Note: Descriptions are shown in the official language in which they were submitted.
The invention relates to new quinolone- and
naphthyridonecarboxylic acid derivatives, processes for
their preparation and also antibacterial agents and feed
additives containing them.
Quinolone- and naphthyridonecarboxylic acids which are
substituted in the 7-position by a bicyclic amine radical
have already been disclosed in EP-A-0,350,733.
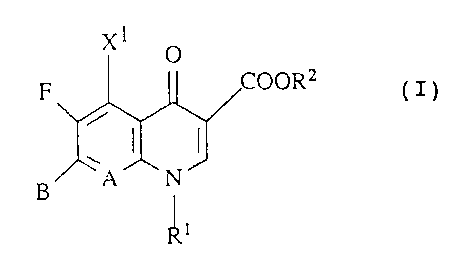
The present invention relates to new compounds of the
formula (I)
OOR2
F (
y
in which
A represents CH, CF, CC1, C-OCH3, C-CH3 or N,
X1 represents H, halogen, NHZ or CH3,
R1 represents C1-C3-a13cy1, FCHZCHZ-, cyclopropyl or
phenyl which is optionally monosubstituted to
trisubstituted by halogen, or A and R1 together can
denote a bridge of the structure C-0-CHz-CH ( CH3 ) -,
xl O
C
~N~
A
L~e A 28 100 - 1 -
RZ represents H, C1-C3-alkyl which is optionally
substituted by hydroxyl, halogen or amino or 5-
methyl-2-oxo-1,3-dioxol-4-y1-methyl,
B represents a radical of the formulae
N N N N N
R3N Y R~ N Y R4 N Y R4 N Y R4 N Y
U a U
in which
Y represents 0 or CH2,
R3 represents C2-CS-oxoalkyl, CHZ-CO-C6H5, CHZCHZCOZR°,
R' OZC-CH= ~-COZR' , -CH=CH-CO2R' or CHZCHZ-C1N,
in which
R' denotes hydrogen or C1-C3-alkyl,
R4 represents H, C1-C~-alkyl, CZ-CS-oxoalkyl, CHZ-CO-
C6H5, CHZCH2CO2R' , R' 0zC-CH=C~-(:OZR' , -CH=CH-COzR ° or
CHZCHZ-CN or represents 5-methyl-2-oxo-1, 3-dioxol-4-
~1-methyl,
in which
R' denotes hydrogen or Cl-C3-alkyl,
Le A 28 x.00 - 2 -
and to pharmaceutically utilisable hydrates and acid
addition salts thereof as well as the alkali metal,
alkaline earth metal, silver and guanidinium salts of the
underlying carboxylic acids. These compounds have a high
antibacterial activity. The compounds according to the
invention are particularly distinguished in that they
have a high activity on dormant and resistant
microorganisms.
Preferred compounds of the formula (I) are those in which
A represents CH, CF, CC1, C-0CH3 or N,
X1 represents H, F, Cl, Br, NHZ or CH3,
R1 represents CZHS, cyclopropyl or 2,4-difluorophenyl,
or A and R1 together can denote a bridge of the
structure C-0-CHZ-CH ( CH3 ) -,
RZ represents H, CH3, CZHS or 5-methyl-2-oxo-1, 3-dioxol-
4-yl-methyl,
B represents a radical of the formulae
H--~~H H 4 .
.. ...,
""~~ H H ""... ~ H H
R3N Y R4 N Y R4 N Y R N Y R N Y
U ~ U
in which
Le A 28 100 - 3 -
s
~~°s~
Y represents 0 or CHZ and
R3 represents CHz-C0-CH3, CHZ-CO-CsHS, GHzCH2-C0-CH3,
CHZCHZCOZR' , R ° 02C-CH=C-COzR' , -CH=CH-COZR' Or
i
CH2CH2-CN,
S in which
R' denotes Ci-CZ-alkyl.
R° represents H, C1-C3-alkyl, 5-methyl-2-oaco-1, 3-dioxol-
4-yl-methyl, CHz-CO-CH3, CHZ-CO~C6H5, CHZCHz-C0-CHa,
CHZCHzCOzR' , R' 02C-CH=C-COZR' , -CH=CH-COzR ° Or CHZCHZ
-CN,
in which
R' denotes C1-CZ-alkyl.
Particularly preferred compounds of the formula (I) are
those in which
A represents CH, CF, CC1, C-OC',H~ or Id,
Xl represents H, F, C1, Hr, NHz or CH3o
R1 represents CZHS, cyclopropyl or 2,4-difluorophenyl,
or A and R1 together can denote a bridge of the
structure C-0-CHZ-CH ( CH3 ) -,
Le A 28 100 - 4 -
CA 02086914 2003-05-15
23189-7450
R2 represents H, CH3 or C2H5,
B represents a radical of the formulae
N N N N
H H H ~,~~,.. "~~~ H H ~~~".. H H ~ H
R4N~Y R4N~Y R4N~Y R4N~Y
in which
Y represents O or CH2 and
R4 represents H, C1-C3-alkyl, 5-methyl-2-oxo-1,3-dioxol-4-
yl-methyl, CH2-CO-CH3, CH2-CO-C6H5, CH2CH2-CO-CH3,
CH2CH2C02R', R~OzC-CH=i-COZR'~ -CH=CH-C02R' OR CH2CH2-CN,
in which
R' denotes C1-C2-alkyl.
Specific compounds of the invention include:
8-chloro-1-cyclopropyl-7-([S,S]-2,8-diazabicyclo-[4.3.0]non-
8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid;
1-cyclopropyl-7-([S,S]-2,8-diazabicyclo[4.3.0]non-8-yl)-6-
fluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic
acid;
1-cyclopropyl-7-([S,S]-2,8-diazabicyclo[4.3.0]non-8-yl)-6,8-
difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid;
1-cyclopropyl-7-([S,S]-2,8-diazabicyclo[4.3.0]non-8-yl)-6-
fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid;
- 5 -
CA 02086914 2003-05-15
23189-7450
1-cyclopropyl-7- ( [S, S] -2, 8-diazabicyclo [4 . 3 . 0] non-8-yl) -5, 6, 8-
trifluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid;
or a pharmaceutically utilisable hydrate or acid addition salt
thereof or an alkali metal, alkaline earth metal, silver or
guanidinium salt of the underlying carboxylic acid.
The compounds of the formula (I)
in which
A, X1, R1 and R2 have the abovementioned meaning, and
B represents a radical of the formula
- 5a -
N
R~~'~ \y
U
in which
R3 and Y have the abovementioned meaning,
are obtained
by reacting a compound of the formula (II)
F , OOR2
N sA~N~
I (zz)
HN Ri
in which
A, Y, Xl, R' and RZ have the abovernentioned meaning,
with a compound of the formula (I2I)
R3~~3 t z z z )
in which a
Le A 28 100 - 6 -
R3 represents Cz-CS-oxoalkyl, CHz-CO-CsHS, CHzCHz-COzR' or
CHZCHz-CN,
in which
R' denotes hydrogen or C1-C3-alkyl, and
X3 represents halogen, in particular chlorine, bromine
or iodine,
if appropriate in the presence of acid binders . [Method A]
Compounds according to the invention of the formula (I)
in which
A, X1, R' and Rz have the abovementioned meaning, and
B represents a radical of the formula
N
R3N Y
in which
Y has the abovementioned meaning and
R3 represents CHZCHx-CO-CH3, CHZCHz-COZR ° ,
Le A 28 100 - 7 -
~~~if.~
R' OzC-CH=C-COzR' , -CH-CH-C02R' or CHZCHZ-CN,
in which
R' denotes hydrogen or C1-C3-alkyl,
can be obtained
by reacting a compound of the formula (II)
Xl
O
F , OOR2
N A"N-
HN~ ~I (II)
with a Michael acceptor such as dialkyl acetylenedi-
carboxylate, alkyl propiolate or a compound of the
formula (IV)
CHZ=CH-R~ ( IV)
in which
RS represents COCH3, COZR' or CN. [Method H]
To prepare enantiomerically pure compounds of the formula
(I), a compound of the formula (V)
Le A 28 100 - 8
XI
O
COOR2
A~N
X
I
R1
in which
A, R1, RZ and X1 have the abovementioned meaning and
XZ represents halogen, in particular fluorine or
chlorine,
is reacted with enantiomerically pure compounds of the
formulae (DTI )
H H H H
N N N N
R4N Y R4N Y R4 N Y R4N Y
~/ ~J ~J ~J
in which
Y represents O or CHZ and
R4 represents H or C1-C3-alkyl,
Le A 28 100 - 9 -
2~~ ~.~t~
if appropriate in the presence of acid scavengers,
and the reaction product is optionally further reacted
with a compound of the formula (IIIa)
R''-X3 ( 2I I a )
in which
X3 has the abovementioned meaning and
R'' represents C2-C5-oxoalkyl, CHZ-CO-CsHs, CHZCH2COZR' or
CHZCHZ-CN,
in which
R' denotes hydrogen or C1-C3-alkyl,
or with a Michael acceptor such as dialkyl acetylene-
diearboxylate, alkyl propiolate or a compound of the
formula (IV)
CHZ=CH-RS ( IV)
in which
R5 represents COCH3, COzR' or CN [Method C] .
If, for example, 8-chloro-1-cyclopropyl-6,7-difluoro-1,4-
dihydro-~-oxo-3-quinolinecarboxylic acid and [S,S]-2,8-
he A 28 100 - 10 -
diazabicyclo[4.3.0]nonane are used as starting compounds,
the course of the reaction can be represented by the
following reaction schemes
0
F COOH H H
N
~~N NH
F
Cl
H
O
F ~ COOH
y
H N \ N
HN C!
'H
If, for example, 6,8-difluoro-1-(2,4-difluorophenyl)-1,4-
dihydro-7-([15,6R)-2-oxa-5,8-diazabicyclo[4.3.0]non-8-
yl)--4~-oxo-3-quinolinecarboxylic acid and diethyl
acetylenedicarboxylate are used as starting substances,
the course of the reaction can be represented by the
following reaction scheme:
O
F ~ COOH
N \ I N fJ
-f- C2HSOZC-C=C-CO2CZH5
HN~~ F i F
~O H ~
F
Le A 28 100 - 11 -
2~~~~.~
0
F , COOH
COZC2H5
H~ N ~ N
CZHSOZC-CH=C
N ~H F ~ F
~O
F
The racemic compounds of the formula (II) used as
starting compounds are mainly known. Enantiomerically
pure compounds of the formula ( TI ) are new and can be
obtained in various ways.
1. A racemic intermediate of the formula (II) is
reacted with an enantiomerically pure auxiliary
reagent, the resulting diastereomers are separated,
for example by chromatography and the chiral auxi-
liary group is removed again from the desired
diastereomer. The following reaction may be shown as
an example:
0
F , COOH
IJ *
N ~ N + ~ ~ CH-NCO ---
HN* F ~ CH3
*cis
Le A 2~ 100 - 12 -
~~~1~~~
O
F , COOH D~astere~r
separation
CH- NH-CO * N F N
\N
CH3
O
F ~ COOH
_ (R> ~. ~ J H~
CH- NH-CO H N F N -
I ~N
CH3 ' H
O
F ~ COOH
N ~' N
HN F
'H
2. The bicyclic amines (VI) are, as enantiomerically
pure compounds, new. They can be prepared by the
following processes:
2.1. The racemic bicyclic amines (~a)
N ~a~
~NH
Y
in vahich
R° represents H Or C1-C3-alkyl,
Le A 28 100 - 13
~~~~v~e
can be reacted with enantiomerically pure acids, for
example carboxylic acids or sulphonic acids such as
N-acetyl-L-glutamic acid, N-benzoyl-L-alanine, 3-
bromocamphor-9-sulphonic acid, camphor-3-carboxylic
acid, cis-camphoric acid, camphor-10-sulphonic acid,
O,0'-dibenzoyl-tartaric acid, D- or L-tartaric acid,
mandelic acid, «-methoxy-phenylacetic acid, 1-
phenyl-ethanesulphonic acid or «-phenyl-succinic
acid, to give a mixture of the diastereomeric salts,
which can be separated by fractional crystallisation
to give the diastereomerically pure salts (see P.
Newman, Optical Resolution Procedures for Chemical
Compounds, Volume 1). The molar ratio between amine
arid enantiomerically pure acid can be varied in a
relatively wide range. By treatment of these salts
with alkali metal or alkaline earth metal
hydroxides, the enantiomerically pure amines can be
liberated.
2.2. In a similar manner, as described in 2.1.,
resolution of the basic intermediates which occur
during the preparation of the racemic bicyclic
amines can be carried out using the abovementioned
enantiomerically pure acids. Examples of basic
intermediates of this type are:
Le A 28 100 - 14 -
~~c~~~l
H H
I
N N
N CE-IZ \ / (b) ~ ~N - CHZ (e)
Y Y
O
CHz \ i Ha \
I
C N ~d) N (e)
~N - CO ~ / ~ NH
O ~/
In the following reaction scheme, the separation of
8-benzyl-cis-2,8-diazabicyclo[4.3,0)nonane into the
enantiomers via the tartrates and conversion thereof
into the enantiomerically pure cis-2,8-
diazabicyclo[4.3.0]nonanes may be shown as an
example of a resolution:
Le A 28 100 - 15 -
~~a~~~.
H
N
N-CHZ-Ph
L(+)-tartaric acid
l.Crystallisation 2. Crystallisation Plother liquor
1 ) NaOH
1) 1 x recrystallisation 1 x recryst. 2) D(-)-
2) NaOH tartaric acid
NaOH
H ~ ~ H H
N-CHZ-Ph N-CH.,-Ph
HZ/ Pd-C
HZ/ pd-C
H H H H
N
N-H ~N-H
e.e.> 99 % e.e.> 99 %
R,R-Configuration S,S-Configuration
Le A 28 100 .- 16 -
~~~~3~i~~
2.3. Both the racemic amines (a) and the basic
intermediates (b) - (e) can be separated by
chromatography, if appropriate after acylation, by
means of chiral support materials (see, for example,
G. Blaschke, Angew. Chem. 92, 14r19~0]).
2.4. Both the racemic amines (a) and the basic
intermediates (b), (c), (e) can be converted by
chemical linkage with chiral acyl radicals into
diastereomer mixtures which can be separated by
distillation, crystallisation or chromatography into
the diastereomerically pure acyl derivatives, from
which the enantiomerically pure amines can be
isolated by hydrolysis. Examples of reagents for
linkage to chiral acyl radicals are: a-methoxy-a-
trifluoromethyl-phenylacetyl chloride, menthyl
isocyanate, D- or L-a-phenyl-ethyl isocyanate,
menthyl chloroformate or camphor-10-sulphonyl
chloride.
2.5. Tn the course of the synthesis of the bicyclic
amines (a), instead of achiral protective groups
chiral protective groups care also be introduced. In
this manner, diastereomers axe obtained which can be
separated. For example, in the synthesis of cis-2, ~-
diazabicyclo[4.3.0]nonane, the benzyl radical can be
replaced by the R- or S-a-phenylethyl radical:
Le A 2B 100 - 17 -
~~~ L
H
0 I 0
~N ~ ~N-CH ~ ~ ---.- N N-CH
C1-I3 CIS CH3
0
2.6. The enantiomerically pure amines (VT) can also be
synthesised from enantiomerically pure precursors,
such as, for example, [R,R]- or [S, S]-3,4-dihydroxy-
pyrrolidine, which should be protected on the
nitrogen by a protective group.
An example of the synthesis of an enantiomerically pure
amine, starting from enantiomerically pure 1-benzyl-3,4-
dihydroxy-pyrrolidine, which may be given is the
following reaction schemes
HO OH HO OH HO OCH2CHZOH
a,b ~~ c,d a
N N N
I I I
Bzl CO CO
R R
TosO OCHZCH20Tos ~ /~
0 N-BzI O N_H
N f ~ a.h
t N ~ N
CO
CO H
R
R
Le A 28 100 - 18 -
~~V~~~~.~
R = for example, ( CH3 ) 3C-O,
a: H2, Pd/A-carbon
b: acylation
c : NaH, HrCHZCOOC2H5 or c : CHz=CH-CH2Br, NaH,
d: LiBH4 d: 03, NaBH4,
a : tosyl chloride, NEt3,
f: benzylamine, xylene, reflex
g: hydrolysis
h: H2, PdJA-carbon
Examples of compounds of the formula (VI) which may be
mentioned are:
cis-2,8-diazabicycla[4.3.0]nonane,
cis-2-oxa-5,8-diazabicyclo[4.3.0]nonane,
trans-2-oxa-5,8-diazabicyclo[4.3.0]nonane,
S,S-2,8-diazabicyclo[4.3.0]nonane,
IR,6S-2-oxa-5,8~-diazabicyclo(4.3.0]nonane,
IS,6R-2-oxa-5,8-diazabicyclo(4.3.0]nonane,
IR,6R-2-oxa-5,8-diazabicyclo[4.3.0]nonane,
IS,6S-2-oxa-5,8-diazabicyclo[4.3.0]nonane,
The reaction of (V) with (VI), in which the compounds
(VI) can also be employed in the farm of their salts,
such as, for example, the hydrochlorides, is preferably
carried out in a diluent such as dimethyl sulphoxide,
N,N-dimethylformamide, N-methylpyrrolidone, hexamethyl-
phosphoramide, sulpholane, acetonitrile, water, an
alcohol such as methanol, ethanol, n-propanol or
isopropanol, glycol monomethyl ether or pyridine.
Mixtures of these diluents can also be used.
