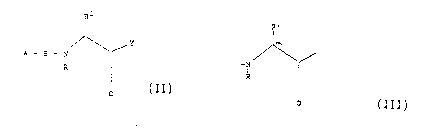Note: Descriptions are shown in the official language in which they were submitted.
WO 92/0437~ 2 ~ 9 ~ 3 ~ 8 PCJ/GB91/0147~
KI~iIliOGENASE INHIBITORS
FIELD OF INVENTION
The invention relates to enzyme in~i~it c-. ?n~ _^
treatment of disease.
BACKG~OUND - KININS
Kinins are natural vasoac~ive pep.ides !iberated n ~:~e
body from high molecular weigh_ precursors (kinincgens) -y ~he
act.ton of selective proteases known as kininoaenases.
There is evidence for the involve~en- of kinir.s ir. _he
following pathological states:
(a) Conditions associated with vasodilatat on and
hypotension, e.g. septic, anaphylact.c and hypovolaemic
shock; carcinoid syndrome and dumpinq syndrome
(b! Condition~ involving inflammation, e.g. acute
ar.hritis, pancreatitis, local thermal injury, c-ush
in -y and brain oedema
(C) Conditions involving bronchoconst-i^~ on,
especially for example the initial, acute aiie_sic
reaction in asthma
(d) Allergic inflammation, particularly allergic
rhinitis and conjunctivitis, together generally known as
hay fever, and the bronchial inflammation and consequent
occlusion found in the non-acute but serious and even
fatal inflammatory phase of asthma.
The kinins (bradykinin, kallidin and Met-Lys-bradyk -.in)
are potent mealators of inflammation. Their main actions a-e
as follows:
(a) They increase capillary permeability which leads to
exudate formation and oedema
(b) They are potent vasodilators in arterioles and
therefore reduce blood pressure and increase blood flow
SIJ'E3~T~,'JT~ SHEET
W092/04371 2 ~ 9 ~ 8 ~ 8 PCT/GB91/0147
' 2
(c) They induce pain
(d) They contract bronchial smooth muscle
(e) They activate pAospholipase A2 and ~hus s~imuiate
the biosynthesis of prostaglandins (PG'C: wni^h
mediate some of their ac~ions.
In reaard to prostaglandins, i. mav ~e noted th2t ~e_~ain
ac~ ons of ~inlns, particularly pain and vascular ~er~eaDil .y
above, are potentiated by PG's, although PG's Ihemselves do
not cause pain nor do thev induce vascular ?erDeabil _y Zt -h2
concen~~ationS found in inflamed tissue. PG's .herefo-e 2C=
as either mediators or potentiators of kinins.
In spite of the above knowledae of ~inins and .he~~
actions, relatively little attention has been paid eo
reduction of their action. In asthma treatment for exampie
clinical attention is primarily directed to the acute
bronchoconstrictiVe reaction, for which there are effective
drugs. Deaths continue to occur f~om the gradually developing
bronchial occlusion, and at present not only are there no
clinically effective inhibitors of kinin release available but
the concept of kinin release inhibition, at least in treating
allergic inflammation, appears to be new. The only substance
that is in fact a kinin release inhibitor and has at_ained
clinical significance is aprotinin ('T-asylol', Bayer, trade
mark), a proteinase inhibitor isolated from bovine tissues
(lungs, lymph nodes and pancreas). It is a strongly basic
protein (pI = 10.5) of MW = 6,500 comprising a single peptide
chain of 58 residues. However, aprotinin is primarily a
trypsin inhibitor (Xi = lO l3M) and is some l06-times less
active against kinin release. It has been found marcinallv
beneflcial in acute pancreatitis, a serious condition, where
it inhibits the activation by trypsin of zymogens or
pançreatic serine proteinases, and in traumatic - haemorrhagic
shock. Aprotinin has to be administered parenterally, and it
freauently produces a painful reaction at the injection site.
CUESTiT'~"_ SnEET
WO92/04371 2 ~ PCT/GB91/~147g
BACKGROUND - KININOGENASES
The kininogenases are serine proteinases, that is to say
proteinases in which the hydroxy group of a serine residue is
the nucleophile involved -in forminq the substrate transition
state. They liberate the ~inins (bradykinin, kallidin) from
the kininogens by limited proteolysis. There are severa
kinds of ~inino~enase:-
(a) Tissue kallikrein (TX, also called glandular
kallikrein GT or urinary kallikrein UX) which is found in
the pancreas, salivary glands, intestines, kidney and
urine. It has MW = 30,000 and acts preferentially on low
molecular weight kinino~en (LMWX) to release the kinin
kallidin (KD). Tissue kallikrein has no potent and fast
acting endogenous inhibitor present in plasma.
(b) Plasma kallikrein (PK) occurs in plasma as an
inac_ive zymogen which is activated by Factor XIIa, and
is part of the intrinsic coagulation cascade. It has MW
= l00,000 and its preferred substrate is high molecular
weight kininogen (HMWK) from which it releases bradykinin
(BK). Plasma kallikrein is rapidly and effectively
inhibited in plasma, by endogenous inhibitors known as
cl-inactivator and ~2-macroglobulin.
(c) Mast cell tryptase which, while not as ac~ive as the
kallikreins in kinin release, we have found to occur in
large amounts in the mast cells of the lung tissue of
asthmatics.
BACXG~OUND - XININOGENS
The kininogens which are the natural substrates for the
kininogenases (they act also as potent inhibi~ors, Xi approx.
ll, of cysteine proteinases such as cathepsins B, H and
L, calpain ~nd papain) occur in two types:
~;UBSTi . 'J I - Sl :,E~
W092/04371 PCT/GB91/0147~
(a) Low molecular weight kininogen (L.~WK~ ~ith moiecula~
weight in the ranae _O,000 - 7G,~00 dependir.g on species
of o-igin and degree of glycosyi~~ion.
(b) High molecular weight .~ininogen (~MWK) Wl-~.
~olecular weight in the ranqe 88,000 ~ ,OOO whic:-, in
addi_ion to serving as an alternative precurso- of ~ .s
and a cysteine proteinase inhibito~, aisc plays ~n
obligatory -ole with plasma ~al~ rein in _he initia~ 0?.
o- ~he ~nt-inisic coaaulation cascade.
The two kininogens, whose mRNA's are transc-i~ed f-om the
same gene, have identicai ?ri~arV secuences throughout -he ~-
ter~inal or heavy chain (H-chain) region, the kinin region ana
the fi_st twelve amino acids of the C-terminal or light chair.
(L-chain). At this point thei- s~ cl~lres dive ge, HMWK
having a lonaer L-chain (MW approximately 45K) than LMWK
(~.8K).
The cleavage of human HMWK by plasma kallikrein is for
example shown schematically in Fig. i, with details of the
sequence at the cleavage sites in Fig. 2 and a ~ore detailed
sequence in Fig. ~ where _he conven~ional nu~Dering _-
-esidues adajcent to a cleavage site is shown for cleavaae
cite I. After excision of one or other kinin sequence, the
H- and L-Chains are held together by a single disulphide
bridge:-
SU5STIIU T ~ SHEET
Wo 92/04371 5 2 ~3 ~ v 8 PCl/GB91/01479
S - S , ~ S -S
H-chain 3K ~ L-erlaIc
~MWK ~1 OOK
P~ i00~;
S - S ----~ - S
H-chain L-cna s
3~, -lK
~- isure ' . Clea~ ce ~~ ~ ir'~ _ . ?~ O~!e__l _^ne.~ne
PK PK
TK ~--Cleavage site II TK
-ile-Ser-Leu-l~et, Lys, Arg-Pro-Pro-Gly-2he-Ser-Pro-Phe-Arg,Ser-Ser-.~rg-Iie-Gly-
~' 380 ~" Bradykinin ~' 3~
Cleavage I , C;eavage site I
site III ~ ~ Kallidin .'
Figure 2. Cleavage of human kininogens by PK and TK: De~ails of
sequence
PK,TK
Pj P4 P3 P2 P~?2 P3 P~ P~
-Phe-Ser-Pro-Phe-Arg Ser-Ser-.~rg-Ile--Gly-
38 j 389 390 394
r igure 3 . Sequences flanking ciea~, age site I i
human H.~IWK
~iU~TlTUTE SHEET
W092/04371 2 ~ S ~ ~ 5 8 P~r/GBgl/0l47~J
As shown, plasma kallikrein and tissue kallikrein act at
a single site to free the kinin C-terminal site, cleaving
between residues 389 and 390, but at sites one residue apar~,
either side of residue 380, to free the N-terminal of
bradykinin (by PK) or kallidin (by TK).
The role of PK and HMWK as clotting factors in _he
intrinsic cascade does not involve the enzymatic release of
Xinins. However many of the ef fects of PX and all those o' TK
do involve such release, being mediated by the kinins released
from the respective substrates HMWK and LMWX through selective
proteolysis.
INDICATIONS
The main clinical indications for kininogenase inhibitors
are inflammatory conditions, particularly allergic
inflammation (e.g. asthma and hay fever). A fuller list of
indications is given below:
(l) Allergic inflammation (e.g. asthma, rhino-conjunctivitis
[hay fever~, rhinorrhoea, ur icaria
(2) Inflammation (e.g. arthritis, pancreatitis, gas~ritis,
inflammatory bowel disease, thermal injury, crush
injury, conjunctivitis)
(3) Smooth muscle spasm (e.g. asthma, angina)
(4) Hypotension (e.g. shock due to haemorrhage, septicaemia
or anaphylaxis, carcinoid syndrome, dumping syndrome)
(5) Oedema (e.g. burns, brain trauma, angioneurotic oedemc
whether or not as a result of treatment with inhibitors
of angiotensin converting enzyme)
SyBCTl~u,_ SHe:ET
W092/04371 2 ~ 8 PCT/GB91/0147~
(6) Pain and irritation (e.g. burns, wounds, cuts, rashes,
stings, insect bites)
STATEMENT OF INVENTION
In one aspect the invention provides a method Gf
treatment (including prophylactic treatment) of an
inflammatory or other condition se~ out 'n the indicat ons
above, particularly an allergic inflammatory condition,
wherein an effective amoun~ of a pep~ide or peptide-anaiogue
kininogenase inhibitor is administered topically or
systemically to a patient suffering from or at risK of the
condition. It is believed that for optimum activity,
administrability and stability in the body the compounas
should not exceed the size of a hexa~eptide, that is to say
should not comprise more than six amino acid or amino acid
analogue residues; the presence of further residues,
particularly in a pro-drug from which residues are cleaved in
the body to give the compound primarily exerting the desired
effect, is however not excluded.
Particularly, the invention provides a method of
_reatment of the allergic inflammatory phase of asthma,
wherein an effective amount of a kininogenase inhi'oitor sucn
as a mast cell tryptase inhibitor is administered topically or
systemically to a patient suffering -rom or at risk of the
condition.
