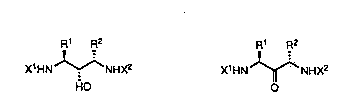Note: Descriptions are shown in the official language in which they were submitted.
Wo 92/00750 2 ~ ~ 6 ~ ~ ~ Pcr/u5gl/04757
:r~
RETROVIRAL PROTEASE INH~ITORS
BACKGROll~ OP TEI~I~I~O~
This invention relates to compounds which are inhibitors of aspartic proteases,
particularly of retroviruses.
Retroviruses, that is, viruses within the family of Retrovindae, are a class of viruses
which ~ransport their genedc mate~ial as ribonucleic acid rather than deoxyribonucleic acid.
Also hlown as RNA-tumor viruses, their presence has been associated with a wide range
of diseases in humans and animals. They are believed to be the causative agents in
pathological states associated with infection by Rous sarcoma virus (RSV), murine
leukemia virus (MLV), mouse mammary tumor virus (MMTV), feline leukesr~ia virus
(FeLV), bovine leukemia virus (BLV), Mason-Pfizer monkey virus (MPMV), simian
sarcoma virus (SSV), sirnian acquired immunodeficiency syndrome (SAD:)S), human T-
lyrnphotropic virus (HTLV-I, -II) and human immunodeficiency virus (HIV-I, HIV-2),
which is the etiologic agent of AII)S (acquired imrnunodeficiency syndrome) and AIDS
related complexes, and many others. Although the pathogens have, in many of these cases,
been isolated, no satisfactory method for treating this type of infection has been developed.
Among these viruses, the Hll~V and HIV have been especially well characterized.
Critical to the replication of retroviruses is the production of functional viral
proteins. Protein synthesis is accomplished by ~anslation of the open reading frames into
polyprotein conslructs, corresponding to the g~g, pol and enY reading frames. The g~g and
~Ql precursor proteins, are processed by a viral protease into the filnctional proteins. The
WO 92t007~0 2 0 ~ 6 ~ ~ ~ 2 - PCI/US91/047~7
HIV-1 protease has been classified as an aspartic acid protease (Meek et al., Proc. NatL
Acad. Sci. ll$A, 8~. 1841 (1989)). The proteolytic activity provided by the viral protease
in processing the polyproteins cannot be provided by the host and is essential to the life
cycle of the retrovirus. In fact, it has been demonstrated that retroviruses which lack the
S protease or contain a mutated form of it, lack infecuvity. See Katoh et al., Virolo~y, 145,
280-92(1985), Crawford, et al., J. VirQI., 53, 899-907(1985), Debouck, et al., Proc. Na~l.
Acad~ Sc;. USA, 84, 8903-6(1987). Inhibition of retroviral yrotease, therefore, presents a
method of therapy for retroviral disease.
Methods to express retroviral proteases in E. coli have been disclosed (Debouck, et
al., Proc. Natl. Acad. Sci. USA, 8903-06(1987) and Tomasse}li et al., Biochemistrv,29,
264-9 (1990) and refs. therein).
Inhibitors of recombinant HIV protease have been reported (Dreyer et al., Proc.
Natl. Acad. Sci. USA, 86, 9752-56 (1989); Tomasselli et al. supra; Roberts et al., Science,
24~, 358 (1990~; Rich et al., J~çbçm~ 33. 1285-' '1990); Sigal et al., Eur. Pat.Appl. No. 337 714; Dreyer et al. Eur. Pat. Appl. No. 3' 000). Moreover, certain of these
inhibitors have been shown to be potent inhibitors of viral proteolytic processing in cultures
of HIV-l infected T-lyrnphocytes (Meek et al., Na ure (London), ~, 90 (199n~ and by
Roberts et al. supra ).
The limitations of current s~rategies for aspartic protease inhibition include (1) oral
bioavailability; (2) plasma clearance lifetimes (e.g., through biliary excresion or
degradation); (3) selectivity of inhibition; and (4) in the case of intracellular targets,
membrane perrneability or cellular uptake. The present invention relates to a new inhibitors
of retroviral and aspartic proteases. Un~ike previously descri'oed inhibitors, the
compounds of this invention are not analogues of peptide substrates possessing a scissile
dipeptide rnimetic. They also deviate substantially from peptide substrate-like st;ucture in
that tney do not possess a conventional amino-to-carboxyl terrninus orienta~ion
SUMMARY OF THE INVENTION
This invention comprises compounds having the structures particularly pointed out
in the claims and described hereinafter which bind to retroviral proteases. These
compounds are inhibitors of viral protease and are useful for t;eating disease related to
infec~on by viruses.
This invention is also a pXarrnaceu~,ical composition, which comp;ises an
aforementioned compound and a pharrnaceutically acceptable carrier therefor.
This invention further constitutes a method ~or treating viral diseases, which
comprises adminislering to a mammal in need thereof an effective amount of an
aforemen~ioned inhibitor compound.
WO 92/û07~0 3 2 0 ~ ~ 9L 1 4 PCI/llS9l/04757
DFI'AILED DES(;~RIPTION OF THE INVENTION
The compounds of this invention have the structure I or II:
R1 R2 ,~ ,
X1HN~NHX2 X1HN ~NHX2
HO Q
I rl
wher~in X1 and X2 are the same or different and are A-(B)n- where n = ~2; and
B is, independently, an a-amino arid chosen ~rom the group: Ala, Asn, Cys, Trp,
Gly, Gln, Ile, Leu, Met, Phe, Pro, Ser, Thr, Tyr, Val, His, or trifluoroalanine, wherein the
amino group of B is bonded to A or the carboxy group of the adjacent residue B, whichever
10 is appropriate, and the carboxy group of B is bonded to the amino group of the adjacen
residue B or I or II, whichever is appropriate; and
A is covalently attached to the arnine group of the adjacent residue B or to ~he atnine
group of I or II if n=0, and ;s:
1) tri~yl,
2) hydrog~n,
3) Cl-C6 alkyl,
4) R3-Co- wherein R3 is:
a) hydrogen,
b3 ( 1 - C6 alkyl, unsubstiNted or substituted with one or.more
0 hydroxyl groups, chlorine atoms, or fluorine atoms,
c) phenyl or naphthyl unsubstituted or substituted with one or more
subs~ituents R4, wherein R4 is:
i) C1 - C4 alkyl7
ii) halogen, whrein halogen is F, Cl, Br or I,
iii) hydroxyl,
iv) nitro,
v) Cl - C3 alkoxy, or
i3 -CO-N(R10)2 wherein R10 is, independently, H or Cl-C4 alkyl:
d) a 5-7 member heterocycle such as pyridyl, furyl, or benzisoxazolyl:
. 30 5) phthaloyl wherein the aroma~c ring is unsubstituted or substituted widl one
or more substitutents ~
6) R5(R6R7C)m-Co- wherein m - 1-3 and R5, R6, and R7 are independently:
a) hydrogen,
b) chlorine or fluorine,
c) Cl - C3 alkyl unsubstituted or subs~tuted with one or r;nore chlonne
or fluorine atoms or hydroxyl groups,
Wo 92/00?~0 PCrJUS91/04757
~08~4~ 4
d) hydroxyl,
e) phenyl or naphthyl unsubsti~uted or substituted with one or more
substitutents R4,
f) Cl - C4 alkoxy,
g) a 5-7 member heterocycle,
h) R5, R6, and R7 may be independently joined to form a monocyclic,
bicyclic, or tricyclic Iing system each ~ing of which is C3-C6 cycloaLIcyl;
7) R5(R6R7C)mW- wherein m = 1-3 and W is OCO or S02 and R5, R6, and
R7 are as defined above, except R5, R~, and. R7 are not chlorine, fluorine or hydroxyl if
they are adjacent to W;
8) R8-W- wherein R8 is a 5-7 member he~erocycle such as pyridyl, furyl, or
benzisoxa~oyl;
9) R9-W- wherein R9 is phenyl or naphthyl unsubstituted or substi~uted with
one or more subs~ituents R4;
10) R5-(R6R7C)m-P(o)(oR1 l) wherein Rl 1 is Cl - C4 alkyl or phenyl;
11) R8-P(O)(OR11)-; or
12) R9-P(o)(oR1 1)-;
Rl and R2 are the same or different and are:
1) -CH2R12 wherein R12 is
a) NH-A wherein A is defined as above;
b) R5-(R6R7C)m-;
c) R5-(R6R7C)mV- wherein V is O or NH, except R5, R6 and R7 are
not hydroxyl, chlorine or fluorine if they are adjacent to V,
d) R5-(R6R7C)m-S(o)n- wherein m = 1-3 and n = 0-2 and R5, R6,
and R7 are as defined above except R5, R6, and R7 are not hydroxyl, chlorine or fluorine if
they are adjacen to sulfur,
e) R8-S(O)n-,
f) R9-s(o)n-~
g) (R13O)P(o)(oRl4)- wherein R13 and R14 are, independently:
i) C1 - C6 alkyl,
ii) C3 - C6 cycloalkyl,
iii) H,
iv) R9,
v) R8,
h) R 1 3P(o)(oR 1 4)-
i) N(R 1)2,
W092/007~0 PCr/US9i/04757
- s 2086414
j) NR1SR16 wherein R15 and R16 are joined tO form a 4-6 mernbered
saturated nitrogenous heterocycle ncluding:
i,~ azetidinyl,
ii) py.~olidinyl,
iii) piperidinyl,
iv) motpholinyl,
k) R170C~H20 wherein R17 is:
i) Cl-~6alkyl,
ii) R9,
iui) CH2Ar wherein Ar is phenyl, naphthyl or a 5-7 membered
heterocycle,
1) ~170cH~cH2ocH2~
m) N-imidazolyl where the imidazole ring is unsubstituted or substituted
by a substituent R4,
n) N-Benzirnidazolyl where the fused benzene ring is unsubstituted or
substituted by one or more substituents R4;
) C2 - C6 alkynyl, optionally substituted with one or more groups R9;
or
P) C2 - C6 alkenyl, optinally substituted with one or more gropus R9;
2) hydrogen,
3) Cl - C6 alkyl, unsubstituted or substituted with one or more chlorine or
fluorine atoms or hydroxyl groups,
4) C3 - q cycloalkyl; and phannaceutically acceptable sal~s thereof.
Peptide compounds of the foregoing description are preferred which are C2
symmetric wherein x1=x2, and Rl=R2.
Suitably the compound has structure I and Rl - R2 and Xl = X2.
Suitably Rl and R2 are Cl-C6alkyl. Preferably Rl and R2 are benzyl.
Suitably Xl and X2 are AlaAla, Val, Cbz-Val, Cbz or hydrogen. Preferably Xl
and X2 are Cbz-Val.
The compounds of this invention are useful in the manufacture of a medicament, in
pa~icular, for a medicarnent for treating infection by ret~druses.
C2 sym¢netric peptide compounds wherein Rl and R2 are Cl-C6 alkyl or aralkyl
and x1 and X2 are single amino acids or mono- or dipeptides; these groups may betenninally subsitute~ by commorl acyl groups or blocking groups commonly used inpeptide synthesis, such as t-Boc or Cbz, are also preferred.
,
- : . . . ..
WO 92/0075~ 0 ~ 6 41~ - 6 - PCI/US91/047~7
Also included in this invention are pharrnaceutically acceptable addition salts,complexes or prodrugs of the compounds of this invention. Prodrugs are considered tO be
any covalently bonded carriers which release the parent drug.
As used herein except where noted, the term "alkyl" refers to a strai~ht or br~nched
5 chain alkyl radical of the indicated number of carbon atoms including, but not limited to,
methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, n-pentyl, I-methylbutyl,
2,2-dimethylbutyl, 2-methylpentyl, 2,2-dimethylpropyl,`n-hexyl, and the like; "alkoxy"
represents an alkyl group of the indicated number of ~arbon atoms attached thr~ugh a
bridging oxygen atom; "cycloalkyl" is intellded to include staurated ring groups, such as
lO cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl; "alkenyl" is meant to
include either straight or branched hydrocarbon chains containing one or more carbon-
carbon double ~onds which may occur at any stable point along the chain, such as ethenyl,
propenyl, butenyl, pentenyl, 2-methyl propenyl, and the like; "alkynyl" refers to either a
straight or branched hydrocarbon chain or the indicated number of carbon atoms which
15 contains a carbon-carbon triple bond which may occur at any stable piont along the chain,
such as ethylyl, 2-propynyl, 2-butynyl, 4-pentynyl, 2-methyl-3-propynyl, and the like.
As used herein except where noted, the terrn "heterwycle" represents a stable 5- to
7-membered mono- or bicyclic heterocyclic ring, which is eitder satureated or unsaturated,
and which consists of carbon atoms and from one to three heteroatoms selected from the
20 group consisting of N, I and S, and wherein the nitrogen and sulfur heteroatoms rnay
optionally be oxidized, and the nitrogen heteroatom may optionally 'oe quatemized, and
including any bicyclic gTOUp in which any of the above-defined heterocyclic rings is fused
to a 'oenzene ring. The heterocyclic rings may be attached to any heteroatom or carhon a~om
which results in tne creation of a stable structure. Examples of such he~erocyclic elements
25 including piperidinyl, piperazinyl? 2-oxopine~zinyl, '~-oxopiperidinyl, 2-oxopyrrolodinyl,
2-oxoazepinyl, azepinyl, pyrrolyl, 4-piperidonyl, pyrrolidinyl, pyræolyl, pyrazolidinyl,
irnidazolyl, pyridyl, pyraz~nyl, pyrirnidinyl, prydiazinyl, oxazolyl, isoxazolyl, morpholinyl,
thiazolyl, quinuclidinyl, indolyl, quinolinyl, isoquinolinyl, benzitnidazolyl, benzopyranyl,
benzoxazoyly, furyl, tetrahydrofuryl, tetrahydrophyranyl, thienyl, thiamorpholinyl
30 sulfoxide, thiarno~pholinyl sufone, and oxadiazolyl.
