Note: Descriptions are shown in the official language in which they were submitted.
TIT~ '
Heterobicyclic Compounds
DESCRIP~ION
The invention relates to heterobicyclic compounds and to
pharmaceutical compositions containing themO
Flavoxate, which is 8-(2-piperidinoethoxycarbonyl)-3-methyl-4-
-oxo-2-phenyl-4H-l-b~nzopyran, and has the formula
O , .:
rT~
COOCH2CH2 N
/
is used as a pharmaceutical agent for urinary tract disturbances
as it possesses a smooth muscle relaxing activity attxibutable
to its calcium antagonist activity. This activity is exerted on
~he bladder dome smooth muscles or can be related to the
micturition centre in the cen~ral nervous system.
The compounds of the invention, described below, essentially
include more complex amino moieties in place of the piperidino
group. Further changes include alternatives to the
ethoxycarbonyl group which spaces the amino moiety from the
benzopyran ring, alternative 2-, 3-, 6- and 7-substitution
patterns in the benzopyran ring, replacement of the ring
h~teroatom by a sulphur atom or by a sulphinyl, sulphonyl or
imino group, and/or 2,3 dihydrogenation of the benzopyran ring.
These structural variations give the new compounds a capability
of interacting with different biological systems, as supported
, ... , . ; .. , . .. ,~., ... ;. ... . . .
-2- 20~Q~
by the affinity of the new compounds for the ~1-adrenergic and
5HT1A-serotoninergic receptors. Flavoxate is practically devoid
of affinity for these receptors.
COMPOIJNDB O~ THE INVENTIO~Xi .
' :'
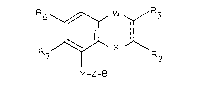
The compounds of the invention have the general formula I
~ ;. ,.
R7~ X ~\R
Y-Z-8 , ~ ;
i '
wherein
~ reprPsents a single or double bond; ~::
X represents an oxygen or sulphur atom or an imino,
alkylimino, sulphinyl or sulphonyl group;
W represents a valence bond or a carbonyl, thiocarbonyl,
methylene or hydroxymethylene group;
R2 represents a hydrogen atom or an alkyl, substituted alkyl,
alkenyl, substituted alkenyl, al}cynyl, substituted alkynyl, :
carbocyclyl, substituted carbocyclyl, heterocyclyl,
su~stituted heterocyclyl or aroyl group; the substituentq
for the a~oresaid substituted groups being one or more
halogen atoms and/or one or more alkyl, cyano, hydroxy,
alkoxy, phenyl, phenoxy, trifluoromethyl, nitro, amino,
alkylamino, dialkylamino, acylamino, alkylsulphonylamino or
benzoyl groups;
R3 xepresents a hydrogen atom or an alkyl, hydroxyalkyl, -
alkoxyalkyl, aralkoxyalkyl, phenyl, hydroxy, alkoxy or
aralkoxy group;
R6 represents a hydrogen or halogen atom or a nitro, amino,
alkylamino, dialkylamino, acylamino, alkylsulphonylamino,
cyano, hydroxy, alkoxy or alkyl group;
represents a hydrogen atom or an alkoxy group;
Y represents one of the following groups, each of which is
, " ., . . ' ~ ,' ,,,. ' ' 'i ', ,,, ' ', , ,'~ , ~,, .
'. ' '' ' ' . ;,, ~., .': "' ' ',', ,'.' ' ' ' ' ',', : , ' ' . ,.. ,. '.. ~ " , ,., ',., ,' ,'' ' . ' ';. ;'
~3~5~
,,
--3--
depicted with its le~t hand end being the end which
attaches to the heterobicyclic ring and its right hand end
being the end which attacheFi to the ~roup Z:
(Yl) -CO-,
(Y2) -Co~-,
(Y3) -CONH-,
~Y4) ~CON(CH3)-,
(Y5) -CON(OH)-,
(Y6) -CH(OH)-,
(Y7) -CH(OAlkyl)-,
(~8) -CH=CH-,
(Y9) -CH=CH-Coo-,
(Y10) -CH=CH-CONH-,
(Y11) -CH=NO-,
(Y12) -CH2-~
(Y13) -CH2COO-,
: (Y14) -CH2CONH-,
(Y15) -CH2NH-~
(Y16) -CH2N(CH3)-~
(Y17) -CH2N(CO~H3)-~
(Y18) CH2N~CONH2) l
(Y19) -CH2NHCO-, -
(Y20) -cH2N(cH3)co-~ `~
(Y21) -CH2NH-CONH-,
(Y22) -CH2NHS02-
(Y23) -CH20-,
(Y24) -CH2S-,
(Y25) ~CH2SO-,
(Y26) -CH2SO2-~
(Y27) -CH2SO2NH-~
(Y28) -CH2SO2N(CH3)-,
(Y29) -NH-,
(y3o) -N(CH3)-,
(Y31) -N(COCH3)-,
(Y32) -N(CONH~
(Y33) -NHCO-,
(Y34) -N(CH3)CO-, .
(Y35) -NH-CONH-,
(Y36) -NHSO2-,
:,:
2 0 9 0
r
4-
~Y37) -o-, :
(Y3~) -S ,
(Y39) -SO-,
(Y40) -SO2-, -
(Y41) -SO2NH-,
(Y42) -SO2N(CH3)-~
(Y43) -CONHO~
(Y44) -CON(COCH3)-,
(Y45) -CSNH-, -
(Y4~) -CSN(CH3)-, :
(Y47) -CON(O r
: ~ ....
(Y4~ HCOO-, and ~. :
(Y49) -COS-;
: ~ ~ Z represents a linear or branched chain alkylene group having
: from 1 to 6 carbon atoms and optionally having one hydroxy .-
substituent; and -
B represents one of the following groups~
: (Bl)
~: .
N N - A
Q
~: ~wherein Q represents a methylene or ethylene group and ~ .
~ A represents one of the following groups~
: ~ (A1) a phanyl group substituted by one or more halogen
:~ atoms andlor one or more alkyl, alkoxy or hydroxy
groups, ~ .
(A2) a 2-pyrimidinyl group, and
(A3) a group of the general formula
~ E
wherein _ is as above defined and E represents
an oxygen atom or a valence bond, ::
''/ .'. "'",' ;''' ,'' , "' ''' ,'" '' ''''`~'" '' ' ' '''' .'
2 ~ 3 r~
( B2 ) ~ L
/~L2
wherein each of L1 and L2 independently represents a
hydrogen atom, a phenyl, 4-fluorohenzoyl or 2-oxo-
-l-benzimidazolinyl group, or a group of the general
formula (CH2)n-0-A wherein n is 0, 1 or 2 and A is as
above defined, with the proviso that L1 and L2 do not
: both represent hydrogen atoms,
: (B3)
12 Rll
wherein each of Rlo and Rll independently represents a
hydrogen atom or an alkoxy or alXylthio group, R12 ~:
represents a hydrogen atom or an alkyl group, and n is ~ -
2 or 3,
: (B4)
R13
2~ ~
O ~ :
wherein R12 is as above defined and R13 represents a ~:~
hydrogen atom or an alkoxy group, and
(B5) ~ :`
R12 ~
wherein R12 is as above defined.
--6--
When = represents a double bond, W represents a carbonyl
group and X represents an imino group, the ring may undergo
tautomerization to give a 4-hydroxy-quinoline structure; such
tautomeric compounds are included within the invention.
The invention also includes prodrugs, enantiomers,
diastereoisomers, N-oxides and pharmaceutically acceptable salts
of the compounds of the general formula I.
The term "prodrug" is used herein to mean a derivative of a
compound of the general formula I in which reactive groups such
as amino, imino or hydroxy groups (particularly as or within the
W, R2, R3, R6, Z, B~ and B2 moieties) or acidic imino groups
(e.g. those present in Y3, Y10, Y19, Y27, Y36 and Y41) have been
masked; the derivative releasing the compound itself within a
living body and ~hus having the same pharmacological effect on
the body as the compound itself. Prodrugs may be prepared so as
to alter the pharmacokinetic properties of the compound, for
instance so as to prepare a delayed release or sustained release
form of the compound, or otherwise modify the metabolism,
adsorbtion, distribution or plasmatic half-life of the compound.
The compounds prepared in Examples 114 and 120 to 122 below are
examples of prodrugs.
The group
R6 ~ w~R3
will be abbreviated hereinafter as Fl. The preferred values of
the substituents in the group Fl are as follows:
~ a double bond,
X: an oxygen atom,
W: a carbonyl group,
R2: a phenyl group,
R3: a methyl group,
R6: a hydrogen atom, and
; ` ,-, , ,': - .,, ~ ,' ' ' " .: ' '' '; ''; ' ' ', ~', ' - '
. ., ., ~ . ~ , . , . . . .~ . .
2 0 ~
--7--
R7: a hydrogen atom.
The group having all these preferred substituents is the
3-methyl-4-oxo-2-phenyl-4H-1-benzopyran-8-yl group.
The preferred groups which Z may represent are trimethylene and
tetramethylene. Y preferably represents one of the groups Y2,
Y3, Y37~ Y40, Y41 or Y42. B preferably represents one of the
groups Bl or B3, especially the 1-(2-methoxyphenyl)-piperazinyl
groupO
Conventional abbreviations used herein include Me for methyl, Et
for ethyl, Ac for acetyl, Alk for alkyl, THF for
tetrahydrofuran, DMF for dimethylformamide, and DMSO for
dimethylsulphoxide.
The adrenergic antagonist activity of tha compounds of the
invention renders them useful as agents acting on body tissues
particularly rich in ~1-adrenergic receptors (such as blood
vessels, prostate and urethra). Accordingly, anti-adrenergic
compound within the invention established as such on the basis
of their receptor binding profile, may be useful therapeutic
agents for the treatment, for example, of hypertension and
micturition troubles associated with obstructive disorders of
the lower urinary tract, including but not limited to those
caused by benign prostatic hypertrophy.
The serotoninergic activity of the compounds of the invention
renders them useful as agents acting on tissues, particularly in
the central nervous system, where 5HT1A receptors are
functioning. 5H~1~ receptors are believed to regulate the action
and release of serotonin as well as the release of other
neuromediators and are found both pre- and post-synaptically.
The compou~ds of the invention have biological acti~ity in
blocking binding between these receptors and their various
specific ligands (e.g. serotonin). Accordingly, the compounds of
the invention which interact with the 5Hr1A receptor
(established as such on the basis of their receptor binding
profile) may be use~ul therapeutic agents for the treatment of
anxiety disorders and depression.
. .
~ ~ "
~: ,:,, . .,, :
, i ' ' ,' , ' 1';. , , ! ' ' ,
--8--
Surprisingly, the compounds of the invention (especially those
displaying affinity for both the ~l-adrenergic and the
5HT1A-serotoninergic receptors) show high selectivity for the
mammalian lower urinary tract, i.e. they are substantially more
active in antagonizing urethral contractions than in lowering
blood pressure. On the contrary, known ~1-antagonists, such as
prazosin, which is 1-(4-amino-6,7-dimethoxy-2-quinazonlinyl)-
-4 (2-furoyl)-piperazine (GB 1156973), do not exhibit such
selectivity (and in fact cause hypotension as a most common side
effect) while flavone derivatives structurally similar to
flavoxate, such as terflavoxate, which is
8-(1,1-dimethyl-2-piperidino-ethoxycarbonyl)-3-methyl-4-oxo-2- -~
-phenyl-4H-1-benzopyran hydrochloride (EP 0072620), have no ~
effect on urethral contractions. Naturally, those compounds of ,~ ;-
the invention that are nst selective for the lswer urinary tract
are preferred as antihypertensive agents, but even the selective
compounds can often be used as antihypertensives because of
their low tsxicity.
. ..
Compounds of the invention have also shswn a good antagonist
effect against contractions of rat bladder strip induced by -
pstassium chloride. This effect can be attributed to a calcium
antagsnistic activity, and renders the new compounds useful as
spasmolytics of the lower urinary tract (i.e. useful in the
treatment of urinary incontinence, urge syndrome and other
similar disorders). ~-~
~ . .
The majority of the compounds of the invention exhibit low
toxicity. Thus they can be used in higher amounts, an advantage
that often more than compensates for a relatively lower level of
activity that some of these compounds have. Naturally, those
compounds exhibiting both high activity and low toxicity are
preferred.
The invention further provides a pharmaceutical composition
compri~ing a compound according to the invention, or a prodrug,
enantiomer, diastereoisomer, N-oxide or pharmaceutically
acceptable salt of such a compound, in admixture with a
'' ' : .' ' .' ' : .' . ': ' ' : ' .
.. . . .. .
9 2~9~ 5~
pharmaceutically acceptable diluent or carrier.
8YNT~BE3I~ OF T~3 CONPOUNDB OF THE INYEMTI03!1
The compounds according to the invention may generally be
prepared (except when the groups R6 and the substituents at R2
are OH, N~2 or aminoalkyl and Y = Y15 or Y29) as follows:
Path a:
By condensing compounds Fl-Y-Z-L, wherein L represents a halogen
atom or a leaving group such as a tosyloxy group, with a
compound H-B. The condensation is preferably, but not
necessarily, carried out at a temperature within the range of
20-140C in a polar solvent such as dimethylformamide or
methanol, usually in the presence of a base such as potassium
carbonate. Such condensations are illustrated in Examples 1 to
3, 7 to 9, 11, 13 to 16, 21, 23 to 31, 38 ~o 42, ~6 ~o 49, 54 to
59, 69, 73, 77, 78 and 84 below. See also Gibson's chapter in
Patai , The Chemistry of the Amino Croup, p.45 et seq., Wiley
Interscience, New York, 1968.
An alternative method for the preparation of the compounds of
the invention is condensation (under the same conditions
described in the preceding paragraph) of a compound Fl-Y-H with
a compound L-Z-B wherein L is as above defined. This
condensation is illustrated in Examples 5, 6, 66, 79 and 81
below. By this route, compounds having Y = Yl5 or Y29 can also
be prepared (see Gibson's chapter in Patai, supr~).
Compounds of formula (I) bearing a NH2 group in R6 or as
sub tituent in R2, may be prepared by reduction of the
corresponding compounds (I) wherein R6 or the substituent in R2
are NO2 groups. Such reduction can be carried out:
- with Ni-Raney catalyst in a protic solvent selected ~rom
methanol, ethanol, isopropanol, water and mixtures of them;
or
- with SnCl2, H2O, optionally in presence of hydrochloric acid,
either in a protic solvent such as methanol, ethanol,
isopropanol, water, acetic acid and mixtures of them, or in
an aprotic solvent such as ethyl acetate; or
- with Fe and aqueous hydrochloric acid in a protic solvent
such as methanol, ethanol, isopropanol, water and mixtures of
- 2 0 ~
-10
them.
The temperatures of the above reactions will be chosen in a
range b~tween 20C and 100C. (J. March, Advanced Organic
Chemistry, III Ed., page 1103, Wiley Interscience, 1985).
Examples of this reduction are given in Examples 94 and 124.
Compounds of formula (I) having a NHAlk group as a R6
substituent can be prapared by monoalkylation, starting from the
corresponding parent compounds (I) where R6 = NH2. For example,
this may be done by first reacting the amino compound (I~ with
an excess of trifluoroacetic anhydride, then reacting the
obtained trifluoroacetyl derivative with an alkyl-L reagent and
finally unprotecting the thus-obtained trifluoroacetyl-alkylated
derivative by treatment with potassium carbonate in methanol or
with sodium borohydride in methanol or dimethylsulphoxide. These
reactions are clescribed in Examples 32 and 33, where they were
carried out on Y groups. -
Alternatively, compounds of formula I having a NHAlk or N(Alk)2
groups as a R6 substituent or as a substituent on the phenyl
group in R2 can be obtained by alkylation of the corresponding
parent compounds (I) where R6 = NH2 with the appropriate
alkanals in presence of a reducing agent, such as sodium
cyanobsrohydride. Descriptions of these reactions are gi~en in
Examples 96 and 97 below.
Compounds bearing a OH group as R6 or as a substituent in R2 may
be prepared starting from the corresponding parent compounds (I)
alkoxy-substituted at said positions. This can be accomplished
by treating the parent compounds for example, with BBr3 in
dichloromethane at 0-40C (T~ W. Greene, Protective Groups in
Organic S~nthesis, page 87, Wiley Interscience, 1981) or
according to other methods described in the same reference.
Compounds o~ khe general formula I in which ---- represents a
single bond can be obtained either by selective hydrogenation of
the corresponding compounds in which ~ represents a double
bond or by conversion of appropriate starting materials in which
the 2,3-bond is already saturated, such starting materials being
ob~ainable according to Reaction Schemes 4, 6 to 9, 11, 12 and
14 below. The latter route is illustrated in Example 87 below,
and is particularly preferred when the compound concerned
, ' ,, '', ' ' , . , ' '.~ ' ' ''' ' ', ' ,~,' ' ' ; ' ' ~
%o~al~
contains a nitro group, as hydrogenation could convert the nitro
group to an amino group. The selective hydrogenations can ~e
carried out using alternatively:
- hydrogen in presence of a metal or metal oxide catalyst (e.g.
palladium on charcoal, or platinum dioxide) in a protic
solve~t at 20-120C (E. H. Rodd, Chemistry of Carbon
Compound~, Vol. IVB, page ~03, Elsevier, 1959); or
- di(isobutyl)aluminium hydride in an aprotic solvent (e.g.
tetrahydrofuran and/or dichloromethane) at -70 to 0C (H.
Sarges et al., J. Med. Chem., 33, 1859, 1990).
Compounds in which W represents a hydroxymethylene group and the
2,3-bond is saturated can be obtained by reduction with sodium
borohydride o~ corresponding compounds in which W represents a
carbonyl group and the 2,3-bond i~ saturated, as reported in
Example 123 below~
In some cases, the compounds of th~ general formula I may be
prepared by conversion o~ other (parent) compounds of the
invention. Such conversions include: .
Path b_ Fl~CO-Z-B - Fl-CH(OH)-Z-B
by reduction as illustrated in Examples 17 to 20 below,
Path c- Fl-CH(OH)-Z-B ~ Fl-CH~OAlkyl)-Z-B
by etherification as illustrated in Example 22 below,
Path d: Fl-(CH2)~-NH-Z-B ~ Fl(CH2)n-N(CH3)-Z-B : -
where n = 0 or 1, by N-methylation as illustrated in
Ex~mple 35 below,
Path e: Fl-(CH2~n-NH~Z-B Fl-(cH2~n-N(cocH3~ Z B
where n = 0 or 1, by N-ace-tylation as illustrated in
~xample 36 below,
Path f: Fl-(CH~)n-NH-Z-B ~ Fl-(CH2)n-N(CONH2)-Z-B :
where n = 0 or 1, by reaction with potassium isocyanate
as illustrated in Example 50 below,
Path a: Fl-CH(OH)-Z-B ~ Fl-CO-Z-B
by oxidation, as illustrated in Example 51 below,
Path h Fl-Y-Z-B - Fl-Y-Z-B(N-oxide) . :
by oxidation as illustrated in Exampl~s 43 and 122
below,
H2N Fl-Y-Z-B ~ CH3CONH-Fl-Y Z-B
(wherein H2N-Fl represents a Fl group i.n which R6 is an
~ ;' ' .
.. . . .
2 ~
. -12-
amino group or R2 includes an amino group) using the
N-acylation method described in Examples 36 and 95
below.
Path j: Fl(R6 = NH2)-Y~Z-B ~ Fl(R6 = C~3SO2NH)-Y-Z-B
by amidification using the method described in Example
112 below.
Path k: Rl R
Fl-Y Z-NH-(cH2~n~o ~ ~ Fl-Y-Z-N(Alk)-(CH2)n-O
or Rll Rll
:: ~l3 ~13 ~:
Fl Y z NH - CH2 r ~ Fl-Y-Z-N(Alk)-CH2 ~ ~ ~
o~ , .
Fl-Y-Z-NH--CNa'~-- Fl-Y-Z-N(Alk)-CE12~ ;
by N-alkylation using th~ procedure described in ~--
Examples 35 and 62 below.
Some compounds may be prepared by addition reactions. For ~ ;
example those in which Z contains a hydroxy substituent may be
prepared by addition across an epoxy group.
~E~h_L~ Fl--Y--CH2--CH--CH2 + H--B ~ Fl-Y-CH2-CH(OH)-CH2-B
as illustrated in Example 45 hereinbelow.
,~ ':: . .
Addition across a double bond is also possible, e.g.:
Path m: Fl-Y-CH=CH2 + H-B ~ Fl Y-CH2-C~2-B
as illustrated in Examples 37, 63 and 82 below. :~
Other ~ynthetic schemes include the formation of ~, Z or B
during the reaction, for example: :
Path n:~ Fl-(X)-(Q)-Cl + A-HN-Z-B ~ Fl-(X)-(Q)-N(A) Z-8
(wh~rein X = bond, CH2 or CH=CH, Q = CO or SO2, and A =
H, alkyl or Pr wherein Pr is a protective group) as
illustrated in Example 12 (particularly preferred) and
.
j . .; ; ;,;
~Q~ ~g
in Examples 60, 61, 64, 67, 68, 72, 87, 88, 93, 98,
116, 129 and 130.
The same compounds may also be prepared by other routes
including:
Fl-(X)-COOH + A-NH-Z-B in presence of a coupling agent
(e.g. dicyclohexylcarbodiimide, N,N'-carbonyl-
-diimidazole or diethyl cyanophosphonate) optionally in
the presence of a promoting agent ( 2 . g .
4-dimethylaminopyridine or N-hydroxybenzotriazole) in
an aprotic or a chlorinated solvent (e.g.
dimethylformamide, chloroform) at -10/140C (Albertson,
Org. React., 12, 205-218, 1962; Doherty et al., ~.
Ned. Chem., 35, 9, 1992; Staab et al., Newer Methods
Prep. Org. Chem., 5, 61, 1968; Ishihara, ChemO Pharm.
Bull., 39, 3236, 1991); as illustrated in Examples 80,
86, 89, 90, 92, 99 to 111~ 113 to 115, 117 to 119 and
128.
Fl~(X)-COOH t A-NH-Z-B without a solvent at 150-220C
(Mitchell et al., J. Am. Chem. Soc., 53, 1879, 1931) or
in high-boiling ether2al solvents (e.g. diglyme~;
Fl-(X)-COO-Alk + A-NH-Z-B optionally in the presence of
a coupling agent ~e.g. trimethylaluminium) in an
aprotic and/or a chlorinated solYent (e.g. hexan~,
dichloromethane~ at - 10/80''C, or without solvents at
80-180C (S. M. Weinreb et al., Tetr~h~dron, 4171,
1977); M. F. Lipton et al., Org. S~nth. 59, 49, 1979);
Fl-(X~-COOH + alkylchloroformate in the presence o~ a
tertiary amine (e.g. triethylamine) followed by
addition of A-NH-Z-B at 0-80C; optionally a promoting
agent (e.g. 1-hydroxypiperidine) may be added before
the amine addition (Albertson, org. React., 12, 157,
1962).
Path o Fl-COCl + HS-Z-B -~ Fl-Y49-Z-B
Path p: Fl-COCl ~ HO-Z-B ~ Fl Y2-Z-B
as illustrated in Example 10 below.
Path q: FlCHO + H2NO-Z-B ~ Fl-Yll-Z-B
as illustrated in Example 70 below.
Path r: Fl-CHO + A-HN-Z-B - Fl-CS-N(A)-Z-B
(where A = H or CH3) in the presence of sulphur in an
2 0 ~
-14-
aprotic solvent, e.g. DMF or pyridine, at 60-120C (M.
Carmack et al., Org. Reaction., 83, 1947; R. Benassi et
al., Org. MagnO Res., 15, 25, 1981), as illustrated in
Example 83 below. -
Path s. Fl-N~2 + HCO~Z-B ~ Fl-Y29-Z-B
as illustrated in Example 34 below.
Path t: Fl-Y-CH3 + HO-CH2-~ ~ Fl-Y-CH2-CH2-B
as illustrated in Example 4 below.
Path u Fl-CH=CH-CONH2 ~ HOCH2-B ~ Fl-Y10-CH2-B.
Path v:
Fl-Y-2-N3 + HCO-CH2-O ~ ~ Fl-Y-Z-B3
Rll -
under reducing conditions as illustrated in Example 44
below.
Path w: R~
Fl-Y-Z~NH2 + L~(C~2)n~ ~ - Fl-Y-Z-B3
: ~ -, '.
Rll
as illustrated in Examples 74, 75 and 76 below.
~13
Fl-Y-~-NH2 + L-CH2~ ~ I Fl-Y-Z-B4
as illustrated in Example 52 below. ;
Path x: -
_~_æ_NH2 + C~ ~ ~ Fl-Y-Z-B5
as illustrated in Example 65 below.
Path y Fl-Y-Z-CHO + HB - Fl-Y-Z-B
as illustrated in Example 53 below.
Persons skilled in the art will be aware that all the above
synthetic pathways b) to y) might be simplified provided that
the reacting intermediate does not bear further groups sensitive
to the same reactants (for example: CO, NH2, NHAlk or OH
groups). Compounds of Pormula I bearing such reactive groups can
be prepared through paths b) to y) provided that the reactive
groups present in the starting materials are protected
beforehand and then deprotected after the reaction as
illustrated in Example 71. Several examples of protection and
, , ~, , , , . ~" :: . , ,. ~ ::
2~9~
-15-
deprotection for various reactive groups can be found in T.W.
Green, Protective Groups in Organic Synthesis, Wiley
Interscience, 1981 (2nd Edition 1991).
Alternatively, unreactive groups (e.g. NO2) can be left
unconverted during the first reaction and then convert d to
reactive ones (e.g. NH2) as a final step of the pathway. See
for example path a).
Which synthetic technique will be pre~erred depends on the
compound desired to be synkhesized, but path n) is generally
preferred for the compounds that can be made by it. Additional
synthetic methods will be apparent to those skilled in the art.
~AR~ING ~A~RIAL8
The starting materials (Fl-Y-Z-L and Fl-Y-H and others~ used in
the preparations described above, may themselves be prepared
from simple compounds such as Fl-COOH, Fl-CHO, Fl-COCl, Fl-NH
and Fl-OH by transformations known to those skilled in the art.
Numerous such transformations are described in detail
hereinbelow. Many of these simple compounds (Fl-COOH, Fl-CHO,
Fl-COCl, Fl-NH2 and Fl-OH) are commercially available or their
synthesis has been reported in the literature. Those which are
not available may be synthesized according to one or more of the
following Reaction Schemes 1 to 16.
' ' ''
Reaction Scheme 1 leads to compounds in which W represents a
carbonyl group and X represents an oxygen atom. ;
.
2~9~
`` -16-
'.
SCHEME l ` ~ ~
: ~ o `
F.,~OH R~R,
A 1 b ~ B 6
; A ~ 3
\ 1 ,
o
R ~ R3 d Fl; \~ R2
4~ ~;
:
A _ CO2CH3. CO2C2H5, NO2, GH=CHCH3,
B = co2H1 NH2
,.: ` '':
,. :., ~ ,,
:
~' .
--17--
Ste~ la
Procedure without isolation of the intermediate phenyl ester:
- R3CH2COCl or (R3CH2CO)2O and a Lewis acid (e.g. AlCl3 or
ZnCl2), without solvent or in an aprotic solvent (e.g.
nitrobenzene or chlorinated solvent) at 20-180C.
Procedure with isolation of the intermediate phenyl ester: ~-
- R3CH2COCl or (R3CH2CO)2O heated with the starting material or
other esterification methods, such as the Schotten-Bauman ;~
procedure. The isolated ester is then heated in nitrobenzene
or another non protic solvent (e.g. a chlorinated solvent~,
or without any solvent, at 20-180C, in the presence of a
Lewis acid such as AlC13 or ZnC12 (A.M. Blatt, Org. React.,
1, 3~2, 1942).
Ste~ lb
- R2COCl or (R2CO)2O and R2COONa alone or in a high-boiling
non-protic solvent (such as o-dichlorobPnzene) at 150-220C;
this reaction also allows the direct transformation of
compounds (2) to compounds (6), when compounds (2) have ;~
A=COOH;
- R~C(OAlk~3 in the presence of HCl04 at 20 40C or in pyridine -
in the presence of piperidine at 60-80C;
- R2COCl or (R2CO)2O in a chlorinated solvent at -10 to 120C
in the presence of a base such as 1,8-diazabicycloundecene
(DBU~.
Step lc
R2COCl in pyridine at 20-100C or in a non-protic solvent at
0-~80C, optionally in the presence of a base such as NEt3 or
4-dimethylaminopyridine.
Ste~ ld
- K2CO3 in acetone or methyl ethyl ketone at 20-80C;
- NaH in DMSO or THF at 0-40OC;
- KOH or potassium t-butoxide in pyridine at 20-100C.
Step le
- HCl or H2SO4 in AcOH at reflux or in an alcohol (MeOH, EtOH,
isopropanol) at 20C to reflux temperature;
- CF3COOH in dichloromethane at 20-40C;
- p-toluenesulphonic acid in benzene or toluene at reflux.
St~ lf ~,
- R2COCl and K2CO3 or KOH in water and a phase transfer
'; , ,,', ",'' .' ':' ~'~'
-18-
catalyst in benzene or toluene at reflux;
- R2COOAlk and lithium bis(trimethylsilyl)amide or lithium
diisopropylamide in THF at -78 to 0C. . . .
steP lq
When A is a COOCH3 or COOC2H5 group~
- NaOH in aqueous EtOH at 0-75C;
- LioH in aqueous DMF, MeOH or THF or a mixtur thereof at :,
1~100C;
- ~Cl in an aprotic solvent such as dioxane at 60-120C.
When A is NO2:
- Reduction with Ni Raney catalyst in a protic solvent (e.g.
isopropanol) or a mixture of protic solvents at 20-100C;
- Reduction with hydrogen and a catalyst (e.g. Ni-Raney or
Pd/C) in a protic solvent (e.g. MeOH, EtOH, isopropanol or a
mixture thereof) at 20-100C; .
- Reduction with SnC12 in the presence of aqueous HCl in a
protic solvent (e.g. AcOH) at 20-100C;
- Reduction in the presence of Fe and aqueous HCl in a protic
solvent at 20 100C.
When A is a CH=CHCH3 group:
- Oxidation with Na2Cr2O7 or other oxidizing agents such as
KMnO4 in acetone/H~SO4 at 0-100C.
Reaction Scheme 2 leads to compounds in which X xepresent~ a ~ :
sulphur atom or a sulphinyl or sulphonyl group and W represents
a carbonyl group. The starting o-mercaptobenzoates (1~ are
commercially available or can ba prepared by known methods, for
example by trans~ormation of the corresponding o-alkoxycarbonyl-
-benzenediaæonium salts upon treatment with potassium
ethylxanthate (M.S. Cohen et al., J. Org. Chem ., 18, 1394,
1953).
,, - . . , , : , "~ , ,:, ;
,. " , ~ , ,
; ; , , ~, ~ ~
, . .. . : ~ - . .
, , .,, ~ , ,
- --1 9-- ~ .
::
: :
SCH~ 2
C''2 ''}~ co2 ,. 3
~ ~ .
o :
, I ~ r.~ ., .
C C~ .. k 2
/ ~ ~ ,~'i.' `
f~
C o .:
~~5 ~ ~~ 3 ~ ~;
11 Il I l! - ::
''.iYC2 C . .~, c ~ ~
4 - .~ ~ ',b !o ;~
,-
; , . ; .... . . . . ; ..
o ~ ~ " ... ..
'-7 1 !1 ."~\ ~
~G, c ~ ur. r~ G
v ,; .
2 ~
-20-
Ste~ 2a
- R2COCH(R3)CN or R2COCH(R3)COOAlk in polyphosphoric acid at
50 120C;
- R2CaC-COOAlk and A12O3 in an aprotic solvent (e.g. Et2O) at
0-40OC;
- R2C-C-COOAlk and a base in an aprotic solvent (e.g. THF or
DMF~ at 20-140C.
The last two options are both followed by treatment with
polyphosphoric acid at 50 120C.
Step 2b
- NaOH in aqueous EtOH at 40-75C;
- LioH in aqueous DMF at 40-100~C.
S~e~ 2c
- Stoichiometric 30% H22 in AcOH at 25-60OC;
- m-chloroperbenzoic acid in chloroform at 0-30C. .
Step 2d
.
~ 30% H22 in AcOH at 50-80C.
Reaction Scheme 3 leads to compounds (2) in which R7 represents .: -
a methoxy group, W represents a carbonyl group and X represents
an oxygen or sulphur atom. Compounds (1~ can be prepared
according to Reaction Schemes 1 and 2 starting from the ~ ~.
appropriate phenols or thiophenols (not substituted at position
2 or 6 with COOAlk or NO2). -
SCHEME 3
R6\1~ 3 a ~ R3
CH30--~\ X R2 CH30 /~
Cl 2 .
',
:
:, ,` ~ : .
5 ~
-21-
Step 3a
- HCHO and gaseous HCl in AcOH containing aqueous HCl (d=1.18)
at 50-100C (P. Da Re et al.l Ann. ~him., 46, 904, 1956).
Thi~ method can be used when R3 is other than H or CH2OH.
Simple 2,3-dihydro intermediates ( ---- = single bond) can be
prepared according to Reaction Scheme 4 provided that other
reactive groups possibly present (e.g. NH2, OH) have been
previously protected as described before. The Compounds ~4) thus
obtained can be converted to the corresponding derivatives
having A-COOH or NH2 according to th~ method of Step lg.
'. ..' :'
" ' .'
,
2~9~
--22--
SCHEME ~
o o
R ~D~ R3 a ~ 1
~ I OTMS o
j' Rs~,R3R6 ``~XR3
~ Fi2
: : 5 . .
O
Rs ~R6 ~q R3 xl~t H
R7/~XH R /~X R2
A A
'. ,, ., ,.
:, . ~. . . , , ~. .
,: , .
2 ~
-23-
Step 4a
- R2-CHO, aqueous NaOH in EtOH or another protic solvent;
- R2-CHO, NaH or potassium t-butoxide in THF (or another
dipolar aprotic solvent) at 0-15noc.
Step 4b -
- Mineral acid (e.g. HCl or H2S04) in water or another protic
solvent (e.g. EtOH, AcOH) at 0-100C.
Step 4c
- R2-CHO, O.lN to lN aqueous NaOH or another suitabl~ base in a
protic solvent;
- R2-CHO, pyrrolidine in a protic (e.g. MeOH) or polar aprotic
solvent at 5-100C (H.J.Kabbe, Synthesis, 1978, p.886).
Step 4d
- Lithium diisopropylamide in THF at 0-20C; then
trimethylsilyl chloride and an organic base (e.g. NEt3) (S.E.
Kelly et al., J. Org. Chem., 56, 1325, 1991). -~
Step 4e
- R2-CHO in a chlorinated solvent (e.g. dichloromethane) at
-78QC, then TiC14 or other Lewis acid (S.E. Kelly et al., J.
Org. Chem., 56, 1325, 1991).
Step 4f
- Lithium dii~opropylamide in THF at -78C, then R2-CHO (A.
Banerij et al., Tetrahedron Letter, 1979, 3685).
Step_4q
- R2-CH=CR3COCl, a Lewis acid (e.g. AlC13) in a suitable
solvent (e.g. nitrobenzene) or without solvent at 20-180C~
Step 4h
- R2-C~=CR3COOAlk, triethylbenzylammonium hydroxide in an
aprotic solvent (e.g. benzene) or without solvent at
50-150C; then aqueous NaOH in MeOH at 20-50C or LioH in
aqueous DMF. (In this case compounds having A=COOCH3 or
COOC2H5 are also hydrolyzed to compounds having A=COO~
Step 4i
- Concentrated EI2S04 or P20s or polyphosphoric acid or a Lewis
acid in nitrobenzene or toluene or without solvent at
0-180C. (Also in this case hydrolysis of A=COOAlk to A=COOH
occurs).
.: , ,-. .
' ' , . ': . ,,: . ' . ' ! . ~ " .' ' ' '; ' . , ' ,
2~15~
-24-
Simple starting materials having R3=OH or OR8, where R8 is alkyl
or aralkyl, may be prepared according to Xeaction Scheme 5 where
A has the same meaning as in Reaction Scheime 1. Compounds (1)
and (2) (which are the same as (2) and (4~ in Reaction Scheme 4
but with R3=H) can be prepared according to Reaction Scheme 4
starting ~rom the appropriate phenols or thiophenols having
R3=H. The compounds (4) used in Reaction Scheme 5 can be
prepared by known methods ~rom the corresponding salycilates or
thiosalycilates (see J.March, Advanced Organic Chemistry, 486,
John Wiley and Sons, New York, 1985; L. Ren~ et al., Eur. J.
Med. Chem. - Chim. Ter., 4, 385, 1977 and references cited
therein). The substituent A in compounds (3) and (6) of Reactisn
Scheme 5 can be transformed by the processes of Step lg into a
substituent B as defined in Reaction Scheme 1.
' "~'
. .
, ~-:., ,., , , ' ,, . ' ` ', , , ' :
. .
: . , . , . ~ .
2 ~ X ~ ~
` -25- ~ ~
'-~ ' .
SCHEME 5 ~ :
o o ' ` :~
R6~ R2 R5 ~ ;:
R7~XH F~7~X R2
A \ a 2 : : :
b ~
o
Rs~ ~ Rs~OH
A A
3 ~-
c ¦ e ¦ `~
RS~ H R~ORs ~
R XH R X R2 .
A A
4 6 : `
,.. ..
f
O
R6 y~L--OR8
R,~XH . ` ~ -
7 ~ :
, ' .: .
-26-
Step 5a
- Aqueous NaOH in an alcoholic solvent (e.g. MeOH or EtOH)
followed by 30% H22 at 10 to -78 C. (No D~ Meyer et al., J.
Med. Chem., 34, 736, 1991 and references cited therein). (Not
when A is CH=CH-CH3; when A=COOR it is simultaneously :
transformed into COOH).
SteP 5b
When ~ = single bond: ,.
- Amyl nitrite or other alkyl nitrite without solvent or in a
suitable solvent (e.g. EtOH or b~nzene) in the presence of a
catalyst such as 37% HCl (Org. React., 7, 327, 1953 and
references cited therein); then aqueous H2SO4 in a protic
solvent ~e.g. AcOH~ at 10-100C (Acheson R.M., An
Introduction to the Chemistry of Heterocyclic Compounds, 347,
John Wiley and Sons, New York, 1976).
When ---~ = double bond: .:
- lithium diisopropylamide in dry THF at -78C; then AcOH and l:
30% H22 (B.D.M. Cunningham ek al., Anti-Cancer Drug Design,
7, 365, 1992).
- R2-CH=CH NO2 (1 to 1.5 equivalent) in a suitable solvent
(e.g. diisobutyl ether, DMSO or DMF) in the presence of a
base te.g. KOH or NaOH) in catalytic or stoichiometric
quantity at 20-150C (see L. Ren~, supra, and T. Sakakibara
et al., Bull. Chem. Soc. Jpn., 51, 3095, 1978).
Step_5d
- 15% H22l NaOH or another base ~e.g. NEt3) in a protic
solvent such as MeOH at 20-100C (S.R. Deshpande et al.,
Synthesis, 835, 1983) or photolysis and alkaline hydrolysis -:~
(Rao T.S. et al., Heterocycles, 22, 1377, 1984), or K02 in
benzene containing 18-crown-6 ether at 20-100C (Rao l'.S.,
Neterocycles, 26, 2117, 1987). (Not when A is CH=CH-CH3; when
A=COOR it is simultaneously transformed into COOH).
Ste~ 5e
- R8L, where L represents a leaving group (e.g. alkylsulphate,
halogen, tosyl) and a base (e.g. K2C03, NaH, KOH, NaOH or
LioH) in a suitable solvent (e.g. THF, DMSO, DMF, benzene) in
the presence or otherwise of a phase transfer catalyst (e.g.
benzyltriethylammonium bromide) at 0-180C.
- ~ . . ,:,
, " , " ; ~ ,: ,
:, ,
: ' ' : ,. . .
27- 209~
step 5f
- By the methods of Step lh. ~
Reaction Scheme 6, in which A has the same meaning as in
Reaction Scheme 1, leads to compounds in which W represents a
thiocarbonyl group. The compounds (1) and (2) of Reaction Scheme
6 can be prepared according to Reaction Schemes 1, 2, 4 and 5.
The substituent A in compound (4~ of Reaction Scheme 6 can be
transformed by the processes of Step lg into a substituent B as
de~ined in Reaction Scheme 1.
SCHEME 6
o o
R6 ~J R2
R,/~ R3
A -I :
a ~ :
' ..; .. ...
S -~
R2~XR' ~ :
A : .
~ 4 ~ ;~
R6~R3 R6"~f ~F~3
R7/~X~R2 R7/~x R2 :
2 3
,
-28-
Step 6a
- P2S5 in pyridine at 50-100C (Stavaux et al., Bull. Soc.
Chim. Fr., 2082, 1967).
Step 6b
- P2S5 or B2S3 or SiS2 or ~awesson's reagent in a chlorinated
solvent (e.g. chloroform) or in an aromatic solvent (e.g.
benzene, toluene, xylene) at reflux (Dean et al., J. Chem.
Soc. C, 2192, 1963; R.K. Razdan et al., J.Med. Chem., 2~
643, 1978; K. Clausen et al., Tetrahedron, 37, 3635, 1991). ::
Step 6c :
- COCl2 without solvent or with an inert solvent (e.g. benzene)
at 40-90C (A. Schonberg et al., Chem. Ber., 101, 701, 1968).
Step 6d
- Thioacetic or thiobenzoic acid or potassium
diethylxantogenate in a ~uitable solvent (e,g. benzene) at
reflux (A. Schonberg, supra).
Reaction Scheme 7, in which A has the same meaning as in
Reaction Scheme 1, leads to compouncls in which W represents a
methylene or hydroxymethylene group. The compounds (1), (2) and
(4) of Reaction Scheme 7 can be prepared according to Reaction ~ :;
Schemes 1, 2, 5 and 6. The substituent A in compounds (7) of
Reaction Scheme 7 can be transformed by the processes of Step lg
into a substituent B as defined in Re,action Scheme l.