The invention extends further to a method of preparation
of a medicament for the topical or systemic treat~ent
(including prophylactic treatment) of conditions as above
particularly for allergic inflammatory conditions and
especially for asthma as above, wherein a kininogenase
inhibitor is associated with a pharmaceutically acceptable
diluent or carrier to constitute said medicament.
In the a~ove, the kininogenase inhibitor is conveniently
SU~STITUTE SHEET
W092/04371 2 0 ~ O ~ ~ 8 PCT/GB91/0147g
but not essentially of the novel kind now described in which
in another aspect, without limitation to any particular
clinical indication, the invention provides synthetic, low
molecular weight compounds that selectively in~ibit
kininogenases and thus block the release of kinins from
kininogens. The inhibitors are peptide analoaues, desirably
(as above) not exceeding the size of a hexapeptide in terms of
amino acid or analogue residues, based on the known amino acid
sequence of the kininogens at cleavage site I, wnich analogues
have sufflcient similarity to the cleavage site sequence tO
bind to the active site of the kininogenase but are no
hydrolysable and therefore remain bound, inactivating the
enzyme.
The inhibitors are essentially of the structure below, in
which A represents the P3 residue, B the P2 residue, C the
Pl residue and '' a carbonyl-activating or binding group the
structure being:-
A - B - C - Y
where A, B and C are amino acyl or amino acyl analogue groups
linked by peptide bonds or conformational analogues thereof
giving a peptide mimic. Other residues in addition to these
essential ones may of course be present, including amino acyl
or amino acyl analogue residues.
In more definitive terms the compounds are represented by
SUBS~E,l,;lE SHEET
WO92/04371 2 ~ 5 ~ PCT/GBg1/0147~
~ `~ Y
A - B - ~ , /
F~ I
C
wherein
A and B = amino acyl (including amino acyl analogue) the
same or different forming a dipeptide group the
amino acid of A optionally carrying a terminal
group (other than hydroqen) and being any amino
or imino-acid residue (but preferably of D-
configuration) and of B being a lipophilic
amino-acid residue of ~- or L-configuration but
not proline or a proline analogue, or a
conformationai analogue of said dipeptide group
wherein the peptide link is replaced by
-CH2-NH- ('reduced'), -CH(OH)-CH2-
('hydroxy'), -CO-CH2- ('keto'), -CH2-CH2-
('hydrocarbon') or other conformational mimic
of the peptide link
and in:-
SUBS T ITUTE Sl IEET
WO92/04371 2 3 ~ O 8 ~ 8 PCT/GB91/0147~ -~
the side chain Rl is that of a basic amino acid
or amino acid analogue (preferably o L-
configuration) and R is H or lower alkyl (cl -
C4) or C~ or the peptide link comprising
-N(R)- is replaced leading to a conformational
mimic as above. For example C~ may be replaced
by nitrogen.
Y = a binding enhancing or carbonyl activating
group for example selected from H (but only if
A or B is cyclohexylalanine, preferably D if a~
A or L if at B) or alkyl (Cl - C20) or
fluoroalkyl (C2 - Cl2); substituted
oxymethylene; thiomethylene; sulphoxy-
methylene; sulphonylmethylene; aminomethylene;
hydrazino-m~thylene; -CH2-Het (where Het = a
substituted or unsubstituted heterocycle);
substituted amino (but when the resulting
compound is a secondary alkylamide B must not
be phenyl-alanine); an amino-acid group or its
ester or amide; a car~oxylic secondary amide
or primary amide, when B must be a bulky
lipophilic, non-aromatic amino-acid e.g.
cyclohexylalanine, adamantylalanine (not Ala
Leu Ile Val Nva Met Nle Phe Tyr Trp Nal (l));
CUBSTITUTE S:~EET
WO92/04371 ? ~ PC~/CB91/014~3
tertiary-carboxamide: carboxy-alkyl group or
its ester or amide.
_n the above context substituents are suitably c~mon
functional groups that increase binding af4inity t3 the enzyme
and/o~ improve pharmacological pro~ertles. ~urth~r in
considering conformational analogues or mimics a dipeptide
mimic ~s a structure containing non-natural amino acid ~amino
acid analogue) residues or which is non-peptidic and which in
I holds the side-chains of A and B o- B and C or all o4 them
in a c~nformation simila- to Ihat ~resent ln the parent
peptide when bound to ~he ac~ive site cf he enzyme. T~ may
also contain features favourable for other interactlons ~ith
the enzyme, e.g. hydrogen bonding. A mimic may be chosen f~om
he published work on such analogues.
Fo- example, the following are mimics of the dipeptide
DPro-Phe (Ph may be rerlaced by -CH2Ph)~-
ZH ~ ~ CO H ~ ~ COzn
(i~ '?~e^~cea' mimi^ 'i ) 'Hydrcxv' or 'ii , 'Ket^' ^-
hyc.cxyetr.y!^r.= ri~ ersmethylene
Ph Ph Ph
H ~ .~" CO2H / ~ .~" CO,H H (CH2)nC02n
O o n=1-4
(iv) ~v) (vi) ('~ydrccar~cn'
mimic
Figure ~, DPro-Phe m~ cs
SUBSTITU T r Si-icET
w092/04371 2 ~ ~ O ~ ~ 8 PCT/GB~1/0147~ _
The preferred compounds represented by the above general
formula are now considered.
~ referred residues for A are imino-acids, (e,g. 3-proline
or an analogue of proline e.g. pipecoiinic acid, azetidine
carboxylic acid etc.); lipophilic amino acids (e.g. DPhe,
DCha, DChg); strongly basic amino acids (e.g. D-Arg cr a
guanicinopAenylalanine) and for B they are L-Phe, -Cha,
L-~Nal, L-Tal, L-(4F)Phe L-(NMe)Phe or other substituted
phenylalanines. A and B may also be the N-alkyl (cl-c~J or
C~-alkyl (Cl-C7 e.g. methyl, benzyl) anaiogues of these amino
acids. Suitable terminal groups for A include lower aikyl
(preferred) or acyl (not excluding amino acyl), alkyl
sulphonyl (straight chain or branched or cyclic), amino-alkyl,
carboxy alkyl, hydroxy alkyl or any other common protecting
group encountered in peptide chemistry.
Groups suitable as group Y are specific to the present
invention in that they are part of the structure giving the
required binding to the active site and are not merely non-
interfering end groups. They form a binding group which
increases affinity to the enzyme and/or a group which
activates the adjacent carbonyl by rendering it more
electrophilic. Specific groups are included in the following
formula:
- /
X 11 ,
SUBSTITUTE Sl~,EET
WO92/04371 2 ~ ~ a ~.J 8 PCT/GB91/~147g
where in a peptide link t~ residue B the ~-nit~ogen may ~e
free or substituted for example by ~ethyl or other C1-C4 alkyl
and thus Rl and R are as before but particularly Rl =
3-guanidinopropyl or other guanidinoalkyl group or an ami~ino-
alkyl or aminoalkyl group, also ~ara- or me~a substit~ted
guanidino or amidino-benzyl or pro~ected forms of the aDove
(the basic nitrogens may zlso be alXyl~ted e.g. with Me, Et~,
and where
Y = groups as aiven below, rirst in more aenerzl
terms and then in terms of ~ore det~iied
preferences, subjec~ in both cases tO ~he
provisos expressed in defining the compounds of
the invention earlier. The de~ailed
preferences are given in groups under roman
numerals, which are also indicated, in
brackets, with the first listing wAich is:-
(I) Y = H (representing aldehydes) or alkyl
including fluoroalkyl (representing ketones)
(II-III-IV) Y = -CH2Q
where Q = -oR2 or -SR~ or -SOR2 or -S0222 or
-NHR2 or - ~: or -D1
\ R'
(note that -~ represents a ring in which D is an ato~ O r
that ring)
wherein R2, R~ and R~ are as below
SUBSTITUTE SHEET
WO 92/043~1 2 ~ 8 1~ Pcr/GB91/0147g
( V-VI ) Y = -rH,~ '`ON O--
~ .~ _
-C.~-~ C~6 C0~~
wnereln R~, R' and R~ are as be!ow
(~'l--VIII) Y = amino acyl or group 'orming a
substituted amide or hydrazide
(IX-X-XI) Y = a group forming an ~-keto amide e.g.
- COR 9 or -CO-D or
--~o.~
anc ln ~hich further:-
R2 = alkyl or substituted alkyl includingaryl or aryl alkyl and -CH~R3 where R3 =
fluoroalkyl
R4 and R5 the same or different but not both
hydrogen = H or C,-C20 alkyl (which may be
further substituted), acyl or alkyl sulphonyl
-~ is a heterocyclic ring (D = nitrogen or
carbon in Group IV and N in Groups VI and X)
optionally unsaturated and optionally with
further hetero atoms and substituents
SUBSTITUTE SHEET
W092/04371 ~ ~,3 ~ PCT/GBgl/0l479
R6 = hydrogen, alkyl, hydroxy~lXyl,
aminoalkyl, alkylaminocarbonyl
R3 = -NH2 as sucn o- alkylated, or amino acvl
~ he listing of more detailed preferences, again ~ithin
the provisos expressed earlier, is -
Group TY = H; alkyl including brancned alkyl (C.-C 0,; zryi
alkyl: or cycloalkyl (Cl~c20); perfluoroalkyl or
partially fluorinated alkyl (C2-Cl~); [e.g. Y = Me;
-CH(CH2CH2CH2CH3)2; -CH(CH~CH2C~C~3)CH2 -cyclohexyl;
-CH2CF2CF2CF~; -CF~CH~CH CH3;.
Group ~I
Y = -CH2QR2 where Q = O, S, SO, SO2, NH and where
R' = Alkyl, branched alkyl or acyl (Cl-Cl2); or
cycloalkyl (Cl-C20); or aryl or aryl alkyl; or -CH2-R3
where R3 = perfluoroalkyl or partially ,luorinated alkyl,
branched or not (Cl-Cl2).
Group __
Y = -CH2N
R'
R~, R5 the same or different = alkyl, branched alkyl,
cycloalkyl, acyl, alkylsulphonyl, carboxyalkyl (the
carboxyl group may be further derivatized to form an
ester or amide with an amino-acid or dipeptide),
carbamoyl, sulphamoyl, N-dialkylamino-, arylalkyl,
haloalkyl including fluoroalkyl, cyanoalkyl, alkoxyalkyl,
SUBSTITUTE SHEET
W092/04371 2 ~ 9 0 ~ ~ 8 PCT/CB91/0147g
1~
hydroxyalkyl, mercaptoalkyl, aminoalkyl and derivatives
thereof e.g. esters, amides and thioeslers; or one of R~
or R~ = hydrogen
Group T~'
Y = -CH~ - ~
where D = nit~ogen or carbon and -~ ~s a saturated or
unsa~urated heterocyclic -_ng or a bicycli_ -ing system,
each is ~ - 8 membered, where there may be other hetero-
atoms (N, S, O) and carbons or nitrogens may op~ionally
be substituted by alkyl, branched alkyl, cycloalkyl,
carboxyalkyl, carboxy (attached lo carbon), amino,
alkoxy, alkoxymethyl or (carbon) as carbonyl or other
groups beneficial for interaction with the enzyme.