When any variable (e.g., A, B, Rl, R2~ R3, ..., R17, heterocycle, substituted
phenyl, etc.) occurs more than one time in any constituent or in formula I or II, its
definition on each occurence is independent of its defini~ion at every other occurence. Also,
combination of substituents and/or vanables are pennissible only if such combinations
35 result in stable compounds. By convention used herein, a geminal diol, for example when
R6 and R7 are simultaneiously hydrowyl, is meant to be equivalent with a carbon-oxyge
double bond.
WO 92/007sO PCr/US91/0~7~7
-7- 2~8~
Other abbreviations and sytnbols comrnonly used in the art used herein to describe
the peptides include the following: -
Am~no acidthree letter code Amino acidthree letter code
Alanine Ala Leucine Leu
Argin~ne Arg Lysine Lys
Asparag~ne Asn Methionine Met
Aspartic Acid Asp PhenylalaninePhe
Cysteine Cys Proline Pro
Glutamine Gln Serine Ser
Glutaminic AcidGlu Threonine Thr
Glycine Gly Tryp20phan Trp
Histidine His Tyrosine Tyr
Isoleucine Ile Valine Val
Asparagine or Aspartic Acid Asx
Glut~nine or Glutamic Acid Glx
In accordance with conventional representation, the amino terminus is on the left
and the carboxy terrninus is on the right. All chiral amino acids (AA) can occur as
racemates, racemic mixtures, or individual enantiomers or diasteriomers, with all isomeric
forms being included in the present invention. ~-AIa refers to 3-~nino propanoic acid. Boc
refers to tbe t-butyloxycarbonyl radical, Cbz refers to the carbobenzyloxy radical, i-Bu
refers to isobutyl, Ac refers to the acetyl, Ph refers to phenyl, DCC refers to
. dicyclohexylcarbodiitnide, DMAP refers to dimethylaminopyridine, HOBT refers to 1-
hydroxybenz~triazole, NMM is N-methylmorpholine, Dl~ is dithiothreitol, EDTA is
ethylenediamine tetraacenc acid, DEA is diisopropyl etllylarnine, DBU is 1, 8 diazobicyclo
[5.4.0] undec-7-ene, DMSO is dimethylsulfoxide, DMF is dimethyl folmamide and THF is
tetrahydrofuran. HF refers to hydrofluoric acid and TFA refers to trifluoroacetic acid.
The peptide moieties denoted by X I and x2 are generally dipeptides or smaller.
However, longer pepndes which encompass the residues defined herein are also believed to
be active and are considered within the scope of this invention.
The selection of residues or end groups may be used to confer favorable
biochemical or physico-chemical properties to the compound. The use of hydrophilic
residues may be used to confer desirable solubility properties or D-amino acids at the
carboxy terminus may be used to confer resistance to exopepddases.
Synthesis of componds I in which Rl = R12CH2, R1 = R2, and xl = x2 is
achieved ~rom D-(+)-arabitol (Schemes 1-2). Thus, D-(+)-arabitol is converted to 1,2:4,5-
Dianhydro-3-(~benzyl)-D-t+)-arabitol (10) as descnbed by S.L. Schreiber, T. S~nnakia
and D.E. Uehling, J. C)r~e. Chem. 54, 15-16 (19B9). The diepoxide 10 can th~n be reacted
widl NaN3 in DMF to provide the resulting dihydroxy terminal diazide, which is converled
to the protected diaziridine, 1.2:4,5-Di-(N-benzyloxycarbonylimino)-3-(0-benzyl)pentanol,
by dimesylation of the dihydroxy terminal diazide followed by reduction with LiAlH4 with
WO 92/00750 2 0 8 6 ~ 1 4 - 8 - PCIlUS9l/û4757
concom,nitant diaziridine formation followed by reaction with benzylchloroformate. The
resulting diaziridine is reacted with appropliate nucleophiles such as (CH3)2CuLi, to
introduce the side-chain groups Rl (Scheme 1). This procedure is especially suited to the
preparation of compound I where R1 = CH2R12 where R12 is hydrogen or is a group that
forrns a staMe and reactive cuprate reagent, such as methyl, butyl, isopropyl, or other alkyl,
aLI~enyl or aryl which is optionally substituted, for exatnple with fluorine or alkoxy or
protected hydroxyl. ~ `
Alternatively, compounds represented by forrnula I in which R1 = R12CH2, Rl =
R2, and Xl = x2 can be prepared from the diepoxide 10 by reaction with appropriate
10 carbon nucleophiles such as cupra~e reagents (R12)2CuLi or alkynylalurninum reagents to
introduce the side-chain groups R1 (Scheme 2). This procedure is also especially suited to
the preparation of compound I where R1 = CH2R12 where R12 is a group that forrns a
stable and reactive cuprate reagent, as described above. The resulting diol product is
converted to the corresponding diatnine with inversion of configuration at the alcohol
15 carbons. One way in which this is accomplished is via conversion of the diol to the
dimesylate, by reaction with methanesulfonyl chloride and triethylamine, followed by
displacement with NaN3 in DMF to provide the substituted 2,4-diazido-3-
benzyloxypentane; conversion to the 2,4-diatnino denvative follows by reduction with a
hydride reagent such as LiAlH4 or by catalytic hydrogenation with a catalyst such as Pd(O)
20 or Raney-Ni to provide the core structure I (Scheme 2). Introduction of the groups X 1 _
x2 is accomplished by standard condensation reactions as are well known in the art.
Compound I in which R12 is NH-A can be prepared from the diepoxide 10 by
reaction with NaN3 in DMF to provide the resul~ing dihydroxy terminal diæide, which is
conver~ed to the colTesponding tetraazide with inversion of configuration at the alcohol
25 carbons as described a'oove, and subsequently to the colTesponding tetraamine. Selective
reaction of the terrninal amines with groups A or with protecting groups such as Boc or Cbz
is then followed by introdution of the groups X 1 = X2. In a related fashion, groups R l =
R2 in compounds I in whicn Rl is N(Rl0)2, NR15R16, R5-(R6R7C)mV- or
R5(R6R7C)m-S(o)n- can be introduced by reaction of diepoxide 10 with the appropriate
30 oxygen, nitrogen, or thiol nucleophile, with subsequent thiol oxidation as necessary;
reaction of diepoxide 10 with the appropriate phosphorus nucleophile in an Arbuzov or
Michaelis-Arbuzov reac~ion allows introduction of gropus R 1 = R2 which are
(R13O)P(o)(oRl4) or R13P(o)(oRl4)-
Altematively, compounds represented by I can be prepared from protected alpha-
35 a nino aldehydes P2NHCH(R l )CHO. The required N-protected alpha-arnino nldehydes are
readily prepared from the respective N-protected alpha-amino acids P2NHCH(Rl)CO~H,
for example by reduction of the corresponiding esters with diisobutyl alurllinum hydride, by
P~-rtUS9~/04757
W 0 92/007~0 9 ~ 0 8 6 ~ 1 ~
reduction of the derived N-methyl, N-methoxy amides P2NHCH(Rl)CONme(OMe) with
LiAlH4 (Fehrentz and Castro, ~h~ 676 (1983)), or by reduction to ~he N-protectedalpha-amino alcohol followed by oxidation with DMSO-(COC1)2 or S03-pyridine (Review:
Jurczak and Golebiowski, Chem Rev. 89, 149 (1989)). Generally the amino protecting
5 group, p2, is t-Boc-, Cbz-, p-toluenesulfonyl or another standard protecting group chosen
as well known in the peptide art. The synthesis of I proceeds via preparation of an
intelTnediate P2NH(Rl)CH(OH)CH(R2)COQ by aldol condensation with an acyl
derivative, R2CH~COQ, under the conditions of E~rans et al. (Evans, Ennis and Mathre, L
~rn. ~kçm~ Soc. 104, 1737-39 (1982); Review: Evans, Nelson and Taber, in Topics in
Stereochemis~, Vol. 13; Allinger, Eliel, Wilen, eds.; Wiley, 1982; pp 1-114.), where Q is
a chiral auxiliary used to direct the stereochemical outcome of the aldol reaction and is often
the oxazolidinone derived from valinol, norephedrine, or phenylalanol. Hydroxyl
protection with a protecting group pl such as TBDMS or benzyl and subsequent hydrolytic
removal of the group Q yields the interrnediate P2NH(R1)CH(OP1)CH(R2)C02H, which
15 is subjected to Curtius rearrangement (Reviews: Banthorpe, in Patai, "The Chemistry of the
Azido Group." pp. 397-405, Interscience Publishers, NY, 1971; Smith, Org~React. 3,
337-449 (1946)) to provide the compound P2NHtR1)CH(OPl)CH(R2)NH2 which is an
tmsymmetrically protected form of I. This route is versatile in that it allows access to all
steroisomers of compounds I and to unsyrnmetrical compounds I, in which ~l, R2 are
20 different and Xl, x2 are different.
Synthesis of compounds represented by formula II is achieved by oxidation of thècentral hydroxyl group within the corresponding compounds I, as is well known in the art.
Useful oxidation reagents include, but are not limited to, Jones Reagent, (COCI)2-DMSO,
pyridinium dichromate, and pyridinium chlorochromate.
WC) 92/~07~0 2 0 8 ~ 4 ~ o PCI/US9~/~757
Eilher enantiomer of compounds of structures I and II can be prepared from the
respec~ve enantiomer of arabitol by the procedures shown in Schemes 1-2.
Scheme 1
S Svnthesis OI I
1) p-TsCI
HO OH
2) NaH. DMF O,~ C
HO` ~C~H 3) Bn-Br ~~
HO BnO
D~+)-Arabito! Bn = benzyl
1) NaN3,DMF
2) MsCl, Et3N Cbz-N~ N-Cbz (R')2CuLi
3) UAIH4
BnO
4) Cbz-Cl
/
R' R' . R1 R2
X1HN ~NHX2 ~ X1HN ~NHX2
. OBn HO
I
Scheme 2
Svnthesis of I
R' R' 1) MsCl, Et3N
'~ "R'" ~OH 2) NaN3,DMF
BnO BnO 3) LiAlH4
1 0 Bn = benzyl
R~ R~ R1 R2
X1 HN ~ NHX2 ~ X~ HN ~ NHX2
OBn HO
I
WO 92t00750 2 0 8 6 414 PCl[/lJS91/04757
Accordingly, in another aspect, this invention is a process for preparing a
compound of the formula:
R~ R~
RiVR...N ~ NR~.RiV
OR~
wherein R' is
1) a) NH-A wherein A, R5- R10 and m are as defined for forrnula I;
b) R5-(R5E~7C)m-;
c) R5-(R6R7C)m V- wherein V is O or NH, except R5, R6 and R7 are no~
hydroxyl, chlorine or fluorine if they are adjacent to V,
d) R5-(R6R7C)m-S- wherein m = 1-3, and R5, R6, and R7 are as defined
above except R5, R6, and R7 are not hydroxyl, chlorine or fluorine if they are adjacent to
sulfur,
e) R8-S-,
f) R9-S-,
g) (R13O)P(o)(oRl4)- wherein R13 and R14 are, independently:
i) Cl-C6alkyl,
ii) C3-C6cycloa}kyl,
iii) H,
iv) R9,or
v) ~8,
h) R13P(o)(oRl4)
i) N(RlO)2~
j) NR15R16 wherein R15 and R16 are joined to form a 4-6 membered
saturated nitrogens heterocycle including:
i) azetidinyl,
ii) pyrrolidinyl,
iii) piperidinyl, or
iv) morpholinyl,
k) R17OCH~o wherein R17 is:
i) c1-c6 alkyl,
ii) R9, or
iii) C~12Ar wherein Ar is phenyl, naphthyl or a 5-7 membered
heterocycle,
1) R170CH2CH20CH2,
WO 92/007gO PCr/US91/04757
~,~8~ 12-
m) N-imidazolyl where the imidazole Ting is unsubstituted or substituted by
a substituent R4,
n) N-~enzirnidazolyl where the fused benzene ring is unsubstituted or
substituted by one or more substituents R4;
S o) C2-C6 aLkynyl, optionally substituted with one or more groups R9; or
p) C2-C6 alkenyl, optionally substituted with one or more groups R9;
2) hydrogen,
3) Cl-C6 alkyl, unsubstituted or substituted with one or more chlorine or iluorine
atoms or hydroxyl groups, or
4) C3-C7 cycloalkyl,
R" is a hydroxyl protecting group, and
R"' and R~v are hydrogen, an arnino-protecting group or talcen together are N2,
which comprises
1) reacting a compound of the formula:
~C
with a compound R'-Z, wherein Z is a moiety which renders R' nucleophilic, and R" is a
hydroxyl protecting group,
20 2) converting the resulting hydroxy groups to displaceable groups,
3) reacting the displaceable groups with a nitrogen nucleophile.
