
The commercial3y available 3-hydroxypicolinic acid is converted
to an alkyl ester using an acid catalys~ and the corresponding alkanol.
S This is followed by alkylation under standard conditions with, for
example l-idododecane or a similar 1-halo compound. This is best
done using a weak base such as K2C03 in dimethylformamide. This
gives the 3-alkoxy derivative. Oxidizing the pyridine nitrogen and
rearranging the resulting N-oxide provides the 2-pyridone. Oxidation
10 is readily effected with a peroxy acid such as 3-chloroperoxybenzoic
acid or similar oxidizing agent. The N-oxide (~a) rearrangement can
be accomplished using trifluoroacetic anhydride in an appropriate
solvent such as dimethylformamide.
Forming the trifluoromethanesulfonate is effected by means of
15 trifluoromethanesulfonic anhydride and a base such as pyridine.
Nucleophilic displacement with sodium azide gives the 2-azido
pyridine derivative (Sb). Reducing the azide to the amine is
accomplished by catalytic hydrogenalion. Reducing the alkyl ester to
the aldehyde is done with a hydride, for example diisobutylaluminum
2 0 hydride. A Wittig reaction is then used to make the 2-amino pyridine
acrylate (Sc). For example methy](triphenylphosphoranylidene)-
acetate may be used. Acylating the amine (me2hyl isophthalolyl
chloride~ followed by hydrolysis of the esters with base (LiOH,
tetrahydrofuran, methanol) yields the target amide. These
2 5 compounds oan be further converted to an ester, amide, salt or
similar compound as defined by formula I by means illustrated
herein or generally lcnown in the art.
Form u 1 ati on s
Pharmaceutical compositions of the present invention comprise
3 0 a pharmaceutical carrier or diluent and some amount of a compound
of the formula (I). The compound may be present in an amount to
effect a physiological response, or it may be present in a lesser
amount such that ~he user will need to take two or more units of the
compositon to effect the treatment intended. These compositions may
35 be made up as a solid, liquid or in a gaseous form. Or one of these
WO 91/18X83C~ 3 PCI/US91/03940
three forms may be transformed to another at the time of being
administered such as when a solid is delivered by aerosol means. or
when a liquid is delivered as a spray or aerosol.
The nature of the composition and the pharmaceutical carrier or
5 di}uent will, of course, depend upon the intended route of
administration, for example parenterally, topically, orally or by
inhalation .
~ or parenteral administration the pharmaceutical composition
will be in the form of a sterile injectable liquid such as an ampule or
10 an aqueous or non-aqueous liquid suspension.
For topical administration the pharmaceutical composition will
be in the form of a cream, ointment, liniment, lotion, pastes, and
drops suitable for administration to the eye, ear, or nose.
For oral administration the pharmaceutical composition will be
15 in the form of a tablet, capsule, powder, pellet, atroche, lozenge~
syrup, liquid, or emulsion.
When the pharmaceutical composition is employed in the form
of a solution or suspension, examples of appropriate pharmaceutical
carriers or diluents include: for aqueous systems, water, for non-
2 0 aqueous systems, ethanol, glycerin, propylene glycol, corn oil,cottonseed oil, peanut oil, sesame oil, }iquid parafins and mixtures
thereof with water; for solid systems, lactose, kaolin and mannitol;
and for aerosol systems, dichlorodifluoromethane,
chlorotrifluoroethane and compressed carbon dioxide. Also, in
2 5 addition to the pharmaceutical carrier or diluent, the instant
compositions may include other ingredients such as stabilizers,
antioxidants, preservatives, lubricants, suspending agents, viscosity
modifiers and the like, provided that the additional ingredients do not
have a detrimental effect on the therapeutic action of the instant
3 0 compositions.
The pharmaceutical preparations thus described are made
following the conventional techniques of the pharmaceutical chemist
as appropriate to the desired end product.
In general, particular}y for the prophy}actic treatment of
3 5 asthma, the compositions will be in a form suitable for administration
by inhalation. Thus the compositions will comprise a suspenslon or
solution of the acti~e ingredient in water for administration by means
of a conventional nebulizer. Alternatively the compositions will
comprise a suspension or solution of the active ingredient in a
WO 91tl8883 2 0 ~ 3 9 5 ~ Pcr/usgl/03940
conventional liquified propellan~ or compressed gas to be
administered from a pressurized aeTosol container. The composi~ions
may also comprise the solid active ingredient diluted with a solid
diluent for administration from a powder inhalation device. In the
5 above compositions, the amount of carrier or diluent will vary but
preferably will be the major proportion of a suspension or solution of
the active ingredient. When the diluent is a solid it may be present in
Iesser, equal or greater amoun~s than ~he solid active ingredient.
Usually a compound of formula I is administered to a subject in
10 a composition comprising a nontoxic amount sufficient to produce an
inhibition of the symptoms of a disease in which leukotrienes are a
factor. When employed in this manner, the dosage of the composition
is selected from the range of from 50 mg to 10~0 mg of active
ingredient for each administration. For convenience, equal doses will
15 be administered I to S times daily with the daily dosage regimen
being selected from about 50 mg to about 5000 mg.
Included within the scope of this disclosure is the method of
treating a disease mediated by LTB4 which comprises administering
to a subject a therapeutically effective amount of a compound of
2 0 formula I, preferably in the form of a pharmaceutical composition.
For example, inhibiting the symptoms of an allergic response
resulting from a mediator release by administration of an effective
amount of a compound of formula I is included within the scope of
this disclosure. The administration may be carried out in dosage
2 5 units at sui~able intervals or in single doses as needed. Usually this
method will be practiced when relief of symptoms is specifically
required. However, the method is also usefully carried out as
continuous or prophylactic treatment. It is within the skill of the art
to determine by routine experimentation the effective dosage to be
3 0 administered from the dose range set forth above, taking into
consideration such factors as the degree of severity of the condition
or disease being treated, and so forth.
Pharmaceutical compositions and their method of use also
include the combination of a compound of formula I with Hl blockers
3 5 where the combination contains sufficient amounts of both
compounds to treat antigen-induced respiratory anaphylaxis or
similar allergic reaction. Representa~ive H I blockers useful here
include: cromolyn sodium, compounds from the ethanolamines class
(diphenhydramine), ethylenediamines (pyrilamine), the alkylamine
WO 91/18883 ,~ PCr/US91/03940
class (chlorpheniramine), the piperazine class (chlorcyclizine), and the
phenothiazine class (prometha~ine). Hl blockers such as 2-[4-(5-
bromo-3 -methylpyrid-2-yl)butylamino] -5-[(6-methylpyrid-3 -
yl)methyl]-4-pyrimidone are particularly useful in this invention.
5 Bioassavs
The specificity of the antagonist activity of a number of the
compounds of ~his invention is demonstrated by relatively low levels
of antagonism toward agonists such as potassium chloride, carbacho~,
histamine and P&F2-
The receptor binding affinity of the compounds used in the
method of this invention is measured by the ability of the compounds
to bind to [3H]-LTB4 binding sites on human U937 cell membranes.
The LTB4 antagonist activity of the compounds used in the method of
this invention is measured by their ability to antagonize in a dose
15 dependent manner the LTB4 elicited calcium transient measured with
fura-2, the fluorescent calcium probe. The methods employed were
as follows:
U937 Ce!l l;~ult~lre Conditions
U937 cells were obtained from Dr. John Bomalaski: (Medical
20 College of PA) and Dr. John Lee (SmithKline Beecham, Dept. of
Immunology) and grown in RPMI- 1640 medium supplemented with
10% (v/v) heat inactivated fetal calf serum, in a humidified
environment of 5% CO2, 95% air at 37C. Cells were grown both in T-
flasks and in Spinner culture. For differentiation of the U937 cells
25 with DMS0 to monocyte-like cells, the cells were seeded at a
concentration of 1 x 105 cells/ml in the above medium with 1.3%
DMSO and the incubation continued for 4 days. The cells were
generally at a density of 0.7~-1.25 x 105 cells/ml and were harvested
by centrifugation at 800 x g for 10 min.
3 0 Preparation of U937 Cell Membrane Enriched Fraction
Harvested U937 cells were washed with 50 mM Tris-HCI, pH 7.4
at 25 C containing 1 mM EDTA (buffer A). Cells were resuspended in
buf~er A at a concentration of 5 x 107 cells/ml and disrupted by
nitrogen cavitation with a Parr bomb at 750 psi for 10 min at 0 C.
3 5 The broken cell preparation was centrifuged at l,000 x g for 10 min.
The supernatant was cen~rifuged at 50,000 x g for 30 min. The pellet
was washed twice with buffer A. The pellet was resuspended at
about 3 mg membrane protein/ml with 50mM Tris-HCI, pH 7.4 at
25C and aliquots were rapidly frozen and stored at -70C.