Grou~ V
Y = -CH~CH(R6)CoNR'R5
~.~, ?.5 as defined -. Gr_u? ~.
?~6 = hydroaen lower alkyl, branched alkyl, cycloalky ,
hydroxyalkyl, amino-alkyl, alkyla~inoczr~onyl.
Grou? VI
Y = -CH~CH~R6)CO~
p,6 as defined in Group V and - ~
as defined in Group I-i (but with D = N)
SUBSTITUTE SHEET
WO92/04371 2 3 3 ~ 8 PCT/CB91/01479
Group VII
Y = an amino-acid residue cr any amide ~seccnGa-r c-
ter_iary) or ester of _hat ~esidue, ~ 5-
c^nfiguration. Pref2r-ed residues a_e C4 lipoph~
a~ino-acids e.~. no~leu^~e, cyo'onex~Jlal2-.i-.e,
ho~oc~clohexylaianine,cyclohexylclycine,~~r~~utviclv- -.e.
G--cu~ '.---_
R'
Y = - N-R~
R7 = H (when however B is not phenylalanine unless 28 is
carboxylalkyl or derivatized carboxylalkylj; or aikyl,
branched alXyl (Cl - Cl2), cycloalkyl (C1-C20)
car~oxyalkyl or bis'carboxyl)alkyl, which may be
derivatized at the car~oxyl c-oup to form an amide e.g.
with an amino-acid (preferred is z~ainine) ^- a
subs~i~uted a~ine; N'-dialkylamino; N'-~lkylamino-,
2~ = R7 the same or differen_ but excluding H.
Group IX
Y = -CO-R9 but only if 3 is a bulky non-aromat~c
lipophilic amino acid o- its N~ alkyl (c. - _A;
derivative (e.g. cyclohexylal~nine but excludi.. Ala,
- Leu, Ile, Val, Nva, Met, Nle, Phe, Tyr, ~=p, Nal(l) and
their N-methyl derivatives) where R9 = NH2,
N'-alkylamino (where the alXyl groups include brznched
and/or cycloalkyl); an amino-acid residue.
SUBSTITUTE SHEET
W092/04371 PCr/GB91/0147g --
2~ 8 1~
Grou~ X
Y = -CO-~
-3J as defined in Group IV (bu- ~ = N)
Grou~ XI
= -CO-NR~R~
as defined in Grou~ ~~_, bu- no_ ~:
The following examples illustrate the invention. They
are given in the form of:-
- Nine tables of compounds with reference number,
structure, molecular on as deter~ined by FA3 ( fas~
atom bomDardment) spect-omet-y and class of compound
(the same as the 'groups' referred to ea_lier
herein)
- Elght detailed exampies of svnthesis
- Twelve synthesis schemes, as ;efer~ed to ~n ~he
detailed examples
- Table of abbreviations
- Description of in vit_o tests of inhioi'ion of
kininogenases and in vivo tests of ef ficacy aaair.st
asthma
A11 s~ructures of inte-mediates we~e ve~i~iec ~ NMR.
SUBSTI T UTE SHEET
wo 92/04371 ? ~ S ~ PCr/GB91/0147
TABLE 1
Aldehvdes
[M+Hl~ Class
S Me-DPhe Cha Arg-H 473
TABLE 2
1 hiome~hvlene Analo~ues and SulDhomethvlene Analogues
[~I+Hl- Class
17 H-DPro Phe Arg CH,S(CH,)1Me 519 II
H-DPro Phe Arg CH2SnBu 505 II
61 H-DPro Phe DLArg CH,SO,nBu 537 II
ABLE 3
E~hers
[M+Hlt Class
23 H-DPro Phe Arg CH20CH,(CF2)3CHF2 647 II
62 H-Pro Phe Arg CH,OCH,CF3 515 II
63 Boc-DPro Phe DLArgCH20CH,CF3 615 II
64 H-DPro Phe DLArgCH20CH,CF3 515 II
H-DPro Cha DLArg CH20CH,CF3 521 II
66 H-DPr~ Phe Arg CH20CH(Me)CF,CF2CF3629 II
67 H-DPro Phe Arg CH20CH2CF3 515 T~
68 H-DCha Phe Arg CH20CH2CF3 571 II
69 H-DArg Phe ArgCH,OCH,CF3 574 II
H-DChg Phe ArgCH20CH,CF3~ 557 II
54 H-DPro Phe Arg CH20(CH2)5CH3 517 II
H-DPro Phe Arg CH,OPh 509 II
SljJ~ I j, 'J I E SHEE~
wo 92/04371 2 3 ~ 8 3 PCr/GB91/0147~J
TABLE 4
Aminomethvlene and related analo~ues
l~+Hl- aass
3i H-DPro Phe ArgCH2N[(CH.)sMel2 600 III
71 H-DPro Phe DArg CEi2(RSjTha 556 IV
72 H-DPro Phe ArgCH2(RS)Tha 556 IV
73 H-DPro Phe Arg CH2 1-Pip-3-(RS)CO,Et 572 IV
7J, H-DPro Phe DLArg CH, l-Pip-3-(RS)CH,O(CH2)3.~11e 586 IV
H-DPro Phe DLArg CH, 1-Pip-3-(RS)CH~NEt, 585 ~
76 H-DPro Phe DLArgCH, 1-Pip-3-CONEt, 599 IV
77 H-DPro Phe DLArg CH, 1-Pip-3-(RS)CO~Bu 599
78 H-DPro Phe DLArg CH, 1-Pip-3-(RS)CON(nBu)~ 655 l~
79 H-DPro Phe DLArg CH2 1-Pip-4-(1'-Pip) 583 I~
H-DPro Phe Arg CH2(NMe)AhaNHnBu 615 I'v
81 H-DPro Phe Arg CH,-Abn 540 IV
82 H-DPro Phe Arg CH,-Hvp(OnBu)NHEt 629 IV
83 H-DPro Phe Arg CH2-Sar Pro NHEt 628 m
84 H-DPro Phe Arg CH2-tHyp(aBu)ROnBu 644 IV
H-DPro Phe DLArg CH2Pic NEt2 641 IV
86 H-DPro Phe DLArg CH2'DHyp(aBu)BOnBu 644 IV
87 H-DPro Phe DLArg CH2pro~coNEt2 5999 IV
88 H-DPro Phe DLArg CH21-Pip-3(RS)CO2H 544 IV
89 H-DPro Phe Arg CH21-Pip-3(RS)CH,CONEt, 613 IV
H-DPro Phe Arg CH2N(CH,Ch)(CH2)5Me 612 m
91 H-DPro Phc DLArg CH2N(OC)2 657 m
92 H-DPro Phe Arg CH2N(Et)Ch 542 m
93 H-DPro Phc Arg CH2N(Me)(CH2)4N H~ 517 m
94 H-DPro Phe Arg CH2N(Me)nBu 502 m
H-DPro Phe DLArg CH,NnBu2 544 m
96 H-DPro Phe DLArg CH2N[nBu]SO,nBu 594 m
97 H-DPro Phe DLArg CH2NlnBu](CH2)3C ONH, 573 m
98 H-DPro Phc DLArgCH2N[I'Hex~(CH2)~NH2 629.5 m
99 H-DPro Phc DLArg CH2N[llHex](cH2)5NH2 601 m
100 H-DPro Phc DLArgCH2N[nHex](CH2)3NH2 573 m
101 H-DPro Phe DLArg CH2N[nBu~(CH2)4Ph 620 m
SUe~TlTUTE SHEET
WO 92/04371 ~ l 8 PCr/G891/0147
TABLE 4 (cont.)