: , , ",
', ' , ' '' ' ' ' ' ' " "', '
-29- 2
SCHEME 7
/~\)n n _ 1, 2
O ,.
R3~R3 a 3
2 ~:
\ c d / ~
. ~ / .,.,.:.
\~ R6 ~ R3 ~ '. ,.
R7 ~r X R2
e~ 7 \5 :; ~ ~
":
OH
R6 ~ Cloi 1~ R3
~7/~X R2 R~~X R2
4 6
b 1 t h
~ ~. ' ''.
~H R2~R2
A A
3 5 ; ~: -
~,";"'' :"~
'-, ,,~
': -
.', ' '' ' '' ,~', ,'''','';' ',i'' " ` ~
- ~ ~ 9 `~
Step~7a
- 1,2-ethanedithiol or 1,3-propanedithiol in an aprotic solvent
(e.g. dichloromethane or benzene or toluene) at 0-110C in
the presence of a catalyst (e.g. p-toluenesulphonic acid or
borontrifluoride etherat~).
Ste~ 7b
- R2COCH2R3 in a suitable mixture of solvents (e.g. EtOAc or
dichloromethane plus EtOH or MeOH) saturated with gaseous HCl
at 0 40C; then aqueous HCl04 in AcOH at 20-100C (L. Jurd,
~etrahedron, 28, 493, 1972).
Step 7c
- ~iAlH4 in THF at reflux (if A is other than COOR and NO2);
- ZnI2 and sodium cyanohorohydride (6 equivalents) in a
rhlorinated solvent (e.g. 1,2-dichloroethane) at room
temperature to reflux (C.K. Lau et al., J. Org. Chem., 51,
30~3, 198~). ;
SteP 7d
- Raney-Ni in an alcoholic ~olvent (e.g. isopropanol) a~ r.t.
to reflux (Hilton et al., J. Am. Chem. Soc., 90, 6887, 1968).
Ste~ 7e
- NaBH4 in a suitable solvent (e.g. MeOH or EtOH or DMSO) at
-10 to 50C (L. Jurd, supra);
- LiAlH4 in THF (or another suitable solvent) at 0-50C (when A
is other than COOR or NO2) (Degani et al., Ann. Chim., 61,
793~ 1971; Kurosawa, Bull. Chem. Soc. Jpn., 51, 1175, 1978~.
Step 7f
- Trityl perchlorate in acetonitrile at room temperature
(Degani et al., supra).
- Melting with P20s at 80-180C (Hortmann et al., J. Am. Chem.
Soc., 96, 6118, 1974).
Step 7h
- NaBH~ in EtOH or another suitable solvent at OC to re~lux
(K. Anaya, Bull. Chem. Soc. Jpn~, 40, 1884, 1967).
- Hydrogen (1-10 atm) in EtOH (or another suitable solvent) in
the presence of a catalyst such as 5% or 10% Pd/C or Raney-Ni
or PtO2 at r.t. to 80C (K. Hanaya, supra). Not when A is
CH=CH--CH3. When A=NO2 it is simultaneously reduced to NH2.
- Aluminium triisopropoxide in isopropanol at room temperature
. :, . , : : . . ,.; . , . . :
-31-
t~ 92C.
Reaction Scheme 8 shows the preparation of simple starting
materials such as (4), (5), (6) and (9), where A has the same
meanings as in Reaction Scheme 1. Compounds (1), (2), (3), (7),
(8) can be prepared according to Reaction Scheme 1, 2, 4, 5, 7,
9, 11. The substituent A in compounds (4~, (5), (6) and (9) of
Reaction Scheme 8 can be transformed by the processes of Step lg
into a substituent B as defined in Reaction Scheme 1.
'
~.. .
. . . , . " , , ., . , , , "
~ ~ 9
SCI~EME 8
o o
Fl6~J~ R5~OH R6~
R7/~ X R2R7/~ X R2 R7--~ X R2
A A A
OH
R ~XOR1 R ~OH
4 A . e
OH ¦ ~ :
R ~R, Rz~
A 9 A
h ¦ ~ g
R,~ Rz R,X~X ~ R2
8 A
R8 a H, Ac
.: , .` .- ~, . . .
:-`` 2 ~
-33-
Step 8a
Pb(OAC~4 in a suitable solvent (e.g. benzene, toluene) at
reflux (G.A. Russel et al., J. Am. Chem. Soc., 1906, 1975).
Step 8b
NaBH4 in alcohols (see Reaction Scheme 7, step 7a), then
alkaline hydrolysis (when A=COOR it can be simultaneously
converted into COOH);
aluminium isopropoxide as described in Reaction Scheme 7,
step 7~;
diborane in THF at -10C to r.t.; then aqueous H22 in the
presence of NaOH (not when A is CH=CH-CH3; when A=COOR it is
simultaneously converted into C~OH). (Kirkiacharian et al.,
C.R. Hebd. Seances Acad~ sci. Ser. C, 289, 227, 1979);
LiAlH4 and AlCl3 in a suitable solvent ~e.g. THF) at OC to
reflux (not for A=COOR or NO2) (Bokadia et al., J.Chem. Soc.,
4663, 1961)~
Step 8c
Hydrogen (100 atm~, copper chromite in EtOH at 140C, see
M.A. Vickars, Tetrahedron, 20, 2873, 1964. When A=N02 it is
simultaneously converted into a NH2 group.
., .
KMnO4 in t-butanol (or another suitable solvent) in the
presence of aqueous NaOH at -10 to 0C. (K. Hanaya, Bull.
Chem. Soc. Jpn., 40, 1884, 1967). (Not when A is CH=CH-CH3).
(See al~o A.H. Haines, Methods for the Oxldation of Organic
Compounds, Academic Press Inc, (London), 1985, chapter
3 .2 .2) .
Osmium tetroxide (see A.H~ Haines, supra, chapter 3.2.1) in a
suitable solvent (eOg. Et2O) at room temperature (Baranton et
al ., Bull . Soc. Chim. Fr., 4203, 1968) (not when A is
CH=CH-CH3);
aqueous H22 in formic or acetic acid at -20 to -50C; then
NaOH, ~2~ 45~C (Baranton et al., supra; A.H. Haines, supra,
chapter 3.2.7.~ (not when A is CH=CH-CH3; when A=COOR it is
simultaneously converted into COOH);
silver acetate and iodine in wet AcOH at 0-20C (K. Hanaya,
supra; A.H. Haines, supra, chapters 3.2.3, 3.2.4, 3.2.~ (Not
when A is CH-CH-CH3).
2 0 ~
-34-
Step 8e
~ 30% H22 in the presence o~ NaHC03 in benzonitrile at
0-110C, then LiAlH~ in THF at 0-40C (not for A=COOR and
CH=CH-CH3) (Clark et al., Austr. Journ. of Chem., 27, 865,
1974~.
Step 8~
- Hydrogen (1-50 atm) in a suitable ~olvent (e.g. EtOH) in the
presence of a metalllc catalyst (e.g. PdC12) at r.t. to 78C.
(when A=N02 it is simultaneously converted into NH2). (Bolyer
et al., Tetrahedron, 23, 341, 1967).
Step 8a
- see step Rb (Clark et al., supra~.
Step 8h
- 0.4M cerium trichloride heptahydrate in MeOH, in a suitable
solvent (e.g. MeOH); then NaBH4 at 0 to 78C. (WO 89/06650);
- NaBH4 in diglyme at O'~C to reflux (G.P. Thakar, Indian J.
Chem., 3, 74, 1965) (when A=N02 it is converted into NH2);
NaBH~ and AlC13 in a suitable solvent (e.g. THF or benzene)
at 0C to reflux (not with A=COOR) (G.P. Thakar/ supra);
- diborane in THF at room temperature (not when A is CH=CH-CH3)
~G.P. Thakar, supra ) .
Simple starting materials having WaCH2 and a single bond at
position 2,3 may be prepared according to Reaction Scheme 9
where A has the same meanings as in Reaction Scheme 1. Compounds
~1) of Reaction Scheme 9 may be prepared according to Reaction
Scheme 6. Compounds (1) of Reaction Scheme 9 may alternatively
be obtained from co~pounds (2) by conversion of the latter into
a 4-tcll~enesulphonic acid ester or a methanesulphon~c acid ester
or into a halogenoderivative, which may be transformed into a
thioether derivative (1) by nucleophilic substitution with a
thiol. These simple conversions can be performed by techniques
known to those skilled in the art. C~mpounds (2) of Reaction
Scheme 9 may be prepared according to Reaction Scheme 7.
Comp~unds (~) of Reaction Scheme 9 where P=OC(S~-aryl or
OC(S)-heteroaryl or OCtS)O-alkyl or OC(S)O-aryl or OC(S)i5-alkyl
may be obtained by reacting compounds ( 2 ~ with the appropriate
chlorothioPormate or chlorothiocarbonate or 1,1~-thiocarbonyl-
-diimidazole as described in J. Org. Chem., 55, 924, 1990 and
. : ;. ,. ,, . i, ". , . , ,.. , ~ ,. .. .
~`o`~
--35--
Synthesis, 362, 1991 and references cited therein. Compounds (4)
may be obtained from compounds (1~ ox (3) by simpl~ elimination
reactions with bases . Compound~ ( 5 ) may be obtained according to
Reaction Scheme 4 . The substituent A in compounds ( 6 ) of
:Reaction Scheme 9 can be transformed by the processes o~ Step lg .
into a substituent B as def ined in Reaction Scheme 1.
--36--
SCHEME 9
OH S (R8 )
R6 ~ ~R3 R6 ~R3
R7~X R2 R7J~ XlR2
A
p ~ ~.
~R,
3 d ~ 6
j
o
'~2 ~2~ XFI2
A A
4 5 . :`
~ '
R8 = Alkyl, aryl, heteroaryl, H or nothing . -~
P = halogen, O-tosyl, O-mesyl, OC(S)aryl, OC(S) heteroaryl,
1-imida~olyl, OC(S)O-aryl, OC(S)O-heteroaryl .
' ;; '
; 2~9~5~ ~
-37- .
Step 9a
- Raney-Ni in a suitable solvent (e.g. isopropanol) at r.t. to
100C. When A is N02 it i~ simultaneously converted to NH2;
- triethyltin hydride in benzene or another aromatic solvent at
30-150C. For other methods like nickel chloride and NaBH4 in
MeOH or ~orane-pyridine complex in tri~luoracetic acid or in
dichloromethane in the presence of AlCl3, see J. March, ~. .
Advanced Organic Chemistryl page 728, J. Wiley and Sons, New
York, 1992 and references cited therein. (not when A is ~:
CH=CH-CH3).
Step 9b
- Hydrogen with a catalyst according to Reaction Scheme 8, step
8f. When A is NO~ it is simultaneously converted to NH2.
Step 9c -
Where P is an O-C derivative:
- tributyltin hydride or tris(trimethylsilyl)silane in the
presence of azaisobutyronitrile in a suitable solvent (e.g.
toluene) at 80-150C; (M. Drescher, Synthesis, 362, 1991; M.
Sekine, J. Org. Chem., 55, 924, 1990);
- a silane (e.g. triethylsilane or diphenylsilane) in a
suitable solvent (e.g. dichloromethane) at -20C to reflux in
the presence of CF3COOH or BF3 (F.M. Mauser, J. Org. Chem.,
55, 555, 1990);
- triethylchlorosilane, sodium iodide in acetonitrile; then Zn
powder in AcOH and acetonitrile at r.t. to 80C (T.Morita et
al., Synthesis, 32, 1981)
Where P is halogen or an 0-S derivative:
- a reducing agent (e.g. sodium cyanoborohydride in
hexamethylphosphotriamide or NaBH4 in DMSO~ chosen from those
cited in J. March, AdvancO Org . Chem ., J . Wiley, New York,
1992, chapter 0-76, 0-77.
Step 9d
- Hydrogen (1-5 atm) in a suitable solvent (e.g. EtOH) in the
presence o~ a catalyst (e.g. 10% Pd/C at 50-78C) (Sarcevic,
Helv . Chim . Acta, 56, 1457, 1973). (When A=NO2 it is .
simultaneously converted to NH2); ~.
- Zn and gaseous HCl in Et20, or Ac20 in toluene at 0-80C. ~
(Todah, Bull. Chem. Soc. Jpn., 45, 264, 1972) (not for ~.
A=N2 ) :.,
, ,! ' ' ,., ~, ; : ' I '
. ' ' ' " ' , ' ;' " ' ' . , , '; , , ' ,1 ", '1 ~ . , . ,; ' ' ' ,.' ' ,,' '
., ' ' ' ' ',
209~
-38-
Step 9e
- Zn and aqueous HCl in a suitable solvent (e.g. EtOH) at
0-78C;
- according to step 9d above (when A=NO2 it is simultaneously
converted ~o NH2);
- hydrazine, NaOH in ekhane-1,2-diol at 200C (Chemical
Abstracts, 74, (1971): 22699) ~not for A=COOR, NO2) or other
methods cited in J.March, supra (not for A=COOR, NO2);
- according to Step 7c (not for A= NO2).
Reaction Scheme 10 shows routes to compounds in which W
represents a valence bond and X represents an oxygen atom or a
sulphur atom:
2 ~
-39- ~
SCHEME 10 ~:
R6 ~ : '
R7/~ XH
A ~:~
~ 1 ~
O
R6~R R,~R ~;
2 3 : ~ -
d / : ~
, C ~ o
6~R3 F~6~ R3 ; ;~
R7/~ R2 R7/~--X R2
A A . -
4 : 7
19 If
R~ R3 F~6 ~R3
R~/~ R2 R7/~Hal
B A . : .
6 ~ :
',~ .,
A _ COOAlk, NO2, CH3
E~ = COOH, NH2
~ ' ' ~ , ''
2 ~
-40
Step lQa
according to Step ~a, but utilizing R3COCl or (R3CO)20
inst~ad of R3CH2COCl or (R3CH2CO)20, wi~h or without
isolation o~ the intsrmediate phenyl ester;
hexamethylenetetramine in CF3COOH at reflux ollowed by
addition of aqueous HCl. If A=COOAlk, it may be hydrolysed to
COOH in such strong acid conditions, and thus need
re-esterification with the appropriate alcohol ~e.g. using
thionyl chloride at reflux temperature) before Step lOc. .
Step lOb
R3COCH(R2)Hal in acetone or methyl ethyl ketone or -
dichloromethane or chloroform in the presence of a suitable
base such as K2C03, NEt3 or NaH, at 20-80C.
Step lOc
R2CH(Hal)COOAlk in an aprotic solvent, e.g. DMF, in the
presence of a base, e.g. K2C03, at 70-100C followed by
hydrolysis of the crude intermediates with a strong base
(e.g. KOH) in a protic solvent such as EtOH at reflux, and ~: :
~inally submitting to decarboxylation - dehydration : : :
conditions using a non-protic solvent (e.g. xylene) and an ~- .
iacid catalyst (e.g. p-toluenesulphonic acid) at reflux or ~ -
simply heating at 240C in quinoline; :~
R2CH2Hal and KOH in refluxing EtOH followed by cyclization of ~ -
the isolated intermediate phenyl(thio)ether with sodium
methoxide in a boiling D~F/MeOH mixture. When A is COOAlk,
intermediates (4) having A=COOH can be obtained;
Using ArCOCH~Br and K2C03 in acetone at reflux, Compounds ::
(4), having R2-ArCO, are obtained.
Step lOd :.
Vigorous stirring in preheated polyphosphoric acid at
90-140C; '
Lewis acid (e.g. AlC13) in chlorobenzene at 70-90C. The :. .
cyclizations carried out on Compounds (3) having R3=Cl with a
Lewis acid (e.g. AlCl3) in o-dichlorobenzene at 45C or with
BF3 in Et20 at 20-25C gives the Compounds (4) where R3=OH, :-
as reported by K.Davies, J. Chem. Soc. P.T., 1, 2624, 1957,
for compounds having X=S and R2=H. ~:
' ''.
sodium alcoholate (1 equivalent) in the same alcohol at
-4~-
0-90C; when A=COOAlk it may be suitable to use the
corresponding AlkOH ai reaction solvent;
- When R2=COOAlk and X=S, compounds (4) can be hydrolyzed to
the corresponding R2=COOH with sulphuric-acetic acid mixture,
(if A=COOAlk is present, it also can give A=COOH), and can be
selectively decarboxylated with copper in anhydrous quinolin~
at 210-220C, to give Compounds (4) where R2=H according to
J.Cooper et al., J. Chem Soc. (C) 1971, 3405.
Step_lOf
- R2CH2X~ and one equivalent of sodium in EtOH at reflux or
with NaHC03 in EtOH:water mixture at 60-90C.
Step 10~ -
- When A=COOAlk or N02, the methods described in Step lg can be
used. It must be noted that reduction of the N02 group to the
NH2 group by catalytic hydrogenation can simultaneously
afford hydrogenation of the double bond at position 2-3, as :
reported by S.L~ Meisel et al., Heterocyclic Compounds / Ed.
Interiscience Publ.: "Compounds with Condensed Thiophene
Rings", page 34, (1954) and M. Ahmed, i~idem, Ed. - . .
Wiley-Interscience: '~Benzofurans", p.56, (1974).
When A=N02 and R2=COAr the reduction carried out with
hydrogen in the presence of Pt on carbon as catalyst gives
the 2,3-dihydro compounds (5) where B=NH2 and R~-CH2Ar as ::
reported in WO 86/07056; :
:: :
when A=CH3 and R2, R3, R6 are not CH3 or R2 does not bear a
CN3 group, the compounds can be transformed into the
corresponding:
A=CH2Br by reaction with N-bromosuccinimide in CCl4 and
2,2'-azobisisobutyronitrile or benzoyl peroxide as catalysts
at reflux;
A=CHO by reaction of the above Compounds with
hexamethylenetetramine in refluxing chloroform followed by
acid hydrolysis o~ the salt in boiling AcOH or by reaction of
compounds having A=CH3 with tetrabutylammonium dichromate in
refluxing chloro~orm according to Valenti et al~ Arzneim. . ~.
Forsch., 40, 122 (1990);
A=COOH by oxidation of the above Compounds ~A=CHO) with
: : : :, ,, , ,. ,..... .: , .. . . . , .. . .,, ... : . ~ -
~ , ~ : .:, : . , ; :. : , , ;
.
:
20~al~
-42-
silver oxide in a mixture o~ protic aqueous solvent (e.g.
EtOH-D~F at 0-70C according to H.R. Rodriguez et al.
Tetrahedron 24, 6587, 1968~ or with KMnO4 in t-butanol in the
pre6ence of NaH2PO4 aqueous solution at 70-75C according to
S. Maruzama et al., Tetrahedron Letters 27, 4537 (1986).
Compounds (4) of the above Reaction Scheme 10 having R3=C6H5
or t-butyl, R2=H and X=O can be transformed into the ~ .
corresponding intermediates having R2=C6Hs or t-butyl and
R3=H by rsacting with polyphosphoric acid at 132C according ~:
to Davies et al. J. Chem. Soc., 1958, 822. :
When X represents an imino or alkylimino group and W is as above :~
defined, other than a valence bond, the simple starting .
materials may be prepared according to the following Reaction
Scheme 11~
''''' ~,'' ,
', ' ''
'' , ' ' ' ~; ~ ,, ' ., , ' ', . ,' ', ', ': ,' :. , ' . ' ' . ' ' ., ' : ' 1 . ., ', .', , ", ' ,
2 ~ .5
.:
--43--
SCHEME 11 :
R6 ~q a R6 ~OOC~COOE~
R7~\~.H R7--~--N R2
A 1 2
b ~ c
r ~r O
R~ C)OC~R3 d Fi6 X 3 ~.
~; R7 I NR R2 R7/~N R2 `;
e ~ :
~ ~ ~ .
Rc~ R6 \f~ R3 :~
R~ NH ~r R7/~ N R2
A A
g ¦ \h j
o \~
R8 ~XF~3 R6~R3
R,/~ N R2 R7~ N R2
6 7
A = COOAlk. NO2
R= H, alkyl
.. . . . . . ..... .... . .. . . .
,, ' ,, ': . i' ~ , ' ! ' .' . , i . i ':; , . .
9 ~
-~4-
Step lla -
~ EtOC(R2)=C(COOEt)2 at 80-140C without solvents or in a polar
solvent (e.g. isopropanol).
Step 11b
- R2COC(R3)COOAlk and p-toluenesulphonic acid or
methanesulphonic acid in a chlorinated solvent (e.g.
chloroform or dichloromethane) or aprotic solvent (e.g. :~
benzene) at reflux under azeotropic conditions. ::
S~ep llc -
- heating in Ph2O in the presence of p-toluenesulphonic acid or
phosphoric acid or ZnO as catalysts at 245-255C according to
Hung. Teljies 6251 (Chemical Abstracts, 79, 92026v, 1973~;
- heating in a high boiling solvent (e.g. Ph2O) followed by :
hydrolysis of the not isolated Compounds (4) (R3=COOEt) with
a strong acid (e.g. HCl) in a protic solvent (e.g. acetic
acid) at reflux to give Compounds (4) where R3=COOH. The
above isolated acids can be decarboxylated by heating in a
high boiling solvent (e.g. Ph2O) to give Compounds (4) where
: R3=~ according to R. Albrecht et al. Ber. 10~, 3118 (1972);
Step lld ~.
- heating in a high-boiling solvent (e.g. Ph2O~ at 255C;
- when R=Alk Compounds (4) are obtained directly from Compounds ~ :
(1), without isolation of Compouncls (3), by condensation with
R2COCH(R3)COOAlk in polyphosphoric acid at 90-150C according
to F. Piozzi et al., Gazz. Chim. It., 100, 678, 1970.
Ste~ lle
- Al/Hg amalgam in aqueous EtOH solution at reflux followed by
acidification with a strong acid ~e.g. HCl) and treatment
with FeCl3 at reflux according to W.A. Denny ek al., J. Med.
Chem., 32, 396, 1989. :
- When A=COOAlk Compounds (4) shculd be hydrolyzed to the
corresponding A=COOH before performing Step lle. ~
- When A=N02, intermediates (5) having A=NH2 are obtained; .-
Step llf . ~ ~ .
- R2CH-CHCHO and arsenic acid in a strong acidic medium (e.g.
concentrated H2SO4) and water at 105-115C according to EP :
0206802.
When A=COOAlk, Compounds (1~ should be hydrolyzed to the :.
corresponding A=COOH before performing Step f. All Compounds
-, ...~ ;- :
"' ', . . . ' ', '
', ' ' . . :' ' ,, ' , ., ,, :
2 ~
-45-
(1~ have R=H and the obtained Compounds ~5) have R3=H.
Step llg
- R2CH(Hal)-CH(R3)COOH in a protic solvent (e.g. water) in the
presence of a strong base such as NaOH at 100-125C followed
by cyclization of the isolated Compounds B-anilinopropionic
acids with preheated polyphosphoric acid at 120-125C or with
phosphorous pentoxide in a high-boiling aprotic solvent (e.g.
xylene) at 120-140C. In some cases, it is use~ul to start
from Compounds (1), where R=tosyl or other suitable
protective groups; the obtained Compounds (6), where R=tosyl,
can be easily converted into Compounds (6), where R=~, by
hydrolysis with a strong acid (e.g. HCl~ in a protic solvent
(e.g. AcOHj at reflux.
When A=COOAlk, Compounds (6) having AeCOOH, are obtained.
Step llh
- R2CHO and ethylene in AcOH and HCl at 25-30C according to
K~D. Hesse, Liebigs Ann. Chem. 741, 117 (1970). Where
Compounds (1) have R=H starting material~ (7), having R=R3=H
are obtained.
- Epichlorohydrin followed by cyclization o~ the isolated
anilinopropanol derivatives in refluxing N,N-diethylaniline
or o-dichlorobenzene in the presence of a proton acceptor
(e.g. NEt3) according to S.D. Boyd et al., J. Org. Chem. 30,
2801 (1965). In this case, Compounds (7) having R=R2=H and
R3=OH are obtained;
S~
- by hydrogenation in the presence of a catalyst ~e.g. platinum
oxide) in a protic solvent (e.g. EtOH) at 20-30C and 2-4 atm
according to G.M. Coppola, J~ Heter. Chem. 15, 645, 1978.
When A=N02, Compounds (7) having A=NH2 are obtained.
Compounds (4), (6) and (7) thus obtained can be converted to
the corresponding derivatives having A=COOH or NH2 according
to the methods of Reaction Scheme 1, step lg.
The synthesis of the simple Compounds (7) of Reaction Scheme 11
having R=H and A=COOH can be also pursued using the method
showed in the Reaction Scheme 12:
,. , . , : ::;
: '
.. ..
'' '' :', . ..
" . ~ :' " ~ ', ' ' . ' :~
5 ~
-46-
SCHEME 12 -
. ~ ~
R6 ~ X a ~ ~/ ~ b ~W~ R3 : :
R7 N R2 R7/~N R2 R7~N~
0~ COOH
W = CH2 or bond 2 3
Ste~ 12a . . .
- oxalyl chloride in a polar solvent (e.g. THF) at re~lux
followed by internal Friedel-Crafts acylation of the crude
chlorooxalylamide with a Lewis acid (e.g. AlCl3) in a non~
-polar solvent (e.g. CS2) at reflux, according to EP 0402859; --~
Step 12b
- 30-35% aqueous H22 and a strong base (e.g. NaOH) in a polar
solvent (e.g. water) at 20-30C followed by addition of a
strong acid (e~g. HC1), as reported in EP 0402859.
Reaction Schemes 13 and 14 lead to simple starting materials in
which X represents an imino group and W represents a valence
bond. In these two Reaction Schemes, A has the same meanings as
in Reaction Scheme 1.
':
: '' ~ .
, : .
.
, .' "
,
' ~ . . . ~.,
',~,''~' '
~;,''''
'`'~' "'
, . . .. . .. . ... . . ... . . . . . ... .
5 ~ ~
-47-
SCHE~IE 1 3
R6~ch 3~ R6~q Cl~c~lz R6~9 : '
R,~NH2 F~7~N R7~\~NH2
2 3
¦ b ¦c ¦d . ::
R,~ Cl ~ R6~ R6~qCH3X 3 ~;
~7 ~f HN R2R7~N R2 R7/~HN R2
4 ~; 6 :: ~
~g '' " " .:
Ig \ ' ,~;
R2 ,.,
R5~COOH R6y~R2 R
R7/~NOz ~h ~ R /~NQ2 R7/~ NO2
7 8 9
, , .
i, . i ,.
~6~NH2 R6~CHO
A R /~
. .
,,: . , . . : ., . , , . , . ;: . - . : ... :.
.: ,
.,, ., : , ., ~;.... ...
~ - ` 2 ~ 3
-48-
Step 13a
- ClCH2C(Cl)=CH2 in thP presence of K2CO3 at 40-80C according
to L~ Purdie, J.Chem. Soc. (C) 1970, 1126.
Step 13b
- R2COHal in pyridine or in a chlorinated solvent (e.g.
dichloromethane) in the presence of proton acceptor (e.g.
NEt3~ at 20-100C or in a polar solvent (e.g. acetone) in the
presence of K2CO3 at 20-80C.
St~p ~3c
- BF3 in MeOH at 130 to 155C;
- heating at 100 to 110C.
The compounds (5) obtained by Step 13c always have R2=C~3.
Step 13d
:
- R2COCH(OAlk)2 in a non polar solvent (e.g. toluene) in the
presence of iodine as catalyst at reflux in azeotropic
conditions followed by reduction of the isolated ~or not
isolated) imino Compound with NaBH4 in a polar solvent (e.g.
MeOH) in the presence of NaOH as catalyst at reflux. If
A-COOAlk, it will be hydrolysed to COOH.
Step 13e
- sodium amide in a high boiling solvent (e.g. ~ -
N,N-diethylaniline) at 220-250C according to F. Piozzi et
al., Gazz. Chim. It., 93, 1382, 19l63;
- potassium t~butoxide in a polar solvent (e.g. DMF~ at
20-100C according to EP 0042298.
Step 13f
- BF3 in an apolar solvent (e.g. benæene) at 5 to 10C.
Step 13~
- Zn or Fe dust in an acidic medium (e.g. AcOH) and water at -
70-100C. If A=NO2, it will be reduced to NH2. ,
Step 13h
- thionyl chloride at reflux. The resultant acyl chlorides are
isolated, and reacted with sodium azide in an acidic medium
(e.g. AcOH) at 10-20C, subsequently heating at 50-70C.
Step~3'
- diazotation with NaN02 in concentrated H~S04 followed by
aqueous ZnCl2 addition at 5-10C and by reaction oÆ the
isolated diazonium salts with CH2=C(R2)COOH in a polar
solvent (e.g. acetone) in the presence of a copper salt (e.g.
-49-
CuCl2) at 25-30C. Examples of steps 13g, 13h and 13i are
given by A. Allais et al., Eur. J. Med . Chsm ., 10, 187, 1975.
Step 13~
- R2CH2NO2 in a polar solvent (e.g~ EtOH~ in the presence of a
base ~e.g. n-butylamine) and catalytic amounts of an acid
(e.g. AcOH) at reflux.
The Compounds (5) thus obtained can be converted to the
corresponding derivatives having A=COOH or NH2 according to the
methods of Reaction Scheme 1 step lg.
With regard to Reaction Scheme 14, it is to be intended that
Compounds (4) having R3=H correspond to Compounds (5) of
Reaction Scheme 13.
o
,. : , ' , ;: ' ' ' '.
,, : .
.. . . .. .
2~
-50
SCHEME 14 - - -
R~NHz F~,~N--I ~,~ NH-NH
A A A
2 3
\ c d /
e fl;~R3
'''~
1 g
R6~;~R3 h Re~ R3 ~ -
R.--~\N, R2 R7~N R2 :
6 7
,;
R _ Alkyl ~:
', '' "
.. .
.
-51
Step 14a
NaNO2 in aqueous acidic medium (e.g. HCl) at -5 to ~5C;
isoamyl nitrite in a polar solvent (e.g. EtOH) at 5-10C;
l~ . ,
aqueous solution of SO2 at 0-10C according to Pfannstiel et
al ., Ber. 75, 1096, 1~42;
triphenylphosphine and heating of the isolated phosphonium
salt in an aqueous-alcoholic HCl solution at reflux according
to Horner et al., Ber. 85l 1073, 1953.
Step 14c
R2COCH(R3)Hal in a basic high boiling solvent (e.g.
N,N-diethylaniline) at 160-180C or by simply heating without
solvents at 180C;
R3COCH(R2)Hal in a polar solvent (e.g. acetone) in the
presence of a suitable proton acceptor (e.g. K2CO3) at reflux
followed by cyclization of the isolated B~anilinoketone ~ '
intermediates with freshly melted ZnCl2 in a protic solvent
(e.g. EtOH) at re~lux; :
R2CH(Hal)CN in the presence of BCl3 and a Lewis acid (e.g.
TiCl4) in an apolar solvent (e.g. benzene) at reflux followed
by cyclization of the isolated 2-amino-~-haloacetophenones ~ -
Compounds with a suitable reducing agent (e.g. NaBH4) in a
polar medium (e.g. dioxane-water) at reflux, according to T.
Sagusawa et al., J. Org. Chem., 44, 578, 1979. By the above
method, Compounds (4), having ~3=H are obtained;
R2COCH(R3)Hal (hal~ an equivalent) in a polar solvent (e.g.
MeO~) at reflux followed by cyclization o~ the isolated
Schiff base intermediates with a strong acid (e.g. CF3COOH)
at 20-30C;
Step 14d
R2COCH2R3 by heating at 100C without solvents or at reflux
in a polar solvent (e.gO MeOH) followed by cyclization of the
isolated hydrazone Compounds with polyphosphoric acid at
100-130C or by simply heating in ethyleneglycol or aqueous
~ormic acid or ethanolic formic acid.
Cyclization can be also carried out by heating in ethanolic
HCl at re~lux or in an AcOH/HCl mixture at re~lux or in
orthophosphoric acid at 95-105C or by simply heating with
anhydrous ZnC12 at 100-220C. When A=COOAlk, Compounds (4)
.., , , .,~. . . .. .
. ~ , .-,
:, ,.. . .. . .. :. .. .
--` 2~9~
-52-
having A=COOH can be obtained.
Step 14e
borane-pyridine complex at 0-30C followed by a protonating
agent addition (e.g. HCl);
tin or zinc and aqueous HCl at 50-100C;
NaBH4 in the presence of a Lewis acid (e.g. AlC13) in
pyridine at 0-30C or alternatively in the presence of a salt
like cobalt or zinc chloride; -
sodium borocyanohydride in AcOH at 20-80C;
hydrogen in the presence of a catalyst (e.g. Pt) in a polar
solvent (e.g. EtOH) at 20~0C..
Other general methods are reported by Houlih~n in
Heterocyclic Compounds, part one, Ed. Wiley-Interscience:
"Indoles", page 462 (1972). When A=NO2, Compounds (4~ can be
reduced to the corresponding Compounds (5) having A=NH2;
NaH and RHal in an anhydrous polar solvent (e.g. DMF) at
20-80C;
RHal in the presence of potas~ium carbonate in a polar
solvent (e.g~ acetone) at reflux;
sodium amide and RHal in a polar anhydrous solvent (e.g. THF~
at low temperature (-70C).
Compounds (4), bearing other reac:tive groups such as NH2 or
OH, have to be protected using suitable protective groups ~,
which can be selectively cleaved at the end by deprotecting ~;
methods;
Step 14q
RHal in the presence of alkaline metal carbonates (e.g.
potassium carbonate) as reported by Houlihan in Neterocyclic
Compounds~ part two, Ed. Wiley-Interscience: "Indoles", page
90 (1972) and references cited therein.
Compounds (5), bearing other reactive groups such as NH2 or
OH, have to be protected as reported above; ~ -
Step 14h
tetrachloro [1,4]-benzoquinone in a polar solvent (e.g. -
eithylene glycol monomethyl ether) at re~lux;
copper (II~ chloride in pyridine at reflux according to
Kikugawa et al. J. Heter. Chem., 16, 1325, 1979.
Compounds (6) having R2 and R3 other than H, can be reduced
:
2 ~
-53-
to the corresponding starting materials (7) by lithium
aluminium hydride according to ~.C. Printy et al., J. Am.
Chem. SocO, 71, 3206, 1949.
Compounds (4) of Reaction Scheme 14 having R2=H and R3=OH may
be obtained from Compounds (7) of Reaction Scheme 11 having
R=R2=H and R3=OH by ring contraction using an oxidant ~e.g.
~odium periodate) and a base (e.g. NaOH) in aqueous EtOH at
reflux according to S.D. Boyd et alO, J. Org. Chem ., 3 0 ,
2801, 19~5.
Starting materials (4), (5), (6) and (7) can be converted
into the corresponding A=COOH or NH2 according to the method
of Reaction Scheme 1, step lg, and ~rom these into the
alternative final products. When NH is present and might
interfere on the following reactions, it can be protected as
reported by T.W. Green in Protective G~oups in Organic
Synthesis, Wiley Interscience, 1981. Alternatively,
unreactive groups (e.g. NO2) can be converted to reactive
ones (e.g. NH2) as a final step of the pathway.
Starting materials in which W represents a valence bond, X
represents an imino group and the 7-substituent is a
carboxymethyl group can be obtained according to Reaction
Scheme 15.
' ' ':. , , . . ; . , ' " '~
1' ',
~, ' ' '.,' ' '.' , '' .,
2~9U1~6
--~i4--
,
-'
SCHEME15
: R7 ~7
R8~`COOH R5
:; ~No2 a N O
NH2
t 2
~ - .' ~ .
b `:-
, ,.
: R,3~,¢R, G R~
AlkOOC 4 3 ~
R3 ~.
. ~,
d
R6~_ ,~R3 ~ ~
R7~ I R2
HOOC 5
, ~ . .
i, ~, :.. , . ... " ,, ., . ,,, .. ,,., . . , , . . ,; ~, . . .
20~
-55-
Step 15a
- Hydrogen in the presence o~ 10% Pd/C as catalyst at 45 lbs in
water containing one equivalent of NaOH, followed by
diazotation with sodium nitrite in HC1 at 0-5C and stannous
chloride. Cyclization is per~omed during acidification of the
tin salt with H2S and completed by re~luxiny in xylene, see
H~ Eo Baumgarten et al., J. Am. Chem . Soc., 82, 3977, 1960.
Ste~ 15b
- R3CH2COR2 in the presence of an acid (e.g. acetic acid) in a
polar solvent (e.g. EtOH) at reflux as reported by W~J.
Welstead et al., J. Ned.Chem., 22, 1074 (1979) for R2=CH3 and
R3=C6H5, where also step 15c and 15d are reported;
Step lSc
- Lower alkanol (e.g. MeOH, EtOH) at reflux in the presence of
a hydrogen chloride stream;
5tep l~d
- a strong base (e.g. KOH) in a polar solvent (e.g. water) at
re~lux.
The preparation of simple starting materials having R3
hydroxyalXyl andlor the corresponding ethers can be carried out
by reacting either Compounds (3) of Reaction 5cheme 1, Compounds
(2)/ (4) or (5) of Reaction Scheme 2, Compounds (4) of Reaction
Schemes 6, 10, 11 and 15, Compounds (5) of Reaction Scheme 13
and Compounds (4) and (6) of Reaction Scheme 14 having R3 = H,
CH3 according to the Reaction Scheme 16, wherein A and B have
the same meanings as in Reaction Scheme 1, R4 represents an
alkyl or aralkyl group and R5 represents an H or alkyl group:
, -. . . . ............. . , , . ,.;~ ; .. , " ., " ;. ~ ; . .... ..
, ' :: '' . ' . ,' ' : , ::: , '; ;, " ~, ~ . , : . "; ;.. . . .
2 ~ 5 ~
-56- ;
'
SCHEME16
:
R6~W~R3
R7/~X R2
A 1 ::
a/ \b
- - :.
5~W~H i 'R6~,~W~CHO ' 1~,,
R~X R2 R7/\~X R2 : ~ .
A 2 A 3
c ¦d :
r R5 ~;
~ OAc ~ e =--~OH
: A 4~ A 5 :
~ : .
: : : f / g , -' :
. ~ ~ / ' F~5
~ O-H, R4 ~ R6~WX~
: R7/~X Rz ~ R7/ I X R2 ``:
B 6 A 7 ,
- _ _ h ~ :-
R4 - Alk, Aralk
R5 -- H, Alk
~. .: . .
2a~0~5~
--57--
Step 16a: -
R3 - H, W = CO, CS (and no activated phenyl rings present):
- Formaldehyde and HCl in water, EtOH or AcOH at 50-100C;
- Chloromethyl methyl ether and fuming sulphuric acid at
50-70C (H. Nakarumo et al., Bull. Chem. Soc. ~ap~, 57/ 2323,
1984);
R3 = CH3, W = CO, CS, bond, and no other methyl groups in the
molecule:
- N-bromosuccinimide in presence of benzoyl peroxide or
2,2l-azobisisobutyronitrile in CCl4 at 50-80C;
Step 16b
R3 = H, W = hond, X = O, S, NH or N-Alk and no electrondonating
groups are present on the other rings of the molecule:
- Phosphorous oxychloride and DMF at 50-140C, or other
Vilsmeyer - Haack reagents ~see Jutz, Adv. Org. Chem., 9,
225, ~976~;
R3 = CH3, W = bond, X = O, S, NH or N-Alk and no other CH3
groups are present: ; .
- Irradiation with a Hg high-pressure lamp in a protic sol~ent
(e.g. AcOH) at 20 100C as reported by Frasca et al.,
Tetrahedron, 23, 603, 1973.
Step 16c
- Sodium or potassium acetate in aprotic solvents (e.g.
acetone, DMF) at 40-120.
Step 16d
in Compound~ (5) = H:
- A reducing hydride (e.g. NaBH4) in a polar solvent (e.g. MeOH
or EtOH or dioxane) at 0-80C;
R5 in Compounds (5) = alkyl:
- Alkyl magnesium bromide in aprotic solvents (e.g. Et20, THF~
at 0-60C;
Step 16e
- NaOH or LiOH in protic solvents (e.g. alcohols, water) or
mixture thereof at 25-50C. (In this case, when A = COOAlk it
can be simultaneously hydroliæed to COOH);
Ste~ 16f
The same methods reported in step lg of Reaction Scheme 1,
but the oxidation of CH = CHCH3 to COOH for compounds (5);
Step 16a
"' ' . '. . ' ' ' , ' ' ~ ,, '' 'I'' ' ' ,'' . . ' ' ~', ' ,,, ".' ' ' ,;
,' ' ,' . : ;', ; ' '.'' ;~ '' "' ` ,
~ ~ ~ 9 ~ ~
-58-
- A strong base (e.g. NaH) and an R4-L reagent (where L is an
halogen atom or a tosyloxy group) in anhydrous aprotic
solvents ~e.g. DMF or THF) at 20-140C;
Step 16h
- R40H and a base (e.g. Na, NaH) in excess R40H or in aproti~
solvents (e.g. DMF or THF) at 20-140C.
The simple Compounds (6) having an hydroxyalkyl group at
position 3, obtained in this way, can be reacted as such or,
alternatively, derivatized at the hydroxymethyl yroup with known
reagents and methods, so that said group does not interfere in
the further reaction steps necessary to prepare those compounds
of formula (I) which bear a protected hydroxyalkyl group such as
R3-
The protected final compounds are finally converted by
deprotecting methods to compounds of formula (I) having R3 =
hydroxalkyl group.
.; .
Prodrugs, as above defined, may be prepared from the
corresponding hydroxy compounds by Method 1 below or from the
corresponding amide compounds by Method 2 below.
Method 1
:, ~.
- by reacting with a chloroformate, an isocyanate or
isothiocyanate, a carbonyl chloride or bromide or another
activated acid derivative (e.g. anhydride) in a suitable solvent
(e.g. chlorinated solvent, DMF, THF, dioxane, acetonitrile,
pyridine) in the presence or absence of a base (for instance,
NEt3, pyridine, 4-dimethylaminopyridine, NaOH, potassium
carbonate or 1,10-diazabicycloundecene or others not specified)
at -20/100C;
- by reacting with a carboxylic acid in the same solvants as
above, in the presence of a condensing agent such as
N,N' carbonyldiimidazole, carbodiimides or o~hers known to the
people skilled in the art;
- by reacting with a dialkyl or diaryl chlorophosphate or
2~0~
-59-
dialkyl cyanophosphonate in the same conditions described above
(for examples o~ such derivatization methods see Example 114
below and S.O. Thorberg et al., J. Med. Chem., 30, 2008, 1987).