Le A 28 100 .- 19 --
~c~~3~i.Lt.~
Acid binders which can be used are all the customary
inorganic and organic acid binding agents, These
preferably include the alkali metal hydroxides, alkali
metal carbonates, organic amines and amidines. Those
which may be mentioned specifically as being particularly
suitable are: triethylamine, 1,4-diazabicyclo-
[2.2.2]octane(DABCO),1,8-diazabicyclo[5.4.0]under-7-ene
(DEU) or excess amine {vI).
The reaction temperatures can be varied within a
relatively wide range. In general the reaction is carried
out between about 20 and 200°C, preferably between 80 and
180°C.
The reaction can be carried out at normal pressure, but
also at elevated pressure. In general, it is carried out
at pressures between about 1. and 100 bar, preferably
between 1 and 10 bar.
When carrying out this process, 1 to 15 mol, preferably
1 to 6 mol of the compound (VI) are employed per mol of
the compound (V).
Examples of compounds of the formula (II) which can be
used both as race~mates and as enantiomerically pure or
diastereomerically pure compounds which may be mentioned
are:
Le A 28 100 - 20 -
XI
O
F / COORZ
H N ~,\ ~N~ (II)
I.I\ ~y~
,~ R i
N \
~ H
~Y
Ri R2 X1 Y A
Cyclopropyl CZHS H CH2 C-H
F-CHzCH2 H H CHZ C-F
Cyclopropyl C2H5 H CH, C-Cl
Cyclopropyl H H CH2 C-OCI-I3 .
Cyclopropyl H H CHZ C-CH3
Cyclopropyl CZHS H CHZ N
Cyclopropyl H Br CHZ C-F
Cyclopropyl H CI CHZ C-F
Cyclopropyl H CH3 CH2 C-F
Cyclopropyl C2H5 NHZ CHZ C-F
Cyclopropyl H H O C-H
Cyclopropyl C2HS H O C_F
C2H5 H H O C-CI
CH3 H H O C-OCH3
Cyclopropyl H H O C-CH3
Cyclopropy H H O ' N
l
Cyclopropyl H Br O C-F
Cyclopropyl H Cl O C-F
Cyclopropyl H CH3 O C-F
Cyclopropyl H NHZ O C-F
Le A 28 100 - 21 -
~~~~~~1~
Xl
0
F / COORZ
N \p ~ N ~ (II)
H H' I
\ N '''~ .:/"" R 1
~y H
RZ X1 y p
Cyclopropyl CH3 H CH2 C_H
Cyclopropyl CHZCH20H H CH2 C-F
cyclopropyl CHZCH20H H Cl-i2 C-Cl
Cyclopropyl H H CH2 C-OCH3
Cyclopropyl H H CH2 C-CH3
Cyelopropyl H H CH2 N
Cyclopropyl H gr CH2 C-F
Cyclopropyl H F CH2 C-F
Cyclopropyl H CH3 CH2 C-F
Cyclopropyl H NH2 CH2 C_I~
Cyclopropyl H 1-1 ~ C-H
Cy~lopropyl CH3 H O C-F
C2H5 H H ' p C-Cl
Cyclopropyl H H 0 C-OCH3
Cycloprop~'1 H ' ~-I- a' C-CH3
Cyclopropyl H H O ' N
Cy~lopropyl H Br 0 C-F
Cyclopropyl H Cl O C-F
Cyclopropyl 1-1 CH3 O C-F ,
C2H5 H NHZ Q C_F
Le A 28 100 _ 22 _
~7 ~ .r
~Oc~~~i~.
xl
0
F , COOR2
H N ~A ~ N ~ (II)
H\ ,,,
~Y-~H
R1 RZ X1 Y A
Cyclopropyl H H Cl-I2C-H
Cyclopropyl H H CHZ C-F
Cyclopropyl H H CHZ C-Cl
Cyclopropyl H H CHZ C-OCH3
Cyclopropyl H H CH2 C-CH3
Cyclopropyl H H CH2 N
Cyclopropyl H Br CHZ C-F
Cyclopropyl H F CH2 C-F
Cyclopropyl H CH3 CHZ C-F
Cyclopropyl H NHZ CHZ C-F
Cyclopropyl H H O C-H
Cyclopropyi H H O C-F
:;:"
Cyclopropyl H H O C-Cl
Cyclopropyl H H O C-OCH3
Cyclopropyl H H O C-CH3
Cyclopropyl H H O N
Cyclopropyl H Br O C-F
Cyclopropyl H F O C-F
Cyclopropyl H CH3 O C-F
Cyclopropyl H NHZ O C-F
Le A 28 100 - 23 -
~~~~~1~
xl
0
F / COORZ
H N \A ~ N ~ (ii)
H\ N~~ R t
~Y ,H
Rt RZ xl Y A
Cyclopropyl H H CH2 C-H
Cyclopropyl H H CHZ C-F
Cyclopropyl H H CH2 C-Cl
Cyclopropyl H H CHZ C-OCH3
Cyclopropyl H H CHZ C-CH3
Cyclopropyl H H CHZ N
Cyclopropyl H Br CH2 C-F
Cyclopropyl H F CI-i2 C-F
Cyclopropyl H CH3 C:H2 C-F
Cyclopropyl I-1 NH2 C:HZ C-F
Cyclopropyl H H O C-H
Cyclopropyl H H O C_F
Cy~lop~opyl H H O C-Cl
Cyclopropyl H H O C-OCH3
Cy~lopropyl H H O C-CH3
Cyclopropyl H H O N
Cyclopropyl H ~r O C-F
Cyclopropyl H F O C-F
Cyclopropyl H CH3 O C-F
Cyclopropyl H NH2 O C-F
Le A 28 100 - 2~
X1
0
F COORZ
N A N
HN R1
~Y
R1 RZ ~C1 Y A
2,4-DifluorophenylH Cl CH2C-F
2,4-DifluorophenylH CH3 CH2C-F
2,4-Difluo=ophenylH H CH2C-CII~
2,4-DifluorophenylH H O C-F
2,4-DifluorophenylH H O C-CI
4-Fluorophenyl H H 0 CH
2,4 DifluorophenylH H O N
2,4-DifluorophenylI-I H O C-OCH3
2,4-DifluorophenylH H O C-CHg
2,4-Difluorophenyll-i H CHZC-F
2,4-DifluorophenylH F CH2C-F
..>.~
?,4-Difluorophenyll-I H CHzC-Cl
~,4-Difluorophenyll-~ H O C-Cl
2,4-Difluo=ophenylH H CHZN
2,4-Difluorophenyll--I H O N
2,4-DifluorophenylH H O C-H
2,4-DiflucrophenylC2~--15 H O C-F
Le A 28 100 - 25 -
The starting compounds of the structures (III) and (IV)
are known. Examples which may be mentioned are:
chloroacetone,4-chloro-2-butanone,5-chloro-2-pentanone,
1-bromo-2-butanone, phenacyl chloride, methyl acrylates,
ethyl acrylates, acrylonitrile, methyl vinyl ketone,
dimethyl acetylenedicarboxylate, diethyl acetylene-
dicarboxylate, methyl propiolate and ethyl propiolate.
The reaction of (II) with (III) is preferably carried out
in a diluent such as dimethyl sulphoxide, N,P7-dimethyl-
formamide, N-methylpyrrolidone, hexamethylphosphoramide,
sulpholane, acetonitrile, water, an alcohol such as
methanol, ethanol, n-propanol or isor.wopa.nol, glycol
monomethyl ether or pyridine in the presence of an acid
binder. Mixtures of these compounds can also be used.
Acid binders which can be used are all the customary
inorganic and organic acid binders, These preferably
include the alkali metal hydroxides, alkali metal
carbonates, organic amines and am:idines. Those which may
be specifically mentioned as being particularly suitable
are: triethylamfne, 1,4-diazabicyclo[2.2.2]octane
(DABCO), 1,8-diazabicyclo(5.4.0]undec-7-ene (DBU) or
excess amine (VI).
The reaction temperatures can be varied within a
relatively wide range. In general the reaction is carried
out between about 20 and 200°C, preferably between 60 and
130°C.
Le A 28 100 - 26 -
~~~~~i.r~
The reaction can be carried out at normal pressure, but
also at elevated pressure. In general, it is carried out
at pressures between about 1 and 100 bar, preferably
between 1 and 10 bar.
When carrying out this process, 1 to 15 mol, preferably
1 to 6 mol, of the compound (III) are employed per mol of
the compound (II).
The reaction of (II) with the Michael acceptors (IV)
according to method ~ is preferably carried out in a
diluent such as acetonitrile, dimethyl sulphoxide,. N,A1-
dzmethylformamide, an alcohol such as methanol, ethanol,
propanol or isopropanol, or glycol monomethyl ether.
The reaction temperatures can be varied within a
relatively wide range. In general, the reaction is
carried out between about 20°C and about 150°C,
preferably between 40°C and 100°C.
The reaction can be carried out at normal pressure, but
also at elevated pressure. In general, the reaction is
carried out at pressures between 1 and 100 bar,
preferably between 1 and 10 bar.
When carrying out the process according to the invention,
1 to 5 mol, preferably 1 to 2 mol, of the compound (IV)
are employed per mol of the compound (II).
Le A 28 100 - 27 -
2~~~~~.~
The preparation of the acid addition salts of the
compounds according to the invention is carried out in a
customary manner, for example by dissolving the betaine
in aqueous acid and precipitating the salt with a water-
s miscible organic solvent such as methanol, ethanol,
acetone or acetonitrile. Equivalent amounts of betaine
and acid can also be heated in water or an alcohol such
as glycol monomethyl ether and then evaporated to dryness
or the precipitated salt filtered off with suction.
Pharmaceutically utilisable salts are understood as
meaning, for example, the salts of hydrochloric acid,
sulphuric acid, acetic acid, glycolic acid, lactic acid,
succinic acid, citric acid, tartaric acid,
methanesulphonic acid, 4-toluenesulphonic acid,
galacturonic acid, gluconic acid, embonic acid, glutamic
acid or aspartic acid.
The alkali metal or alkaline earth metal salts of the
carboxylic acids according to the invention are obtained,
for example, by dissolving the betaine in excess alkali
metal or alkaline earth metal hydroxide solution,
filtering off undissolved betaine and evaporating the
filtrate to dryness. Pharmaceutically suitable salts are
sodium, potassium or calcium salts. Py reacting an alkali
metal or alkaline earth metal salt with a suitable silver
salt such as silver nitrate, the corresponding silver
salts are obtained.
Apart from the active substances mentioned in the
examples, for example, the compounds listed in the
Le A 28 100 - 28 -
following tables (optionally in the cis- or traps-form)
can also be prepared by the processes describeda
X~
O
F / COOH
H N ~p~N~
R~
N
-H
lZ3 - X 1 A
C2H502C-CH2-CHZ- H C-F
CH302C-CH=CH- H C-F
NC-CH2-CH2- H C-F
5-Methyl-2-oxo-1,3-dioxol-4-yl-methyl- H C-F
CH3-CO-CH2- H C-CI
5-Methyl-2-oxo-1,3-dioxol-4-yl-methyl- H C-Cl
CH3-CO-CH2-CH2- H C-H
CH3-CO-CHz- H C-H
C2H502C-CH2-CI-Iz H C-H
C2Hs02C-CH=C_COZCZHs H C-H
CH30zC-CH=CF-I- H C-H
CaHsO2C-CH=CH- F C-F
CH3-CO-CH2CHz- NHZ C-F
CZHSOzC-CHzCH2- NHz C-F
CH3OZC-CH=C-CO2CH3 NHZ C-F
C2HSOZC-CH=C-COzC2Hs ~THZ C_F
i
Le A 28 100 - 29 -
R3
X1
O
F / COON
H N \A' _NJ
R3
~N
'H
X1 A
C2HSp2C_CH=CH- NH~ C-F
CH3-CO-CH2CH2- H N
CZH502C_CH2-CH2- H N
NC-CH2CHz- H N
C2HSOZC-CH=C-COZCZHS H N
CH302C-CH=CH- H N
CH3-CO-CHzCH2- CH3 C_H
CH3-CO-CH2_ CHI3 C-H
C2H502C-CHZCHZ- CH3 C-H
C2HSO2C-CH=C-CO2CZH5 CH3 C_H
CH30zC-CH=C-COZCH3 CI-I3 C_H
C2HSO2C-CH=CH- CH3 C-H
CH302C-CH=CH- CH3 C-F
C2HSOZC-CH=C-COZC2H5 CH3 N
Le A 28 100 - 30 -
C~ ~ e) .~
Xt
O
F / COOH
H N ~A~y~
R \ N''l,I , "r~~ H
R3 X1 A
CH3-CO-CI-i2CHz- H C-F
CH3-CO-CHZ- H C-F
C2H502C-CHZCHZ- H C-F
NC-CH2CH2- H C-F
CH302C-CH=CH- H C-F
CH302C-CH=C-COzCH~ H C-F
C2HSO2C-CH=C-CO2C2H5 H C-F
5-Methyl-2-oxo-1,3-dioxol-4-yl-methyl- H C-F
CH3-CO-CHZCH2- H C-C1
CH3-CO-CHZ- H C-Cl
- C2H5O2C-CH2CH2- H C-Cl
NC-CH2CH2- H C-CI
CH30zC-CH=CH- H C-CI
CH3O2C-CH=C-CO2CH3 H C-Cl
C2Hs02C-CH=C-CO2C2E15 H C-C1
5-Methyl-2-oxo-1,3-dioxol-4-yl-methyl- H C-Cl
)Je A 2>3 100 - 31 -
XI
0
F / COOH
N A~N
H
N~~~" H
R3 Xl A
C2H502C-CHI C-C02CZH5 H C H
C2H502C-CH=CH- H C-H
CH302C-CH=CH- F C-F
C2I-ISOZC-CH2-CH2_ F C-F
C2HSOZC-CH=CH- NHZ C-F
CZHSO~C-CH=C-COZCZHS NHZ C-F
CEI3-CO-CH2CH2- CHI C-H
C2HSOZC-CH=CH- CH3 C-H
(.zH5O2C-CH=C-CO2C2H5 CH3 C-H
I
CH3O2C-CH=C-CO2C2H5 CH3 N
C~I3:C0-CH2CH2- H C-OCH3
C2H502C-CH=CH- H C-OCH3
C2Hg02C-CH=CH H N
NC-CH2CH2 H N
CH3-CO-CH2CH2 H N
Le A 28 100 - 32 -
CA 02086914 2003-05-15
23189-7450
A specific process of the invention is a process
for preparing 1-cyclopropyl-7-([S,S]-2,8-
diazabicyclo[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-8-methoxy-
4-oxo-3-quinolinecarboxylic acid which comprises reacting 1-
cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-
quinolinecarboxylic acid with (+)-[S,S]-2,8-
diazabicyclo[4.3.0]-nonane, followed, if required, by
converting the obtained compound into a pharmaceutically
utilisable hydrate or acid addition salt thereof or an
alkali metal, alkaline earth metal, silver or guanidinium
salt of the underlying carboxylic acid.
The invention also provides pharmaceutical and
antibacterial compositions comprising a quinolone- or
naphthyridone-carboxylic acid compound of the invention or a
pharmaceutically utilisable hydrate or acid addition salt
thereof or an alkali metal, alkaline earth metal, silver or
guanidinium salt of the underlying carboxylic acid; and a
pharmaceutically acceptable carrier, diluent or excipient.
The invention also provides uses of the compounds
and compositions of the invention for: preventing,
controlling or curing an infectious disease; as an additive
for the preservation of an organic or inorganic material; or
for the preparation of a medicament for the prevention,
control or cure of an infectious disease.
The invention also provides a commercial package
comprising a compound or composition of the invention
together with instructions for use thereof in the treatment
of an infectious disease.
- 32a -
The compounds according to the invention have potent
antibiotic activity and exhibit, together with low
toxicity, a broad antibacterial spectrum against gram-
positive and gram-negative microorganisms, in particular
against enterobacteria; especially even against those
which are resistant to various antibiotics, such as, for
example, penicillins, cephalosporins, aminoglycosides,
sulphonamides and tetracyclines.
These useful properties make possible their use as
chemotherapeutic active substances in medicine and also
as substances for the preservation of inorganic and
organic materials, in particular of organic materials of
all types, for example polymers, lubricants, dyes,
fibres, leather, paper and wood, of foodstuffs and of
water.
The compounds according to the invention are active
against a very broad spectrum of mircoorganisms. With
their aid, gram-negative and gram-positive bacteria and
bacteria-like microorganisms can be controlled and the
diseases produced by these pathogens can be prevented,
ameliorated and/or cured.
The compounds according to the invention are
distinguished by increased activity on dormant and
resistant microorganisms. In the case of dormant
bacteria, e.g. bacteria which show no detectable growth,
the compounds act at concentrations which are far below
those of substances known hitherto. This relates not only
Le A 28 100 - 33 -
to the amount to be employed, but also to the rate of
destruction. It was possible to observe results of this
type with gram-positive and gram-negative bacteria, in
particular with Staphylococcus aureus, Pseudomonas
aeruginosa, Enterocaccus faecalis and Escherichia coli.
The compounds according to the invention also show
surprising increases in activity against bacteria which
are classified as less sensitive to comparable
substances, in particular resistant Staphylococcus
aureus, Escherichia coli, Pseudomonas aeruginosa and
Enterococcus faecalis.
The compounds according to the invention are particularly
active against bacteria and bacteria-like microorganisms.
They are therefore particularly highly suitable for the
prophylaxis and chemotherapy of local and systemic
infections in human and veterinary medicine which axe
caused by these pathogens.