Aminomcthvlene Ketones and reiated analoc~ues
lM+Hl~ Cl~ss
102 H-DPro Phe DLArgCH1~[nHex](cH~)6cO~H~ 643 ~I
103 H-DPr~ Phe DLArg CH,N[~He%i(CH,j6.~H~ 615 iII
104 H-DPro Phe DLArg CH~NrnHexl(CH2)7OH630 m
105 H-DPro Phe DLArg CH~NrnHex](CH2)~NHAc 671 m
106 H-DPro Phe DLArg CH,~lrnHex](CH,)6CONHE~ 671 III
107 H-DPro Phe DLArg CH~N[nHex](CH2)6CO,.\~le 658 III
108 H-DPro Phe DLArg CH,NrnHex](CH,)6CO,H 644 III
109 H-DPro Phe DLArg CH,~'rnBu](CH,)3CO~Et. 6'9 lII
110 H-DPro Phe DLArg CH,i~:'Bu](CH,)3CO.~HE~ 601 lII
TABLE 5
Keto Isostere Ccetainin~ Analo~ues
[M+H]+ Class
H-DPro Phe DLArg~GlyPro-NHEt 599 VI
111 H-DPro Phe Arg~Gly P~NHE~ 599 VI
112 H-DPro Phe Arg~Gly Arg-NH2 530 V
113 H--~ Phe DLArg~JlyAla-NH2 545 V
114 H-DPTo Phe i .~rg~Gly Aha-NH, 587 V
115 H-DPro Phe DLArg~lyAha-NH~Bu 643 V
TABLE 6
Subs~ate Analo~ues
[M+H~t Class
45 H-DPro Phe ArgChg-NH2 557 VII
116CPr-CO Phe ArgChg-NH, * VII
117~IeSO2 DPro Phe Arg Chg-NH2 635 VII
118MeCO DPro Phe Arg Chg-NH2 599 VII
119H-DArg Phc ArgSer-NH, * VII
120H-DCha Phe Arg Ser- NH2 * VII
121H-DPhe Phe ArgSer- NHi ~ V~
122H-DPic Phc Arg Ser- NH2 * VII
SUBSTITUTE SHEET
WO 92/0~371 2 0 ~ 8 - pcr/GB91/o1479
TABLE 6 (cont ~
Substra~e Analo ues
[MfHl Class
1~3 H-DPic Phe Arg Chg-NH~ ~ VII
1~1 H-DPro Phe ArgSer-NH~ ~ VII
1~5 H-DPro Cha Arg Ser-NH~ * VII
126 H-DPro Cha ArgGly-NH, * VII
127 H-DPro Phe Arg Ser-Arg-lNH. ~ VII
1~8 H-DPro Phe Arg Lys-N~ VII
1~9 H-DPro Phe Arg Aha-NH. ~ VII
130 H-DPro Phe Arg Phe-~H, 565 VII
131 H-DPro Phe Arg Leu-NH, 531 VII
13~ H-DPro Phe ArgIle-NH, 531 VII
133 H-DPro Nal Arg Ser-NH, ~ VII
131 H-DPro Phe Arg DAha-NH, 531 VII
135 H-DPro Phe Arg Aha-NH¢CH,)3.Ue 587 VII
136 H-DPro Phe Arg Nleucinol 518 VII
137 H-DPro Phe ArgSesOnBu NH2 561 VII
138 H-DPro Phe Arg Cha-NH, 571 VII
139 H-DPro Phe ArgAda-NH2 623 VII
140 H-DPro Phe ArgHch-NH, 585 VII
141 H-DPro Nal ArgCha-NH, * VII
142 H-DPro Cha Argcha-NH2 VII
143 H-DPro PhepNO,ArgSer-NH, ~ VII
144 H-DPro Nal Arg Ile-NH2 * VII
145 H-DPro Phe ArgIlePr~NH, * VII
146 H-I)Pro Phe ArgAhaPro-NH2 VII
147 H-DPro Nal ArgAha-NH, * VII
148 H-DPro Cha ArgNpg-NH, * VII
149 H-DPro Cha ArgHch-NH, * VII
150 H-DPro Nal ArgHch-NH, * VII
151 H-DPro Phe ArgNpg-NH2 545 VII
152 H-DPro Phe ArgChg-1-Pip 625 VII
153 H-DPIo Phe ArgChg-NH(CH2)5Me 641 VII
154 H-DPro 4-Fph Arg Chg-NH2 575 V~
Sa~sfactory am~no acid analysis obtained
SUBC T ITU I E SHEET
WO 92/0437l 2 ~ rj g PCI/C~9l/01479
TABLE 7
Arnides
~I+Hlt Class
47 H-DPro Phe Arg N[(CH.)5~1e](C~ Ch ~ 5 YrrI
15i H-DPro Phe Arg N(Me)nBu 488 VIII
1i6 H-DPro Phe Arg NH(CH2)3CO Arg-NH. * V~I
157 H-DPro Phe Arg NH(CH2)3NH. 475 vm
1i8 H-DPro Phe Arg NH(CH.)4CO Arg-NH, ~ VIII
li9 H-DPro Phe Arg NH(CH2)4NH~ ~ Y~I
160 H-PTO Phe Arg NH(CH.)sCO Arg-NH~ 687 VIII
161 H-DPro Phe Arg NH(CH.)5NH. 503 ~v'lII
162 H-DPro Phe Arg NH(CH2)6cO Arg-NH2 701 VIII
163 H-DPro Phe Arg ~H(CH,)7CO Arg-NH. 715 vm
164 H-DPro Phe Arg NH(CH2)7CONH(CH2)3l\/le615 VIII
165 H-DPro Phe Arg NH(CH.)7l~AC 573 VIII
166 H-DPro Phe Arg NH(CH,)7NH, 53l VIII
167 H-DPro Phe Arg NH(CH2)7CONH2 559 vm
168 H-DPro Phe Arg NH(CH2)7CO-GlY Gly ATg-~H2 829 vm
169 H-DPro Phe ArgNH(CH2)7CO-GlyArg-NH 772 VIII
170 H-DPro Phc ArgNH(CH2)7CO-GlyGly-GlyArg-NH2 886 vm
171 H-DPro Phe Arg N[nHex]2 586 VIII
172 H-DPro Cha Arg NHCH2Ch 520 VIII
173 H-DPro c~.~al Arg NHCH2Ch 564 VIII
174 H-DPro ,BNal Arg NHCH2Ch 564 vm
175 H-DPro His ArgNHCH2Ch 504 vm
176 H-DPro(4Me)Phe Arg NHCH2Ch 528 vm
177 H-DPro Phe NarNHCH2Ch 500 vm
178 H-DPic Phe Arg NHCH2Ch 528 V~I
179 H-Drlc Phe Arg NHCH.Ch 576 vm
180 H-DThi Phe Arg NHCH.Ch 570 vm
181 nBu DPTo Phc Arg NHCH2Ch 570 vm
* Satisfacto~y amino acid analysis obtaincd
SUBSTlTUrE SHEET
wo 92/04371 2 () ~ O ~ ~ 8 2 PCI/GB91/01479
TABLE 8
Ketones
[M+H~- Class
~9 H-DPro Phe ArgCH3 117
48 H-DPro Phe Arg(CF2)2CF3 **
53 H-DPro Phe ArgCH~CH~nHex~. ***
** Molecular ion not de ^:ed
TABLE 9
a-Ketoarnides
[M+H]T Class
11 H-DPro Phe DLLys CON[nBu]2 530 XI
H-DPro ChaDLArgCON[T~Bu]~ *** XI
*** [M+Hl~notavailable
SUBSTITUTE SHEET
wo 92/0437I 2 ~ PCr/GB91/0147~
,
EXAMPLE I
~le-DPhe-Cha-Arg-H
The synthesis of 5 was carried OUt according to Scheme I. Arabic nume~als
underlined e.g. 1 refer to structures in these schemes. Roman numerals in
parentheses e.g. (i) reter to reaction steps.
(i) Isobutvl chloroforrnate (10.'~ mmol) was added to a solution of
Boc^Arg(Z.)OH (9.23 mmol) and N^methvlmorpholine (11.08
mmol) in dry l~F (25 cm3) at -20C. After ^0 mins the solid was
filtered off and the filtrate added to a solution of sodium
~orohyd~ ;e (10.3 mmol) in water (10 cm3) at 0C. After 3 hours
0.3 M K}.S04 was added. the crude product extracted wi~n EtOAc
and purified by flash chromatography on silica with EtOAc - petrol
(4:6~. The alcohol 1 was isolated as a white solid (97%).
(ii) The Boc ~oup of I (4.75 mmol) was removed with sa~. HCl/Dioxan
and the product acylated with Boc-Cha-ONSu (9.5 mmol) in
CH~CI2 (20 cm3) at 0C in the presence of N-methylmorpholinc.
After two hours the reaction was worlced up using standard
procedures and the crude product purified by flash chromatography
on silica with EtOAc - petrol (4:6). The pure alcohol was isolated
as a colourless oil (90%).
(iui) The Boc group of 2 (4.27 mmol) was removcd with sat. HCI~lDioxan
and she producs reacted with Z(NMe)DPhe-OH (5.12 mmol) in she
presence of HOBt (10.2 rnmol), water soluble carbodiirnide (6.1
mmol) and N-methylmorpholine in DMF (20 cm3) at 0C. Afser 18
hours the reaction was wor~ed up using ssandard procedures and the
product purified by flash chrornatography on silica with EtOAc -
pesrol (1:1). The pure alcohol 3 was isolased as a colourless oil
- (52%).
SUBSTi T UTE SHEET
wo 92/0~371 pcr/Gs91/o:47~
2f~9~8
26
(ivj The alcohol 3 (2.22 mmol) was dissolved in CH,C12/AcOH ~30:1)
and Dess-Mamn Per;odinane (4.5 mrnol) added. After 2~ nours a~
room temperature the reaction muxture was diluted with EtOAc and
poured into a solution of sodium thiosulphate (32 mmol) and sat.
NaHC03. The crude product was purified by flash chrornatography
on silica with EtOAc-petrol (3:7). The pure aldehyde _ was isoia~ed
as a colourless oil (75%).
(v) The aldehyde _ (1.6~ mmol) was dissolved in MeOH/H,O/AcOH
(90:9:1, 50 cm3) and hyarogenated over 59O Pd/C. The crude
material was purified by mplc on *Vydac Cl8 (15-25 Il) using
~eCN/H,O/TFA to give pure ~ (CH-851) as a white solid (780
mgj. Tlc EtOAc-Pv-AcOH-H,O (30:20:6:11), RF 0.66 on silica.
After hydrolysis at 110C/2' hrs with 6N HC1 peptide content based
on Cha was 40%. FAB mass spec [M+H]+ = 473 (Calc. mJz = 472).
EXAMPLE II
11 H-DPro-Phe-Lys-CONnBu2 (see Scheme 11)
(i) TcbocONSu (14.8 mmol) was added to a solution of
H-Lys(Z)-OMe. HCl (12.2 mmol) and triethylamine (14.8 mmol) in
CH,CI2 (50 cm3). After 3 hours at room temperature the reaction
was worked up using standard procedurcs and the product purified
by flash chroma~ography on silica using EtOAc - petrol (7:13). The
purc estcr 6 was isolatcd as a colourless oil (100%).
(ii) Diisobu~ylaluminium hydride (1.5 M solution in toluene, 50 rnmol)
was added to a solution of 6 (12.2 rnmol) in dry toluene (100 crn3)
at -78C over a period of 20 minutes. After a further 15 minutcs
methanol (10 cm3) was added followed by a saturated solution of
Rochellc's salt (100 cm3). After 2~ hours the reaction was wor~ed
up using standard procedurcs and thc product purificd by flash
chrornatography on silica using EtOAc - petrol (3:7). The pure
ald~hyde Z was isolated as a colourless oil (49%).
Tradc narnc
~U~3~ H 7''LJ'i E SHEET
wo 92/04371 2 ~ J ~ PCI'/GB91/01479
(iii) Potassium cyanide (18 mrnol~ and 1 M h~drochlorie aeid ~30 em;)
were added to 2 solution of Z (5.98 mmol) in ethyl acetate (30 em3)
After 18 hours at room temperature the reaetion was worked up
using standard procedures and the produet purified by flash
chromatography on silica using EtOAc - petrol (4:6). The pure
cyanohydrin 8 was isoiated as a colourless oil (88%).
(iv) A 4 M solution of HCl in dioxan (50 cm3) was added to a solution
of 8 (5.28 mmol) in dry me~hanol (15 cm3) at OC. After 18 hours
at room temperature an ice/water mixture (15 cm3) was added.
After 3 days at 4C soiid KHC03 wæ added. The reaetion was
worked up using standard proeedures and the product purifîed bv
flash chromatography on silica using EtOAc - petrol (11:9). The
pure ester 8b was isolated as a yellow oil (59%).
(v) Aetivated zine dust was added in srnall portions to a solution of 8b
(3.1 mrnol) in AeOHJH2O (9:1, 25 em3). After 1~ hours at room
temperature the zine was filtered off, the filtrate evaporated in
vaeuo and the residue wæ taken up in EtOAe. This solution was
washed with sat. ~JaHCO3, water, brine, dried (Na,SO4) and
evaporated in vaeuo. The amine 2 was isolated as a colourless oil
(85%).
(vi) The amine 2 (2.63 mmol) was aeylated with Boe-Phe-ONSu (3.04
mrnol) in CH2Cl2 (30 em3) at OC in the presenee of N-methyl
morpholine. After 3 hours the reaction was worked using standard
proeedures and the erude produet purified by flash ehromato~aphy
on siliea with EtOAelPet Ether (6:4). The pure ester 10a was
isolated æ a eolourless oil (92%).