Hydroxyl protecting groups are those groups which are commonly used in the art to
mask the reactivity of the hydroxyl group, while also capable of being selectively removed
tO regenerate the hydroxyl group. Typically, the oxygen-hydrogen bond is replaced by an
25 oxygen-carbon bond. Useful hydroxyl protecting groups are described in Greene, T.W.,
Protective Gr~ups in Or~anic Svnthesis, John Wiley & Sons, New York (1981), ~ut many
others are well known in the art. The arylmethyl ethers, subsdtuted or unsubstituted, are
one particularly useful class of groups for protecting the hydroxyl group. The benzyl
protecting group, optionally with substituents upon the aryl ring, is useful.
30 Typically Z is hydrogen, an alkali metal, such as Li, Na or K, or an earth metal,
such as magnesium, or a transition metal, such as copper, alurninum, titanium, zinc or
cadmiurn, or a species derived therefrom. Representative of R'-Z are optionally substituted
alkyl, aryl or heteroaryl lithium, alkyl, aryl or heteroaryl magnesium halides ~eg. Gngn~rd
reagents), li~hium diallcyl cuprate, lithium diaryl cuprate, or the alkali metal salts of
35 optionally substituted alkyl alcohols, phenols or benzyl alsohols. Lithium diphenyl cuprate
is especially useful.
2~8~41~
WO 92/~07~0 PCr/VS9~/04757
- 13-
The hydroxyls are converted to suitable displaceable groups, such as mesylate,
tosylate, brosylate, benzoate, acetate and halide, by methods common in the art. The tosyl
group is especially suitable and is forrned by reac~ing the hydroxyl groups with tosyl
chloride, for instance.
S Suitable nitrogen nucleophiles are those which are able to react with a displaceble
group. Unhindered organic arnines or heterocyles, metal salts of amines, heterocycles or
azide are useful. Generally an nitrogen containing group of the forrnula R"'R'VN-7,
whenein Z is as defined above and R"' and R~v are hydrogen, an amino-protecting group or
taken together are N2 (eg. azide) are useful. A metal a~de, such as sodium or potassium
azide, is preferable. Subsequent reduction of the azido groups provides arnino groups.
Particularly useful intelmediate compounds of this invention are:
N3~N, HO~OH
OR" ORU RUO
III IV V
wherein R and R" are as defined above.
The compounds of this invention are prepared by the solid phase technique of
. Merrifield (J. Am. Chem~Soc., 85, 2149 (1964), or preferably by solution methods
known to the art. A combination of solid phase and solution synthesis may be used, as in a
csnvergent synthesis in which di-, tri-, or tetra-peptide fragrnents may be prepared by solid
phase synthesis and either coupled or further modified by solution synthesis. The methods
of peptide synthesis generally set forth in J. M. Stewar;t and J. D. Young, "Solid Phase
Peptide Svnthesis". Pierce Chernical Company, Rockford, n (1984) or M. Bodonsky,Y.A. Klauser and M. A. Ondetti, "Peptide $ynthesis", John Wiley & Sons, Inc., New
York, N.Y. (1976), or "The Peptides" gross and Meienhoffer, eds.; Acad. Press, 1979,
Vols I-III, may be used to produce the peptides of this invention and are incorpora~ed herein
by reference.
Each amino acid or peptide is suitably protected as known in the peptide art. For
example, the Boc- or carbobenzyloxy-group is preferred for protection of the amino group,
especially at the position. A benzyl group or suitable substituted benzyl group is used to
protect the mer~apto ~roup of cysteine, or other thiol containing amino acids; or the
hydroxyl of serine or threonine. The tosyl or nitro group rnay be used ~or protection of the
guanidine of Arg or the imidazole of His, and a suitably subsutu~ed carbobenzyloxy group
or benzyl group may be used for the hydroxyl group of Tyr, Ser or Thr, or she E-arnino
group of lysine. Suitable substitution of the c~rbooenzyloxy or benzyl protecting groups is
ortho and/or para substitution with chloro, bromo, nitro or methyl, and is used to modify
WO 92/00750 P~r/US91/04757
2o8~ 4 -14-
the reactivity of the protective group. Cysteine and o~her sulfur-containing amino acids
may also be protected by forma~ion of`a disulfide with a thioalkyl or thioaryl group. Except
for the Boc group, the protective groups are, most conveniently, those which are not
removed by mild acid treatment. These protective groups are removed by such methods as
S catalytic hydrogenation, sodium in liquid arnrnonia or HF treatment as known in the art.
If solid phase methods are used, the peptide is built up sequentially starting from the
carboxy serrninus and working toward the amino telminus of the peptide. Solid phase
synthesis is beglm by covalently attaching the C terminus of a protected amino acid to a
suitable resin, such as a benzhydrylamine resin (BHA), methylbenzhydrylarnine resin
(MBHA) or chloromethyl resin (CMR), as is generally set forth in U.S. Patent No.4,244,946. A BHA or MBHA support resin is used for the carboxy terminus of the
product peptide is to be a carboxamide. A CMR support is generally used for the carboxy
term~nus if the produced peptide is to be a carboxyl group, although this may also be used
to produce a carboxamide or ester.
Modification of the terrninal amino group of the peptide is accomplished by
alkylation or acetylation as is generally known in the art. These modifications may be
carned out upon the amino acid prior to incorporation into the peptide, or upon the peptide
after it has been synthesized and the terrninal amino group liberated, but before the
protecing groups have been removed.
Typically, acetylation is calr.ed out upon the free amino group using the acyl halide,
anhydride or activated ester, of the colTesponding alkyl acid. in the presencé of a tertiary
amine. Mon~allcylation is canied out most conveniendy by reductive alkyla~ion of the
amino group with an appropriate aliphatic aldehyde or ketone in the presence of a mild
reducing agent, such as lithium or sodium cyanoborohydride. Dialkylation as well as
quatemization may be callied by treating the amino group with an excess of an alkyl halide
in the pt~sence of a base.
Solution synthesis of peptides is accomplished using conventional methods used to
form amide bonds. Typically, a protected Boc-arnino acid which has a free carboxyl group
is coupled to a protected amino acid which has a free amino group using a suitable
earbodiimide coupling agent, such as N, N' dicyclohexyl carbodiimide (DCC), optionally
in the presence of catalysts such as l-hydroxybenzotriazole (HOBT) and dimethylamino
pyridine (DMAP). Other methods, such as the formation of activated esters, anhydrides or
acid halides, of the free carboxyl of a protected Boc-arnino acid, and subsequent reaction
with the free amine of a protected arnino acid, optionally in the presence of a base, are also
3~ suitable. For example, a protected Boc-amino acid or peptide is treated in an ~nhydrous
solvent, such as methylene chloride or te~hydrofuran (THF), in the presence of a base,
such as N-methyl morpholine, or a trialkyl amine, with isobutyl chloroformate to form the
WO 92/00750 2 0 8 ~ 4 1 ~ PCr/US91/04757
- 15-
mixed anhydride, which is subsequently reacted with the free amine of a second protected
amino acid or peptide. The peptide formed by these methods may be deprotected
selectively, using conventional techniques, at the amino or car~oxy terminus and coupled to
other peptides or arnino acids using similar techniques. After the peptide has been
S completed, the protecting gTOUpS may be removed as hereinbefore described, such as by
hydrogenation in the presence of a palladium or platinurn catalyst, treatment with sodium in
liquid ammonia, hydrofluoric acid, trifluoroacetic acid or alkaLi.
Esters are often used to protect the telminal carboxyl group of peptides in solution
synthesis. They may be converted to carboxylic acids by treatrnent with an aLkali metal
10 hydroxide or carbonate, such as potassium hydroxide or sodium carbonate, in an aqueous
alcoholic solution. l`he acids may be converted to other esters via an activated acyl
interrnediate as previously described.
The arnides and substituted amides of this invention are prepared from carboxylic
acids of the peptides in much the same manner. Thus, arnmonia or a substituted arnine may
15 be reacted with an activated acyl interrnediate of an amino-protected a-arnino acid or
oligopeptide to produce the arnide. Use of coupling reagents, such as DCC, is convenient
for forming substituted amides from the carboxylic acid itself and a suitable arnine.
ln addition, the methyl esters of this invention may be converted to the amides, or
substituted-amides, directly by treatment with arnmonia, or a subsdtuted amine, in methanol
20 solution. A methanol solution of the methyl ester of the peptide is saturated with amrnonia
and stirred in a pressurized reactor to yield the simple carboxamide ~ the peptides.
Procedures for the determination of the inhibition constant (Ki) by Dixon analysis are
described in the art, e.g., in Dreyer, et al. Proc. Natl. Acad. Sci. U.S.A., 86, 9752-9756
(1989). A peptidolytic assay is employed using the substrate Ac-Arg-Ala-Ser-Gln-Asn-
25 Tyr-Pro-Val-Val-NH2 and recombinant HIV protease as in Stricker, et al., Proteins, ,
134-154 (1989). The lower Ki value indicates a higher bindin~ a~finity.
Pharmaceutical compositions of the compounds of this invention, or derivatives
thereof, may be formulated as solutions or lyophilized powders for parenteral
administration. Powders may be reconstituted by addition of a suitable diluent or other
30 phannaceutically acceptable carrier prior to use. The liquid formulation is generally a
buffered, isotonic, aqueous solution. Examples of suitable diluents are normal isotonic
saline solution, standard 5% dextrose in water or buffered sodium or ammonium aceta~e
solution. Such formulation is especially suitable for parenteral adminis~alion, but may also
be used for oral adminis~ation or contained in a metered dose inhaler or nebulizer for
35 insufflation. It may be desirable to add excipient such as polyvinylpyrrolidone, gelatin,
hydroxy cellulose, acacia, polyethylene glycol, mannitol, sodium chloride or sodium
ci~ate.
WO 92/0075~l PCr/US91/04757
2~8~4~ - 16-
A preferred composition for paren~eral administration may additionally be comprised
of a quantity of the compound encapsulated in a liposomal carrier. The liposome may be
forrned by dispersion of the compounds in an aqueous phase with phospholi~ids, with or
without cholesterol, using a variety of techniques, including conventional handshaking,
S high pressure extrusion, reverse phase evaporadon and microfluidization. A suitable
method of making such compositions is more fully disclosed in copending Application
Serial No. 06f763,484 and is incorporated herein by reference. Such a caTrier may be
optionally directed toward its site of action by an imm~moglobulin or protein reactive with
the viral particle or infected cells. The choice of such proteins would of course be
10 dependent upon the antigenic deterrninants of the infecting virus. An example of such a
protein is the CD-4 T-cell glycoprotein, or a denvative thereof, such as sCD-4 (soluble CD-
4), which is reactive with the glycoprotein coat of the human imrnunodeficiency virus
(HIV). Such proteins are disclosed in copending Applica~ion Serial No. 07/160,463,
which is incorporated herein by reference. Similar targeting proteins could be devised, by
15 methods known to the art, for other viruses and are considered within the scope of this
mvention.
Alternatively, these compounds may be encapsulated, tableted or prepared in a
emulsion or syrup or oral administration. Pharmaceutically acceptable solid or liquid
carriers rnay be added to enhance or stabilize the composition, or to facilitate preparation of
20 the composition. Liquid carriers include syrup, peanut oil, olive oil, glycerin, saline and
water. Solid carriers include starch, lactose, calcium sulfate dihydrate, terra alba,
rnagnesium steaIate or s~earic acid, talc, pectin, acacia, agar or gelatin. The carrier may also
include a sustained release material such as glycerol monostearate or glycerol dis~earate,
alone or with a wax. Tl~e amount of solid carrier varies but, preferably, will be between
25 about 20 mg to about 1 g per dosage unit. The phannaceutical preparations are made
following the conventional techniques of pharrnacy involving rnilling, mixing, granulating,
and compressing, when necessary, for tablet forms; or milling, mixing and filling for hard
gelatin capsule forms. When a liquid carrier is used, the preparation will be in the form of a
syrup, elixir, emulsion or an aqueous or non-aqueous suspension. Such a liquid
30 foqmulation may be administered directly p.o. or filled into a soft gelatin capsule.
For rectal adminiscration, a pulverized powder of the compounds of this invention
may be combined with excipient such as cocoa butter, glycerin, gelatin or polyethylene
glycols and molded into a suppository. The pulverized powders may also be compounded
with an oily preparation, gel, crearn or emulsion, buffered or unbuffered, and administered
3~ through a transdermal patch.
I~is invention is also a method for treating viral infection, particularly infection by
retr~viruses, which comprises adrninistering a compound of fomlula I to a patient infected
W~ 92/00750 2 0 ~ 6 ~ 1 4 PCI/l~S9~/~4757
with a susceptible virus. The method is particularly applicable tO infection by the Human
Immunodeficiency Virus, type 1. When the compounds of this invention are used to induce
anti-viral activity in patients which are infected with susceptible viruses and require such
treatment, the method of treatrnent comprises the administration orally, parenterally,
S bucally, trans-dermally, intravenously, intrarnuscularly, rectally or by insufflation, of an
effective quantity of the chosen compound, preferably dispersed in a pharrnaceu~ical camer
I~osage uni~s of the active ingredient are selected from the range of 0.05 to 50 mg/kg of
body weighL Dosage units will typically be from 50 to 1000 mg. These dosage units may
be administered one to ten drnes daily for acute or c'n~onic infection. The dosage will be
readily deterimined by one skilled in the art and will depend upon the age, weight and
condition of the patient, and the route of adrninistration. Combination therapy as described
in Eur. Pat. Appl. No. 337 714 a~ pages 42-47 are included herein.