WO 91/18883 1 7 2 ~ 8 3 ~ ~ ~ PCr/~S91/03940
~LL~4 to U397 Membrane Rece~Q~
[3H]-LTB4 binding assays were performed at 25 C, in 50 mM
'I`ris-HCI (pH 7.5) buffer containing 10 mM CaC12, 10 rnM MgC12, [3H]-
LTB4, U937 cell membrane protein (standard conditions) in the
presence or absence of varying concentrations of LTB4, or SK&F
compounds. Each expenmental point represents the means of
triplicate determinations. Total and non-specific binding of [3H]-LTB4
were determined in the absence or presence of 2 ~,lM of unlabeled
LTB4, respectively. Specific binding was calculated as the difference
between total and non-specific binding. The radioligand competition
experiments were performed, under standard conditions, using
approximately 0.2 ,LM [3H]-LTB4, 20-40 ~g of U937 cell membrane
protein, increasing concentrations of LTB4 (0.1 nM ~o 10 nM) or other
competing ligands (0.1 IlM to 30 ~M) in a reaction volume of 0.2 ml
and incubated for 30 minutes at 25 C. The unbound radioligand and
competing drugs were separated from the membrane bound ligand
by a vacuum filtration technique. The membrance bound
radioactivity on the filters was determined by liquid scintillation
spectrometry.
2 0 Saturation binding experiments for U937 cells were performed,
under standard conditions, using approximately l 5-50 llg of U937
membrane protein and increasing concentrations of [3H]-LTB4 (0.02-
2.0 mM) in a reaction volume of 0.2 ml and incubation at 22C, for 30
minutes. LTB4 (2 ~lM) was included in a separate set of ineubation
2 5 tubes to determine non-specific binding. The data from the
saturation binding experiments was subjected to computer assisted
non-linear least square curve fitting analysis and further analyzed by
the method ~f Scatchard.
Uptake of Fura-2 by Differentiated U937 Cells
3 0 Harvested cells were resuspended at 2 x l06 cells/ml in Krebs
Ringer Hensilet buffer containing 0.1% BSA (RlA grade), 1.1 mM
MgSO4, 1.0 mM CaC12 and 5 mM HEPES (pH 7.4, buffer B). The
diacetomethoxy ester of fura-2 (fura-2/AM) was added to a final
concentration of 2 nM and cells incubated in the dark for 30 minutes
at 37 C. The cells were centrifuged at 800 x g for 10 minutes and
resuspended at 2 x 1 o6 cells/ml in fresh buffer B and incubated at
37C for 20 minutes to allow for complete hydrolysis of entrapped
ester. The cells were centrifuged at 800 x g for 10 minutes and
resuspended in cold fresh buffer B at 5 x 106 cells/ml. Cells were
wogl/l8~83 ~Q~ 6 1 8 Pcr/us91/03940
maintained on ice in the darl; until used for fluorescent
measurements .
Fluorescent Measurem~:~obilization
The fluorescence of fura-2-containing U937 cells was measured
5 with a fluorometer designed by the Johnson Foundation Biomedical
Instrumentation Group. Fluorometer is equipped with temperature
control and a magnetic stirrer under the cuvette holder. The wave
lengths are set at 339 nm for excitation and 499 nm for emission. All
experiments were performed at 37C with constant mixing.
U937 cells were diluted with fresh buffer to a concentration of
1 x 106 cells/ml and maintained in the dark on ice. Aliquots (2 ml) of
the cell suspension were put into 4 ml cuvettes and the temperature
brought up to 37C, ~maintained in 37C, water bath for 10 min).
CuYettes were transferred to the fluorometer and fluorescence
15 measured for about one minute before addition of stimulants or
antagonists and followed for about 2 minutes post stimulus. Agonists
and antagonists were added as 2 1ll aliquots.
Antagonists were added first to the cells in the fluorometer in
order to detect potential agonist activity. Then after abbut one
2 0 minu~e 10 nM LTB4 (a near maximal effective concen~ration) was
added and the maximal Ca2+ mobilizatibn [Ca2+]i was calculated using
the following formula:
[Ca2+]i = 224{FmaX-F}
F was the maximum relative fluorescence measurement of the
sample. Fmax was determined by Iysing the cells with 10 ~1 of 10%
Triton X-100 (final Concentration 0.02%). After Fma~ was determined
67 ~1 of 100 mM EDTA solution (pH 10) was added to totally chelate
30 the Ca2+ and quench the fura-2 signal and obtain the Fmin. The
lCa2+]i level for lO nM LTB4 in the absence of an antagonist was 100%
and basal [Ca2+]i was 0%. The ICso concentration is the concentration
of antagonist which blocks 50% of the lOnM LTB4 induced lCa2+]i
mobilization. The ECso for LTB4 induced increase in [Ca2+]i
3 5 mobilization was the concentration for half maximal increase. rhe K;
for calcium mobilization was determined using the foranula:
1 9 ;~ 6
wo gl/18~83 PCr/US91/03940
K ICso
LTB4]
+ [EC
With the experiments described, the LTB4 concentration was 10 nM
and the ECso was 2 nM.
Results of compounds tested by these methods are given in
Figure III.
Figure XII
~3inding~ IC~ tKj~. ~lM Ca Mol~i]i~a~ion
U-937 PMN U-937 P M N
Structure ~m~a~ Whole Cell Whole ~ell ~ % A~onist
Ex 1 4-0(14) 2.0 2.4 3.7 0 0
Ex 2 23(8.0) 4.7 - 3.0 0
Ex 3 47(17) 5.8 0.65 0.58 0 0
Ex 4 6.5(2.2)3.4 2.2 8.5 0 0
- Ex 5 41(14) 1.1 2.2 0.72 0 0
Ex 6* 6.1(2.0)0.68 0.14 0.74
* Title compound.
Exam~les
The following set of examples are given to illustrate how to
make and use the compounds of this invention. These Examples are
just that, examples, and are not intended to circumscribe or otherwise
limit the scope of this invention. Reference is made to the claims for
defining what is reserYed to the inventors by this document.
Example~ A
~-(4-MethoxvphenvlLoctan-l -(4-toluenesulfonate)
A(1 ! 7-Octyn-l-ol
35% XH in mineral oil (27g, 240mmol) under an argon
atmosphere was washed with hexane and treated dropwise with 1,3-
diaminopropane. The mixture was sIirred at room temperature until
it became homogeneous The flask was cooled to O~C and 3-octyn-1-
ol (lOg, 79mmol, Lancaster Synthesis~ was slowly added. The
2 5 reaction was then stirred at room temperature for 18 hours. The
reaction was quenched with H20 (50mL) and the product was
extracted into ether. The organic layer was washed with 10% HCI
~3XlSmL) and ~rine and dried (MgS04). Evaporation gave the title
product which was used without further purification: 1H NMR
3 0 (9OMHz, CDC13) ~ 3.65 (t, J=~Hz, 2H, OCH2), 2.23 (m, 2H, CH2~. 2-0 (m,
Wo gl/18883 ~ ,3~$ ~ 2 0 PCT/llS91/03940
lH, acetylenic), 1.7-1.2 (m, 8~, (CH2)4); IR (neat) ~max 3350, ~930,
2125 cm~l.
A(2) 7-Octvn-1-t-butYldiphenYlsilvl ether.
7-Octyn- I -ol (3 .8g, 30mmol) was dissolved in dimethyl-
formamide (lOmL) and treated with t-butylchlorodiphenylsilane
(10.2mL, 33mmol) and imidazole (3.65g, 45mmol) at 0C. The
reaction was stirred at 0C for 10 minutes and at room temperature
for 3 hours. Water was added and the product was extraceed into
l O ethyl acetate. The ethyl acetate extract was washed with H20 and
brine and dried (Na2S04). The solvent was evaporated and. the
residue purified by flash column chromatography (silica, hexanes) to
give a yellow oil: IH NMR (2~0MHz, CDC13) ~ 7.7 (d, 4H, aryl), 7.4 (m,
6H, aryl), 3.63 (t, 2H, OCH2), 2.23 (m, 2H, CH2), 1.97 (t, lH, acetylenic),
1.6-1.3 (m, 8H, (CH2)4), 1.05 (s, 9H, t-butyl); IR (film) l)max 3321,
2940, 2125 cm- I .
A(3! _8-~4-Methoxyphenvl!-7-octyn -1 -t-butv]diphenylsilYl _ether
To a flame-dried flask under an argon atmosphere was added
4-iodoanisole (5.34g, 22mmol) in triethylamine (50mL) followed by
2 0 the addition of 7-octyn-1 -t-butyldiphenylsilyl ether (9.84g, 27mmol),
(Ph3P)2PdC12 (35ûmg, 0.44mmol), and CuI (200mg, 0.88mmol). The
resulting mixture was heated at 50C for 4 hours. Upon cooling to
room temperature the reaction mixture was filtered and the solven~
evaporated. l`he residue was partitioned between ethyl acetate and
2 5 H20 and the organic layer was collected and washed with brine and
dried (Na2S04). The solvent was evaporated and the residue was -
purified by flash column chromatography (silica, l % ethyl acetate in
hexanes) to give an oil: lH NMR ~250MHz, CDC13) ~ 7.7 (d, 4H, aryl),
7.4 (m, 6H, aryl), 7.35 (d, 2H, aryl), 6.8 (d, 2H, aryl3, 3.8 (s, 3H, OCH3),
3 0 3.7 (t, 2H, OCH2), 2.4 (t, 2H, CH2), 1.7-1.3 (m, 8H, (CH2)4), 1.05 (s, 9H,
t-butyl).
A(4) 8-(4-Methoxyphenyl!octan-l-t-butvldiphenvlsilvl ether.