Method 2
Prodrugs derivatives of "acidic" NH groups as discussed above
can be synthesized from the compounds o~ formula I by preparing
an N~hydroxy(substituted)methyl derivative and reacting that in
the same described above for oxygen derivatization.
The intermediate N-hydroxy(substitutad)methyl derivative can be
isolated or directly reacted to give the desired compound.
N-hydroxy(substituted3methyl derivatives of the type Ny-CH(Rl)OH
where R1 = H or CCl3 can be obtained by reacting the appropriate
compounds of formula X with formaldehyde or CCl3CHO as described
in H.E. Zaugg, Organic Reactions, 14, Chapter 2, 52 J. Wiley and
Sons New York, 1965 or in J. P. Chupp, J. O~g. Chem., 28, 2592,
19~5.
In the case where R1 = phenyl said compounds can be synthesized
by reacting with benzaldehyde and cyclic amine (e.g. morpholine~
in MeOH or dichloromethane:MeOH 1 1 at 0C to reflux and
hydrolyzing the intermediate with 0.~N HCl at pH 4. ~O.
Jacobseen, ~nnalen, 157, 243, 1884; E~. Bundgaard et al. Int. J.
Pharm., 22, 45~ 1984).
All the above described reaction pathways and steps are to be
intended as examples and are not limiting the scope o~ the
invention. Those skilled in the art are aware that said chemical
transformations are performed on polyfunctional substrates and
that the reagents used might inter~ere, reacting also with other
groups present in the molecules. For example, catalytic
hydrogenation can transform nitro groups into amino groups as
desired, but also isolated double bonds might be hydrogenated
and halogen ato~s removed; lithium aluminium hydride can reduce
conjugated ketones to alkanes as desired (e.g. step 7c in
~eaction Scheme 7) but also COOAlk groups to CH2OH or NO2 groups
to ~N=N- etc.. The undesired side reaations can be avoided or
minimized by choosing the appropriate conditions or by using
5.'~
-60- -
alternative reagents or different synthetic pathways. Should
this "alternative" approach give negative results, the undesired
obtained intermediates must be transformed into useful ones .~
using methods known to those skilled in the art. :~
, ~ .
-61-
D~TAIL~D PRBP~R~IO~ OF IN~R~DIATE~
8-t3-bromopropoxycarbonyl)-3-methyl-4-oxo-2-phenyl-4H-1-
~benzopyran ~Intermediate I!
g o~ 1,3-dibromopropane was added dropwise at ambient
temperature to a suspension of 30 g of sodium
3-methyl 4-oxo-2-phenyl-4H-1-benzopyran-8-carboxylate in 150 ml
of dimethylformamide and 35 ml of water. The reaction mixture
was ~tirred at ambient temperature for 5 days. 100 ml of water
was added and stirring was continued for a ~urther 15 minutes.
The precipitate was filtered off by suction, washed with water
and purified by flash chromatography on silica gel, eluting with
chloroform:ethyl acetate 95:5. The collected fractions were
evaporated to dryness in vacuo and the residue was
recrystallized from ethanol to give 27.7 g of the title
compound, m.p. 114-115C.
The benzopyran carboxylate salt used in the ~oregoing synthesis
was prepared by dissolving 104 g oP the corresponding acid in
560 ml of hot methanol and adding 280 ml of an aqueous solution
of 31 g of sodium hydrogen carbonat:e. 850 ml of acetone was
added to the solution to precipitat~ the desired salt, which was
collected by suction filtration (62 g, m.p. > 280C). The
corresponding acid was prepared according to Da Re, P. et al.,
J. Med. Pharm. Chem., 2, 263, 1960.
8-hvd ox~me_hyl-3-methyl-4-0X-2-PhenY~ r~9:b~
(Intermediate II)
467 ml o~ a 1.48N solution of sodium borohydride in anhydrous
dimethylformamide was added over a period of 30 minutes, under
stirring at ambient temperature, to a solution of 100 g of
3-methyl 4-oxo-2-phenyl-4H-1-benzopyran-8-carbonyl chloride in 1
litre of anhydrous dimethylformamide. The reackion mixture was
stirred for 2~ hours at ambient temperature. 88 ml of 2N
hydrochloric acid was added while maintaining the temperature at
0-5C. 102 ml of 12.7N sodium hydroxide solution was then
added. The mixture was poured into 6 litres of water, stirred
for 3 hours, and filtered on a Buchner funnel. The filter cake
was washed with 4N sodium hydroxide solution and then with
water. The resultant white solid was crystallized from methanol
2 ~
-62-
to give 50 g of the title compound, m.p. 145-147C.
E-8-~2-carboxYvinyl)-3-methvl-4-oxo-2-~henvl-4H-1-benzopYran
(Intermediate III)
A mixture of 7.92 g of 8-formyl-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran (prepared as described in Uney~ma, K. et al., Bull .
Chem. Soc. Jap., 58, 2361, 1985), 3.75 g of malonic acid and
0.46 ml of piperidine in 15 ml of anhydrous pyridine was stirred
at 100C for 3 hours. After cooling to 20-25C the reaction
mixture was poured into a mixture of 90 g of crushed ice and 33
ml of hydrochloric acid (d~1.18). The resultant precipitate was
collected by suction filtration, washed with water and
crystallized twice from 95% ethanol to give 5.5 g of the title : :
csmpound, m.p. 226-229C.
-:.'"~ ''.
E-8-(2-chlorocarbonYlvinYl)-3-methyl-4-oxo-2-phen~l-4H
-benzopyran (Intermediate IV~ :
A solution of 9.2 g of Intermediate III and 7.8 g of thionyl
chloride in 75 ml of toluene was refluxed for 3 hours. After ~
cooling to 20-25C the resultant crystals were collected by --
suction ~iltration, washed with acetone and dried in vacuo to
give 6.8 g of the title compound, m.p. (l90) 196-lg8C after
recrystallization from toluene.
~. -
8-a~etyl-3-methyl-4~oxo- -phen~1-4H-1-benzopy~ran
tIntermedia e V~
1.17 g of magne~ium turnings, 7.4 ml of anhydrous ethanol and
0.~ ml o~ anhydrous carbon tetrachloride were placed in a round
bottomed flask under a stream of nitrogen. When the temperature
began to rise, 7.5 ml of anhydrous chlorobenzene was added,
followed by the slow dropping (25 minutes) of a solution of 5.28
ml of anhydrous diethylmalonate and 3.5 ml of anhydrous
chlorobenzene in 16 ml of anhydrous ethanol. The reaction flask
was heated to 75C for two hours, cooled to 25C and a solution
o~ 8.8 g o~ 3-methyl-4-oxo-2-phenyl-4H-1-benzopyr~n-8~carbonyl
chloride in 88 ml of anhydrous chlorobenzene was slowly added,
without exceeding 35C. The reaction mixture was further
stirred for two hours at 35C and then cooled to 0C. 13 ml of
water and 1.9 ml of sulphuric acid (d = 1.84) were added. The
2 ~
-63-
solution obtained was decanted from the insoluble inorganic
matter and stripped in vacuo.
The crude acylmalonate obtained was refluxed for six hours with
10.4 ml of acetic acid, 7 ml of water and 1.3 ml of sulphuric
acid (d = 1.84). After cooling, the solution was poured into
iced water and the precipitate was collected by suction
filtration and washed with aqueous sodium carbonate.
Crystallization from 90% ethanol gave 6.5 g of the title
compound, m.p. 159-161C.
8-bromoacetyl-3-methyl-4-oxo-2 phenyl-4H-1-benzopyran
(Intermediate VI)
A solution of 11.2 g of bromine in 250 ml of chloroform was
added, over a period of two hours at 20~25C, to a solution of
19.5 g of the Intermediate V in 700 ml of chloro~orm. After
~tirring for 1 hour at 20-25C, the solution was washed with 400
ml of 2N aqueous sodium hydroxide solution and then repeatedly
with water, dried with anhydrous sodium sulphate and stripped in
vacuo. The crude product was treated with diethyl ether,
collected by suction filtration and crystallized from acetone,
yielding 16 g of the title compound, m.p 134-135C.
8-(2-hYdroxyethylcarbamoyl)-3 meth~l-4-oxo-2-phenyl-4H-1-
-benzopyran ~Intermediate VII)
The title compound was prepared in the same manner as
Intermediate XXXVI, but using 2-aminoethanol instead of
3-aminopropanol. M.p. 206-208C. -
3-methyl-4-oxo-2-phenyl-4H-1-benzo~Yran-8-sul~honyl chloride
Untermediate VIIIl
A solution of 4.55 g of sodium nitrite in 12 ml of water was
added dropwise to a stirred mixture of 15.1 g of 8~amino-3-
-methyl-4-oxo-2-phenyl-4H-1-benzopyran [prepared as described in
Da Re, P. et al., Il . Farmaco (Ed. Sci.), ll, 670J 1956] in 150
ml of hydrochloric acid (d-1.18) at -5C. Stirring was continued
at 0C for 30 minutes. The solution was poured, over a period of ~ `
10 minutes and at -5 to 0C, into 120 ml of a 30% by weight
solution of sulphur dioxide in acetic acid containing 1.53 g of
cupric chloride dihydrate and 13 ml of water. After 1 hour at
': '
' '
,. , : , ,, . . .; . . ,, ,.,; . : : : : , :
-64-
0C and 1 hour at 20-25C, 300 ml of iced water was added to the
mixture. A precipitate formed and was collected by suction
filtration, washed with watex and dried in a desiccator over
sodium hydroxide until of constant weight to give 18 g of crude
title product, m.p. 165-170C, for use without further
purification.
8-(3-chloropro~oxy)-3~methyl-4-oxo-2-}?henyl-4H-l-benzoPyran
(Intermediate IX)
This compound was prepared in the samie manner as Intermediate
XI, but using l-bromo-3 chloropropane instead of 1-bromo-
-2~chloroethane. m.p. 98-102C after washing with petroleum
ether:diethyl ether 7:3. -
8-acr~lamido-3-methyl-4-oxo-2-pheny_-4H-1-benzo~vrgn
(Intermediate XL
A solution of 1.75 ml of acryloyl chloride in 15 ml of anhydrous
tetrahydrofuran was added dropwise at -10C to a stirred mixture
of 5 g of 8-amino-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran and 3
ml of triethylamine in 60 ml of anhydrous tetrahydrofuran.
After stirring at 0C for 1 hour and at ambient temperature for
1 hour, the reaction mixture was poured into water and filtered
under suction. The filter cake was washed with water.
Desiccation gave 5.5 g of the title compound, m.p. 229-230C.
8-~2~chloroethox~)-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran -
(Intermediate XI)
A mixture of 7.52 g of 8-hydroxy-3-mPthyl-4-oxo-2-phenyl-4H-l-
-benzopyran (prepared as described in Da Re, P. et al., Ann.
Chim., 1962, p.506 et seq.), 6,22 g of anhydrous potassium
carbonate and 25.5 ml of 1-bromo-2-chloroethane in 70 ml of
dimethylformamide was stirred at 60~C for 25 hours. The mixture
was cooled to 20-25C and poured into 600 ml of water. The
organic solution, obtained by extraction with dichloromethane,
was washed with aqueous sodium chloride solution and dried on
anhydrous sodium sulphate. The solvents and excess
1-bromo-2-chloroethane were evaporated off in vacuo to yield 8.8
g of the title compound, m.p. 141-142C after crystallization
from chloroform:hexane.
, ' j,; ~:' :, . ~ ', ;
o~
-65-
8-~2-azidoethoxy)-3-methYl-4-oxo-2-~henyl 4H-1-benzopyran
(Intermediate XII)
A mixture of 15.2 g of Intermediate XI and 6.24 g of sodium
azide in 150 ml of anhydrous dimethylformamide was stirred at
70-75OC for 12 hours. After cooling to 20-25~C, the reaction
mixture was poured into 1~ litres of water and extracted with
dichloromethane. The organic solution was washed with aqueous
sodium chloride solution and dried on anhydrous sodium sulphate.
The solvents were evaporated off in vacuo~ The residue was taken
up in water, collected by suction filtration and dried to give
14 g of the title compound, m.p. 119-120C.
iB-[N-(2-hy~roxyethyl!-N-methyl-carbamoyl]-3-methyl-4-oxo-2-
-phen~1-4H-î-benzopvran LIntermediate XIII)
A solution of 1.6 ml of 2-methylamino-ethanol in 10 ml of water
was added dropwise over a period of 5 minutes to a suspension of
6 g of 8-chlorocarbonyl-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran
and 1.52 g of potassium carbonate in 60 ml of acetone. After
stirring for 2~ hours at 20-25C, the solvent was removed in
vacuo and the residue was taken up in 150 ml of acetone. The
mixture wa~ refluxed for 15 minutes, and was then filtered. The
solvent was evaporated from the filtrate and the residue was
dissolved in 20 ml of dimethylformamide, treated with 14 ml of
1.~% sodium carhonate solution, stirred for 30 minutes at
20-25C and diluted by addition of 150 ml of wiater. The mixture
was extracted with chloroform and the organic layer was washed
with 0.5N hydrochloric acid and then with water. The solution
was dried over anhydrous sodium sulphate and the chloroform was
evaporated off. The resulting oil was taken up in 200 ml of
diethyl ether and stirred for 2 hours at 20-25C. The solids
were collectad by filtration and crystallized from ethyl acetate
to give 4.97 g of the title compound~ m.p. 128-130C.
8-(2-chloroethYlcarbamoyl)-3-methyl-4-oxo-2-phenyl-4H-l-
~benzopyran (Intermediate XIV)
The title compound was prepared in the siame manner as
Intermediate XXXVII, but using Intermediate VII in place of
Intermediate XXXVI and carrying out the reaction at ambient
. , , ' , ~ . . . -
' "
2 a ~
-66-
temperature. ~.p. 181-182C (ethyl acetate).
8-tN-methyl-2-chloro-ethylcarbamoylL-3-methyl-4-oxo-2-ph-e-n
-4H~1-benzopvran (Intermediate XV)
A solution of 1.1 ml o~ thionyl chloride in 2 ml of
dichloromethane was added to a solution of 3.37 g of
Intermediate XIII in 20 ml of dichloromethane, and the mixture
was stirred for 4 hours at ambient temperature. Removal of the
solvent gave an oil which was taken up in diethyl ether. The
title compound precipitated as a white solid, collected by
filtration ~or use without further purification. M.p. (118)
126-128C (diethyl ether).
-(4-bromobutoxy?-3-me,thYl-4-oxo-2-phenyl-4H-1-benzopyran
(Intermediate XVI ?
A mixture of 5 g of 8-hydroxy-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran, 4.2 g of anhydrous potassium carbonate and 43.6 g
of 1,4-dibromobutane in 45 ml of dimathylformamide was stirred
at 75C for 2 hours. The mixture was cooled to 20-25C, poured
into 100 ml of water and extracted with dichloromethane. The
organic solution was washed with aqueous sodium chloride
solution and dried on anhydrous sodium sulphate. The solvents
and excess 1,4-dibromobutane were evaporated off in vacuo. The
residue was rinsed with 55 ml of petroleum ather:diethyl ether
7:4 and collected by suction filtration to yield 5.6 g of the
title compound, m.p. 91-92C.
8-(5-bromo~entyloxy)-3-methy1-4-oxo-2-phenyl-4H-1-benzop~ an
(Intermediate XVII)
This compound was prepared by the method described ~or the
preparation of Intermediate XVI, but using 1,5-dibromopentane in
place of 1,4-dibromobutane and puri~ying the crude product by
column chromatography on silica gel (elution with
dichloromethane:ethyl acetate 99:1). m.p. 75-76C, after rinsing
with petroleum ether:diethyl ether 30:4.
8-(2-chloroethoxymethyl)-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran (Intermediate_XVIII~
6 ml of thionyl chloride in 18 ml of chloroform was added at 0C
-- 2 0 ~
-67-
to a stirred solution of 23 g o~ Intermediate XXII and 11 ml of
triethylamine in 185 ml of chloroform. The reaction mixture was
warmed to 70C and stirred for 2 hours. After cooling to
ambient temperature, it was poured into water. The organic
layer was separated, washed with sodium chloride solution, dried
on anhydrous sodium sulphate and evaporated to dryness in vacuo.
Yield: 24 g of the title compound. A sample crystallized from
ethanol had a melting point of 102-103C.
8-chloEomethyl-3-methyl-4-oxo-2-~henYl-4H-1-benzo~yran
(Inte~ediate XIX)
53.4 g of Intermediate II and 38.8 ml of anhydrous triethylamine
were dissolved in 440 ml of chloroform. Into this solution,
maintained at -10 to -2C, there was dropped a solution of 19.8
ml of thionyl chloride in 80 ml of anhydrous chloroform. ~he
reaction mixture was stirred at ambient temperature for 4 hours,
and ~hen diluted with 400 ml of water. The aqueous phase was
extracted with chloroform, and the extracts were added to the
chloroform phase. The chloroform solution was washed with
brine, dried on anhydrous sodium sulphate and evaporated to
dryness in vacuo. Yield: 56 g of the title compound, which on
recrystallization from ethanol was shown to have a melting point
of 112-113C.
4-oxo-2-phenyl-4H-l-benzopyran
(Intermediate XX)
A solution o~ 15.1 g of anhydrous zinc chloride and 14.5 ~g of
sodium cyanoborohydride in 400 ml of anhydrous methanol was
added dropwise at O~C into a stirred mixture of 58.8 g of
8-formyl-3-methyl-4 oxo-2-phenyl-4H-1-benzopyran, 60.7 g of
methylamine hydrochloride and 125 ml of triethylamine in 600 ml
of anhydrous methanol. After stirring for 5 hours at 20 25C,
the solvent waæ evaporated off in vacuo and the residue was
taken up in 200 ml of water and collected by suction filtration.
The crude product was dissolved in ~queous acetic acid, washed
with ethyl acetate and reprecipitated by addition of cold 6N
sodium hydroxide solution. 49 g of the title compound was
obtained. m.p. 97-99C, after crystallization from 75% ethanol.
~ ,' ; . , " ': , ,,, ' '
~- ~ o ~
-68-
8-(2-chloroethylthiomethyl~-3-methyl-4~oxo-2-phenyl-4H-1- -
-benzopyran ~Intermediate XXI)
A solution of 37 g of Intermediate XIX and 10.5 g of thiourea in
370 ml of ethanol was refluxed for 1 hour. The reaction mixture
was cooled to ambient t2mper~ture, and 42 g of
8-amidinothiomethyl-3-methyI-4-oxo 2-phenyl-4H l-benzopyran
spontaneously crystallized. A sample recrystallized from
ethanol had a melting point of 233-235C.
48 ml of 35% aqueous sodium hydroxide solution was added to a
vigorously stirred suspension of 35 g of the compound thus -
prepared and 1.05 g of benzyl triethylammonium chloride in 440
ml of 1,2-dichloroethane. The mixture was stirred for 2~ hours
and then poured into 300 ml o~ water. The aqueous layer was
extracted with 1,2-dichloroethane and the extracts were added to
the oryanic layer which was washed with sodium ~hloride
solution, dried on anhydrous sodium sulphate, and evaporated to
dryne~s in vacuo. The residue was crystallized from methanol,
giving 22 g of the title compound, m.p. 82-83C.
8-(2-hydroxvethoxvmethyl?-3-methyl-4-oxo-2-~henYl-4H-l-
-benzopyran (Intermediate XXII)
A solution of 2.5 g of Intermediate XIX in 25 ml of xylene and 3
ml of dioxane was prepared. 0.15 g of sodium was dissolved in
3.10 ml of anhydrous ethylene glycol, and this solution was
added dropwise at ambient temperature to the solution of
Intermediate XIX. After refluxing for 5~ hours, the reaction
mixture was cooled to ambient temperature and poured into 50 ml
of water. It was extracted with dichloromethane, and the extract
was washed with sodium chloride solution, dried on anhydrous
sodium sulphate and evaporated to dryness in vacuo. The solid
residue was crystallized from ethanol, giving 2.1 g of the title
compound, m.p. 132-133C.
8 trifluoroacetamido-3-methvl-4-oxo-2-~henY1-4H-1-benzopyran
(Intermediate XXIII)
A solution of 9.5 ml of trifluoroacetic anhydride in 20 ml of
anhydrous dichloromethane was added dropwise at -5 to 0C to a
solution of 5 y of 8-amino-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran in 50 ml of anhydrous dichloromethane. The reaction
20~a~
-69-
mixture was stirred for 2 hours at 20-25C and then poured on to
crushed ice. The organic solution obtained by extraction with
dichloromethane was washed with cold 5% aqueous sodium
bicarbonate solution and with water, and was dried on anhydrous
sodium sulphate. The solvent was removed in vacuo and the
residue was crygtallized from ethanol to give 5.2 g o~ the title
compound, m.p. 175-176C.
8~aminomethvl-3-methYl-4-oxo-2-phen~1-4H-1-benzo~Yran
(Intermediate XXIV)
A mixture of 21 g of Intermediate XXIX and 19 g of
triphenylphosphine in 160 ml of tetrahydrofuran was stirred at
ambient temperature for 8 hours. Thin layer chromatography
showed the disappearance of Intermediate XXIX. 3 ml of water
was added, and stirring was continued for a further 24 hours.
The solvents were removed on a rotary evaporator, and the
residue was dissolved in water as its acetate. The aqueous
solution was washed with ethyl acetate, made basic with 37%
sodium hydroxîde solution and filtered on a Buchner funnel. The
filter cake was washed with water and desiccated to give 18 g of
the title compound. The hydrochloride, recrystallized from
ethanol, had a melting point of 256-258C.
8-(2-chloroethylsulphonylmethyll-3-methyl-4-oxo-2-phenyl-4H-l-
-benzopvran (Intermediate XXV3
41.6 ml of aqueous 30% hydrogen peroxide was added dropwise at
40C over a period of 20 minutes to a solution of 26.2 g of
Intermediate XXI in 300 ml of glacial acetic acid. The mixture
was heated to 60C, stirred at that temperature for 4~ hours,
cooled to ambient temperature and poured into 60 ml of water.
Filtration on a Buchner funnel gave a filter cake which was
washed with water and desiccated, yielding 29.4 g of the title
compound. A sample was crystallized from ethanol. m.p. (89
159-161C.
8~2-chloroethylsulphinylmethyl)-3-_P_hyl-4- _o 2-phenyl-4H-l-
-benzopyran (Intermediate XXVI)
36 ml of aqueous 30% hydrogen peroxide was quickly added
dropwi~e at 10C to a solution of 12 g of Intermediate XXI in 84
; ~ , .'.
", , ,, , . ' ' ' . , . : . ' . ' . ;.:' ' ,. ', : ' '', , . ' ': '
2 0 ~
-70-
ml o~ glacial acetic acid. The reaction mixture was stirred for
4 hours at ambient temperature, and then poured into 220 ml of
water. The title compound was collected by suction filtration,
washed with water and desiccated. Yie~d 12.4 g, m.p. 142~145C
(methanol). '~
. . . .
8-rN-methYl-N-(2-chloroethyl)-aminomethyll-3-methvl-4-oxo-2-
-phenyl-4H-1-be~zopyran (Intermediate XXVII)
A mixture of 22 g of Intermediate XX, 66 ml of 1-bromo-2-
-chloroethane and 11 g of anhydrous potassium carbonate in 88 ml
of dimethylformamide was stirred at 20-25C for 12 hours. The
reaction mixture was then poured into 600 ml of water and :~
extracted with dichloromethane. The organic layer was washed
with water, dried on anhydrous sodium sulphate and acidified
with eth~nolic hydrogen chlorideO The solvent and the excess
1-bromo-2-chloroethane were distilled off in vacuo at 70-80C.
The residue was taken up in cold lN aqueous sodium hydroxide
solution and extracted with dichloromethane. The organic
solution was washed with water, dried on anhydrous sodium
sulphate and evaporated to dryness in vacuo at 2i5-30C. The
crude title product was purified by flash chromatography on -~
silica gel, eluting with ethyl acetate:petroleum ether 7:3, to
give 18 g of the title compound melting at 118-120C a~ter
crystallization ~rom ethanol.
:
:
(Intermediate XXVIII~
A mixture of 7 g of 1-(2-methoxyphenyl)-piperazine, 7.33 g of
anhydrous potassium carbonate, 1.75 g of potassium iodide and
5.6 ml of 1-chloro-2-methyl-2-propanol was stirred for 90
minutes at 70C and for a further 6 hours at 90C. The reaction
mixture was poured into iced water and extracted with ethyl
acetate. The organic layer was washed with aqueous sodium
chloride solution, dried on anhydrous sodium sulphate and
evaporated to dryness in vacuo. The title product was obtained ~ - -
as an oil, and was characterised as its dihydrochloride
crystallized from ethanol, melting at 225-227C.
," ~ 0 ~
-71~
8-azidomethyl-3-methvl-4-oxQ 2-~henyl-4H-1-benzo~vran
(Intermediate XXIX)
A mixture of 22.8 g of Intermediate XIX and 6.8 g of sodium
azide in 110 ml of dimethylformamide was stirred ~or 3 hours at
100C. After cooling to ambient temperature, 130 ml of water
and 88 ml of ethanol were added to the reaction mixture. After
1 hour, the crystals were collected by vacuum filtration, washed
with water, and desiccated. Yield: 22 g of the title product. A
sample recrystallized from ethanol had a melting point of
132-134C.
y~roxyethvl~-aminomethyll-3 methyl-4-oxo-2 ~henyl-4~-1-
~benzopyran (Intermediate XXX~
A solution of 2.38 g of anhydrous zinc chloride and 2.30 g of
sodium cyanoborohydride in 71 ml of anhydrous methanol was added
dropwise under stirring to a mixture of 9.24 g of 8-formyl-
-3-methyl 4-oxo-2-phenyl-4H-1-ben~opyran and 9.12 g of
ethanolamine in 90 ml of anhydrous methanol. Stirring was
continued at 20-25C for 5 hours, before removal of the solvent
in vacuo. 250 ml of water was added to the residue, and the
insoluble matter was collected by suction filtration and washed
with water. The crude product was dissolved in lN acetic acid
and the solution was washed with ethyl acetate. The aqueous
solution was then made alkaline by addition o~ 2N sodium
hydroxide solution and the precipitate was collected by suction
filtration and washed with water to give 8.5 g of the title
compound, m.p. 117-121C after drying at 60C.
8-~N-methvl-N-chloracet~yl-aminomethy~-3-methvl-4-oxo-2-~henyl-
-4H-1-benzopvran (Intermediate XXXI~
A solution of 6 ml of chloracetyl chloride in 60 ml of
1,2-dichloroethane was added dropwise at 5 to 0C to a solution
of 20 g of Intermediate XX and 10 ml of triethylamine in 200 ml
of 1,2-dichloroethane. After stirring at 20-25C for 2 hours,
150 ml of water was added to the reaction mixture and the phases
were separated. The organic phase was washed with water and
dried on anhydrous sodium sulphate. The solvent was removed in
vacuo, and the residue was crystallized from ethanol to give
22.5 g of the title compound, m.p. 146-148C.
'. .; '
.. . ,.i , j ",,,, : , . , ,, : . . .. . ...
, ': ' ' ' . ''' ' , ' , ' ~ ' ' . ' .'.. , ' ' ,.; : ' ~' " : ' ',
5 ~ ~
-72-
8-chloracetamidomethyl-3-methyl-4-oxo-2-~henYl-4H-l-benzoPvran
~Intermediate XXXII)
A solution of 3.2 ml of chloracetyl chloride in 32 ml o~
1,2 dichloroethane was added dropwise, under stirring at -5C,
to a mixture of 10 g of Intermediate XXIV and 5.5 ml o~
triethylamine in 80 ml of 1,Z-dichloroethane. The reaction
mixture was stirred at ambient temperature for 1 hour and then
150 ml of water was added. The phases were separated; the
aqueous phase was extracted with 1,2-dichloroeth~ne and the
extracts were added to the organic phase which was then washed
with a cold saturated solution of sodium bicarbonate, washed
with water, dried on anhydrous sodium sulphate and evaporated to
dryness in vacuo. The residue was crystallized from ethanol to
give 10.7 g of the title compound, m.p. 152-155C. -
phenyl-4H-1-benzo~yran ~Intermediate XXXIII~
8.65 g of Intermediate XXX and 4.15 ml of triethylamine were
dissolved in 70 ml of tetrahydrofuran. To this solution, at
-10C, there was added dropwise over a period of 40 minutes a
solution of 2.35 ml of acetyl chloride in 23 ml of
tetrahydrofuran~ After stirring for 3 hours at 0-10C and for 2
hours at 20-25C, the solvent was evaporated off in vacuo. 100
ml of water was added to the residue, and extraction with
dichloromethane was effected, pooling the successive organic
extracts and then removing the solvent in vacuo. The residue
was dissolved in 50 ml of methanol and 3 g of potassium
carbonate and 10 ml of water were added. After stirring at 50C
for 20 minutes, to hydrolyse the N,0-diacetyl derivative which
had formed, the solvent was removed in vacuo and the residue was
treated with water and dichloromethane as above described. The
dichloromethane solution was again evaporated to dryness, and
5.9 g of 8-[N-acetyl-N-(2-hydroxyethyl)-aminomethyl]-3-methyl-
-4-oxo-2-phenyl-4H-1-benzopyran, m.p. 171-172C, was obtained.
3.6 ml of thionyl chloride in 30 ml of dichloromethane was added
dropwise at 0C to a solution of 6.1 g of the compound thus
prepared in 70 ml of dichloromethane. After stirring for 90
minutes at 20-25C, the reaction mixture was washed with water
' ' ', '' ", ' " ' ' ' ' ' ' ''''" ', '"', '" ' ' ' '
' :, ' ', ' , ' ' ., ' '
'. ' ' . , ' ' "' ~ ' " , " ' '; '' ~' , ' " ' ' ' ' ", ' ' '. . ' , '
2 ~
-73-
and dried. The solvent was removed in vacuo to gi~e the crude
title product for use without further purification.
8-(~-chloropro~ylthio~-3 methyl-4-oxo-2-phenyl-4H-1-benzopyran
(Intermediate XXXIV)
A solution o~ 20.1 g of stannous chloride dihydrate in 18 ml of
hydrochloric acid (d=1.18~ was added over a period o~ 5 minutes
at 65C to a solution of 6 g of Inte~mediate VIII in 70 ml of
acetic acid. After 10 minlltas, the reaction mixture was cooled
to 30-35C and the solvent was removed in vacuo. The residue was
taken up in water, and the insoluble matter was collected by
suction filtration, washed with water and dried. Yield 3.2 g of
8-mercapto-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran, melting at
115-118C after crystallization from ethanol.
A mixture of 8 g of the compound so prepared, 27 ml of
1-bromo-3-chloro-propane, 0.2 g of tPtrabutylammonium bromide
and 6.2 ml of 35% sodium hydroxide in 80 ml of benzene was
vigorously stirred for 4 hours at 20-25C. 100 ml of water and
40 ml of dichloromethane were added. The organic layer was
separated o~, washed with water and dried on anhydrous sodium
sulphate. The solvents and excess l-bromo-3-chloro-propane were
removed in vacuo. The residue was purified by column
chromatography on silica gel, eluting with petroleum ether:ethyl
ac0kate 9:1, and 5.7 g of the title compound was obtained. After
crystallization from methanol, it showed a melting point of
84-86C.
8- ! 3-chloro~ro~vlsulhonyl~-3-meth~1-4-oxo-2-~henyl-4H-1-
-benzoEyran (Intermediate XXXV)
7 ml of 30% hydrogen peroxide was added at 20-25C to a solution
o~ 3.45 g of Intermediate XXXIV in 35 ml of acetic acid. After
stirring for 4 hour~ at 60C, the reaction mixture was cooled to
20-25C. 30 ml of water was added. A precipitate formed, and was
collected by suction filtration, washed with water and dried,
yielding 3.4 g of the title compound. After crystallization from
acetone, it showed a melting poink of 160-163C.
-74-
8-(3-hydroxypEo~ylcarbamoyl)-3-methyl-4 oxo-2-phenyl~4H-l-
A solution of 7.6 ml of 3-aminopropanol in 50 ml of water was
added dropwise over a period of 30 minutes to a suspension o~ 30
g of 3 methyl-4 oxo-2-phenyl-4H-l-benzopyran-8-carbonyl chloride
and 15.2 g of potassium carbonate in 400 ml of acetone. The
thick suspension was stirred for 3 hours at 20-25C. The
æolvents were removed i~ vacuo and the residue was taken up in
300 ml of water. After stirring for 1 hour, the precipitate was
collected by suction filtration and washed with water. The crude
product was purified by cry~tallization from 95% ethanol and
23.8 g of the tile compound were obtained, m~p. 191-193C. An
additional 4.7 g o~ the title compound was obtained by
concentration in vacuo o~ the crystallization filtrate.
.,
8-(3-chloroPropylcarbamoyl3-3-methvl-4-oxo-2-~henyl-4H
-benzopyran ~Intermediate XXXVII)
A solution of 1.1 ml of thionyl chloride in 2 ml of chloroform
was added to a boiling solution of 3~37 g of Intermediate XXXVI
in 20 ml oP chloroform. After stirring for 90 minutes under .
reflux, the solvent was removed in vacuo and the residue was :
crystallized from acetonitrile to give 3 g of pure title
compound, m.p~ (188) 193-194C.
~ .
8- r 1-hydroxy~4~gthyL~h~ honyloxv~-but~11-3-methYl-4- -
-oxo-2-~enyl-4H-1-benzo~y~ran (Intermediate XXXVIII)
1.12 g of sodium cyanide in 3 ml of water wa~ added at 20-250C
to a stirred mixture of 3.96 g of 8-formyl-3-methyl-4-oxo~
-2-phenyl-4H-1-benzopyran, 2.61 g of morpholine and 4.48 g of
p-toluenesulphonic acid in 20 ml of tetrahydrofuran and 30 ml of
1,2-dichloroethane. The reaction mixture was refluxed for 4
hours, and then 10 ml of cold water was added. The
tetrahydrofuran was distilled off at no~mal presqure, and 10 ml
of 1,2-dichloromethane and 10 ml of chloroform were added. The
organic phase was separated, washed with aqueous sodium chloride
501ution, dried over anhydrou~ sodium sulphate and evapora~ed to
drynes~ in vacuo. The residue was ~uspended in diethyl ether,
Piltered off, and crystallized from chloroform:ethyl acetate.
Yield: 3.55 g of 8-(morpholino-cyanomethyl)-3-methyl-4-oxo-
.
, ' . ..................... ,i , ~ . ~ .,, , , , ,;
,, , ,. ' :'.
--~ 2~0 ~ 5~
-75-
-2-phenyl 4H-l-benzopyran, m.p. 236-238~C.
3.5 ml of a 30~ solution of potassium hydroxide in anhydrous
methanol was added under stirring at ambient temperature to a
suspension oP 22.~ g of the compound thus prepared in 520 ml of
anhydrous tetrahydro~uran. 6.3 ml of acrylonitrile in 20 ml of
tetrahydrofuran was dropped into this suspension, and the
reaction mixture was stirred at ambient temperature ~or 1 hour.
The solvents were evaporated oPf in vacuo. Crystallization of
the residue from methanol gave 23.22 g of
8-(1,3-dicyano~1-morpholino-propyl~-3-methyl-4-oxo-2-phenyl-4H-
-1-benzopyran.
23.2 g of the compound thus prepared was dissolved in 250 ml o~
dioxane. 250 ml of 6M hydrochloric acid was added and the
mixture was refluxed for 2~ hours. After cooling ~o ambient
temperature, the mixture was poured into 700 ml of aqueous
sodium chloride solution and extracted with ethyl acetate. The
extracts were washed with aqueous sodium chloride solution and
treated with 700 ml of lM sodium hydroxide solution. The
aqueous layer was washed with ethyl acetate and acidified with
37% hydrochloric acid. The precipitate was collected by suction
filtration and crystallized from ethanol to give 10.2 g oP 8-(3-
-carboxy-1-oxopropyl)-3-methyl-4-oxo~2-phenyl-4H-l-benzopyran,
m.p. 191-192C.
Diborane, generated by dropping a solution of 2.1 ml of freshly
distilled boron trifluoride diethyl etherate in 10 ml of
anhydrous diglyme into 19 ml of a 0.66 M solution of sodium
borohydride in diglyme, was bubbled into a suspension of 2.28 g
of the compound thus prepared in 23 ml o~ anhydrous
tetrahydrofuran, stirred at 0C under nitrogen flux. Stirring
was continued for 20 minutes at 0C and for a further 20 minutes
at ambient temperature. Methanol was cautiously dropped into
the mixture at 0C to quench the reaction. The solvents were
removed by evaporation in vacuo. The residue was purified by
flash chromatography on silica gel, eluting with petroleum
ether:ethyl acetate 3:7. The collected fractions were
evaporated in vacuo to leave 2 g of 8-~1,4-dihydroxybutyl)-3-
-methyl-4-oxo-2-phenyl-4H-1-benzopyran, m.p. 133-134C.
2.8 g oP p-toluenesulphonyl chloride was added at 0C to a
stirred solution of 3.17 g o~ the compound thus prepared in 32
-76-
ml of anhydrous pyridine. The mixture was stirred for 6 hours
at O~C and stood overnight at -4C without stirring. It wa~
then poured into 200 ml of aqueous sodium chloride solution,
acidified with 10 ml of 12N hydrochloric acid and filtered under
suction. The filter cake was dissolved in chloroform, and the
solution was washed with aqueous sodium chloride solution and
dried on anhydrous sodium sulphate. The solvent was distilled
off in a rotary evaporator. ThP residue was purified by flash
chromotography on silica gel, eluting with petroleum ether:ethyl
acetate 1:1. The collected fractions were evaporated to dryness
in vacuo, yielding 3.04 g of pure title product, m.p. 123-124C.
4~r4-(2-methoxyphenyl)~ erazinyll-butyraldehyde
(Intermediate XXXIX)
A solution of 5.4 g of 2-(3-chloropropyl)-dioxolan and 15.9 g of
1-t2-methoxyphenyl)-piperazine in 60 ml of dimethylformamide was
stirred at 80C for 4 hours. After cooling to 20-25C, the
reaction mixture was poured into 500 ml of ice cold 0.5N sodium
hydroxide solution and ~xtracted with dichloromethane. The
organic phase was washed with water and dried on anhydrous
sodium sulphate. The solvent was removed in vacuo, and the
residue was purified by flash chromatography on silica gel,
eluting with dichloromethane:ethanol 95:5. 9.8 g of
2-{3-[4-(2-methoxyphenyl)-1-piperazinyl]-propyl}-dioxolan was
obtained as an oil.
~NR CDC13 ~6~
1.5-2.0 (4H, m, CH2CH2CH)
2.2-3.2 (lOH, m, 5 x CH2N)
3.7-4.0 (7H, m, OCH3 and 2 x OCK2)
4.8 (lH, t, OC~0)
6.7-6.9 (4H, m, aromatic protons)
A solution of 12.8 g of the compound thus prepared in 200 ml of
tetxahydrofuran and 420 ml of lN hydrochloric acid was
maintained at 20-25C for 24 hours. It was then made alkaline
with 5N sodium hydroxide solution and immediately extracted with
dichloromethane. The organic layer was washed with water and
dried on anhydrous sodium sulphate. The solvent was evaporated
off in vacuo, and the residue was purified by flash
, . . ~ . . .. ' ' . ,. ': , .; . .. , , , ,: .
20~01~6
-77-
chromatography on sili a gel, eluting with
dichloromethane:methanol 97:3. 6.4 g of the title compound was
obtained as an oil.
N~ CDCl3 ~
l.g-2.0 (2H, m, CH2CH~CH2)
2 . 2-2 . 8 (8H, m, 3 x CH2N and CH2CHO)
2.9-3.2 (4H, m, 2 x CH2NAr)
3.8 (3H, s, OCH3~
6.8 (4H, s, aromatic protons)
9.3 ~lH~ s, CHO)o
8- 1 2, 3 -epoxypropoxy~ -3-methyl-4-oxo-2-phenyl-4H-1-benzopyran
(Intermediate XL)
7 ml of 2, 3-epoxypropyl chloride was aclded dropwise at 20-25~
to a stirred mixture o~ 5 g of 8-hydroxy-3-methyl-4-oxo-2-
-phenyl-4H-1-benzopyran and 9 . 7 ml ~f 2N sodium hydroxide in 10
ml of ethanol. After 6 hours at 20-25C, the reaction mixture
was poured into 100 ml of water and the precipitate which ~ormed
was collected by suction filtration. After drying and purifying
by flash chromatography on silica gel (eluant petroleum ether:
ethyl acetate 65:35), there was obtained 4.45 g of the title
compound, m.p. 128-129C.
8-rN-methyl-2-(4-meth~l~henvlsul~honvloXy)-ethvlsUlphamoYll-
-3 methyl-4-oxo-2-Phenyl-4H-l-benzopyran LIntermediate XLI~
A solution of 5 g o~ Intermediate VIII in 60 ml of
dichloromethane and 20 ml of tetrahydrofuran was added dropwise
at 0C to a mixture of 2.5 ml of 2-methylaminoethanol and 2.1 ml
of triethylamine in 20 ml of dichloromethane. After stirring for
2 hours at 20-25C, 100 ml of water and 100 ml of dichlormethane
were added to the reaction mixture. The phases were separated
and the organic solution was dried on anhydrous sodium sulphate.
The solvent was removed in vacuo, and the residue was purified
by column chromatography on silica gel, eluting with petroleum
ether:ethyl acetate 3:7. There was thus obtained 4.5 g of
8-(N-methyl-2-hydroxyethylsulphamoyl)-3-methyl-4-oxo-2-phenyl-
-4H-l-benzopyran, melting at 146-147C after crystallization
from ethanol.