The compounds are also suitable for controlling
protozoonoses and helminthoses.
The compounds according to the invention can be used in
various pharmaceutical preparations. Preferred
pharmaceutical preparations which may be mentioned are
tablets, coated tablets, capsules, pills, granules,
suppositories, solutions, suspensions and emulsions,
pastes, ointments, gels, creams, lotions, powders and
sprays.
Le A 28 100 - 3~ -
The table below confirms the surprising advantages of the
compounds according to the invention compared with
ciprofloxacin in the Staphylococcus aureus-infected mouse
models
Tablet Activity in Staph. aureus infection
in the mouse (mg/kg)
Substance p.o. s.c.
Ciprofloxacin 80 80
Example 27 10 2.5
Example 29A 5 5
Example 31 10 10
Example 33 10 5
Example 35 2.5 2.5
The compounds according to the invention, compared to
known structurally similar compounds, show an improved
antibacterial action, in particular with anaerobic
microorganisms. O
F COOH
I
N
CI
Component according to the invention as in Ex.ampls 2Bs A
CH3
R = N , disclosed in EP-A-0,350,733s B
~~N -
Ciprofloxacin C
Le A 28 100 - 35 -
~~~~~:~.<.~
'1'F3D1 -
~
Compound
Species Strain A B C
Bacteroides ES 25 0,25 1 8
fragilis DSM 2151 0.25 0.5 4
Clostridium 1024027 0.125 0.5 0.5
perfringens
Bact. DSM 2079 0.5 2 $
thetaiotaomicron ~
(MIC values in ~sg/ml~ agar dilution test ir. the
multipoint inoculator (Denley); isosensitest agar).
Preparation of the.precursors
Example A
[S:S]I-2,8-Diazabicycloj4.3.Olnonane
H
H
N
NH
H
1) [S,S]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonane
Method T:
a) Separation of the diastereomeric salts:
Le A 28 100 - 36 -
3.0 g (20 mmol) of D-(-)-tartaric acid are dissolved
in 10 ml of dimethylformamide by heating to 80°C and
the solution is treated with a solution of 2.16 g
(10 mmol) of cis-8-benzyl-2,8-diazabicyclo[4.3.0]-
nonane in 3 ml of dimethylformamide. The mixture is
stirred at 0°C for 1 hour, and the product is
filtered off with suction and washed with
dimethylformamide and methoxyethanol.
Yield: 1.93 g,
Melting point: 146-151°C,
[a]D3 = -19.3° (c = 1, 1H20) . .
Diastereomerically pure [S,S]-8-benzyl-2,8-
diazabicyclo[4.3.0]nonane D-tartrate is obtained by
a single recrystallisation from methoxyethanol.
[«]p3 = -22.7° (c = 1, HZO) .
Melting point: 148-154°C.
b) Liberation of the base:
40 g of [S,S[-8-benzyl-2,8-diazabicyclo[4.3.0]nonane
D-tartrate are dissolved ire 250 ml of water and
treated with 32 g of 45 ~ strength sodium hydroxide
solution. The precipitated oil is taken up in 150 ml
of tart-butyl methyl ether, the aqueous phase is
extracted again with 150 ml of tent-butyl methyl
ether and the comba.ned organic phases are
concentrated after drying over sodium sulphate. The
Le A 28 100 - 37 -
residue is then distilled in vacuo.
Yield: 18.5 g of [S,S]-8-Benzyl-2,8-diazabicyclo-
[4.3.0]nonane,
Boiling point: 107-109°C/0.1 mbar,
[~]o° = 17.3° (undiluted).
Method 3I:
75.0 g (0.5 mol) of L-(°~)-tartaric acid are dissolved in
250 ml of di.methylformamide at 80°C and 54.1 g (0.25 mol)
of cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane are added
dropwise as a solution in 75 ml of dimethylformamide. The
mixture is slowly cooled to 20°C and the crystal
suspension is stirred for 1 hour. The crystals ([R,R]-8-
benzyl-2,8-diazabicyclo[4.3.0]nonane L-tartrate) are
filtered off with suction and the filtrate is
concentrated on a rotary evaporator. The residue is
dissolved in 500 m1 of mater and wor3ced up as described
in Method I using 63 g of 45 ~ strength sodium hydroxide
solution.
Yield: 25.2 g of [S,S]-8-benzyl-2,8-diazabieyclo[4.3.0]-
nonane;
the product contains 3.6 ~ of the R,R-enantiomer
(determined by gas chromatography after derivatisation
with menthyl chloroformate).
The compound can be reacted with D-(-)-tartaric acid
Le A 28 100 - 38 -
according to Method I to give diastereomerically pure
[S,S]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane D-tartrate.
Recrystallisation in this case is not necessary.
Method III:
73.6 g (0.34 mol) of cis-8-benzyl-2,8-diazabicyclo-
[4.3.0]nonane are added dropwise at 80 to 90°C as a
solution in 111 ml of dimethylformamide to a solution of
102 . 9 g ( 0 . 685 mol ) of L ( * ) -tartaric aeid in 343 ml of
dimethylformamide. The mixture is seeded with [R,R]-8-
benzyl-2,8-diazabicyclo[4.3.0]nonane L-tartrate and
slowly cooled to an internal temperature of 18°C. The
crystals are filtered off with suction, and the filtrate
is seeded with [S,S]-8-benzyl-2,8-diazabicyelo[4.3.0]-
nonane L-tartrate and stirred until it has crystallised
completely. (After concentration and liberation of the
base as described in Method :C, [S,S]-8-benzyl-2,8-
diazabicyclo[4.3.0]nonane D-tartrate can be obtained from
the mother liquor by purification with D-tartaric acid).
The product is then filtered off with suction, washed
20. with dimethylformamide and isopropanol and dried in air.
The crystals are recrystallisecl from 88 ~ strength
ethanol. 52 g of [S,S]-8-benzyl-2,8-diazabicyclo[4.3.0]-
nonane L-tartrate trihydrate are obtained.
Melting points 201-204°C,
[«]D3 = +5.2° (c = 1, Hz0) .
The salt can be processed as described in Method I
Le A 28 100 -'39 -
(liberation of the base) to give enantiomerica~.ly pure
[S,S]-8-benzyl-2,8-diazabicyclo[4.3.0]n~anane.
Method IV:
a) Separation of enantiomers of cis-8-benzyl-7,9-dioxo
2,8-diazabicyclo[4.3.0]nonane to give jlS,GR]-$
benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane.
The procedure is analogous to Example H (Method
II/a), using I~-(-)-tartaric acid as the chiral
auxiliary reagent, or the procedure is as follows:
Mother liquor and washing liquor from [1R,6S]-8-
benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane L-
tartrate (from Example B, Method IT/a) are con-
centrated together, the residue is taken up in water
and the solution is extracted three times with
toluene. The toluene phases are discarded. The
aqueous phase is treated with saturated sodium
hydrogen carbonate solution until a pH of 7 to 8 is
obtained, then extracted four times with methylene
chloride. The combined methylene chloride phases are
dried over magnesium sulphate and concentrated.
Yield: 14.4 g (60 ~ of theory of the originally
employed racemic cis-8-benzyl-7,9-dioxo-2,8-diaza-
bicyclo[4.3.0]nonane).
ja]D3 = -4.5° (c = 5, ethanol).
Le A 28 100 - 40 -
These 14.4 g (59 mmol) axe crystallised from 120 ml
of ethanol analogously to Example B (Method II/a)
using 8.6 g (57 mmol) of D-(-)-tartaric acid. .
Yield: 8.9 g (77 ~ of theory) of [1S,6R]-8-benzyl-
7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane D-tartrate.
[c~JD3 = _46.2° (c = 0.5, 1N HC1);
after recrystallisation from an ethanol/glycol
monomethyl ether mixture a further purification is
carried out:
[a]D3 = -59.3° (c = 0.5, aP1 HC1) .
5.0 g (12.7 mmol) of the diastereomerically pure
tartrate obtained in this manner were converted, as
described in Example B, Method II/a, into the free
amine:
Yield: 3.0 g (96 ~ of theory) of [1S,6R]-8-benzyl-
7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane,
Melting point: 60-61°C,
[a]n3 = _22.2° (c = 5, ethanol).
An enantiomer excess of 96.6 ~ ee was determined by
gas chromatography after derivatisation with menthyl
chloroforxnate.
b) Reduction of[1S,6R]-8-benzyl-7,9-dioxo-2,8-diazabi-
cyclo[4.3.0]nonane to [S,S]-8~benzyl-2,8-diazabi-
cyclo[4.3.0]nonane.
Le A 28 100 -- 41 -
~Q~~a~
The procedure is analogous ~to Example s (Method zx,
b), [1S,6R]-8-benzyl-7,9-dioxo-2,8-diazabicyclo-
[4.3.0]nonane being, however, employed as the
starting material.
The crude product obtained after working up proved
to be [S,S]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane
on derivatisation with menthyl chloroformate.
Racemisation was thus observed during the reduction.
2) [S,S]-2,8-Diazabicyclo[4.3.0]nonane
28.4 g (0.131 mol) of [S,S]-8-benzyl-2,8-diazabi-
cyclo[4.3.0]nonane are hydrogenated at 90°C and 90
bar in the course of 5 hours over 5.8 g of palladium
on active carbon (5$) in 190 ml of methanol. The
catalyst is then filtered off with suction and
washed with methanol, and the filtrate is
concentrated on a rotary evaporator. The residue is
distilled without fractionation.
Yield: 15.0 g (90.5 ~ of theory) of [S,S]-2,8-
diazabicyclo[4.3.0]nonane,
Boiling point: 44-59°C/0.18 mbar,
[«]DZ = _2.29° (undiluted),
ee > 99 $ ( determined by gas chromatography after
derivatisation with Mosher's reagent).
Method V:
3.75 g (25 mmol) of Z-(+)-tartaric acid are
Le A 28 100 - 42 -
~~lor~~~.~~.
initially introduced in solution in 50 ml of
dimethylformamide at 80C and 10.82 g (50 mmol)
of
cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane are
added
dropwise as a solution in 15 ml of dimethylformamide.
The mixture is seeded with [R,R]-8-benzyl-2,8-diaza-
bicyclo[4.3.0]-nonane L-tartrate and stirred for
one
hour at about 72 C to complete seed crystal formation.
This is then slowly cooled to 15C, and the crystals
are filtered off with suction and washed twice
with
13 ml of dimethylformamide in each case. The
combined filtrates are heated to 80C and treated
with a further 3.75 g (25 mmol) of L-(+)-tartaric
acid. The mixture is additionally heated to 119
C
until a clear solution is formed, and again slowly
cooled to room temperature with seeding with [S,S]-_
8-benzyl-2,8-diazabicyclo[4.3.0]-nonane L-tartrate.
The crystals are filtered off with suction, washed
successively with dimethyl-formamide, 2-methoxy-
ethanol and ethanol and dried in air.
Yield: 9.59 g
Melting point: 188 to 192C.
The crystals are recrystallised from 95 ml of 80
strength ethanol. 8.00 g of [S,S]-8-benzyl-2,8-
diazabicyclo[4.3.0]nonane L-tartratetrihydrate(76
of theory) are obtained which melts at 112 to 118C
with foaming, then resolidifies and melts again
at
199 to 201C.
[ a ] D3 4 . 5 ( c = 1, water )
ee: 98.0 ~ (determined by gas chromatography after
derivatisation with menthyl
Le A 28 100 - 43 -
2~~~~~~.~
chloroformate).
Example B
[R,R1-2.8-Diazabicycloj4.3.OLnonane
H
H
N
NH
H
1) [R,R]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonane
Method I:
The crystals of [R,R]-8-benzyl-2,8-diazabicyclo[4.3.0]-
nonane obtained according to Example A, Method II
(4~.2 g) are washed with dimethylformamide and_
methoxyethanol and recrystallised from 300 ml of
methoxyethanol. 45.6 g of enantiomerically pure [R,R]-8-
benzyl-2,8-diazabicyclo[4.3.0]nonane L-tartrate are
obtained (enantiomer purity determined by gas
chromatography after derivatisation with menthgl
chloroformate).
I5 Melting point: 121-I24°C,
[a]D~ = +22.3° (c = 1, Ha0) .
The salt (44.5 g) is converted into the free base as
described in Example A, Method Ib. 20.2 g of [R,R]-8-
benzyl-2,8-diazabicyclo[4.3.0]nonane are obtained.
Boiling point: 107-111°C/0.04 mbar,
Le A 28 100 - 44 -
[a]D° _ _17.5° (undiluted).
Method II
a) Separation of enantiomers of cis-8-benzyl-7,9-dioxo-
2,8-diazabicyclo[4.3.0]nonane to give [1~t,6S]-8-
benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane
24.1 g (98.8 mmol) of cis-8-benzyl-7,9-dioxo-2,8-
diazabicyclo[4:3.0]nonane are heated to reflux with
stirring in a mixture of 410 ml of ethanol and 25 m1
of acetonitrile in a three-necked flask. 14.8 g
(98.8 mmol) of L-(+)-tartaric acid are then added at
once. After all the tartaric acid has completely
dissolved, the heating is first turned off, but the
flask is left in the oil bath. When the system has
cooled until the solution no longer boils, the
stirrer is turned off. Crystallisation and addition
of seed crystals takes place at a temperature of
50°C. After standing overnight and cooling to room
temperature, the precipitated crystals are filtered
off with suction and washed with a little
ethanol/petroleum ether (1:1) and dried at 80°C for
2 hours.
Yields 9.8 g (50 ~k of theory) of [1~,6S]-8-benzyl-
7,9-dioxo-2,8-dl.azabicyclo[4.3.0]nbnane
L-fiaxtrate, [«]p3 = +47.7° (c = 0.5, lId HCl).
the compound can be further purified by
Le A 28 100 - 45 -
recrystallising twice from a mixture of ethanol and
glycol monomethyl ethers
_ +58.6° (c = 0.5, 1N HC1).
1H-NMR (DMSO): 7.22-7.35 (2m, 2H, aryl-H); 4.55 (s,
2H, benzyl-CHZ); 4.28 (s, 2H, tartaric acid-CH); 3.91
(d, 1H, 1-CH); 2.97 (dd, 1H, 6-CH); 2.53-2.66 (m,
2H, 3-cH2) ;, 1. 78 and 1. 68 ( 2m, 2H, 5-CHZ) ; 1.42 and
1.28 ppm (2m, 2H, 4-CHZ).
C18H22N2~8 ( 394 )
Calculated: C 54.4 H 5.6 N 7.1 0 32.5
Found: C 54.7 H 5.8 N 7.1 0 32.4
The determination of the absolute configuration was
carried out by means of an X-ray structural.
analysis:
H
H O HOw'C02H
HO'~ COZH
H O
3.6 g (9.1 mmol) of the diastereomerically pure
tartrate obtained in this manner are dissolved in
water to liberate the base and treated with
saturated sodium hydrogen carbonate solution until
a pH of 7 to 8 is obtained. The aqueous solutzon is
extracted four times with 20 ml of methylene
chloride each time. The combined methylene chloride
phases are dried over magnesium sulphate and
Le A 28 100 - 46 -
~~ ~~:~~.
Concentrated.
Yield 2.2 g (99 ~ of theory) of jlR,6S]-8-benzyl-
7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane
Melting points 60-61°C,
[ « ] D3 = +21. 8 ° ( c = 5 , ethanol ).
An enantiomer excess of 93.8 ~ ee was determined by
gas chromatography after derivatisation with menthyl
chloroformate.
b) Reduction of [18,65]-8-benzyl-7,9-dioxo-2,8-
diazabicyclo[4.3.0]nonane to jR,R]-8-benzyl-2,8-
diazabicyclo[4.3.0]nonane
In a heated flask, 0.34 g (9 mmol) of lithium
aluminium hydride is introduced under NZ in 18 ml of
anhydrous tetrahydrofuran and 0.73 g (3 mmol) of
jlR,6S]-8-benzyl-?,9-dioxo-2,8-diazabicyclo[4.3.0]-
nonane is added drnpwise as a solution in 3 ml of
anhydrous tetrahydrofuran. The mixture is then
boiled for 16 hours with reflux condensation.
Woxking up is carried out by dropwise edition of
0.34 ml of water in 10 m~ of tetrahydrofuran,
0.34 ml of 10 ~S strength sodium hydroxide solution
and 1.02 ml of water. The precipitate is filtered
off with suction and washed with tetrahydrofuran,
and the filtrate is concentrated. 0.? g of crude
[R,R]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane remains
(GC purity: 99 ~k).
Le A 28 100 - 47 -
~i~~ ~r~
It was not possible to determine any racemisation
during the gas chromatographic determination of the
enantiamer purity using menthyl chloroformate.
2) [R,R]-2,8-diazabicyclo[4.3.0]nonane
19.4 g (0.09 mol) of [R,R]-8-benzyl-2,8-diaza-
bicyclo[4.3.0]nonane are hydrogenated according to
the procedure of E~cample A, 2.
Yield: 9.61 g (85 ~ of [R,R]-2,8-diazabicyclo-
[4.3.0]nonane,
Boiling point: 45-58°C/0.08 mbar,
[,a]D3 = +2.30° (undiluted).