The Exarnples which follow selve to illustrate this invention. The Examples are
intended to in no way lirnit the scope of this invention, but are provided tO show how to
make and use the compounds of this invention.
In the Exarnples, all temperatures are in degrees Celsius. Amino acid analyses were
perforrned upon a Dionex Autoion 100. Analysis for peptide content is based upon Amino
Acid Analysis. FAB mass spectra were performed upon a VG Aab mass spectrometer
using fast atom bombardment. NMR spectra were recorded a~ 250 MHz using a BrukerArn 250 spectrometer. Multiplicities indicated are: s=singlet, d-doublet, t-tIiplet, q-quartet,
m-multiplet and br indicates a broad signal.
rification of Recombinant EIIV Protease
Melhc~s for expressing recombinant HIV protease in lE.coli have bee described byDebouck, et al., Proc. Natl. Acad. Sci. USA, 84, 8903-6 (1987). The enzyme used to
assay the compounds of this invention was produced in this manner and purified from the
cell pellet as previously described by Stickler et al. Proteins, _, 139-154 (1989).
EXAMPLES
Exarnple I
Preparation of (3S. 5S)-3~5-diamino-4-hvdroxvheptane 1
Preparation by the Procedure of Scheme 1:
a) 1.2:4~:~nhvdr~D^(+)-arabitol 9
Benzyl trichloroacetimidate (29.1 mL, 201 mrnol) was added to a solution of D-(+)-
arabitol (13.9 g, 91.4 mrnol; azeotropically dried with toluene) in dry acetonitrile (200 rnL)
WO 92/00750 PCr/lJS91/04757
- 18-
208~4~
under Ar, and the mixture was stirred overnight. The solution was concentrated by rotary
evaportion, dissolved in ethyl acetate (500 mL), washed with 5% ~aHC03 (2x30 mL) and
brine (30 rnL), dired over Na2SO4 and concentrated. The residue was dissolved in dry
THF (500 mL) and colled to -10C, and solid sodiurn methylate (11.1 g, 205 mmol) was
S addc-d with mechanical stirnng under Ar. After 20 min the mixture was poured into ether
(2.5 1~, filtered through glass fiber filter paper to remove sodium methylate, and
concentrated by rotary evaporation at 30C. The residue was purified by flash
chromatography (silica gel, 3:2 ether:pentane) tO provide the titled compound 9 (3.96 g,
34.1 mmol, 37% yield).
b~ 1~2:4.5-dianhvdro-3-~O-benzvl)-D-t+~-arabito~
Benzyl bromide (8.92 mL, 75 mmol) was added to a slurry of NaH (1.8 g. 75
mmol) in ~IF (9 mL). The mixture was cooled to -10C under Ar, and compouncl 9
(3.90 g, 34 rnmol) in l~IF (9 mL) was added dropwise with stirring. The ice bath was
removed, and the mixture was allowed to warm briefly to 40C, then was recooled. The
rnixture was diluted with 1% acetic acid (100 mL), and extracted with ethyl ace~te (2x250
mL). The organic extracts were washed with 5% NaHCO3 (7S mL) and bAne (75 mL),
dried over Na2SO4, and concentrated. Flash chromatography of the residue (gradient, 0-
4% ethyl acetate in pentane) provided the titled compound 10 (3.88 g, 17.2 IT~nol, 55%
yield). lHNMR (C.DC13): o 7.4-7.1(5H, m) 4.75(1H, d; J = 12 Hz), 4.65(1H, d, J = 12
Hz}, 3.2(1H, m), 3.05(1H, m), 2.9~1H, t; J = 7 Hz), 2.8(2H, m), 2.65(2H, m).
c) (2S .4S)- I ,5-Diazido-2.4-dihvdroxv-3-benzvlox~pentane 11
To 1.65 g (8.0 mmol) of compound 10 in 50 mL water and 50 rnL dioxane was
added 6.5 g (100 mmol) NaN3 and 340 mg (1 rnrnol) tetra-n-butylammonium bisulfate.
The reaction tnixture was heated under reflex for 4 hr, cooled, and concentrated to ca. 50
rnL volume by rotary evaporation, and extrac~ed with ethyl acetate (3xS0 rnL). The
combined organic extracts were dried (Na2SO4) and concentrated to an oil, which was
combined with ether:hexane (1:1) and allowed to crystallize at 4C to provie 1.76 g of titled
compound 11~69% yield). IHNMR (CDC13): o 7.5-7.3(5H, m), 4.6(2H, dd; J = 12 Hz),
4.0(2H, m), 3.6-3.0(6H, m).
d~ (2S.4S)-1~5-diazido-2.4-di-(methanesulfonvloxv)-3-benzvloxvpentane.l2
To 630 mg (2.15 rnmol) of bisazide diol 11 in 6.0 mL of pyridine was ndded 350
35 llL (4.5 mmol) methanesulfonyl chloride at 0C. The reaction mixture wns allowed to wnm
to 25C and stirred for 20 hr, then was diluted with 12 rnL 6 N HCI, and extrncted with lO0
mL methylene chloride. The organic layer was washed with 3% NaHCO3, dried (Nn~SO~)
,. . ~
WO 92/00750 2 0 8 6 ~14 PClfUS91/047~7
,
and concentrated. Flash chrotnatography of the residue (9S:5 CH2C12:ether) provided 867
mg (90% yield) of the titled compound 12. IHNMR (CDC13): ~ 7.4-7.2(5H, m), 5.0-
4.7(4H, m), 4.02(2H dd, J = 3 Hz), 3.9-3.5(4H, m), 3.15(3H, s), 3.10(3H, s).
S e) (2S~4S):~:4~5-di-(N-carbQnbenzvloxvimino)-3-benz~!oxvpentane 13
To 310 mg (0.69 mmol) of the compound 12 of step (d) in 2 rnL dry TH~ at 0C
was added 1.5 rnL (1.5 mmol) 1 M LiAlH4 in THF. The rnixture was allowed to walm to
25C and stirred overnight. Water (0.1 mL) was added, followed by 0.1 mL 15% NaOh
and 0.4 mL water. The n~ixture was sti~red vigorously with lû mL ether and filtered.
Concentra~on of the ether layer provided teh crude bisaz:iridine, which was dissolved in 5
mL CH2C12 and combined with 200 ~LL (1.4 mmol) tnethylamine and 200 ~LL (1.4 mmol)
of benzyl chloroformate. The mixture was stirred at 25C for 3 hr, then f~ltered. The
filtrate was concentrated and the residue was purified by flash chromatography (ethyl
acetate:hexanes 1:5) to provide 89 mg (28% yield) of the titled compound 13. IHNMR
(400 MHz; CDC13): ~ 7.4-7.2(15H, m), 5.0(1H, d; J = 12 Hz), 4.93(1H, d; J = 12 Hz).
4.88(1H, d; J = 12 Hz), 4.84(1H, d; J = 12 Hz), 4.74tlH, d; J = 12 Hz), 4.39(1H, d; J =
12 Hz), 2.82(1H, t; J = 6 Hz), 2.6(1H, m), 2.45(1H, m), 2.25(1H, d; J = 6 Hz),
2.15(1H, d; J = 6 Hz), 2.04t1H, d; J = 3 Hz), 2.02(1H, d; J = 3 Hz). MS (DCI, NH3):
Il-Jz 473.1(M+)-
(3A.~S)-~S-di-(carbonbenzylQx~lamino)-4-benzvloxvheptane 1~
To a suspension of CuI (72 mg, 0.375 mmol) in 1.5 mL ether at -25C was added
0.5 rnL 1.5 M CH3Li in ether. The resulting colorless solution was cooled to -45C and a
solution of bisazindine 13 (10 mg, 0.02 mmol) in 0.5 mL ether was added. After stirling ~t
-45C for 1 hr, the mixture was allowed tO walm to 10C over a period of 6 hr, then was
stirred for an additional 2 hr. The mix~ure was diulted with 2 mL saturated aqueous NH4CI
and 1 rnL saturated aqueous NH3, then was extracted with ether. The organic layer was
washed with bnne, dried (Na2SO4) and concentrated to provide the titled compound 14
(10 mg). IHNMR (CDC13): ~ 7.2(15H, m), 5.2-4.9(4H, m), 4.93(1H, d; J = 12 Hz),
4.88(1H, d; J = 12 Hz), 4.84(1H, d; J = 12 Hz~, 4.7,~2H, d; J = 12 Hz), 4.4(2H, d; J = 12
Hz), 3.7(2H, m), 3.4(1H, d; J = 2 Hz), 1.6(4H, m), 1.0(3H, t; J = 7 Hz), 0.g(3H, t; J = 7
Hz).
~ t3S.5$2-3~5-diamino-4-hvdroxvheptane
The product of step (f) is st*ed with 20% Pd(OH)2 on carbon (50% by weight) in
0.1 N methanolic HCI under an ~tmosphere of hydrogen for 24 hr. Filtra~ion ancl remov~l
WO 92/0075~ PCr/US91/047~7
2~8~414 -20-
of solvents provides the ti~led compound 1, as the dihydrochloride salt wherein X1 and X~
are hydrogen and Rl and R2 are ethyl.
Preparation by the Procedure of Scheme 2:
a~ (3R~5R~-3.5-dihydroxv-4-benzyloxvheptane 1~
To a suspension of CuI (143 mg, 0.75 mrnol) in 3 mL e ther at -35C was added
methyllithium (1 rnL, 1.5 M in ether; 1.5 rnmol). The resulting colorless solution was
stirred at -30C for 30 min, then cooled to -78C. A solu~ion of bisepoxide 10 (78 mg,
0.37 mmol) in 2 mL ether was added. The reaction was allowed to warm to 25C over 4
hr, the saturated aqueous NH4Cl and concentrated aquaous NH3 were added. The mixture
was extracted with ether, the organic layer was dried over Na2SO~ and the solvent was
removed to furnish the titled compound (93 mg, 100% yield). IHNMR ~CDC13): ~ 7.4-
7.1(5H, m), 4.7(1H, d; J = 12 Hz), 4.55(1H, d; J = 12 Hz), 3.9-3.7(2H, m), 3.~(1H,
dd), 2.5(2H, b), 1.6-1.4(4H, m), 1.0(3H, t; J = 7 Hz), 0.9(3H, I; J = 7 Hz).
b). (3R,SR)-3,5 methanesulfonvloxv-4-benzvloxvheptane 16
Methanesulfonyl chloride (0.3 mL) was added dropwise to diol 15 (93mg) in
pyridine (lmL) at 0C. The mixture was allowed to warm to 25C. After 12 hr the mixture
was dilutçd with cold 6N HCI ( 10 mL) and extracted with CH2C12. The organic extract
was washed with 3% NaHCO3, dried over MgSO4, and concentrated. The residue was
pulified by flash chromatography to provide the titled compound (83 mg, 56% yield).
lHNMR (CDC13): ~ 7.4-7.2(5H, m), 4.75 (lH, m), 4.7(1H, d; J = 12 Hz), 4.6(1H, d; J =
12 Hz), 4.58(1H, m), 3.9(1H, dd; J = 2.6 Hz), 3.0(3H, s), 2.9(3H, s), 2.1-1.5(4H, m),
1.05(3H, t; J = 7 Hz), 1.0(3H, t; J = 7 Hz).
c). (3S. SSl-3.5-diazido-4-benzvloxvheptane 17
A mixture of bismesylate 16 (83 mg. 0.21 mmol) and sodium azide (0.5 g 7.7
mlllol) in 1.5 mL dimethylfotmatnide was heated to 70C for 12 hr. After cooling, ethyl
acetate (20 mL) was added and the rnixture was filtered and concentrated. Flash
chromatogr~phy of the residue provided the titled compound (54 mg, 9n% yield). IHNMR
(CDC13): ~ 7.4-7.2(5H, m), 4.65 (2H, s), 3.5-3.2(3H, m), 2.0-1.5(4H, m), 1.1(3H, t; J
- 6 Hz), 1.05(3H, t; J = 6 Hz).
d) 13$ 5Sl-3~5-diamino-4-benzvloxyheptane l~
To 328 mg (1.14 mmol) bisa~ide 17 in 5 rnL THF at 0C was added 200 mg
LiAlH4. The nuxture was allowed to warrn to 25C and was stirred for S hr. The reaction
was quenched by addition of 0.5 mL 15% NaOH, stirred for 15 min~ dilu~ed wi~h 150 rnL
.
2~8641~
WO 92/007~0 PCr/US91/04757
- 21 -
ether and filtered. Concentration of the filtrate provided the titled compound (278 mg, 100%
yield). IHNMR (CDC13): â 7.26(5H, m), 4.5 (2H, dd; J = 12Hz), 3.1 (lH, dd; J = 4.6
Hz), 2.9(1H9 m), 2.75(1H, m), 1.8-l.O(lOH, m), 0.9(3H, t; J = 7 Hz), 0.85(3H, t; J =
Hz).
s
e) (3S. 5~-~m!~roxvheptane 1
To the diamine product 18 (52 mg) in 4 mL methanol was added 50 mg 20%
Pd(OH)2 on carbon and 2 drops concen~ated aqueous HCl. The mixture was stirred under
an atmosphere of H2 for 16 hr, then was filtered and concentrated ~o provide the titled
compound 1 (53 mg) as the dihydrochlonde salt. IHNMR (CD30D): ~ 3.75(1H, dd; J =4.7 Hz), 3.2-3.0(2H, m), 1.8-1.4(4H, m), 0.9(6H, t; J = 7 Hz).