The compound thus prepared was converted to the title compound
.: '
2 ~
-78-
by p-toluenesulphonylation according to the second step of the
procedure described below for the preparation of Intermediate
XLII. The title compound was used without further purification.
~=I7 I__methylphenylsulPhonvloxv)-ethylsulph~moyll-3-methyl-
-4-oxo-2-phenyl~4H-1-benzopyran (Intermediate XLII)
A solution of 5 g of Intermediate VIII in 37 ml of
tetrahydrofuran was added dropwise at 0C to a mixture of 2.5 ml
of ethanolamine and 2.5 ml of triethylamine in 25 ml of
tetrahydrofuran. After stirring at 20-25C, the reaction mixture
was poured into 400 ml of water. A precipitate formed, and was
collected by suction filtration, washed with water and air
dried, yielding 4.6 g of 8-(2-hydroxyethylsulphamoyl)-3-methyl-
-4-oxo-2-phenyl-4H-1-benzopyran, melting at 186-187C after
crystallization from ethyl acetate.
2.1 g of p-toluenesulphonyl chloride was added portionwise at
0C to a solution of 3.6 g of the compound thus prepared in 25
ml of pyridine. After 6 hours at 20-25C, the reaction mixture
was slowly poured on to crushed iee containing a slight excess
of hydrochloric acid. A precipitate formed and was collected by
suction filtration and washed with water. 4.9 ~ of the title
compound was obtained, melting at (163) 166-169C after
crystallization from ethyl acetate.
8-(3-aminopropylcarbamoyl~-3-methYl-4~oxo-2-phenyl-4H-1-
-benzopyran hYdrochloride ~Intermediate XLIII)
A solution of 21.6 g o~ 3-methyl-4-oxo-2-phenyl-4H-1-benzopyran-
-8-carbonyl chloride in 250 ml of anhydrous tetrahydrofuran was
dropped at 0-10C into a stirred solution of 17 g of
3-(2-methyl-2-propoxycarbamoyl)-propylamine (prepared as
described in Saari, W.S. et al., J. Med. Chem., 33, 97, 1990)
and 13 ml of triethylamine. After stirring ~or 2 hours at
ambient temperature, the reaction mixture was poured into water
and filtered to recover 12.3 g of 3-(2-methyl-2-
-propoxycarbamoyl)-propyl 3-methyl-4-oxo-2-phenyl-4H-l-
-benzopyran-8-carboxamide which was recrystallized from ethanol.
M.p. 178-180C.
A solution of 4.3 ml of trifluoroacetic acid in 15 ml of
anhydrous dichloromethane was added dropwise at -5C under
- . , , , ': . ,.
, ' "" " '', ~ , .'. ' ' '" ' '.
. ' , . , . : ,
~, ,, . , . , , : '' .:', ,,' '
, , , : . ... ... . ..
,~ : ..; ~ ~ ,
2 0 ~
-79-
stirring to a solution of 3.3 g of the compound thus prepared in
35 ml of anhydrous dichloromethane. After warming to ambient
temperature, the mixture was stirred for 3 hours. The
dichloromethane and the excess trifluoroacetic acid were
evapora ed off at 20-25C using a rotary evaporator. The oily
residue was dissolved in dichloromethane and lN sodium hydroxide
solution was added. The organic layer was washed with water,
dried on anhydrous sodium sulphate and filtered. Excess
ethanolic hydrogen chloride was added to the filtrate, and the
solvent was removed in vacuo. The residue was crystallized from
ethanol to give 1.5 g of the title compound, m.p. 253-255C.
3-t2-chloroethvlureido)-3~methYl-4-oxo-2-~henvl-4H-l-benzo~ran
(Intermediate XLIV)
4 ml of 2-chloroethyl isocyanate were added, under stirring at
ambient temperature, to a solution of 3.9 g of 8-amino-3-methyl-
-~-oxo-2-phenyl-4H-1-benzopyran in 52 ml of anhydrous
dimethylformamide. Stirring was continued at 70C for 5 hours.
Water was added to the reaction mixture, which was then
extracted with ethyl acetate. The organic phase was evaporated
to dryness in vacuo. The residue was suspended in diethyl ether
under stirring. The title product was then filtered off and
recrystallized from methanolO Yield 3.74 g, m.p. 213-214C.
~Z,E)-8~4- r 2-~,3-dioxan~l)l-1-butenyl~ 3-methyl-4-oxo-2-
-phenyl-4H-1-benzopvran (Intermediate XLV~
1.6 ml of 2.5N butyllithium in hexane was added dropwise at
-20C to a solution of 1.53 g of 2-[2-tl,3-dioxanyl)]-ethyl
triphenylphosphonium bromide in 10 ml of anhydrous
tetrahydrofuran. The mixture was stirred for 20 minutes at
-20C. A solution of 0.8 g of 8-formyl-3-methyl-4-oxo-2-phenyl- ~-
-4H-l-benzopyran in 11 ml o~ anhydrous tetrahydrofuran was
dropped into the mixture, which was then warmed to 0C over a
period of 90 minutes and then to ambient temperature over a
period of 30 minutes. The reaction was quenched by addition of
methanol. The solv~nts were evaporated off in vacuo and the
residue was purified by flash chromatography on silica gel,
eluting with ethyl acetate:petroleum ether 3:7, to give the
title compound, as a mixture of diastereoisomers E and Z, m.p.
~ '
: ~,
-80- .
(93) 98-100C. The ratio of the two isomers was determined by
NMR spectroscopy and resulted in E:Z = 65:35.
~NR, CDCl3 (~)
8.1-8.2 (m, lH, CH in position 5 of the benzopyran ring)
7.2-7.8 (m, 7H, other aromat~c C~ groups of $h~ benzopyran and
phenyl rings)
6.9 (dt, lH, F1-CH of the E isomer)
6.8 (dt, lH, Fl-CH of the Z isomer)
6.4 (dt, lH, Fl-CH=CH of the E isomer)
5.9 (dt, lH, Fl-CH=CH of the Z isomer)
4.6~4.7 (m; lH, OCHO)
3.6-4.2 (m, 4~I, OCH2O of the dioxane ring) ::
2.4 2.7 (m, 2H, CHC~2CH)
1.9-2.3 (m, 5H, CH3 and CH2 in position 5 of the dioxane ring~
8-~4- r 2-tl.3~dioxanYl~l-butvl~-3-meth~1-4-oxo-2-~henyl--4H-l-
-benzopyran ~Intermediate XLVI
A mixture of 0.2 g of 10% palladium-on-carbon catalyst and of 1 ~-
g of Intermediate XLV in 24 ml of methanol was hydrogenated in a
Parr apparatu~ at ambient temperature with a hydrogen pressure
of 1.5 atmospheres. After the theoretical hydrogen consumption,
the catalyst was filtered off and the solvent was removed by ::
evaporation in vacuo. The residue was crystallized from
cycloh~xane to give the title compound, m.p. 11~ 119.5C.
8-carboxymethyl 3 ~ethyl-4-oxo-2-phenyl-4H-1-benzopyran
(Inter ediate XLVII~
4 . 5 g of potassium permanganate was added, portionwise over a
period of 1% hours under stirring at 0-10C, to a ~ixture of
2.76 g o~ 8-allyl-3-methyl~4-oxo~2-phenyl-4H-1-benzopyran (P. Da
Re, US 33504113, 0.17 g of Aliquat 336, 1.12 ml of acetic acid,
56 ml of dichloromethane, 3.2 ml of sulphuric acid (d=1.84) and
60 ml of water. Stirring was continued at ambient temperature
for 5 hours. 3.4 g of sodium metabisulphite were added
portionwise at 0-5C within 15 minutes. The organic lay~r was .
separated, washed with water and extracted with 60 ml of lN
aqueous sodium hydroxide solution. The aqueous phase was
acidified by addition of dilute hydrochloric acid and extracted
2 ~
,
-81-
with ethyl acetate. The organic phase was washed with water,
dried on anhydrous sodium sulphate and, after filtration,
evaporated to dryness in vacuo. The residue was treated with
carbon tetrachloride and the solid was collected by suction
filtration to give 1 g of the title compound, m.p. 191-192C
(acetonitrile~.
8-(4-chlorobutyramido)-3-methyl-4-oxo-2-phenyl-4H-1-benzopyran
(Intermediate XLVIII~
The title compound was prepared in the same manner as
Intermediate X, but using 4-chlorobutyryl chloride instead of
acryloyl chloride. The solid obtained, filtered from water and
dried, was rinsed with hot diethyl ether and collected by
suction to give the title compound. A sample, crystallized from
50% aqueous ethanol and washed with diethyl ether, melted at
(153) 162-164C.
8-methylamino-3-methyl-4-oxo-2-~henyl-4H-1-benzopyran
(Intermediate XLIX)
A solution of 0.5 g of Intermediate XXIII in 1.5 ml of anhydrous
dimethylformamide was added dropwise under stirring, at -5~C, to
a suspension of 0.045 g of sodium hyclride (80% in mineral oil).
After stirring at room temperature for 1 hour, 0.092 ml of
methyl iodide in 0.6 ml of anhydrous dimethylfo~mamide was added
dropwise. Then the reaction mixture was stirred at 50C for 1
hour, cooled to 20C, poured into water, filtered by suction and
dried at 60C for 3 hours to recover 0.6 g of
8-(N-methyltrifluoroacetamido)-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran.
~MR ~CD~13, ~)
8.15 (dd, lH, benzopyran CH in 5)
7.10-7.60 (m, 7H, other benzopyran and phenyl CHs)
3.30 (s, 3H, CH3-N)
2.10 (s, 3H, benzopyran CH3 in 3)
A mixture of 0.44 g of the above compound and 0.05 g of sodium
~orohydride in 4 ml of ethanol and 1 ml of dimethylsulphoxide
was stirred at room temperature for 1 hour, and then quenched
with an excess of 4 N hydrochloric acid. After removal of
ethanol by evaporation in vacuo, the residue was rinsed with
. . , ' , ', ; . : '. ,: . ~ ~
2 ~
--82--
water, then with 3N sodium hydroxicle and extracted with ethyl
acetate. The organic layer was washed with water, dried on
anhydrous sodium sulphate and evaporated to dryness in vacuo.
The solid residue was crystallized from ethanol to give O . 22 g
of the title compound, melting at 143-146C.
8-~N-methylacrylamido)-3-methyl-4-oxo-2~phenyl-4H-1-benzopvran
¢Intermediate L)
This oompound was prepared in the same manner as Intermediate X,
but using Intermediate XLIX instead of 8-amino-3-methyl-4-oxo-2-
phenyl-4~ benzopyran. Instead of diluting with water, THF
was removed by evaporation in vacuo and the crude residue was
dissolved in ethyl acetate and washed with water. The organic
solution was dried on anhydrous sodium sulphate and evaporated
to dryness in vacuo to give the title compound. A sample,
purified by column chromatography on silica gel (eluting with
ethyl acetate:petroleum ether 4:6) and crystallized from
cyclohexane, melted at 136-137C.
1- r 2 - ! 1. 3-dihydro-1~3-dioxo-2H-isoindol-2-yloxy)-ethyll-4-
-(2-methoxvphenYl)-piperazine l~termediate LI)
A mixture o~ 6.73 g of N-hydroxyphthalimide, 3.73 g of sodium
acetate and 10 g of 1-(2 chloro~ethyl)-4-(2-methoxyphenyl)-
-piperazine in 100 ml of anhydrous dimethylsulphoxide was
stirred at 100C for 4 hours. The reaction mixture was then
cooled to room temperature, poured into water and extracted with
ethyl acetate. The collected organic layers were washed with lN
sodium hydroxide, dried on anhydrous sodium sulphate and
evaporated to dryness in vacuo to give 7.58 g of the title
compound. A sample was crystallized from cyclohexane and found
to melt at (76) 80-83C.
.
1-~2-aminooxYethvl)-4-(2-methoxyphenyl)-piPerazine hydrochloride
~Intermediake LII)
A solution of 6.59 g of Intermediate LI and 1.10 ml of 85%
hydrazi~e hydrate in 130 ml of 95% ethanol was re~luxed for 4
hour~. Ethanol was removed by evaporation in vacuo. The
residue was rinsed with water and then with an excess of 37%
hydrochloric acid and ~iltered. The acid aqueous solution was
" '
: , . . : .. , . . - ., . ~ ~
~83-
made basic with 5% sodium hydroxide and extracted with
chloroform. The organic layer was dried on anhydrous sodium
sulphate and evaporated to dryness in vacuo to give 4.3 g of the
title compound as an oil. A sample was converted to the
hydrochloride by salification with ethanolic hydrogen chloride
in dichoromethane~ The solvents were removed by evaporation in
vacuo and the crude residue was crystallized from ethanol,
giving the title compound, m.p. 208-209C.
8-f4-chlorobutylthio2-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran
~ntermediate LIII)
Tha title compound was prepared in the same manner as
Intermediate XXXIV, but using 1-bromo-4-chlorobutane instead of
1-bromo-3-chloropropane. ~.p. 81-84C (ethanolj.
. ' '~ ."
8-(4-chlorobutylsulphinyl)~3-methyl-4-ox~ æ_enyl-4H~
-benzopyran (Intermediate LIV~ -
The title compound was prepared in the same manner as
Intermediate XXVI, but using Intermediate LIII instead of
Intermediate XXI. A sample, crystallized ~rom
cyclohexane:benzene 0.5:1, melted at 124-125C~
'- ~
~-carboxy-4-oxo-3-phenyl-4H-1-benzopxran
(Intermediate LV)
A solution of 38.22 g of silver nitrate in 75 ml of water was
added dropwise, under stirring, at 2Q-25C, to a solution of
22.5 g of 8~formyl-4-oxo-3-phenyl-4H-l-benzopyran (prepared as
described by G. Atassi et al., Eur. J. Med. Chem. - Chim. Ter.,
20~ 393 (lg85)) in 150 ml of 85% ethanol and 450 ml of
dimethy]formamide. Then, a solution of 32.67 g of 85% potassium
hydroxide in 195 ml of water was added dropwise under stirring
at 15-20C. After additional stirring at room temperature,
the reaction mixture was filtered ~y suction; the mother liquor
was acidified with 37% hydrochloric acid and diluted with 1.2
litres of water. Filtration by suction and washing with water
to neutrality gave t~e title compound as a crude product. This
was suspended in 150 ml of ethyl acetate and stirred with 444 ml
of 0.3 M sodium hydrogen carbonate until clear layers were
obtained. The aqueous layer was washed with 75 ml of ethyl
2 ~
.,
-84-
acetate, then made acidic with 37% hydrochloric acid, filtared
and dried at 60-65C to give 19.12 g of the title compound,
melting at (215) 218C. A sample, crystallized ~rom ethanol,
had the same melting point.
~-chlorocarbonyl-4-oxo-3-phenyl-4H-l-benzol~yran
rIntarmediate L~
A mixture of 15.97 g of Intermediate LV and 15.6 ml of thionyl
chloride in 75 ml o~ anhydrous toluene was stirred at 80-85 C
for 4 hours. After removal of the solvent under vacuo, the
residue was rinsed twice with 20 ml of toluene and evaporating
to dryness in vacuo to give, after drying, 16 g of the title
compound melting at ~126) 138-140C which was used without
further purification. M.p. (130) 138-140C (toluene).
8-(N-acetylcarbamoyl~-3-meth~1-4-oxo-2-~henyl-4H-1-benzopyran
(Intermediate LVII)
A mixture of 3.5g of 8-carbamoyl-3-methyl-4-oxo-2-phenyl-
-4H-1-benzopyran (described in JP 61-238783), 4.8 ml of acetic
anhydride and 0.25 ml of sulphuric acid (d=1.098~ was stirred at
140C for 3 minutes. The reaction was cooled to ambient
temperature, diluted with water and filtered by suction to givP,
after washing with water and dessication, 3.88g o~ the title
compound
N~ ~CDC13, ~) :
10.50 (bs, lH, imidic NH)
8.35-8~70 (m, 2H, CH in position 5 and 7 of the benzopyran ring)
7.45-8.00 (m, 6H, other aromatic CHs)
2.60 (s, 3H, CH3C0)
2.20 (s, 3H, CH3 in position 3 of the bPnzopyran ring)
2-(2-methylthiophenoxy)-acetaldehyde diethyl acetal
tIntermediate LVIII)
A mixture of 15.2 ml of 97% 2-bromoacetaldehyde diethyl acetal,
14 g o~ 2-(m~thylthio)-phenol, 13.7 g of anhydrous potassium . .
carbonate and 3.13 g of tricaprylmethylammonium chloride in 140
ml of anhydrous dimethylformamide was stirred at 95C for 38
hours. At the end of this period, the reaction mixture was
cooled to ambient temperature, poured into 1 litre of water and
".,~. ... , ~,~"/, , , , ;.;' . . .
', " ' ', ' "'.`" "': " .
:' , ' ' ' ' , ' , , . :. .. ' .. '',
-85-
extracted with diethyl ether. The organic layer was washed with
water, dried on anhydrous sodium sulphate and evaporated ko
dryness in vacuo. The oily residue was purified by column
chromatography on silica gel eluting with petroleum ether:ethyl
acetate ~9 1. Evaporation in vacuo of the collected fractions
yielded 12.9 g of pure title compound. A sample was
crystallized from n-hexane and melted at 50-52~C.
2-(2-methylthiophenoxy)-acetaldehyde (Intermediate_LIX) -
A mixture of 10.5 g of Intermediate LVIII and 140 ml of 2N
hydrochloric acid in 85 ml of anhydrous tetrahydrofuran was
stirred at 50C for 2 hours. The organic solvent was then
removed by evaporation in vacuo, and the aqueous residue was
extracted with ethyl acetate. The organic layer was washed with
water~ dried on anhydrous sodium sulphate and evaporated to -~
dryness in vacuo giving 9.5 g of the title compound as a solid
which was used without further purification. A sample was
crystallized from cyclohexane yielding the pure title compound,
m.p. 102~104C.
8-(4-chlorobutylsulphonyl)-3-methyl-4-oxo-2-phenyl-4H-l-
-benzop~ran (Inte~mediate LX)
. .
The title compound was prepared by the same method as
Intermediate XXV, but using Intermediate LIII instead of
Intermediate XXI. It was crystallized fr~m diisopropyl ether
and melted at 112-~15C.
8-ethoxycarbonyl-4-oxo-4H-1-benzopyran~(Intermediate ~XI~
4.35 g of sodium metal was added in pieces at amblent
temperature to a solution of 9.85 g of ethyl 3-acetyl-2-hydroxy
benzoate (synthesized from 3-acetyl-2-hydroxybenzoic acid
(prepared as described in R.E. Ford, J. Med. Chem., 29, 538
(1986)) re~luxing in 6N ethanolic hydrogen chloride ~or 1.5
hours, evaporating to dryness in vacuo and purifying the crude
by column chromatography on silica gel (eluant ethyl
acetate-petroleum ether 8:2) - m.p. 47C (hexane)) in 98 ml of
ethyl formate.
The reaction mixture went spontaneously to re~lux for 20
minutes; then it was stirred at ambient temperature for 4 hours
2 ~
-86-
and the ethyl formate was removed by evaporation to dryness in
vacuo. The crude solid obtained was rinsed with 120 ml o~
ethanol and 67 ml of 5.6M ethanolic hydrogen chloride. The
mixture was stirred at reflux for 30 minutes; after this period
it was cooled to ambient temperature and evaporated to dryness
in vacuo~ The residue was purified by column chromatography on
silica gel eluting with ethyl acetate:petroleum ether 3:7 to
6:4 to give 8.31 g o~ the title compound. A sample crystallized
from cyclohexane melted at 88-89C.
8-carboxy-4-oxo-4H-1-bjenzopyran (Intermediate LXII)
30 ml of 6N hydrochloric acid was added to a solution of 4.0 g
of Intexmediat~ LXI in 30 ml of dioxane and the resulting
mixture was stirred at reflux for 5 hours. The reaction
mixture was cooled to ambient temperature and poured into 200
ml o~ water. After 12 hours at 0 to 5C, tha title compound was
filtered off under suction. Washing with water and diethyl
ether yielded, after desiccation, 2.8 g of the title compound,
used without further purification. A ~ample washed with boiling
acetonitrile:methanol 25:1, filtered and crystallized from
acetic acid, melted at 253-254C.
,
8-carboxy-6-hydroxy-3-methyl-4-oxo-2-phenvl-4H-l-benzopyran
(Intermediate LXIII ?
A mixture of 1.5 g of 8-carboxy-6-methoxy-3-methyl-4-oxo-
-2-phenyl-4H-l-benzopyran (prepared as described in JP 61-15880)
and 28 ml of 57% hydroiodic acid in 47 ml of acetic acid was
stirred at reflux for 18 hours. The reaction mixture was cooled
to ambient temperature and poured into water; lN sodium
hydroxide was added to adjust the pH to 4 to 5. 2 g of sodium
thiosulphate was added and stirring was continued for 15
minutes. After this period, the crude title compound was
filtered off under suction and dissolved in 0.5M sodium
hydroxide; the basic solution was washed with ethyl acetate and
acidified to pH 1 by adding 37~ hydrochloric acid. The title
compound wa~ collected under suction and de~iccated. Yield: 1.12
g of the title compound, used without further purification and
melting at 2/9-281C after crystallization from 50~ ethanol.
: : `
2~9~5~
-87-
2-hYdroxy-5-nitro-3-~ropionyl-benzoic acid (Intermediate LXIV)
97.1 g of 2-hydroxy-3-propionyl-benzoic acid, prepared as
described in Brit 1, 343, 119 (1974), were added in 5 minutes to
500 ml of sulphuric acid (d=1.84) stirred at -25~C. A mixture of
40 ml of 65% nitric acid and loo ml of sulphuric acid (d=1.~43
was added in 40 minutes maintaining the temperature of the
reaction mixture between -20 and -13C. The mixture was stirred
at -18C for an additional 30 minutes. After this period, it
was cautiously poured into a mixture of 2.0 kg of crushed ice
and 500 ml of water, stirred for lo minutes and filtered to
give the title compound, after washing with water and drying at
50C for 6 hours. Crystallization of this solid from 50%
ethanol yielded 91.5 g of title compound melting at 186-189C,
used without further purification. A sample was recxystallized
from 50% ethanol and melted at 189-191C.
,'.
ethyl 2-hydroxy=~-nitro-3-2ropionyl-benzoate (Intermediate LXV~
A solution of 93.3 g of Intermediate LXIV and 25 ml of sulphuric
acid (d=1.84) in 490 ml of ethanol was refluxed for 17 hours.
After cooling to ambient temperature, 47.7 g of sodium carbonate
was added portionwise and th~ ethanol was evaporated off in
vacuo. The residue was rinsed with 1.2 litres of water, made
alkaline by adding 3~% sodium hydroxide and stirred for 15
minutes. 37% hydrochloric acid was added to thii~ suspension to
adjust the pH to 6. Filtration yielded 85.4 g of the title
compound used without further purification (m.p. 75-77C). A
sample was crystallized twice from ethanol and melted at
76-77C.
8-ethoxycarbonyl-3-methyl-6-nitro-4-oxo-2-phenyl-4H-1-benzopyran
(Intermediate LXVI)
A mixture of 48.1 g of Intermediate LXV, 63 ml of benzoyl
chloride and 85.6 g of sodium benzoate was stirred at 180C
(bath temperature) for 8 hours. The pasty mixture was cooled to
60-70C; 700 ml of 50% ethanol was added and the resulting
mixture was stirred again at 50C for 30 minutes. 60 ml o~ 35%
sodium hydroxide was added at 5C, ensuring that the temperature
did not rise above 15c. Filtration by suction, followed by
washing with 50~ ethanol and water afforded a crude product,
~ ' ~
~9~ 5~
-88-
which was purified by double passage on column chromatography
on silica gel eluting first with dichloromethane:petroleum
ether graduat~d 8:2 to 9:1, then with dichloromethane and
finally with dichloromethane:ethyl acetate 95:5. Evaporation in
vacuo of the collected fractions gave the title compound, which
was washed with 140 ml of ethanol. Yield 43 g, m.p. 132-133C
(ethanol).
8-carboxy-3-methyl-6-nitro-4-oxo-2-phenyl-4H-l~benzopyran
(Intermed~iate LXVII~
~ mixture of 15.9 g of Intermediate LXVI and 48 ml of lN sodium
hydroxide in 320 ml of ethanol was stirred at reflux for 30
minutes. The organic solvent was removed by evaporation in vacuo
and the resulting suspension was diluted with 200 ml of water
and made acidic with 37% hydrochloric acid. Filtration and
washing with diethyl ether yielded 11.1 g of the title compound
melting at (258) 286-292C and used without further
purification. After crystallization from ~imethylformamide:water
6:4 the title compound exhibited the same melting point.
. .. .
8-chlorocarbonyl-3-methyl-6-nitro-4-oxo~2-phenyl-4H-l-benzopyran
(Intermediate LXVIII)
A mixture of 6.2 g of Intermediate LXVIII 5.2 ml of thionyl
chloride and 0.1 ml of anhydrous dimethylformamide in 60 ml of
toluene was ætirred at 90C for 2 hours. Evaporation to dryness
in vacuo and desiccation yielded 6.5 g of the title compound,
m.p. 161-162C, used without further purification. A sample was
crystallized from toluene and had the same melting point.
8-carboxy-7-methoxy-3-methyl-4-oxo-2-~henyl-4H-1-benzopyran
(Intermediate LXIX)
216 ml of a 0.3M solution of potassium permanganate in water was
added dropwise over 40 minutes to a mixture of 7.94 g of
8-formyl-7-methoxy-3-methyl-4-oxo-2-phenyl-4H-1-benzopyran
(prepared as described in Da Re et al., J.Org.Chem., 25, 1097,
1960) and 54 ml of 5~ sodium dihydrogen phosphate in 162 ml of
t-butanol, stirred at 75C. After a furthex 2~ hours stirring
at the same temperature, the reaction mixture was cooled to
ambient temperature and 81 ml of lM sodium dithionite was
. .. . . . . . ........................ .. .
.:
.
~- ` 2 ~ 5 ~ :
-89-
dropped slowly therein. The mixture was extracted with ethyl
acetate. The organic layer was washed four times with 160 ml of
0.5N sodium hydroxide; the collected basic aqueous layers were
washed with diethyl ~ther and made acidic with 37% hydrochloric
acid. The title compound precipitated. It was filtered o~ and
washed with water, yielding a~ter desiccation 3.3 g, used for
further reactions without further purification and melting at
180-181C after crystallization from 95% ethanol.
Ethyl 3 ~ropionyl 2-(4-tri~luoromethylbenzoyloxy)-benzoate
(Intermediate LXX~ -
A solution of 6.7 g of 4-trifluoromethylbenzoyl chloride
(prepared from the corresponding benzoic acid and thionyl
chloride in benzene at reflux and used without purification) in
50 ml o~ chloro~orm was added dropwise to a solution of 7.13 g :
of ethyl 2-hydroxy-3-propionyl-benzoate and 4.9 ml of
triethylamine in 50 ml of chloroform. The mixture was stirred
at ambient temperature for 2 hours; the solvent was then
evaporated off in vacuo and the resiclue was purified by column
chromatography on silica gel sluting with petroleum ether:ethyl
acetate 85:15. ~vaporation in vacuo to dryness o~ the collected
fractions yielded 7.4 g of the title compound as on oil.
~NR spectrum ~t 60 ~Hz (CDCl3, ~)
7.6-8.5 (m, 6H, aromatic CHs)
7.5 (t, lH, phenol ring, CH in 5)
4.2 (q, 2~, COOCH2) -.
2.9 (q, 2H, COCH2)
1-1.3 ~2t ,6H, 2xCH3)
8-ethoxvcarbonvl-3-methyl-4-oxo-2-~4-trifluoromethvlphen~l!-
-4H-l-benzopyran (Intermedlate LXXI)
A mixture of 6.96 g of Intermediate LXX and 2.58 g of potassium
t-butoxide in 35 ml of pyridine wa~ stirred ~t 100C for 2
hours. After this period, the reaction mixture was cooled to
ambient temperature, poured into a solution o~ 50 ml of acetic
acid in 600 ml of water and extracted with ethyl acetate. The
organic layer was washed with 10% hydrochloric acid and with
water, dried on anhydrous sodium sulphate and evaporated to
dryness in vacuo to give 6.9 g of 1-(2-hydroxy-
, . , , ~
go 2~1~0~
-3-ethoxycarbonyl~-2-methyl-3-(4-trifluoromethylphenyl)-
-1,3~propanedione. A solution of the compound thus prepared and
2.2 ml of 37% hydrochloric acid in 35 ml of glacial acetic acid
was stirred at 100C for 1~ hours, After cooling to ambient
temperature, the mixture was poured into 630 ml of lN sodium
hydroxide and extracted with ethyl acetate. The organic layer
was washed with water, dried on anhydrous sodium sulphate and
evaporated to dryness in vacuo. The crude was purif ied by
column chromatography on silica gel eluting with petroleum
ether:ethyl acetate 85:15. Evaporation in vacuo to dryness
yielded 2.95 g of the title compound, melting at 111-113 C
after crystallization from cyclohexane.
8-carboxy-3-methyl-4-oxo-2-(4-trifluoromethylphenyl)
-4H-l-benæopyran (Intermediate LXXII)
A mixture of 2.95 g of Intermediate LXXI and 0.43 g of lithium
hydroxide mon~hydrate in 12.5 ml o~ methanol and 12.5 ml of
tetrahydrofuran containing 8 ml of water was stirred at ambient
temperature for 1~ hours. The mixture was poured into a solution
of 30 ml of lN hydrochloric acid in 300 ml of water and filtered
by suction to give 2.47 g of the title compound used without
further purification. A sample was crystallized from 60% ethanol
and melted at 253-254C.
8-ethoxycarbonyl-2-(4-benzoYlPhenYl)-3-methyl-4-oxo-
-4H-l-benzopyEan ~Intermediate ~XXIII)
The title compound was synthesized ~ollowing the procedure of
Intermediate LXX and LXXI in the established order but starting
from 4-benzoylbenzoyl chloride, instead of 4-trifluoromethyl-
-benzoyl chloride and reacting this compound in 1,2-dicloro-
ethane instead of chloroform and in the presence o~
4-dimethylaminopyridine instead of triethylamine. After the
usual workup, the residue was purified by column chromatography
on silica gel eluting with dichloromethane:ethyl acetate 9:1.
Evaporation in vacuo to dryness of the collected fractions
yielded the title compound used without further puriPication. A
sample was crystallized from cyclohexane and melted at
125-136C (dec.).
s:
,
.
2 0 ~
--91-- :
8-carboxy-2-(4-benzoYlphenyl)-3-methyl-4-oxo-4H-l-benzo~yran
(Intermediate LXXIV)
The titl~ compound was prepared in the same manner as
Intermediate LXXII but ~tarting from Intermediate LXXIII instead
o~ Intermediate LXXI. It was purified by dissolving the crude in
0.5M sodium hydroxide, washing the aqueous layer with ethyl
acetate and precipitating the pure title compound by addition of
37% hydrochloric acid. A sample was crystallized from acetic
acid ~nd melted at 260-262C.
ethyl 2-(4-~henoxvbenzovloxY)-3-propionYl-benzoate
~Intermediate LXXV)
The title compound was prepared by the procedure described for
Intermediate LXX, but starting from 4-phenoxybenzoyl chloride
instead of 4-trifluoromethylbenzoyl chloride. Evaporation of the
solvent yielded the pure title compound. -
BpeatrUm ~t 200 N~ ~CDCl39 ~)
8.17 (dd, 3H, phenyl CHs in position ortho to the
carboxylate groups)
7.92 (dd, lH, phenyl CH in position ortho to the C0
group)
7.38-7.48 (m, 3H, phenyl CHs in position meta to the
carboxylate groups) ~-
7.25 (d, lH, CH in position 4 of the phenoxy ring)
7.05; 7.10 (2d, 4H, other C~s of the phenoxy ring) ~ :
4.25 (q, 2H, CH20)
2.90 (q, 2H, CH2C0)
1.05~1.20 (m, 6H, 2xCH3)
8=ethoxycarbonvl-3_meth~1-4-oxo-2-r4-phenoxY~henYl?-4H-l-
-benzopyran (Intermediate LXXVI) .
The title compound was prepared in the same manner as
Intermediate LXXI, but starting from Intermediate LXXV instead
o~ Intermediate LXX. Pur.ification was by column chromatography .,
on silica gel eluting with petroleum ether:ethyl acetate 6:4.
Evaporation in vacuo yielded the pure title compound, m.p.
98-100C.
'' " . ",. ,: ' , , ' ,: ,~ . ,,
-g2~
8-carboxy-3-methyl-4-oxo-2-f4-~henoxyphenylL-4H-1-benzopyxan
(Intermediate ~XXVII~
This compound was obtained in the same manner as Intermediate
LXXII, but starting from Intermediate LXXVI instead of
Intermediate LXXI. M.p. 216-218C.
8-carboxy-2-(t-butyl)-3-methyl-4-oxo-4H-l-benzopYran --
(Tntermediate LXXVIII~
6 ml of pivaloyl chloride was added dropwise into a stirred
solution of 8.9 g of ethyl 2-hydroxy-3-propionyl-benzoate in 20
ml of anhydrous pyridine. The reaction mixture was stirrad at
80C for 6 hours, cooled at ambient temperature and poured into
a mixture of 200 g of crushed ice and 30 ml of lON hydrochloric
acid. After extraction with diethyl ether, the organic phase
was washed with brine, dried on anhydrous sodium sulphate and
evaporated to dryness in vacuo, yielding 11.4 g of crude ethyl
2-pivaloyloxy-3-propionyl-benzoate.
2.4 g of this compound were dissolved in 4 ml of anhydrous
pyridine and 1 g of anhydrous potassium t-butoxide was added.
The resultant mixture was heated for 15 minutes at ~00C, cooled
to ambient temperature and poured into 50 g of iced water
containing 8 ml of lON hydrochlori acid. After extraction with
diethyl ether, the organic phase was washed with brine, dried on
anhydrous sodium sulphate and evaporated to dryness in vacuo,
giving 2.1 g of crude ethyl 2-hydroxy-3-(2-pivaloylpropionyl)-
-benzoate, which was used without purification in the next step.
2 g of the compound thus prepared were heated at 100C for 15
minutes after dissolution in a mixture containing 15 ml of
acetic acid and 1.5 ml of 37% hydrochloric acid. Ater cooling
to ambient temperatllre, the mixture was poured into 100 ml of
water and extracted with diethyl ether. The organic phase was
washed with 5% aqueous sodium hydrogen carbonate followed by
water, dried on anhydrous sodium sulphate and evaporated in
vacuo, yielding 1.6 g of crude 8-ethoxycarbonyl-2-(t-butyl)-
-3-methyl-4-oxo 4H-1-benzopyran.
~.5 g o~ the above e5ter was dissolved in 20 ml o~ methanol. 3
ml o~ lON sodium hydroxide was slowly added, maintaining the
temperature between 25 and 35C. After 1~ hours at ambient
temperature, the reaction mixture was diluted with 100 ml of
~. . :;
'; , :;: .: '.
. r . ~
'"' ' ', ' ' ~ ' ' ' '' '
~, ' ~ ' '. ' " ", "
; "' ~ . '.:. ' ' '
2~015~ ~
, .
-93~
water and extracted with ethyl acetate. The aqueous layer was
acidified with 3N hydrochloric acid. The precipitate was
collected by suction, washed with water and crystallized from
ethanol, yielding 0.8 g of the title compound, melting at
225-228~.
8-carboxv 2-cYclohexyl-3-methyl-4-oxo-4H-1-benzopyran
(Intermediate LXXIX)
This compound was prepared according to the reaction sequence
and methods described for Intermediate LXXVIII, but starting
from cyclohexylcarboxylic acid chlsride instead of pivaloyl
chloride and with other minor differences. In particular, ethyl
2-cyclohexylcarbonyloxy-3-propionyl-benzoate was obtained after
8 hours stirring in pyridine at ambient temperature and
transposed to ethyl 2-hydroxy-3-(2-cyclohexylcarbonyl-
~ propionyl) benzoate upon heating with potassium t-butoxide for
2~ hours at 100C. Cyclization to 8-ethoxycarbonyl-
-2-cyclohexyl-3-methyl-4-oxo-4H-l-benzopyran was carried out by
heating in a mixture of acetic and hydrochloric acids at 100C
~or 1~ hours and the hydrolysis to the title compound was
performed in 20 minutes at ambient temperature. The title
compound melted at 224C after crystallization from 40%
ethanol.
8 e~hoxvcarbonyl-2-f2-furyli-3-methvl--4-oxo-~H-l-benzopyran
fIntermediate LXXX~
A mixture o~ 3.2 g of Intermediate XC and 1.3 g of anhydrous
potassium t-butoxide in 8 ml of anhydrous pyridine was stirred
at 60C for 15 minutes, cooled to ambient temperature and poured
into 60 ml of iced water containing 15 ml of lON hydrochloric
acid. After extraction with ethyl acetate, the organic phase was
washed with 5% aqueous sodium bicarbonate and water and dried on
anhydrous sodium sulphate. Upon evaporation in vacuo 2.5 g o~
crude ethyl 3-(2-furoyl propionyl)-2-hydroxy-benzoate was
obtained.
2.5 g o~ the compound so obtained was stirred at 100C ~or 30
minutes with 10 ml of acetic acid and 0.7 ml of 37%
hydrochloric acid. After cooling to ambient temperature, the
mixture was poured into 180 ml of water. The title compound
2~9~
-94-
precipitated and was collected by suction filtration, washed
with water and crystallized from isopropanol. Yield 1.5 g, m.p.
~37-139~.
8-carboxy-2-(2-furYl~-3-methyl-4-oxo-4H-1-benzopyran
(Intermediate LXXXI)
A mixture of 3.5 g of Intermediate LXXX and 6 ml of lON sodium
hydroxide in 40 ml of methanol was stirred at ambient
temperature for 1 hour and poured into 500 ml of water. After
extraction with ethyl acetate, the aqueous layer was acidified
with 3N hydrochloric acid. The title compound precipitated and
was collected by suction filtration, washecl with water and
crystallized from a 7:3 mixture of methanol:chloroform. Yield
2.55 g, m.p. 272-277C.
8-ethoxvcarbonYl-3-methyl-4-oxo-2-(2 thienvl)-4H-l benzopyran
(Intermediate LXXXII~
This compound was prepared in two steps according to the methods
reported for Intermediate LXXX, but u-~ing Intermediate XCI
instead of Intermediate XC. The title compound melted at
116-118C after crystallization from isopropanol.
8-carboxY-3-m-ethyl-4-oxo-2-(2-thienyl~-4H-l-benzopyran
LIntermediate-LxxxIIIl
This compound was prepared according to the method described for
Intermediate LXXXI, but using Intermediate LXXXII instead of
Inkermediate LXXX. Melting point 287-294C after crystallization
from a 7:3 mixture of methanol and chloroform.
8-carboxY-4-oxo-2-phenYl-4H-l-benzothio~yran
~Intermediate LXXXIV)
A mixture of 1 g of 8-methoxycarbonyl-4-oxo-2-phenyl-4H-1-
-benzothiopyran (Intermediate XCII), 2.2 ml of 12.5 N sodium
hydroxide, 15 ml of methanol and 5 ml of dioxane was stirred at
ambient temperature for 2% hours. After evaporation in vacuo,
water was added until complete solution and this solution was
extracted with chloroform. Th~ separated aqueous phase was
acidified with dilute hydrochloric acid until complete
precipitation of the crucle, which was filtered off and purified
': ' '~'" '
-95-
by crystallization from acetic acidO Yield 0.62 g, m.p. 302C.
(E~-8-ethoxycarbonYl-3-methYl 4-oxo-2-(2 styrvl),-4H-1-benzo~vran
(Intermediate LXXXV!
This compound was prepared in three steps according to the
methods described for Intermediate XC (first step) and
Intermediate LXXX (second and third steps). In the first step,
(E)-cinnamoyl chloride was used instaad of 2-furoyl chloride and
the obtained (E)-ethyl 2-hydroxy-3-(2-styryl-propionyl)-
-benzoate was used without purification for the second step.
~he title compound melted at 129-130C after crystallization
from isopropanol.
-:
-8-carboxy-3-methyl-4-oxo-2-st~ryl-4H-1-benzopyran
(Intermedi,ate LXXXVI)
Thi~ compound was prepared according to the method described for
Intermediate LXXXI, but starting from Intermediate LXXXV instead
of Intermediate LXXX~ and maintaining the reaction at ambient
temperature for 10 hours. The title compound melted at 284-286C
after crystallization from ethanol.
8-carboxy-3-,methyl-2-(4-methylPhen~1)-4-oxo-4H-1-benzo~yran
(Intermediate LXXXVII)
A mixture of 1.9 g of 2-hydroxy-3-propionyl-benzoic acid
(prepared as described in Brit. l, 343, 119, 1974), 5.2 g of
anhydrous sodium 4-methylbenzoate and 3.9 ml of 4-methylbenzoyl
chloride wa~ stirred at 185-195C for 8~ hours. After cooling
to ambient temperature, the solidi~ied mass was stood overnight
with 100 ml of chloroform. The mixture was then shaken with 5~
aqueous sodium carbonate solution, added until the pH of the
aqueous phase reached 8.9. The organic phase was extracted
again with 3~ sodium carbonate and the aqueous phases were
pooled, repeatedly extracted with diethyl ether and acidi~ied
with lON hydrochloric acid. The precipitate was purified by
flash chromatography on silica gel eluting with
chloroform:methanol 100:2 to 100:20. The title compound,
obtained by evaporating in vacuo the pooled fractions containing
it, melted at 249-251~C, after crystallization from ethanol.
~ -,: : , : , , , . , . .~ ,.: , , - -, : , , .
~1~9~'3~
96
8-ethoxvcarbonYl-2-(4-fluoro~henyl)-3-meth~l 4-oxo-4H-1-
benzopyran LIntermediate LXXXVIII)
This compound was prepared in three steps according to the
methods described for Intermediate XC (first step) and
Intermediate LXXX (~econd and third steps). In the first step,
4-fluorobenzoyl chloride was used instead of 2-furoyl chloride
and the reaction lasted 20 hours at ambient temperature,
yielding ethyl 2-(4-fluorobenzoyloxy)-3-propionyl-benzoate~ This
compound was used without puri~ication for the second step. The
title compound melted at 128-130C after rinsing with diethyl
ether and crystallization from ethanol.