Example C
iS.S]I-2-Methyl-2.8-diazabicyclo~4.3.0]nonane
CH3
IH
N
C~NH
H
1) [S,S]-8-Benzyl-2-methyl-2,8-diazabicyclo[4.3.0]-
nonane
X3.2 g (0.2 mmol) of [S,S]-8-benzyl-2,8-diazabi-
cycla [ 4 : 3 . 0 ] nonane are treated with 20 ml of 37 ~
formaldehyde solution, 40 ml of water and 24 g of
acetic acid and the mixture is hydrogenated over
Le A 28 100 - 48 -
2 g of palladium on active carbon (5 $) at 20°C and
20 bar fox 10 hours. The catalyst is then filtered
off with suction, the filtrate is rendered alkaline
with potassium carbonate and the product is
extracted with tart-butyl methyl ether. After drying
over sodium sulphate, the mixture is concentrated
and the residue is distilled in vacuo.
Yield: 14.8 g,
Boiling point: 114-124°C/0.14 mbar.
2) [S,S]-2-Methyl-2,8-diazabicyclo[4.3.0]nonane
12.9 g (56 mmol) of [S,S]-8-benzyl-2-methyl-2,8-
diazabicyclo[4.3.0]nonane are hydrogenated over
1.1 g of palladium on active carbon (5 ~) at 90°C
and 90 bar in 90 ml of methanol. The mixture is then
filtered, the filtrate is concentrated on a rotary
evaporator and the residue is distilled in vacuo.
Yield: 5.5 g of enantiomeyically pure [S,S]-2-
methyl-2,8-diazabicyclo[4.3.0]nonane (detection by
derivatisation with Mosher's reagent),
Boiling point: 78-81°C/14 mbar.
Le A 28 100 - 49 -
Example D
jR.Rl1-2-Methyl-2r8-diazabicvcloj4.3.0],nonane
CH3
IH
N.
C~NH
H
The compound is prepared by the v~~orking instructions
described in Example C, starting from 43.2 g (0.2 mol) of
[R,R]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane.
Yield: 4.9 g of [R,R]-2-methyl-2,8-diazabicyclo[4.3.0]-
nonane.
Boiling point: 30-33°C/0.12 mbar.
Example E
cis-7 , 9-nioxo-8-t j 1S,L-~-_phenyl-ethyl l-2~, 8-
diazabicyclo~4.3.0]nonane
O
~N - CI-I- Ph
CH3
H
1) N-([1S]-1-Rhenyl-ethyl)pyridine-2,3-diearboximide
74.5 g (0.5 mol) of pyridine-2,3-dicarboxylic
anhydride are initially introduced at 20°C in
solution in 500 ml of dioxane and 60.5 g (0.5 mol)
T.e A 28 100 - 50 -
~0~~~1~
of S-(-)-1-phenyl-ethylamine are added dropwise,
whereupon the temperature rises to 33 ° C . The mixture
is stirred for a further 1 hour and then
concentrated on a rotary evaporator, and residual
solvent is removed at 40°C10.1 mbar. The residue is
taken up in 245 g (2.4 mot) of acetic anhydride, and
the solution is treated with 4.9 g (0.06 mol) of
anhydrous sodium acetate and stirred at 100°C for
1 hour. After cooling, the mixture is poured onto 1
1 of ice-water while stirring well, and the
precipitate is filtered off with suction, washed
with cold water and hexane and dried in air.
The crude product (114 g, Melting points 112-114°C)
is recrystallised from 285 ml of methanol.
Yields 96.3 g (76 ~),
Melting point: 115-117°C,
[a]p2 - _46.9° (c = 2, ethanol).
2) cis-7,9-Dioxo-8-([1S]-1-phenyl-ethyl)-2,8-
diazabicyclo[4.3.0]nonane
79.7 g (0.316 mol) of N-([1S]-1-phenylethyl)-
pyridine-2,3-dicarboximide are hydrogenated over
l0 g of palladium on active carbon (5 $ strength) at
90°C/100 bar in 600 ml of tetrahydrofuran. The
catalyst is filtered off after completion of the
absorption of hydrogen and the filtrate is
completely concentrated. 83.7 g of a viscose residue
are obtained. v
Le A 28 100 - 51 -
,t
~~~u i.~~~
Puritys 95 ~ strength,
1H-~tMR (CDC13, 200 MHz ) : 1. 4-1. 7 (m, 3H) ; 1. 82 and
1.83 (2d, 3H); 1.9-2.05 (m, 1H); 2.28 (broad s, 1H);
2.54-2.86 (m, 3H); 3.77 (d, 1H); 5.39 (q. 1H); 7.24
7.48 ppm (m, 5H).
Example F
cis-2-Oxa-5,8-diazabicvclof4.3.Olnonane
H
I
N cis
0
1) trans-1-Benzoyl-3-bromo-4-(2-hydroxyethoxy)-
pyrrolidine
95 g (0.55 mol) of 1-benzoyl-3-pyrroline are
dissolved in 380 g of ethylene glycol and 101 g
(0.57 mal) of N-bromosuccinimide are added in 5 g
portions in the course of 2 hours. The mixture is
then stirred overnight at room temperature, poured
into water and extracted with methylene chloride,
and the solution is dried over. magnesium sulphate
and concentrated. The residue (188 g) was
chromatographed on silica gel using ethyl acetate.
Yields 136.5 g (78 ~ of theory),
Purity by GCs 99 ~.
Le A 28 100 - 52 -
2) traps-1-~enzoyl-3-bromo-4-(2-tosyloxyethoxy)-
pyrrolidine
92 g (0.239 mol) of traps-1-benzoyl-3-bromo-4-(2-
hydroxyethoxy)-pyrrolidine, 32 g (0.316 mol) of
triethylamine and 1 g of ~-dimethylaminopyridine are
dissolved in 750 ml of toluene and 60 g (0.31 mol)
of tosyl chloride in 450 ml of toluene are added
dropwise. The mixture is stirred at room temperature
for two days, water is added, and the aqueous phase
is separated off and extracted with toluene. The
toluene solutions are washed with 10 ~S strength
hydrochloric acid, dried over magnesium sulphate and
concentrated, the residue is dissolved in ethyl
acetate and the solution is filtered through silica
gel. The filtrate is concentrated.
Yield: 125 g (91 ~ of theory).
The thin layer chromatogram shows a homogeneous
compound.
3) cis-8-Benzoyl-5-benzyl-2-oxa-5,8-diazabi-
cyclo(4.3.0]nonane
124 g (0.265 mol) of traps-1-benzoyl-3-bromo-4-(2-
tosyloxyethoxy)-pyrrolidine are heated under reflux
overnight with 86 g (0.8 mot) of benzylamine in
1.5 1 of xylene, the salts of benzylamine are
filtered off with suction and the filtrate is
concentrated.
Le A 28 100 - 53 -
~~s~~l.~~
Crude yield: 91.2 g.
4) cis-5-~enzyl-2-oxa-5,8-diazabicyclo[4.3.0]nonane
91 g (0.265 mol) of cis-8-benzoyl-5-benzyl-2-oxa-
5,8-diazabicyclo[4.3.0]nonane are heated under
reflux overnight with 200 ml of concentrated
hydrochloric acid and 140 ml of water. After
cooling, the benzoic acid is filtered off with
suction, the filtrate is concentrated to half the
volume, the solution .is rendered alkaline with
ZO potassium carbonte and extracted with chloroform,
the extract is dried over potassium carbonate and
concentrated, and the residue is distilled.
Yield: 30.7 g (48.8 ~ of theory),
foiling point: 134-142°C/0.6 mbar,
Purity by GC: 92 ~.
5) cis-2-Oxa-5,8-diazabicyclo[4.3.0]nonane
dihydrochloride
26 g (0.11 mal, 92 ~ strength) of cis-5-benzyl-2-
oxa-5,8-diazabicyclo[4.3.0]nonane in 180 ml of
ethanol and 19 ml of cancentrated hydrochloric acid
are hydrogenated in 3 g of palladium/active carbon
(10 ~ Pd) at 100°C and 100 bar. The catalyst is
filtered off with suction, the filtrate is
concentrated and the separated crystals are dried in
a dessicator over phosphorus pentoxide.
Le A 28 100 - 54 -
Yield: 17.1 g {77 ~ of theory),
Melting point: 244-250°C.
Example G
Separation of enantiomers of cis-5-benzyl-2-oxa-5,8-
diazabicyclo[4.3.0]nonane
150.1 g (1 mol) of D-(-)-tartaric acid are initially
introduced into 700 ml of methanol at 60 to 65°C and
218.3 g (1 mol) of cis-5-benzyl-2-oxa-5,8-diazabicyclo-
[4.3.0]nonane are added dropwise as a solution in 300 ml
ZO of methanol. The mixture is then slowly allowed to cool
to about 49°C until the solution becomes cloudy, and is
seeded with crystals of 1R,6S-5-benzyl-2-oxa-5,8-diaza-
bicyclo[4.3.0]nonane D-tartrate obtained in a prior ex-
periment, stirred for 30 minutes at this temperature for
seed crystal formation and then slowly cooled down to
0 to 3°C. After filtering off with suction, the solid is
washed with ~ mixture of 200 m1 of-_ ethanol and 100 ml of
methanol cooled to 0°C and then 3 times with 300 ml of
ethanol in each case and the product is then dried in
air.
Yield: 160.3 g of 1R,6S-5-benzyl-2-oxa-5,8-diazabicyclo-
[4.3.0]nonane tartrate (87 ~ of theory)
Melting point: 174.5 to 176.5°C
ee > 97 ~ (after derivatisation with 1-phenyl-ethyl
isocyanate and HPLC analysis).
Le A 28 100 - 55 -
[ cx ] D3 = +24 . 0 ° ( c = 1, methanol ) .
156.9 g of the 1st crystallisate are recrystallised from
1,500 ml of methanol.
Yield: 140.0 g (89 ~ recovered)
Melting point: 1?6 to 177°C
[«]D3 = +25.2° (c = 1, methanol).
The methanolic mother liquor from the 1st crystallisation
is concentrated on a rotary evaporator. The syrupy
residue (236 g) is dissolved in 500 ml of water, adjusted
to pFi 12 to 13 with 250 ml of 6N sodium hydroxide
solution, extracted 3 times with 350 ml of toluene each
time, and the extracts are dried over sodium carbonate
and concentrated in vacuo. The residue, 113.1 g of a
brown oil which, according to gas chromatographic
investigation, contains 97 ~ of cis-5-benzyl-2-oxa-5,8-
diazabicyclo[4.3.0]nonane, is employed without purifica-
tion for the preparation of the 1S,6R-enantiomer.
113.1 g (0.518 mol) of crude concentrated 1S,6R-5-benzyl-
2-oxa-5,8-diazabicyclo[4.3.0]nonane axe dissolved in
155 ml of methanol and added dropwise to a boiling
solution of 77.8 g (0.518 mol) of L-(+)-tartaric acid in
363 m1 of methanol. A crystal magma is gradually formed
during the dropwise addition. The mixture is stirred at
60°C for 1 hour and then slowly cooled to 0°C in the
course of 2 hours. The crystals are filtered off with
suction and washed with a 2:1 mixture of ethanol and
methanol cooled to 0°C and then 3 times with ethanol. The
Le A 28 100 - 56 -
product is then dried in air.
Yield: 145.5 g of 1S,6R-5-benzyl-2-oxa-5,8-diazabi-
cyclo[4.3.0]nanane L-tartrate (79 ~ of theory)
Melting point: 174.5 to 176.5°C
ee > 97 ~ (after derivatisation with 1-phenyl-ethyl
isocyanate and ~IPLC analysis)
[a]D3 ~ -24.0° (c = 1, methanol).
Liberation of the enantiomerically pure bases:
144 g (0.39 mol) of 1S,6R-5-benzyl-2-oxa-5,8-diazabi
cyclo[4.3.0]nonane tartrate are dissolved in 250 ml of
water and 175 ml (1.05 mol) of 6 N sodium hydroxide solu
tion are added. The deposited oil is taken up in 500 ml
of toluene, the organic phase is separated off and the
ac;ueous phase is extracted a further 3 times with 250 ml
of toluene in each case. The combined organic phases are
dried over sodium carbonate, filtered and cancentxated on
a rotary evaporator. The residue is distilled through a
cm Vigreux column under a high vacuum.
Yield: 81.6 g (96 ~ of theory) of 1S,6R-5-benzyl-2-oxa-
20 5,8-diazabicyclo[4.3.0]nonane
Boiling point: 120 to 139°C/0.04 to 0.07 mbar
Purity: 100 ~ determined by gas chromatography
Density: b "' 1.113 g/ml
- _60,9° (undiluted).
Distillation residue: 0.12 g
In the same manner, 76.0 g (93 ~ of theory) of 1R,6S-5-
benzyl-2-oxa-5,8-diazabicyclo[4.3.0]nonane are obtained
from 139.2 g (0.37b mot) of IR,6S-5-benzyl-2-oxa-5,8-
diazabicyclo[4.3.0]nonane tartrate.
Le A 28 100 - 57 -
[a]D3 = +61.2° (undiluted).
The separation of enantiomers described for cis-5-benzyl-
2-oxa-5,8-diazabicyclo[4.3.0]nonane can also be carried
out analogously with traps-5-benzyl-2-oxa- 5,8-
diazabicyclo[4.3.O~nonane to give R,R- and S,S-5-benzyl-
2-oxa-5,8-diazabicyclo[4.3.0]nonane.
Example H
1) tart-Butyl 3S,4S-4-allylnxy-3-hydroxypyrrolidine-1-
carboxylate
16.5 g (0.55 mol) of 80 ~ strength I~aH are initially
introduced into 500 ml of absolute dioxane and a
solution of 107,5 g (0.53 mol) of tart-butyl S,S-
3,4-dihydroxypyrrolidine-1-carboxylate
(DE-A-3,403,194) dissolved hot in absolute dioxane
is added dropwise at 60C. The mixture is stirred at
60C for 1 hour and 64 g (0.53 mol) of allyl bromide
are then added dropwise . The mixture is then stirred
at 60C for three haurs. It is concentrated and the
residue is dissolved in 200 ml of water and 600 ml
of methanal. The solution is extracted three times
with 200 mi ~f pentane each time, the methanol is
stripped off on a rotary evaporator, the residue is
diluted with 200 ml of water and the mixture is
extracted with methylene chloride. The methylene
chloride solution is dried over MgSC74 and
concentrated, and the residue is dissolved in
Le A 28 100 - 58 -
tent-butyl methyl ether (200 ml). 9 g of starting
material (44 mmol) crystallised out overnight. The
ether solution is concentrated and distilled.
Yield: 83 g ( 80 ~ of theory relative to recovered
starting material and diallyl ether)
Boiling point: 149°C10.7 mbar to 159°C/0.9 mbar.
The distillate contains 5 ~ of the starting material
and 4 ~ of diallyl ether.
The pentane extract yielded 17 g of a mixture of
15 $ desired product and 84 ~ of diallyl ether.
(a]D3 - -10.5° (c = i, methanol).
2) tart-Butyl 3S,4S-3-hydroxy-4-(2-hydroxyethoxy)-
pyrrolidine-1-carboxylate
64 g (0.24 mol, 91 ~ strength) of tart-butyl 3S,4S-
4-allyloxy-3-hydraxypyrrolid9_ne-~.-carboxylate are
dissolved in 250 ml of methanol and cooled to 0°C,
and ozone is passed through the solution until a
washing bottle containing potassium iodide solution
and connected in series indicates the emergence of
ozone and thus complete reaction. Residues of ozone
are carried out by means of a stream of nitrogen,
then the resulting ozonide is reduced at 0°C using
18 g of sodium borohydride, which is added in 1 g
portions. The mixture is then stirred overnight at
room temperature and concentrated, the residue is
diluted with water, and the mixture is treated with
Le A 28 100 - 59 -
~~t~~~~~
20 g of potassium carbonate and extracted five times
with 100 ml of methylene chloride each time. The
organic solutions are dried over magnesium sulphate
and concentrated.
Yields 65.8 g (100 ~ of theory)
The product is 91 ~ strength by gas chromatography.
[«]D~ - _15.2° (c = 0.97, methanol).
3) 35,48-1-tent-Butoxycarbonyl-3-tosyloxy-4-(2-
tosyloxyethaxy)-pyrrolidine
2.7 g (10 mmol, 91 ~ strength) of tent-butyl 35,48-
3-hydroxy-4-(2-hydroxyethoxy)-pyrrolidine-1-
carboxylate axe initially introduced into 30 ml of
methylene chloride, 6 ml of 45 ~ strength radium
hydroxide solution and 0.1 g of benzyltriethyl-
ammonium chloride are added and a solution of 2.86 g
(20 moral) of tosyl chloride in 10 ml of methylene
chloride are then added dropwise with cooling. The
mixture is then stirred for a further hour at room
temperature and poured into 20 ml of water, the
organic phase is separated of:~ and the aqueous phase
is extracted with methylene chloride. The organic
phases are dried over magnesium sulphate and
concentrated.
Yielda 5 g (90 ~ of theory).
The product is homogenous by thin layer
chromatography.
Le A 28 100 - 60 -
4) tert-Butyl 1S,6R-5-benzyl-2-oxa-5,8-diazabicyclo-
[4.3.0]nonane-8-carboxylate
87 g (156 mmol) of 3S,4S-1-tert-butoxycarbonyl-3-
tosyloxy-4-(2-tosyloxyethoxy)-pyrrolidine are heated
under reflex overnight with 58 g (0.54 mol) of
benzylamine in 1 1 of xylene. The mixture is cooled,
precipitated salts of benzylamine are filtered off
with suction and the residue is concentrated.
Xield: 43 g (58 ~ of theory).
The product is 67 ~ strength by gas chromatography.