Example 2
Preparation of (3S. ~S)-3.~di-~alanvlalanyl)amino-4-hYdroxYheptane 2.
a) (3S.SS)-3.5-di-(carbobenzvloxYalanvlalanvl)am~no-4-benzvloxvheptane 19
To the diamine product 18 of Example 1, step (k) (87 mg, 0.37 mmol) in 6 mL
DMF was added 221 mg (0.75 mmol) carbobcn~yloxyalanylalanine, 115 mg (0.75 rnmol)
HOBT, and 154 mg (0.75 mrnol) DCC. The mixture was st~rred overnight, then was
concentrated, taken up in ethyl acetate, filtered, washed with water and brine and dired
(MgS04). Removal of solvent followed by MPLC (silica; 2% methanol in CH2C12)
provided the titled compound (109 mg). IHNMR (OMSO-d6): o 8.0-7.2(21H, m),
5.0(4H, bs), 4.6(2H, dd; J = 12 Hz), 4.2(2H, m), 4.0(2H, m), 3.8(1H, m), 3.7(1H, m),
3.5(1H, dd; J = 4.7 Hz), 1.7-1.3(4H, m), 1.1(12H, m), 0~75(6H, t).
b) ~3$.5S)-3.5-di-~alanylalanvl)amino-4-hYdrox~heptane 2
To the product 19 of step (a) (4.5 mg) in 1 mL DMF was added 10 mg to 20~o
Pd(0~2 on carbon. The mixture was stirred under 1 atmosphere of ~2 for 6 hr, then was
filtered and concentrated to provide the titled compound 2 (3 mg) wherein X1 and X2 are
AlaAla and R1 and R~ are ethyl. IHNMR (CD30D): o4.3(2H, m)t 3.8(2H, m), 3.7(1H,
m~, 3.5(1H, m), 1.8-1.2(16H, m), 0.8(6H, dt).
Example 3
Preparation of (3S~5S)-3.5-di-!carbobenzvl_~v Ivl~amino4-hvdroxyheptane 3.
To 133 mg (O.S mrnol) Cbz-Yal in 2 mL TE~ at -40~C was added 65 ~L (0.5
mmol)of isobutylchloroformate. ~fter stirring for 10 rn~n a solution of 25 mg (0.17 mmol)
diamine hydrochloride 1 and 50 IlL NMM in 1 mL DMF was added. The mixture was
w~ 92'0~7~0 ~'~,6 4~ 4 22 - Pcr/US9l/04757
slowly warmed to 20C and stirred overnight, then was diluted with ethyl acetate, washed
with 5% HCI, 5~ NaHCO3, and brine and the organic layer was concentrated. The
residue was purified by flash chromatography (ethyl acetate:hexanes~ to provide the titled
compound 3 (18 mg) wherein Xl and x2 are Cb~-Val and Rl and R2 are ethyl. IHNMR
(CDC13): ~ 7.5-7.3(10H, m), 6.9(1H,br d), 6.35(1H, br d3, .5.5(H, br d), 5.25(1H, br d),
5.1(4H, br s), 4.0-3.1(6H, m), 2.4-2.1(2H, m), 2.0-1.4(4H, m), 1.0-0.8(18H, m). MS
(~;AB): rnlz 613.2(M+H~+.
Example 4
Prepara~ion of (2S~ 4~)-2.4-di-~alanylalanvl)amino-3-hvdrox~- 1 .5-diphenvlpenta
a). (2Rt4R~-2.4-dihydroxv-3-benzvloxv-1~5-diphenvlpentane 20
To a suspension of CuI (191 rng, 1 mmol) in ether (5 mL) at -60C was added
phenyllithium (4.0 mL, 2.0 mmol; 0.5 M in ether, freshly prepared from bromobenzene and
15 lithium wire). The mixture was walmed to -5ûC, then recooled to -78C. A solu~ion of
bisepoxide 10 (40 mg, 0.19 mmol) in ether (1 rnL) was added. The mixture was allowed to
warm to 25C over 5 hr with sti~Ting. After an additional 12 hr, the m~xture was diluted
with ether and washed with 20 rnL of 1:1 concentrated aqueous arnmonia:saturatedNH4CL. The organic layer was dried over MgSO4 and concentrated. The residue was
20 purified by medium-pressure liquid chromatography (1:4 ethyl acetate:hexanes) to provide
~e tided compound 2Q (22 mg, 30% yield) as a colorless solid. IHNMR (CDCL3): S 7.5-
7.0)15H,m), 4.75(1H, d; J=12 Hz), 4.55(1H, d; J=12 EIx), 4.2(2H, m), 3.3(1H, m), 3.0-
2.7(6H, m).
b) f2S.4S)-2~4-di-(alanvlalanvl~amino-3-hvdroxv-1.5-diphenylpentane 4
The ~itled compound 4 wherein Xl and x2 are AlaAla and Rl and R2 are PhCH2 is
prepared from compound 20 by the procedures of Exarnple 2.
Example S
Preparation of (4R~6R~-4.6-diamino-5-hvdroxv-2.8-dimethvl- 1 .8-nonane' 5,
a). (4R.6R~ -dihvdroxv-5-benzvloxv-2.8-dimethvl-1.8-nondiene 21
To a suspension fo CuI ( 192 mg, 1.0 mmol) in ether (2 rnL) a~ -60C was added
isopropenyllithium (5.2 mL, 2.0 rs~nol; 0.38 M in ether, freshly prepared from 2-
35 bromopropene and lithium wire). The mixture was warmed to -45C, then recooled to -
78C. A solution of bisepoxide 10 (60 mg, 0129 mmol) in ether (5 mL) was added. The
rnixture was allowed to warrn to 0C over 2 hr with stirring. The n~ixture was diluted with
208~414
WO 92/007S0 - 23 - PCI/US91/047~7
ether and washed with S mL of l: l concentrated aqueous ammonia:saturated NH4CI. The
organic layer was dned over MgSO4 and concentra~ed to provide the titled compound 71
(81 mg> 96% yield) as a colorless solid. IHNMR (CDCL3): ~ 7.3(5H, m), 4.9(1H, bs),
4.85(1H, bs), 4.80(1H, bs), 4.65(1H, d; J=12 Hz), 4.58(1H, d; J=12 Hz), 4.0(7H, m),
3.3(1H, dd; J=2.5 Hz), 2.55(1H, d; J=6 Hz), 2.55(1H, d; J=4 Hz), 2.5-2.1(4H, m),1.7(1H, s).
b) L4R.~R)-4.6-dihydroxv-5-b~nxyloxv-2.8-dimethYlnonanej~
To the product 21 of step (a) (105 mg, 0.38 mmol~ in CH2C12 (1 mL) was added
55 mg Ir(COD)Py(PCy)3PF6 (Crabtree catalyst). The mixture was sti~red for 6 hr under I
atmosphere H2, then was filtered and concentrated to provide the ~itled compound 22 (110
mg, 100% yield). IHNMR (CDC13): o 7.3(5H, m), 4.6(1H, dd; J=12 Hz), 4.0(2H, m),
3.1(1H, br s), 2.7(2H, br s), 2.0-1.1(6H, m), 1.0-0.8(12H, m).
c~ ~4R.6R)-4.6-di-(methanesulfonvloxv)-5-benxvloxv-2.8-dimethylnonane 23
Methanesul~onyl chloride (0.25 tnL) was added dropwise to diol 2Z (93 mg) in
pyridine (1 mL) at 0C. The mixture was allowed to warm to 25C. After 10 hr the mixture
was diluted with cold 6N HCl (10 mL) and extracted with CH2C12. The organic extract
was washed with 3% NaHCO3, dried over MgSO4, and concentrated. The residue was
purified by flash chromatography to provide the titled compound 23 (210 mg). IHNMR
(CDC13): ~ 7.4(5H, m), 5.0(1 EI, d; J=12 Hz). 4.7(1H, m), 4.6(1H, d; J=12 Hz),
3.85(1Hx dd; J-4.7 Hx), 3.0(3H, s), 2.0(3H, s), 2.0-1.1(6H, m), 1.0-0.9(12H, m).
d) (4R.6R)-4 6-dia~ido-5-benzvlox~2,8-dimethvlnonane 24
To the product 23 of step (c) (210 mg) in 2 mL DMF was added 870 mg (15 tnrnol)
NaN3. The mixture was heated lo 70C for 7 hr, then was cooled and diluted with ethyl
acetate. The filtrate was concentrated and the residue was purified by MPLC (ethyl
acetate:hexanes 1:20) to provide the titled compound 24 (52 mg). lHNMR (CDC13): o
7.3(5H, m), 4.55(2H, dd; J-12 Hz), 3.4-3.15(3H, m), 2.0-1.1(6H, m), 1.0-0.75(12H,
30 m)-
e). (4$.6S)-4~6-diamino 5-benzvloxv-2.8-dimeth~nonane _2~
To the product 24 of step (d) (52 mg, 0.15 mrnol) in THF (3 mL) was added 80 mg
LiAlH4 (2 mmol) at 0 C. The mixture was sti~red at 25 C overnight, then was quenched
with lN NaOH and diluted with ether (50 mL). Filtration and concentration provided the
titled compound 25 ~44 mg) as a colorless oil. IHNMR (CDC13): ~ 7.3(5H~ m), 4.6(2H,
dd; J = 12 Hz), 3.1(1H, m), 3.05-2.95(2H, m), 1.9-1.1(6H, m), 1.0-0.8(12H, m).
WO 92/00750 PC~/US91/04757
24 -
f) ~4R~6R~4~6-diami_o-5-hYdroxv-2.8-dimethyl-1~8-nonane diahydrochloride
To 165 mg of diamine 25 from step (e) in 10 mL methanol containing 5 drops of
conc. HCI was added 100 mg 20% Pd(OH)2 on carbon. The mixtllre was s~rred overnight
under 1 atrn H2, then was filtered and concen~rated to provide the titled compounds 5 (75
5 mg) wherein X1 and X2 are hydrogen and Rl and R2 are isobutyl. IHNMR (CD30D):
3.7(1H, m), 3.2(1H, m), 1.8-1.0(6H, m), 0.99-0.8(12H, m).
Example 6
Preparation of (4R,6R)4,6-di-(alanylalanyl)amin~hydroxv-2 8-dimethyl-1 .8-nonane
a) (4R,6R~-4~6-di-(carbobenzvloxvalanvlal~anvl)amino-5-benzvloxy-2.8-dimethvl-1~8-
nonane ~
To the product 25 of Example 5, srep (e) (44 mg, 0.15 mmol) in 2 rnL DMF was
added 110 mg Cbz-AlaAla (0.375 mmol), 58 mg (0.375 mmol HOBT, and 72 mg (û.375
mmol) DCC. The mixture was stirred for 48 hr at 25 C, then was diluted with 20 rnL ethyl
acetate and filtered. The filtrate was concentrated and the residue was purified by MPLC
(gradient, 0-5% methanol in CH2C12) to provide the titled compound 26 (24 mg).
IHNMR (CD30D): ~ 8.0-7.2 (21H, m), 5.0 (4H, overlapping dd), 4.2 (2H, m), 4.0 (4H,
m), 3.5 (lH, br s), 1.7-1.15( 18H, m), 0.9-0.7(12H, br t).
(b) (4R. 6R~-4~6-Di-falanvlalanvl)amino-5-hvdroxY-2.8-dimethvl -1.8-nonane 6
To the product 26 of step (a) (12 mg) in 2 mL DMF was added 50 mg of 2Q%
Pd(OH)2 on carbon. The mixturç was stirred under 1 a~mosphere of H2 for 10 hr, then
was diluted with methanol, filtered and concentrated to provide the titled compound 6 (7.5
mg) wherein Xl and x2 are AlaAla and Rl and R2 are isobutyl. IHNMR (CD30D): ~
4.25 (2H, m), 3.8 (lH, m), 3.65 (2H, m), 3.1 (lH, br d), 1.6-1.1 18H, m), 0.7 (12H, br
d~.
Ex~mple 7
Preparation of (4R.6R~-4~di-carbobenzvlo'zy)amino-5-hvdroxy~2.8-dimethvl-1,8-nonane
To 6.0 mg of the bis-arnine hydrochloride product 5 in 0.5 rnL CH2C12 at 20C
werc added S mL ~iethylamine and 10 mL benzyl chloroforma~e. After 3 hr sti~ring, the
mixnure was applied tO a silica column and eluted with CH2CI~ followed by ether to
provide the titled compound 7 (4.6 mg) wherein Xl and X2 are Cbz and Rl and R2 are
isobutyl. IHNMR (CDC13): ~ 7.3 (IOH, bs), 5.1-4.9~6H, m), 3.9-3.7('~H, m), 3.5-
2086~14
WO 92/007~0 PCr/US91/04757
3.35(2H, m; lH exchangeable with D2O), 1.7-1.5(4H, m), 1.3-1.2(2H, m), 1.1-0.8(12H,
m).
eparation of (4R.6R)-4,6-di-(carbobenzvlo~walvl)am-ino-5-hydroxv-2,8-dimethvl-1~8-
~n~n~
The titled compound 8 wherein Xl and X2 are Cbz-Val and Rl and R2 are isobutyl
was prepared by the procedure of ExamDle 3, except using compound 5 in place of
compound 1. INMR (250 MHz,CDC13) 7.5-7.28 (m, 10H), 6.8 (br s, lH), 6.3 (br d,
lH), 5.6 (br s, lH), 5.25 (br d, lH), 5.1 (br s, 4H), 4.5-3.5 (m, 6H), 1.5-2.5 (m, 6H),
0.6-l.û (m, 24 H).