8-carboxy-2-(4-fluorophenyl)-3-methyl-4-oxo-4H-l benzopyran
(Intermediate LXXXIX~
A solution of 3.3 g of Intermediate LXXXVIII and 0.6 g of
lithium hydroxide hydrate in 50 ml of tetrahydrofuran, 10 ml of
methanol and 10 ml of water was maintained at ambient
temperature ~or 5 hour~ and then poured into 300 ml of lN
hydrochloric acid. The precipitate formed was collected by
suction, washed with water and dried, yielding 2.3 g of the
title compound, melting at 249-250C after crystallization from
95% ethanol.
ethyl 2-(2-furo~loxy)-3-~roE~ ~EUoate
~1
4.35 ml of 2-furoyl chloride were added dropwise at 10-15C
into a stirred mixture of 8.9 g of ethyl 2-hydroxy-3-propionyl-
-benzoate and 5.4 g of 4-dimethylaminopyridine in 25 ml of
dichloromethane. After 2 hours at ambient temperature, the
reaction was quenched with 200 ml of water. The organic layer
was washed with 5~ sodium bicarbonate, dried on anhydrous
sodium sulphate and evaporated in vacuo. The residue was
puri~ied by flash chromatography on silica gel eluting with
petroleum ether:ethyl acetate 4:1, obtaining 9.4 g of the title
compound as a low melting solid, used without further
purification in the next step.
NMR ~peotrum at 60 M~z (CDC13, ~)
8.2 (lH, dd, CH in position 4 of the benzene ring)
8.0 (lH, dd, CH in position 6 o~ the benzene ring)
5 :~
-97- : .
7.7-7.8 (lH, dd, CH in position 5 of the furan ring)
7.43 (lH, t, CH in position 5 o~ the benzen~ ring)
7.45 (lH~ s, CH in position 3 o~ the furan ring)
6.6-6.8 (lH, m, CH in position 4 of the furan ring)
4.3 (2H, q, COOCH2CH3)
2.9 (2H, q, COCH2CH
0.95-~.35 (6H, m, 2xCH3)
~yl 3-Propionyl-2-(2-thienylcarbonyloxy)-benzoate
~Intermediate XCI~ ~
This compound was prepaxed by the method described for ..
Intermediate XC, but using 2-thienylcarboxyli~ acid chloride ~. ;
instead of 2-furoyl chloride.
N~R 8p~ctr~m ~t 60 N~2 (CDC13, S~
7.1-B.35 (6H, m, aromatic CHs) :
4.25 (2H, q, COOOEI2CH3) .:
2.9 (2H, q, COCH2CH3)
0.95-1.3 (6~, m, 2xCH3) ~ ;
8-methoxycarbonyl- 4-oxo-2-phenyl-4H-l-benzothiopyran - .
(Intermediate XCII)
A mixture of 16.8 ml of methyl thiosalicylate, 25.6 ml of ethyl .:
benzoylacetate and 360 g of polyphosphoric acid was stirred at ;~
90C ~or 3 hours. After cooling to ambient temperature, the
mixture was poured into crushed ice and the crude was collected
by filtration, washed with water and purified by
crystallization from ethanol. M.p. 170-171C.
~lemontal ~n~1~8i~ ~or C~7~123~
Calculated (~) : C, 68.90; H, 4.08; S, 10.82.
Found (~) : C, 68~59; H, 4.13; S, 10.69.
N~ ~psatru~ at 200 N~z (CDC13, ~)
8.83-8.95 (dd, lH,benzothiopyran CH in 5)
8.45-8.53 (dd, lH,benzothiopyran CH in 7)
7.68-7.80 (m, 2H, 2-phenyl CHs in 2 and 6)
7.55-7.65 (t, lH, benzothiopyran CH in 63 .
7.45-7.55 (m, 3H, 2-phenyl CHs in 3,4 and 5)
7.24 (s, lH, benzothiopyran CH in 3)
4.00 (s, 3H, COOCH3)
~ ` ! ; , " '~' i ' " ' ' ' ' ;~ ' ' " ;
2 D ~ 6
,
-98-
8-ethoxycarbonyl-3-bromometh~1-4-oxo-2-phenyl-4H-1-benzopyran
(Intermediate XCIII)
A mixture of 9.2 g of 8-ethoxy~arbonyl 3-methyl-4-oxo-
-2-phenyl-4H-1-benzopyran (prepared as described by Da R~ P. e~
al ., J. Ned . Pharm. Chem., 2, 263, 1960), 6.4 g of
N-bromosuccinimide and 0.04 g of benzoylperoxide in 80 ml of
anydrous carbon tetrachloride was stirred at reflux for 1%
hours. After cooling to ambient temperature the formed
succinimide was collected by suction and washed with cold
carbon tetrachlorid~. The mother liquors were evaporated to
dryness in vacuo and the residue was rinsed with diethyl ether
and collected by suction filtration yielding 9.2 g of the title
compound, melting at 133-134C after crystallization from
ac~tone:n-hexane.
8-ethoxycarbonyl-3-acetoxymethyl-4-oxo-2-phenyl-4H-1-benzopyran
(Intermediate XCIV)
A solution of 10.2 g of sodium acetate trihydrate in 30 ml of
water w~ added dropwise at ambient temperature to a solution of
29 g of Intermediate XCIII in 300 ml of dimethylformamide.
After stirring at 50C for 1~ hours, the reaction mixture was
poured into 2 litres of water and the precipitated title
compound was collected by suction filtration and crystallized
from acetone yielding 20 g (two crops collected), m.p.
151-152C.
8-carboxy-3-hvdroxymethyl-4-oxo-2-phenyl-4H-l-benzo~yran
(Intermediate XCV)
116 ml of lN sodium hydroxide were added over 10 minutes to a
stirred suspension of 14.8 g of Intermediate XCIV in 300 ml of
95% ethanol. The reaction mixture was then heated at ~0-65~C for
15 minutes, obtaining a clear solution which was maintained at
ambient temperature for a further hour. After evaporation in
vacuo, the residue was dissolved in 200 ml of water and the
solution acidified by slow addition of lO ml of hydrochloric
acid (d31.18). After stirring for 1 hour at ambient temperature
the title compound was collected by suction filtration, washed
with water and crystallized from isopropanol, yielding 9.3 g,
m.p. (225) 237~240C.
:, .
. .
: ., . , . , . , .:: ,, .... ~ :, : , , , , ,.~ .. . . . . .
-~ 2~9~
99
~th~1_2-~4-nitroben~o~loxy)-3-propionyl-benzoate
(Intermediate XCVI~
The title compound was prepared according to the procedure
described for Intermediate XC~ but using 4-nitrobenzoyl chloride
instead o~ 2-furoyl chloride. The product was obtained as a low
melting solid (m.p. (40) 78-80C).
N~R ~poctrum ~t C0 M~s (CDC13,
7.85-8.50 (m, 6H, aromatic CHs)
7.50 (t, lH, CHs in position 5 of the phenol ring)
4.25 (q, 2H, CH2O)
3.95 (q, 2H, CH2)
0.95-1.30 (m, 6H, CH3)
-benzopyran (I~termedia~e XCVIIL
A mixture of 29.7 g o~ Intermediate XCVI and 10.18 g of
anhydrous potassium t-butoxide in 89 ml of anhydrous pyridine
was stirred at 100C for 13 hours. The reaction mixture was
cooled to ambient temperature, poured into 400 ml of 4N
hydrochloric acid and extracted with di~hloromethane. The
organic layer was washed repeatedly with water, then with 2,5%
sodium hydrogen carbonate, dried on anhydrous sodium sulphate
and evaporated to dryness in vacuo. The crude product was
purified by column chromatography on silica gel eluting with
hexane:ethyl acetate 7:3. Evaporation in vacuo of the collected
~raction yielded 7 g of the title compound, melt~ng at (130)
145-1~8C.
8 carbox~-3-~ethyl~ nitroPhenyl)-4-oxo-4H-1-benzopy~an
~Intermediate XCVIII)
A suspen~ion o~ 0.38 g of Intermediate XCVII in 4.75 ml of
dioxane and 4.75 ml of methanol was stirred at 50C~ 1O29 ml of
lN sodium hydroxide was added and stirring was continued for 3
hours at tha same temperature. The reaction mixture was cooled
to ambient temperature and 3N hydrochloric acid was added until
the pH was 1. The suspension was filtered by suction giving 0.13
g of the title compound, which melted at 320-321C after
crystallization from dioxane.
. .
'. : ; . ; ,':, ~
.
.. . . .
.
-loo- 2 ~
8-ethoxvcarbonvl-3-methvl-4-oxo-2-trifluoromethyl-4H~
-benzopyran (Intermediate XCIX)
3.16 ml o~ 1,8-diazabicyclo[5.4.0]undec-7-ene was added dropwise
ak 0C by a syringe to a stirred mixture of 3 g of ethyl
2-hydroxy 3~propionyl-benzoate and 5.53 ml of trifluoroacetic
anhydride. The reaction mixture was stirred at 60C for 4 hours;
after this period it was cooled to ambient temperature and
diluted with ethyl acetate and water. The organic layer was
washed with lN sodium hydroxide and water, dried on anhydrous
sodium sulphate and evaporated to dryness in vacuo. The residue
was purified by column chromatography on silica gel ~luting
with petroleum ether:ethyl acetate 95:5 yielding 0.8 g of the
title compound.
N~ i~peotrum 21t zoo~æ !CDCl3~ ~)
8.41;8.37 (2dd; 2H, CHs in position 5 and 7 of the benzopyran
ring)
7.51 (t, lH, CH in position 6 of the benzopyran ring)
4.46 (q, 2H, COOCH2)
2.22-2.27 (m, 3H, JH_F=2.16Hz, CH3 in position 3 of the
benzopyran ring)
1.39 (t, 3H, CH2CH3)
8-carboxy-3-methyl-4-oxo-2-trifluoromethyl-4H-1-benzoPyran
(Intermediate C~
The title compound was prepared by the same method as
Intermediate LXII but using Intermediate XCIX instead of
Intermediate LXI and, after dilution with water, extracting with
ethyl acetate instead of filtering. After drying on anhydrous
sodium sulphate and evaporating the organic layer to dryness in
vacuo, the title compound was obtained as a solid which melted
at 175-178C.
3- r 4-(2-methoxyDhenyl)-1-piperazinyl~~N~methyl-~ropYlamine
LIntermediate CI)
42 ml of 35% aqueous methylamine was added to a solution of 8.2
g of 3-[4-(2-methoxyphenyl)-1-piperazinyl]-propyl chloride in
48 ml of dimethylformamide. The mixture was heated at 60C in a
closed ves~el ~or 5 hours, and then cooled to 30C. The solvent
2 ~
--101-- -
was evaporated off in vacuo, and the residue was stirred for 30
minutes with 100 ml of diethyl ether. The solids, collected by
suction ~iltration, were dissolved in 200 ml of chloroform:5N
methanolic ammonia 100:3. After 30 minutes stirring at ambient
temperature, the solution was adsorbed on a chromatographic
column, which was eluted with chloroform:5N methanolic ammonia
graduated 100:5 to 100:15. The fractions containing the title
compound were pooled and evaporated in vacuo, yielding 3 g of
Intermediate CI as a thick oil.
~M~ ~p~ctru~ at ~ON~z IDN80-d6,
6.80 ~s, 4H, aromatic CHs~
3.75 (s, 3H, OCH3)
3.20-2.75 (m, 4H, piperazinic CH2s at pos. 3,5)
2.75-2.10 (m, 8H, piperazinic CH2s at pos. 2,6 and CH2CH2CH2)
2.40 (s, lH, NH)
2.30 (s, 3H, NCH3)
1.80-1.40 (m, 2H, CH2CH2CH2)
ethyl 2-benzoyl-3-ethylbenzorbLfuran-7-carboxylate
(Intermediate CII)
A mixture of 11.1 g of ethyl 2~hydroxy-3-propionyl-benzoate, 9.9
g of phenacyl bromide, 6.9 g of anhydrous potassium carbonate
and 200 ml of acetone was stirred at: refluxing temperature for
7 hours. After cooling to ambient temperature, the inorganic
salts were separated by filtration and the solution was
evaporated in vacuo. The residue wais purified by flash
chromatography on silica gel eluting with toluene. The title
compound, obtained by evaporating in vacuo the pooled fractions
containing it, was crystallized from 90% ethanol. Yield 9.8 g,
m.p. 64-66C.
7-carboxy-2-benzoyl-3-ethyl-benzorblfuran (Intermediate CIII)
A mixture of 7 g o~ Intermediate CII, 275 ml of 0.95N sodium
hydroxide and 400 ml of dioxane was stirred at refluxing
temperature for 4 hours. After cooling to ambient temperature,
the dioxane was evaporated in vacuo and replaced with the same
volume of water. APter ~iltering with charcoal, the solution
was acidified with dilute hydrochloric acid and the precipitate
was filtered and purified by crystallization from acetone.
,, ,, , ~
-102-
Yield 4.94 g, m.p. 193-195C.
8-methoxycarbonyl-3-methYl~2-l4-methYlPhenyl~-4-oxo-4H-1-
-benzopyran (Intermediate CIV)
This compound was prepared in three steps according to the
methods described for Intermediate XC (first step) and
Intermediate ~XXX (second and third step). In the first step,
4-methylbenzoyl chloride was used instead of 2-furoyl chloride
and methyl 2 hydroxy 3-propionyl-benzoate was used instead of
ethyl 2-hydroxy-3-propiionyl-benzoate. The reaction lasted 4
hours at ambient temperature, yielding methyl
2-(4-methylbenzoyloxy)-3-propionyl-benzoate. This compound was
used without purification for the s~cond step which lasted 1%
hours at 100C. In the third st~p 96~ sulphuric acid was used
instead of 37% hydrochloric acid. The title compound melted at
174-175C after crystallization from ethanol.
8-ethoxvcarbonYl-2-(4-bi~henvl~l ?- 3-methvl-4- xo-4H-1-benzoPvran
(Intermediate CV3
This compound was prepared in three steps according to the
methods described for Intermediate XC (first step) and
Intermediate CIV (second and third steps). In the first step
4-phenylbenzoy~ chloride was used instead of 2-furoyl chloride
and the reaction lasted 20 hours at room temperature and 13
hours at reflux. Purification was performed by column
chromatography on silica gel eluting with petroleum ether:ethyl
acetate graduated lOO:S to 100:10, yielding ethyl
2-(4-biphenylyl)3-propionyl-benzoate. The title compound melted
at 1~5-167C after rinsing with 95% ethanol.
8-carboxy-2-(4-biphenvlYl)~3-methyl-4-oxo-4H-l-benzopyran
(Intermediate CVI)
A mixture o~ 4.3 g 0~ Intermediate CV and 35 ml of 35~
hydrochloric acid in 50 ml of 1,4-dioxane and 15 ml of water was
stirred at reflux for 16 hours. After cooling, the mixture was
poured into 200 ml of water and extracted with ethyl acetate.
::,. ,. : .
2 t~
,; '
-103-
Th2 organic layer was separated and extracted with 20% aqueous
sodium carbonate. The precipitate formed after acidifying the
aqueous layer with dilute hydrochloric acid was collected by
suction, washed with water and dried, yielding 2.5 g of the
title compound~ melting at 242.5-244C.
,3-carboxy-2-(4-hyd~oxyphenyl)-3-methyl-4-oxo-4H-l-benzopyran
.
(Intermediate CVII !
A mixture of 3 g of 8-ethoxycarbonyl-2-(4-methoxyphenyl)-
-3-methyl-4-oxo-4H-1-benzopyran (prepared as described in JP
58-225083) and 60 ml of 48% hydrobromic acid in 80 ml of acetic
acid was stirred at re~lux for 8 hours. After cooling, the
mixture was poured into 500 ml of water and the precipitate was
collected by suction and washed with water. The crude product
was purified by ~lash chromatography eluting with chloroform~
isopropanol graduated 9:1 to 7:3 followed by methanol elution,
yielding 1 g the title compound, melting at 300C.
4- r 4_(2-methoxv~henyl)-1-piperazinyll-N-methyl-butylamine
(Intermediate CVIII)
A solution of 3.8 ml of trifluoroacetic anhydride in 25 ml o~
anhydrous dichloromethane was added Idropwise under stirring at
0C to a solution of 2.53 g of 4-[4-(2-methoxyphenyl)-
-l-piperazinyl]-butylamine in 25 ml of anhydrous
dichloromethana. After 2 hours stirring at room temperature, the
reaction mixture was diluted with dichloromethane and washed
with water. The organic layer was dried on anhydrous sodium
sulphate and evaporated to dryness in vacuo, yielding 3.3 g of
pure 4-[4-(2-methoxyphenyl)-1-piperazinyl]-N-trifluoroacetyl-
-butylamine
NMR ~peetru~ at 60 M~z ~CDC13, ~)
7.70-8.00 (bs, lH, NH)
6.80-7.20 (m, 4H, aromatic CHs)
3.85 (8, 3H, CH30)
2.90-3.80 (m, 12H, piperazine CH2s, CH2N and CH2NHC0)
1.50-2.05 (m, 4~, C-CH2CH2-~)
, . . .
-104
0.88 g Of 50% sodium hydride was added portionwise under
stirring at 0C to solution of 3.3 g of the ahove intermediate
in 46 ml of anhydrous dimethylformamide. After 1 hour stirring
at the same temperature, 0.57 ml of methyl iodide was added. The
reaction mixture was stirred at 0C for additional 1~ hours;
then it was poured into water and extracted with ethyl acetate.
The organic layer was washed with water, dried on anhydrous
sodium sulphate and evaporated to dryness in vacuo, yielding
1.13 g of crude 4-[4-(2-methoxyphenyl)-1-piperazinyl]-
-N-trifluoroacetyl-N-methyl-butylamine used in the following
step without further purification. 0.18 g of sodium borohydride
was added to a solution of 1.13 g of this intermediate in 30 ml
of ethanol and the resulting mixture was stirred at 60C for 1
hour. After cooling to room temperature, th~ reaction mixture
was poured into water, and extracted with dichloromethane. The
organic layer was washed with water, dried on anhydrous sodium
sulphate and evaporated to dryness in vacuo, yielding 0.82 g of
pure title compound.
~NR ~otr~m at 60 ~z (CDC13, C)
6.80-7.20 (m, 4H, aromatic CHs)
3.85 (~, 3H, CH30)
2.90-3.20 (m, 4H, piperazine CH2s, positions 3 and 5)
2.30-2.80 (m, 8H, piperazine CH2s, positions 2 and 6; 2 CH2N)
2.50 (s, 3H, CH~N)
1.80 (s, lH, NH)
1.40-1.80 (m, 4H, C-CH2-CH2-C)
1-(3-amino-2,2-dimeth~lProE~ 4-(2-methoxyphenyl)-piperazine
The title compound may be prepared by treating
1-(2-methoxyphenyl)-piperazine with isobutyraldehyde, 37~
formaldehyde in water and acetic acid at 90-150C or with the
same in ethanolic hydrogen chloride (Mannich reaction) to give
1-(2-formyl-2-methylpropyl)-4-(2-methoxyphenyl)-piperazine,
which is then aminated by treatment with excess ammonia under
reducing conditions. The latter may be hydrogen and a catalyst
(for example, palladium-on-charcoal, Raney nickel or platinum
dioxide) in a solvent (for example, ethanol, mekhanol,
isopropanol, dichloromethane, chloroform or dimethylformamide)
at between ambient temperature and 80C, or alternatively a
2 ~
-105-
metal hydride (for example, sodium borohydride, sodium or
potassium cyanoborohydride, or sodium triacetoxyborohydride) in
a solvent (for examplej methanol~ ethanol, chloroform, benzene
or 1,2-dichloroethane) in the presence of an acid (for example,
gaseous hydrogen chloride or acetic acid).
It may be reacted with 8-chlorocarbonyl-3-methyl-4-oxo-2-phenyl-
-4H-1-benzopyran in the manner described in Example 12
hereinbelow to give 8-(2,2-dimethyl-3 [4-(2-methoxyphenyl)-1-
-piperazinyl]-propylcarbamoyl)-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran.
8-trifluroacetamidomethvl-3-methyl-4-oxo-2-ehenYl-4H-l-
-benzopyran
This compound can be prepared according to the procedure
described for Intermediate XXIII but using Intermediate XXIV
instead of 8-amino-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran. It
can be used as starting material, instead of Intermediate
XXIII, in the reaction described in Example 32 to yield
8-{2-[4-(2-metho~yphenyl)-1-piperazinylJ-ethylaminomethyl}-3 -~
-methyl-4-oxo-2-phenyl-4H-l-benzopyran.
.' :'.
8-L~-chloroethvlureidomethYl)-3-methYl-4-oxo-2-ph~enyl-4H-1-
~b~ ,
This intermediate can be prepared by the method described for
the preparation of lntermediate XLIV, but using Intermediate
XXIV instead of 8-amino-3-methyl-4-oxo-2~phenyl-4H-1-
-benzopyran. It can be reacted with a compound o~ formula H-s,
according to Path (a) to give the desired final compounds.
8-ethenvlsul~honY-laminomethyl-3-methyl-4-oxo-2-phenvl-4-H
-benzopyran
Thi~ compound can be prepared by reacting Intermediate XXIV
with 2-chloroethylsulphonyl chloride in a halogenated solvent
such as dichloromethane in the presence of triethylamine at
0-40C, according to A.A. Goldberg, J. Chem. Soc., 464, 1945.
It can be reacted with the appropriate compounds H-~, according
to Path (m) to yield the corresponding final compounds.
Operating as described above, but starting from
8-amino-3-methyl-4 oxo-2-phenyl-4H-1-benzopyran the final
2~3913
-106-
compounds Fl'-Y36-(CH2)2-B can be obtained.
8-chlorosulphonylmethyl-3-methvl-4-oxo-2-~henyl-4H-l-benzopyran
This intermediate can be prepared by reacting
8-amidinothiomethyl-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran
~whose synthesis is described in Intermediate XXI~ with
chlorine gas in water at -10 to +10C according to T.B. Johnson
et al., J. Chem. Soc., 61, 2548, 1939. By reaction of this
inter~ediate with the appropriate compounds A-NH-Z-B according
to Path n, the desired final compounds can be obtained.
--107
ESAllPli}ll;/
pl~ ~
8-{2-t4-(2-metho~yp~e~yl~ l-piperazinyl]-1-o~oethyl}-3- :
-methyl~ oxo-2 phenyl-4~-1-b~opyx~ hyaro~hlori~e ~ :
solution of 11.5 g of 1-(2-methoxyphenyl)-piperazine in 30 ml
of methanol was added dropwise at 20-25C to a stirred mixture :: :
consisting of 21.4 g of lnterm diate VI and 4.1 g of potassium :-
carbonate in 120 ml of methanol. After 4 hours stirring at the
same temperature, the reaction mixture was stripped in vacuo.
The residue was extracted with chloroform and the organic ~::
solution was washed with water, dried on anhydrous sodium
sulphate/calcium chloride, filtered and stripped in vacuo. The
obtained crude product was dissolved in acetone and a slight
excess o~ ethanolic hydrogen chloride was added. After
collection by suction filtration and recrystallization from 95%
ethanol, 16.3 g of the title compound was obtained, m.p. (189)
195-199C.
' :
~xa~pl~ 2
8r{2-~4~ methylph~yl~-1 pip~razinyl3-1-oxo~thyl~-3-mQthyl-
-4-oxo-2-phe~yl-4~ be~zopyra~ hydroohloride
This compound was prepared according to Example 1, but using
1 (2-methylphenyl)-piperazine instead of 1-(2-methoxyphenyl)-
-piperazine and carrying out the reaction in dimethyl~ormamide
for 1 hour instead of in methanol for 4 hours.
M.p. (194) 203-206C (isopropanol).
~x~m~le .
8 {2-~4-(2-etho~y~e~yl)-1-piper~zinyl]-~-oxoethyl}-3-methyl-
-~-oxo-2-phe~yl~ be~zopyran hydro~hlorido
' ' `
This compound was prepared according to Example 1 but using
1-(2-ethoxyphenyl)-piperazine instead of 1-(2-methoxyphenyl)-
-piperazine and carrying out the reaction in dimethylformamide
~or 2 hours instead of in methanol for 4 hours.
M.p. 208-210C (isopropanol).
-108- ~ ~ 9~ 5
~ le 4
8-{3-[4-l2~etho~h0~ piperazi~yl~ o~opropyl} 3-
-~thyl-~o~o 2-phe~yl~4~-1 bonzopyr~n ~ydroohlori~e
A solution of 10 ml of 37% ~ormaldehyde in 15 ml o~ methanol was
dropped, over a period of 3 minutes at 0C, into a solution of
~.75 g o~ 1-(2-methoxyphenyl)-piperazine in 10 ml of methanol.
After 12 hours at 0C, the mixture was stripped in vacuo and
redissolved in 15 ml of methanol. 20 ml of 3.6N hydrogen
chloride in diethyl ether was added at 0C. After stripping in
vacuo, the residue was suspended in 15 ml of 1,4-dioxane. A
solution of 8.3 g of Intermediate V in 100 ml of 1,4-dioxane was
added under stirring at 20-25C. After stirring for 8 hours at
reflux the reaction mixture was cooled to 30-40C. 50 ml of
methanol was added and the mixture was refluxed for a further 2
hours. After cooling to 20-25C, the resultan-t solution was
diluted with 300 ml of diethyl ether. Stirring was continued
for a further 3 hours at the same temperature. The title
compound was collected by suction filtration and recrystallized
from ethanol. Yield 4 g, m.p. 209-210C.
E~a~l~ 5
~-{3-[~-~2-metho~yphe~yl) 1-pip~razinyl]-~ropoxy~srbo~yl~-
3-methyl ~-o~o-2-phenyl 4~-1-benzopyr~ dihydroohloride
A mixture of 4.24 g of 8-carboxy-3-methyl-4-oxo-2-phenyl-
-4H-1-benzopyran and 6.3 g of anhydrous potassium carbonate in
60 ml of dimethylformamide was stirred at 80C for 30 minutes.
5.23 g of 1-(3-chloropropyl)-4-(2-methoxyphenyl)-piperazine was
then added and stirring was continued at 8QC for 3~ hours. The
reaction mixture was cooled to ambient temperature, poured on to
iced water and extracted with ethyl acetate. The organic
extracts were washed with aqueous sodium chloride solution,
dried on anhydrous sodium sulphate, and evaporated to dryness in
vacuo. The residue was taken up in ethanol and excess ethanolic
hydrog~n chloride was added to the solutionO Yield: 8.16 g of
the title compound, m.p. 198 203C.
. ,~
--109-- ' '
Exa~Pl~ 6
8 ~2-t4-~2-~ethoxyp~e~yl)~1-pip~ra~i~yl] 0thoxyoarbo~yl}-
-3-methyl-4-o~o-2-ph~yl~ 0~30~yr~ hy~roohlori~e
Operating as described in Example 5, but using -
1-(2-chloroethyl)-4-(2-methoxyphenyl)-piperazine instead of
1-(3-chloropropyl)~4-(2-methoxyphenyl~-piperazine, the title
compound was o~tainedl m.p. 200-203C from ethanol.
Bx~m~le 7
8-{3-t4-(2-~hlorophe~yl)-1-pip~ra~inyl]-propo~yo~rbo~yl}-
3-~ethyl 4-oxo-2-phonyl~ banzopyran dihydro~hlorid~ -
A mixture o~ 2.8 g of 1-(2-chlorophenyl~-piperazine
hydrochloride and 4.2 g of anhydrous potassium carbonate in 25
ml of dimethylformamide was stirred at ambient temperature for
15 minutes. 4.81 g of Intermediate I was added, and stirring
was continued for 2 days. The reaction mixture was then poured
into 200 ml of cold water, and extracted with diethyl ether and
ethyl acetate. The organic extracts were wa~hed in turn with
aqueous sodium chloride solution, O.lN acetic acid, aqueous
sodium chloride solution, aqueous 4% sodium carbonate fiolution
and water, and were then dried on anhydrous sodium sulphate.
After evaporation to dryness in vacuo, the residue was dissolved
in 160 ml of acetonitrile and exc:ess hydrogen chloride in
diekhyl ether was added. ~hs insoluble title compound was
recrystallized from acetonitrile. Yield 3.6 g, m.p. 138-143C.
Ex~m~lo
8-t3~ pho~yl-1-piperazi~yl~-propoxycarbo:llyl~-3-m~thyl-~-oxo-
-2-phe~yl-4~ 1-ben~opyra~ ~ihy~rochloride
The title compound was prepared by the method described in
Example 7, but using 1-phenyl-piperazine in place of
1- (2-chlorophenyl) -piperazine hydrochloride. Recrystallization
was from methanol; the melting point was 229-231C. ::
- , . , . . , . ........... , ~ , ..... . . .. . . . . .
... . , . .. ... ~ . .. . .. . .
-110~
~a~le 9
8-{3-~4-~2-~ethcxyp~e~yl) l;pip~razi~yl~-propo~y¢arbonyl}-
-3-~et~yl-4-oxo-2-ph~yl-4~ be~opyran ~ihydro~hlori~e
Operating as described in Example 7, but using
1-(2-methoxyphenyl)-piperazine hydrochloride instead of
1-(2-chlorophenyl)-piperazine hydrochloride, the kitla compound
was obtained. This represents an alternative route to the
product of Example 5.
~ ~ .
8-~3-t~-l2-~tho~yphonyl)-l-piper~zi~yl]-2-~thyl-2-
-propoxyoarbo~yl3-3-~e hyl-4-oxo-2-p~e~yl-~ be~zopyra~
~ihydroe~lorid~
5.29 g of Intermedi~te XXVIII in 25 ml of 1,2-dichloroethane was
added dropwise at 60C to a solution of 6 g of
8-chlorocarbonyl~3-methyl-4-oxo-2-phenyl-4H-1-benzopyran in 22
ml of 1,2 dichloroethane. The reaction mixture was refluxed for
16 hours, and then cooled to ambient temperature and pollred into ~-
cold 0.5N sodium hydroxide solution. Water and dichloromethane
were added. The organic phase was separatsd off, washed with
aqueous sodium chloride solution and dried on anhydrous sodium
sulphate. The solvents were evaporated o~f and the oily residue
was purified by flash chromatography on silica gel, eluting with
petroleum ether:ethyl acetate 85:15. The collected fractions
were evaporated to dryness in vacuo and the residue was
dissolved in ethanol. Excess ethanolic hydrogen chloride was
added to give 6.71 g of the title compound, m.p. 203-204C.
' '.'
Bx2m~1~ 11
8r~3-t~-~2-~et~oxyphenyl)-1-piperazi~yl~-propylcarb~moyl}-
-3-~eth~1-4-oxo-2-phe~yl ~ be~opyr~n ~ihy~rochloride ~ ~-
hem~hyd~ata -
.: .
A mixture o~ 6.28 g of 1-(2-methoxyphenyl)-piperazine and 5.34 g
of Intermediate XXXVII was heated at 180C for 5 hours. After
cooling, the dark mass was purified by flash chromatography on
silica gel, eluting with dichloromethane:methanol 100:3. The
fractions containing the title compound were pooled. The
solvents were removed in vacuo and the residue was dissolved in
~oiling ethanol. The solution was ~iltered, acidified with
ethanolic hydroyen chloride, and stood overnight at 20 25C.
The crude product was collected by ~iltration and crystallized
from ethanol to give 5 g o~ the title compound, m~p. (177)
182-186C.
~ ,
{3~ 2~ethoxypheny~ pipera~inyl~-propyla~ba~oyl}
3-~ethyl-~-o~o-2-phe~yl-4~ banzopyran aihy~roohlorid~
hemihydr~te
solution of 4.48 g of 8-chlorocarbonyl-3-methyl-4-oxo-
-2-phenyl-4H-1-benzopyran in 40 ml of chloroform was added
dropwise ov~r a period of 10 minutes at ambient temperature to a
solution o~ 3.74 g of 3-[4-(2-methoxyphenyl)-1-piperazinyl]-
-propylamine, prepared as described in GB 2161807, and 1.97 g of
triethylamine in 50 ml of chloro~orm. After stirring for 2
hours, the solution was washed first with O.SN hydrochloric
acid, secondly with a saturated aqueous sodium bicarbonate
solution and finally with water. The chloroform solution was
dried on anhydrous sodium ~ulphate and the solvent was
evaporated off in vacuo. The residue was worked up as described -
in Example 11 to give 6~67 g of the title compound, m.p. (177)
182-186C. This represents an alterhative route to the product
of Example 11.
The following salts were al~o prepared:
monohydrochloride hydrate, m.p. 151-154C,
monomethanesulphonate, m.p. 162-164C, and
~ hemimalate hydrate, m.p. 110-112C.
This example has described the condensation of the amine,
3-[4-(2-methoxyphenyl)-1-piperazinyl]-propylamine, with the
carbonyl chloride, 3-methyl-4-oxo-2-phenyl-4H-l~benzopyran-8-
-carbonyl chloride. It should be noted that the amine can be
condensed with the corresponding ~ree acid or the corresponding
ethyl ester by heating equimolar amounts thereo~ with or without
:' ' ,' ' ' : ' ' "' ' ' ............ , : ;' ~ ,
~, , .
-112-
a solvent. If a solvent is used, a high boiling point
hydrophilic or hydrophobic solvent is appropriate. The amine
can also b~ condens d at ambient temperature with an equimolar
amount of the corresponding free acid in the presence o~
N,N'-dicyclohexylcarbodiimide and 4-dimethylaminopyridine in a
solvent such as dichloromethane, chloroform, tetrahydrofuran or
dimethylformamide.
~x~mPle 13
8-~2-[~-~2~methogy~hs~yl)-1-pi~erazinyl]-othylo~rb~moyl} 3-
-~thyl-4-oxo-2-phe~yl 4~ l-be~zopyra~ ~onohydroohloride
h~ihydrat~
The title compound was prepared by the method described in
Example 16, but using Intermediate XIV instead of Intermediate
XV and heating at 55-60C for 32 hours. Also, work up was varied
as follows. After collecting the base by filtration,
purification was by flash chromatography on silica gel, eluting
with chloroform:methanol 100:0.5 and then 100:1. The fractions
containing the title compound were pooled and the solvents were
removed in vacuo. The residue was crystallized from ethanol.
After filtration, the solids were taken up in boiling water and
suf~icient dilute hydrochloric acid was added to effect
solution. The crystalline salt separated on cooling and was
collected by suction filtration. M.p. 119-123C.
P~
~-{3-[2-(2-metho~yphe~o~)-ethylami~o~-propylcarb~moyl}-3-
~methyl-4-oxo-2-ph~yl-4~ 1-be~20pyr~ hy~ro~hlori~e
.
Operating as described in Example 11, but using ~ -
2-(2-methoxyphenoxy)-ethylamine (prepared according to Augstein,
J. et al, J. Med. Chem., 8, 356, 1965) instead of
1-(2-methoxyphenyl)-piperazine, heating for 2 hours instead of 5
hours, and using dichloromethane:methanol 100:5 as eluant, the
title compound was obtained. M.p. 200-Z02C (eth~nol).
; . ~ . , . : . ., . , . . : , . . , ,~: . , . . . : . . .. . : ,
, . . " .
- 2 ~
-113-
B~ample 15
8-[3~ ph~yl-1-piperazinyl) ~ropylcarb~oyl]-3-~ethyl 4-o~0-2-
-ph~1-4~-1 b~n~opy~n ~o~o~y~ro~hlori~s h~ihyd~te
operating as described in Example 11, but using
1-phenylpiperazine instead of 1-(2-methoxyphenyl)-piperazine,
heating for 2 hours instead o~ 5 hours, and using
dichloromethane methanol 100:4 as eluant, the title compound was
obtained. ~.p. (251) 255-258C with decomposition (87%
ethanol).
~ampl~ 16
~-t~-~et~Yl-2-t4-(2-methoxypho~y~ piporazi~yl]-
-ethylcarba~oyl}-3-methyl-4-o~o-2-ph~yl-4~-1be~zopyr~n
~o~ohydrochloride
A mixture of 3.56 g of Intermediate XV, 2.35 g of
1~(2-methoxyphenyl)-piperazine, 2.76 g of anhydrous potassium
carbonate and 1~66 g o~ potassium iodide in 25 ml of
dimethylformamide was stirred at 100C for 6 hours. After
cooling, the solvent was removed in vacuo and the residue was
taken up in 50 ml o~ water, stirred for 1 hour at ambient
tempsrature, collected by ~iltration, washed with water and
crystallized from 95% ethanol in the presence of a small amount
of activated charcoal ~for decolo~xing). The base was
dissolved in 105 ml of boiling 0.086 N hydrochloric acid. After
cooling, the crystallized salt was collected by filtration,
giving 4.3 g of the title compound (m.p. 201-203C).
Example 17
8-{1-~ydroxy-2-t4-~2-methoxyphenyl)-1~pipernzin~ ethyl}-3-
-methyl-~-oxo-2-phenyl-4~ benzopyran hydro~hlori~e
: : .
1.36 g of sodium borohydride was added portionwise at 0 to 5C
to a solution of 15.5 g of the compound prepared in Example 1 in
1500 ml o~ methanol. A~ter stirring for 90 minutes at 0 to 5C,
3~ hydrochloric acid was added in order slightly to acidify the
reaction mixture, which was then stripped in vacuo. The residue
was shaken with 2N aqueous sodium hydroxide solution and
,
':' . ~ :" :
"~,............ .
:2 ~
-114-
extracted with chloroform. The organic layer was dried on
anhydrous sodium sulphate/calcium chloride, filtered, acidified
with ethanolic hydrogen chloride and stripped in vacuo. After
washing with diethyl ether, the crude product was crystallized
from ethanol to give 9.5 g of title compound, m.p. 248-249C.
~a~l~ B
8-{1-hy~ro~ry-2-t4-~2-~sthylph~nyl~ piperazinyl]-e~hyl~-3
-~thyl-~-oxo 2-phe~yl~4~ be~zopyra~ hyarochloride ;~
This compound was prepared according to Example 17, but starting
from the compound prepared in Example 2 rather than that
prepared in Example 1~ M.p. 257-258C (ethanol). '~:
EXEIDIP~0 lg ' . .
8-{1-~ydro~y-2-[4-~2-etho~yphe~yl~-1 piperazi~yl]-ethyl}-3-
-methyl-4-o~o-2-pha~yl-4H-l-be~zopyra~ hydrochloride
This compound was prepared according to Example 17, but starting
from the compound prepared in Example 3 rather than that
prepared in Example 1. M.p. 241-242C (methanol). : :
. '
~a~le ~0 .
8 {1-~ydroxy-3-t~-~2-~ethoYyphe~yl)-l~-piperaæinyl] pro~yl~3-
-~ethyl-~-o~o-2-phe~yl-4~1-benzopyra:~ hydro~hlori~e
This compound was prepared according to Example 17, but starting
~rom the compound prepared in Example 4 rather than that
prepared in Example 1. The crude base was purified by flash :chromatography (silica gel, eluant - ethyl acetate:chloroform
4:1). The fractions c~ntaining the pure base were pooled,
acidified with ethanolic hydrogen chloride and stripped in
vacuo. The residue was crystallized from ethanol. M.p. (126)
156-160C.
, . ' , . ,, ~ , ' ,, , , ,, , . ,, 1" , " "" ," ., ,",, ., , , . ~ ," ' " , ~ ,
-115-
~ampl~ 21
~ {~:by~ro~ 4-l4-~2-m~tl~o~phe~y~ -piE~razinyl]-bu~yl}-3
-~ethyl~oxo-2-phenyl~ b~nzopyran hy~r~ablorid~ monohydrate
A solution of 3.04 g of Intermediate XXXVIII and 2.45 g of
1-(2-methoxyphenyl)-piperaæine in 21 ml of anhydrous
dimethylformamide was stirred for 5 hours at ambient
temperature. A further 1.22 g of 1-(2-methoxyphenyl)-piperazine
was added, and the mixture was stirred for 4 hours, poured into
300 ml of water, and extracted with ethyl acetate. The combined
organic extracts were washed with aqueous sodium bicarbonate
solution and then with aqueous sodium chloride solution, and
evaporated to dryness in vacuo. The residue was purified by
flash chromatography on silica gel, eluting with ethyl
acetate:methanol 95:5. The collected fractions were stripped on
a rotary evaporator, and the residue wa~ dissolved in 0.81M
ethanolic hydrogen chloride and stripped again in vacuo. The
solid residue wa~ crystallized from water:ethanol 9:1 to give
2.43 g of the title compound, m.p. 144-146C.
E~a~lo 22
~-{~-ethoxy 2-~4-~2-methoxyphenyl)-1-pip~r~zinyl] ethyl}-3
-~ethyl-~ oxo-2-phonyl-4~ benzopyran hyaroahloride
6 ml o~ anhydrous dimethylsulphoxide was added to 6.55 g of
sodium hydride (50% in mineral oil, repeatedly washed with
hexane) under nitrogen. A solution of 3 g of the compound
prepared in Example 17 in 50 ml of anhydrous dimethylsulphoxide
was added to the mixture at 20-25C. After stirring for 1 hour
at 20C, 0.66 g of ethyl bromide was added. The reaction
mixture was stirred for an additional 20 minutes at the same
temperature and then poured into iced water. The crude product
obtained after suction filtration was purified by flash
chromatography (silica g~l, eluant - chloroform:ethyl acetate
8:2). The fractions containing the pure title compound were
pooled, acidified with ethanolic hydrogen chloride, and stripped
in vacuo. The residue was crystallized from chloroform:diethyl
ether and dried in vacuo at 140C to give 1.6 g of the title
compound, m.p. (155) 209C.
. , , , ~ :
,
- ~ .
. .
.~ :
-116- -.
~a~pl~ 23
~ethyl-2~t~-(2-m~thoxyph~yl)-l-pip~razi~yl~
-~thyl~mi~omethyl}-3~ethyl-~-o~o-2-ph~yl-4~1 benzopyra~
~ihy~ochlorid~ h~mihydxat~
A mixture of 5.2 g of Intermediate XXVII, 3.1 g of :
1-(2-methoxyphenyl)-piperazine and 2.2 g of anhydrous potassium
carbonate in 50 ml of dimethylformamide was stirred at 70C for
7 hours. After cooling to 20-25C, the reaction mixture was .
poured into 500 ml of water, and extracted with dichloromethane.