5) 1S,6R-5-Benzyl-2-oxa-5,8-diazabicyclo[4.3.0]nonane
43 g (90 mmol) of tert-butyl 1S,6R-5-benzyl-2-oxa-
5,8-diazabicyclo[4.3.0]nonane-8-carboxylate are
heated under reflex in 35 ml of concentrated
hydrochloric acid and 35 ml_ of water until the
evolution of carbon dioxide is complete. The mixture
is rendered alkaline with pcatassium carbonate and
extracted with chloroforrn, the: organic solutions are
dried over MgS04 and concentrated, and the residue is
distilled twice through a 20 cm 'Vigreux column.
Yield: 11.1 g (55 ~ of theory)
Boiling point: 108 - 115°C/0:07 mbar
[«]D6 = ~58.3° (undiluted).
Le A 2s loo - sl -
Example T
1) tart-Butyl 3R,4R-4-allyloxy-~-hydroxypyrrolidine-1-
carboxylat~:
The reaction is carried out analogously to Example
H1) using tart-butyl R,R-3,4-dihydroxypyrrolidine-
1-carboxylate:
Boiling point: 145°C/0.1 mbar
[«]D3 = +9.5° (c = 1.0, methanol).
The product is 95 ~ strength by gas chromatography.
ZO 2) tart--Butyl 3R,4R-3-hydroxy-4-(2-hydroxyethoxy)-
pyrrolidine-1-carboxylate
The reaction is carried out analogously to Example
H2) using tart-butyl 3R,4R-4-allyloxy-3-hydraxy-
pyrrolidine-1-carboxylate:
Yield: 99 ~ of theory (0.175 molar batch)
[«]D -- +16.5° (c = 0.94, methanol).
3) 3R,4R-1-tart-Butoxycarbonyl-3-tosyloxy-4-(2-
tosyloxyethoxy)-pyrrolidine
The reaction is carried out analogously to Example
H3) using tart-butyl 3R,4R-3-hydroxy-4-(2--
hydroxyethoxy)-pyrrolidine-l~.carboxylate
Yield: quantitative (0.11 molar batch).
4) tart-Butyl 1R,6S-5-benzyl-2-oxa-5,8-
diazabicyclo[4.3.0]nonane-8-carboxylate
Le A 28 100 - 62 -
The reaction is carried out analogously to Example
H4) using 3R,4R-1-tert-butoxy-carbonyl-3-tosyloxy-
4-{2-tosyloxyethoxy)-pyrrolidine:
Yield: 40 ~ of theory (0./1 molar batch).
5) 1R,6S-5-Benzyl-2-oxa-5,8-diazabicyclo[4.3.0]nonane
The reaction is carried out analogously to Example
H5) using tent-butyl 1R,6S-5-benzyl-2-oxa-5,8-
diazabicyclo[4.3.0]nonane-8-carboxylate:
Yield: 63 ~ of theory (40 mmolar batch)
Roiling point: 120°C/0.06 mbar
The product is 95 ~ strength by gas chromatography
(a]D3 = +58.5° (undiluted).
Example J
1) 1S,6R-2-Oxa-5,8-diazabicyclo[4.3.0]nonane
dihydrochloride
7.5 g (34.4 mmol) of 1S,6R-5-benzyl-2-oxa-5,8-
diazabicyclo(4.3.0]nonane arse hydrogenated at 100°C
and 100 bar on 1 g of palladium/active carbon (10
Pd) in 200 ml of ethanol with the addition of 7 m1
of concentrated hydrochloric acid. The catalyst is
filtered off with suction and washed sevezal times
with water. The aqueous filtrate is concentrated,
whereupon the residue crystallises. The crystals are
thoroughly saturated with ethanol, filtered off with
suction and dried in air.
Le A 28 100 - 63 -
~J~~~.~~
Yield: 4.6 g (66.5 ~ of theory)
Melting point: 233-235°C.
2) 1S,6R-2-Oxa-5,8-diazabacyclo[4.3.0]nonane
59 g (0.27 mol) of 1S,6R-5-benzyl-2-oxa-5,8-
diazabicyclo[4.3.0]nonane are hydrogenated at 120°C
and 120 bar on 5 g of palladium/active carbon (10
Pd) in 500 m1 of ethanol. The catalyst is faltered
off with suction, the filtrate is concentrated and
the residue is distilled.
Yield: 32.9 g (95 ~ of theory)
Boiling point: 65°C/0.03 mbar
Rotation : [a]De = +8.2° (undiluted).
ee values >_ 99.5 ~ (by derivatisation with Mosher
reagent).
Exam lp a K
1) 1R,6S-2-Cxa-5,8-diazabicyclo[4.3.0]nonane
dihydrochloride
The reaction is carried out analogously to Example
J1) using 1R,68-5-benzyl-2-oxa-5,8-diazabicyclo
[4.3.0]nonane:
Yield: 77 ~ of theory (23.8 mmolar batch)
Melting point: 230-232°C.
2) 1R,6S-2-Uxa-5,8-diazabicyclo[4.3.0]nonane
Le .A 28 100 - 64 -
The reaction is carried out analogously to Example
J2) using 1R,6S-5-benzyl-2-oxa-5,8-diazabicyclo-
[4.3.0]nonane:
Yield: 93.3 ~ of theory (1.58 molar batch)
Boiling point: 63 - 65°x/0.03 mbar
Rotation: [oc]p~ = -8.4° (undiluted).
ee value: >_ 99.5 ~ (by derivatisation with Mosher
reagent).
1R,6R- or 1S,6S-2-Oxa-5,8-diazabicyclo[4.3.0]nonane
can be obtained analogously.
Example L
1R,6S-2-oxa-5,8-diazabicyclo[4.3.0]nonane dihydro-
bramide
1) 1R,6S-5-(1R-Phenylethyl)-8-tosyl-2-oxa-5,8-diaza-
bicyclo[4.3.0]nonane
101.8 g (0.196 mol) of traps-3-brom-1-tosyl-4-(2-
tosyloxyethoxy)-pyrrolidine and 72 g (0.584 mol) of
R-(+)-1-phenylethylamine in 900 ml of xylene are
heated under reflux overnight. The cooled solution
is washed with 2N sodium hydroxide solution and
dried over potassium carbonate, the drying agent is
removed and the solvent is concentrated. Qn cooling,
crystals are deposited from the residue which are
filtered off with suction and recrystallised from a
mixture of 750 ml of petroleum ether and 200 ml of
Le A 28 100 - 65 - -
~)a.~~l
n-butanol.
Yields 15 g (39.6 $ of theory of optically pure
material.
Melting point: 188°C,
Rotation : [a]oe = +103.7° (c = 1, CHC13) .
2) 1R,6S-8-Tosyl-2-oxa-5,8-diazabicyclo[4.3.0]nonane
13 g (33.6 mmol) of 1R,6S-5-(1R-phenylethyl)-8-
tosyl-2-oxa-5,8-diazabicyclo[4.3.0]nonane are hydro-
genated at 100°C and 100 bar on 2.5 g of palladium/
active carbon (10 ~ Pd) in 200 ml of ethanol. The
catalyst is filtered off with suction, the filtrate
is concentrated and the residue is recrystallised
from 30 ml of toluene.
Yield: 7.5 g (79 ~ of theory),
Melting point: 160-161°C,
Rotation: [«]03 = +17.5° (c = 1.21, CHC13).
3) 1R,6S-2-Oxa-5,8-diazabicyclo[4.3.0]nonane dihydro-
bromide
7 g (24.8 mmol) of 1R,6S-8--tosyl-2-oxa-5,8-diaza-
bicyclo[4.3.0]nonane axe dissolved in 25 ml of 33 ~
strength hydrogen bromide solution in glac~.al acetic
;..,..~
acid, 5 g of phenol are added and the mixture is
stirred overnight at room temperature. It is diluted
with diisopropyl ether, and the crystallised salt is
filtered off with suction and dried in air.
Yield: 5.5 g.
Le A 28 100 - 66 -
U ZA 'v
Derivatisation with Mosher reagent and gas
chromatographic analysis shows only one detectable
enantiomer (ee > 99.5 ~).
Example M
5-Bromo-1-cyclopropyl-6,7,8-trifluoro-1,4-dihydro-4-oxo-
3-quinalinecarboxylic acid
Br O
F ~ COOH
J
F ~ ~:I
F
1) 2-Bramo-3,4,5,6-tetrafluoro-benzoyl chloride
365 g (1.33 mol) of 2-bromo-3,4,5,6-tetrafluoro-
benzoic acid [Tetrahedron 23, 4719 (1967)] are
introduced into 2 1 of thionyl chloride and the
mixture is heated under refl.ux for 11 hours until
the evolution of gas stops. F;xcess thionyl chloride
is stripped off in vacuo and the residue is
distilled.
Yield: 330 g (85 ~ of theory),
Boiling point: 81-85°C/3-5 mbar.
2) Diethyl (2-broma-3,4,5,6-tetrafluoro-benzoyl)-
malonate
15.9 g (0.167 mol) of magnesium chloride are
Le A ?.8 100 - 67 -
introduced into 150 ml of anhydrous acetonitrile
(dried over zeolite) and 26.9 g (0.167 mol) of
diethyl malonate are allowed to drop in with
cooling. The mixture is cooled to 0°C, 46~ ml
(33.7 g - 0.33 mol) of triethylamine are added
dropwise and the mixture is stirred for 30 minutes.
48.9 g (0.168 mol) of 2-bromo-3,4,5,6-tetrafluoro-
benzoyl chloride are then added dropwise, and the
mixture is stirred for a further 1 hour at 0°C and
brought to room temperature overnight. It is treated
with 100 ml of 5 N hydrochloric acid and extracted
three times with methylene chloride, and the
extracts are dried with NaZSOa and concentrated in
vacuo.
Crude yield: 62.7 g.
3) Ethyl (2-bromo-3,4,5,6-tetrafluoro-benzoyl)-acetate
60 g of crude diethyl (2-bromo-3,4,5,6-tetrafluoro-
benzoyl)-malonate are introduced into 150 m1 of
water and treated with 0.6 g of 4-toluenesulphonic
acid, and the mixture is heated under reflex for 6
hours. It is extracted with methylene chloride, and
the extract is washed with water, dried with Na2S04
' a and concentrated.
Crude yields 46 g,
Boiling point (sample distillation in a bulb tube):
150-160°C(oven)/3 mbara
Mass spectrum: m/B 342 (M+) , 297 (M+-OCZHS) , 263 (M+-
Br), 257, 255 (M+-CHzCO2C2HS), 235 (263-28) .
Le A 28 100 - 68 -
4) Ethyl 2-(2-bromo-3,4,5,6-tetrafluoro-benzoyl)-3-
ethoxy-acrylate
45 g of crude ethyl (2-bromo-3,4,5,6-tetrafluoro-
benzoyl)-acetate are introduced into 32.2 g
(0.31 mol) of acetic anhydride and 28.4 g (0.19 mol)
of triethyl orthoformate and the mixture is heated
under reflux for 2 hours. Excess reagent is first
stripped off in vacuo, then under a high vacuum
(bath up to 120-130°C) and the crude product is
reacted to the next step.
Crude yield: 50.7 g
5) Ethyl 2-(2-bromo-3,4,5,6-tetrafluoro-benzoyl)-3-
cyclopropylamino-acrylate
50.7 g of crude product from Step 4) axe treated
dropwise with 8.6 g (0.15 mot) of cyclopropylamine
in 90 ml of ethanol with ice-cooling, the mixture is
stirred at room temperature, allowed to stand
overnight and again well cooled, and the
cry5tallisate is filtered of;f with suction, washed
with cold ethanol and dried.
Yield: 29 g (42 ~ over 4 steps),
Melting point: 103-105°C (from ethanol).
6) Ethyl 5-bromo-1-cyclopropyl-6,7,8-trifluora-1,4-
dihydro-4-oxo-3-quinolinecarboxylate
Le A 28 100 - 69 -
L3 =I .~. ~~
28 g (68 mmol) of ethyl 2-(2-bromo-3,4,5,6-
tetrafluoro-benzoyl)-3-cyclopropylaminoacrylate axe
heated under reflux for 6 hours with 6.9 g
(164 mmol) of sodium fluoride in 88 ml of DMF. The
mixture is poured into water after cooling, and the
deposited precipitate (red) is filtered off with
suction, washed with plenty of water and dried at
80°C in a recirculating air oven.
Crude yield: 27.3 g,
Melting point: 150-175°C;
after recrystallisation from glycol monomethyl
ether:
Melting point: 187-191°C.
7) 5-Bromo-1-cycloprnpyl-6,7,8-trifluoro-1,4-dihydro-
4-oxo-3-quinolinecarboxylic acid
26.7 g (68 mmol) of crude ethyl 5-bromo-1-
cycloprapyl-6,7,8-trifluoro-1,4-dihydro-4-oxo-3-
quinolinecarboxylate are introduced into a mixture
of 165 ml of acetic acid, 110 ml of water and 18 ml
of concentrated sulphuric ~~cid and heated under
reflux for 2 hours: The cooled xeaction mixture is
poured into ice-water, and the deposited precipitate
is filtered off with suction, washed with plenty of
water and dried in a recirculating air oven at 80°C.
Yield: 19.7 g (80 ~ of theary),
Melting point: 208-210°C (with decomposition);
after recrystallisationfrom glycol monomethyl ether:
Melting points 212-214°C (with decomposition);
Le A 28 100 - 70 -
,~ n
:~~.az~~~
NMR, li-I (DMSO) : 8. 73 s ( 1H at C-2 ) , 4, 10' m ( 1H, cyclo-
propyl), 1,2 m (4H, cyclopropyl) [ppm].
Mass spectrum: '°/e 361 (M''-H20) , 317 (M-COZ) , 41
( 100 ~, C3H5) .
preparation of the final compounds
Examt~le 1
O
F , COOH
J
N ~ N
F
N
H
A. 1-~clopropyl-7- ~[l S . S ] -2 . 8-diazabic~,rclo [ 4 . 3 . 0~ non-8-
~1L 6,8-difluoro-1.4-dihydro-4-oxo-3-guinolinecarboxylic
ac id
141.5 g (0.5 mol) of 1-cyclopropyl-6,7,8-trifluoro-1,4-
dihydro-4-oxo-3-duinolinecarboxylic acid are heated under
reflux for 1 houx with 69.25 g (0.55 mol) of (+)-[S,S]-
2,8-diazabicyclo[4.3.0]nonane (ee> 99.5 ~, GC 99.8
strength) in a mixture of 1500 ml of acetonitrile and
750 ml of dimethylforariamide in the presence of 55 g
(0.5 mol) of 1,4-diazabicyclo-[2.2.2]octane. The
suspension is cooled, and the precipitate is filtered off
with suction, washed with water and then additionally
stirred with 1 1 of water (pH 7). The product is filtered
off with suction and dried at 60°C in a recirculating air
Le A 28 100 - 71 -
~J~J1~
oven,
Yield: 163.4 g (84 ~ of theory),
Melting point: 249-251°C (with decomposition)
B. (-)-1-Cyc.loproPyl-7-~.IS.S]-2.8-diazabicycloj4.3.0]non=
8-yl)-6,8-difluoro-1.4-dihydro-4-oxo-3-quinoline-
carboxylic acid hydrochloride
6.0 g (15.4 mmol) of 1-cyclopropyl-7-([S,S]-2,8-diaza-
bicyclo[4.3.O~non-8-yl)-6,8-difluoro-1,4-dihyriro-4-oxo-
3-quinolinecarboxylic acid are dissolved in 40 ml of
half-concentrated hydrochloric acid at 60°C and the
solution of the hydrochloride is filtered. The filtrate
is concentrated to one half, cooled in ice and treated
with 40 ml of ethanol. The yellow crystallisate is
filtered off with suction, washed with ethanol and dried
at 60°C in a high vacuum, where the colour lightens.
5.51 g (84 $ of theory) of the hydrochloride are
obtained, which is already very gore.
For further purification, it is dissolved in 50 ml of
water in the presence of heat. 'The yellow solution is
treated with 5 ml of half-concent:cated hydrochloric acid
and cooled in ice, and the deposited crystallisate is
filtered oft with suctian, washed well with ethanol and
dried first at room temperature and then under a high
vacuum at 100°C.
Yield: 4.64 g (70.8 ~ of theory),
Le A 28 lU0 - 72 -
~~~u~~.~
Melting point: 324--325°C (with decomposition),
TLC (silica gel, dichloromethane/methanol/17 ~ strength
aqueous ammonia = 30:8:1): homogeneous, Rf value: 0.3,
[«]D5: -256° (c = 0.5, H20),
Purity (HPLC): 99.4 ~ strength,
G20H21~2N3~3 ~ HC1 ( 425 . 5 )
Calculated: C 56.4 H 5.2 N 9.9 G1 8.3
Found: C 56.3 H 5.4 N 9.8 C1 8.3
Example 2
O
F , COOH
i J
H N ~' N
~ Cl
N[ J'H
A. 8-Chloro-1-cyclopropyl-7-(fS,S~ -2,8-diazabicyclo-
L4.3.Olnon-8-vl~-6-fluoro-1.4-dihydro-4-oxo-3-guinoline-
carboxylic acid
2 batches of the following size are run in parallel and
worked up together:
180 g (0.6 mol) of 8-chloro-1-cycloprapyl-6,7-difluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylic acid axe heated
under reflux for 1 hour with 84 g (0.67 mol) of (+)-
(S,S]-2,8-diazabicyclo(4.3.0]nonane in a mixture of 1.8 1
of acetonitrile/900 ml of dimethylformamide in the
Le A 28 100 - 73 -
presence of 99 g (0.88 mol) of 1,4-diazabicyclo[2.2.2]-
octane (DABCO) (internal temperature: 90.5°C). The yellow
solution is cooled and treated with seed crystals
(obtained from a 5 ml sample which was concentrated;
residue stirred with acetronitrile). The mixture is
stirred far 2 hours at about 3°C, and the deposited
precipitate from both batches is rapidly filtered off
with suction, washed with acetronitrile and introduced
into 1.5 1 of ice-water. The initially thin, well-
stirrable suspension after about 10 minutes becomes a
poorly stirrable mass, which is diluted with a further
150 ml of water. The precipitate is filtered off with
suction, washed with water and dried at 80°C in a
recirculating air drying oven.