To 8-(4-methoxyphenyl)-7-octyn- 1 -t-butyldiphenylsilyl e~her
3 5 (2.2g, 4.6mmol) in ethanol (lOmL) and ethyl acetate (lOmL) was
added 5% PdlC (lOOmg). The mixture was subjected to 75 psi of H2
for 4 hours. The reaction was filtered through Celite and the solvent
evaporated to give an oil: lH NMR (250MHz, CDC13) ~ 7.7 (d, 4H. aryl),
7.4 (m, 6H, aryl), 7.05 (d, 2H, ary]), 6.8 (d, 2H, aryl), 3.8 (s, 3H, OCH3).
wo 91/18883 ~ ~3 ~ ~ ~ Pcr/us9l/o394o
3.6 (t, 2H, OCH2), 2.5 (t, 2H, benzylic), 1.75-1.3 (m, 12H, (CH2)6)~ 1-0
(s, 9H, t-butyl).
A(5~ (4-Methoxyphenvl)octan-1-ol
8-(4-Methoxyphenyl)octan- I -t-butyldiphenylsilyl ether (2.2g,
4.6mmol) in tetrahydrofuran (20mL) was cooled to 0C and treated
with tetrabutylammonium fluoride (14mL, 14mmol, lM in
tetrahydrofuran). The cooling bath was removed and the reaction
was stirred at room temperature for 24 hours. The reaction was
10 diluted with ethyl acetate and was washed with H20 and brine and
dried (Na2S04). The solvent was evaporated and the residue was
purified by flash column chromatography (silica, 0-20% ethyl acetate
in hexanes) to give a white solid: m.p. 47-49C; 1H NMR (250MHz,
CDC13) ~ 7.15 (d, 2H, aryl), 6.86 (d, 2H, aryl), 3.85 (s, 3H, OCH3), 3.6
(t, 2H, OCH2), 2.62 (t, 2H, ben~ylic),- 1.75-1.3 (m, 12H, (CH2)6).
Af6) 8-~4-Me~hoxyphenvl?octan-1-(4-toluenesulfonate~.
6-(4-Methoxyphenyl)octan-1-ol (5.9g, 25mmol) was dissolved
in dry CH2C12 (lOOmL) under an argon atmosphere and cooled to 0C.
2 0 To this was added pyridine (2.5mL, 30mmol) and 4-toluenesulfonyl
chloride (5.4g, 28mmol). The reaction was stirred at 0C for 20
minutes and at room temperature for 24 hours. The reaction solution
was washed with H20 and brine and dried (Na2S04). The solvent was
evaporated and the residue purified by flash column chromatography
25 (silica, 0-10% ethyl acetate in hexanes) to give a white solid: IH NMR
(25ûMHz, CDC13) ~ 7.79 (d, 2H, aryl), 7.35 (d, 2H, aryl), 7.09 (d, 2H,
aryl), 6.82 (d, 2H, aryl), 4.04 (s, 2H, OCH2), 3.8 (s, 3H, OCH3), 2.55 (t,
2H, benzylic), 2.46 (s, 3H, CH3), 1.75-1.15 (m, 12H, (CH2)6~-
3 0 Example B
- 6-(4-Methoxvphenvl)hexan-1-(4-toluenesulfonate)
B(I! ~-Hexvn-1-t-butvldiphenylsiIvl ether
3 5 5-Hexyn-1-ol (3g, 30mmol, Aldrich) was dissolved in
dimethylformamide (1 OmL) and treated with
t-butylchlorodiphenylsilane (10.2mL, 33mmol) and imidazole (3.65g,
45mmol) at 0C. The reaction was stirred at 0C for 10 minutes and
at room temperature for 3 hours. Water was added and the product
WO 91/18883 ~ 6 2 2 Pcr/US91/03940
was extracted into ethyl acetate. The ethyl aGetate extract was
washed with li20 and brine and dried (Na2S04). The solvent was
evaporated and the residue purified by flash column chromatography
(silica, hexanes) to give a yellow oil: IH NMR (250MHz, CDC13) ~ 7.7
(d, 4H, aryl), 7.4 (m, 6H, aryl), 3.65- (t, 2H, OCH2), 2.2 (m, 2H, CH2), 1.9
(t, lH, acetylenic), 1.7 (m, 4H, CH2-CH2), 1.05 (s, 9H, t-butyl).
B(2~ 6-(4-Methoxvphenyl)-5-hexvn-]-t-butyldiphenylsilyl ether.
To a flame-dried flask under an argon atmosphere was added
4-iodoanisole (5.34g, 22mmol) in triethylamine (50mL) followed by
the addition of 5-hexyn-1-~-butyldiphenylsilyl ether (8.83gi
27mmol)~ (Ph3P)2PdC12 (350mg, 0.44mmol), and Cul (200mg,
0.88mmol). The resulting mixture was heated at 50C for 4 hours.
Upon cooling to room temperature the reaction mixture was filtered
and the solvent evaporated. The residue was partitioned between
ethyl acetate and H20 and the organic layer was collected and washed
with brine and dried (Na2S04). The solvent was evaporated and the
residue was purified by flash column chromatography tsilica, 1%
ethyl acetate in hexanes) to give an oil: 1H NMR (250MHz, CDC13) ~
7.7 (d, 4H, aryl), 7.4 (m, 6H, aryl), 7.35 (d, 2H, aryl), 6.8 (d, 2H, aryl),
3.8 (s, 3H, OCH3), 3.7 (t, 2H, OCH2), 2.4 (t, 2H, CH2), 1.7 (m, 4H, CH2-
CH2), 1.05 (s, 9H, t-butyl).
B(3! 6-f4-Methoxyphenyl)hexan-l-t-butyldiphenylsilYI ether.
To 6-(4-methoxyphenyl)-5-hexyn-1-t-butyldiphenylsilyl ether
(2.0g, 4.6mmol) in ethanol (lOmL) and ethyl acetate (lOmL) was
added 5% Pd/C (1 OOmg). The mixture was subjected to 75 psi of H2
for 4 hours. The reaction was filtered through Celite and the solvent
evaporated to give an oil: 1H NMR (250MH7, CDC13) S 7.7 (d, 4H, aryl),
7.4 (m, 6H, aryl), 7.0~ (d, 2H, aryl), 6.8 (d, 2H, aryl), 3.8 (s, 3H, OCH3),
3.6 (t, 2H, OCH2), 2.5 (t, 2H, ben2ylic), 1.55 (m, 4H, CH2-CH2), 1.3 (m,
4H, CH2-CH2), 1.0 (s, 9H, t-butyl).
Bf4! 6-(4-Methoxyphenyl!hexan-l-ol.
3 5 6-(4-Methoxyphenyl)hexan-1 -t-bu~yldiphenylsilyl ether (2.0g,
4.6mmol) in tetrahydrofuran (20mL) was cooled to 0C and treated
with tetrabutylammonium fluoride (14mL, 14mmol, lM in
tetrahydrofuran). The cooling bath was removed and the reaction
was stirred at room temperature for 24 hours. The reaction was
WO 91/18883 2 ~' Pcr/ussl/03940
diluted with ethyl acetate and was washed with H2 0 and brine and
dried (Na2S04). The solvent was evaporated and the residue was
purified by flash column chromatography (silica, 0-20% ethyl acetate
in hexanes) to give a white solid: IH NMR (250MHz, CDC13) ~ 7.05 (d.
2H, aryl), S.8 (d, 2H, aryl), 3.8 (s, 3H, OCH3), 3.65 (t, 2H, OCH2), 2.55 (t,
2H, benzylic), 1.6 (m, 4H, CH2-CH2), 1.4 (m, 4H, CH2-CH2).
B(SL 6-(4-Methoxyphenyl)hexan-1-~4-toluenesulfonatel.
6-(4-Methoxyphenyl)hexan-l-ol (5.36g, 25mmol) was dissolved
in dry CH2C12 ~IOOmL) under an argon atmosphere and cooled to 0C.
To this was added pyridine (2.5mL, 30mmol) and 4-toluenesulfonyl
chloride (~.4g, 28mmol). The reaction was stirred at 0C for 20
minutes and at room temperature for 24 hours. The reaction solution
was washed with H20 and brine and dried (Na2S04). The solvent was
evaporated and the residue purified by flash column chromatography
(silica, 0-10% ethyl acetate in hexanes) to give a white solid: lH NMR
(250MHz, CDC13) ~ 1.6-1.3 (m, 8H, (CH2)4), 2.4 (s, 3H, CH3), 2.5 (t, 2H~
benzylic), 3.8 (s, 3H, OCH3), 4.0 (t, 2H, OCH2), 6.80 ~d, 2H, aryl), 7.0 (d,
2H, aryl), 7.3 (d, 2H, aryl), 7.8 (d, 2H, aryl).
Example C
E-6-(4-Methoxvphenvl)-1 -f4-toluenesulfonate)-5-hexene
C(l 2 E-6-(4-Methoxvphenyl)-5-hexenoic acid.
2 5 To a freshly prepared solution of lithium hexamethyldisilazide
(64mmol) in tetrahydrofuran (30mL)7 under an argon atmosphere,
was added a suspension of (4-carboxybutyl)triphenylphosphonium
bromide (17.6g, 30mmol) in tetrahydrofuran (45mL) at room
temperature. The reaction was stirred for 1~ minutes during which
3 0 time the orange-red color of the ylide developed. A solution of
4-anisaldehyde (4.5g, 30mmol) in tetrahydrofuran (30mL) was added
dropwise and stirring was continued for an additional 20 minutes.