Example 9
Pre~2a,ation of (2S.4S~ 5-di~henvl.3-hvdroxv-2.4-bis(benzvloxvcarbonvlaminovalinvl-
15 ~m~nS~
a) 2R,4R)-1,2,4,5-dianhydro-3-benzyloxyarabitol 33
To a solution of 15.2 g (100 mmol) of D(+)-arabitol in 350 tnL of pyridine cooled in an ice
bath was added 38.8 g (203.5 mmol) of p-toluenesulfonyl chloride in small portions.
20 StilTed for 3 h, wanned to room temperature, poured to 500 rnL of ether. Ether layer
separated, aqueous layer extracted with 800 mL of ether. The combined organic layers were
washed with 40û mL of 3~b sodium bicarbonate, dried over anhydrous magnesium sulfate
and solvents removed in vacuo to give 34.17g (74%) of the ditosylate 32. IHNMR
(CD3COCD3) o 7.75 (d, 4 H, J= 7 Hz), 7.25 (d, 4 H, J= 7 Hz), 4.4-3.5 (m, 7 H), 3.2
(b, 3 H), 2.5 (s, 6H).
To 10 g (55-60% in oil, 230 mmol ) of sodium hydride in 300 mL of THF was
added at 0 a solu~ion of ~he bistosylate 32 in 200 rnL of THF and stirred vigorously for I
h. The reaction mixture was treated with dropwise addition of 12 rnL of benzyl bromide in
10 ~ of THF and stirled at 0 for 1 h. Allowed to warm to room temperature and stirred
overnight. Quenched with 50 rnL of water. dropwise at 0. Extracted with ether, washed
with water, dried over anhydrous sodium sulfate and solvents removed in vacuo. The
residual oîl was fileered thro silica gel (fIrst eluted with hexane to remove unreacted benzyl
b~omide and then ethyl acetate Hexane ,1:4) to yield 9.0 g of the bisepoxide as a slight oil.
Further p~ifica~ion was acheived by flash chromato~aphy (silica, ethyl acela~e,hexane
1:10) to give 6.10 g of the diepoxide 33. IHNMR (CDCl3, 250 MHz) o 7.4-7.1 (m,
SH), 4.75 (d, lH, J= 12 Hz), 4.65 (d, 1 H, J= 12 Hz), 3.2 (m, IH), 3.05 (m. 1 H), ~.9
(t,lH,J=7Hz),2.8(m,2H),2.65(m,2H).
WO 92/007~0 PCI/US91/~4757
- 26-
~,~8~4~4
b) t2R,4R)- 1 ,S-diphenyl,3-benzyloxy-2,4-dihydroxypentane 34
To a suspension of 2.99 g (15.5 mmol) of of copper(1) iodide in 30 mL of THF
w~s added 18 mL of 1.8 M Phenyl lithium in cyclohexane ( (freshly opened bottle). The
rea~tion m~xture was wanned to -50 and recooled tO -78 and a solution of l.Olg (4.9
tnmol) of the bisepoxide 33 in 10 mL of TH~ was added and allowed to warrn to room
temperature and s~T~ed overnight. Processed as usual to give 1.75 g of of the diol 34 as an
oil. Trituration with ether/hexane gave 1.45 g ~82 %) of a colorless solid. Anal Calcd for
C24H2603: C(79.53), H(7.23); Found: C(79.25), H(7.18); MS(DCI, NH3) (M+H)+
364.5; IHNMR ~CDC13, 250 MHz) ~ 7.4-6.8 ~m, 15 H), 4.62 (d, 1 H, J= 12 Hz), 4.5
(d, 1 H, J= 12 Hz), 4.2 (m, 2 H), 3.3 (dd, 1 H, J= ), 2.8 (m, 4 H).
c) (2R,4R)-1,5-diphenyl-3-benzyloxy-2,4-bis(methanesulfonyloxy)pentane 3~ and
(2S,4S)-1,5-diphenyl-3-benzyloxy-2,4-diazidopentane 36
To 400 mg of the diol in 5 mL of pyridine at 0 was added 1 mL of rnethane
sulfonyl chloride and stirred for 18 h at room temperature. Poured into 20 mL of ice cold3
N hydrochloric acid extracted with 50 mL of methylene chloride, washed with 3 % sodium
bicarbonate dried over anhydrous sodium sulfate and solvents removed in vacuo to give
1.20 g of 3~ as an oil.
The a~ove crude product was dissolved in ~ mL of DMSO and added 1.05 g ( 16
mmol) of sodium azide. The reaction mix~ure was heated at 80 for 6 h and then at 100 for
an additional 8 h. The reaction was cooled, diluted with ether and unreacted sodium azide
was filtered off. I'he combined solvents were removed in vacu and subjected to flash
chromatog~aphy to give 285 mg of an inseparable mixture of monoazide 36, (2S,4S)- 1,5-
diphenyl-3-benzyloxy^4-azido-pent- l-ene, and bisazide 37 in the ratio 70: 30 as caclulated
from lHNMR.
d) (2S,4S)-l,S-diphenyl,3-benzyloxy-2,4-diaminopentane 38
To 279 mg of the mixture of azide products (36+37) obtained above in 10 mL of
diethyl ether at 0 was added 3 mL of a lM soluion of lithium aluminium hydride in THF
over 10 n~n. Sn~red at 0 for 30 min w~rmed to room temperature and sti~red for 3 h.
Cooled in an ice bath and quenched with 1 mL of 10% sodium hydroxide, diluted with
ether and stirred for 2 h. The precipitate was filtered off through celite and washed with
ether. Removal of solvents followed by chromatography on 10 g florisil (hexane, eshy
acetate:hexane 1:4, then methano) gave 112 mg of pure diamine 38. IHNMR (CDCl3,
250 MHz) ~ 7.2 (m, 15 H), 4.7(d, 21 H, J= 12 Hz), 4.5 (d, 21H, J= I H), 4.1(m, lH),
2.3-3.2 (m, 6 H3.
2086~14
wo 92/00750 27 PCI/US91/04757
,
e) (2S,4S)-1,5-diphenyl-3-hydroxy-2,4-diaminopentane 39
62 mg of the diamino compound was subjected to hydrogenation in 10 rnL of
methanol containing 75 mg of conc. hydrochloric acid over 25 mg of Pd/C. Stirred for 8 h,
S catalyst was fil~ered off and washed with methanol. Removal of solvents gave 68 mg of a
solid which on trituration with hexane ether provided 48 mg of the pure diamine
as the hydrochloride. IHNMR ~CD3OD, 250 MHz) ~ 7.4-6.9 (m, 10 H), 4.1 (bd, lH,
J= 6 Hz), 3.5-3.7 (m, 2 H), 2.5-3.4 (m, 4 H~.
f) (2S,4S)-1,5-diphenyl,3-hydroxy-2,4-bis~benzyloxycarbonylaminovalinyl-
am~no)pentane 40
The tided product was prepared by the mixed anhydride method from 37 mg (0.107
mmol) of the diamine hydrochloride, 150 mg of Cbz-Val, 98 ~L of N-methyl morpholine
and 80 ~IL of isobutyl chlorofomlate 68 mg of a white solid. Analytical samples were
prepared by flash colurnn chromatography t silica, 10% MeOHlCH2C12). MS(ES/MS)
(M-H)+ 735; IHNMR (CDC13, 250 MHz) ~ 7.4-7.0 ( m, 20 H), 6.1 (d, lH, J= 7 Hz),
5.5 (d, lH, J= 7 Hz), 5.0 (m, 8 H), 4.0 (m, 2 H), 3.6 (m, 2 H), 2.8-3.4 (m, 4 H), 2.2
(m, lH), 1.85 (m, lH), 0.9 (d, 3 H, J= 7 Hz), 0.86 (d, 3 H, J= 7 Hz), 0.7 (d, 3 H, J= 7
Hz), 0.55 (d, 3 H, J= 7 Hz).
Exam~le 10
Preparadon of (3$. 5S)-3.~-di-(carbobenzYloxYalanvlalanyl)amino-4-hvdroxvheptane ~IL
a) (2R,4R)-1,5-diazido-2,4-dihydroxy-3-benzyloxypentane 41
To 1.65 g (8.0 mrnol) of bisepoxide 33 in 50 mL water and 50 mL dioxane was
added 6.5 g (100 mmol) NaN3 and 340 mg (1 rnmol) tetra-n-butylarnmonium bisul~ate.
The reaction mixture was heated unde~ reflux for 4 hr, cooled, and concentrated to ca. 50
rnL volume by rotary evaporation, and extracted with ethyl acetate (3x50 mL). The
combined organic extracts were dried (Na2S04) and concentrated to an oil, which was
combined with with ether:hexane (1:1) and allowed to crystallize at 4C to provide 1.76 g of
the titled compound (69 % yield). IHNMR (CDC13): o 7.5-7.3(5H, m), 4.6(2H, dd; J =
12 Hz), 4.0(2H, m), 3.6-3.0(6H, m).
b) (2R,4R)- 1 ,5-diazido-2,4-di-(methanesulfonyloxy)-3-benzyloxypentane 42
To 630 mg (2.15 mmol) of bisazido diol 41 in 6.0 rnL of pyridine w~s added 350
llL (4.5 mmol) methanesulfonyl chloride at 0C. The reaction mixture was allowed to
wann to 25C and stirred for 20 hr, then was diluted with 12 rnL 6 N HCI, and extracted
2~ 4 - 28 -
with 100 mL methylene chloride. The organic layer was washed with 3% NaHCO3. dried
(Na2S04) and concentra~ed. Flash chromatography of the residue (95:5 CH2C12:e~her)
provided 867 mg (90% yield) of Ihe tilled compound. lHNMR (CDC13): ~ 7.4-7.2(5H,m), 5.0-4.7(4H, m), 4.02(2H, dd, J = 3 Hz), 3.9-3.5(4H, m), 3.15(3H, s), 3.10(3H, s).
c) (2S,4S)-1,2:4,S-di-(N-carbobenzyloxyimino)-3-benzyloxypen~ane 43
To 310 mg (0.69 mmol) of the compound 42 in 2 mL dry THF at 0C was added
1.5 rnL (1.5 mmol) 1 M LiAlH4 in THF. The rnixture was allowed ~o walm to 25C and
sti~red overnight. Water (0.1 rnL~ was added, followed by 0.1 rslL 15% NaOH and 0.4 rnL
10 water. The mixture was sti~ed vigorously with 10 mL ether and filtered. Concentration of
the ether layer provided the crude. bisaziridine, which was dissolved in S rnL CH2C12 and
combined with 200 ~L (1.4 rnrnol) triethylamine and 200 ~L (1.4 mmol) of benzyl
chloroformate. The rnixture was stirred at 25C for 3 hr, then filtered. The filtra~e was
concentrated and the residue was purified by flash chromatography (ethyl acetate:hexanes
1:5) to provide 89 mg (28% yield) of the titled compound. IHNMR (400 MHz; CDC13):
7.4-7.2(15H, m), 5.0(1H, d; J = 12 Hz), 4.93(1H, d; J = 12 Hz), 4.88(1H, d; J = 12
Hz), 4.84(1H, d; J = 12 Hz), 4.74tlH, d; J = 12 Hz), 4.39(1H, d; J = 12 ~z), 2.82(1H,
t; J = 6 Hz), 2.6(1H, m), 2.4S(lH, m), 2.25(1H, d; J = 6 Hz), 2.15(1H, d; J = 6 Hz),
2.04(1H, d J = 3 Hz), 2.02(1H, d; J = 3 Hz). MS (DCI, NH3): rn/z 473.1~M+).
d) (3S,5S)-3,5-di-(carbobenzyloxyamino)-4-benzyloxyheptane 44
To a suspension of CuI (72 mg, 0.375 mmol) in 1.5 mL ether at -25C was added
0.5 rnL 1.5 M CH3Li in ether. The resulting colorless solution was cooled tO -45DC and a
solution of bisaziridine 43 (10 mg" 0.02 mmol) in 0.5 mL ether was added. After stimng
at ~45C for 1 hr, the rnixture was allowed to warm ~o 10C over a period of 6 hr, them was
stirred for an additional 2 hr. The mixture was diluted w~th 2 mL saturated aqueous NH4CI
and ~ mL saturated a~queous NH3, then was extracted with ether. The organic layer was
washed with brine, dried (Na2S04) and concentrated lo provide the titled compound (10
mg). ~HNMR (C~DC13): ~ 7.2(15H, m), S.2-4.9(4H, m), 4.93(1H, d; J = 12 Hz),
4.88(1H, d; J = 12 Hz), 4.84(1H, d; J = 12 Hz), 4.7(2H, d; J = 12 Hz), 4.4(2H, d: J =
12 Hz), 3.7(2H, m), 3.4(1H, d; J = 2 Hz), 1.6(4H, m), 1.0(3H, t; J = 7 Hz), 0.9(3H, t; J
= 7 ~}z). MS(DCI,NH3)
(M+H)+ 505. 1
e) (3R,5R)-3,5-dihydroxy-4-benzyloxyheptane 4;
To a suspension of CuI (143 mg, 0.75 mmol) in 3 rnL ether at -35C was added
methyllithium (I mL, 1.5 M in ether; 1.5 mmol). The resulting colorless solution was
'~: " ' '. '
2~86~4
WO 92/007~0 Pcr/US9l/04757
- 29 -
stirred at -30C for 30 min, then cooled to 78C. A solution of bisepoxide 33 (78 m~"
0.37 mmol) in 2 mL ether was added. The reaction was allowed to warm to 25C over 4
hr, the saturated aqueous NH4Cl and concen~ated aqueous NH3 were added. The rr~ixture
was extracted wi~h ether~ the organic layer was dried obver Na2S04 and the solvent was
removed furnish the titled compound (93 mg, 100% yield). 1HNMR (CDCl3): ~ 7.4-
7.1(5H, m), 4.7(1H, d; J = 12 Hz), 4.55(1H, d; J = 12 Hz), 3.9-3.7(2H, m), 3.2(1H,
dd), 2.5(2H, b), 1.6-1.4(4H, m), 1.0(3H, t; J = 7 Hz), 0.9(3H, t; J = 7 Hz);
MS(DCI,NH3) (M+H)~ 239.2
f) ~3R,SR)-3,5-Methanesulfonyloxy-4-benzyloxyheptane 4S
Methanesulfonyl chloride (0.3 mL) was added dropwise to diol 45 (93 mg) in
pyridine (1 mL) at 0C. The mixture was allowed to warm tO 25C. After 12 hr the mixture
was diluted with cold 6N HCI (10 mL) and extracted with CH2C12. The organic extract
was washed with 3% NaHC03, dried over MgS04, and concentrated. The residue was
purified by flash chromatography to provide the titled compound (83 mg, S6% yield).
