Note: Descriptions are shown in the official language in which they were submitted.
WO 92/01870 rCl/US91/-7--~
2~9231~
PLATELET AGGREGATION INHIBITORS
Fleld ot the Inventlon
The present invention relates to inhibitors ot platelet aggregation. Specitically, the
5 InvenUon is dlrected to peptides comprising the tripeptide sequence -Lys-Gly-Asp- capable ot
actin~ as antagonlsts o1 the 1inal common pathway o1 platelet aggregation and that act as
potent antithrombotics. The invention 1urther relates to therapeutic applications of these
inhibltors in diseases 1Or which blocking platelet aggregation and intracellular adhesbn is
indicated.
Background of the Inventlon
Platelets are particles ~ound in whole biood known to participate in thrombus forrnatbn
and bbod coagulation. A membrane spanning glycoprotein receptor, GP llb Illa, is present on
the surface of platelets and is known to be invoived in the coagulation ;orocess. GP llb Illa is a
non-covalent, calcium ion dependent heterodimer complex composed ot alpha and beta
1 5 subunits (Jennings, et aL, J. Biol. Chem. (1982) 257, 10458) capable of binding protein
li~ands. This glycoprotein receptor contributes to normal platelet tunctbn through
interactions with protein ligands containing the tripeptide amino acid sequence Arg-Gly-Asp
~RGD).
One protein ligand known to be important 1Or thrombus formation and containing the
2 0 RGD sequence is tibrinogsn. Fibrinogen contains two RGD sequences bcatect at Arl95-97
and Au572-574 (Doolinle, R. F., Watt, K. W. K., Colnrell, B. A., Stron~, D. D. and Riley, M.
(1979) Nature 280, 464468) that have been shown to interact with the GP llbllla receptor
(Hawiger,et aL, Biochemistty, 28, 2909-2914 (1989). A third regbn ol tibrinogen,corresponding to the 1inal 1 2 residues ,400-411, o1 the gamma chain carboxy terminus and
2 5 having the sequence HHLGGAKQAGDV, has also been demonstrated to bind with GP llb Illa
(Kioczewiak, M., Tlmmons, S., Lukas, T. J., and Hawber, J. (1984) Biochemistry 23,1767-
1774). Evldence ior the Involvement ot both the RGD and gamma chain regions in binding
with GP llbll4 Is largely derived trom binding and inhibitbn data and 1rom studies with
synthetic RGD pep~ides (Gartner, T. K. and Bennen, J. S. (1985) J. Biol. Chem. 260, 11891-
3 0 11894; Pbw, E. F., Pierschbacher, M. D., Ruoslahti, E., Marguerie, G. A., and Ginsberg, M. H.
(1985) P~oc. Nat. Acad. &L USA 82, 8057-8061; Haverstick, D. M., Cowan, J. F., Yamada, K.
M., and Santoro, S. (1985) Blood 66, 946-952; and DSouza, S. E., Ginsberg, M. H., Lam, S.-
C. T., and Pbw, E. F. (1988) J. Bbl. Chem. 263, S943-3951) and gamma chain carboxy
terminus anabgs (Kioczewiak, M., Timmons, S., i3ednarek, M. A., Sakon, M., and Hawiger, J.
3 5 (1989) Biochemistry28, 2915-2919). In this latter strdy, all amino acid replacements in the
gamma chain carboxy terminal dodecapeptide reduced inhibitory activity o1 the analog except
replacement ot Ala408 (ie. immediately preceding GD) with Arg which increased inhibitory
potency 6-told (see also Timmons, etal.., Biochemis~ry 28 2919-2923 [1989]).
The interaction o1 GP llb Illa with 1ibrinogen is stimubted by certain 1actors released or
4 0 exposed when a blood vessel is injured. Multiple 1actors, including a variety o1 physioiogic
WO 92/07870 , PCI'/US91/07809
~ )92~1~ 2 ~
stimuli and soluble mediators initiate platelet activation via several pathways. These pathways
have æ a common final step the activation of the GP llb "la receptor on the piatelet surface
and its subsequsnt binding ~o ~ibrinogen tollowed by aggregatbn and thrombus tormation.
By vlrtue of these interartions GP llb Illa is an important component of the platelet aggregation
syslem (Pytela e~ al. Scicnce (1986) 231 1559). Therefore inhibition of the interaction of
GP llb Illa with Arg-Gly-Asp containing pro~eins such as fibrinogen is one way o~ modulating
Ihrombus ~ormation. An inhibitor which prevents this binding interaction wouid antagonize
platelet activatbn by any stimulus and therefore wouid have important antithrombotic
properties.
1 0 It is known however that proteins and peptides containing the RGD sequence are
also recognized as ligands for a number of other cell adhesion receptors in additbn to GP llb
Illa. These cell adhesion receptors comprise a tamily of heterodimeric protein receptors
known as the integrins (Ginsberg M. H. Loftus J. C. and Pbw E. F. (1988) Thrombosis and
Haemostasis 59, 1-6; and Hynes R. O. (1988) Cell 48 549-554). Among the other
1 5 receptors shown to bind RGD containing ligands are the vitronectin recep~ors (VnR) and the
1ibronectin receptors (FnR) (Pytela etal. (1985) Proc. NatL Acad. ScL, USA 82 5766-5770;
Pytela et aL (1985) Cell 40 191-198; and Sanchez-Madrid et aL (1983) J. Exp. Med. 158
1785-1803). Furthermore it is believed that other integrin receptors may be discovered that
also interact with RGD containing ligands. Thus a iS believed that a partkulariy useful
2 0 antithrombolk wouid ioe one thal specifkally inhibited the interactbn between RGD
rontaining proteins and the platelet GP llbllla receptor while not effecting the interaclion
between the other integrins and their endogenous ligands.