Yield: 402 g (82.7 ~ of theory), faintly yellow product;
Melting point: 193-196°C (with decomposition),
Rf value (silica gel; methylene chloride/methanol/17
strength aqueous NH3 = 30:8:1): 0.4.
B 8-Chloro-1-cyclogro~~yl-7- ( j S , S ] -2 , 8-
diazabicyclo _P 4 . 3 . 0 )non-8-yl ~ 6-fluoro-1, 4-dihvdro-4-oxo-
3-quinolinecarboxvlic acid h~yidrochloride
13.1 g (32 mmol) of 8-chloro-1-cyclopropyl-7-([S,S]-2,8-
diazabicyclo[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-
3-quinolinecarboxylic acid are suspended in 50 ml of
water and brought into solution by addition of 50 ml of
half-concentrated hydrochloric acid. The mixture is
filtered through a glass frit, the filtrate is
Le A 28 100 - 74 -
i ~1~~~_~~
concentrated in vacuo and the residue is stirred with
about 300 ml of abs. ethanol. The suspension is cooled in
ice, the precipitate is filtered off with suction,, washed
with ethanol and dried first at room temperature and then
at 100°C in vacuo.
Yield: 13.4 g (93.8 ~ of theory);
Melting point: 328-330°C (with decomposition);
Rf value (silica gel, methylene chloride/methanol/17
strength aqueous NH3 = 30:8:1): 0.4;
Purity (HPLC): 99.9 ~ strength,
[a]D4: -164.4° (c = 0.45, H20),
C20H21C1FN3o3. HC1 ( 442 . 3 )
Calculated: C 54.3 H 5.0 N 9.5 C1 16.0
Found: C 54.3 H 5.0 N 9.5 C1 16.0
C. The following salts, for example, can also be
prepared analogously:
8-chloro-1-cyclopropyl-_7-([~i,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl),-6-fluoro-1,4-dihydro-4-oxo-3-
quinoline carboxylic acid methanesulphonate
- 20 8-chloro-1-cyclopropyl-7-([S,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl).-6-fluoro-1,4-dihydro-4-oxo-3-
quinoline carboxylic acid toluenesulphonate
8-chloro-1-cyclopropyl-7-([S,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-
Le A 28 100 - 75 -
quinoline carboxylic acid sulphate
8-chloro-1-cyclopropyl-7-([S,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-
quinoline carboxylic acid acetate
8-chloro-1-cyclopropyl-7-([S,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-
quinoline carboxylic acid lactate
8-chloro-1-cyclopropyl-7-([S,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-
quinoline carboxylic acid citrate
8-chloro-1-cyclopropyl-7-([S,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-
quinoline carboxylic acid embonate
Example 3
O
COOH
J
y ~
H
N'
H
25 Analogously to Example 1, the following are obtained
with 1-cyclopropyl-6,7-difluoro-1,4-dihydro-4-oxo-3-
quinolinecarboxylic acids
T~e A 28 100 - 76 -
A. 1-Cyclopropyl-7-([S,S]-2,8-diazabicyclo[4.3.0]non-
8-yl)-6-fluoro-1,4~-dihydro-4-oxo-3-quinoline-
carboxylic acid
Melting point: 256-258°C (with decomposition).
B. 1-Cyclopropyl-7-([S,S]-2,8-diazabicyclo[4.3.0]non-
8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinoline-
carboxylic acid hydrochloride,
Melting point: > 320°C (with decomposition),
[a]D6: -90.6° (c = 0.48, H20) .
Example 4 F O
F , COOH
J
N \ N
H N
'H
A. 6 g (20 mmol) of 1-cyclopropyl-5,6,7,8-tetrafluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylic acid are
heated under reflux for 1 hour with 2.7 g (21.4
mmol) of (+)-[S,S]-2,8-diazabicyclo[4.3.0]nonane in
40 ml of acetonitrile/20 ml of I~-methylpyrrolidone
in the presence of 2.2 g (20 mmol) of 1,4
diazabicyclo[2.2.2]octane. 2'he suspension obtained
is cooled, and the precipitate is filtered off with
suction, washed with acetonitrile and dried at
100°C/12 mbar.
Le A 28 100 - 77 -
~t,1?~~...~ ~'~.
Yield: 6.7 g (82.3 ~ of theory) of 1-cyclopropyl-7-
([S,5]-2,8-diazabicyclo(4.3.0]non-8-y1)-5,6,8-tri-
fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid,
Melting point: 257-259°C (with decomposition); after
recrystallisation from glycol monomethyl ether:
Melting point: 260-265°C (with decomposition).
B. 1.5 g (3.7 mmol) of the product from Step A are
introduced into 6 ml of 1 N hydrochloric acid. After
a short time, the hydrochloride deposits, and is
filtered off with suction, washed twice with 5 ml of
ethanol each time and dried at 100°C/12 mbar.
Yield: 1.4 g (85.7 ~ of theory) of 1-cyclopropyl-7
([5,5]-2,8-diazabicyclo[4.3.0]non-8-yl)-5,6,8
trifluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic
acid hydrochloride,
Melting point: > 310°C (with decomposition),
[a]D6: -272° (c = 0.5, H20) .
Example 5
NH2 O
F , COOH
~J
N ~ N
H~N F ~ x HCl
'H
5.2 g (13 mmol) of the product from Example 4A are
treated with 15 ml of liquid ammonia in 80 ml of pyridine
in an autoclave and heated at 130 °C for 12 hours . The
Le A 28 100 - 78 -
mixture is then cooled, the autoclave is let down, the
mixture is concentrated and the residue is treated with
acetonitrile in an ultrasonic bath. The undissolved
precipitate is filtered off with suction, the residue is
dissolved in about 150 ml of water in the presence of
heat, the solution is filtered and the hydrochloride is
precipitated using 10 ml of half-concentrated hydro-
chloric acid, filtered off with suction and dried at
100°C in a recirculating air drying oven. The product
obtained is suspended in 100 ml of glycol monomethyl
ether at 110-115°C and brought into solution by addition
. . of 38 ml of half-concentrated hydrochloric acid. The
solution is filtered hot through a glass frit, cooled and
the cooled yellow crystallisate is filtered off with
suction, washed with ethanol and dried at 120°C~12 mbar.
Yield: 2.5 g (44 ~ of theory) of 5-amino-1-cyclopropyl-
7-([S,S]-2,8-diazabicyclo[4.3.0]non-8-yl)-6,8-difluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylic acid hydro-
chloride,
Melting point: > 335°C (with decomposition; dark
colouring already below 335°C),
[a]De: -2$0.8° (c = 0.53, H20) .
Exam~gle 6
O
COOH
Ha/-'N N N
x HCl
N
J H
Le A 28 100 - 79 -
~fl ~~~.i~:~.
1.4 g (5 mmol) of 7-chloro-1-cyclopropyl-6-fluoro-1,4-
dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid are
stirred at room temperature for 1 hour with 1.3 g
(10.3 mmol) of (+)-[S,S]-2,8-diazabicyclo[4.3.0]nonane
with exclusion of water in 15 ml of acetonitrile. After
standing overnight, the precipitate is filtered of.f with
suction, washed with acetonitrile and chromatographed on
silica gel for purification (eluent: methylene chloride/-
methanol/17 ~ strength aqueous ammonia 30x8:1; Rf value:
0.4). The 1-cyclopropyl-7-([S,S]-diazabicyclo[4.3.0]non-
8-yl)-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-
carboxylic acid obtained is dissolved in 15 ml of half-
concentrated hydrochloric acid, the solution is
evaporated and the residue is stirred with ethanol. The
precipitate is filtered off with suction, washed with
ethanol and dried at 120°C/12 mbar.
Yield: 960 mg (47 ~ of theory) of 1-cyclopropyl-7-([S,S]-
2,8-diazabicyclo[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-
oxo-1,8-naphthyridine-3-carboxylic acid hydrochloride
Melting point: 345-346°C (with decomposition),
[a]p°: +5.4° (c = 0.5, H20).
Example 7
O
COOH
~J
H, N ~ N
..", H
H\N//'.
Le A 28 100 - 80 -
Analogously to Example 1, the fallowing are obtained
using-(~)-[R,R]-2,8-diazabicyclo[4.3.0]nonane:
A. 1-Cyclopropyl-7-([R,R]-2,8-diazabicyclo[4.3.0]non
8-yl)-6,8-difluoro-i,4-dihydro-4-oxo-3-quinoline
carboxylic acid
Melting point: 247-249°C (with decomposition).
H. 1-Cyclopropyl-7-([R,R]-2,8-diazabicyclo[4.3.0]non-
8-yl)-6,8-difluoro-1,4-dihydro-4-oxo-3-guinoline-
carboxylic acid hydrochloride,
Melting point: 322-326°C (with decomposition),
Purity (HPLC): 99.4 ~ strength, ee: 98.6 $,
[«]D4: +250° (c = 0.5, H20) .
Example 8
O
F ~ COOH
H ,,, N w N
Cl
N .. ..." H
Analogously to Example 2, the following are obtained
~,5 using (-)-[R,R]-2,8-diazabicyclo[4.3.0]nonane:
A. 8-Chloro-1-cyclopropyl-7-([R,R]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-
quinolinecarlaoxylic acid,
Le A 28 100 - 81 -
~ a
W' '1:5 a .:. ~,;r.
Melting point: 192-195°C (with decomposition).
B. 8-Chloro-1-cyclopropyl-7-([R,R]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-
quinolinecarboxylic acid hydrochloride,
Melting point: 323-324°C (with decomposition).
Purity (HPLC): 99.9 ~ strength,
[a)D°: +164.5° (c = 0.53, HZa).
CzDHzIC1FN303. HC1 ( 442 . 3 )
Calculated: C 54.3 H 5.0 N 9.5 C1 16.0
Found: C 54.2 H 5.0 N 9.5 C1 16.1
Example 9
O _
F ~ COOH
J
H F.i ,,,.
N ~ N
N . ..,,
'H
Analogously to Example 1, the following are obtained from
1-cyclopropyl-6,7-difluoro-1,4-dihydro-4-oxo-3
quinolinecarboxylic acid and (-)-[R, R]-2,8-diazabicyclo
[4.3.0)nonane:
A. 1-Cyclopropyl-7-([R,R)-2,8-diazabicyclo[4.3.0]non-
$-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinoline-
carboxylic acid,
Melting point: 25~-258°C (with decomposition).
Le A 28 100 - 82 -
~~~~~1~~
B. 1-Cyclopropyl-7-([R,R]-2,8-diazabicyclo[4.3.0]non-
$-yl)-6-fluoro-1.,4-dihydro-4-oxo-3-quinoline-
carboxylic acid,
Melting point: decomposition above 320°C,
[a]p°: +92.5° (c = 0.53, HZO) .
Example 10
O
F , COOH
J
H
cis
~O
A. 1-Cyclopropyl-6,8-difluoro-1,4-dihydro-7-(cis-2-oxa-
5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid:
1.43 g (5 mmol) of 1-cyclopropyl-6,7,8-trifluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylic acid are
heated under reflux for :1 hour with 0.74 g
(5.4 mmol) of 93 ~ strength cis-2-oxa-5,8-diaza-
bicyclo[4.3.0]nonane in the presence of 0.67 g (6
mmol) of 1,4-.diazabicyclo[2.2.2]octane in a mixture
of 15 ml of acetonitrile/75 ml of dimethylformamide.
The suspension is concentrated, the residue is
stirred with water, and the precipitate is filtered
off with suction and dried in vacuo at 80°C.
Yield: 1.67 g (85.4 ~ of theory),
Melting point: 210-212°C (with decomposition).
Le A 28 100 - 83 -
B. 1-Cyclopropyl-6,8-difluoro-1,4-dihydro-7-(cis-2-oxa-
5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid hydrochloride: .
1.6 g (4 mmol) of the product from Step A are
dissolved in 120 m1 of half-concentrated hydro-
chloric acid at 60°C, the solution is concentrated,
the residue is stirred with ethanol and the preci-
pitate is filtered off with suction and dried at
90°C iri vacuo.
Yield: 1.57 g,
Melting points 300-303°C (with decomposition),
Purity (FiPLC): 97 ~ strength.
C. Analogously to Example 10A, 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-7-(1R,6S-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid of melting point 204-206°C (with
decomposition) is obtained 'using 1R,6S-2-oxa-5,8-
diazabicyclo[4.3.0]nonane.
D, Analogously to Example 10B, 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-7-(1R,6S-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid hydrochloride of melting point 324-
325°C (with decomposition) is obtained using the
betaine from Example lOC.
[a]o°: -241° (c = 0.59, H20) .
Le A 28 100 - 84 -
E. Analogously to Example 10A, 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-7-(1S,6R-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid of melting point 204-206°C (with
decomposition) is obtained using 1S,6R-2-oxa-5,8-
diazabicyclo[4.3.0]nonane.
[a]D5; +248° (c = 0.57, DMF).
F. Analagously to Example IOB, 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-7-(1S,6R-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid hydrochloride of melting point 323°C
(with decomposition) is obtained using betaine from
Example 10E.
[a]DSt +238° (c = 0.5, HZO) .
Example 11
O
F , CO03-I
y w y
H~N~ C!
cis
~O
Analogously to Example l0, the following are obtained
using 8-chloro-1-cyclopropyl-6,7-difluoro-1,4-dihydro-4-
oxo-3-quinolinecarboxylic acids
Le A 28 100 - g5 -
A. 8-Chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-7-(cis-
2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl-4-oxo-3-
quinolinecarboxylic acid,
Melting point: 180-185°C (with decomposition),
B. 8-Chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-7-(cis-
2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-
quinolinecarboxylic acid hydrochloride,
Melting point: 227-232°C (with decomposition).
C. 8-Chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-7-
(1R,6S-2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-
3-quinolinecarboxylic acid,
Melting point: 186-188°C (with decomposition).
[«]D6: -269° (c = 0.5, DMF).
D. 8-Chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-7-
(1R,6S-2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-
3-quinolinecarboxylic acid hydrochloride
Melting point: 278-280°C (with decomposition),
[«]D4: -208° (c = 0.5, HZ~) .
E. 8-Chloxo-1-cyclopropyl-6-fluoro-1,4-dihydro-7-
(1S,6R-2-oxa-5,8-diazabicyclo[4.3.0]nori-8-yl)-4-oxo-
3-quinolinecarboxylic acid
Melting point: 188-190°C (with decomposition).
+270° (c = 0.5, DMF).
Le A 28 100 - 86 -
A (.1 n
F. 8-Chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-7-
(1S,6R-2-oxa-5,8-diazabicycla[4.3.0]non-8-yl)-4-oxo-
3-quinolinecarboxylic acid hydrochloride,
Melting point: 292-294°C (with decomposition).
[a]D': +193° (c = 0.5, H20) .
Example 12
O
F ~ COOH
J
N \ N
cis
H\
~O
Analogously to Example 10A, the following are obtained
using 1-cyclopropyl-6,?-difluoro-1,4-dihydro-4-oxo-3-
quinolinecarboxylic acid:
ZO A. 1-Cyclopropyl-6-fluoro-1,4-dihydro-7-(cis-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid,
Melting point: 246-249°C (with decomposition) (from
glycol monomethyl ether).
B. 1-Cyclopropyl-6-fluoro-1,4-dihydro-?-(1R,6S-2-oxa-
5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid,
Melting point: 243-245°C (with decomposition)
Le A 28 100 - 8? -
2~~~a.~:~
C. 1-Cyclopropyl-6-fluoro-1,4-dihydro-7-(1R,6S-2-oxa-
5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid hydrochloride,
Melting point: 300°C (decomposition)
[a]p3: -99° (c = 0.5, Hz0) .
Example 13
F O
COOH
J
N ~ N
~y~s
F
~o
Analogously to Example 10A, the following are obtained
using 1-cyclopropyl-5,6,7,8-tetrafluoro-1,4-dihydro-4-
oxo-3-quinolinecarboxylic acid:
A. 1-Cyclopropyl-5,6,8-trifluoro-1,4-dihydro-7-(cis-2-
oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-
quinolinecarboxylic acid,
Melting point: 210-216°C (with decomposition).
B. 1-Cyclopropyl-5,6,8-trifluoro-1,4-dihydro-7-(1R,6S-
2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-
quinolinecarboxylic acid,
Melting point: 234-237°C (with decomposition).
[a]D4: -287° (c = 0.5, DMF).
Le A 28 100 - 88 -
C. 1-Cyclopropyl-5,6,8-trifluoro-1,4-dihydro-7-(1S,6R-
2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-
quinolinecarboxylic acid,
Melting point: 236-237°C (with decomposition).
[«]D°: +282° (c = 0.5, DMF) .
Examlale 14
NHZ O
COON
J
-N ~ N
H\N
~'~s
O
A. 4.1 g (10 znmol) of the product from Example 13A are
treated with 5 ml of liquid ammonia in 40 ml of
pyridime and heated at 130°C for 10 hours in an
autoclave. After cooling, the precipitate is
filtered off with suction, washed with water and
dried at 100°C in a recirculating air drying oven.