SUBS, ITU T _ SHEET
WO 92/04371 2 0 ~ 0 8 5 8 pcr/Gs91/ol47~ ",
28
(vii) The Boc group of 10a (2.41 mmol) was removed using sat.
HClJDioxan and the product acylated with Boc-DPro-ONSu ~2.g'~
mmol) in CH2C12 (30 cm3) at OC in the prescnce of
N-methyl-morpholine. After 3 hours the rcaction was worked up
using standard procedures and the product purified by flash
chromatography on silica using EtOAc/Petrol (3:1). The pure ester
10b was isolated as a colourless oil (64%).
(viii) Li~hium h,vdroxide (1.6 mmol) and water (3 cm3) were added to a
solution of 10b (I.S4 mmol) in THF (30 cm3). After 4 hours a~
room temperature the THE was removed in vacuo, the pH of the
residue adjusted to pH 4 with 1 M citric acid and extracted with
CHCI3. The organic extracts were washed with brine, dried
(Na,SO4) and evaporaled in vacuo. The pure acid 10c was isolated
as a colourless oil (70%).
(ix) Pentafluorophenol (1.3 mmol) and water soluble carbodiimide (1.3
mmol) were added to a solution of 10c (1.07 mmol) in CH2CI2 (20
cm3) at OC. After 2~ hours dibutylatnine (2.1 mrnol) was added to
this solution,at OC and the pH adjusted to pH 9 with DEA. After
18 hours at room temperature the reaction was worked up using
standard procedures and the product purified by flash
chromatography on silica using EtOAc/Petrol (7:3). The pure
arnide 10d was isolated as a colourless oil (48%`).
(x) Dess-Martin Pcriodinanc (0.97 mmol) was added to a solution of
10d (0.52 mmol) in CH2C12 (100:1, 40 cm3). After 2 hours at room
temperature further Dess-Mamn Periodinane (0.52 mmol) was
added. After a further 3 hours the reaction mLsture was diluted with
EtOAc and poured into a solution of sodium thiosulphate (7.3
mmol) in water and sat. NaHCO3 were added. The crude product
was purified by flash chroma~tography on silica with EtOAc/pcsrol
(9:11). The pure keto amide 10e was isolated as a colourless oil
(57%).
SUBSII,~)E S~lEi~T
WO 92/04371 2 ~ 9 ~ 8 Pcr/cB91/01479
2c~
(xij The Boc group of 10e (0.24 mmol) was removed using sat.
HCUDioxan. The resultar.; produc; was dissolved in A.,OH~I-,O
(9:1) and hydrogenated over 5% Pd/C. The crude material was
purified by mplc on *Vydac Cl8 (15 - 25 ~1) using MeCNlH20~IFA
to give 11 (CH-1463 89.7 mg). Hplc, *Novapak C~8, 4 11 (8 x 100
mrn), linear gradient 20 -- 80% 0.1% TFA/MeC~' into 0.1 S~
TFAIH,O over 25 min at 1.5 ml min-l indicates the presence of two
epimers D-Arg (40%) ~t 11.2 min and L-Arg (60%) at 12.6 min.
After hvdrolysis at 110C/'2 h with 6N HCI. amino acid analvsis
Phe 0.93, Pro 1.07.
EXAMjPLE III
17 H-DPro-Phe-Arg-CH,S(CH,)4CH3 (see Scheme m)
(i) Boc-Arg(Z2)OH (46.1 mmol) was dissolved in dry THF (200 cm3).
N-methylmorpholine (50.85 mmol) and isobutyl chloroformate
(50.73 mrnol) were added at -20C. After 20 min. this mixture was
added to a solution of diazomcthane (0.1 mole) in Et2O at -5C.
After 2 hours the diazoketone 12 was isolated as a yellow solid.
(ii) The diazoketone 12 (46.1 mrnol) in dry THF was treated with HBr
(69.15 mmol) in EtOAc at -20C followed by addition of sat.
NaHCO3 after 45 mins. The crude product was extracted with
EtOAc and crystallised fTom EtOH to give pure
Boc-Arg(Z2)CH2Br, 13, (85%).
(iii) l-Pentanethiol (1.27 mmol) in dry DMF (5 cm3) was treated with
sodium hydride (1.4 mmol). After 30 rnins Boc-Arg(Z2)CH2Br 13
(1.27 mmol) was added-40C for 20 mins and -5C for 2~ hours.
After addition of 0.3 M KHS04 and extraction of the crude product
with EtOAc, flash chrornatograpy or silica with EtOAc - pet~1
(15:85) yielded tlA_ pure thiomethylene compound 14 as a colourless
oil (74%).
Trade name
SUBS, ITUTE ~HEET
WO 92/0437l 2 U 3 ~ 8 ~ 8 PCr/GB9~ 479
.,
(iv) The Boc pro~ecting group of 1~ (0.94 rnmol) was removed using sat
HCl/~ioxan and the resulting prociuc~ was acyiated with
Boc-Phe-OPfp (1.13 mmolj in CH2CI2 at OC in the presence of
DEA. The crude product was purified by flash chromatography on
silica with EtOAc - petrol (3:7) yielding the pure thiomethylene
analogue 15 as ~ colouriess oil (S5%).
(v) The Boc protecting group of 15 (0.5~ mmol) was removed using sat.
HCI/Dioxan. The resulting product was dissolved in D~IF and
treated with Boc-DPro-OH (0.63 mmol) in the presence OI- HOBt
( 1.05 mmol), water soluble carbodiirnide (0.76 mmol) and
N-methylmorpholine. After a standard work-up the crude material
was purified by flash chromatography on silica with EtOAc - petrol
(4:6) yielding the pure thiomethylene compound 16 as a colourless
oil (74%).
(vi) The Boc protecting group of 16 (0.38 mmol) was removcd using sat.
HClfDioxan. The resultant product was dissolved in AcOH/H2O
(9:1) and hydrogenated over 5% Pd/C. The crude material was
purified by mplc on ~Vydac C18 (15-25 ~), using MeCN/H20~ A
to give pure 17 tCH-574, 41 mg). Hplc, ~Novapak Cl8, 4~L (8 x 100
mm~, linear gradient 20 ~ 80% 0.1% TFA/MeCN into 0.1%
TFA/H20 over 25 min at 1.5 ml min-l indicates the presence of two
epimers D-Arg (<5%) at 9.8 min and L-Arg (>95%) at 11.1 min.
After hydrolysis at 150C/1.5 h with 6N HCl, amino acid analysis
Phe, 0.80; Pro, 1.00.
All analogucs in Table '' were synthesised by the descTibed method.
61 was synthesised by the oxidation of 60 with meta-
chloroperoxybenzoic acid.
Trade name
SUBSTITU T t- SHEET
wo 92/04371 21~ ~ O ~ ~ 8 PCr/GB91/0l479
EXAMPLE IV
'3 H-DPro-Phe-Arg-CH~OCH~(CF~)3CHCF~ (see Scheme IV)
(i) IH, IH, 5H-Octafluoro-l-pentanol (2.45 mmol) in dry DMF (8 cm3)
was treated with sodium hydride (1.83 mmol). After 30 mins the
bromoketone 13 (1.65 mmol) was added at -40C and left at this
temperature for 30 mins and -5C for 2~ hours. Addition of 0.3 M
KHSO4 and extraction with EtOAc gave the crude product which
was purified by flash chromatographv on silica using EtOAc - pe~rol
(15:85). The pure fluoroether 18 was isolated as a colouriess oil
(69%).
(ii) The fluoroether 18 (1.13 mmol) was dissolved in MeOH (40 cm3),
sodium borohydride (1.18 mmol) was added to this solution at 0C.
After 15 min 0.3 M KHSO4 was added and the m.ixture extracted
with EtOAc giving the pure compound 19 as a colourless oil (88%).
(iii) The Boc pro~ecting group of 19 (1.0 mmol) was removed with sat.
HCUDioxan. The resulting pr~duct was dissolved in CH2Cl~ and
acylated with Boc-Phe-OPfp (1.2 mmol) in the presence of DE~A at
0C. After a standard work up the crude product was purif1ed by
flash chroma~ography on silica with EtOAc - petrol (3:7) yielding
the pure product ~0 as a colourless oil (55%).
'iv) 20 (0.55 mrnol) was deprotected ~ith sat. HCI/Dioxan and acylated
with Boc-DPn~OPfp (1.63 mmol) in CH2C12 at 0C in the prcsence
of DEA. After a standa~d work up the crude product was purified
by flash chroma~ography on silica with EtOAc - petrol (4:6)
yielding the pure product 21 as a colourless oil (48%).
SUBSTI' UTc SIJ,EET
wo 92/04371 2 0 9 ~ 8 ~ 8 pcr/GBg1/o1479
(v) ' 1 (0.2~ mmol) was dissolved in CH,Cl,/AcOH (30:1 ) and
Dess-Martin Perio~linane (0.48 rnmoij was aaaed. Afier 2 hours a~
room temperature the reaction mixture was diluted with EtOAc ana
poured into a solution of sodium thiosulphate (3.5 mmol) in water
and sat. NaHCO3. The crude product was purified by flash
chromato~raphy on silica with EtoAc - petrol (7:13) yielding the
pure fluoroether '~ as a colourless oil (64%).
(vi! The fluoroether ~ (0.16 mmol) was deprotected and purified as
described in Example III (vi). Pure ~3 (CH-619) was isolated as a
white solid (50.9 mg). Hplc, *Novapak Cl8 411 (8 x 100 mrn), linear
gradient 20 ~ 80% 0.1% ~ .VMeCN into 0.1ao TFA/H,O over '5
mins at 1.5 ml min l indicated a single product (TR = 11.5 min).
After hydrolysis at 150C/1.5 hr with 6N HCl, ~mino acid analysis
Phe, 1.00; Pro, 1.2.
All analogues in Table 3 were synthesised by the described medhod
except 54 and 55 which were synthesised by methods oudined in
Schemes xm and XIV respectively.
EXAMPLE V
31 H-DPro-Phe-Arg-CH,N[(CH2)5CH3~2 (see Scheme V)
DL
(i) Boc-Arg(Z2)OH (18.5 mmol) was dissolved in CH2C12 (50 cm3).
To this solution at 0C was addcd trichloroethanol (20.35 mmol)~
water soluble carbodiimide (22.2 mmol) and
dimethylaminopyridine (0.93 mmol). After 3 hours the reaction
was worked up using standard procedures giving the pure
~ichloroethyl derivative 24 as a colourless oil (100%).
Trade name
SUeSTl i l,' I c SnEET
WO 92/04371 2 0 S ~ 8 pcr/GB9l/ol479
(iij ~ (18.1 mmol) was depro~ec~ed with sa~. HCVDioxan and acylated
with Boc-Phe-ONSu (27 2 mmol) in CH2CI~ a~ 0C in ~he presence
of N-methylmorpholine. After 3 hours the reaction mixture was
worked up using stanaard procedures and the crude producl was
purified by flash chromatography on silica with E~OAc - petrol (2:8)
vielding rhe pure product 25 as a white solid (97%).