The reaction was quenched with H20 (50mL) and diluted with ether
(30mL). The aqueous layer was acidified to pH 1.0 with 3N HCI and
3 5 the product was extracted into ethyl acetate (3X50mL). The
combined organic layers were dried (MgS04) and the product was
purified by flash column chromatography (silica, 1% methanol in
CH2C12) to yield the E-olefin as a solid: IH NMR (200MHz, CDC13) ~ 7.3
WO 91/18~83 '~ 2 4 PCr/US91/03940
(d, 2H, aryl), 6.8 (d, 2H, aryl), 6.3 (d, I H, olefin), 6.0 (m, I H, olefin), ~.8
(s, 3H, OCH3), 2.3 (m, 4H, allylic CH2 and CH2C02), 1.8 (q, 2H, CH2).
C(2) E-6-(4-Methoxvphenvl!-5-hexen-1-ol.
E-6-(4-Methoxyphenyl)-5-hexenoic acid (1.1 g, 5.0mmol) in dry
ether (lOmL) was slowly added to a suspension of LiAlH4 (240mg,
6.0mmol) in ether (lOmL) under an argon atmosphere. The reaction
mixture was refluxed for 45 minutes. Upon cooling to room
temperature the reaction was quenched with H20 (lOmL) followed by
I O 6N H2S04 (7mL). Ethyl acetate (20mL) was added and the organic
layer was separated and dried (MgS04); evaporation gave a white
crystalline solid: mp. 65-66C; IH NMR (200MHz, CDC13) ~ 7.2 (d, 2H,
aryl), 6.8 (d, 2H, aryl), 6.3 (d, IH, olefin), 6.1 (m, lH, olefin), 3.8 (s, 3H,
OCH3), 3.6 (t, 2H, OCH2), 2.2 (q, 2H, allylic), I.S (m, 4H, CH2- CH2);
I S Anal. Calcd. for C13HIgO2: C, 75.65; H, 8.80, found: C, 75.45; H, 8.95;
MS (CI): 207 (M~H).
C(3! _E-6-~4-Methoxyphenyl)-1 -~4-toluenesulfonate)-S-hexene.
E-6-(~-Methoxyphenyl)-S-hexen-l-ol (1.6g, 7.0mmo]) was
2 0 dissolved in dry CH2C12 (SOmL) under an argon atmosphere and
treated with 4-toluenesulfonyl chloride (7.0g, 36mmol) and pyridine
(3mL). The reaction solution was stirred at room temperature for 3.5
hours. Water (40mL) was added to the reaction and the organic layer
was separated and dried (MgS04). The product was purified by flash
2 S column chromatography (silica, 10% ethyl acetate in hexane) to give
an oil: lH NMR (200MHz, CDC13) ~ 7.8 (d, 2H, aryl), 7.3 (d, 2H, aryl),
7.2 (d, 2H, aryl), 6.8 (d, 2H, aryl), 6.2 (d, lH, olefin), 6.0 (m, lH, olefin),
4.1 (t, 2H, OCH2), 3.8 (s, 3H, OCH3), 2.4 (s, 3H, CH3), 2.1 (q, 2H, allylic),
1.6 (m, 4H, CH2- CH2); MS (Cl): 361 (M+H).
Example I
N-(3-Carboxyphenyl!-6-(E-2-carboxvethenvl!-5-decvloxv-2-
picolinamide, disodium salt
3 S I (a! 3-Decyloxv-2-(hydroxvmethv1)pvridine.
3-Hydroxy-2-(hydroxymethyl)pyridine hydrochloride (~OOmg,
3.09mrnol, Aldrich, 85%) was dissolved in dry dimethylformamide
(lOmL) and trea~ed sequentially with anhydrous K2C03 (1.30g,
9.27mmol) and 1 -iododecane (0.80mL, 3.71 mmol). The reaction was
wo 91/t8883 2 ~ ~ 3 ~ ~ ~ pcr/us91/o394o
vigorously stirred under an argon atmosphere at 90C for 1.5 hours
Upon coolin~ tO room temperature the reaction mixture was diluted
with ethyl acetate (lOOmL) and washed with H20 (5X20mL) and
brine and dried (MgS04). The compound was purified by flash
5 column chromatography (silica, 20% ethyl acetate in petroleum ether)
to give the captioned product: IH NMR ~25ûMHz, CDC13) ~ 8.17 (m,
lH, 6-pyridyl), 7.2 ~m, 2H, 4-pyridyl, 5-pyridyl), 4.78 (s, 2H, CH2),
4.48 (broad singlet, lH, OH~, 4.05 (t, J=6.6Hz, 2H, OCH2), 1.9-0.90 (m, I
1 9H, aliphatic).
1 0
1 (b) ~g~2~carboxaldehyde.
3-Decyloxy-2-(hydroxymethyl)pyridine from 1 (a), (560mg,
2.1 lmmol) in dry CH2Cl2 (7mL) was treated with MnO2 (1 .80g,
20.7mmol) and was stirred at room temperature for 24 hours. The
15 reaction was filtered through a pad of Celite and the solvent was
removed in vacuo giving the aldehyde as a pale yellow oil. The
aldehyde was used directly in the next step without further
purification .
2 0 1 (c~ _ 2-(E-2-Carboxvmethylethenyl?-3-decyloxypYridine.
3- Decyloxy-2-pyridine carboxyaldehyde from the preceeding
step (429mg, 1.63mmol) was dissolved in dry toluene (3.5mL) under
an argon atmosphere and treated with methyl (triphenyl-
phosphoranylidene)acetate (820mg, 2.4$mmol). The reaction mixture
2 5 was heated at 45~C, at which point the reaction became homogeneous,
for 30 minutes. Upon cooling to room temperature the reaction was
diluted with ethyl acetate (lOOmL) and washed with H20 (2X20mL~
and brine and dried (MgS04). The product was purified by flash
column chromatography (silica, 10: 5: 85, e~hyl acetate: CH2C12:
30 petroleum ether) to give the product as a pale yellow solid: IH NMR
(200MHz, CDC13) ~ 8.25 (m, IH, 6- pyridyl), 8.1 (d, J=16.2Hz, lH,
olefin), 7.25 (m, 2H, 4-pyridyl, 5-pyridyl), 7.05 (d, J=16.2Hz, lH,
olefin), 4.05 (t, J=6.6Hz, 2H, OCH2), 3.85 (s, 3H, C02CH3), 1.95-0.90 (m,
1 9H, aliphatic).
I(d! 2-(E-2-(:arboxvmethvlethenyl)-3-decvloxvpvridine N-oxide.
2-(E-2-Carboxymethylethenyl)-3-decyloxypyridine (390mg,
1.22mmol) was dissolved in dry CH~C12 (6mL) under an argon
atmosphere, cooled to O~C, and treated with 85%
WO 91/1888~ i 2 6 PCI'/US91/03940
3-chloroperoxybenzoic acid (278mg, 1.34mmol). Following the
addition, the cooling bath was removed and the reaction was stirred
at room temperature for 24 hours. The reaction solution was diluted
with CH2Cl2 (50mL) and poured into saturated aqueous NaHC03
5 (SOmL). The aqueous phase was extracted with CH2C12 (3XSOmL)
and the combined CH2C12 extracts were washed with brine and dried
(MgS04). Flash column chromatography (silica, 10% CH2Cl2 in ethyl
acetate~ gave the N-oxide as a pale yellow solid: lH NMR (250MHz,
CDC13) ~ 8.18 (d, J=16.2Hz, lH, olefin), 7.97 (d, J=6.5Hz, lH, 6-pyridyl),
10 7.58 (d, J=16.2Hz, lH, olefin), 7.11 (dd, J=~.6, 6.5 Hz, lH, S-pyridyl),
6.82 (d, J=8.6Hz, lH, 4- pyridyl), 4.08 (t, J=6.6Hz, 2H, OCH2), 3.82 (s,
3H, C02CH3), 1.93- 0.88 (m, 19H, aliphatic).
I (el 6-(E~-2-Carboxymeth~LlethenYl)-S-decYloxY-2-pYridone
l S 2-~E-2-Carboxymethylethenyl)-3-decyloxy-pyridine N-oxide
(180mg, 0.537mmol) was dissolYed in dry dimethylformamide
(2.2mL) under an argon atmosphere and cooled to 0C. To this was
slowly added trifluoroacetic anhydride (0.76mL, 5.38mmol) followed
by removal of the cooling bath. The reaction was stirred at room
2 0 temperature for 18 hours. The reaction solution was diluted with
ethyl acstate (75mL) and slowly poured into saturated aqueous
NaHC03 (30mL). The organic layer was washed with NaHC03 (20mL)
and brine and dried (MgS04). The product was obtained as a yellow
solid and was used without further purification: IH NMR (250MHz,
25 CDC13)~7.75 (d, J=16.3Hz, IH, olefin), 7.40 (d, J=9.8Hz, lH, 3-pyridyl),
7.01 (d, J=16.3Hz, lH, olefin)9 6.73 (d, J=9.8Hz, lH, 4-pyridyl), 3.95 (t,
J=6.6Hz, 2H, OCH2), 3.82 (s, 3H, C02CH3), 1.~2-0.88 (m, l9H, aliphatic);
MS (CI): 336 (M+H).