The organic phase was washed with water and dried on anhydrous .:::
sodium sulphate. The solvent was removed in vacuo. The residue : .
was purified by flash chromatography on silica gel, eluting with :
ethyl acetate:petroleum ether 98:2. The title compound was
obtained by salification with ethanolic hydrogen chloride. M.p. :~
217-219C. -:-:
B~2mple 24
8-{~-~cetyl 2-t4-(2-~tho~yph~nyl)~1-pip~r~zi~yl]-
-~thylamino~othyl}-3-methyl 4-0~0-2-phenyl-~H-l-ben20pyran
hyarochloride -.
A mixture of 5 g of Intermediate XXXIII and 5.3 g of
1-(2-methoxyphenyl)-piperazine in 75 ml of dimethylformamide was
~tirred at 95C for 2 hours. Aftex cooling to 20-25C, the
reaction mixture was poured into 200 ml of water, made alkaline
with potassium carbonate and extracted with ethyl acetate. The
organic phase was washed with water and dried on anhydrous :
sodium sulphate. ~he solvent was removed in vacuo. The residue
was purified by flash chromatography on silica gel, eluting with
dichloromethane:methanol 100:0.2. Saliication of the pure base
with ethanolic hydrogen chloride and recrystallization from
methanol gave 4.4 g of the title compound, melting at (200)
227-228C and containing one equivalent of methanol.
i''~'~` :' '' '
" ' ' . " ' ' ' " ' " ~ . ' ' ' " " ,' . . ' ", ; ' . ', , ' , . '
2 ~ -$~
~117-
~a~pl~ 25
8-[4-(2-~tho~yp~yl~ piper~zinyl~oeta~ido~ethyl~-3-~athyl-4-
-o~o~2-phonyl-4~ b~opyra~ hy~ro~hlorid0
mixture of 3.42 g of Intermediate XXXII, 2.74 g of
1-(2-methoxyphenyl)-piperazine and 0.71 g of anhydrous potassium
carbonate in 34 ml o~ anhydrous dimethylformamide was stirred at
0C for 2 hours. The reaction mixture was poured into water and
filtered under suction. The resultant solid was puri~i~d by
flash chromatography on silica gel, eluting with ethyl
acetate:petroleum ether 6:4. The collected fractions were
evapsrated to dryness in vacuo, and the residue was crystallized
from ethyl methyl ketone. The base obtained was treated in
ethanolic solution with a molar eguivalent of aqueous 2.25N
hydrochloric acid to give the title compound, m.p. 168-170C.
=1~
8~{X~ th~l-N-[~-~2-m~thoxyph~yl~ iperazinyl~
-aoHt3midomothyl~~3-~thyl-4-0~0~2-phl3nyl-4~ ben~opyran -~-
hy~ro~hlorid~ hydrate
~ . .
A mixture o~ 5 g of Intermediate XXXI, 2.9 g of
1-(2-methoxyphenyl)-piperazine and 2 g of anhydrous potassium
carbonate in 50 ml of dimethylformamide was stirred at 20-25C
for 3 hours. The reaction mixture was then poured into 500 ml of
wa~er and extracted with dichloromethane. The organic phase was
washed with water and dried on anhydrous sodium sulphate. The
solvent wa~ removed in vacuo. The residue was purified by flash
chromatography on silica gel, eluting with ethyl
acetate:petroleum ether 6:4, and crystallized from acetone to
give 3.6 g of the base of the title compound, melting at
144-145C. The base was dissolved in ethanol and 8N hydrochloric
acid and water were added, yielding the title compound, m.p.
218-220C, after drying at 100C in vacuo.
. ~ ~ - , : . ; , " ,~ ",
,
.:, ', ~ ', .
, , '
i.
B~amplo 27 - -
8 {2-t4-(2~ t~o~yphenyl)-l-pip~r~ziIlyl]-~tho~ymothyl}~3-
-mothyl~-oxo-2-phe~yl~4~ benzopyr~n dihy~roG~lori~e
A mixture of 4 g of Intermediate XVIII, 2.4 g of
1-(2-methoxyphenyl)-piperaziner 1.96 g of potassium iodide and
1.65 g of anhydrous potassium carbonate in 40 ml of anhydrous
dimethylformamide was stirred at 90C ~or 7 hours. After cooling
to ambient temperature, the mixture was poured into water and
extracted with dichloromethane. The combined extracts were
washed with aqueous sodium chloride solution, dried on anhydrous
sodium sulphate and evaporated to dryness in vacuo. The residue
was crystallized from ethyl acetate and the collected crystals
were dissolved in ethanol and treated with excess ethanolic
hydrogen chloride to give 5.21 g of the title compound, m.p.
199-201C.
~am~l~ 28 `
8 ~2-~2-~2-etho~y~honoxy)-ethylami~o]-~thoxy~thyl}-3-~et~yl-4-
-o~o-2 phenyl~ b0~zopyran hyaroohlori~ ;
Operating as described in Example 27, but using
2-(2-ethoxyphenoxy)-ethylamine in place of 1-(2-methoxyphenyl)-
-piperazine and including a pur:ification step of flash
chromatography on silica gel eluted with ethyl acetate:methanol
97:3, 4.25 g of the title compound was obtained. M.p~ 191-193C. ~-
~a~ple 29
8-~2-t4-~2-methoxyph~ y~ pip~axazi~ly~ thyllthioDl~thyl}-3
-methyl-4-o~ 2-phe~yl~ bQn~opyra~ hy~rochloriae
2.5 g of potassium carbonate, 2.13 g of potassium iodide and
3.15 g of 1-(2-methoxyphenyl)-piperazine were added to a
solution of 5 g of Intermediate XXI in 50 ml of dimethyl-
formamide, and the mixture was stirred at 90C for 4~ hours.
After cooling to ambient temperature, the reaction mixture was
poured into 450 ml o~ water and extracted with ethyl acetate.
The organic extracts were washed with water, dried on anhydrous
sodium sulphate and evaporated to dryness in vacuo. A solution
--119--
of the residue in acetane was treated with a molar equivalent of
3.8N hydrogen chloride in diethyl ether, filtered and
recrystallized ~rom ethanol to yield 6,15 g of the title
compound, m.p. (218) 223-224C.
~Q
8-{2-t4-~2-~ethoxyph~yl)-1-pi~e~azinyl3-ethyl~ulphi~yl~ethyl}-
-3-methyl-4-oxo-2-p~e~1-4~-1-benzopyra~ ~ihydroc~lori~e
h~mihyarat~
The title compound was prepared by the method described in
Example 29, using Intermediate XXVI instead of Intermediate XXI
and stirring for 2~ hours rather than for 4~ hours. After the
usual work up, the residue was purified by flash chromatography
on silica gel, eluting with ethyl acetate:methanol 97:3. The
collected fractions were acidified with excess ethanolic
hydrogen chloride, evaporated to dryness in vacuo. The residue
wa~- crystallized from ethanol, giving 5.2 g of the title
compound, m.p. 170-172C. This compound contains 1 equivalent o~
ethanol.
~-e~
8-{2-[~-~2-~thoxyphenyl)-1-pîperazi~l]-ethylsulpho~yl~ethyl}-
3 ~ethyl-~-oxo 2-pho~yl-4~ benzopyrau hydroshlori~e
A mixture of 4.5 g of Intermediate XXV, 2.36 g of
1-(2-methoxyphenyl)-piperazine and 0.84 g of potas~ium carbonate
in 45 ml of anhydrous dimethylformamide was stirred at ambient
temperature for 2~ hours. The reaction mixture was poured into
300 ml of water and filtered under suction, washing with water.
The solid base was crystallized from ethanol and had a melting
point of 143-146C. The crystallate was dissolved in
1,2-dichloroethane and acidified with ethanolic hydrogen
chloride. 4.4 g of the title compound, m.p. 229-233C, was
obtained by recrystallization from methanol:water 1:3.5.
,. , -., ., , ~,j . .
: '-., ~, ' ' ' ' ~
J
': '' ' :
: . ' ' -
': ' ' " ' .:
-120
~a~pl~ 32
~-~2-r4 (2-~etho~yph~yl)~ iperazi~yl] ~thylamino}-3-methyl-
-4-o~o-2-phe~yl-~ ben~opyr ~ ~ihy~xo~hlori~e
A solution of 3.7 g of Intermediate XXIII in 10 ml of
dimethylformamide was added dropwise at 0C to a suspension of
0.9 g of sodium hydride (50% in mineral oil) in 9 ml o~
dimethylformamide. The cooling bath was removed, and after 30
minutes at 20-25C a solution of 4.1 g of 1-(2-chloroethyl)-4-
-(2-methoxyphenyl)-piperazine in 10 ml of dimethylformamide was
added. The mixture was stirred at 90 for S hours and then
coolPd to 20-2SC. A further addition of 0.25 g of sodium
hydride (50% in mineral oil) followed by 1.36 g of
1-(2~chloroethyl)-4-(2-methoxyphenyl)-piperaæine in 5 ml of
dimethylformamide was made. The mixture was stirred at 90C ~or
8 hours and ag~in cooled to 20-25C. 200 ml o~ water was
cautiously added, and the mixture was extracted with ethyl
acetate. The organic layer was washed with water and dried on
anhydrous sodium sulphate. The solvent was evaporated off in
vacuo and the residue was purified by flash chromatography on
silica gel, eluting with n-hexane:ethyl acetate 3:2. This gave a
mixture of the base of the title compound and the corresponding
N-trifluoroacetyl compound.
3.8 g o this mixture was dissolved in 35 ml of ethanol and 35
ml of dimethylsulphoxide. ~o this solution, 0.55 g o~ sodium
borohydxide was added portionwise at 20-25C. The mixture was
stirred for 3 hours at this temperature, and then poured into
200 ml of water and extracted with ethyl acetate. ~he organic
extracts were washed with water, dried on anhydrous sodium
sulphate and evaporated to dryness in vacuo. The residue was
dissolved in dichloromethane. 2 equivalents of ethanolic
hydrogen chloride were added to give the title compound, which
was recrystallized from ethanol. Yield 3.8 g, m.p. 231-234C.
,. , . . .,, ,, "
.. . . . . . .. .
2 ~
-121-
~x~el~ 33
8-~3-[4~2 ~ethoxyphe~yl) l piperazi~yl~ propylami~o~-3-~ethyl-
-4-o~o 2 phe~yl ~ b~o~yra~ hydroehlori~ 2.75 hydr~te
Using 1-(3-chloropropyl)-4-(2-methoxyphenyl)-piperazine instead
of 1-(2-chloroethyl)-4-(2-methoxyphenyl~-piperazine, but
otherwise operating as described in Example 32, the title
compound was obtained. M.p. 206-208C (10% ethanol).
~ample 34
8-{~-[4-~2-m0tho~yphenyl)~ ipera2inyl~-butyl~mi~o}-3-met~yl-
-4-o~o-2-phenyl~ 1-be~zopyxan hydro~hloride hemihydrate
A mixture of 4.5 g of Intermediate XXXIX, 3.9 g sf
8-amino-3-methyl-4-oxo-2-phenyl-4H-1-benzopyran, 8.3 g of sodium
triacetoxyborohydride and 3.4 ml of acetic acid in 40 ml of
1,2-dichloroethane was stirred ~or 6 hours at 20-25C. 15 ml of
5% aqueous sodium bicarbonate solution was then added, and the
mixture was stirred for 10 minutes. The mixture was then made
alkaline by addition of 0.5N sodium hydroxide solution and
extracted with dichlormethane. The organic extracts were washed
with water and dried on anhydrous sodium sulphate. Th solvent
was evaporated off in vacuo and the residue was purified by
flash chromatography on silica gel, eluting with ethyl
ace~ate:p~troleum ether 9:1. The base obtained was dissolved in
dichloromethane and 1 equivalent of ethanolic hydrogen chloride
was added. After removing the solvent in vacuo, the residue was
crystallized from 50% ethanol to give 1.6 g of the title
compound, m.p. (140) 151-153C.
Bxa~lo 35
8-{~-m~thyl-3~[4-(2-~Qkho~yphenyl)-l-pipsrzzi~yl]-propyla~iuo}-
-3-~ethyl-~-o~o-2-phe~yl-4~ benz~pyr ~ hy~roohlorida
ho~ihy~rata
A mixture of 4 ~ of the compound prepared in Example 33, in the
form o~ its base, 4.35 ml of 37% aqueous formaldehyde and 1.15 g
of sodium cyanoborohydride in 25 ml of acetonitrile was stirred
at 20-25C, maintaining the pH in the range 5-6 by the addition
;;
... ,, .': ; : ,~ :
~- j
-122-
of acetic acid during the reaction. After 4 hours the solvent
was evaporated off in vacuo. 80 ml of ethyl acetate and 200 ml
of ice cooled lN sodium hydroxide solution were added to the
residue. The organic phase was washed with water, dried on
anhydrous sodium sulphate and evaporated to dryness in vacuo.
The residue was purified by flash chromatography on silica gel,
eluting with ethyl acetate:petroleum ether 3:1. The pure base
which was obtained was dissolved in diethyl ether. 1 equivalent
of ethanolic hydrogen chloride was added and the solvent was
removed in vacuoO The residue was crystallized from water to
give 2 g of the title ~ompound, m.pO 186-187C
36
8-{~ tyl-3-l4-(2-~ethoxyphenyl)~1-pipera2inyl]-propylamino~
~3 m~thyl-~-o~o-2-phe~yl~ be~zopyr~ hy~xohlori~e hy~r~te
A mixture of 4.8 g of the compound prepared in Example 33, in
the ~oxm of its base, 2.8 ml of acetic anhydride and 33 ml of
pyridine was stirred at 80C for 4 hours. After cooling to
20-25C, the reaction mixture was poured into 200 g of iced
water, acidified with lON hydrochloric acid and extracted with
dichloromethane. The organic extracts were washed with water,
dried on anhydrous sodium sulphate and evaporated to dryness in
vacuo. The residue was purified by flash chromatography on
silica gel, eluting with ethyl acetate:methanol 95:5. The pure
base which wa~ obtained was dissolved in dichloromethane.
equivalent of ethanolic hydrogen chloride was added and the
solvent was removed in vacuo~ The residue was crystallized from
acetonitrile to give 3 g of the title compound containing 0.33
equival~nts of acetonitrile, m.p. 208.5-210.5C.
. .
Xxam~le 37
8-{3-t~-(2-methoxyphenyl) 1 pipera~inyl]-pro~ionamido}-3-~ethyl- `
-4-oxo-2-phe~yl-4~-1-bon~opyxan hydrochloride
.- .: .
A mixture of 3.97 g of Intermediate X and 3.07 g of ~;~
1-(2-methoxyphenyl)-piperazine in 40 ml of dimethylformamide was
stirred at 60C for 6 hours. The reaction mixture was then
cooled to ambient temperature and poured into water. Following
5 ~
,~
.
-123-
extraction with dichloromethane, the organic phase was washed
with water and dried on anhydrous sodium sulphate. The solvent
was removed in vacuo. The crude residue was crystallized from
ethanol to give the base of the title compound, which was then
dissolved in hot ethanol. 1 molar equivalent of 0.81M ethanolic
hydrogen chloride was added to the solution. 4 g of the title
compound, m.p. 255 257C, was obtained.
~xampl~ 38
8-{2-[~-~2-methoxyphenyl)-1-piperaæinyl3 ethylurei~o}-3 methyl-
~ o~o-2-ph~yl ~H~ e~zopyran metha~e~ulpho~t~
.,
A mixture of 3.34 g of Intermediate XLIV and 7.22 g of
1-(2-methoxyphenyl)-piperazine was stirred at 100C for 5 hours.
An additional 1.8 g of 1-(2-methoxyphenyl)-piperazine was then
added and stirring was continued for a further 2 hours at 100Co
After cooling to ambient temperature, the reaction mixture was
poured into water and extracted with ethyl acetate. The organic
phase was washed with aqueous ~odium hydroxide solution, dried
on anhydrous sodium sulphat~ and evaporated to dryness in vacuo.
The crude residue was purified by flash chromatography on silica
gel, eluting with ethyl acetate:methanol 98:2. The collected
fractions were evaporated to dryness in vacuo and crystallized
from water:ethanol 4:6. The crystals were redissolved in
dichloromethane and treated with 1 molar equi~alent of
methanesulphonic acid. The crude methanesulphonate obtained by
evaporation in vacuo was crystallized from ethyl acetate:ethanol
1:1 to yield 2.35 g of the title compound, melting at 191-193C.
~am~l~ 39
8-{2-~4-~2 ~et~oxyphe~yl) 1-p~perazinyl]-otho~y}-3-msthyl-
-~-oxo-2-phenyl~ benzopyr~ h~ro~hlori~e hy~rate
A mixture of 6.61 g of the Intermediate XI, 8.34 g of
1-(2-methoxyphenyl)-piperazine and 1.26 g of sodium iodide in 70
ml o~ dimethylformamide was stirred at 80C for 17 hours. After
cooling to 20-25C, the reaction mixture was poured into 600 ml
of water, made alkaline with 5~ aqueous sodium bicarbonate and
extracted with dichloromethane. ~he organic extracts were washed
-124-
with aqueous sodium chloride solution, dried on anhydrous sodium
sulphate and evaporat d to dryness in vacuo. The residue was
purified by flash chromatography on silica gel, eluting with
dichloromethane:methanol 99:1, then 98:2. The fractions
containing the base of the title compound were pooled, and the
~olvent was removed in vacuo. The residue was dissolved in
ethanol and ethanolic hydrogen chloride was addedO The title
compound crystallized and was collected by suction filtration.
It was recrystallized from 95~ ethanoln Yield 6.5 g, m.p.
224-225C.
. . .
El~e~tal ~alysis: ~ :
Found % C=66.38, H=6.34, N=5.35, Cl=6.76, H20=3.35
Calculated % : 66.34, 6.14, 5.33, 6.75, 3.43
NNR Sp~tru~ at 60 M~ [CD~13-CD30~)
7.8-7.1 (m, 8H, aromatic protons of the benzopyran ring)
7.1-6.6 (m, 4H, aromatic protons of the 2 methoxyphenyl group)
4.8-4.4 (m, 2H, OCH2)
4.4-4.1 (m, 3H, H20 and N+H) ~.
3.9-300 (m, lOH, 5 x CH2N)
3.8 (s, 3H, O~H3)
2.1 (s, 3H, CH3) ~ :
B~a~pl~ 40
8-t3-~4-(2-m~thoxyphe~ pipera~in~yl]-propoxy}-3-methyl- ~
-4-oxo-2-ph~yl-~K-1-b~zopyr~ dihy~rochlori~e :
. .: .
This compound was prepared by the method described in Example
39, but using Intermediate IX instead of Intermediate XI.
Purification by flash chromatography was omitted as unnecessary
in this case. M.p. 226-227C.
": .
Bxample 41
8-{~-[4-(2-motho~yphenyl)-1-piperaz~yl]-butoxy}-3-methyl-
-4-oxo-2-phenyl~ benzopyran aihydrochlori~e
A mixture of 7.75g of Intermediate XVI, 4.7 g of
1-(2-methoxyphenyl)-piperazine, 3.3 g of potassium iodide and
: ' . , ' , ' ' `;'~
2 11 ~
-125
2.8 g of anhydrous potassium carbonate in 78 ml of
dimethylformamide was stirred at 75C for 2 hours. After cooling
to 20-25C, the reaction mixture was poured into 600 ml of water
and extracked with dichloromethane. The organic extracts were
washed with water and dried on anhydrous sodium sulphate, and
the solvent was then evaporated off in vacuo. The residue was
purified by column chromatography on silica gel, eluting with
ethyl acetate. The pure title compound thus obtained as its base
was transformed into its dihydrochloride by treatment with
ethanolic hydrogen chloride. After crystallization from ethanol,
6.5 g of title compound was obtained. m.p. 217-219C.
E~mple 42
8-{5-t4~2-~etho~yphenyl~ 1-piperazinyl]-pantyloxy~-3-methyl-
-~-oYo-2-phenyl~ be~zopyr~n hydxo~hlorid~
This compound was prepar d by the method described in Example
41, but using Intermediate XVII instead of Intermediate XVI.
M.p. 173C (ethanol). The corresponding base melts at 117-118C
(ethanol).
~xamPl~_~3
8-{3 [~-~2-~etho~yphe~yl)-1-o~o-1-piperazinyl~-propo~y}-3
-met~yl-4~oxo 2-phenyl ~ benzopyra~ 1.75-hyarate
2.93 g of magn~sium monoperoxyphthalate in 10 ml of water was
added dropwise at -15C to a solution of 4.34 g of the compound
prepared in Example 40 and 0.1 g of benzyl(triethyl)ammonium
chloride in 20 ml of dichloromethane and 20 ml of methanol. The
mixture was stirred for 2 hours at 0C and then warmed to
ambient temperature. It was poured into water and made basic by
additisn of aqueous sodium hydroxide solution. Extraction with
dichloromethane gave~ after the usual work up, a solid which was
purified by flash chromatography, eluting with
dichloromethane:methanol 9:1. The collected fractions containing
the pure compound were evaporated to dryness in vacuo and the
residue was cry~tallized from acetonitrile to give 0.5 g of the
title compound, m.p. 89-92C.
-126
2~a~ple 44
.
8-{2-t2-~2~6-~i~etho~ypheno~y)-~th~la~ni~o3-et~o~y~3-~ethyl-4
-o~o-2~phe~yl~4~ l-be~opyran hydroahlori~Q
A mixture of 4.5 g o~ Intermediate XII, 3.7 g of triphenyl-
phosphine and 2.85 g of 2,6-dimeth,oxyphenoxyacetaldehyde
(prepared according to Nelson, W.L. et al., J. Med. Chem., 22,
1125, 1979) in 45 ml of benzene was stirred at 20-25C for 18
hours and at reflux for 5 hours. The solvent was evaporated off
in vacuo and the residue was dissolved in 80 ml of anhydrous
methanol. 3A molecular si~ve was added. Next, 0.61 g of sodium
borohydride was added at 0C. The mixture was stood for 1 hour
at 0C and for 1 hour at 20-25C, and then poured into iced
water and extracted with dichloromethane. The organic extracts
were washed with water and dried on anhydrous sodium sulphate.
The solvent was removed in vacuo and the residue was purified by
flash chromatography on silica gel, eluting with
dichloromethane: methanol 49:1. The base obtained was treated
with ethanolic hydrogen chloride. After crystallization from
ethanol, the title compound was obtained. Yield 40%, m.p.
200-202C.
:
~xa~pl~ 5
8-~2-hydro~y-3-t4-~2-m~thoxyphenyl)-1~-piper~zinyl]-propoxy}-3-
-methyl-~-oxo-2-phe~yl-~ 1 be~zopyra~ hydrochloride ~'
A solution of 3.7 g of Intermediate XL and 4.64 g of
1-(2-methoxyphenyl)-piperazine in 40 ml of dimethylformamide was
stirred at 80C for 3 hours. After cooling to 20-25C, the
reaction mixture was poured into 400 ml of water and extracted
with dichloromethane. The aqueous phase was made alkaline with
lN sodium hydroxide solution and extracted with ethyl acetate.
The combined organic extracts were washed with water and dried `~
on anhydrous sodium sulphate. The solvents were evaporated off
in vacuo. The residue was purified by flash chromatography on
silica gel, eluting with ethyl acetate. The fractions containing
the title compound in the form of its base were pooled, and
stripped in vacuo. The residue was dissolved in dichloromethane
and one equivalent of ethanolic hydrogen chloride was added. The
' ' ,:': ' '' , ' ~ ' i l" " . " , " , " ~ . " ,.,, ~, . . . .
2V~Ol~
-127-
solvents were removed in vacuo and the residu~ was crystallized
from ethanol. 5 g of the title compound containing one molar
equivalent of ethanol were obtained. M.p. (122) 126-128C with
decomposition.
~x~mpl~ 46
.
8 ~3-[~ 2-~thoxyphenyl3-1-piper~zi~yl~-propylthio~-3-~et~yl-
-~-oxo-2-p~#~yl~4~-1-be~æopyr~
A mixture of 4.4 g of Intermediate XXXIV, 2.5 g of
1-(2 methoxyphenyl)-piperazine, 1 g o~ potassium iodide and 1.8
g of anhydrous potassium carbonate in 40 ml of dimethylformamide
was ~tirred at 100C for 3 hours. After cooling to 20-25~, the
reaction mixture was poured into 350 ml of water and extracted
with dichloromethane. The organic extracts were washed with
water and dried on anhydrous sodium sulphate, and the solvent
was then evaporated off in vacuo. The residue was purified by
column chromatography on silica gel, eluting ~rith ethyl acetateo
petroleum ether 3:2, and by crystallization from ethanol,
yiPlding 3.9 g of the title compound, m.p. (70) 96-9~C.
~am~le 47
8-{3-t4-~2-methogyph~yl)-l-pipera~inyl]-propyli~ulphonyl~ 3-
-mathyl-~ oxo-2-ph~yl-4~ e~æopyra~ hydrochlori~e
A solution of 3.8 g of Intermediate XXXV and 4 g of
1-(2-methoxyphenyl)-piperazine in 40 ml of dimethylformamide was
heated at 60C for 7 hours. After cooling to 20-25C, the
reaction mixture was poured into 500 ml of water and extracted
with dichloromethane. The organic e~tracts were washed with
water and dried on anhydrous sodium sulphate, and the solvent
was then evaporated off in vacuo. The residue was purified by
flash chromatography on silica gel, eluting with ethyl acetate:
petroleum ether 1:1. The base o~ the title compound was
obtained. It was dissolved in ethanol and one equivalent of
ethanolic hydrogen chloride was added to give 4.5 g of the title
compound, m.p. (215) 226-228C.
: .~
'' ~. ''
; i j, : : ! . , . ~ '
~':" ' ' ' ' ' ,,', ' '' ' , ., " ' ' :: . ''', ; ,,: .; . ' ,.', " . I ' :, , '" , ' '
-128-
~mpl~ ~8
8-~2-~4-~2-~etho~yph~yl)-1 pip~ra~i~yl]-et~yl8ulpha~0yl}-3
-~ethyl-4-o~o 2-phe~yl~4~ be~zopyr~ hy~ro~hlori~e
A solution of 4.5 g of Intermediate XLII and 3.8 g of
1-(2-methoxyphenyl)-piperazine in 40 ml of dimethylformamide was
heated at 70C for 7 hours. After cooling to 20-25C, the
reaction mixkure was poured into 150 ml of water and extracted
with dichloromethane. The organic solution was washed with water
and dried on anhydrous sodium sulphate, and the solvent was then
evaporated off in vacuo. The residue was purified by flash
chromatography on silica gel, eluting with ethyl asetate:
petroleum ether 3:7, and the title compound was obtained by
salification with ethanolic hydrogen chloride. Yield 2.9 g, m.p.
236-238C.
ple
8-tN ~ethyl-~ [~2-metho~yph~nyl)-1-piper~inylJ-
-~thyl3ulph~moyl}-3-~ethyl-4-oxo-2-phonyl~ -be~zopyran
hydro¢hlori~
The title compound was obtained by the method described in
Example 48, but using Intermediate XLI instead of Intermediate
XLII. M.p. 194-198C (ethanol3.
~x~le 50
8-{~-c~rbamoyl-3~ (2-m0thoxyphe~yl)-l-piperazi~yl]-
ropyla~i~o} 3-~athyl-4-o~or2-ph~nyl-4~ benzopyran
~tha~e~ulphonat~ he~ihydrate
A mixture of 4.06 g of the compound of Example 33 and 1.5 g of
potassium cyanate in 42 ml of glacial acetic acid was stirred at
50C for 4 hours. The rection mixture was poured into iced water
and made alXaline. The precipitate was collectPd by suction
filtration, dried and purified by flash chromatography using a
silica gel column eluting with ethyl acetate:methanol 98:2. The
~ractions, containing the title product as a base, were
evaporated to dryness in vacuo and one equivalent of
methanesulphonic acid was added to the residue after dissolution
.. . . . .. . . . . . .. .. . . . . .
129- 2~15~
in 30 ml of dichloromethane. The solvent was evaporated off in
vacuo and the residue wa~ crystallized from ethanol to give 3.1
g of the title compound (m.p. 157-160C, with decomposition).
This compound contained one molar equivalent of ethanol.
~ ple 51
8-{~-t~a-(2~ thc~acyph~ yl3 l-pip~r2~ yl]Dl oacobutyl}~3-
-~thyl-4-o~o-2-phe~yl-4~ opyran ~ethanesulpho~ate
A ~olution of 1.33 ml of anhydrous dimethylsulphoxide in 9 ml of
dichloromethane was added at -70C to a solution of 0.74 ml of
oxalyl chloride in 6 ml o~ dichloromethane. After stirring at
-70C for 15 minutes, a solution of 2.8 g of the compound of
Example 21 (as a base) in 14 ml of dichloromethane was added.
After 15 minutes at the same temperature, 4.7 ml of anhydrous
triethylamine was added and the temperature was raised to -30C
over a period of 30 minutes. Stirring was continued at -30C
for another 30 minutes. After letting the temperature rise to
0C, the mixture was diluted with 120 ml of water and extracted
with dichloromethane. The organic phase was washed with water,
dried on anydrous sodium sulphate and evaporated to dryness in
vacuo. The residue was purified by flash chromatography in a
silica gel column, eluting with ethlyl acetate:dichloromethane
9:1. The fractions, containing the title product as a base, were
evaporated to dryness in vacuo and one equivalent of
methanesulphonic acid was added to the residue after dissolution
in 30 ml o~ dichloromethane. The solvent was evaporated o~f in
vacuo and the residue was crystallized from ethanol to give 2.9
g of the title compound, m.p. 194-195C.
-
8-{3-t2-~1,4-benzodioxanyl)m~thylami~o]propyl~arbamoyl} : :
-3-methyl-4~oxo-2-phenyl 4~-1-ben~opyran methan~ulpho~ate ~ ;
A mixture of 5.56 g of Intermediate XLIII, as a base, 4.58 g of
2-(p-toluenesulphonyloxymethyl)-1,4-benzodioxane and 1.9 g of :
anhydrous potassium carbonate in 80 ml of anhydrous :
dimethylformamide was stirred at 110C for 5 hours. The
reaction mixture was cooled to ambient temperature, poured into :~
-130-
water and extracted with dichloromethane. The organic phase was
washed with water, dried on anhydrous sodium sulphate, .filtered
and evaporated to dryness in vacuo. The residue was purified by
flash chromatography using a silica gel column, eluting with
ethyl acetate:methanol 95:5. The fractions containing the title
compound a~ a base were evaporated to dryness in vacuo. The
residue was dissolved in ethanol and one eguivalent of
methanesulphonic acid dissolved in ethyl acetate was added. 'rhe
crystallized product was ~iltered off and recry~tallized from
ethanol to give 2.4 g o~ the title compound, m.p~ 172 174C.
5. 3
8-{4-L~- ~2~;~et~0~phe~yl)-l-piper~zinyl]-butyl}~3-mathyl-
-4-o~o 2-p~enyl-4~ b~zopyran methane~ulpho~ato
A solution of 2.8 g of Intermediate XLVI and 0.13 g of
p-toluenesulphonic acid in 150 ml of methanol was refluxed for 5
hours. After cooling to 20-25C, 0.8 g of anhydrous potassium
carbonate was added and stirring was continued for 3 hours.
After filtration, the reaction mixture was evaporatad to dryness
in vacuo to give 2.5 g of 8-(4,4 dimethoxybutyl~-3-methyl-4-oxo-
2-phenyl-4H-1-benzopyran.
~ (CD~13, ~)
1.6-1.9 (4H, m, CHCH2CH2CH)
2.2 (3H~ s, flavone CH3)
2.9 (2H, t, Fl-CH2)
3.3 (6H, s, 2 x OCH3)
4.4 (lH, t, CH(OCH3)2)
7.3 (lH, dd, flavone CH in 6)
7.5-7.8 (6~, m, flavone CH in 7, and 5 x phenyl CH)
8.1 (lH, dd, flavone CH in 5)
A solution of 2.5 g of the compound thus prepared in 10 ml of
water and 30 ml of acetic acid was heated at 50C for 2~ hours.
The reaction mixture was cooled to ambient temperature, diluted
with iced water, basified with aqueous sodium carbonate, and
extracted with chloroform. The organic phase was dried on
anhydrous sodium sulphate, filtered and evaporated to dryness in
vacuo. The residue was puri~ied by flash chromatography on
silica gel, eluting with petroleum ether:ethyl acetate 3:1. 2.1
131-
g of 8-(4-oxobutyl)-3-methyl 4-oxo-2-phenyl-4H-1-benzopyran was
obtained ~>75% yield) and was used with no further purification
in the next step.
~MR t~C13, ~)
1.9-2~1 (2H, dd, CH2CH2CH~CH0)
2.2 (3H, s, flavone CH3)
2.5 (2H, t, CH2CHO)
2.9 (2H, t, Fl-CH2)
7.3 (lH, dd, ~lavone CH in 6)
7.5-7.7 (6H, m, flavone CH in 7, and 5 x phenyl CH)
8.1 (lH, dd, flavone CH in 5)
9.7 (lH, s, CHO)
2.3 ml of 6N hydrochloric acid in ethanol, a solution of 2.1 g
of the compound thus prepared in 40 ml of methanol and 0~45 g o~
sodium cyanoborohydride were added in succession to a solution
of 8 g of 1-(2-methoxyphenyl~-piperazine in 30 ml of methanol.
After stirring the reaction mixture at ambient temperature for
24 hours, it was poured into 500 ml of iced water and extracted
with dichloromethane. The organic phase was washed with water,
dried on anhydrous sodium sulphate and evaporated to dryness in
vacuo. The residue was purified by f:Lash chromatography using a
silica gel column, eluting with ethyl acetate:petroleum ether
9:1. The ~ractions, containing the title product as a base,
were evaporated to dryness in vacuo. The residue was dissolved
in 30 ml of dichloromethane ~ and one equivalent of
metha~esulphonic acid was added. The solvent was evaporated off
in vacuo and the residue was crystallized from acetone to give
2.35 g of the title compound (m.p. 141-143C).
: .: .
~xa~p~ 5~
8-~3-(4-phenyl-1-pip~riainyl)-~ropylcarb~oyl~ 3-~thyl-4-
-o~0-2-phenyl-4E-1-b~n~opyran methansulpho~æte
',
This compound was prepared by the method described in Example
11, using 4-phenylpiperidine instead of. 1-(2-methoxyphenyl)-
-piperazine and conducting the reaction for l hour instead of 5
hours. Purification was carried out by flash chromatography
using a silica gel column, eluting with dichloromethane:methanol
100:5. M.p. 157-159C (ethyl acetate). The respective base
-
-132-
melted at (127) 147-149C (ethanol). ~-.
~ample 55
8 [3-(4,~ ~iphe~yl-1-piperidi~yl~ propyl~arbi~oyl]-3-~ethyl
~-o~o-2-phenyl~ benzopyra~ methansulpho~ te
This compound was prepared by the method described in Example
11, using 4,4-diphenylpiperidine instead of 1-(2-methoxyphenyl)-
-piperazine and conducting the reaction for 2 hours instead of 5
hours. N.p. 221-223C (ethyl acetate).
~xampl~ 56
~-{3-t~-~4-~luorohen~oyl)-1-pi~eridlnyl~-propyl-ci~rba~oyl}-3-
-~ethyl-~-o$o-2-p~enyl-4~ b~n~opyr~ .
This compound was prepared by the method described in Example
11, using 4-(4-~luorobenzoyl)-piperidine instead of
1-(2-methoxyphenyl)-piperazine and conducting the reaction for .
30 minute instead of 5 hours. Puri~ication was carried out by
flash chromatography using a silica gel column, eluting with
dichloromethane:5N methanolic ammonia graduated 100 1 to 100:20.
M.p. 181-183C (ethanol).
:
~ampl~ 57
~-{3-~4-(2-o~o-1-be~imi~azolinyl)-1-piperiainyl]-propyl-
-o2rba~oyl}-3 ~othyl-~-oxo 2~ph~nyl-4~ benzspyra~
This compound was prepared by the method described in Example
11, using 4-(2-oxo-1-benzimidazolinyl)-piperidine instead of
1~(2-methoxyphenyl)-piperazine. Purification was carried out by
flash chromatography using a silica gel column, eluting with
chloroform:5N methanolic ammonia 100:3.
M.p. 238-241C (ethanol).
B~am~ls 5fl
8-{3 t~-~2-pyri~iai~yl)-1-pipera~inyl]-pro~yl~arbamoyl~-3-
-methyl 4-oxo-2-phenyl-4~ benzopyran metha~esulphon te
The title compound was prepared by the method described in
",
.. . . , i . .. . . . .
-133-
Example 11, using 1-(2-pyrimidinyl)-piperazine instead of
1-(2-methoxyphenyl)-piperazine and conducting the reartion for 2
hours. The product was purified by flash chromatography using a
silica gel column, eluting with chloroform:methanol 100:3. The
desired fractions were dissolved in dichloromethane and one
equivalent of methanesulphonic acid was added to the solution.
After evaporation of the solvent in vacuo, the residue was
boiled for 1 hour with ethyl acetate and then collected by
filtration. M.p. 209-210C. The product contained 0.2
equivalents of ethyl acetate and 0.1 equivalents of water. The
respective base melted at 178-180C (ethanol~.
E~l~ 59
8~3-~4-(2~ rox~phe~yl3-1-pip~ra~i~yl~ propylsarba~oyl}-
-3-~ethyl-~-oxo-2-phe~yl-4~ be~zopyran
Operating as described in Example 11, but using 1-(2-
-hydroxyphenyl)-piperazine instead of 1-(2-methoxyphenyl)-
-piperazine, heating for l~ hours instead of 5 hours, and using
dichloromethane:methanol 100:3 to 100~10 as eluant for column
chromatography, the ~itle compound was obtained. M.p. 118-120C
(ethanol 95%).
E~pl~ 60
8-{4~t~-~2-~etho~yphe~yl)-1-piperazi~yl~-butylcarba~oyl}-
-3-~ethyl-~-oxo-2-p~e~yl-4~ b~zopyra~ methan~ulpho~at3
. . ,. ~
This compound was prepared by the method used in Example 12, but
using 4-[4 (2-methoxyphenyl)-1 piperazinyl]-butylamine instead
of 3-[4-(2-methoxyphenyl)-1-piperazinyl]-propylamine. The
reaction mixture was stirred at room temperature for 22 hours,
diluted with water and ~iltered by suction~ washing the
insoluble solids with water. The crude residue was dried and
purified by column chromatography on silica gel, eluting with
ethyl acetate:methanol 9:1. The fractions contining the pure
product as a base were collected, evaporated to dryness in vacuo
and dissolved in dichloromethane. Methanesulphonic acid was
added to the solution and the salt was precipitated by adding 2
volumes of ethyl acetate, filtered and recrystallized from
1 5 ~
,,
-134- :
ethanol to give the title compound, m.p. Z30-232C. This product
contained 0.3 molar equivalent of ethanol.
~mpl~ 61
8-{3-[4-~2~etho~yph~yl~ piper~zi~yl]-propyl~ulpha~oyl}-
-3-mst~ 4-oxo~ he~yl-4~ be~zopyxan ~etha~esulpho~ate
This compound was obtained operating as described in Example 12
but using Intermediate VIII instead of 8-chlorocarbonyl-
-3-methyl-4-oxo-~ phenyl-4H-1-benzopyran and stirring for 24
hours instead of 2~ hours. The crude product was puri~ied by
column chromatography on silica gel, eluting with ethyl acetate:
methanol 98.5~1.5. The collected fractions containing the pure
product as a base were evaporated to dryness in vacuo and
dissolved in dichloromethane. Methanesulphonic acid was added
to the solution and the solvent was removed by evaporation in
vacuo. The crude salt was crystallized from ethanol to give the
title compound, m.p. (196) 198-200C.
a~ 2 .:
8-~3-[~ thyl-2-~2-msthoxyphe~o~y)-et;hylamino]-
-propylcarbamoyl}-3 ~othyl-~ oxo-2-phe~yl~ b~nzopyr~
hydro~hlorîd~
: ''
A solution of 10.5 ml of 40% formaldehyde in water was added to
a suspension of 6.66 g of the compound prepared in Example 14 in
55 ml of acetonitrile and 20 ml of water. After stirring for 15
minutes at room temperature, 2.70 g of sodium cyanoborohydride
95% was added to the red solution, and after an additional 15
minutes in the same conditions 1.38 ml of acetic acid was added.
After stirring or 3 hours, the solvent~ were removed in vacuo
and the residue was rinsed with 250 ml of water and 250 ml of
chloroform. After addition of 3N sodium hydroxide, the organic
phase was separated of~ and the aqueous phase was extracted
twice with chloroform. ~he solvent was re~oved from the
collected organic phases by evaporation in vacuo and the residue
was puri~ied by flash chromatography on silica gel, eluting with
chloro~orm:5.2N methanolic ammonia 100:0.5 to 100:2. The
collected fractions containing the pure title compound as a base
., , -, ~ , ,.. :: , ..
.
.: :.': ' ~ .- :: . ' . : : :
~ . - . ; ,:
:
, , : , . : ~ :~
. .
2Q9015~
-135-
were evaporated to dryness in vacuo and the residue was
dissolved in hot ethanol. The solution was acidified with
ethanolic hydrogen chloride and, after evaporation of the
solvent in vacuo, the residue was rinsed with diethyl ether and
stirred at room temperature. The crude product was collected by
filtration and crystallized from acetonitrile to give 3.1 g of
the title compound, m.p. 14~-148C.
~ pla 63
8-~N-~thyl-3-14 (2 m3thoxypho~yl)rl-pip0razinyl3-propio~mi~o~-
-3 mothyl 4-oxo-2-phe~yl 4H 1-be~zopyra~ metha~s~ul~ho~at~
Operating as described in Example 37, but using Intermediate L
instead of Intermediate X and stirring at 90C or 4 hours
instead of 60C for 6 hours, the title compound was obtained as
a crude base. After purification by column chromatography on
silica gel, eluting with ethyl acetate:methanol 95:5, a crude
methanesulphonatei was obtained as described in Example 61 and
crystallized from acetone to give the title compound, m.p.