(iii) '5 (17.1 mmol) was deprotecsed with sat. HCVDIoxan and acylated
wi~h Boc-DPro-ONSu (26.2 mmol) in CH,CI. a~ 0C in ~he
presence of N-methylmorpnoline. Af~er 3 hours the reaction was
worked up using standard procedures and the crude product purified
by flash chromatography on silica with EIOAc - petrol (35:65)
giving the pure protected tripeptide 26 as a colourless oil (94%).
(iv) Activated zinc powder was added to a solution of 26 (16.47 mmol)
in glacial acetic acid. After 3 hours at room temperature the zinc
was filtered off, the filtrate evaporated and the crude product
purified by flash chromatography on silica with EIOAc - petrol -
AcOH (74:25:1) giving the pure tripeptide 27 as a white solid
(91%).
(v) The prolected tripeptide 27 (15 mrnol) was dissolved in dry THF
(40 cm;), N-methylmorpholine (18 mmol) and isobutvl-
chloroformale (16.6 mmol) were added at -20C. After 20 mins the
mixture was added to a solution of diazomethane (35 mmol) in Et10
at -5C. After 2~ hours the diazoketone 28 was isolated as a yellow
oil.
(vi) The diazoketone 28 (15 mmol) in dry TE~ was treated with HBr
(22.5 mmol) in EtOAc at -20C followed by addition of sat.
NaHCO3 after 45 mins. The cmde product was extracted with
EtOAc and purified by flash chmmatography on silica with EtOAc -
petrol (1:1). The pure bromoketone 29 was isolated as a white solid
(72%).
SUESTi, '~ H c SHEET
WO 92/04371 2 ~ ~1) 8 5 8 PCr/GB91/0147~
(vii) I)ihexylarnine (1.25 mmol) and NaHCO3 (0.8 mmol) were added to
a solution of bromoke~one 29 (0.23 mmol) in dr,v l~lF (5 cm3).
After 18 hrs at room temperature 0.3 M KHS04 was added to the
reaction mixture and the crude product was pu~ified by flash
chromatography on silica with EtOAc - petrol (35:6~). The
protected aminomethylene ketone 30 was isolated as a yellow oil
(54%).
(viii) The aminomethvlene ketone 30 (0.1' mmol) was depro~ec~ed and
purified as described in Example III (vi). Pure 31 (CH-694) was
isola~ed as a white solid (41 mg). Hplc, linear gradient 20 ~ 80%
0.1~7o TFA/MeCN into 0.1ao TFA/H,O over 25 mins at 1.5 ml min~;
indicated the presence of ~wo epirners D-Arg (50%) at 11.2 min and
L-Arg (50%) at 12.5 mins. After hydrolysis at 110Cl2' hrs with
6N HCI, arnino acid analysis Phe, 0.91; PTO, 1.09.
A11 analogues in Table 4 were synthesised by the describcd metho~
The required arnines were synthesised by standard synthetic
methods sucn as reductive amination and ~he Curtius rearrangemcnt.
EXAM.DLE ~tl
40 H-DPro-Phe-Arg~Gly-Pro-NHEt (see SchemeVI)
(i) H2C(CO2Tce)2 (6.81 mmol) was treated with sodiurn hydride (5.67
mmol) in dry THF (30 cm3). After 45 mins the bromoketone 13
(4.52 mmol) was added at -5C. After 2t hours 0.3 M KHS04 was
added. the crude product extracted with EtOAc and purified by flash
chromatography on silica with EtOAc - petrol (2:8). The pure
Boc-Arg(Z2)CH,CH(CO2Tce)~ 32 was isolated as a colourless oil
(83%).
(ii) Achvated zinc was adided to a solution of 32 (3.65 mmol) in glacial
acetic acid. After 2t hours at room temperature the zinc was
filDd off, the filtrate evaporated and the diacid 33 isolated.
SU''ST,TUT-- SHEET
wo 92/04371 2 ~ pcr/GB~ 47~
(iii) A solution of the diacid 33 in toluene was heated at reflux for 45
mins. The solvent was evaporated and the crude produc~ purified by
flash chromatography on silica with EtOAc - petrol - AcOH
(60:39:1). The Boc-Arg(Z2)~Gly-OH 34 was isolated as a
colourless oil (70% from ~).
(iv) Trichloroethanol (2.82 mmol), wa~er soluble carbodiimide ( '.~1
mmol) and dimethylaminopyridine (0.117 mmol) were added to a
solution of 31 (2.34 mmoi) in CH~CI, (50 cm;) at 0CC. After 2.
hours the reaction was worked up using s~andard procedures and the
crude product purified by flash chromatography on silica with
E~OAc - petrol (85:15). The trichloroe~hyl derivative 35 was
isolated as a colourless oil (83~o).
(v) The Boc pro~ecting group of 35 (1.85 mmol) was removed using sat.
HClJDioxan and the resulting product acylat~ ' with Boc-Phe-OPfp
(6.04 mmol) in CH~CI, in the presence of DEA. After 2 hours the
reaction was worked up using standard procedures and the crude
product purified by flash chromatography on siiica with E~OAc -
petrol (2:8). The pure product 36 was isolated as a colourless oil
(85%).
(vi) The Boc protecting g~up of 36 (1.58 mrnol) was removed using sat.
HCl/Dioxan and the resulting product acylated with
Boc-DPro-OPfp (5.12 mmol) in C~2Cl2 in the presence of DEA.
After 2 hours the reaction was wor~ed up using standard procedures
and thc crude product punfied by flash chromatography on silica
with EtOAc - petrol (35:6~i). The pure produc~ 32 was isolaud as a
colourless oil (79%).
(vii) Activated zinc dust was added to a solution of 37 (1.13 rnmol) in
glacial acetic acid. After 2~ hours at room temperature the zinc was
filtered off, the filtrate cvaporated and thc crude product purificd by
flash chrornatography on silica with EtOAc - petrol - AcOH
(70:29:1). The product 38 was isolatcd as a colourless oil (76%).
SUB~ T ITUTE SHEET
wo 92/04371 2 ~ 9 ~ 8 ~ 8 pcr/GBg1/~1479
(viii) The protected keto isostere 38 (0.26 mrnol) was converted to its Pfp
ester by treatment with Pfp-OH (0.29 mmol) and water soluble
carbodiimide (0.31 mmol) in CH,C12 (8 cm3) at 0C for 2~ hours.
This Pfp ester was coupled at 0C to H-Pro NHEt . HCl salt (0.78
mrnol) in the presence of DEA. After 18 hours the reaction was
worked up using standard procedures and the product purined bJ
~lash chromatography on silica with CHCl3-~leOH-AcOH (97:2:1).
The product 39 was isolated as a colourless oil (91 C~o).
(ix) The protected keto isostere containing anaiogue 39 (0.23 m.mol) was
deprotected as described in Example m (vi). Pure 40 (CH-595) was
isolated as a white solid (40 mg). Hplc, linear gradient 10 ~ 50%
0.1% TFA/MeCN into 0.1% TFA/H,O over 25 mins at 1.5 ml min l
indicated the presence of two epimers D-Arg (46%) at 10.1 min and
L-Arg (54%) at 11.7 min. After hyarolysis at 150C/1.5 hrs with
6N HCl amino acid analysis Phe, 0.91; Pro, 1.09.
All analogucs in Table 5 were synthesised by the described method.
EXAMPLE VII
45 H-DPro^Phe-Arg-Chg-NH, (see Scheme VII)
(i) Boc-Phg-OH (19.9 mmol) was dissolved in AcOH/H~O (9:1, 100
cm3) and hydrogenated over Rh/C at 60 p.s.i. for 3 days. The
catalyst was filtered off and the solvent removed to give
Boc-Chg-OH 41 (100%).
(ii) Water soluble carbodiimide (4.1 mmol) and HOBt (4.3 mmol) were
added to a solution of 41 (3.9 mmol) in CH~Cl,IDMF (2:1, 60 cm3)
at room temperature. After 30 mins 35% arnmonia solution (0.8
cm3) was added. After a further 3 hours at room temperature the
reaction was wor~ed up using standard procedures and the product
recrystallised from EtOH to give the pure arnide 42 as a whitc solid
(60%).
SUE~STiTl,' T E SHEET
WO 92/04371 ~ .3 8 Pcr/cB91/0l47~
(iii) The Boc group of 42 (0.77 mmol) was removed with sa~.
HCVDioxan to give the amide 43 as a white solid (100%).
(iv) The protected tripeptide 27 (0.38 mmol) was dissolved in DMF (5
cm3). 43 (0.76 mmol), HOBt (0.76 mmol) water soluble
carbodiimide (0.46 mmol), and N-methylmorpholine were added at
0C. After 18 hours at room temperature the reaction was worked
up using stand~rd procedures and the product purified by flash
chromatography on silica with CHCI3/MeOH (99:l). rne pure
pro~ected tetrapeptide 44 was isolated as a white solid (69%).
(v) The protected tetrapeptide 41 (0.27 mmol) was deprotected and
purified as described in Example III (vi). Pure 45 (CH-640) was
isolated as a white solid (58 mg). Hplc, linear gradient 10 1 50%
0.1% TFA/MeCN into 0.1% TFAJH20 over 25 rnin at 1.5 ml min-l,
single peak detected at 14.2 tnin. After hydrolysis at 110C/22 hrs
with 6N HCl, amino acid analysis, Arg, 0.96; Phe, 1.00; Pro, 0.95.
All analogues in Tablc 6 were synthesised by the described method
or by other standard peptide coupling methodology. (M. Bodansky
& A. Bodansky, The Practic~ of Pep~ide Synthesis, Springer-Verlag,
1984)
EXAMPLE vm
47 H-DPro-Phe-Arg-N[(CH2)sCH3](CH2)3Ch (see Scheme Vm)
(i) Thc protccted tripcptidc 27 (0.32 mmol) was dissolved in DMF (5
cm3), HN[(CH2)5CH3](CH2~3Ch (0.96 rnrnol), HOBt (0.64 col),
water soluble carbodumide (0.38 rnmol) and N-methylmorpholine
were added al OC. After 18 hours at room temperature the
reaction was worked up using standard proccdures and thc product
purified by flash chrornatography on silica using EtOAclHexanc
(4:6). The amide 46 was isolated as a colourless oil (32%).
SU5~,TH 1_:-, c SHEET
W O 92/04371 2 ~ 9 ~ ~ 3 8 PC~r/GB~1/01479
38
(iij The pro~ec~ed tripeptide arnide 46 (0.1 mmol) was deprotected and
purified as described in Example I~ (vi). Pure 47 (CH-~85) was
isola~ed as a white solid (17 mg). Hplc, linear gradient 40 - 90%
0.1% TFAtMeCN into 0.1% TFAJH70 over ~5 min at 1.5 rnl min-l,
single peak detected at 10.7 min. After hydroiysis at 110C for 22
hrs with 6N HCI, ~nino acid analysis Phe, 1.01: P~, 0.99.