3 0 1 (f) 6-(E-2-CarboxvmethvlethenYl~-S-decvloxv-2-trlfluoro-
methylsulfonate.
To a cooled (0C) solution of 6-(E-2-carboxymethylethenyl)-S-
decyloxy-2-pyridone (200mg, 0.596mmol) in dry CH2C12 (3.0mL)
under an argon atmosphere was added dry pyridine (0.48mL,
3 5 5.96mmol) and trifluoromethanesulfonic anhydride (0.30mL,
1.78mmol). The reaction was stirred at 0C for lS minutes. l'he
reaction was diluted with e~hyl acetate (~OmL) and washed with H20
(2ûmL), 2% HCI (lOmL), saturated NaHC03 (20mL), and brine and
dried (MgS04). Purification by flash column chromatography (silica.
wo 91/lB883 2 ~ 8 3 9 ~a ~ PCr/US9i/03940
5% ethyl acetate in petroleum ether) gave the sulfonate as a colorless
oil: IH NMR (250MHz, CDC13) ~ 7.97 (d, J=15.8Hz, lH, olefin), 7.36 (d,
J-8.8Hz, lH, 3-pyridyl), 7.11 (d, J=8.8H[z, IH, 4-pyridyl), 6.96 (d,
J=15.8Hz, lH, olefin), 4.05 (t, J=6.5Hz, 2H, C)CH2), 3.83 (s, 3H, C02CH3).
1.92-0.88 (m, 19H, aliphatic).
I (g! N-(3-~(E-2-carboxymethylethenvl)-~-
decvloxY-2-picolinamide.
6-(E-2-Carboxymethylethenyl)-S-decyloxy-2-trifluoromethyl -
1 0 sulfonate (160mg, û.342mmol) was dissolved in dry
dimethylformamide (1.25mL) and treated sequentially with methyl
3-aminobenzoate (775mg, 5.13mmol, Lancaster), Pd(OAc)2 (4.5mg,
0.020mmol), and 1,1 '-bis(diphenylphosphino)ferrocene (22mg,
0.040mmol). S~arbon monoxide was gently bubbled through the
1 5 solution for 5 minutes. The reaction was then heated at 90C under a
CO-atmosphere (balloon pressure) for 4 hours. Upon cooling to room
temperature the reaction was diluted with ethyl acetate (75mL) and
washed with 2~o HCI (5XlOmL), H20 (lSmL), saturated NaHC03
(lSmL), and brine and dried (MgS04). Purification by flash column
2 0 chromatography (silica, 10:20:70, ethyl acetate:CH2C12:petroleum
ether) gave the arnide as a colorless solid: 1H NMR (250MH2, CDC13)
9.85 (s, lH, NH), 8.29 (s, lH, 2-phenyl), 8.27 (d, J=8.7Hz, lH, 3-
pyridyl), 8.14 (d, J=7.9Hz,lH, 4-phenyl), B.10 (d, J=15.8Hz, lH, olefin),
7.84 (d, J=7.9Hz, 1H, 6-phenyl), 7.48 (dd, J=7.9Hz, lH, 5-phenyl~, 7.38
(d, J=8.7Hz, lH, 4-pyridyl), 7.08 (d, J=15.8Hz, lH, olefin), 4.12 (t,
J=6.6Hz, 2H, OCH2), 3.95 (s, 3H, CO~CH3), 3.88 (s, 3H, C02CH3), 1.96-
0.88 (m, l9H, aliphatic); Anal. Calcd. for C28H366N2: C, 67-72; H~
7.31; N, 5.64, found: C, 67.50; H, 7.27; N, 5.57; MS (CI): 497.5 (M+H).
3 0 1 fh)_ N-f3-Carboxyphenvl!-6-(E-2-carboxyethenvl!-5-decvloxv-2-
picQlinamide~ disodium salt
N-(3-Carboxymethylphenyl)-6-(E-2-carboxymethylethenyl)-5 -
decyloxy-2-picolinamide (60mg, 0.121mmol) was dissolved in
tetrahydrofuran (1.25mL) and MeOH (0.50mL) and treated with lM
LiOH (0.50mL). The reaction was stirred at room temperature for 6
hours. The reaction was made mildly acidic by the addition of 2% HCl
(0.75mL), it was then diluted with ethyl acetate (50mL) and washed
with H20 (3XlOmL) and brine and dried (MgS04); the solvent was
removed in vacuo. The diacid was dissolved in saturated aqueous
wo 91/1888~ ~ ~ 2 8 pcr/us91/o394o
Na2C03 (3-5mL) and purified by Reversed Phase MPLC (RP-18 sill~d.
10-65% MeOH in H20) and isolated by lyophilization and was
obtained as a white amorphous solid: lH NMR (250MHz, CD30D) ~
8.22 (s, 1 H, 2-phenyl), 8.13 (d, J=8.7]Hz, I H, 3-pyridyl), 7.90-7.70 (m,
2H, 4-phenyl, 6- phenyl), 7.73 (d, J=15.8Hz, lH, olefin), 7.65 (d,
J=8.7Hz, IH, 4- pyridyl), 7.48 (dd, J=7.~Hz, IH, S-phenyl), 7.17 (d,
J=15.8Hz, IH, olefin~, 4.26 (t, J=6.6Hz, 2H, OCH2), 1.98-0.82 (m, 19H,
aliphatic); FAB-MS: (+ve), 513.1 (M+H); (-ve), 489.0 (M-Na).
I O Example 2
N-(3-(; arboxyphenyl)-6-(2-carboxYethYl)-S-
decvloxy-2-picolinamide
2(a! N-(~ arboxymethvlphenvl!-6-(2-carbQxvmethylethvl)-5-
1 S decv}oxv-2-picolinamide.
N-(3-Carboxymethylphenyl)-6-(E-2-carboxymethylethenyl)-S-
decyloxy-2-picolinamide (70mg, 0.141mmol) was dissolved in ethyl
acetate (lmL), treated with 5% Pd/C (lOmg), and stirred under an
atmosphere of H2 (balloon pressure) for 4 hours. The reaction could
not be followed by TLC and the product vvas not soluble in ethyl
acetate. The precipitated product was dissolved by the addition of
CH2C12 (SmL) and the solution was filtered through a pad of Celite.
The product was purified by flash column chromatography (silica, 5%
ethyl acetate in CH2C12) to give captioned picolinamide as a white
2 5 solid: 1 H NMR (250MHz, CDC13) ~ 10.02 (s, lH, NH), B.48 (s, 1 H, 2-
phenyl), 8.18 (d, J=7.9Hz, lH, 4-phenyl), 8.11 (d, J=8.5Hz, lH, 3-
pyridyl), 7.81 (d, J=7.9Hz, lH, 6-phenyl), 7.46 (dd, J=7.9Hz, IH, 5-
phenyl), 7.20 (a, J=8.5Hz, lH, 4-pyridyl), 4.05 (t, J=6.4Hz, 2H, OCH2),
3.94 (s, 3H, C02CH3), 3.68 (s, 3H, C02CH3), 3.24 (t, J=6.9Hz, 2H, CHz),
2.88 (t, J=6.9Hz, 2H, CH2), 1.88-0.86 (m, l9H, aliphatic); Anal. Calcd.
for C28H386N2~ 7-45; H, 7.68; N, 5.62, found: C, 67.26; H, 7.76; N,
5.54; MS ~CI): 499 ~M+H).
2~b! N-(3-Carboxyphenyl!-6-l2-carboxyethvl!-S-decyloxy-2-
3 5 picQlinamide, dipotassium salt.
N-(3-Carboxymethylphenyl)-6-(2-carboxymethylethyl)-5 -
decyloxy-2-picolinamide (54mg, 0.108mmol) was suspended in
tetrahydrofuran (I.lmL) and methanol (0.70mL) and treated with lM
LiOH (0.45mL, 0.45mmol). The reaction was stirred at room
wo 91/18883 ~ 5f~ PCr/USs1/03940
temperature for 30 hours. The reaction mixture was diluted with
ethyl acetate (~OmL) and poured into 2% HCI (lSmL). The ethyl
acetate layer was washed with H20 (3X20mL) and brine and dried
(MgS04). The solvent was removed in v~c~Q and the solid diacid was
- S dissolved in an aqueous KHC03 solution (3-SmL). Purification by
Reversed Phase MPLC (RP-lB silica, 10-65% methanol in H20) and
isolation by Iyophilization gave the salt as a white amorphous solid:
lH NMR (250MHz, CD30D) â 8.49 (s, lH, 2-phenyl), 8.00 (d, J=8.5Hz,
lH, 3-pyridyl), 7.88 (d, J=7.9Hz, lH, 4-phenyl), 7.72 (d, J=7.9Hz9 lH, 6-
phenyl), 7.36 (m, 2H, 4-pyridyl, 5-phenyl), 4.11 (t, J=6.4Hz, 2H, OCH2),
3.l9 (t, J=6.9Hz, 2H, CH2), 2.66 (t, J=6.9Hz, 2H, CH2), 1.92-0.87 (m,
l9H, aliphatic); FAB-MS: (+ve), 547.4 (M+H); (-ve), 507.3 (M- K).
1~ Example 3
N-(3 -Carboxvphenvl )-6-(E-2-carboxyethenYl)-5 -tetradecvl oxy -2-
picolinamide~ dilithium salt
3(a~ 3-Hy~r~y-2-pvridine carboxyaldehYde.