The crude product (2 g) is purified by recrystalli
sation from glycol monomethyl ether: yellow
crystallisate.
Yields 1.3 g (31 ~ of theory) of 5-amino-1-
cyclopropyl-6,8-difluoro-1,4-dihydro-7-(cis-2-oxa-
S,8-diazabicyclo[4.3.0]non-8-y1)-4-oxo-3-quinoline-
carboxylic acid,
Melting point: 233-240°C (with decomposition).
Le A 28 100 - 89 °
2~~~~~.~.~
B. 5-Amino-1-cyclopropyl-6,8-difluoro-1,4-dihydro-7-
(1R,6S-2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-
3-quinolinecarboxylic acid is obtained analogously
using the product from Example 13C,
Melting point: 212-214°C (with decomposition),
[a]D5: -260° (c = 0.5, DMA') .
C. 5-Amino-1-cyclopropyl-6,8-difhuoro-1,4-dihydro-7-
(1S,6Ft-2-oxa-5,8-diazabicyclo(4.3.0]non-8-yl)-4-oxo-
3-quinolinecarboxylic acid is obtained analogously
using the product from Example 13C,
Melting point: 213-215°C (with decomposition),
(a]D6: +261° (c = 0.5, D1H:F') .
Mass spectrum: m/e 406 (M+,95 ~), 346, 249, 98, 41,
28 (100 ~).
Example 15
O (S)
F ~ COO- CHZ- CH- CHZ CI-I3
cH3
cis xCF3COOH
H\N F
A. 7-(2-tart-Butoxycarbonyl-2,8-diazabicyclo(4.3.0]non-
8-yl)-1-cyclopropyl-6,8-difluoro-1,4-dihydro-4-oxo-
3-quinolinecarboxylic acid
L,e A 28 100 - 90 -
~fl
7.8 g (20 mmol) of 1-cyclopropyl-7-(2,8-diaza-
bicyclo[4.3.0]non-8-yl)-6,8-difluoro-1,4-dihydro-4-
oxo-3-quinolinecarboxylic acid are dissolved in a
mixture of 60 ml of dioxane/water (2:1) and 20 ml of
1 N sodium hydroxide solution and the mixture is
treated with ice-cooling and stirring with 5.24 g
(24 mmol) of di-tart-butyl pyro-carbonate. The
mixture is stirred at room temperature for 1 hour
and allowed to stand overnight. The deposited
precipitate is filtered off with suction, washed
with 250 ml of water and dried overnight at 50°C in
a recirculating air drying oven.
Yield: 9.34 g (95.5 ~ of theory),
Melting point: 216-219°C {with decomposition).
B. 2S-Methyl-1-butyl 7-(2-tart-butoxycarbonyl-2,8-
diazabicyclo[4.3.0]non-8-yl)-1-cyclopropyl-6,8-
difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylate
2 .15 g ( 4 . 4 mmol ) of the product from Step A are
susper:ded. in 60 ml of tetrahyctrofuran/water ( 1:1 ) at
room temperature and 1.65 g (5 mmol) of cesium
carbonate are added. The mixture is allowed to react
at about 40°C in an ultrasonic bath for 20 minutes,
about 40 ml of the solvent are distilled off at
40°C/12 mbar) and the solution which remains is
lyophilised, the slightly soluble crude caesium salt
being obtained. 3.3 g of this crude salt are
dissolved in 40 ml of dimethylformamide and treated
with 1.4 g of S(+)-1-bromo-2-methyl-butane and
Le A 28 100 ° 91 -
p n
~;~~;.~~:~
reacted overnight in the ultrasonic bath at 40-50°C.
The suspension obtained is concentrated, and the
residue is treated with water and extracted with
methylene chloride. After drying with sodium
sulphate, the solution is concentrated and the
residue is purified by chromatography (silica gel,
eluent: methylene chlaride/methanol 95:5).
Yield: 950 mg (38 ~ of theory),
Melting point: 72-83°C (with decomposition).
C. 2S-Methyl-1-butyl 1-cyclopropyl-7-(2,8-diazabicyclo-
[4.3.0]non-8-yl)-6,8-difluoro-1,4-dihydro-4-oxa-3-
quinolinecarboxylate trifluoroacetate
570 mg (1 mmol) of the product of Step D are
dissolved in 3 ml of trifluoroacetic acid at roam
temperature and the solution is concentrated at
60°C/12 mr..r. The viscous oil obtained is stirred
with 5 ml of ether, a solid product being obtained.
This is filtered off with suction, washed with ether
and dried at 80°C in a high vacuum.
Yield: 450 mg (78 ~ of theory),
Melting point: 214-216°C (with decomposition),
[oc]D5: +2.8° (C = 0.5, DMF).
Example 16
O
F ~. COOH
cR) I J
/ \ N \ N
CH- NH- CO F
N cis
CH3
Le A 28 100 - 92 -
390 mg (1 mmol) of 1-cyclapropyl-7-(2,8-diazabicyclo-
[4.3.0]non-8-yl)-6,8-difluoro-1,4-dihydro-4-oxo-3-
quinolinecarboxylate are dissolved in a solution of 40 mg
of sodium hydroxide in 3 ml of water at room temperature
in an ultrasonic bath and the solution is treated with
ice-cooling with a solution of 160 mg (1.1 mmol) of R-
(+)-a-methyl-benzyl isocyanate. The deposited precipitate
is filtered off with suction, washed with dioxane and
dried at 100°C in a high vacuum.
Yield: 530 mg (99 ~ of theory) of 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-4-oxo-7-(2-[1R-phenyl-ethyl-amino-
carbonyl]-2,8-diazabicyclo[4.3.0]non-8-yl)-3-quinoline-
carboxylate,
Melting points 208-210°C (with decomposition),
[a]D5: -23.2° (c = 0.5, DMF).
The reaction product can be separated into the
diastereomers by chromatography and the carbamoyl radical
removed again by acidic hydrolysis, the compounds of
Examples 1 and 7 being obtained.
Example 17
O
F ~ COzC2H5
H N ~ N
H~ F
N~ H
Le A 28 100 - 93 -
~~ y~~~~
1.52 g (5 mmol) of ethyl 1-cyclopropyl-6,7,8-trifluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylate are reacted with
550 mg (5 mmol) of 1,4-diazabicyclo[2.2.2]octane and 760
mg (6 mmol) of (+)-[S,S]-2,8-diazabicyclo-[4.3.0]nonane
in 30 ml of acetonitrile for 2 hours at 50°C and for 2
hours at 60°C. After cooling, the suspension obtained is
filtered off with suction, and the precipitate is washed
with water and dried at 90°C in vacuo.
Yields 0.99 g (47.5 ~ of theory) of ethyl 1-cyclopropyl-
7-([S,S]-2,8-diazabicyclo[4.3.0]non-8-yl)-6,8-difluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylate,
Melting points 194-195°C (from acetonitrile),
[a]D~s -188.9° (c = 0.51, CHC13) .
Example 18
O
F , COOH
J
H N ~ N
H .. O
N H ~ CHs
1.4 g (5 mmol) of 9,10-difluoro-2,3-dihydro-3-methyl-7-
oxo-7H-pyrido[1,2,3-de][1,4]-benzoxacine-6-carboxylic
acid are reacted with 0.85 g (7.7 mmol) of 1,4-
diazabicyclo[2:2.2]octane and 0.7 g (5.6 mmol) of (+)-
[S,S]-2,8-diazabicyclo[4.3.0]nonane in 15 ml of
acetonitrile/7.5 m1 of dimethylformamide analogously to
Example 1:
Yield: 1.24 g (64 ~ of theory) of 10-([S,S]-2,8-
diazabicyclo[4.3.0]non-8-yl)-9-fluoro-2,3-dihydro-3-
Le A 28 100 - 94 -
s~9 ~ L~ ?' ~.: .:i
~t~~ti~.:;.v
methyl-7-oxo-7H-pyr5.do[1,2,3-de][1,4]benzoxacine-6-
carboxylic acid,
Melting point: 265-268°C (with decomposition),
[a]D: -232,2° (c = 0.58, CHC13)
3S-10-([S,S]-2,8-~7iazabicyclo[4.x.0]non-8-yl)-9-fluoro-
2,3-dihydro-3-methyl-7-oxo-7H-pyrido[1,2,3-
de][1,4]benzoxacine-6-carboxylic acid is also obtained
analogously.
Example 19
O
COOH
~ ~~ NJ
H
H~N _
'H
OCH3
ZO 1-Cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo
3-quinolinecarboxylic acid is reacted analogously to
Example 1 and the reaction product is purified by
chromatography (silica gel, eluent: methylene
chloride/me~thanol/17 ~ strength aqueous ammonia
30:8:1).
1-Cyclopropyl-7-([S,S]-2,8-diazabicyclo[4.3.0]non-8-yl)-
6-tluoro-1,4-dihydro-8-methoxy-4-oxo-3-
quinolinecarboxylic acid of melting point 203--208°C (with
decomposition) is obtained.
[a]D3: -193° (c = 0.4, CHC13) .
Le A 28 100 - 95 -
Example 20 O
F ~ COOH
J
H N ~ N
H~ F
N ~H C2Hs
The reaction is carried out analogously to Example lA
using 1-ethyl-6,7,8-trifluoro-1,4-dihydro-4-oxo-3-
quinolinecarboxylic acid and 1-ethyl-7-(jS,S]-2,8-
diazabicyclo[4.3.0]-non-8-yl)-6,8-difluoro-1,4-dihydro-
4-oxo-3-quinolinecarboxylic acid of melting point 236-
239°C (with decomposition) is obtained (recrystallised
from glycol monomethyl ether);
[«]D3c -186.3° (c = 0.3, CHC13) .
ZO Example 21
O
F ~ COOH
~ N N N
H .F
N~H I \
x HCl
F
A. Ethyl 7-([S,S]-2,8-diazabicyclo[4.3.0]non-8-yl)-1-
(2,4-difluorophenyl)-6-fluoro-1,4-dihydro-4-oxo-1,8-
naphthyridine-3-carbo~cylate
1.9 g (5 mmol) of ethyl 7-chloro-1-(2,4-difluoro-
phenyl)-6-fluoro-1,4-dihydro-4-oxo-1,8-
naphthyridine-3-carboxylate are stirred at 10°C for
Le A 28 100 - 96 -
~~~~~.~
3 hours with 680 mg (5.4 mmol) of [S,S]-2,8-
diazabicyclo[4.3.0]nonane in the presence of 560 mg
(5 mmol) of 1,4-diazabicyclo[2.2.2]octane in 20 m1
of acetonitrile. The suspension is filtered oft with
suction, washed with water and dried. 0.35 g of
product is obtained. By concentrating the mother
liquors, stirring the residue with water, isolating
the undissolved product and purifing by
chromatography (silica gel, eluent: dichloro-
methane/methanol/17 ~ strength aqueous ammonia), a
further 0.7 g of product is isolated.
Total yield: 1.05 g (44 $ of theory),
Melting point: 184-185°C (with decomposition)
[a]D3; +6.8° (c = 0.46, CHC13) .
B. 7-([S,S]-2,8-Diazabicyclo[4.3.0]non-8-yl)-1-(2,4-
difluorophenyl)-6-fluoro-1,4-dihydro-4-oxo-1,S-
naphthyridine-3-carboxylic acid hydrochloride
0.8 g (1.7 mmol) of the product from Step A are
heated under reflux for 4 hours in a mixture of
10 ml of acetic acid and 8 ml of half-diluted
hydrochloric acid. The mixture is concentrated, the
residue is stirred with a little water, and the
precipitate is filtered off with suction, washed
with ice-sold ethanol and dried.
Yield: 0.67 g (83 $ of theory),
P3el.ting point: 324-326°C (with decomposition),
[a]D5: +10.8° (c = 0.37, 17MF') .
Le A 28 100 - 97 -
Example 22 CH3 O
F ~ COOH
H N \ N
I-IN H
0.56 g (2 mmol) of 1-cyclopropyl-6,7-difluoro-1,4-
dihydro-5-methyl-4-oxo-3-quinolinecarboxylate are heated
at 120°C for 2 hours with 0.38 g (3 mmol) of [S,S]-2,8-
diazabicyclo[4.3.0]nonane and 0.45 g (4 mmal) of
1,4-diazabicyclo[2.2.2]octane in 3.5 ml of dimethyl
sulphoxide. After cooling, the solvent is removed in a
high vacuum. The residue is taken up with acetonitrile.
The solid is separated off, washed with acetonitrile and
dried at 60 to 80°C.
Yield: 0.5 g (65 ~ of theory) of 1-cyclopropyl-7-([S,S]-
2,8-diazabicyclo[4.3.0]non-8-yl)-fi-fluoro-1,4-
dihydro-5-methyl-4-oxo-3-quinolinecarboxylate
Melting point: 217-219°C (with dec:omposition),
[a]D: -119° (c = 0.5, DMF)
Example 23
CH3 O
F , COOH
y N ~ N
NN
Le A 28 100 - 98 -
~~~i
A. 837 mg (3 mmol) of 1-cyclopropyl-6,7-difluoro-1,4-
dihydro-5-methyl-4-oxo-3-quinolinecarboxylic acid
are heated under reflex for 2 hours with 1.1 g
(10 mmol) of 1,4-diazabicyclo[2.2.2]octane and
665 mg (3.3 mmol) of 1R,6S-2-oxa-5,8-diazabi-
cyclo(4.3.0]nonane-dihydrochloride in a mixture of
m1 of acetonitrile and 5 ml of dimethylformamide.
The mixture is evaporated, the residue is stirred
with 30 ml of water, and the precipitate is filtered
10 off with suction and dried at 80°C in vacuo.
Yield: 400 mg (34 ~ of theory of 1-cyclopropyl-6-
fluoro-1,4-dihydro-5-methyl-7-(1R,6S-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid,
Melting point: 213-214°C (with decomposition).
$. 0.4 g of the betaine from Step A is dissolved in
5 ml of half-concentrated hydrochloric acid at room
temperature, the solution is concentrated and the
residue is stirred with about 3 ml of ethanol. The
precipitate is filtered off with suction and dried
at 80°C/12 mbar.
Yield: 290 mg (66 ~ of theory) of 1-cyclopropyl-6-
fluoro-1,4-dihydro-5-methyl-7-(1R,6S-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-quinoline-
carboxylic acid hydrochloride,
Melting point: 305-308°C (with decomposition),
[~]D3: -79° (c = 0.52, H20)
Le A 28 loo - 99 -
Example 24
Br O
F , COOH
J
H N W N
F ,
IAN H
~0
362 mg (1 mmol) of 5-bromo-1-cyclopropyl-6,7,8-trifluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated
under reflux for 1.5 hours with 220 mg (2 mmol) of 1,4-
diazabicyclo[2.2.2]octane and 220 mg (1.1 mmol) of 1S,6R-
2-oxa-5,8-diazabicyclo[4.3.0]nonane dihydrochloride in a
mixture of 3 ml of acetonitrile and 2.5 m1 of dimethyl-
formamide. The suspension is cooled, and the precipitate
is filtered off with suction, stirred with 30 ml of water
and dried at 90°C in a high vacuum.
Yield: 320 mg (68 ~ of theory) of 5-bromo-1-cyclopropyl-
6,8-difluoro-1,4-dihydro-7-(1S,6R-2-oxa-5,8-diaza-
bicyclo[4.3.0]non-8-yl)-4-oxo-3-quinolinecarboxylic acid,
Melting point: 263-264°C (with decomposition),
[a]n~: -t~251° (c = 0.3, CHZC12].
Example 25
O
F ~ COOH
H N \ N
CHS\N F
Le A 28 100 - 100 -
Analogously to Example 1, the following are obtained
using [S,S]-2-methyl-2,8-diazobicyclo[4.3.0]nonane:
A: 1-Cyclopropyl-6,8-difluoro-1,4-dihydro-7-([S,S]-2
methyl-2,8-diazobicyclo[4.3.0]non-8-yl)-4-oxo-3
quinolinecarboxylic acid,
melting point: 230-233°C (with decomposition)
(recrystallised from glycol monomethyl ether);
B. 1-Cyclopropyl-6,8-difluoro-1,4-dihydro-7-([S,S]-2-
methyl-2,8-diazobicyclo[4.3.0]non-8-yl)-4-oxo-3-
quinolinecarboxylic acid hydrochloride,
melting point: 258-260°C (with decomposition),
[«]D5: -216.3° (c=1, x2o) .
Example 26
o -
F , COOH
H N ~ N
CHI \ F
N~~~" H
Analogously to Example 1, the following are obtained
using [R,R]-2-methyl-2,8-diazabieyclo[4.3.0]nonane:
A: 1-Cycopropyl-6,8-difluoro-1,4-dihydro-7-([R,R]-2-
methyl-2,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-
quinolinecarboxylic acid,
melting point: 228-230°C (with decomposition)
(recrystallised from glycol monomethyl ether):
B: 1-Cyclopropyl-6,8-difluoro-1,4-dihydro-7-([R,R]-2-
methyl-2,8-diazabicyclo[4.3.0]non-8-yl)-4-oxo-3-
Le A 28 100 - 101 -
qu:inolinecarboxylic acid hydrochloride,
melting point: 258-260°C (with decomposition),
[alDS: +213.8° (c=1, Hz0) .