200-202C.
~xample 6~
8-{3 ~ 2-~e ho~yph~yl3-1-piper~ yl]-propyl~rbamoyl}
-3-phenyl-4oxo-4X-1-b~nzopyr~ di~atha~ulphon~te
The title compound was prepared operating as described in
Example 12, but using Intermediate LVI instead o~
8-chlorocarbonyl-3 methyl-4-oxo-2-phenyl-4H-1-benzopyran and
stirring for 24 hour~ instead of 2% hours. The crude product
was purified by column chromatography, eluting with ethyl
acetate:methanol 92:8, and the pure base, obtained by
evaporation in vacuo of the collected fractions, was dissolved
in dichloromethane. Two equivalents of methanesulphonic acid
were added. The crude dimethanesulphonate, obtained after
evaporating off the solvent, was recrystallized from acetone,
m.p. 153--156 (200)C.
~o~a~
-136-
B~m~l~ 65
8-~3~l(3,~-~ihy~ro-1-oxo-2~aphthyl)-~thylami~o]-
ro~ylcarb~oyl} 3 methyl-4 0~0-2-ph~yl-~H-l ben20pyran
~tha~o~ulphonate
A mixture of 6 g of Intermediate XLIII, 2.4 g of 2-methylene-
-~-tetralone (prepared as described in Org. Synth ., 60, 88,
1981) and 3.14 ml of triethylamine in 48 ml of anhydrous
dimethylformamide was stirred at room temperature for 6 hours,
and then at 50C for 1 hour. The reaction mixture was diluted
with water and extracted with dichloromethane. The organic
layers were washed with water, dried on anhydrous sodium
sulphate and evaporated to dryness in vacuo. The crude residue
was purified twice by column chromatography eluting first with
dichloromethane:methanol 95:5 and then with dichloromethane:
methanol:5.8N methanolic ammonia 98:2:0.2, to give 1.74 g o~ the
title compound as a base. The base was converted into the
methanesulphonate by the procedure described in Example 61. The
salt was recrystallized first from acetone and then from
acetonitrile to give the title compound, m.p. (69) 157-159C.
~a~ 66
8-{2-[4-~2-m2~ho~yphe~yl)-l-pipera~i~yl]-ethoæyG~rbonyl~othyl}-
-3-~eth~ o~o-2-~ho~yl-~H-l-benzopyr~ dihy~roohlorid~
'~''
The title compound was prepared by the method described in
Example 5, but using Intermediate XLVII in place of 8-carboxy-
-3-methyl-4-oxo-2-phenyl-4H-1-benzopyran and 1-(2-chloroethyl)-
-4-(2-methoxyphenyl)-piperazine instead o~ 1-(3-chloropropyl)-
-4-(2-methoxyphenyl)-piperazine. M.p. 193-196~C from ethanol: -
diethyl ether.
Bx~pl~ 67
8-{4-t~-(2-~ethoxypheu~ piperazinyl]-butyl~ulphamoyl}-
-3-~ethyl-4-oxo-2-phenyl-4~ be~zopyran dimeth~esulpho~ate
The title compound was prepared as described in Example 61 but
using 4-[4-(2-methoxyphenyl)-1-piperazinyl]-butylamine instead
of 3-~4-(2-methoxyphenyl)-1-piperazinyl]-propylamine. The crude
-,, - ., - ;, . . . . . . ,. . , ~ -
- -
,
: ~ . , ,. .... ~ ,
; ~ .
:: : : , . ,
.
2~9~ 5~
-137-
dimethanesulphonate was crystallized first from acetonitrile and
then from ethanol. M.p. 172-174C.
~x~le 68
8~ 62-t~tr~hy~ropyra~yloxy)-3-[~ (2-~ethoxy~h~yl)-
~l-piper~zi~yl~-~ropylearb~moyl}-3-m~thyl-4-o~o-2-p~e~yl
-4~ bea~opyræn ~ethane~ulphonats he~ihyarat~
, ,:.
A solution of 3.6 g of 1-(3-chloropropyl)-4-(2-methoxyphenyl)-
-piperazine in 30 ml of anhydrous dimethylformamide was added
dropwise, under stirring at 0C, to a mixture of 3.92 g of
0-(2-tetrahydropyranyl)-hydroxylamine (prepared as described by
R.N. Watrener et al., Angewandte Chem. Int. Ed., 5, 511, 1966).
Stirring was continued for 2 hours at 0C, and then for 12 hours
at 110C. The reaction mixture was cooled to room temperature
and dimethylformamide was removed by distillation in vacuo. The
residue was rinsed with water and extracted with ethyl acetate.
The combined organic layers were washed with water and dri~d on
anhydrous sodium sulphate. The solvent was evaporated of~ in
vacuo to give 4.39 g of 1-[3~ tetrahydropyranyloxyamino)
-propyl]-4-(2 methoxyphenyl)-piperazi:ne.
l~_~NR (CDCl3~ ~)
6.50-6.75 (m, 4H, aromatic protons)
5.20 (bs, lH, NH)
4.60 (~, lH, O-CH-O)
3.30-4.00 (m, 5H, OCH3 and tetrahydropyran CH20)
2.80-3.20 (m, 6H, piperazine 2 x CH2, alkyl chain CH2N)
2.20-2.80 (m, 6H, piperazine 2 x CH2, alkyl chain CH2N)
1.30-2.00 (m, 8H, tetrahydropyran 3 x CH2, alkyl chain C-CH2-C~
A solution of 2.79 g of 8-chlorocarbonyl 3-methyl-4-oxo-
-2-phenyl-~H-1-benzopyran in 47 ml of chloroform was added
dropwise at room temperature to a mixture of 3.26 g of the
compound thus prepared and 1.42 g of potassium carbonate in 47
ml of chloroform. The reaction mixture was stirred for 3 hours,
~nd then diluted with 75 ml of chloroform and washed three times
with lM sodium hydroxide. The organic layer was washed with
water, dried on anhydrous sodium sulphate and evaporated to
dryness in vacuo. The crude residue was purified by column
.. . . .
~. .
,, ,' ' ' ", . . " ''"'' ' '
2 ~
138
chromatography on silica gel eluting with ethyl acetate:
methanol 98:2. The collected fractions were evaporated to
dryness in vacuo to give 2.99 g of pure title compound as a
base. The base was dissolved in dichloromethane, and methane-
sulphonic acid was added to the solution. The solvent was
removed by evaporation in vacuo and the crude salt was ~ .
crystallized from ethyl acetate to yield the title compound, ~ -:
m.p. 1~9-160C.
: ',
~æampla 69 ~
8-{4-t4-~2-~etho~yp~nyl);~l-piperaziayl]-butyra~ido}- :
-3-~ethyl-4-o~o-2-ph0~yl-4~ be~zopyran methanesulphonat~
h0~ihy~r~t~ ,
The title compound was prepared by the method described in
Example 38, but using Intermediate XLVIII instead of
Intermediate XLIV and stirring for 1 hour at 70C and 2 hours at
130C instead of 7 hours at 100C. After the usual workup, the
crude residue was purified by column chromatography on silica
gel eluting with ethyl acetate:methanol 95:5. The fractions
containing the pure title compound as a base were collected and
evaporated to dryness in vacuo. The residue was dissolved in
dichloromethane and one equivalent of methanesulphonic acid was
added to the solution. After evaporation of the solvent to
dryness in vacuo, the crude salt was crystallized from acetone, ~
m.p. 175-176C. ' :
!~Q ,
~-8-{2-[~-~2-m~tho~yphe~yl~ pip~az~nyl]-etho~yimi~o-methyl}-
-3-m~thyl-4-oxo-2 phenyl-4~-1-benzopyra~ -
A solution of 5.4 g of 8-formyl-3-methyl~4~oxo-2-phenyl-4H-l-
-benzopyran and 5.13 g of Intermediate LII in 10 ml of
chloroform, containing 3A molecular sieve, was stirred at reflux
for 6 hours. The molecular sieve was removed by filtration and
the solution was evaporated to dryness in vacuo. The crude
product was purified by column chromatography on silica gel,
eluting with ethyl acekate~petroleum ether 7:3. Two groups of
fractions were collected and evaporated to dryness in vacuo.
. . . . . . . ..
: , . .
..
., :, : . . .
, ~
2 ~
-139- ::
The firsk eluted group of fractions (less polar) contained
almost pure title compound; the second group ~more polar) was a
~:1 mixture of the E and Z diastereomers, as determined by NMR. ;~ .
~ R ~D~13, ~)
8i.75 (dd, 0~5H, benzopyran CH in 7, Z)
8.65 (s, O.SH, iminic CH, E) : :
8.30 (dd, lH, benzopyran CH in 5, E+Z)
8.15 (dd, 0.5H, benzopyran CH in 7, E~
8.00 (s, 0.5H, iminic CH, Z) ~:
7.60-7.75 (m, 2H, phenyl CH in 2' and 6'/ E~Z)
7.50-7.60 ~m, 3H, phenyl CH in 3', 4' and 5', E+Z) :
7.45 (dd, 005H, benzopyran CH in 6, Z)
7.41 (dd, 0.5H, benzopyran CH in 6, E) -.
6.70-7.10 (m, 4H, phenyl protons, B+Z)
4.41 (t, 2H, CH20, E+Z) ~:~
3.86 (s, 3H, CH30, E~Z)
3.05-3.20 (m, 4H, piperazine 2 x CH2, E+Z)
2.70-2.90 (m, 6H, piperazine 2 x CH2 and CH2N, E+Z)
2.20 (s, 1.5H, benzopyran C~3 in 3, ~) .
2.18 (s, 1.5H, benzopyran CH3 in 3, E) ~ -
The E diasteromer was crystallized from ethanol:water 2:1 to .
give 2.5 g of pure title compound, m.p. 107-109C. `:
~x~ple 71 .
8-~N-hy~ro~y 3-~4-~2-methox~phe~yl)-1-~piperazinyl]-
ipropyloarb~moyl}-3-met~yl-~-o~o-2-pho~yl~ 1-b~opyra~ :
m~thian~sulpho~at~ 9.25 ~20 :
:,
A solution of 2.04 g of the compound of Example 68 as a base in
104 ml of 1.6N ethanolic hydrogen chloride was stirred for 12 ;
hours at ambient temperature. Ethanol was removed by ~.
evaporation in vacuo and the residue was rinsed with lN sodium
hydroxide and dichloromethane. The organic layer was collected, "~
washed with water, dried on anhydrous sodium sulphate and ~.
ev~porated to dryness in vacuo. One molar equivalent of :.
methanesulphonic acid was added to a solution of the residue in
dichloromethane. The solvent was removed and the crude :. :
methanesulphonate crystallized from acetone to give 1.02 g of
the title compound, m.p. 211-213C. The product contained 0.25
`: ' ., . '~ . .! ";` l .: ` !~ . ., .; .
2~9~
-140-
mole of water.
. . .
E~am~le 72
~-8-~2-{2-t4-~2~m~tho~yphe~yl)-1 piper~ yl~-ethylcarb~moyl}- : :
-ethenyl~-3-~ethyl-4-o~o-2-phe~yl-4H-1 ~e~zopyra~
~tha~e ulphona~9 1-2 ~2~
The title compound was obtained by the method described in
Example 61 but using Intermediate IV instead of Intermediate
VIII and 2-[4-(2-methoxyphenyl)-1-piperazinyl] ethylamine
instead of the corresponding propylamine, in 1,1,2,2-
-tetrachloroethylene as solvent. At the end, the reaction
mixture was diluted with water and chloroform and washed with lN
aqueous sodium hydroxide, then with water. To the organic
layer, after drying on anhydrous sodium sulphate, was added
methanesulphonic acid and the solvents were evaporated off in
vacuo. The crude product was crystallized twice from
isopropanol to give the title compound containing 1.2 molar ;~ .
equivalents of water. M.p. 124-127C.
~ampl~ 73
~-{~-~4-(2-metho~yphe~yl) 1 pip~razinyl]-butyl~ulphi~yl}
-3-~ethyl-~-o~o-2-phenyl~ 1 benzopyr~ meth~ne~ulphonate
The title compound was prepared according to Example 38, but
using Intermediate LIV instead of Intermediate XLIV, stirring at
70C for 3 hours and again at 90C for 3 hours after adding a
catalytic quantity (0.01 equivalents) of potassium iodide.
Purification by column chromatography on silica gel, eluting
with ethyl acetate:methanol 9:1, gave the title compound as a
base. To the crude base, dissolved in dichloromethane, was
added one molar equivalent of methanesulphonic acid. After
removal of the solvent by evaporation in vacuo, the resulting
salt was crystallized from acetone to give the title compound,
m.p. 183-184C.
,
- : : . -
,
-141-
~a~ple 748-{3~[3~(2 ~e$ho~yph~o~y)-propylamino~-propylaarb~oyl}-
-3 ~e~h~ o~o~r2-ph~yl-4~ ba~20pyran ~eth~esulphonats
h~mihy~r~t~
The title compound was prepared according to the method
described in Example 76 but using 3-(2-methoxyphenoxy)-propyl
~hloride (prepared as described in B. Willhalm, Tetrahedron,
20, 1185, 1964) instead of 2-(2,6-dimethoxyphenoxy)-ethyl
bromide. ~he residue from dichloromethane extraction was
purified hy column chromatography on silica gel eluting with
dichloromethane:methanol:5N methanolic ammonia 9:1:0.3; the pure
base was converted into the methanesulphonate, which was
crystallized twice from ethyl acetateoacetonitrile 9:1 to give
the title compound, melting at (60) 87-90C.
Ex~pl0 75
8-~3-t2-(2-methylthiophe~o~y)-ethylamino3-propyloiarb~moyl}-
-3-mothyl-4-o~o-2-pho~yl 4~ be~zopyra~ ~thanesulpho~te
1.85 g of 95% sodium borohydride was added to a solution of 7 g
of Intermediate LIX in 70 ml of methanol stirred at 0C. After
stirring for 1 hour at the same temperature, the solvent was
removed by evaporation in vacuo. The r~sidue was diluted with
water and 2N hydrochloric acid and extracted with ethyl
acetate. The organic layer was washed with watar, dried on
anhydrous sodium sulphate and evaporated to dryness in vacuo,
giving 6.6 g of pure 2-(2-methylthiophenoxy)-ethanol as on oil.
8.57 g of p-toluenesulphonyl chloride was add~d portionwise to
a solution of the compound thus prepared in 35 ml of pyridine
stirred at 0C. After stirring for 14 hours at ambient
temperature, the reaction mixture was poured into cold 2N
hydrochloric acid and extracted with dichloromethane. The
organic layer was washed twice with water, dried on anhydrous
sodium sulphate and evaporated to dryness in vacuo yielding 7.8
g of a 3:1 mixture o~ 2-(2-methylthiophenoxy)ethyl
p-toluenesulphonate and 2-(2-methylthiophenoxy)-ethyl chloride
~assessed by NMR) as a low melting solid which was used without
further purification.
.., " , . . . . . . . . .
.,. ~.. , ,. . ;; , . i. . .
, , ., " . , ~ , , ; . :, :, .. . .
2~01~
-142
A homogeneous mixture of 3.3 g of the above mixture and 8 g of
Intermediate XLIII was kept in an oil bath at 140C for Z0
minutes. After this period the molten mass was cnoled to ambient
temperature and solidified. The solid residue was rinsed with
dichloromethane and 4N sodium hydroxide. The organic layer was
washed with water, dried on anhydrous sodium sulphate and
evaporated to drynes~ in vacuo.
The crude product was purified by column chromatography on
silica gel eluting with dichloromethane:methanol 9:1 giving
2.07 g of the title compound as a base. This was converted by
the usual method into a crude methanesulphonate, which was
crystallized first from acetone and then from acetonitrile.
M.p. 143-146C.
~pl~ 7Ç
8-{3-~2-~2~C-di~ethoxyph~oxy)-~thyla~ino]-~ropyl~ar~amoyl}-
~3-~thyl 4-o~o-2-phenyl~ benzopyran hydro~hlori~e
A homogeneous mixture o~ 3.3 g of 2-(2,6-dimethoxyphenoxy)-ethyl
bromide (prepared as described in J. Augstein et al., J. Med.
Chem., 8, 356, 1965) and 8.4 g of Intermediate XLIII was heated
in an oil bath at 150C for 10 minutes. The molten mass was
cooled to ambient temperature and solidified. The solid residue
was rinsed with ethyl acQtate and 2N sodium hydroxide. The
oryanic layer was washed with water, dried on anhydrous sodium
sulphate and evaporated to dryness in vacuo. The oily residue
was purified twice by column chromatography on silica gel
eluting firstly with ethyl acetate:methanol:5N methanolic
ammonia 97 3:0.3 and then with dichloromethane~methanol:
triethylamine 90~10:0.3. This gave 3.3 g of the pure title
compound as a base. The crude hydrochloride, obtained by the
usual method, was crystallized from acetone followed by
acetonitrile. M.p. 179-181C.
~amDlo 77
a- ~3-t~5-chloro-2-metho~yphenyl)-1-piper~zinyl]-propyl-
¢axbamoyl}-3-methyl-4-oxo-2-phenyl-4~-1-be~opyra~.
This compound was prepared by the method described in Example
, ,
: ' , . ~ . :' .
.. , , . ~;
0 9 0 1 5 6
-143-
11, but using 1-(5-chloro-2-methoxyphenyl)-piperazine instead of
1-(2 methoxyphenyl)-piperazine and carrying out the reaction for
6 hours instead of 5 hours. Puri~ication was carried out by
flash chromatography on silica gel, eluting with chloroform : 5N
methanolic ammonia 100:1. The title compound melted at 163-166~C
a~ter crystallization from 95% ethanol.
.:
ple 78
(B)-8-{~ [4-(2-~ethoxy~enyl)-1-pipera~i~yl] l-but~yl}-
-3~ethyl-~ o~o-2-phe~yl~ be~ o~yra~
33.4 ml of a 1 molar solution of lithium
bis(trimPthylsilyl)amide in anhydrous tetrahydrofuran was added
dropwise over a period of 15 minutes to a suspension of 6.4 g of
3-hydroxypropyltriphenylphosphonium bromide in 60 ml of .~:
anhydrous tetrahydrofuran cooled to -15C. Then a solution of 4
g of 8-~ormyl 3-methyl-4-oxo-2-phenyl-4H-l-benzopyran in 40 ml
of tetrahydrofuran w~s added dropwise. The reaction mixture was
stirred at 0C for 30 minutes, then at ambient temperature for
1~ hours. :
Quenching with met~anol, followed by evaporation to dryness in
vacuo, gave a residue which was purified by column
chromatography on silica gel eluting with ethyl
acetate:petroleum ether 6:4. The collected fractions were
evaporated in vacuo giving 4.17 g of
8-(4-hydroxy-1-butenyl)-3-methyl-4-oxo-2-phenyl-4H-1-benzopyran
as a E-Z diastereoisomer mixture having a 3.5:1 ratio determined
by NMR.
l~_NN~, 200~8 ~CDC13, ~)
8.10-8.20 (d, lH, benzopyran CH in 5, E+Z)
7.30-7.80 ~m, 7H, other aromatics, E~Z) :
6.80j7.00 (2d, lH, aryl-CH=, E+Z)
6.41 (dt, 0.78H, CH-CH2, E)
5.90 (dt, 0.22H, CH-CH2, Z) :
3.60-3.80 (m, 2H, CH20, E+Z) .:
2.45-2.60 (m, 2H, CH C_2, E+Z) ~ .
2.18 (s, 3H, benzopyran CH3 in 3, E+Z)
1.60-1.90 (sa, lH, OH, E+Z)
.. .. . . . . ....... .. . . .... . . . . ..
, . " , , , ., , .. . , : : :. . , : : .. . .
&
~.
-144- - -
1.65 g of p-toluenesulphonyl chloride was added to a solution of
2.2 g of the above mixture in 24 ml of anhydrous pyridine was
stirred at 0C. Stirring was continued for 48 hours at the same
temperature, and the reaction mixture was then poured into cold
lN hydrochloric acid and filtered hy suction. The gummy solid
was washed with water and rinsed with dichloromethane. ~he
solution was dried on anhydrous sodium sulphate and evaporated
to dryness in vacuo to give 2.30 g of (E,Z)-4-{8-[3-methyl-
-4 oxo-2-phenyl-4H-1-benzopyranyl}-3-butenyl p-toluenesulphonate
having the same diastereoisomer composition as the above
Int~rmediate.
A solution of 2.85 g of the p-toluenesulphonic acid ester above
and of 2.98 g of 1-(2-methoxyphenyl)-piperazine in anhydrous
dimethylformamide was stirred at ambient temperature for 48
hours. After this period, the mixture was poured into 250 ml of
water and extracted with ethyl acetate. The organic layer was
washed with water, dried on anhydrous sodium sulphate and
evaporated to dryness in vacuo giving a residue which was : -
purified by column chromatography on silica gel eluting with
ethyl acetate:petroleum ether 6:4. The collected fractions were
evaporated to dryness in vacuo and the crude product was
crystallized from 70% ethanol yielding 1.~8 g o~ the title
compound that melted at 119-121C.
l~_N~R, 200M~8 ICDC13, ~)
8.14 (dd, lH, benzopyran CH in 5)
7.85 (dd, lH, benzopyran CH in 7
7.41-7.70 (m, 5H, phenyl CHs)
7.34 (dd, lH, benzopyran CH in 6)
6.70-7.10 (m, 5H, aryl-CH= and methoxyphenyl CHs)
6.30-6.50 (dt, lH, J~rans=16.5Hz, CH-CH2)
3.86 (s, 3H, CH30)
3.00-3.15 (m, 4H, 2 piperazine CH2)
2.50-2.80 (m~ RH, 2 piperazine CH2, CHC_2CH2N)
2.18 (s, 3N, benzopyran CH3 in 3)
" . , . ~
', , , ': . ' :. : .: . ' :: ~ ." ' :.. ' ~ , : :;, , ~:
,,, ~ ,.............. . .
: . ,. : ,
2~9~5~
`
-145-
E~am~ls 79
(~) 8 ~2~2-~ (2-~etho~yph0~yl)~1~piper zi~yl]-ethosya~rhonyl}
~t~e~yl~-3 ~thyl-4-oxo-2-phe~yl-4H-1-ben~opyra~
~ethan~sulpho~at~ -
The title compound was prepared according to Example 6, but
using Intermediate III instead of 8-carboxy-3-methyl-4-oxo-
-2-phenyl-4H-1-benzopyran. After the usual workup, the residue
was crystallized twice frsm ethanol; the solid obtained was
purified by column chromatsgraphy sn silica gel eluting with
chloroform:ethyl acetate 8:2 to give the pure base, which was '~
dissolved in chloroform:ethansl 1:1. Methanesulphonic acid was
added to the solutisn and the sslvents were removed by
evapsration in vacuo. The crude salt was crystallized from
isopropanol yielding the title compsund, melting at 193-195C.
This compound contained 0.33 equivalent of isopropanol and 0.25
e~uivalent of water.
~. .
~ pl~ 80
8-t2-~4-~2-~thoxy~he~yl)-1-piper~zinyl~-ethylcarbamoylm~thyl}-
-4-o~o 2-ph~yl-4~-1-b~zopyran ~etha~ulpho~ate hydrate
A mixture of 2.8 g of Intermediate XLVII and 1.28 g of
1-hydroxybenzotriazole in 20 ml of anhydrous dimethylformamide -,
was stirred at 0-5C for ~5 minutes. A solution of 1.96 g of
dicyclohexylcarbodiimide in 20 ml of anhydrous dimethyl-
formamide was added dropwise to the mixture over a period of
about 40 minutes. After stirring for 8 hours at ambient
temperature, a solution of 2.24 g of 1-(2-aminoethyl)-
-4-(2-methoxyphenyl)-piperazine in 15 ml of anhydrous dimethyl-
formamide was added. After 5 hours stirring and overnight
standing at the same temperature, the insoluble matter was
filtered off and the filtrate was poured into about 300 ml of
water and made alkaline by addition of lN sodium hydroxide. The
mixture was extracted with dichloromethane and the organic
layer was separated, dried on anhydrous sodium sulphate and ;
evaporated in vacuo. The crude product was purified by f lash
chromatography on silica gel eluting with chloroPorm:methanol
95:5. one molar equivalent of methanesulphonic acid was added to
9-~
-146-
the solution of the crude base in ethanolt Diethyl ether was
added until the salt crystallized. The salt was then filtered
off ar~d recrystallized from ethanol:diethyl ether 1:2 to give
1.15 g of the title compound, m.p. 160-162C.
~mpls 81
8-{~ ~c~tyl-3-[4-~2-met~o~yph0~yl)-1-piperAzl~yl]-
-p~opylc~rb~moyl}-3-~ethyl-4 oxo-2-phenyl-4~ be~zopyra~
A solution of 2.86 g of Intermediate LVII, 5.04 g of
1-(3-chloropropyl)-4-~2-methoxyphenyl)~pipera~ine and 2.58 g of
anhydrous potassium carbonate in 50 ml of dimethylformamide was
stirred at 90C for 7 hours. After cooling to ambient
temperature, the reaction mixture was poured into 500 ml of
water and extracted with dichloromethane. The organic layer was
washed with water, dried on anhydrous sodium sulphate and
evaporated to dryness in vacuo. The residue was purified by
column chromatography on silica gel eluting with ethyl
acetate:petroleum ether 7:3 to give 1.89 g of the title
compound melting at (55) 62-63C.
~am~le 82
8-{2-[4-t2-~etho~yphenyl)-1 ~iporazin~yl]-~thyl~ulpho~ylamino}-
-3-~thyl-~ oxo-2-phe~yl-~H-l-bo~o~yr~ ~ethan~sulpho~ate
1.05 ml of 2-chloroethanesulphonyl chloride was added dropwise
~o a solution of 5 g of 8-amino-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran and 1. 4 ml of triethylamine stirred at 0C. The
reaction mixture was stirred at ambient temperature for 2 days.
A~ter filtering off the precipitated solids, the solution was
evaporated to dryness in vacuo to give a crude residue
containing 8-(ethenylsulphonylamino)-3-methyl-4-oxo-2-phenyl-
-4H-1-benzopyran used without further purification. A mixture
of 7.54 g of this residue and 5.8 g of 1-(2-methoxyphenyl)-
-piperazine and 4.15 g of potassium carbonate in 100 ml of
dimethylformamide was stirred at ambient temperature for 4
hours, poured into 600 ml of water and extracted with ethyl
acetate. The organic layer was evaporated to dryness in vacuo
and the residue was purified by column chromatography on silica
" ~ . ,
2~o~ ~
-147-
gel eluting with petroleum ether.acetone 8:2. The collected
fractions were evaporated to dryness in vacuo and crystallized
from 70% ethanol to give 0.75 g of the title compound as a
base. This solid was dissolved in dichloromethane and one
equivalent of methanesulphonic acid was added to the solution.
The crude methanesulphonate, obtained by evaporation in vacuo,
was crystallized from acetone, yielding 0.6 g o~ the title
compound melting at 202-203C. ~:
xa~l~ 83
8-{3-~4-~2-m~t~oxyphellyl)-1 piperaginyl] propylthiocarba~moyl}
-3-~athyl-4 oxo-2 phenyl-4~ 1-benzopyra~ ~th~esulpho~ato
A stirred mixture of 0.8 g 8-formyl-3-methyl-4-oxo-
-2-phenyl-4H-1-benzopyran, 0.75 g of 1-(3-aminopropyl)-
-4-(2-methoxyphenyl)-piperazine and 0.14 g of sulphur in 5 ml . :~
of pyridine was re~luxed for 6 hours. After solvent evaporation
in vacuo, the residue was purified by flasih chromatography on
silica gel, eluting with chloroform. The obtained title ~:
compound (as the base) was dissolved in dichloromethane and one
equivalent of methanesulphonic acid was added. The title
compound was obtained by evaporation in vacuo and -
crystallization of the residue from acetonitrile. Yield 0.7 g,
m.p. 189-190C. .
2~mpl~ 84
~-{~ (2-Dle~ho~yphe~y~ pip~ri~i~yl~ ~utylsulpho~iyl}-3~
-mothyl 4-oxo-2-ph~yl-4~1-be~20pyra~ mQth ~isulpho~ate ~ % ~2
~he title compound was prepared according to Example 73, but
using Intermediate LX instead of Intermediate LIV. Its
purification was performed by column chromatography on silica
gel, eluting with ethyl acetate:petroleum ether 7:3. One molar
equivalent of methanesulphonic acid was added to a solution of
the ~rude base in dichloromethane. A~ter removal o~ the solvent
by evaporation in vacuo, the resulting salt was crystallized
~rom acetone to give the title compound, m.p. 212-214C.
., , ' ` , ' : ' ~' , ~ : ' ,
20~n~S~
-1~8-
~mpl~ 85
8-{3-[~-~2 me~oxyp~e~yl)~l piperazi~ylJ-propyl~rba~oyl}-
-3-hydro~y~thyl~4-o~o-2-phe~yl-4~ be~æopyran. 0.4 ethanol
A mixture o~ 3.6 g of Intermediate XCV and lo 65 g of
1-hydroxybenzotriazole in 35 ml of anhydrous dimethylformamide
was stirred at 0~5C for 15 minutes. A solution o~ ~.5 g of
dicyclohexylcarbodiimide in 35 ml of anhydrous dimethyl-
formamide was added dropwise. A~ter stirring for 1 hour at the
same temperature, a solution of 1-(3-aminopropyl)-4-
-(2-methoxyphenyl)-piperazine was added. After stirring for 2
hourc at the same temperature and standing overnight at ambient
temperature, the reaction mixture was evaporated to dryness in
vacuo. The residue was purified by ~lash chromatography eluting
with a mixture of dichloromethane:methanol 100:3, ~ollowed by
crystallization from ethanol. 2.5 g of the title compound was
obtained, m.p. 152-154C.
~x~ple 86
8-~3-t4-(2-methoxyphe~yl)-1 piper~i~yl~-propyl¢~rb~oyl}-
-~-o~o-2-phe~yl-~ b~zopyran ~e h~n~Yulphonate . ~ ~2
3.6 ml of diethyl cyanophosphonate were added dropwise at 0-5C
into a ~tirred solution of 4 g of 8-carboxy-4-oxo-2-phenyl-
-4H 1-benzopyran (prepared as described in Da Re et al., Ber.,
99, 1962, 1966) and 3.75 g of 1-(3-aminopropyl)-
-4-(2-methoxyphenyl)-piperazine in 35 ml of anhydrous dimethyl-
formamide. Immediately afterwards, 2.5 ml of triethylamine was
added dropwise at the same temperature. After 30 minutes
stirring at 0-5C and 1 hour at ambient temperature, the
reaction mixture was poured into 350 ml of 2.5% aqueous sodium
carbonate. The precipitate formed was stirred for 1 hour at
ambient temperature, collected by suction filtration and
crystallized from ethanol. The base of the title compound, thus
obtained, was dis601ved in dichloromethane and one equivalent
o~ methanesulphonic acid was added. After evaporation in vacuo,
a glassy solid was obtained, which was crushed and refluxed for
1 hour in acetone, yielding 5 g of the title compound melting
at 191-194C.
-149-
Exampl~ 87
8-~3~t~-(2~ hoxyphe~yl~ l-pipera~i~yl]-propylo~rb~oyl};
-2,3-~ihy~ro-4-o~o-4H-1-b~n~opyr~ metha~e~ulphonate
A solution of 0.84 ml of thionyl chloride in 17 ml of anhydrous
dichloromethane was added dropwise to a solution of 2.Q g of
8-carboxy 2, 3-dihydro-4-oxo-4H~1-benzopyran (prepared according
to Lichtenber~er et al., Bull.Chem.Soc.Fr., 275, 1963) and 1.75
ml of tri~thylamine in 17 ml of dichloromethane stirred at
ambient temperature. Stirring was continued for 1~ hour at the
same temperature; after this period the reaction mixture was
evaporated to dryness in vacuo to give crude s-chlorocarbon
-2, 3-dihydro-4-oxo-4H-1-benzopyran. This was used in the method
of Example lo instead of 8-chlorocarbonyl-3-methyl-4-oxo-
-2-phenyl-4H-1 benzopyran to prepare the base of the title
compound, which was puri~ied by column chromatography eluting
with ethyl acetate:methanol 85:15. The yield was 1.91 g o~ the
pure base. After dissolution in dichloromethane, acidification
with methanesulphonic acid, and evaporation to dryness in vacuo,
the resultant crude salt was crystallized from acetonitrile
giving 1.57 g of the title compound, melting at 175-177C.
E~ampl~ 88
~-{3-t~ (2-~thoxyphe~yl)-1-pip~ra%in~yl]-propyl~rb~oyl}
-~oxo~ b~zopyran ~ethane~ulpho~t~ . 1.25 H20
The title compound was prepared by the method described in
Example 87 but using Intermediate LXII instead o~
8-carboxy-2,3-dihydro-4-oxo-4H-1-benzopyran. The crude methan~-
sulphonate was rinsed with diethyl ether, filtered and
repeatedly crystallized from acetonitrile. M.p. 155-157C.
Bx~mple 89
8-{3-t~ ~2-ms~ho~phenyl)-l-pipera~inyl]-propyla2rb~moyl}-
-6-bro~o;3~ethyl-4-oxo~2-phenyl-4H-1-ben$opyra~
This compound was prepared according to Example 86 but using
8-carboxy-6-bromo-3-methyl-4-oxo-2-phenyl-4H-l-benzopyran
,.... . ,. ... , . . , .: ,. " . . . . .. . . . ....
2~90l~6
-150-
(prepared as described in EP 107~04~ i~stead o~ 8-carboxy-
-4-oxo-2-phenyl-4H-1-benzopyran. It was purified as a base by
flash chromatography on silica gel eluting with
dichloromethane:methanol lOQ:3, and crystallized from 95%
ethanol. M.p. (150) 1~4-159C.
B~m~l~ 9O
S~{3-[~-(2-nethoxyphenyl)-1-piperazinyl]-propylGarb~moyl}-
-6-m~t~oxy-3-m~thyl~4-o~o-2-phe~yl~4~ be~op~ra~
~eth~e~ulpho~te
1.01 ml of diethyl cyanophosphonate and 0.85 ml of triethylamine
were added to a solution of 1.7 g of 8-carboxy-6-methoxy-3-
-methyl-4-oxo-2-phenyl-4H-1 benzopyran (prepared as described in
JP 61-15880) and 1.51 y 1-(2-methoxyphenyl)-4-(3-aminopropyl)-
-piperazine in 20 ml of anhydrous dimethyl~ormamide stirred at
0C. After stirring for 1 hour at oC to ambient temperature,
the reaction mixture was poured into a mixture of 100 ml of
water and 10 ml of lN sodium hydroxide. The base of the title
compound precipitated; it was filtered off and washed with
water. After desiccation it was converted in the usual way to
the methanesulphonate, which was crystallized from
acetonitrile. Yield 1.7 g; m.p. 185~186C.
E~ample 91
~-~3~ (2-metho~yph~yl)-1-pipera~inyl]-propylc~Fbamoyl}-
6 hydro~y3-methylN4-0~0-2-ph~yl;4~ b~zopyran
mDth ne~ulphon~te
0.8 g of the compound pr~pared in Example 114 and 5.8 ml of lN
sodium hydroxide in 10 ml of methanol were stirred at ambient
temperature for 4 hours. After standing overnight, 15 ml oP lN
sodium hydroxide and 15 ml of methanol were added and the
mixture was stirred at ambient temperature for 1 hour. Methanol
was evaporated off in vacuo a~d water was added to the residue.
The æuspension was filtered by suction to give 0.48 g of the
base of the title compound. This was converted by the usual ~- -
procedure into its methanesulphonate salt, recrystallized from
acetonitrile. M.p. 200-202C.
: ''
, ~ , , , . , :: : : .. ,. ~ .":, . . .
2~9~
. ~
-151-
~ 2 :-
8-~3 C4-~2-~ths~yph~yl)-1-pip~razi~yl3-pro~yl~rba~oyl}-
-3,6~ th~1-4-oxo-2 phe~yl~ benzop~r~ mothane~ulphonste
The title compound was prepared according to Example 90 but
using 8-carboxy-3,6-dimethyl-4-oxo-2-phenyl-4H-1-benzopyran
(prepared as described in Da Re et al., Arch.Pharm., 296, 714, : ~ .
1963) instead of 8-carboxy-6~methoxy-3-methyl-4-oxo-2-phenyl-
-4H-1-benæopyran. The crude methanesulphonate was crystallized
from acetonitrile and melted at 196 197C.
~ample ~3
8-~3-[4-(2-DIet~o:~yphenyl~ piperazi~yl]-propyla2rbamoyl}- -,.
-3 ~et~ 6~itro-~-o~o-2-phe~yl-~ benzopyran :~
.
~his product was obtained operating as described in Example 12
but using Intermediate LXVIII instead o~ 8-chlorocarbonyl-
-3 methyl-4-oxo-2-phenyl-4H-l-benzopyran and 1,1,2-trichloro-
ethane instead of chloroform. After the usual workup, the crude
product was purified by column chromatography eluting with
dichloromethane:methanol 98:2. Evaporation in vacuo to dryness
of the collected ~ractions and crystallization from ethanol
yielded the title compound, m.p> 159.5-161C.
~a~pl~ ~4
B-~3-r4-~2-~tho~yphenyl)-1 piper~zinyl~ propylc rba~oyl}-
-6-~i~o-3-~ethyl~oxo-2 ph~yl~ l b~xopyr~
A mixture o~ 33 g of the compound prepared in Example 93, 109 ml
o~ lN hydrochloric acid, 105 ml of water and 8.78 g of
Raney-Nickel in 950 ml of ethanol was hydrogenated in a Parr
apparatus, hydrogen pressure 2 atmospheres, stirring at 40C for
12 hours. After this period, the catalyst was filtered o~ and
washed with 80% ethanol. The mother liquors were evaporated in
vacuo to a volume o~ 80 ml and filtered. The crude product was
washed with water, and suspended in water; 37% hydrochloric acid :
was added until the pH was 1. The insoluble matter was filtered
of~ by ~uction and the filtrate was alkalinized by adding 35% ~ ::
: .
'. , ," ."~.'. ' .. ' '~ ',' ' ' '. ' , .,' . . ';. "', "" . "'
! " ', ' , ', ,' ' ~ ! , ' ', '
~ 2~9~
-152-
sodium hydroxide. The title compound precipitated. It was
recovered by filtration and washed with water. Des~iccation
yielded 26 g melting at (108) 215 217.5C for use in Example 95
without further purification. For characterization, 4.7 g of
the product were crystallized first from ethanol and then from
85% ethanol giving 3 g of the title compound, m.p. 218-219C.
~x~mpl~ 95
8-{3-[~-t2-m~thoxyph~2yl)-l-pip~razinyl]-propylG~rbamoyl}-
-6-~o~t~mido-3-m0thyl-4-oxo 2-phanyl~4H~l~be~zopyra~
The title compound was obtained according to the procedure
described in Example 36, but using the compound prepared in
Example 94 instead of that prepared in Example 33. The reaction
mixture was diluted with water and filtered by suction, washing
the solid with watsr. After desiccation at 80C, this solid was
purified by column chromatography on silica gel eluting with
chloroform:methanol 95:5. Evaporation in vacuo of the collected
fractions and crystallization from 95% ethanol of the residue
gave the title compound, m.p. (150) 218-220C.
~a~le 96
~-{3-[~ thoxyphenyl)-1 piperazin~yl]-propylc~rb~moyl}
-6 e hyl~mi~o-3-~t~yl-~ oxo-2-phenyl~ be~zo~yr~
A mixture of 8.42 g of the compound of Example 94, 0.45 ml of
acetaldehyde, 0.59 g of 85% sodium cyano~orohydride and 3.3 ml
of 4.85N ethanolic hydrogen chloride in 73 ml o~ methanol was
stirred at ambient temperature for 5 days. After this period,
the reaction mixture was poured into cold 1.5N sodium hydroxide;
the sucpension was diluted with water and filtered by suction.
After desiccation, the residue wa~ purified by column
chromatography on silica gel eluting with chloroform~methanol
100:3. Evaporation in vacuo of the collected fractions yielded
6 g of the compound of Example 94 and 2.67 g of the title
compound, which melted at 198-201C after recrystallization
from ethanol.
., ~
~ 2~
-153-
pl~ 97 :~
8-it3-[~-~2-metho~yph~yl) 1-pip2razi~yl]-propylo~rb~oyl~ :
-6-~i~ethyl~0-3-~thyl-4-o~o-2-phenyl~ be~opyr~n
The title compound was prepared according to Example 35 but
using the compound prepared in ~xample 94 instead of that
prepared in Example 33, reacting 10 molar equivalents of 40%
formaldehyde instead of 7 molar equivalents and 3 moles of ::
sodium cyanoborohydride instead of 2 moles and stirring at
ambient temperature for 18 hours instead of 4%j hours. After the
usual workup and purification by column chromatography on silica : .
gel eluting with chloroform:methanol 97:3, evaporation in vacuo :-.
of the collected fractions and crystallization of the residue
from ethanol yielded the title compound which melted at
183-186C.
Bxa~le g8 :
8-{3-[4-52-~t~o~yphenyl)-1-pip~razinyl]-propylcarb~moyl}-
-~-m~ oxy-3-~th~1-4 oxo-2-phonyl-4~-1-benzopyr~
The title compound wai prepared according to the procedure
descxibed in Example 87 but using Inteirmediate LXIX instead of
8-carboxy-2,3-dihydro-4-oxo-4H-1-benzopyran. After the usual
workup, the solid residue was purified by column chromatography
on silica gel eluting with ethyl acetate:methanol 8:2. The
collected fractions were evaporated to dryness in vacuo and thei
residue was crystallized from acetonitrile to give the title
compound, melting at 151-152C.