3-Hydroxy-2-(hydroxymethyl)pyridine hydrochloride (1.32g,
6.9mmol, Aldrich, 85%) was dissolved in dry CH2C12 (35mL) and
treated with triethylamine (1.lmL, 7.89mmol) and MnO2 (6.0g,
69mmol~. The reaction was stirred at room temperature for 18 hours,
filtered through a pad of celite, and concentrated in vacuo. The crude
2 S aldehyde was used directly in the next step without further
purification.
~(b~ 3-Tetradeçyloxv-2-p~yridine carboxvaldehvde.
3-Hydroxy-2-pyridine carboxyaldehyde obtained above (appx.
3 0 6.9mmol) was dissolved in dry dimethylformamide (lOmL) and
treated sequentially with anhydrous K2C03 (2.86g, 20.7rnmol) and
1-iodotetradecane (2.00mL, 7.59mmol). The reaction was vigorously
stirred under an argorI atmosphere at 90C for 4.5 hours. Upon
cooling to room temperature the reaction mixture was diluted with
3 S ethyl acetate (lOOmL) and washed with H20 (SX20mL) and ~rine and
dried (MgS04). Purification by flash column chromatography (silica,
30% ethyl acetate in petroleum ether) gave the carboxyaldehyde as a
pale yellow oil: lH NMR (250MHz, CDC13) ~10.43 (s, lH, CHO), 8.38
WO 91/18883 ~ 3~ 6 pcr/lJs91/o394o
(dd, J=4.1, l.5Hz, lH, 6- pyridyl), 7.42 (m, 2H, 4-pyridyl, 5-pyridylj.-
4.10 (t, J=6.5Hz, 2H, OCH2), 1.91-0.88 (m, 27H, aliphatic).
3(c) 2-(E-2-Carboxvmethvlethenvl)-3-tetradecvlox~,~pvridine.
Prepared according to the procedure described for 2-(E-2-
carboxymethylethenyl)-3-decyloxypyridine: 1 H NMR (250MHz,
CDC13) ~ 8.22 (dd, J=4.0, 1.8Hz, lH, 6-pyridyl), 8.10 (d, J=15.8Hz, lH,
olefin), 7.21 (m, 2H, 4-pyridyl, 5-pyridyl3, 7.02 (d, J=15.8Hz, I H,
olefin), 4.02 (t, J=6.5Hz, 2H, OCH2), 3.81 (s, 3H, CO2CH3), 1.88-0.88 (m,
1 0 27H, aliphatic).
3(d! 2-(E-2-CarbQxymethvlethenyl~-3-tetradecYloxypyridine N-
oxide.
This compound was prepared according to the procedure
1 5 described for 2-(E-2-carboxymethylethenyl)-3-decyloxypyridine
N-oxide: 1H NMR (250MHz, CDC13) ~ 8.18 (d, J=16.2Hz, lH, olefin),
7.95 (d, J=6.5Hz, IH, 6-pyridyl), 7.58 (d, J=16.2Hz, lH, olefin), 7.10
(dd, J=8.5, 6.5 Hz, lH, 5-pyridyl), 6.80 (d, J=8.5Hz, lH, 4-pyridyl), 4.08
(t, J=6.6Hz, 2H, OCH2), 3.82 (s, 3H, CO2CH3), 1.88-0.88 (m, 27H,
2 0 aliphatic).
3(e~ 6-¢E-2-Carboxvmethylethenvl~-5-~etradecvloxy-2-pvridone.
This compound was prepared according to the procedure
described for 6-(E-2-carboxymethylethenyl)-5-decyloxy-2-pyridone:
IH NMR (250MHz, CDC13)~.7.75 (d, J=16.3Hz, lH, olefin~, 7.40 (d,
J=9.8Hz, lH, 3-pyridyl), 7.01 (d, J=16.3Hz, IH, olefin), 6.73 (d, J=9.8Hz,
IH,-4-pyridyl), 3.95 (t, J=6.6Hz, 2H, 0CH2), 3.82 (s, 3H, C02CH3), 1.82-
0.88 (m, 27H, sliphatic).
3(fl 6-(E-2-Carboxvmethvlethenyl!-5-tetradecyloxv 2
trifluoromethylsulfonate.
This compound was prepared according to the above procedure
for preparing 6-(E-2-carboxymethylethenyl)-5-decyloxy-2- ;
trifluoromethylsulfonate: lH NMR (250MHz, CDC13)~.7.96 ~d,
J=15.7Hz, lHt olefin), 7.35 (d, J=8.8Hz, lH, 3-pyridyl), 7.10 (d, J-8.8Hz,
lH, 4-pyridyl), 6.96 (d, J=15.7Hz, IH, olefin), 4.04 (t, J=6.5Hz, 2H,
OCH2), 3.82 ~s, 3H, CO2CH3), 1.85-0.88 (m, 27H, aliphatic).
wo 91/l8883 2 ~ ~ 3 9 ~ ~ PCr/US91tO394~)
3(g! N-(3-Carboxymethvlphenvl!-6-LE-2-carboxYmethYIethenv
tetradecvloxy-2-picolinamide.
The method of Example I (g) was used to prepare N-(3-
carboxyme~hylphenyl)-6-(E-2-carboxymethylethenyl)-5-decyloxy-~-
picolinamide: lH NMR (250MHz, CDC13) ~ 9.86 (s, IH, NH), 8.29 (s, lH,
2-phenyl), 8.27 (d, J=8.7Hz, lH, 3-pyridyl), 8.13 (d, J=7.9Hz, lH,
4-phenyl), 8.09 (d, J=15.8Hz, IH, olefin), 7.84 (d, J=?.9Hz, IH,
6-phenyl), 7.48 (dd, J=7.9Hz, IH, 5-phenyl), 7.38 (d, J=8.7Hz, IH,
4-pyridyl), 7.08 (d, J=15.8Hz, lH, olefin), 4.12 (t, J=6.6Hz, 2H, OCH2),
1 0 3.95 (s, 3H, C02CH3), 3.88 (s, 3H, C02CH3), 1.94-0.88 (m, 27H,
aliphatic); MS (Cl): 553.4 (M+H).
3(h~ N-(3-C~,rboxyphenyl)-6-(E-2-carbo~yethenyl!-5-tetradecYloxv-
2-picolinamide~_dilithium salt
1 5 N-(3-Carboxymethylphenyl)-6-(E-2-carboxymethylethenyl )-5 -
tetradecyloxy-2-picolinamide (173mg, 0.313mmol) was dissolved in
tetrahydrofuran (4.0mL) and methanol (I.OmL) and treated with IM
LiOH (I.OmL). The reaction was stirred at room temperature for 48
hours. The resulting gel was disso]ved in H20 (3mL) and the
tetrahydrofuran and methanol were removed in vacuo. The product
was purified by Reversed Phase MPLC (RP-18 silica, 10-65% methanol
in H20) and isolated by ~yophilization to give the salt as a colorless
amorphous solid: lH NMR (250MHz, CD30D) ~ 8.32 (s, IH, 2-phenyl),
8.12 (d, J=8.7Hz, lH, 3-pyridyl), 7.85 (d, J=15.7Hz, lH, olefin), 7.83 (d,
2 5 J=7.9Hz, lH, 4-phenyl), 7.76 (d, J=7.9Hz, IH, 6-phenyl), 7.52 (d,
J=8.7Hz, lH, 4-pyridyl), 7.38 (dd, J=7.9Hz, lH, 5-phenyl), 7.26 (d,
J=15.7H~, IH, olefin), 4.16 (t, J=6.6Hz, 2H, OCH2), 1.94-0.89 (m, 27H,
aliphatic); FAB- MS: (+ve), 537 (M+H); (-ve), 529 (M-Li).
3 0 Example 4
N-(3-Carboxyphenvl)-6-(E-2-carboxyethenv]!-5-
dodecyloxy-2-picolinamide. (dilithium salt)
N-(3 -Carboxyphenyl)-6-(E-carboxyethenyl)-5-dodecyloxy-2-
picolinamide, dilithium salt, was prepared according to the procedure
3 5 described for N-(3-carboxyphenyl)-6-(E-carboxyethenyl)-5-
tetradecyloxy-2-picolinamide, dilithium salt by substituting
1-iodododecane for 1-iodotetradecane (See Example 3).
WO 91/t88~3 ~ '?~ ` 3 2 PCI/US91/03940
4(a! 3-Dodecv_oxY-2-pvridine carboxyaldehyde: I H NMR (250MHz,
CDC13) ~ 10.43 (s, lH, CHO), 8.38 (dd, lH, 6-pyridyl), 7.42 (m, 2H, 4-
pyridyl, 5-pyridyl), 4.1 (t, 2H, OCH2), 1.91-0.88 (m, 23H, aliphatic).
4(b! _2-(E-2-Carbo~$yrnethylethenyl!-3-dodecvloxy~yridane: 1 H NMR
(250MHz, CDC]3~ ~ 8.22 tdd, IH, 6-pyridyl), 8.1 (d, lH, J=15.8Hz,
olefin), 7.21 ~m, 2H, 4-pyridyl, S-pyriclyl), 7.02 (d, lH, J=15.8Hz,
olefin), 4.02 (t, 2H, OCH2), 3.81 (s, 3H, CO2CH3), l.B8- 0.88 (m, 23H,
aliphatic).