IHNMR (CDC13): ~ 7.4-7.2(5H, m), 4.75 (lH, m), 4.7(1H, d; J = 12 Hz), 4.6(1H, d; J
= 12 Hz), 4.58(1H, m), 3.9(1H, dd; J = 2.6 Hz), 3.0(3H, s), 2.9(3H, s), 2.1-1.5(4H,
m), 1.05(3H, t; J = 7 Hz), 1.0(3H, t; J = 7 Hz).
g) (3S, SS)-3,5-diazido~-benzyloxyheptane 47
A mixture of bismesylate 46 (83 mg, 0.21 mmol) and sodium azide (O.SO g, 7.7
mrnol) in 1.5 mL dimethylformamide was heated tO 70C for 12 hr. After cooling, ethyl
acetate (20 rnL) was added and the mixture was filtered and concentrated. Flash
chroma~ography of the residue provided the titled compound (54 mg, 90% yield). lHNMR
(CDC13): ~ 7.4-7.2(5H, m), 4.65 (2H, s), 3.5-3.2(3H, m), 2.0-1.5(4H, m), 1.1(3H, t: J
= 6 H~), 1.05(3H, t; J - 6 Hz).
h) (3S, SS)-3,5-diamino-4-benzyloxyhept~ne 48
To 328 mg (1.14 mmol) bisazide 47 from step (j) in 5 mL THF at 0C was added
200 mg LiAlH4. The mixture was allowed to warrn to 25C and was stilTed for S hr. The
reac~ion was quenched by addition of 0~5 mL 15% NaOH, stirred for 15 min, diluted with
150 mL ether and filtered. Concentration of the fil~ate provided the titled compound (278
mg, 100% yield). lHNMR (CDC13): ~ 7.26(5H, m), 4.5 (2H, dd; 3 = 12Hz), 3.1(1H,
dd; J = 4.6 Hz), 2.9(1H, m), 2.75(1H, m), 1.8-l.O(lOH, m), 0.9(3H, ~; J = 7 Hz),0.85(3H, t; J = 7 Hz).
WO 92/0~7~0 ~ s PCrtUS91/04757
i) t3S, 5S)-3,S-diarnino-4-hydroxyheptane 49
52 mg of the diamine product 48 in 4 mL methanol was added 50 mg 20%
Pd(OH)2 on carbon and 2 drops concentrated aqueous HCI. The mixture was stirred under
an atmosphere of H2 for 16 hr, then was filtered and concentrar.ed to provide the titled
compound (53 mg) as the dihydrochloride salt. lHNMR (CD30D): ~ 3.75(1H, dd; J =
4.7 H~), 3.2-3.0~2H, m), 1.8-1.4(4H, m), 0.9(6H, t; J = 7 Hz).
j) (3S,5S)-3,5~ (carbo~enzyloxyalanylalanyl)amino-~hydroxyheptane 50
To the diamine product 49 (87 mg, 0.37 mmol) in 6 mL D~: was added 221 mg
(0.75 mmol) carbobenzyloxyalanylalanine, 115 mg (0.75 rnrnol) HOBT, and 154 mg (0.75
mrnol) DCC. The mixture was stirred overnight, then was concentrated, taken up in ethyl
acetate, filtered, washed with water and brine and dried (MgSO4). Removal of solvent
followed by MPLC (silica; 2~o methanol in CH2cl2) provided the titled compound (109
mg). IHNMR (DMSO-d6): ~ 8.0-7.2(21H, m), 5.0(4H, bs), 4.6(2H, dd; J = 12 Hz),
4.2(2H, m), 4.0(2H, m), 3.8(1H, m), 3.7(1H, m), 3.5(1H, dd; J = 4.7 Hz), 1.7-1.3(4H,
m), 1.1(12H, m), 0.75(6H, t). MS(FAB) (M+H)+ 789.3
Example 1 1
Preparation of (3S. SS)-3.5-di-talanvlalanvl)amin~4-hvdroxvheptane ~1
To the product 19 t4.5 mg) in 1 mL DMF was added 10 mg of 20% Pd(OH)2 on
car~on. The mixture was stirred under 1 atmosphere of H2 for 6 hr, then was filtered and
concentrated to provide the titled compound (3 mg). IHNMR (CD30D): ~ 4.3(2H, m),3.8(2H, m), 3.7(1H, rn), 3.5(1H, m), 1.8-1.2(16H, m), 0.8(6H, dt).
Example 12
Preparation o~S. 5S)-3.5-di-fcarbobenzyloxvvalYl)amino-4-hYdroxvheptane 52~
To 133 mg (0.5 rnrnol) Cbz-Val in 2 mL THF at -40C was added 65 ~,IL (0.5
rn nol) of NMM and 65 ~L (0.5 mmol)of isobutylchloroforrnate. After stirring for 10 min
a solution of 25 mg ~0.17 mmol) diarr~ne hydrochloride 1 and 50 ,uL NMM in 1 mL DMF
was added. The mixture was slowly warrned to 20C and stirred overnight, then was
diluted with ethyl acetate, washed with 5% HCI, 5% NaHCO3, and brine and the organic
layer was concentrated. The residue was purified by flash chromatography (ethyl
acetate:hexanes) to provide the titled compound (18 mg~. IHNMR (CDC13): ~ 7.5-
7.3(10H, m), 6.9(1H, bd), 6.35(1H, bd), 5.5(1H, bd), 5.25(1H, bd), 5.1(4H, bs), 4 0-
3.1t6H, m), 2.4-2.1(2H, m), 2.0-1.4(4H, m), 1.0-0.8(18H, m). MS (FAB): rn/z
61 3.2(M+H)~.
20~41~
wo g2/007~0 P~r/U~91/04757
- 31 -
E~ample 13
PreparatiQn ~,, 6~4~-diamino-5-hydroxy~8-dimethvl-1~8-nonane dihvdrochloride
a) (4R, 6R)-4,6-dihydroxy-5-benzyloxy-2,8-dimethyl-1,8-nondiene 53
To a suspension of CuI (192 mg, 1.0 mmol) in ether (2 mL) at -60C was added
isopropenyllithium (5.2 tnL, 2.0 mmol; 0.38 M in ether, freshly prepared from 2-bromopropene and lithium wire). The rnixture was war,ned to -45C, then recooled to -
78C. A soluuon of bisepoxide 33 (60 mg, 0.29 mmol) in ether (5 mL) was added. The
nnxture was allowed to warm to 0C over 2 hr with stining. ~e rnixture was diluted with
ether and washed with 5 mL of 1:1 concentrated aqueous arnmonia:saturated NH4CI. l he
organic layer was dried over MgSO4 and concentrated to provide the ti~led compound (81
mg, 96% yield) as a colorless solid. IHNMR (CDC13): o 7.3(5H, m), 4.9(1H, bs),
4.85(1H, bs), 4.80(1H, bs), 4.75(1H, bs), 4.65(1H, d; J = 12 Hz), 4.58(1H, d; J = 12
Hz), 4.0(2H, m), 3.3(1H, dd; J = 2.5 Hz), 2.55(1H, d; J = 6 Hz), 2.55(1H, d; J = 4 Hz),
2.5-2.1~4H, m), 1.7(1H, s). MS(DCI,NH3) (M+H)+ 291.4
b) (4R, 6R)-4,6-dihydroxy-5-benzyloxy-2,8-dimethylnonane 54
To the product 53 (105 mg, 0.38 mmol) in CH2C12 (lrnL) was added 55 mg
Ir(COD)Py(PCy)3PF6 (Crabtree catalyst). The mixture was sdrred for 6 hr under 1
20 atmosphere H2, then was filtered and concentrated to provide the titled compound (110 mg,
100% yield). IHNMR (CDC13): ~ 7.3(5H, m), 4.6(1H, dd; J = 12 Hz), 4.0(2H, m),
3.1(1H, bs), 2.7(2H, bs), 2.0-1.1(6H, m), 1.0-0.8~12H, m).
MS(DCI,NH3) (M+H)~ 295.4.
c) (4R, 6R)-4,6-Di-(methanesulfonyloxy)-5-benzyloxy-2,8-dimethylnonane 55
Methanesulfonyl chloride (0.25 mL) was added dropwise to diol S4 ~93 mg) in
pyridine (1 rnL) at 0C. The tnixture was allowed to warm to 25C. After 10 hr the mixture
was diluted with cold 6N HCI (10 mL) and extracted with CH2C12. The organic extract
was washed with 3% NaHCO3, dried over MgSO4, and concentrated. The residue was
punfied by flash chromatography to pro~ide the titled compound (210 mg). IHNMR
(CDC13): ~ 7.4(5H, m), 5.0(1H, m), 4.8(1H, d; J = 12 Hz), 4.7(1H, m), 4.6(1~, d; J =
12 Hz), 3.85(1H, dd; J = 4.7 Hz), 3.0(3H, s), 2.9(3H, s), 2.0-1.1(6H, m), 1.0-0.9(12H,
m).
d) (4R, 6R)-4,6-diazido-$-benzyloxy-2,8-dimethylnonane 56
To the product 55 (210 mg) in 2 mL DMF was added 870 mg (15 mmol) NaN3.
The mixture was heated to 70~t:1 for 7 hr, then was cooled and diluted wi~h ethyl acet2te.
The filtrate was concen~ated and the residue was purified by MPLC (ethyl aceute:hex;lnes
WO 92/00750 PCirtUS9l/04757
2 0 ~ 32 - -
1:20) to provide the titled compound (52 mg). IHNMR (CDC13): ~ 7.3(5H, m),
4.55(2H, dd; J = 12 Hz), 3.4-3.15(3H, m), 2.0-1.1(6H, m), 1.0-0.75(12H, m).
e) (4S, 6S)-4,6-diamino-5-benzyloxy-2,8-dimethylnonane 57
S To the product 56 (52 mg, 0.15 mmol) in THF (3 ml) was added 80 mg LiAlH4 (~
mmol) at 0C. The mixture was stirred at 25C overnight, then was quenched with lN
NaOH and diluted with ether (50 ;nL). Filtration and concentration provided the ~i~led
compound (44 mg) as a colorless oil. IHNMR (CDCl3): ~ 7.3(5H, m), 4.6(2H, dd; J =
12 Hz), 3.1(1H, m), 3.05-2.95(2H, m), 1.9-1.1(6H, m), 1.0-0.8(12H, m).
f) (4S, 6S)-4,6-diarnino-5-hydroxy-2,8-dimethyl-1,8-nonane dihydrochloride 58
To 165 mg of diamine 57 in 10 rslL methanol containing S drops of conc. HCI was
added 100 mg 20% Pd(OH)2 on carbon. The mixture was stirred overnight under 1 atm
H2, then was filtered and concentrated to provide the titled compound (75 mg). IHNMR
(CD3OD): ~ 3.7(1H, m), 3.35(1H, m), 3.2(1H, m), 1.8-1;0(6H, m), 0.99-0.8(12H,m).MS(DCI, NH3) tM+H)+ 203.2.
Example 14
Preparation of (4S,6$)-4,6-di-(carbobenzyloxyalanylalanyl)am~no-5-hydroxy-2,8-
dimethyl-1,8-nonane 59
To the prcduct ~8 (44 mg, 0.15 tnmol) in 2 mL DMF was added 110 mg Cbz-
AlaAla (0.37S mmol), 58 rng (0.37~ rnmol) HOBT, and 72 mg (0.375 mmol~ DCC. The
rnixture was stirred for 48 hr at 25C, ~hen was diluted with 20 rnL ethyl acetate and
filtered. The filtrate was concentrated and Ihe residue was purified by MPLC (gradient, 0-
5% methanol in CH2C12) to provide the titled compound (24 mg). IHNMR (CD30D):
8.0-7.2(21H, m), 5.0(4H, overlapping dd), 4.2(2H, m), 4.0(4H, m), 3.5 (IH, br s),
1.7-1.15(18H, m), 0.9-0.7(12H, br t).