Example 27
O
F , COOIi
~J
N ~ N
CH3-CO-CH2CH2 ~ F
N~ H
1.95 g (5 mmol) of the product from Example lA are heated
under reflux for 4 hours with 2.1 g (30 mmol) of methyl
vinyl ketone in 50 ml of ethanol. The mixture is
concentrated, the residue is stirred with water, and the
precipitate is filtered off with suction, washed with
ethanol and dried at 100°C/12 mbar.
yield: 2.1 g (91.5 ~ of theory) of 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-oxo-7-([S,S]-2-[3-oxo-1-butyl]-2,8-
diazabicyclo[4.3.0]non-8-yl)-3-quinolinecarboxylic acid,
Melting point: 181-183°C (with decomp~sition)
(recrystallised from glycol monomethyl ether,
[a]D°:-120.7° (c = 0.57, CHZCIz)
Le A 28 100 - 102 -
Example 28 O
COOH
y
N w N
CH3-CO-CHZ N
N H F
1.95 g (5 mmol) of the product from Example 1A are heated
at 50-80°C for 3 hours with 1.0 g (10.8 mmol) of
chloroacetone and 1.3 g (13 mmol) of triethylamine in
30 ml of dimethylformamide. The solution is concentrated,
the residue is stirred with water (pH 6), and the
undissolved precipitate is filtered off with suction,
washed with water and dried at 100°C in a recirculat.ing
air drying oven (crude yields 1.3 g) and recrystallised
from glycol monomethyl ether:
Yield: 1.12 g (50 ~ of theory) of 1-cyclopropyl-6,8-
difluoropl,4-dihydro-4-oxo-?-([S,5]-2-(2-oxopropyl]-2,8-
diazabicyclo[4.3.0]]non-8-yl)-3-quinolinecarboxylic acid,
Me~.ting point: 181-184°C (with decomposition),
[«]pie-72° (c -- 0.55, CHC13) .
Example 29
F , COOH
H N ~ N
C~-f3-CO-CH2CH2 ~ ~,l
H
Le A 28 100 - 103 -
A. The product from Example 2A is reacted analogously
to Example 27 and 8-chloro-1-cyclopropyl-6-fluoro-
1,4-dihydro-4-oxo-7-([S,S]-2-[3-oxo-1-butyl]-2,8-
diazabicyclo[4.3.0]non-8-yl)-3-quinolinecarboxylic
acid of melting point 107-109°C is obtained.
[rx]D3: -53° (c = 0.67, CHC13),
purity: 99.2 ~ strength (HPLCj.
B. Rac. 8-chloro-1-cyclopropyl-6-fluoro-I,4-dihydro-4--
oxo-7-cis-2-[3-oxo-1-butyl]-2,8-diazabicyclo[4.3.0]-
non-8-ylj-3-quinolinecarboxylic acid of melting
point 224-125°C is obtained analogously using 8-
chloro-1-cyclopropyl-7-(cis-2,8-diazabicyclo[4.3.0]-
non-B-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinoline-
carboxylic acid.
Example 30
O
F , COOH
N w
CH3-CO-CHz ~ F
N C1S
~O
I.56 g (4 mmol) of the product from Example l0A are
treated with 0 . 82 g ( 8 . 8 mol ) of chloroacetone and 1 . 05 g
(10.4 mmol) of triethylamine in 30 ml of dirnethyl-
formamide and the mixture is heated at 50-80°C fox
3 hours. The yellow solution obtained is concentrated at
Le A 28 100 - 104 -
80°C/15 mbar, and the oily residue is stirrad with water
until it solidifies. The solid product is filtered off
with suction, washed with water and recrystallised from
glycol monomethyl ether.
Yield: 830 mg (47 ~ of theory) of 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-4-oxo-7-(cis-5-[2-oxopropyl]-2-oxa-
5,8-diazabicyclo[4.3.0]non-8-yl)-3-quinolinecarboxylic
acid,
Melting points 192-193°C (with decomposition).
Example 31
O
F s COOH
-
N ~ N
CH3-CO-CHZCHZ ~ F
N cis
~O
7..56 g (4 mmol) of the product from Example l0A are
heated under reflux fox 3 hours with 1.8 g (25.6 mmol) of
methyl vinyl ketone in 50 ml of ethanol. The suspension
is concentrated at 70°C/12 mbar, and the residue is
stirred with water and recrystallised from glycol
monomethyl ether.
Yield: 1.33 g (72 $ of theory) of 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-4-oxo-7-(cis-5-[3-oxo-1-butyl]-2-
Le A 28 100 - 105 -
oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-3-
quinolinecarboxylic acid,
Melting point: 188-189°C (with decomposition).
Example 32
O
F ~ COOH
J
H N w N
CZHSOZC-CHz-CHZ ~ C1
N~ H
1.95 g (4.8 mmol) of the product from Example 2A are
heated under reflux for 2 hours with 3 g ( 30 mmol ) of
ethyl acrylate in 30 ml of glycol monomethyl ether. The
mixture is evaporated, the residue is stirred with water,
and the precipitate is filtered off with suction, dried
(Crude yield: 1.9 g) and recrystallised from glycol
monomethyl ether.
Yield: 1.45 g (60 ~ of theory) of 8-chloro-1-cyclopropyl
7-([S,S]-2-[2-ethoxy-carbonyl-ethyl]-2,8
diazabicyclo[4.3.0]non--8-yl)-6-flL~oro-1,4-dihydro-4-oxo
3-duinolinecarboxylic acid,
Melting point: 117-118°C (with decomposition),
[a]pe: °103.5° (c = 0.49, DMF),
Purity: 99.6 ~ strength (HPbC).
Le 1~ 28 100 - 106 -
fi,
Example 33
U
COOH
N w I v~
H
NC-CHI-CHZ ~ C
H
1.95 g (4.8 mmol) of the product from Example 2A are
heated under reflux for 5 hours with 0.8 g (15 mmol) of
acrylonitrile in 30 ml of ethanol. ~'he mixture is
evaporated, and the residue is stirred with water, dried
(crude yield: 1.9 g) and recrystallised from glycol
monomethyl ether.
Yield: 1.6 g (73 ~ of theory of 8-chloro-7-((S,S)-2-[2-
cyanoethyl]-2,8-diazabicyclo(4.3.0]non-8-yl)-1-
cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3-quinoline-
carboxylic acid,
Melting point: 153-255°C (with dec:omposition),
[a]D': -98.6° (c = 0.53, DMF),
Purity: 96 ~ strength (HPLC),
Mays spectrum: m/e 458 (M'') , 250, 149 ( 100 ~, C9H13~2) ,
llQ~ 49.
he A 28 100 - 107 -
Example 34
O
F ~. COOH
CH300C J
H N w N
CH=C, .
N~ H
CH300 '~/C
1.95 g (5 mmol) of the product from Example lA are heated
under reflux for 2 hours with 1.2 g (8 mmol) of dimethyl
~cetylenedicarboxylate in 60 ml of ethanol. The
suspension is concentrated, the residue is stirred with
water, and 'the precipitate is filtered off with suction
and dried. the crude product (2.3 g) is recrystallised
from glycol monomethy~l etherldimethylformamide.
Yield: 2 g (74 ~ of theory) of 1-cyclopropyl-7-[2-(1,2-
me~thoxycarbonyl-vinyl)-1S,65-2,8-diazabicyclo[4,3.0]non-
8-yl]-6,8-difluoro-1,4-dihydrc~-4-oxo-3-quinoline-
carboxylic acid,
Melting point: 262--264°C (with decomposition);
[«]pG: +28.8° (c = 0.24, CH~Clz) .
Fxam~le 35
O
F ~ COOI-I
CH300C
I ~-I N ~ N
CH=C~N CI
H
cH3ooc
be A 28 100 - 108 -
~~~~"~ j;~
The product from Example 2A is reacted with dimethyl
acetylenedicarboxylate analogously to Example 34. 8-
Chloro-1-cyclopropyl-7-(2-(1,2-bis-methoxycarbonyl-
vinyl)-1S,6S-2,8-diazabicyclo(4.3.0]non-8-yl]-6-fluoro-
1,4-dihydro-4-oxo-3-quinolinecarboxylic acid of melting
point 210-212°C (with decomposition) is obtained in 87
yield;
[cx]D'': +16.6° (c = 0.5, DMF).
Example 36
O
F , COOH
J
N ~ N
CZHSOzC - CH= CH
,N
~~cis
?80 mg (2 mmol) of 1-cyclopropyl-7-(cis-2,8-diazabicyclo-
[4.3.0]non-8-yl)-6,8-difluoro-1,4-dihydro-oxo-3-quino-
linecarboxylic acid are heated under reflux for 1 hour
with 500 mg (5 mmol) of ethyl propiolate in 15 ml of
ethanol. The suspension is cooled, and the precipitate is
filtered off with suction, washed with 25 ml of ethanol
and dried at 80°C in a high vacuum.
Yield: 880 mg (90 ~ of theory) of 1-cyclopropyl-7-[2
(traps-2-ethoxycarbonylvinyl)-cis-2,8-diazabicyclo
[4.3.0]non-8-yl]-6,8-difluoro-1,4-dihydro-4-oxo-3
quinolinecarboxylic acid,
Le A 28 100 - 109 -
Melting goint: 244-246°C.
analogously to Examgle 36, the following are obtained
from the corresgonding starting materials:
Le A 28 100 - 110 -
~M
M _M M ~ _C~1
U T U ~"'
.T. ~ U . U U
,
U U U U ~
U
~i W Ui ~n vi
0 0 0 '~?o ~'
J O O
O
a ~ U
J U
~ ~
'
O O O ' o -
O M V'1~ o N o
_ 0
O M d'w0 M
L3 + CV N N
a
a
~
U o
~J 00 .--. M (~
O 00 V (V M N
O ~ ~ .,~ ('7N N N N C'1(V
~ C W ~J O N V'1
1' ~
-I ~W 00 V' _ M ('7
U
~ N r~ c'JN N N N
J
N N N N N N N
Tr ~,~
"'"' ~ U U U U U U O U
~,,
X ~ U x Z .:4~ ra..~
U M
U
U
Q U U V Z O U U
~ w U
~,
~
O H U7
y .,.1
N Id
r1
a ~ (d
a, ,~,1
~
.; U ~
a,
Q N Q ~ M Q
ro N N M tI J ,-...~,-.U
~ N
+~
-- H
O
r~I
pa O
~
M M V ~ d' ~
~e A 28 100 - 111 -
~
+
b
.,.,
b
a~
a~
.'.,
b
a
0
c~ ~ -~.,
U U
U U U
U ~i vi vi
O Q N
p
~d-~ A o ~
~.70
~ 2~ + et'V ;
i
~
~ ~
_---~ b~
p ~ M
- C
O p
- \ ~N N N
~
i~
S-I ~
/
Id
O
,'
~
ri ~.~, '' ~ r~ ,.i.'.:..
,-~-~
'?; i
~ ~U
~~ U U
~ U
U
+? ~
w
a N
~ ,~
N -e-.N d~
~ U ~
a
~ o U a
~
ro
~
~ x
N
~
~ ~
~ ..
V
O ~
ri '~
t0 O
Qt
Le A 28 100 - 112 -
~n r, .a
Example 48
O
F ~ COOH
CHOC
2 ~ / H N \ N
C=C\ CI
H '~I~ H
8-Chloro-1-cyclopropyl-7-([S,S]-2,8
diazabicyclo(4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-4-oxo-
3--quinolznecarboxylic acid is reacted with methyl
propiolate in ethanol or methanol analogously to Example
36 and 8--chloro-l~cyclapropyl-6-fluoro-1,4-dihydro-7-[2-
(trans-2-methoxycarbonyl-vinyl)-[S,S]-2,8-diazabicyclo-
[4.3.0]non-8-yl)-4-oxo-3-quinolinecarboxylic acid of
melting point 220-222°C (with decomposition) is obtained,
(a]D4: +8.2° (c = 0.5; CHC13).
Exa.m~le 49
F , COOH
H ~ ~
N N
C = C\ o~~CI
H ~'~~O'~'~
407:5 g (1 mmol') of 8-chlorc~-1-cyclopropyl-6-fluoro-1,4-
dihydro-7-(15,6R-2-oxa-5,8-diazabicyclo[4.3.0]npn~8-yl)-
Le A 28 100 - 113 -
5
4-oxo-3-quinolinecarboxylic acid (from Example 11E) are
heated under reflux for 1 hour with 210 mg (2.5 mmol) of
methyl propiolate in 10 ml of methanol. The mixture is
concentrated and the isolated crude product (450 mg) is
recrystallised from 4 ml of acetonitrile.
Xielde 8-Chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-7-[5-
(traps-2-methoxycarbonyl-vinyl)-15,68-2-oxa-5,8-
diazabicyclo[4.3.0]non-8-yl]-4-oxo-3-quinolinecarboxylic
acid,
Melting point: 153-156°C (with decomposition),
L«]D8o +36° (c = 0.5, CHC13).
Example 50
F O
F , COOH
H \~ J _
cH3o2c ~ ~ ~ N
C-C~ ~ F
N
H ~ O cis
Reaction with the compound of Example 13A is carried out
analogously to Example 49 and 1-cyclopropyl-5,6,8-
trifluoro-1,4-dihydro-7-[5-(traps-2-methoxycarbonyl-
vinyl)-cis-2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl]-4-oxo-
3-quinolinecarboxylic acid of melting point 169~170°C
(with decomposition) is obtained (from glycol monomethyl
ether).
Le A 28 100 - 114 -
Example 51
O
F COOH
i
H
CH j02C, / H
C C\ ~~ p
H ~O H
Reaction is carried out with the compound from Example
10E analogously to Example 49 and 1-cyclopropyl-6,8-
difluoro--1,4-dihydro-7-[5-(traps-2-methoxycarbonyl-
vinyl)-1S,6R-2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl]-4-
oxo-3-quinolinecarboxylic acid of melting paint 230-234°C
(with decomposition) i obtained (from glycol monomethyl
ether); _
[«]~e= -27° (c = 0.5, CHC13) .
Example 52
L~r O
F COOI-~
CH30zC\ HH N ~ ~
C-C\ F
H~ ~ O N
Reaction is carried out with the compound from Example
24 analogously ~o Example 49 apd 5~bro~o-l~cyclopropyl-
6,8-difluoro-1,4-dihydro-7-[5-(traps--2-methoxycarbonyl-
~e A 28 100 - 115 -
v
vinyl)-1S,6R-2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl]-4-
oxo-?-quinolinecarboxylic acid of melting point 158-160°C
(with decomposition) is obtained (from isopropanol):
(a]De: +8° (c = 0.27, CHC13) .
Example 53
O
F C02CzHs
H
CH302C ~ / H N ~ ~ N
C = C \ ~~~ F
N H
H ~O
Reaction is carried out with the compound from Example
17 analogously to Example 36 and methyl 1-cyclopropyl-7-
[2-(trans-2-ethoxycarbonyl-vinyl)-1S,6S-2,8-
diazabicyclo(4.3.0]non-8-yl]-6,8-difluoro-1,4-dihydro-4-
oxo-3-quinolinecarboxylate of melting point 168-169°C is
obtained.
Example 54
F O
F COOH
i
CO2C~H5
H N N
C.,H502C - CH= C\ ~) F
N ,H
O
Le A 28 100 - 116 -
818 mg (2 mmol) of 1-cyclopropyl-5,6,8-trifluoro-1,4-
dihydro-7-(1R,6S-2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)-
4-oxo-3-quinolinecarboxylic acid (from Example 13B) are
treated with 680 mg (4 mmol) of diethyl acetylene-
dicarboxylate in 15 ml of ethanol and the mixture is
treated in an ultrasonic bath at 30°C for 1 hour. The
suspension is filtered off with suction, and the
precipitate is washed with ethanol and dried at 70°C in
a high vacuum.
Yield: Bg0 mg (77 ~ of theory) of 1-cyclopropyl-7-(5-
(1,2-bis-ethoxycarbonyl-vinyl)-1R,6S-2-oxa-5,8-
diazabicyclo(4.3.0]non-8-yl]-5;6,8-trifluoro-1,4-dihydro-
4-oxo-3-quinolinecarboxylic acid,
Melting points 220-222°C (with decomposition) (tram
glycol monomethyl ether)
(a]a5: -57° (c=0.5, CHC13). _
Example 55
O
COOH
N \ ~N~
CaHsO2C~
~N~ F
traps, rac.
O
The reaction is carried out with 1-cyclopropyl-6,8-
difluoro-1,4-dihydro-7-(traps-2-oxa-5,8-diaza[4.3.0]non-
8-yl)-4-oxo-3-quinolinecarboxylic acid analogously to
Le A 28 100 - 117 -
Example 36 and 1-cyclopropyl-7-[5-(traps-2-ethoxy-
carbonyl-vinyl]-traps-2-oxa-5,8-diaza[4.3.0]non-8-yl]-
6,8-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid
is obtained.
Melting point: 266-268°C (with decomposition) (from
glycol monomethyl ether).
EXample 56
O
F ~ COOH
J
CH300C ~ N
F
N ~rans rac.
t ,
O
1-Cyclopropyl-6,8-difluoro-1,4-dihydro-7-(traps-2-oxa-
5,8-diaza[4.3.0]non-8-yl)-4-oxo-3-quinolinecarboxylic
acid is reacted with methyl propiolate analogously to
Example 36 and 1-cyclopropyl-7-[5-(traps-2-methoxy-
carbonyl-vinyl)-traps-2-oxa-5,8-diaza[4.3.0]non-8-yl]-
~,8-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid
is obtained.
Melting point: 275-277°C (with decomposition).
he A 28 100 - 118 -