4(c!_ 2-(E-2-CarboxymethYlethenvl!-3-dodecYloxypyridine N-oxide:
lH NMR (250MHz, CDC13) ~ 8.15 (d, lH, J =16.2Hz, ole~ln), 7.9 (d, lH,
6-pyridyl), 7.58 (d, lH, J=16.2Hz, olefin), 7.1 (dd, lH, 5- pyridyl), 6.8
(d, lH, 4-pyridyl), 4.08 (t, 2H, OCEI2), 3.82 (s, 3H, C02CH3), 1.88-0.88
1 5 (m, 23H, aliphatic).
4(e~ 6-(E-2-CarboxymethYlethenyl?-S-dodecyloxv-2-pYridone: 1 H
NMR (250MHz, CDC13) ~ 8.0 (s, lH, OH), 7.75 ~d, lH, J=16Hz, olefin), 7.4
(d, lH, 3-pyridyl), 7.0 (d, lH, J=16Hz, olefin), 6.7 (d, lH, 4-pyridyl),
4.0 (t, 2H, OCH2), 3.82 (s, 3H, CO2CH3), 1.85- 0.88 (m, 23H, aliphatic).
4(f! __6-(E-2-CaTboxymethylethenYl)-5-dodecvloxy-2-trifluo~O-
methvlsulfonate: lH NMR ~250MHz, CDC13) ~ 7.95 (d, lH, J=15.9Hz,
olefin), 7.37 (d, lH, 3-pyridyl), 7.1 (d, IH, 4- pyridyl), 6.95 (d, IH,
J=15.9Hz, olefin), 4.1 (t, 2H, OCH2), 3.8 (s, 3H, CO2CH3), 1.89-0.88 (m,
23H, aliphatic).
4(g! N-(~ arboxymethylphenyl1-6-(E-2-carboxymethYlethenyl~
dodecyloxy-2-picolinamide: lH NMR (250MHz, CDC13) ~ 9.86 (s, lH,
NH~, 8.29 (s, lH, aryl). 8.27 (d, lH, 3-pyridyl), 8.13 (d, lH, aryl), 8.09
(d, lH, J=15.8Hz, olefin), 7.84 (d, IH, aryl), 7.5 (t, lH, aryl), 7.38 (d7 lH9
4-pyridyl), 7.08 (d, lH, J=15.8Hz, olefin), 4.15 (t, 2H, OCH2), 3.98 (s,
3H, CO2CH3), 3.88 (s, 3H, CO?CH3), 1.94-0.88 (m, 23H, aliphatic); Anal.
Calcd. for C30H4oN2o6: C9 68.68; H, 7.S9; N, 5.34, found: C, 68.43; H,
7.54; N 5.21; MS (Cl): 525 (M+H).
4(h) N-(3-Carboxyphenvl!-6-(E-2-carboxyethenyl~-5-d~?dçcyloxy-2-
picolinamide~ dilithium sa]t: lH NMR (250MHz, CD30D) ~ 8.37 (s, IH,
aryl), 8.12 (d, lH, 3-pyridyl), 7.85 (d, lH, J=15.7Hz, olefin), 7.83 (d,
WO 9l/18883 2 0 3 3 9 ~ ~ PCI/US9t/0394~
lH, aryl), 7.77 (d, lH, aryl), 7.55 (d, lH, 4-pyridyl), 7.38 (t~ IH, aryl)~
7.26 (d, lH, J=15.7Hz, olefin), 4.16 (t, 2H, OCH2), 1.90-0.88 (m, 23H,
aliphatic); FAB- MS: (+ve), 509 (M+H); (-ve), 501 (M-Li).
Ex~mp!e 5
N~ arboxvphenvl)-6-(E-2-carbQxyethenyl!-5-octyloxY-2
I)icolinamide~ dilithium salt
- N-(3-Carboxyphenyl)-6-(E-carboxyethenyl)-5-octyloxy-2-
picolinamide, dilithium salt, was prepared according to the procedure
1 0 described for N-(3-carboxyphenyl)-6-(E-carboxyethenyl~-5-
tetradecyloxy-2-picolinamide, dilithium salt (Example 3), substituting
1-iodooctane for 1-iodotetradecane.
5(a~ 3-Octvlox -2-py~idine carboxvaldehyde. 1 H NMR (~50MHz,
1 5 CDC13) ~ 10.43 (s, lH, CHO), 8.38 (dd, lH, 6-py~idyl), 7.42 (m, 2H, 4-
pyridyl, 5-pyridyl), 4.1 (t, 2H, OCH2), 1.91-0.88 (m, 15H, aliphatic~.
5(b) 2-fE-2-Carboxvmethylethenyl)-3-octyloxypvridine. 1 H NMR
(2~0MHz, CDC13) ~ 8.22 (dd, IH, 6-pyridyl), 8.1 (d, lH, J-15.8Hz,
olefin), 7.21 (m, 2H, 4-pyridyl, 5-pyridyl), 7.02 (d, lH, J=15.8H,
olefin), 4.02 (t, 2H, OCH2), 3.81 (s, 3H, C02CH3), 1.88- 0.88 (m, 15H,
aliphatic) .
S(c) 2-(E-2-carboxvmethvlethenvll-3-octyloxvpvridine N-oxide. 1 H
2 5 NMR (250MHz, CDC13) ~ 8.15 (d, l H, J=16.2Hz, olefin), ?.9 (d, lH, 6-
pyridyl), 7.58 (d, lH, J=16.2Hz, olefin), 7.1 (dd, lH, ~- pyridyl), 6.8 (d,
lH, 4-pyridyl), 4.08 (t, 2H, OCH2), 3.82 (s, 3H, C02CH3), 1.88-0.88 (m,
l 5H, aliphatic).
3 0 $~d~ 6-(E-2-CarboxYmethvlethenYl~-5-octvloxv-2-pYridone. 1 H NMR
(250MHz, CDC13) ~ 8.0 (s, lH, (:)H), 7.75 ~d, lH, J=16Hz, olefin), 7.4 (d,
lH, 3-pyridyl), 7.0 (d, lH, J=16Hz, ole~ln), 6.7 (d, lH, 4-pyridyl), 4.0 (t,
2H, OCH~), 3.82 (s, 3H, C02CH3), 1.85-0.88 (m, 15H, aliphatic).
3 5 5(e) 6-LE-2-Carboxvmethv!ethenvl~-5-octvloxv-2-
trifluoromethvlsu!fonate. IH NMR (250MHz, CDC13) ~ 7.95 (d, IH,
J=15.9Hz, olefin), 7.37 (d, lH, 3-pyridyl), 7.1 (d, lH, 4-pyridyl), 6.95
(d, lH, J=15.9Hz, olefin), 4.1 (t, 2H, OCH2), 3.8 (s, 3H, C02CH3), 1.89-
0.88 (m, 1 SH, aliphatic).
3 4
WO 91/18883 PCT/US91/03940
2(~8'~9r~ ~
5(f) N-(3-~arboxymethylphenvl)-6-fE-2-carboxvmethvlethenyl)-~-
octvloxy-2-picolinamide. IH NMR (250MHz, CDC13) ~ 9.86 (s, lH, NH),
8.29 (s, IH, aryl), 8.27 (d, IH, 3-pyridyl), 8.13 (d, IH, aryl), 8.09 (d,
lH, J=15.8Hz, olefin), 7.84 (d, IH, aryl), 7.5 (t, IH, aryl), 7.38 (d, lH, 4-
pyridyl), 7.08 (d, IH, 3=15.8Hz, olefin), 4.15 (t, 2H, OCH2), 3.98 (s, 3H,
C02CH3), 3.88 (s, 3H, CO2CH3), 1.94-0.88 (m, 15H, aliphatic).
5(~) 1`1-(3-Carboxyphenyl~-6-(E-2-c~oxvethenvl)-~-octyloxv-2-
picolinamide. dilithium salt 1H NMR (250MHz, CD30D) ~ 8.37 (s, lH,
- aryl), 8.12 (d, lH, 3-pyridyl~, 7.85 (d, lH, J=15.7Hz, olefin), 7.83 (d,
lH, aryl), 7.77 (d, lH, a~yl), 7.55 (d, lH, 4-pyridyl), 7.38 (t, lH, aryl),
7.26 (d, IH, J=15.7Hz, olefin), 4.16 (t, 2H, OCH2), 1.90-0.88 (m, 15H,
aliphatic); FAB- MS: (+ve), 601.3 (M~H); (-ve), 598.9 (M-H).
Example 6
N-f3-~arboxyphenyl!-6-(E-2-carboxyethenyl~-5- ~8-(~
methoxvphenvl!octvloxvl-2-pico!inamide~ dilithium salt
N-(3 -Carboxyphenyl)-6-(E-2-carboxyethenyl)-5 - [ 8 -(4-
2 0 methoxyphenyl)octyloxy]-2-picolinamide, dilithium salt was prepared
according to the procedure described for N-(3-carboxyphenyl)-6-(E-
2-carboxyethenyl)-5-tetradecyloxy-2-picolinamide, dilithium salt
(Example 4)j substituting 8-(4-methoxyphenyl)octan-1-(4-
toluenesulfonate) for 1-iodotetradecane (See Example 3).
2 5 Following the procedures of Example 3(d) et seq, the following
compounds were prepared.