E~ample 99
8-{3~ 52 iethoxyphe~yl) 1-pipor~zinyl]-propylGarbamoyl}-
-3-~2thyl-~-oxo-2-~4-tri~luoromet~ylphe~yl)-4~ benzopy~a~
~etha~esulphianate se~uihydrate
The title compound was prepared following the procedure
described in Example 90, but starting from IntermediatP LXXII ..
instead of 8-carboxy-6-methoxy-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran. The methanesulphonate melted at (85) 90-120C ~:(dec), after crystallization from acetonitrile.
,, , . . ., . : ,. ~ :, . .
, . , , . .. : ., ,....... , .. .: . .
... . , , , . -
: . ~,; . ' . .
.
:
209~1~6
f .
-154-
~a~ple 100
8-{3-[4-(2-~ethox~ph~yl7~ iperazi~yl~propyla2rbamoyl~-
-2~(4-benæoylph~yl~-3-~ethyl-~-o~o-4~;1-ben~opyr~
~ethanoi~lp~o~te hemihydr~to
The title compound wa~ prepared following the procedure
described in Example 90, but starting from Intermediate LXXIV
instead of 8-carboxy-6-methoxy-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran. The crude methanesulphonate was crystallized from
acetonitrile. M.p. 20~ 210C.
~_a~le 101
8-{3~14 (2~ tho~phç~l~y~ pip~ræzillyl]-propylc~rb ~oyl}- :
-3-methyl-~-oxo-2-~4-pho~loxyphonyl)-q~ benzopyrall : -
~eth~e~ulphon~to . 0.25 ~2
The title compound was prepared following the procedure
described in Example 90, but starting from Intermediate LXXVII
instead of 8-carboxy-6-methoxy-3-methyl-4-oxo-2-phenyl-4H-1- :
-benzopyran. The crude methanesulphonate was crystallized from
acetonitrile. M.p. 200-202C.
ampl~ 102
8-{3-[4~(2-m~thoxyphe~yl~ pipera%i~yl] propylc~rba~oyl}-
-2,3-di~sthyl-~-o~o~4~ e~o~yr n
~ ' ' ' - . '
This compound was prepared according to Example 86, but using
8-carboxy-2,3-dimethyl-4-oxo-4H-1-benzopyran (prepared according
to Da Re, Farmaco Ed. Sci., 11, 678, 1956) instead of
8-carboxy-4-oxo-2-phenyl-4H-1-benzopyran and carrying out the ~: .
reaction ak ambient temperature for 5 hours. It was purified by :
flash chromatography on silica gel eluting with chloroform: ~
methanol 98:2, and crystallized from acetone. M.p. 155-158.5C. ~ .
:,..:. ~ '.,.
, . . . : , , , ~:, . ..
-155- 2 ~
~ample 103
8-{3-t~ (2-~tho~yp~e~yl)-1-pipor ~i~yl~-~ropyloarb~moyl}-
2-(t-butyl)-3 ~ethyl-4-o~o-4~ b~zopyra~ ~ihy~ro~hlori~e
~ihy~r~te
~his compound was prepared according to Example 86, but using
Intermediate ~XXVIII instead of 8~carbo~y-4-oxo-2-phenyl-4H~
~enzopyran. The base was purified by ~lash chromatography on
silica gel eluting with chloroform:methanol 49:1 and converted
into the dihydrochloride in methanol:diethyl ether. It melted at
226-229.5C after recrystallization from methanol:diethyl ether
1:1. '
~xampl~ ~04
8-{3-t~ 2 ~etho~yphe~yl)-l-piperazinyl]-prop~lcarbamoyl~-
-2-oycloh0xyl-3-~ethyl-4-o~o-4~ be~æopyr~ . .
This compound was prepared according to Example 86, but using
Intermediate LXXIX instead of 8-carboxy-4~oxo-2-phenyl-4H~
-benzopyran, and ~arrying out the reaction for 5 hours at
ambient temperature. It was purified by flash chromatography on
silica gel, eluting with chloroform:methanol 49:1, and
crystallized from acetonitrile (m.p. 155-157C).
B~a~Plo 105
~ {3- L~- ~2-metho~yphs~yl)-1-piperazi~yl]-~ropylo~rb~oyl}~
-2-~2Q~uryl)-3 ~ethyl-4-o~o-~ ~sopyran
This compound was prepared according to Example 86, but using
Intermediate LXXXI instead of 8-carboxy-4-oxo-2-phenyl-4H-1-
-benzopyran, and completing the reaction at ambient temperature :
for 5 hours. After quenching, the title compound was isolated by
extraction with chloroform and purified by flash chromatography
on silica gel eluting with chloroform:methanol ~9:1, followed
by crystallization from acetonitrile. M.p. lS1-153C.
,. ~ :;
:' ' ;, ' ' ' ' '
, . 2~9~
-156-
E~mpl~ 106
9-{3-[4-~2-m~t~o~yphe~yl)~l-piperazi~yl] propylcarb~moyl}
-3-~eth~1-4-o~o-2-thie~yl-4~ be~zopyra~
This compound was obtained according to Example 86, but using
Intermediate LXXXIII instead of 8-carboxy-4-oxo-2-phenyl-4H-l-
-benzopyran. It was purified by stirring in water (completely
to remove dimethylformamide~, followed by column chromatography
on silica gel eluting with chloroform:methanol 49:1, and
crystallized from acetonitrile. M.p. 174-175C.
~3mple 107
8 {3-[~-~2-methox~phe~yl~ pip~ra~inyl]-propylcarbamoyl}-
-~~oxo-2-pha~yl-4~ be~othiop~ran
.' ,: '
A mixture of 2.8 g o~ Intermediate LXXXIV and 3.4 g of 1,1'
-carbonyldiimidazole in 60 ml of anhydrous dimethylformamide was
stirred under nitrogen at ambient temperature for 1% hours.
2.7 g of 1-(3-aminopropyl)-4-(2-methoxyphenyl)-piperazine were
then added. After further 2 hours stirring at ambient
temperature, the reaction mixture was poured into 300 ml of
water and extracted with chloroform. The organic layer was
dried on anhydrous sodium sulphate and evaporated in vacuo. The
residue was puri~ied by flash chromatography on silica gel
eluting with chloroform:methanol 49:1, washed with water and
crystallized from acetonitrile, yielding 2 g of the title
com~ound melting at 144-146C.
,~
~ample 108
~ 8-{3-~4-~2-~t~oxyphe~yl)-1 piper~inyl]-propylGarbamoyl}- -
-3-~et~yl-4-oæo~2-(2-~tyryl)-4~ be~opyra~
This compound was prepared according to Example 86, but using
Intermediate LXXXVI instead of 8-carboxy-4-oxo-2-phenyl-4H~
-benzopyran. It was purified by crystallization from
acetonitrile. M.p. 191-194C.
901~
-157-
E:xa~pl~ 109
8-~3-[~-~2-~tho~phe~yl)-1 pi~razi~yl~-pro~ylc~rb~moyl~-
~3 ~thyl-2-~-~ethylphenyl)-~;oxo-4~ o~opyr~
This compound was prepared according to Example 86, but using
Intermediate LXXXVII instead of 8-carboxy-4-oxo-2-phenyl-4H-1-
-benzopyxan and completing the reaction at ambient temperature
for 4 hours. After ~uenching, the title compound was isolated by
extraction with ethyl acetate, drying on anhydrous sodium
sulphate and evaporation in vacuo, followed by rinsing with
diethyl ether and crystallization from acetonitrile. M.p.
161-163C.
~ample 110
~-{3-~ (2-~atho~yphenyl~ piper~zi~yl]-propylcarbamoyl}-
2~ thoxyp~e~yl)-3-methyl-4 o~o~ b~zopyran : .
This compound was prepared according to Example 86, but using
8 carboxy-2-(4-methoxyphenyl3-3-methyl-4-oxo-4H-l-benzopyran
(prepared as described in EP 108986) instead of 8-carboxy 4-oxo-
-2-phenyl-~H-l-benzopyran, and completing the reaction at
ambient temperature for 3~ hours. It was purified by flash
chromatography on silica gel eluting with chloro~orm:methanol :
49:1, followed by crystallization from acetonitrile. M.p.
158-161C. -
Bxa~
8-{3~t~-(2-~nethoxyph~nyl)~ pip~r zi~yl]-propylc~rb~unoyl}- ,,
-2-~4-~luoroph~nyl)-3~m~thyl-~-o~o-4~ be~opyran
This compound was prepared according to Example 86, but using
Intermediate LXXXIX instead of 8-carboxy-4-oxo-2-phenyl-4H-1-
-benzopyran. It was purified by flash chromatography on silica
gel eluting with chloroform:methanol 100:2 to 100:6 and
crystallized from 95~ ethanol. M.p. 166-168C.
: : .
:, ' ~ : .
,,,,, . ., " j .
20~ 5~
-15~-
E~a~ple 11~
8-~3-t~-~2-~at~oxxphe~yl~ piper ~i~yl~ propylaarbamoyl}-
~6-metha~sul~ho~yla~i~o-3-~t~yl-4-o~o 2-ph~yl-4~ benzopyra~
hy~rooh~ori~o
0.032 ml o~ methanesulphonyl chloride in 1 ml of dimethyl-
formamide was added dropwise over a period of 10 minutes to a
solution of 0.21 g of the compound of Example 94 and 0.062 ml of
triethylamine in 4 ml of dimethylformamide, stirr2d at -20C. :: ;
Stirring was continued at the same temperature for 3~ hours.
After this period, the reaction mixture was poured into water : ::
and the suspension was filtered by suction, yielding 0.1 g of .
the title compound which was recrystallized from 80% ethanol.
M.p. 272 275C.
~a~le 113 :
8-{3-C~-t2-~etho~phe~yl)-1-pip~ra~i~yl~-~propylcarbamoyl}- '',
-3-~thyl-2 (4-~itroph~yl)-4-o~o-4~ be~opyra~ :
The title compound was prepared ~ollowing the procedure :~ :~
described in Example 90 but startirlg from Intermediate XCVIII
instead of 8-carboxy-6-methoxy-3-methyl-4-oxo-2-phenyl-4H-l-
benzopyran. After stirring for 1 hour at ambient temperature,
the reaction mixture was poured into cold 2~ sodium carbonate
solution and the precipitated solid was collected by suction
filtration. After desiccation and crystallization from ethanol,
the title compound melted at (60) 185-187~C.
. ~
Ex~mple_114
8-~3-t~-~2-met~oxypl~el~yl)-1-piperasci:nyl]-propylcarb~moyl~
-6-~iothoxyphos~honyloxy-3-~e~hyl-4-oxo-2-phenyl~ b~nzopyra~ .:
The title compound was prepared following the procedure
described in Example 90, but starting from Intermediate LXIII
instead of 8-carboxy-6-methoxy-3-methyl-4-oxo-2-phenyl-4H-1-
-benzopyran and using 2 equivalents of diethyl cyanophosphate
instead o~ 1.1 equivalents. Filtration from water yielded the
title compound melting at 48-50C. The title compound may be
regarded as a prodxug o~ 8-{3-[4-(2-methoxyphenyl)-1-
:.
-159- :
-piperazinyl~-propylcarbamoyl}-6-hydroxy-3-methyl-4-oxo-2-
-phenyl-4H-l-benzopyran
8-~3-t4-(2-~ethox~ph~nyl)-1-pi~r~i~yl] ~ropylaarbamoyl}- .
-3-~thyl ~-oxo-2 tri~luoro~ethyl~ be~zopyr~n
th~~ ulphoaults
Operating as described in ~xample 90, but starting ~rom
Intermediate C instead of 8-carboxy-6-methoxy-3-methyl-4-oxo
-2-phenyl-4H-l-benzopyran, the crude base of the title compound
was obtained. After the usu~l workup by extraction with ethyl
acetate~ the residue was purified by column chromatography on `~
silica gel eluting with ethyl acetate:methanol 9:1. It was
converted into its methanesulphonate by the usual procedure and
recrystallized from ethyl acetate. M.p. 145-148C.
~a~ple 116
8~ ~ethy~-3-[4 (2-~ethoxyphe~yl)-l~piperazi~yl]
-propyle~rba~oyl~-3-m~t~yl ~-oxo-2-ph~3~yl-4~ 1-be~zopyra~
h~roohlori~e
This compound was prepared according to Example 12, but using
Intermediate CI instead of 3-[4-(2-methoxyphenyl~-
-1-piperazinyl]-propylamine. The crude base was purified by
flash chromatography on silica gel, eluting with chloroPorm:5N
methanolic ammonia 100:1, and transformed into the hydrochloride
in the usual way. M.p. 195-198C, after crystallization from
acetone.
Bx~mple 117
7-{3~ (2-~tho~yphenyl)-1-piper~zi~yl]-propyl~arb~moyl~-
-~-be~oyl-3-ethyl-b~sotb]~ur~
This compound was prepared according to Example 86 but using
Intermediate CIII instead of 8-carboxy-4-oxo-2-phenyl-
-4H-l~benzopyran-8-carboxylic acid, and completing the reaction ,
at ambient temperature ~or 4 hours. It was purified by
crystallization from ethanol. M.p. 165-166C. ~ :
. .
. , , , . . ~
..
. " ' ! ~ ,
,',,' , ', ' ' , ;., ~ .
-160-
E~m~le 118
8-~3-t4-~ ~etho~ph~yl) 1-piperazinyl]-propyloarb~oyl~-
2 ~4-biphenylyl)~3-~thyl-4-oxo~4~ 1-be~opyra~.
The title compound was prepared according to Example 86 but
using Intermediate CVI instead of 8-carboxy-4-oxo-2-phenyl-
-4H-1-benzopyran. The reaction lasted 20 hours at room
temperature. The base was purified by crystallization from
ethanol (m.p. 164-166C).
~:~c~pl~ 119
8-{ -t4 (2-metho~yphenyl)-1-pipor~zi~yl~-propyla~rba~oyl}-
-3-methyl-4-o~o-2-~3-pyridyl)-4~ 1 be~zopyra~.
A ~ixtur~ of 6.2 g of methyl 3-propionyl-salicylate and 5O8 g of
nicotinoyl chloride hydrochloride in 18 ml of anhydrous pyridine
was stirred and heated at 100C for 2 hours under nitrogen. 16 -~
ml of triethylamine was then added and heating was continued for
1 hour at the same temperature. The reaction mixture was cooled
to room temperature and poured into 600 ml of water. The
precipitate was collected by suction and washed with water,
yielding 5.4 g of methyl 2~hydroxy-3-(2-nicotinoylpropionyl)-
-benzoate, which wa~ used without purification in the next step.
3.4 g of the compound thus prepared was heated at 100C for 1
hours after dis~olution in a mixture containing 15 ml of acetic
acid and 1 ml of 37% hydrochloric acid. After cooling to room
temperature, the mixture was poured into lSo ml of water and
extracted with ethyl acetate. The organic phase was washed with
5% aqueous sodium hydrogen carbonate followed by water, dried on
anhydrous sodium sulphate and evaporated in vacuo, yielding 1.3
g of crude 8-methoxycarbonyl-3-methyl-4 oxo-2-(3-pyridyl)- i
-4H-1-benzopyran.
1 g of this ester was dissolved in 9 ml of methanol and 15 ml of
1,4-dioxan~ a~d slowly added with 1.7 ml of lON sodium
hydroxide, maintaining the temperature between 20 and 25C.
After 1 hour at 50C, the reaction mixture was poured into 150
ml of water and extracted with ethyl acetate. The aqueous layer
was acidified with lN hydrochloric acid. The precipitate was
209~
~;
-161
collected by suction, yielding 0.6 g of 8-carboxy-3-methyl-
4-oxo-2-(3-pyridyl)-4H-l-benzopyran, which was used without
purification in the next step.
The title compound was prepared according to Example 86 but
using the acid thus prepared instead of 8-carboxy-4-oxo-
-2-phenyl-4H-l-benzopyran and carrying out the reaction for 2
hollrs at room temperature. The base was puri~ied by ~lash
chxomatography on silica gel eluting with chloroform:methanol
98:2, followed by crystallization from acetone. Yield: 0.15 g,
m.p. 134.5-137C~
E~ample 120
8-~3-~4-~2-~eto~yphe~yl)-l-piper~zi~yl]-propyl~arb3~oyl}-
-3-~ethyl-4-o$o-2-phenyl 4~ be~zopyran.
1 g of compound of Example 59 and 0.32 g of
4-dimethylaminopyridine were dissolved in 10 ml of
dichloromethane. 0.15 ml of acetyl chloride, was slowly added,
maintaining the temperature between 8 and 10C. After 2 hours at
ambient temperature, the reaction mixture was poured into 70 ml
of water and extracted with dichloromethane. The organic layer
was washed with 5~ aqueous sodium hydrogen caxbonate followed by
water, dried on anhydrous sodium sulphate and evaporated to
dryness in vacuo. The crude base was purified by fla~h
chromatography on silica gel eluting with ethyl acetate: methanol
9;1, followed by crystallization from ethanol. Yield: 0.7~ g o~
the title compoundl m.p. 120-123C.
~mpl~ 121
8-{3-[4-~2-~ethyl~o¢ar~o~yloxyphenyl~ piper~æinyl]-
-propylcaxba~oyl}-3-~at~yl-~-oxo-2-p~enyl~ bs~zopyr~n~
3 g of compound of Example 5~ and 1.8 ml of methyl isocyanate
were dissolved in 30 ml o~ dry dimethylformamide and stirred at
room temperature for 24 hours. The mixture was diluted with
water, stirred for 2 hours, and then filtered under suction. The
crude base was purified by flash chromatogxaphy on silica gel
eluting with chloroform:5N methanolic ammonia 100:3. The title
compound melted at 132-135C after crystallization ~rom ethanol.
, . :. , .................. ... , ,:
.
,: , . .. , . , :~
2~9~1~6
-162-
~pl~ 1~2
8-{3-14 (2-m~thoxypho~yl)~ iper~zi~yl~-propylc~rbamoyl~-
-6-~eto~y-3-~ethyl-~3xo-2-ph~yl-4~-l be~zopyr~
0.17 ml of acetyl chloride was added dropwise over a period of 5
minutes, under stirring at 0C, to a solution of 1 g of the . :-
compound of Example 91 and 0.32 ml of triethylamine in 36 ml of
chloroform. After 2 hours stirring at the same temperature, the
reaction mixture was diluted with dichloromethane and water. The
organic layer was separated, washed with water, dried on
anhydrous sodium sulphate and evaporated to dryness in vacuo.
Crystallization of the residue from acetonitrile yielded 0.8 g
of the title compound, melting at 148-149C. ::
Exa~Ple 123 ::
~R~ 8~3-t4-~2 ~ethoxyphenyl) l-piper~2inylJ-propylcarbamoyl}-
-2,3~dihydro~ yaro~y~ benzopyra~ ~etha~e ulphonate.
The title compound was prepared by the method described in
Example 17 but starting from compound of Example ~7 instead of
the compound of Example 1. The reaction mixture was diluted with
water and stirred for 15 min~tes; it was then extracted with
ethyl acetate. Usual work up gava a crude product, which was
purified by flash chromatography on silica gel eluting with
dichloromethane:methanol 95:5. Evaporation in vacuo of the :
collected fraction yielded the pure base, which was converted
into th~ mathanesulphonate and crystallized from acatonitrile.
M.p. 172-175C.
'~ ' '
~e~
8 {3-t4~g~-~et~oxyph~yl)-1-pipera~i~yl]-propylG~rbamoyl}-
-2~ ami~ophe~yl)-3-~ethyl-~-oxo-4~-1 bon20pyra~
2.22 g of the compound of Example 113 and 0~56 g of Raney-Nickel
in 96 ml of ethanol and 4.8 ml of acetic acid was hydrogenated
in a Parr apparatus (~ydrogen pressure = 1 atm) at room
temperature. A~ter shaking for 6 hours, the catalyst was
~iltered off. The filtrate was made alkaline with 3N sodium
.. .. " . '' ' ; ' . . ~ :'
. . . . . . . . . .
2~9~1~6
'. .
-163-
hydroxide and diluted with water. After standing for 2 days, the
precipitated title compound was collected by suction filtration,
washed with water, desiccated and recrystallized first from
ethyl acetate and then from ethanol. Yield: 1.5 g, m.p.
192-194C). .
xa~pl~ 125
8-{3-[~-(2-m~tho~yph0~yl~ piperazi~yl]-propyla~rb~oyl~-
2~(4 ~cetyl~mi~oph~yl)-3-~othyl-4~-1-be~zopyra~.
The title compound was prepared by the method described in
Example 36~ but usin~ the compound prepared n Example 124
instead of that prepared in Example 33. It was purified by :
crystallization from 95% ethanol. M.p. 207-209C.
.
~a~la 126 .,.
8~-{3-t4 (2-~ethoxyphenyl~ piperazinyll-propyl~arbamoyl}-
-2-(~-hydrosyphenyl)-3-~ethyl-4~ benæopyran ~ihydrochlori~e
~o~ohy~rst~.
The title compound was prepared by the method described in
Example 86 but using Intermediate CVII instead of
8-carboxy-4-oxo 2-phenyl-4H-l-benzopyran, carrying out the :-
reaction for 14 hours at ambient temperature and using :
hexamethylphosphoramide as co-~olvent. The isolated f '
diethylpho~phonyl ester of the title compound was hydrolized by
alkaline treatment followed by neutralization with dilute
hydrochloric acid. The crude base was extracted with chloroform.
The organic layer was washed with water and Pvaporated in vacuo.
The salt was prepared by addition of ethanolic hydrogen chloride
to an acetone solution of the base, evapoxating to dryness and
rinsing with acetone. M.p. 193 205C.
~x~m~le 127
8-{3 [q-(2-met~oxyphonyl)-1-piperazi~yl]-propyl~arbamoyl}-
-2-phe~yl-Ç,~ 4-trloxo~ benzothiopyran ~o~ohy~r~te.
0.32 ml of 30% hydrogen peroxide was added to 0.8 g of the
compound o~ Example 107 in 15 ml of acetic acid. The mixture wa~ .
~ .: ,, : , .: : ,,
.. . .
, ,, , ::
,
~30~ ~6
,
-164- :
stirr~d at 50C for 3 hours. 0.48 ml of 30% hydrogen peroxide
was added to the mixture in three equal portions at 2 hour
intervals. After cooling, the mixture was poured into 240 ml o~
water, neutralized (pH 7) with 5~ aqueous sodium hydrogen ~:
carbonate and extracted with chloro~orm. The organic layer was
washed with water, dried on anhydrous sodium sulphate and
evaporated in vacuo, yielding 0.18 g of khe title compound, ~ -
melting at 172-175C after crystallization from acetonitrile. -.: :
E~lo 128
7-{3-[4-~2-methosyph~yl)-1-piper~zi~yl~-propylaarb~moyl}~
-2-phe~yl--b~nzo[bl furanO
The title compound wa~ prepared by the method described in
Example 86 but using 7-carboxy-2-phenyl benzo[b]furan (prepared
as described in EP 0306226) instead of 8-carboxy-4-oxo-2-phenyl-
-4H-1-benzopyran, and carrying out the reaction for 1~ hours at
ambient temperature. It was purified by crystallization from
carbon tetrachloride. M.p. 132-136C. . :
E~ plG 129
8 {~-~et~yl-3-t4-(2-~etho~yphe~yl)-1 pipera~ l]-propyl
ul~amoyl~-3-~eth~ oxo-2-phenyl-4~1-b~n~opyra~
~eth2n~sulpho~at~.
.: . .
2.29 g of Intermediate VIII was added portionwise under stirring
at ~C to a solution of 1.5 g of Intermediate CI and 0.95 of
triethylamine in 30 ml of chloroform. After stirring for 2 hours
at ambient temperakure, the reaction mixture was diluted with
dichloromethane, water and 0.5 N sodium hydroxide. The organic
layer was washed with water, dried on anhydrous sodium sulphate
and ~vaporated to dryness in vacuo. The residue was purified by
flash chromatography on silica gel eluting with ethyl
acetate:methanol 96:4. Evaporation in vacuo of the collected
fractions yielded the pure title compound as a base. This was
converted by the usual method into its methanesulphonate salt,
which was crystallized from ethyl acetate yielding 2.75 g,
melting at 135-141C (dec.). .
~ ~ .
.; ; ' ', , ' ; ! ' . .,
209a~5~ ~
-165-
B~ 30
8-{~-~th~1 4-[4-~2-~etho~phe~yl)ol-piper~zi~yl] butyl-
~ul~a~oyl~-3-~oth~ oxo-2-phe~yl-4~1 ben~opyr~
~ethan~3ulpho~t~.
The title compound wa obtained by the method described in
Example 129 but using Intermediate CVIII instead o~ Intermediate
CI. It wa~ crystallized from acetonitrile. ~.p. 173-175C.
. .
2~9~
~. ~
-166-
PEARNACOLOGICA~ A
~etho~olo~y
Male Sprague Dawley rats ~Crl: CD' BR~ of 200-300 g b.w., female
Albino Swiss mice [Crl: CD-l (ICR~ BR] 20-30 g ~.w., and male
Beagle dogs (10-12 kg b.w.) were obtained from Charles River,
Italy and Nossan (Correzzana, Milan, Italy), re~pectively.
Animals were housed with free access to food and water and
maintained on forced light-dark cycle at 22-24C until the day
of experiments.
Acute toxicity
The acute toxicity of synthesized compounds was evaluated in
female albino Swiss mice after intraperitoneal and oral
administration. Four logarithmic sci~led doses of the compounds
were dissolved or suspended in 0.5% Methocel and administered in
a volume of 10 ml/Xg to groups of 4 mice/dose. Mortality was
recorded 7 days af~er the administration. Data analysis: the
LDso values and their fiducial limits were calculated according
to the method of Weil (Biometrics, 8, 249, 1952).
....
eceptor Binding studies:
H~prazosin bindinq~ receptors)
Rat cerebral cortices were homogenized in 50 volumes of original
wet weight of ice-cold 50 mM Tris-HCl buffer pH 7O4~ The
homogenates were centrifuged at 48,000 x g for 10 minutes, and
the pellets were resuspended in the same volume of ice-cold
bu~fer, centrifuged and resuspended two more times. The final
pellets obtained were resuspended in the same volume of buffer
and incubated according to the conditions reported in the table
below.
The foregoing receptor binding studies, as well as the
experimental data on dogs reported below, establish compounds of
the invention as 1-blockers, i.e. to be within a class of
substances widely used as antihypertensive and anti-BPH agents.
See, e.g., Frishman, W.H. et al., Medical Clinics of N. ~merica,
72, 427, 1988 and references cited therein.
2~
-167-
- r3Hl8-OH-DPAT bindinq (5HTlA__eceptors~
Rat hippocampi were homogenized in 50 volumes of original wet
weight of ice-cold 50 mM Tris-HC1 buf~er pH 7.4. The
homogenates were centrifuged at 48,000 x g for lO minutes, and
the pellets were resuspended in the same volume of ice-cold
buffer, incub~ted ~or 10 minutes at 37C, centrifuged and
resuspended two more times. The final pellets obtained were
resuspended in the same volume of buffer and incubated
according to the conditions reported in the table below.
These receptor binding studies serve to establish compounds of
the present invention as ligands for the 5HT1A receptor. As
previously reported, compounds that are 5HT1~ ligands exert
anxiolytic and antidepressant effects in animals and humans . .
(Hamon, M. et al., Ann. N.Y. Acad. Sci., 600, 114, 1990; Traber
J. et al., T.I.P.S., 8, 437, 1987) :
Receptor bindina studies
~1 5-HTlA
adrenergic serotonergic
RECEPTOR/LIGAND [3~prazosin r 3H]8-OH-DPAT
Conditions
[nM] ligand 0.35 1.0
preparation (c.m.p) 1 ml 1 ml
10 mg/ml 10 mg/ml
incubation buffer* Tris HCl Tris HCl
50 mM 50 mM
pH 7.4 pH 7.4
non-specific binding prazosin 5-HT
2 ~M 10 ~M
incubation 25C . 25C
30 min 30 min
.. ... ... __ ,
c.m.p. = crude membranes preparation;
* iii= containing ascorbic acid 1% and pargyline 10 ~M
The incubations were terminated after the appropriate time (see
table) by rapid filtration through Whatman GF/B filters using a
Brandel cell harvsster. The filters were washed twice with 15 ml
. .
~::, , ;; , ' '
:, , ~
,: . . , : :
,
2090~6
-16~-
of ice-cold buffer (see table). The radioactivity retained on
the fil$ers was determined by liquid scintillation counting.
Nonspecific binding (which amounted generally to 10-30~) was
evaluated by adding high concentrations of the specific
displacers (see table). All the compounds were initially tested
at lx10-6 M concentration, and in the presence of significant
displacing acti~ity, a complete competition curve was generated
(down to a concentration of 10-11 M~. All the samples were run
in triplicate.
The competition curves were always analyzed (to evaluate the
IC50 values) by non linear ~urve fitting of the logistic
equation according to the method reported by De Lean et al. (~m.
J. Physiol~, 235, E97, 1978), utilizing the ALLFIT program
~publically available from the National Institutes of Health
(N.I.H.) Bethesda, Maryland, USA] written for the IBM PC.
_~-induced contractions of rat_bladder strips:
The whole bladder of the rat was removed and immediately placed
in Krebs solution warmed at 37C. Strips of detrusor muscle ~-
(20-30 mm long, 1-2 mm wide), were cut from the dome of the
bladder. Each strip was placed in a 10 ml organ bath and
con~ected, under a constant load of 1 g, to an isometric strain -
gauge (DY-1 Basile, Comerio, Varese, Italy). Contractions were
recorded by means of a Basile 7070 polygraph. After a 60 minutes
equilibration period the strips were exposed to 80 mM KCl (final
concentration). This produced a rapid phasic contraction
followed by a slow ensuing and sustained tonic component. When
the tonic contraction was stable, the strips were washed and 30
minutes later a new contraction was induced. After having
recorded two or more reproducible responses, one concentration ~`
o~ the tested drugs was added to the bath and 30 minutes later a
new contraction was induced. The experimental groups consisted
of at lea t two preparations taken from different animals for
each concentration of drug tested. The ICso values of inhibition
of agonist-induced contractions were evaluated by linear
regression a~alysis. - ~
-. :. ...
E~ects on Urethral Contractions and Blood Pressure in Dogs
The experiments were performed according to the method of
~090~
-169-
Imagawa et al. (J. Pharmacol. Methods, 22, 103-111, 1989), with
substantial modifications, as follows: Adult male beagle dogs,
weighing 8-10 Kg, were anaesthetized with pentobarbital sodium
(30 mg/Kg i.v. and 2 mg/Kg/h i.v.), intubated and spontaneously
ventilated with air room. In order to monitor systemic blood
pressure (BP), a PE catheter was introduced into the aortic arch
through the right common carotid artery.
A collateral of the left femoral vein was cannulated for
infusion of anaesthetic, and the right femoral vein was
cannulated for administration of drugs. For intrarterial (i.a.)
injection o~ noradrenaline ~NA), a PE catheter was introduced
into the lower portion of abdominal aorta via the right
external iliac artery. Through such procedure, NA was
selectively distributed to the lower urinary tract. Via a
midline laparotomy, the urinary bladder and proximal urethra
were exposed. In order to prevent the filling of the bladder,
the two ureters were cannulated and the urine led outside. In
order to record the prostatic urethral pressure, a Mikro-tip
catheter ~6 F) was introduced into the bladder via the external
urethral meatus, and withdrawn until the pressure transducer
was positioned in the prostatic urethra. A ligature was secured
between the neck of the bladder and urethra to isolate the
respon~e of the latter and avoid any interaction with the
bladder. Another ligature was put around the Mikro-tip catheter
at the external urethral meatus, to secure the catheter itself.
i~fter a stabilizing period followiny surgical procedure (30
min), in which arterial and prostatic urethral pressure were
continuously monitored as basal values, i.a. administration of
NA was made at intervals of 10 min. The dose of NA chosen was
such to produce an increase of at least 100% in urethral
pressure. ~he test compounds were administered i.v. in a
cumulative manner with intervals of 15-20 min between
administrations. I.a. injections of NA were repeated
approximately 5 min. after every dosing of test compound.
Dose response curves were constructed computing the percentage
inhibition to the increase in urethral pressure (NA induced),
and the percentage drop in blood pressure produced by the test
compound. ED25 for diastolic blood pressure (dose inducing a
25% decrease) and IDso (dose inducing a 50% inhibition of
~, :
, ~ ,
.. , ~ , . , :
2~0156
"
-170- ~.
NA-induced increase in urethral pressure) were computed by :
means of linear regression analysis. - :
Resul~s
Compounds as prepared in the Examples were tested according to
the methods reported above, and the results are given in the ~-~
Tables below, together with comparative results for the
reference standards used. Compounds having receptor affinity
(IC50 values) lower than about 500 nM are generally considered
to have good a~finity. Compounds with ICso values less than 100
nM are generally preferred.
.,: ;, ~. .
!
., , . , . . -. . , , - -.. , ,. . . . .. , ~
2~9~
-17 1- . - .
TABL~3 I .
~ _ .
Compound Receptor Bindin~ Acute Toxicity K+ S$imulation ¦
in Mice of_ Rat Bladder ¦
Example IC50 (nM) ~D50 (mg/kg)ICso (~M) l
No . ~1 5-HT1a i . p . p . o . Contractions I . .
Phasic Tonic ¦
. _ . _ _ I :
4 550 55 3461732
19 621>3000 I ,
6 107 1000 233 l :.
7 ~ 155 3841915
8~ 6~ 111 >5001915
11 29 5 247 2g7 2.9 3.0 I .
1~ 68 229 >1000 >3000 10.0 10.0 I .
1~ 61 6 140 559 I . '
8 131 306 496
16 2201050 345 778 1.6 2.2
17 59 910 299 608 8.8 3.8 I . .
18 270>1000 457 3000 I .
19 165 340 >looo >3000
169 85 297 - 594 2.7 2.5
21 17 33 297 566
22 117 48 >500 >2000
24 690 212 >1000 >3000 I . ~.
26 27~>1000 >500 >2000
27 23 124 399 >3000 1.0 0.8 I
28 120 96 Z03 1127 I ::
: 29 86 45 730 >3000 lO. o 10.0 I
32 119 46 301 >2000 10.0 >10 I . .
33 17 3~ 399 >2000 10.0 10.0 I .
34 30 34 >500 2.8 3.6
8 329 959
36 18 54 >500 >2000
37 32 77 ~500 >2000
3~ 20 344 >500 >2000
39 90 170 >1~00 >3000
83 140 349 0.5 0.9
~1 43 53 399 2241 0.7 1.4
42 111 39 459 2163 10.0 10.0
43 166~lono
4~ 685 201 84 399 0.6 0.5
15 106 329 1727
46 86 23 .
47 36 23 330 1047 2.7 5.2
48 104 5 500 1914
49 152 9 432 >2000
39 300 211 299
51 22 84 >500 >2000
52 89 2 127 224
53 7 41 127 1020
54 35 143 106 421.
291~1000 128 >2000
56 25 748 >500
_ _ , .
[N.B. Table 1 is continued on the next page]
.. .. .. . ... . . .
- .
.
.,, , .;
,,:, ..
209~5~
-172-
T~B~ ~co~ti~u~
Com~ound R~ce~tor Bindinq Acute Toxicity Stimulation
in Mice of Rat Bladder I
Example IC50 (nM) LD50 (mg/ky) ICso (~M)
No . ~1 5-HT1a i . p . p . o . Contractions l
Phasic Tonic ¦
_ . . _ _ _ I
57 69 >1000 500 >200~ l
58 >1000 126 258 508 l
59 5 9 >500 278
203 329 l
61 36 11 315 592 I
62 252 278 128 3~4 l
63 19~ 99 309 479
64 ~0 11 l
113 26 I
66 57 56 601
67 4 4 508 1~68
68 2~. 13
~9 16 87 34~ l
53 85 >500 1868 l
71 108 69 l
72 145 111 47g >2000 I
73 6 89 202 462 I
7~ 178 4~2
~47 15
78 18 116 l
79 77 141 >500 >2000 I
2~ 162
81 13 81 l
: 83 30 5 I
84 5 2 ~
: 85 16 14
86 ~8 20 I
87 119 107 237 >500 I
88 216 11
93 47 38
94 39 66
98 103 12
99 44 5
102 56 18
:104 g 15
107 37 16 .
ll0 26 15 .
Flavoxate1000>>1000 385 808 13 13
2~01~6
, ~
-173-
T~Bh8 II
Effects on Urethral Contraçtility and Blood Pressure in Dogs
_ . .
CompoundUrethra ~BP DBP~Urethra ¦
Example No.ED50 (~g/~) ED25 (~g/k~) ratio
. . _ ,. _ _ I :
37.0 1074 29.0 I .
11 1.4 390 278.6 I .
13 16.0 215 13.4
17 10.0 6.2 0.6*
21 6.6 127 19.2
27 3.2 908 3.1* .
~l.o 152 13.8 . .
41 57.0 745 13.1
42 31.0 ~04 13.0
16.4 186 11.3 :
47 35.0 530 15.1 f
~ . , _ _ . . .. .... ._ ..
Prazosin 3.6 6.6 1.8*
¦Terflavoxate>loO00 6060
,
Urethra: active dose in inhi~iting by 50% the
noradrenaline induced contraction of
urethra
DBP: active dose in lowering diastolic blood
pressure by 25~
DBP/urethra: ratio between the active doses
(selactivity index)
* non-selective: substantial effect on
both urethra and DBP
Effective Amounts
The following represent guidelines to effective oral, parenteral
or intravenous dose ranges expressed in mg/kg of body weight per
day for the following uses:
(a) In obstructive disorders of the lower urinary tract:
General 0.001 - 20
Preferred 0.05 - 1
Most Pre~erred** 0.3
(b) As antihypertensives:
General 0.01 - 20
Preferred 0.1 - 5
" , . ~ -;,, , ~, . . .:
., , , . . ;. ~ .. . .
:, - : . . . .
,,
,. .. ; , .
., .
,, , , - , . . .
2 Q ~ 6
. . .`
-174-
Most Preferred** 1
(c) As anxiolytics - antidepres~ants:
General 0.01 - 20
Preferred 0.05 - 5
Most Preferred** 0.5
(d) As bladder spasmolytics:
General 0.01 ~ 20
Pre~erred 0.02 - 10
Most Preferred** 2
** Most preferred values refer to oral dosing. Intravenous
dosages should be 10 to 100 fold lower.
Patients in need of treatment by the present compounds and
compositions also include humans that have one or more
depression symptoms (as defined, e.g. in Harrison's Principles
of Internal Medicine, XII Ed., McGraw-Hill, Inc., p.2124) or
humans that display anxiety symptoms (Harrison's, supra,
pp.2131-2134).
Selective use dosages, i.e. dosages that are active in the lower
urinary tract without a substantial e~`fect on the blood pressure
depend on the par~icular compound employed but, generally, up to
four times the EDso oP a selective compound can be administered
without substantial effect on blood prPssure. Further
refinements and optimization of dosages are possible using no
more than routine experiments.
The active compounds o~ the invention may be orally
administered, for exiample, with an inert diluent or with an
edible carrier, or they may be enclosed in gelatin capsules, or
they may be compressed into tablets. For the purpose o~ oral
therapeutic administration, the active compounds of the
invention may be incorporated with excipiants and used in the
~orm of tablets, troches, capsules, elixirs, suspensions,
syrup~, wafers, chewing gum and the like. These preparations
should contain at least 0.5% of active compounds, but the amount
of active ingredient may be varied depending upon the particular
form and may conveniently be between 5% to about 70% of the
weight o~ the unit. The amount of active compound in such
compositions i~ such that a suitable dosage will be obtained
although the desired dos~ge can be obtained by administering a
plurality of dosage forms. Preferred compositions and
' ` , ;;,, ;.; ~, ~ ~ ' '; ' . ;!. . ' '
-- 2 0 9 ~
~175-
preparations according to the invention are prepared so that an
oral dosage unit form contains between 1.0-300 milligrams of
active compound.
The tablets, pills, capsules, troches and the like may also
contain for example the following ingredients: a binder such as
micro-crystalline cellulose, gum tragacanth or gelatin; an
excipient such as starch or lactosP, a disintegrating agent such
as alginic acid, Primogel, cornstarch and the like; a lubricant
such as magnesium stearate or Sterotex; a glidant such as
colloidal silicon dioxide; and a sweetening agent such as
sucrose or saccharin may be added or a flavouring agent such as
peppermint, methyl salicylate, or orange flavouring. When the
dosage unit ~orm is a capsule, it may contain, in addition to
materials of the above type, a liquid carrier such as a fatty
oil. Other dosage unit forms may contain other various
materials which modify the physical form of the dosage unit, for
example, as coatings. Thus, tablets or pills may be coated with
suyar, shellac, or other enteric coating agents. A syrup may
contain, in addition to the active compounds, sucrose as a
sweetening agent and certain preservatives, dyes, colouring and
flavours. Materials used in preparing these various
compositions ehould be pharmaceutically pure and non-toxic in
the amounts used.
For the purpose of parenteral therapeutic administration, the
active compounds of the invention may be incorporated into a
solution or suspension. These preparations should contain at
least 0.1~ of active compound, but may be varied between 0.5 and
about 30% of the weight thereof. The amount of active compound
in such compositions is such that a suitable dosage will be
obtained. Preferred compositions and preparations according to
the present inventions are prepared so that a parenteral dosage
unit contains between 0.2 to 100 milligrams of active compound.
The solutions or suspensions may also include the following
components: a sterile diluent such as water for injection,
saline solution, ~ixed oils, polyethylene glycols, glycerine,
propylene glycol or other synthetic solvents; antibacterial
agents such as benzyl alcohol or methyl parabens; antioxidants
such as ascorbic acid or sodium bisulphite; chelating agents
such as ethylenediaminetetraacekic acid; bufEers such as
. - . . .
..
: , , .;
- . i , -, .
209~15G
-176-
acetates; citrates or phosphatPs and agents for the adjustmentof tonicity ~uch a sodium chloride or dextrose. The parenteral
multiple dose vials may be of ylass or plastics material.
~: '', ''`'~'; '' ,.'
, ~` '. .'
'
, '.''":"
:
", " " " , ,. . i , !, ,. . s,, . : ,: , . . : " . ' , ; . ,. , ~, . . .