Examplç 15
Preparation of (4S~6S~-4.6-di-ta!anvlalanYl)amino-5-hvdroxY-2~-dimethYl- 1 .8-nonane
~Q
To the product 59 (12 mg) in 2 mL DMF was added 50 mg of 20% Pd(OH)2 on
carbon. The mixture was stilTed under 1 atmosphere of H2 for 10 hr, then was diluted with
methanol, filtered and concentrated to provide the titled compound (7.5 mg). IHNMR
~CD30D): ~ 4.25(2H, m), 3.8(1H, m), 3.65(2H, m), 3.1(1H, bd), 1.6-1.1(18H, m),
0.7(12H, bd). MS(FAB) (M+H)+ 487.
WO 92/007~02 ~ 8 6 ~ 1 ~ PCItUS9~/047~7
- 33-
Exam~le 16
Preparation of !4S 6S1-4.6-di-(carbobenzvloxy)amino-5-hvdroxx-2~8-dimethvl-1~8-
nonane 6L
To 6.0 mg of the bis-amine hydrochloride product ;8 in 0.5 mL CH2C12 at 2()C
S were added S mL t iethylarnine and 10 rnL benzyl chloroformate. After 3 hr stimng, the
mixture was applied to a silica column and eluted with CH2C12 followed by e~er to
provide the dtled compound (4.6 mg). lHNMR (CI:)C13): ~ 7.3(10H, bs), 5.1-4.9 (6H,
m), 3.9-3.7(2H, m), 3.5-3.35(2H, m; lH exchangeab}e with D20), 1.?-1.5 (4H, m),
1.3-1.2 (2H, m)~ 0.8 (12H, m).
Example 17
Preparation of (2S. 4S)-2~4-di-(p-toluenesulfonyl~amin~3-hvdrox~-1.5-diphenvlpentane
I'he titled product was prepared by sulfonylation of compound 40 (Example 9) with
p-toluenesulfonyl chloride in methylene chloride and triethylamine. lHNMR (250 MHz,
CDC13) 8.0-6.8 (m, 18H), 5.5 (d, lH, J=7Hz), 5.2 (d, lH, J=7Hz), 3.2-3.7 D(m, 4H),
2,4-2.7 (m, 4H), 2.5 (s, 3H), 2,45 (s, 3H).
Enz~rne Inhibition
Inhibition of HIV protease activitv
The inhibition assay has been previously described in Dreyer e~ al. ~oc. Natl.
Acad. Sci. USA. 86, 9752-9756 (1989) and Moore et al. Bioch Bioph. Re~Çom., 159,420 (1989). A typical assay contained 10 mL MENDT buffer (50 mM Mes (pH 6.0; 2-(N-
mospholino) ethanesulfonic acid), 1 nM EDTA, lmM dithiothreitol, 200 mM NaC1, 0.1%
Triton X-100); 2, 3, or 6 mM N-acetyl-L-arginyl-L-alanyl-L-seryl-L-glutaminyl-L-asparaginyl-L-tyrosyl-L-prolyl-L-valyl-L-valinamide (Ac-Arg-Ala-Ser-Gln-Asn-Tyr-Pro-
Val-Val-NH2; Km = 7 nM); and micromolar and submicromolar concentrations of
symhetic compounds. Following incubation at 37C for several minutes, the reaction was
initiated witb O.OOI-O.lOmg purified HIV protease. Reaction rnixtures (37C) were
quenched after 10-20 minu~es with an equal volume of cold 0.6 N trichloroacetic acid, and,
- following centrifugation to remove precipitated material, peptidolysis products we.~
analyzed by reverse phase HPLC (Beckman Ultrasphere ODS, 4.5 mm x 25 mm; rsobilephase; 5-~0% acetonitrilelH20 - .1% TFA 915 min.), 20% acetonitrile/H20 - .1% TFA (5
min) at 1.5 mL~min, detection at 220 nm. The elution positions of Ac-Arg-Ala-Ser-Gln-
Asn-Tyr-P~Val-Val-NH2 (17-18 min) and Ac-Arg-Ala-Ser-Gln-Asn-Tyr ~10-l l min)
were confirrned wi~h authen~ic material. Initial rates of Ac-Arg-Ala-Ser-Gln-Asn-Tyr
formation were dete~mined from integration of these peaks, and typically, the inhibitor~
WO 92/007~0 Pcr/ussl/o4757
2, 0 8 6 ~
properties of the synthetic compounds were deterrnined from slope/intercept analysis of a
plot of 1/v vs. [inhibitor] (Dixon analysis). Ki values resulting from this type of primary
analysis are accurate for competitive inhibitors only, and under conditions in which the
Michaelis constant of the subs~ate used is well-determined.
It is desirable for the compounds of this invention to have Ki values less than 50
M, preferably less than 10 ~LM and more preferably less than 1 llM.
Following the procedures set for~h herein and the teachings of the foregoing
exa~s~ples the compounds set forth in the following Table can be prepared having the
structure and dle substituent groups as designated therein.
Inhibition of rHIV- I Protease
Compound IC50
3 50
6 80
8 80
0.123
52 50
62 1,000
TABLE I
R1 R2
Xt HN ~ NHX2
HO
I
~o, ~1 andX2 . B~ B2
H ethyl
2 AlaAla ethyl
3 Cbz-Val ethyl
4 AlaAla PhCH2
H i-Bu
6 AlaAla i-Bu
7 Cbz- i-Bu
8 Cbz-Val i-Bu
101 Cbz-Ala i-Bu
2 ~B Ala i-Bu
2~S~14
WO 92/007~0 35 PCI/US91/04757
3 ~-AIaVal i-Bu
4 Cbz-AlaAla i-Bu
105 BocAla i-Bu
6 AcAlaAsn i-Bu
7 AcGlnAsn - i-Bu
8 Cbz-PheAla i-Bu
9 trifluor~AlaAla i-Bu
110 Cbz-tTiflu~rAlaAla i-Bu
trifluo~oAla i-Bu
2 Cbz-trifluoroAla i-Bu
3 Ph(CH2)2CO i-Bu
4 Boc i-Bu
115 Ac i-Bu
6 PhSo2 i-Bu
7 HCO i-Bù
8 Propionyl i-Bu
9 i-Butyryl i-Bu
120 Ph(CH~)2CO i-Bu
PhSO2Val i-Bu
2 Phenyllactoyl i-Bu
3 Phenyllactoyl-Val i-Bu
4 Cbz-Ala PhCH2
125 Cbz-Val i-Bu~cnyl
6 Cbz-Val 2-Propenyl
7 Cbz-Val 3-Butenuyl
8 Cbz-Val n-Pentyl
9 Cbz-Val Ph(cH2)2-
130 Cbz-Val Cyclohexyl-CH2-
Cbz-Val 2-Napthyl-CH2-
2 Cbz-Val 3-Napthyl-CH2-
3 Cbz-Val 2-Butynyl
4 Cbz-Val 3-Indoylmethyl
135 Cbz-Val trans-3-phenyl-3-propenyl
6 Cbz-Val N-Piperidinyl-CH2-
7 Cbz-Val N-Morpholinyl-CH2
8 Cbz-Val (CH3)2N-CH2
9 Cbz-Val t-ButylNH-CH2-
wo92~u~o 8 ~ 4 - 36 - PCr/US91/04757
140 Cbz-Val N-lmidazoyl-CH~
Cbz-Val PhCONH-CH2
2 Cbz-Val N-IndoYI-CH2
3 ~bz-Val t-ButylCONH-CH2
4 Cbz-Val BocNHCH~
145 Cbz-Yal NH~CH~
6 Cbz-Val N-benzi~dazolyl
7 Cbz-Val PhCH~O-CH2
8 Cbz-Val PhO-CH2
9 Cbz-Val CH3(CH2)20-CH2
150 Cbz-Val CH30-CH~
Cbz-Val (CH3)2CHO-CH~
2 Cbz-Val t-Butyl-O-CH23 Cbz-Val (CH3)2CHClH20-CH~
4 Cbz-Val CH3CH2(CH3)CHO-CH~
155 Cbz-Val Cyclohexyl-O-CH2
Cbz-Val PhCH ~OCH70-CH2
7 Cbz-Val CH30CH20-CH2
8 Cbz-Val CH30CH2CH20CH20CH2
9 Cbz-Val CH3S-CH2
160 Cbz-Val PhS-CH2
Cbz-Val (CH3)2CHS-CH
2 Cbz-Val CH3((: H2) ~S-CH~
3 Cbz-Val CH3(~H2)3s-cH~
4 Cbz-Val CH3S(O)-CH2
1~5 Cbz-Val CH3S(0)2-CH 76 Cbz-Val PhS(0)2-CH~
7 Cbz-V~I i-Propyl-S(0)2-( ~H2
8 Cbz-Val n-Propyl-S(0)2-CH2
9 Cbz-Val n-Butyl-S(O)~-CH7
170 Cbz-Val (Ph~O)~P(O)-CH 7
Cbz-Val (CH30)~P(O)-CH~
2 Cbz-V~l (n-ButylO)~P(O)-CH
3 Cbz-V~ O) ~P(O)-CH~
4 Cbz-Al~ (CH30)~P(O)-CH~
~08~41 4
~O 92/00750 PCI/US91/04757
- 37 -
TABLE II
Rl R~2
X1 HN ~NHX2
II
~1~2 ~,1~2
H ethyl
2 AlaAla ethyl
3 Cbz-Val ethyl
4 AlaAla PhCH2
H i-Bu
6 AlaAla i-Bu
7 Cbz- i-Bu
8 Cbz-Val i-Bu
201 Cbz-Ala i-Bu
2 ,B Ala i-Bu
3 ,~AlaVal i-Bu
4 Cbz-AlaAla i-Bu
205 BocAla i-Bu
6 AcAlaAsn i-Bu
7 AcGlnAsn i-Bu
8 Cbz-PheAla i-Bu
9 trifluoroAlaAla i-Bu
210 Cbz-triflurorAlaAla i-Bu
trifluoroAla i-Bu
2 Cbz-trifluoroAla i-Bu
3 Ph(CH2)2CO i-Bu
4 Boc i-Bu
215 Ac i-Bu
PhSo2 i-Bu
7 HCO i-Bu
8 Propionyl i-Bu
9 i-Butyryl i-Bu
220 Ph~C~2)2co i-Bu
PhSO2Val i-Bu
2 Phenyllactoyl i-Bu
WO ~2/00750 ~, 0 ~3 6 Li 4 - 38 - PCr/VS9~/04757
3 Phenyllactoyl-Val i-Bu
4 Cbz-Ala PhCH2
225 Cbz-Val ` i-Butenyl
6 Cbz-Val ` 2-Propenyl
7 Cbz-Val - 3-Butenuyl
8 Cbz-Val n-Pentyl
9 Cbz-Val Ph(CH2)2-
230 Cbz-Val Cyclohexyl-CH2-
Cbz-Val 2-Napthyl-CH2-
2 Cbz-Val 3-Napthyl-CH2-
3 Cbz-Val 2-Butynyl
4 Cbz-Val 3-Indoylmethyl
- 235 Cbz-Val trans-3-phenyl-3-propenyl
6 Cbz-Val N-Piperidinyl-S~H2-
7 Cbz-Val N-Morpholinyl-CH2-
8 Cbz-Val (CH3)2N-CH2
9 Cbz-Val t-ButylNH-CH2-
240 Cbz-Val N-Imidazoyl-CH2
Cb~-Val PhCONH-CH2
2 Cbz-Val N-Indoyl-CH2
3 Cbz-Val t-ButylCONH-CH2
4 Cbz-Val BocNHCH2
245 Cbz-Val NH2CH2
6 Cbz-~al N-benzimidazolyl
7 Cbz-Val PhCH20-CH2
8 Cbz-Val PhO-CH2
9 Cbz-Val CH3(CH2)20-CH2
250 Cbz-Val CH30-CH2
Cbz-Val (CH3)2CHO-CH2
2 Cbz-Val t-Butyl-O-CH2
3 Cbz ~ ~al (CH3)2cHcH2O-cH2
4 Cbz-Val CH3CH2(CH3)CHO-cH2
255 Cbz-Val (: yclohexyl-O-CH2
6 Cbz-Yal PhcH2ocH2o-cH2
7 Cbz-Val CH30CH20-CH2
8 Cbz-Val CH3OCH~cH2OcH2oc~I2
9 Cbz-Y3} CH3S-CH~
", , .
~0~414
WO 92/00750 39 PCI/US91/04757
260 Cbz-Val PhS-CH2
Cbz-Val (CH3)2CHS-CH2
2 Cbz-Val CH3(CH2)2S-CH2
3 Cbz-VaI CH3(CH2)3S-cH2
4 Cbz-Val CH3StO~-cH2
265 Cbz-Val CH3S(O~2-CH2
6 Cbz-Val PhS(0)2-CH2
7 Cbz-Val i-Propyl-S(0)2-(~H2
8 Cbz-Val n-Propyl-S(0)2-CH2
9 Cbz-Val n-Butyl-$(O)2-CH2
270 Cbz-Val (Ph2O)2P(O)-cH2
Cbz-Val (CH30)2P(O)-CH2
2 Cbz-Val (n-ButylO)2P(O)-CH2
3 Cbz-Val (EtO)2P(O)-CH2
4 Cbz-Ala (C~l3O)2p(o)-cH2