6ta! _2-fF-2-Carboxymethy-lethenvl~-(4-methoxvphenyl)
-octvloxvlpvridine.
The tosylate of, Example A was used to prepare this compound.
lH NMR (250MHz, CDC13) ~ 8.28 (dd, J=4.0, 1.8Hz, lH, 6-pyridyl), 8.17
(d, J=15.8Hz, lH, olefin), 7.28 (m, 2H, 4-pyridyl, 5-pyridyl), 7.12 (d,
J=8.6Hz, 2H, aryl), 7.02 (d, J=15.8Hz, lH, olefin), 6.89 Sd, J=8.6Hz, 2H,
aryl), 4.08 (t, J=6.5Hz, 2H, OCH2), 3.87 (s, 3H, C02CH3), 3.85 (s, 3H,
35 OCH3), 2.61 (t, J=7.5Hz, 2H, benzylic), 1.94-1.38 (m, 12H, aliphatic).
WO 91/18883 2 ~ ~ 3 9 5 ~ Pcr/us91/03940
6(b~ 2-(E-2-5~xvmethvlethenyl)-3-~8-~-methoxvphenYl~-
oct~loxvlpyridine N-oxide.
lH NMR (250MHz, CDC13) ~ 8.02 (d, J=16.2Hz, lH, olefin), 7.80 (d,
J=6.5Hz, lH, 6- pyridyl), 7.39 (d, J=16.2Hz, IH, olefin), 7.00 (m, 2H, 5-
pyridyl, 4-pyridyl), 6.85 (d, J=8.6Hz, 2H, aryl), 6.65 (d, J=8.6Hz, 2H,
aryl), 3.91 (t, J=6.5Hz, 2H, OCH2), 3.68 (s, 3H, C02CH3), 3.62 (s, 3H,
OCH3), 2.37 (t, J=7.5Hz, 2H, benzylic), 1.82-1.10 (mj 12H, aliphatic).
6(c~ 6-(E-2-Car~oxvmethylethenvl)-5-~8-(4- nethoxvphenvl!-
1 0 octvloxvl-2-pvridone.
lH NMR (250MHz, CDC13) ~ 7.75 (d, J=16.2Hz, lH, olefin), 7.40 (d,
J=9.8Hz, lH, 3-pyridyl), 7.10 (d, J=8.6Hz, 2H, aryl), 7.00 (d, J=16.2Hz,
lH, olefin), 6.82 (d, J=8.6Hz, 2H, aryl), 6.70 ~d, J=9.8Hz, lH, 4-pyridyl),
3.95 (t, J=6.5Hz, 2H, OCH2), 3.85 (s, 3H, CO2CH3), 3.82 (s, 3H, OCH3),
1 ~ 2.57 (t, J=7.5Hz, 2H, benzylic), 1.85-1.22 (m, 12H, aliphatic).
6(d! N-(3-Carboxymethvl~henyl!-6-(E-2-~arboxymethylethenvl~-5-
4-me~hoxy~henyl)octvloxvl-2-picolinamide.
Melting point - 70-73C; lH NMR (250MHz, CDCI~) ~ 9.87 (s, lH,
NH), 8.31 (s, lH, 2-phenyl), 8.28 (d, J=8.7Hz, lH, 3-pyridyl), 8.15 (d,
J=7.9Hz, lH, 4-phenyl), 8.08 (d, J=15.8Hz, lH, olefin), 7.85 (d, J=7.9Hz9
lH, 6-phenyl), 7.48 (dd, J=7.9Hz, IH, S-phenyl), 7.36 (d, J=8.7Hz, lH,
4- pyridyl), 7.10 (d, J=8.6Hz, 2H, aryl), 7.08 (d, J=15.8Hz, lH, olefin),
6.85 (d, J=8.6Hz, 2H, a~yl), 4.12 (t, J=6.5Hz, 2H, OCH2), 3.95 (s, 3H,
2 5 CO2CH3), 3.88 (s, 3H, CO2CH3), 3.79 (s, 3H, OCH3), 2.56 (t, J=7.5Hz, 2H,
- benzylic), 1.99-1.28 (m, 12H, aliphatic); Anal. Calcd. for C33H38N27:
C, 68.97; H, 6.67; N, 4.88, found: C, 69.21; H, 6.88; N, 4.46; MS (CI): 575
(M+H).
3 0 6(e! N-f3-Carboxyphenyl~-6-(E-2-carboxvethenvl!-S-r8-(4-
methoxvphenyl~octvloxyl-2-picolinamide. dilithium salt . Melting
point 315C (dec.); 1H NMR (250MHz, CD30D) ~ 8.31 (s, lH, 2-phenyl),
8.12 (d, J=8.7Hz, lH, 3-pyridyl), 7.86 (d, J=7.9Hz, lH, 4-phenyl), 7.85
(d, J=15.8Hz, lH, olefin), 7.76 (d, J=7.9Hz, lH, 6-phenyl), 7.52 (d,
- 35 J=8.7Hz, lH, 4-pyridyl), 7.39 (dd, J=7.9Hz, lH, 5-phenyl), 7.26 (d,
J=15.8Hz, lH, olefin), 7.07 (d, J=8.6Hz, 2H, aryl), 6.80 (d, J=B.6Hz, 2H,
aryl), 4.15 (t, J=6.5Hz, 2H, OCH2), 3.74 (s, 3H, OCH3), 2.53 (t, J=7.5Hz,
2H, benzylic), 1.93-1.37 (m, 12H, aliphatic); Anal. Calcd. for
~3~6 3 6 Pcr/US9i/03940
C31H32N2~)7Li2 5/2 H2O: C, 61.69; H, 6.18; N, 4.64, found: C, 61.6~,
H, 5.91; N, 4.60; FAB-MS: (+ve), 559.4 (M+H); (-ve), 551.4 (M-Li).
Following the same procedure, but substituting for methyl 3-
aminobenzoate the appropriate chloro substituted methyl
3-aminobenzoate, the following compounds were prepared:
N-(3-carboxy-6-chloropheny3)-6-(E-2-carboxyethenyl)-5-[~-~4-
methoxyphenyl)octyloxy]-2-picolinamicle, dilithium salt; and
N-(3-carboxy-4-chlorophenyl)-6-(E-2-carboxyethenyl)-5-[8-(4-
methoxyphenyl)octyloxy]-2-picolinamide, dilithium salt.
Exam~le 7
Salts may be converted to the free acid by dissolving the salt in
an aqueous solution and adding sufficient acid so as to bring the pH to
about neutral (pH 7,0) or thereabouts. Any acid may be used though
it is preferred to use a mineral acid such as HCI or the like. It is
preferred to use a dilute rather than a concentrated acid, for exan~ple
a 1 to 6 normal solution is most useful. Acid may be added at room
temperature or thereabouts; no special conditions are required. Once
the solution reaches a neutral pH or becomes acidic, the :acid will
2 0 precipitate out of solution an may be recovered by crystallization
techniques, or any other technique which may prove useful for a
given acid.
Example
2 5 Formulations for pharmaceutical use incorporating compounds
of the present invention can be prepared in :various forms and with
numerous excipients. Examples of such formulations are given below.
Inhalant ~ormulation
A compound of formula I, I to 10 mg/ml, is dissolved in isotonic
saline and aerosilized from a nebulizer operating at an air flow
adjustd to deliver the desired amount of drug per use.
Tablets: A compound of formula I, I to 10 mg/ml, is dissolved in
3 ~ isotonic saline and aerosolized from a nebulizer operating at an air
flow adjusted to deliver the desired amount of drug per use.
WO 91/t8883 2 ~ 8 3 ~ 5 6 PCr/US91/03940
Ingredients Per Tablet Per 10,000
Tablets
1. Active ingredient
(Cpd of Form. I) 40 mg 400 g
2. Corn Starch 20 mg 200 g
3. Alginic acid 20 mg 200 g
4. Sodium alginate 20 mg 200 g
5. Magnesium stearate1.~ mg 13 g
101.3 mg 1013 g
Procedure for making tablets:
Step 1 Blend ingredients No. 1, No. 2, No. 3 and No. 4 in a suitable
mixer/blender.
15 Step 2 Add sufficient water portionwise to the blend from Step I
with careful mixing after each addition. Such additions of water and
mixin~ until the mass is of a consistency to permit its conversion to
wet granules.
Step 3 The wet mass is converted to granules by passing it
2 0 through an oscillating granulator using a No. 8 mesh (2.38 mm)
screen .
Step 4 The wet granules are then dried in an oven at (60C) until
dry .
Step 5 The dry granules are lubricated with ingredient No. 5.
2 5 Step 6 The lubrieated granules are compressed on a
suitable tablet press.
Supposi tories:
Ingredients Per Supp. Per 1000 Supp.
3 0 1. Formula I compound40.0 mg40 g
Active ingredient
2. Polyethylene &Iycol 13~0.0 mg 1,350 g
1000
3. polyethylene glycol 450.0m~ 4~0 ~
3 5 4000 1840.0 mg1,840 g
Procedure:
Step 1. Melt ingredient No. 2 and No. 3 together and stir until
uniform.
Step 2. Dissolve ingredient No. 1 in the molten mass from Step 1
4 0 and stir unsil uniform.
Step 3. Pour the molten mass from Step 2 into supository moulds
and chill and remove the suppositories from moulds and wrap.