All analogues in Table 7 were syn~hesised by the described method.
The required tripeptide presursors were iynthesised in d similar
manner to 27 or by standard peptide coupling methodology. (M.
Bodansky & A. Bodansky, The Prac~ice of Peptide Synthesis,
Springer-Verlag, 1984.) Required arnines were either cornmercially
available or synthesised via the Curtius rearrangement.
nBu-DPro-OH for 181 was synthesised by reductive arnination.
SU~STI I U-, E SHEET
wo 92/04371 ~ T~ ~ ~ 8 PCr/GBg1/ot47g
Scheme I
(Syn~hesis of comDound O
æ (i) isuococl/~TMMrrHF z,
Boc-Arg-OH Boc-Arg 2 O~
N;IBH~,H~O
(ii) HC!/Dio,Y~n
Boc-Ch2 ON'Su/~i~l.M
CH,CI~
Z~ (iiij l-lCI/Din,Y;~n ~ Z.
Z(~TMe)DPhe-Ch~-Arg ROH ~ Boc-Cha-Ar~ 2OH
Z(NMe)DPhe-OH
3 HOBt/wscd
NMMtDMF
(iv) Dess Martin
Periodinane
CH.CI~tAcOH
Z, (v) H" Pd/C
Z(~Me)DPhe-Ch~-Arg-H ~, Me-DPhe-Cha-Arg-H
MeOHlH~O/AcOH 5
SlJ~S T i T ~, . E Si~EET
WO 92/04371 2 ~ ~ a ~ ~ 8 pcr/GBg~ 47s~
Scheme I I
(Syn~nesls or comwuna ~)
Z ('~ cbocOXSu Z
H-L~,s-O~Vle . ~iCI ~ Tcboc-Lys-~)~le
E:,.',/CH,C;, 5
~ii) DIBALToluenc !
,z iiii., ~c~ ,z
.. boc-LysQ~r~ ~ T~boc-Lys-H
HCI/H,O
Q~ EtOAc
(iVJ ~CIJDioxan !
~leOH/0C
H,0/4C ~
7 (Vj Z.~/AcOH Z
Tcooc Lys~CO,~Ie ~H-Ljs~CO~lc
8b 2
(vi) Boc-Phe ONSu
CHCl2/N~I ~
Z (vii-~ HCllDioxan Z
Boc-DPro-Phe L~s2~CC~Me 4 Boc-Phe-Lys~HCO,.?~e
Boc-DPro-ONSu
VC~.Cl~ 10a
(viiij LiOH/H,O ¦ .
1~ 1
- Z (ix) PWH/wscdJCH,CI, Z
Boc-DP;o-Phe-Lys~rO2H . ~ Boc-DPro-Phe-Lys~CON"Bu~
!Cc nBu,NHlD~ 10d
(x) Dess Maran
Pcnodinane
CHCl,,/AcOH r
(xi) HC~JDioxan ,Z
H-DPro-Phc-Lys-CONnBu, ~ , Boc-DPro-Phe-Lys CONnBu,
H2, PdJC
Ll . AcOH/H20 lOc
SUBSTIIUTE SHEET
WO 92/04371 j . PCI/GB91/0147
Scheme I~T
(Svnthesis of eompound ~)
Z.(i) iBuOCOCI/~MrrHF Z~
Boc-Ar~-OH ~Boe-Ar~-Cr~.
(ii) CH.N~ . ~
-
(ii) HBr/E:OAe
THF I
Z~ (iii) CH~(CH~)S Na æ'
Boe-Arg-S(CH,)4CHt Boe-Ar-CH.Br
D~LF ~.
(iv) HCl~Dioxan
Boe-Phe-OPfp
DIEA/CH2CI,
æ (v) HC.JDiox~n æ
Boe-Phe-Arg-CH~S(CH~)4CH~ ~ Boc-DPro-Phe-Arg-C~I S(Cr.. ).C-~
Boc-DPro-OPfp 16
-- DEA/CH~CI~ --
(vi) HCI/Diox~n
H, Pd/C
AeOH/H.O r
H-DPro-Phe-Ars-CH S(CH~)4CI-I,
17
CU~S-, I I U ~ SI IEET.
WO 92/04371 2 a 9 ~ ~ a ~ PCI/CB91/014
42
Sc;~leme ~V
(Synrhes2s of compound ~)
z.(i) CHF.(CF.)3CH,O ~'~; æ
Boc-Arg-CH ,Br ~ Boc-Arg-CH~OCH~CF~)3C~F~
DMF
(ii) NaBHJ~eOH i
æ (iii) HCl,'Diox2n æ
Boc-Phe-Ar ,HCH~OCH~tCF.)3CHF, --- Boc-.'~rt~HrH.OCH.(CF.)~Cr.r~
~n ~oc-Phe-OPf? 1O
DIEA/CH,CI.
(iv) HCI/Dioxan
Boc-DPro-OPfp
DIEA/CH,Cl,
æ
Boc-DPro-Phe-ArgHCH,OCH2(CF2)3CHF2
21
(v) Dess Martin
Pcriodinane
CH2Cl~!AcOH I
Boc-DPro Phc-Arg-CH20CH2(CFi)3CHF2
22
(vi) HCVDiox~n
H2 PdlC
AcOHIH20 ~
H-DP~Phc-Arg-CH~OCH2(CF2)3C~F2
23
Sl ;Li~ i i I ' J i-_ S~EET
WO 92/04371 ~ ) 8 PCT/CB~1/01474
43
Scheme ~'
(Svn~nesis ot compound l `,
Z~ (i) Tce-OH/wscd Z.
Boc-Arg~-OH ~Bor-Arg-OT-^
D ~IAP~CH.C;. , .
(iij HCUDiox:~n
30c-?he-O~iSu I
CH.C!./~
Z~ (iii`) HClJDiox~n Z.
Boc-DP.o-Phe-Arg-OT.o Boc-?ho-Arg-O 1 C'
~$ Boc-DP o-ONSu ^c
-- CH~CI~
(iv) Zn/AcOH
Z, iv) BuOCOC,/~ I/T.. - ~,
~oc-DP o-Ph~-Arg-OTc ~ Boc-DPro-Phe-Arg-Cr~'
tEt.O ' 8
(vi) HBr/E~OAc
~,F
z (vii) HN[(CH.?sCH312 Z'
Boc-DPro-Ph^-Arg-CH~N[(cH~)scH3]2 ~ -- Boc-DPro-Phe-Arg-CH.Br
~HCO~/l'HF ,,9
HC~lDiox~n
I H" Pd,'C.AcOH H.O
H-DPro-Phe-Ar~-CH~N~(CH2)5cH3l2
3l
~;~JB~T,T~ SH~ET
WO 92/04371 2 ~ 9 ~ ~ ~ 8 Pcr/GB91/~)147~
Scheme VT
(Svn~hesis of comr)o~nd 4())
Z, ;i`~ N~lr CH(CO~TceJ~ Z
Bc~-~rg-C~ r ~~ec- ~rg-ch~cH(cc)~
3 _
(ii') Zn/AcOH
Z. ~ iii) Toiuene / ~ Z~
Boc-Arv'Gl,v-OH ~ -- 3oc~Arg-cH~cH(
(iv) ¦ Tee-OI llw~c~
l D~AP/CH~CI~
z ~ ;v) HCllDiox~n Z~
Boc-Arv!~Gi,v-OTce Boc-Phe-Arg~Giv-OTce
~c Boc-Phe-OPf?
~~ DIEA/CH.C!.
(vi) HCI/Diox~n
Boc-DPro-OPfp
CH~CI,~DIEA ~ r
Z2 (vii) ZntAcOH æ
Boc-DPro-Phe-Arg~Gly-OH - ----Boc-DPro-Phe-Arg~'~Gl,v-OT.e
38
(viii) ¦ PfpOHtwscdtCH.CI.
HPro NHEt/DEA
~ z, (ix) HCltDiox~n
Boc-DPro-Phe-ArgXGly-Pro-NHE~ ~H-DPro-Phe-Arg~Gly-Pro-NHE~
H2, PdtC
39 AcOHlH,O 40
SUBSTITU ~ - SHEET
WO 92/04371 2 3 ~ 8 PCr/GB91/01479
Seheme vn
(Synthesis of compound ~)
(i) H2, Rh/C
Boe-Phg-OH ~ Boe-Chg-OH
AcOH/H~O/60 psi
(ii) HOB~Jwsed
D~IF/CH.Cl,
~H3
(iii) HCVDioxan ~r
H-Chg-NH. HCI ~I Boe-Chg-l~iH.
43 42
Z2
(iv) Boe-DPro-Phe-Arg-OH 27
HOBt/wsedJDMF
Z~ (v) HCl/Dioxan
Boe-DP`To-Phe-Arg-Chg-.~H, -- H-DPro-Phe-Arg-Chg-NH~
44 H,, PdlC 45
SUE3;~-ri, U I E SHEET
WO 92/04371 2 ~ 3 a 8 ~ 8 6 PCl'/GB91/1)147
Scheme Vl~
(Synthesis of compound ~ ~
z" (i) HN~(CH,)5CH~j (C~ Ch z,
Boc-DPro-?he-~rg-OH ~ ~ Boc-DPro-Phe-Ar~-.`.[(C'.i.~5CH3j(C','.;3C:.
HOBt/wscd~lF
(ii`) HCI/Diox~n
H. PdJC
AcOHlH.O
H-DPro-Phe-Arg-N[(CH.)5CH3](CH,)3Ch
,
SUE,STi,UTE SHErT
WO 92/0~371 2 ~ 9 i~ ~ ~ 8 PCI/GB91tO1479
Scheme IIY
;Svn~r,esis o~ com~ou-d 18
Boc-Arg30H
¦ Dcss-Ma,~n
I Penodinane
CH.Cl,;AcOH
~ CF3CF,CF,I Z~
30c-Arg-H ---~Boc-ArgHCF1CF.C~:,
Z. ~ HF,~ souna
HClJDioxan
Boc-Phe-ONSu
~TMMJC~cl2 ~
~ HC'vDioxan Z~
8c~c-DP~o-?h.:-ArgHCF2CF,CF; ~ Boc-Phe Arg~CF.CF,CF,
Boc-DPro-O~Su
~IC~.Cl~
Dess Ma~n
Penoainane
CH2CI,JACOH
æ HCVDioxan
30c-DP o-Phe-Arg-Cr.CF1G3 ~-H DPro-Phe-Arg-CF1CF2CF3
H ,. Pd/C
AcOH/~I.O 1 8
Sl i3S T iTU I E SHEET
wo 92/04371 2 ~ 9 ~ ~ ~ 8 Pcr/GB91/0147g
Scheme X
(Symhesis of compound 49)
~- ZnJAcOH . Z~
Boc-DPro-Phe-Arg-CH,Br ~ Boc-DPro-Phe-Arg-CH~
'9
HCVDiox~n
H,, PdJC
AcOHlH~O i
H-DPro-Phe-Arg-CH3
49
SUBST3TUTE SHEET
WO 92/04371 ~ ~) 'j 8 PCr/CBgl/n147~
Scheme Xl
(SynLncs~s o~ compound S~)
Z, Na~ O~)-OMe Z,
30c-A~g CH2Br DMF Boc-Arg-C}~.0 ~ OMc
(i) .~'aBHJMcOH
(ii) TBDMS TriflaLe
Lu~denelcH2cl2
Z' ~ CcsTic 3mmonium ni~aLc æ
30c Arg ~ C~i~O}~. ~i C~/H O 30c Arg~D~sc~o~ oM~
I ~1) 3~ss Mamn Peno~nane
CH,Cl~/AcOH
(.i) B. 2/McOH/H20/~aHCO3
Z.~
Boc-ArQ~'sCCi,.~,lc ~C~)io~an Z'
Boc Cha4NSu
CH2CI,~NMM
~Cl'Dioxan
Boc DPro4NSu
~MhU~l2Cl2 ~ ~
,- ~'aO~ OH æ
Boc DPro Cha ArglBDMsco~.H Boc DPro-Cha Arg~CO.Me
OC
(i) ~OH~wscdJCH,C;,
(ii) ~u,,~r~
Boc-DP ro Cha-Arg~CON"BU2 ~ Bx DP~-Cha-Arg-CON~Bu2
(u) Dess-Ma~ssn
P~iodinane
CH2CI2/ACOH
(i) HC~ioxan
(ii) H2PdtC
MeOH/H20
HCl ~ ~
H-DPr~Cha An CO2N'Bu,
SO
SUBSTlTUTc ~ -tT
WO 92/04371 2 a 9 a 3 ~ 8 ~ pcr/GB9l/ol47~
~cheme XII
;Svnur:esis or compour.c ~j
iBuOCOC!,~IJl~ Z
On~-OH - ~ ~oc-Om3O~
~aBHJH,O
H~ PdJC
.~cOH/H~O
Bzl
Boc-Om~OH hCHO/CH~C~ /~csO~ 3Oc~ OH
~aBH~eOH
Z ONS u
E~ ~/CH Cl
I
BzL ZDess-:.~arn;l Pe:ioain~ne Bzl.Z
Boc-C;~ ~OH Boc-O~n-H
CH,Cl1,AcOH
H-C(C0713zl)~ N~mD an~exCHfC~ 2
nHexV60C
CO-H)~ H, Pd/C ~Hex.C(CO,Bz!),
MeOH
~,rroluene
n~ RedAl~Toluene
.. cx~CHCO,H ~ nHcx~CHCH,OH
hlsCVEt,.~/C~a, ~
, ~
nHcx~C~CH2Br LiBrlAcctone nHcx ,CHCH~OMs
5''
( cont . )
SUBSTITUTE SHEET
WO 92/04371
2 ~ 5 ~ PCr/GB91/0147~
,
Scheme XII (corlt.)
~ex.~ .C~i,BI -~ Boc OrnC ~.C.~.'.eY.
HCl/Dioxan
Boc-Phe ONSu
CH2Cl~,~MM
30C~ r~e.~ r~ r.C'v'Diox~n Bz!. Z
.. . .... ~.... x. ~ Boc-Phe C~:l~CH.C'.~'-^x
Boc-DP~o-O~'Su/N~
CH,CI,
H. PdlC
.~,cOH/~G
CH,S-C ~ ~ z
Boc-DPro-?hc- irn~HCX~C.~n.~-x. EtOHJ~ ~ Boc2-DP~o Phe-ArgQ~CY.~CY.nHc%,
Dess Manin Penodinane
CH2a2ACOH
H-DP o-?he-~g-CH,C~nH:x, ~ HCllDioxan Boc DPro Phe-Arg-C~l.C~Hex~
H, Pd/C
c, AcOEVH.O
SUE3STITUTE SHEET
2 ~ 9 0 3 5 8 PCI/GB91/0147
Scheme xm
(Synthesis of compound ~)
~ PhCOCO~HtICF æ
Boc-DPro-Phe-Arg-CHqBr ~ ~Boc-DPro-Phe-Arg-CH,OCOCOPh
DMF
'9
~HCOJH,Orl~F
æ CH,(CH,)5VAg,O Z~
Boc-DPro-Phe-Arg-CH,O(CH,)cCHl ~ Boc-DPro-Phe-Arg-CH~OH
CH,CI,
HCU.Dioxan
~ H2, Pd/C AcOH/H2O
H-DPro-Phe-Arg-CH20(CH2)sCH3
54
SUBSTITUTE SHEET
WO 92/04371 2 ~ 9 ~ PC~/GB91/01479
Scheme XlV
(Synthesis of compound ~)
Z^ PhOH/KF Z'
Boc-DPro-Phe-Arg-CH,Br D~IF ~ Boc-DPr~Phc-Arg-CH~OPh
,9
HCVDioxan
H, Pd/C AcOH/H.O ~
H-DPro-Phe-Arg-CH,OPh
SUBSTITUTE SHEET
WO 92/04371 ~ ~ ~ a ~ ~ 8 pcr/GB9l/o147g
ABBREVIATIO\IS l,SED
Abn 3-Az~bicvcio~3 ~ non3ne
Ac ~ce:~ i
' cOH Ace:ic ~cid
Ad~ Ad:lm n ~ i;mine
Ah I ~-Aminone < lnoic ~cid ( ~i'orie~c no
Boc ~ert-Butviox-c~rbonvl
Bu Butvl
Ch Cvciohexvl
Ch~ Cvclohe~,vi3i~nine
Chg C,vclohexvlglvcine
Cpr Cvclopropvl
DIEA Diisopropvlethvl~mine
D~1AP '-Dimerhvl:lmino-pvridine
3~F Dimemvlform~mide
E;OAc E:h,vi 3cet~te
FAB Flst A~om Bomb3rdment
~-Fph 4-Fluorophenvl~l~nine
Hch Homoc,vcloheYvl~l,qnine
HOBt l-Hvdroxvbenzorriqzole
hplc high perrorrn3nce liquid chrom3rogr3pilv
Hvp 4-Hydroxyproiine
'Hvp trlns-4-~lvdroxyprniille
SUBSTI I UTE S~EET
wo 92/04371 2 ~ 8 PCT/GB91/0147
ke:o isoat_r. -COC~ -
.~ Ie Methyi
.\ leCN Ace:oni~rile
.~leOH Meth;lnoi
;npic medium pressure ( ~ree~tive I iicuid .: .om~to~ ?n~
pn~hyi;~ nine
IM i~-~vlethylmorphoiine
.~c, ~leopentyh~iycine
0~: 0~
OH H-droxy isoste.e -CY.O~i-
ONSu hydroxysuccinimide
Petrol Petroieum e~her 60 - ~0C
P.fp Pen~ iuorophenyl
Phe-4NO~ '-Nitrophenyi~l~nine
Ph~ Phenyi~lycine
P.c Pipecolinic~cid
Pip Piperidyl
Py Pyridine
R Reduced isostere -CH~-
S~r S~rcosine (N-methyl ,iycine)
T~3AF Tetr~butyl~mmonium fluoride
TBDMS ~ert-Bu~yldimeshyisilvi
Tcboc ( I -Dime~hyl- I -trichloromeshyl)eshoxy c~rbonyl
Tcc 2.~ ~-Trichloroethyl
SUBSTI-rUTE SHEET
wo 92/04371 pcr/GB9l/ol479
2 1~ 5 8
n~!a i . 3 . ~ -T, .~.c~n ~ lhe x~n- droa~epy l
Tesahyarofuran
I hi Thicnyialanine
.ic ~ 3~'-Te~~h-aroiso~inol:ne-3-car~ox-ii. ac:d
~i. L~in ia-e. cl;:oma~og~pny
wscd water soluble carbodiimide
Z BenzyloxycarDonvl
!; G ; ~
SUB~; ~ ITUTE SHEET
WO 92t04371 _ 2 i3 9 a ~ 5 ~ pcr/GB~l/o147~
BIOLOGICAL AC~I'~'IT~
Compounds were ested n v-l-o ~~ ~he Col owi-._
ac~ivities using stanaard procedures:
(a) Inhibi.ion of human . ssue kallikrein ?lasma kall Xrei~
and mast cell tryptase hydrolysinc ~he ch~omoaeni~
suDstrates 5-2266 S-2302 and S-2266 -espec~:~ely ~e_hs~
s adaDted ~-om t~at of ~ohansen u ~ et 21., ~ n~
ss. Reac. la86 ~ 185-1a2). A series Oc 2easu.emen~s
were car~ied out usina a -.umber o _ e_en.~ -.h_- =^~
concent~ations and a least .wo di feren. subs_~a=e
concentrations. The inhlbitory constan~ Ki was
determined a-zphically usina a Dixo~. -lc. (M. ~ixo-
~iochem. ~. 195~ 5~ ~o)
(b) Inhibi__~~ of kinin release from low and hich ~oiec--ia-
weight kininoqens by tissue and plasma kallikrein
respectively. A series of measurements were carried out
using two subst.ate concent_ations. ~he act vity is
calculated 2s the amount o~ kinen released per m nute
this beina determ-ned by -adioimmu-.oassay usinc
polyclor;al antibodies. The inhibitory ~onstan~ Ki was
deter~ined graphicall-l usina a Dixon plot.
All the examples ln ~ables 1 - 9 have Ki values ln the
range 10 ~ - 10 3 M against one or all of the enzymes in
the chromogenic assay.
In vivo ac~ivity has been tested in well-established
pharmacology models of asthma based on the sensitised guinea
pig. A selection of these hibitors representing -he
different chemical types pro ~ to be highly effect ve in
blocking both the acute phase response and the late phase
reaction their efficacy being comparable or superior to those
of the topical steroids and B2-agonist currently used in
asthma therapy.
SUBSTITU-I E SHEtT
WO92/04371 2 ~ 9 ~ 3 ~ ~ PCT/GB91/0147~
When the compounds of _he present invention are used as a
medicine, there are no critical limitations to ~he
administration methods. The present enzyme inhibitor can 3e
formulated by any conventional method in pharmaceutics. ~or
example, the present enzyme inhibitor may be applied in any
conventicnal manner including int-avenous inject~on,
intramuscular lnjectlon, instlllation, orai administration.
respiratory inhalation, instillation, -hinencnysis, ~na
external sKin .rea~ment. ..lthouah ~here are no c-lt-^zl
limitations to the aàminls_-atlon dosage, ~he suitable doszae
is 1 to 1000 mg/day/person.
SUE~Ti';'u"l E SHEET