Many common human disorders aré characteristkaliy associated with a
hyperthrombotk state leading to in~ravascular thrombi and emboli. These are a mapr cause of
2 5 medkal morbidity leadlns ~o Infarct~on slroke and phlebit~s and o~ mortalay ~rom stroke and
pulmonary and cardiac emboll. Pa~len~s with a~herosclerosis are predisposed to arterial
thromboembolic phenomena tor a variety of reasons. Atherosclerotic plaques form niduses for
platelet plugs and thrombii that lead to vascular narrowing and occiusion resuiting in
myocardial and cerebral ischemb disease. This may happen spontaneoush or tolklwing
3 û procedures such as angioplasty or endarteroectomy. Thrombii that break off and are released
into the circulat~on cause ~n~arctbn of d~fferent organs espec~ally the bra~n extremit~es heart
and kWneys.
In additbn to being invohed in arterial thrombosis platelets may also play a role in
venous thrombos~s. A iarge percentage o~ such pat~ents have no antecedent r~sk ~actors and
3 5 devebp venous thrombophlebitis and subsequent pulmonary emboli wi~hout a known cause.
Other patients who ~orm venous thrombi have underiying diseases known to predispose to
these syndromes. Some ot these patients may have genetic or acquired deficiencies of
lactors that normally prevent hypercoagulability such as antithrornbin-3. ahers have
mechanical obstnJctions to venous tlow such as turnor masses that lead to bw fbw states and
.
..
.
:
, ~
WO 92/07870 PCI/US91/07809 .
3 . 2~231~ ~
thrombosis. Patients with malignancy have a high incidence of thrombotic phenomena tor
unclear reasons. Antithrombotic therapy in this situalion with currently available agents is
dangerous and often ineffective.
It is also known that patients whose biood fbws over artificial surfaces, such as
5 prosthetlc synlhe~ic cardiac valves or through extrar orporeal perfusion devices, are at risk for
the development ot platelet plugs, thrombii anrJ emboli. For example, it is standard practice
with patients having artiticlal cardiac valves to be continuously ar~ti-coagulated. However, in all
instances, platelet activation and emboli ~ommatbn may still occur despite adequate
anticoagulatbn treatment.
1 0 Thus, a large category of patients, including those with atherosderosis, coronary
artery disease, artificial heart vaives, cancer, and a history o~ stroke, phlebitis, or pulmonary
emboli, are candidates ~or limited or chronic antithrombotic therapy. The number ot available
therapeutic agents is limited and these, ~or the most part, act by inhibiting or reducing levels o~
circulating clotting factors. These agents are 1requently not etfective against the patient s
1 S underiylng hematoiogic problem, which often concems an increased propensity ~or platelet
aggregatbn and adhesbn. They also cause the patient to be susceptible to abnommal
bleeding. Available antiplatelet agents, wch as aspirin, inhibd oniy pan ot the platelet
activatbn process and are there~ore onen inadequate ~or therapy.
An agent whkh e~ectiveiy inhibds the tinal common pathway o~ platelet activation,
2 0 nameb 1ibrinogen binding to the GP llb Illa receptor, shouid accordingly be usetul in a large
group ot disorders characterked by a hypenhrombotic state as described above. The present
Inventbn contemplates such an agent whkh is a new composdbn, namely a poiypeptide that
may conslst in pan ot natural amino acids and in part ot unnatural amino acids as well as non-
peptidyl porlbns. This new composdbn is bel~eved to ~ntertere with the interactbn ot Arg-Gly-
2 5 Asp containlng peptides, panicularly ~ibrinogen, wdh the GP llb Illa complex thereby preventingplatel~t awre~atbn. Platele~ aggregalion has been identined as an eariy step in the ~ormation
ol platelet plugs, emboli and thrombli in the circulatory system whkh in tum have been shown
to piay an active rol~ in cardbvascular complicatbns and disease. Inhibdbn ot tibrinogen
blndlng to the GP llb 114 complex has been shown to be an ettective antithrombotic treatrnent
3 û In anlmals (H. K. Gold, et al., Circulatbn ~1988) 77,670-677; T. Yasuda, et aL, J. Clin. Invest.
(1988) 81, 1284-1291; B. S. Coller, etal., Bbod (1986) 68, 783-786.)
A nurnber ot syntheth peptides, including cyclic disulfbes, have been disclosed as
inhibitors ot tibrinogen binrJing to platelets all ot which contain the Arg-Gly-Asp sequence.
See U.S. Patent 4,683,291; W089/05150; EPO O 319 506 A~; EPO O 341 915 A2; Plow et
3 5 aL, Proc Natl. Acad. Sci. USA (1985) 82,8057-8061; Ruggeri etal., Proc. NaM Acad. Sci. USA
(1986) 83, 5708-5712; Haverstick e~ al., Blood (1985) 66, 94~952; Pbw e~al., Bbod (1987)
70, 110-115; F. El F. Ali, e~al., Proc. EleventhAmer. Peptide Symp. (1990) 94-96; M.
Pierschbacher and E. Ruoslahti, J. Biol. Chem. (1987) 262~ 1 7294-17298; anrJ retererlces
- ' . ,' ' . ~:
WO 92/07870 PCT/US91/07809
2092315 4 ~ ~
cited in the above publications. These Arg-Gly-Asp containing peptides are belived to act as
competitive inhibitiors, out competing tibrinogen for the GPllbllla receptor.
Synthetic peptides in which one or more ot the RGD amino acid residues has been
replaced with another amino acid or anabg have also been described. EPO 0 368 486 A2
5 dlscioses a Arg-Tyr-Asp-21mer that Is about 10-~oid less active In a platelet aggregation assay
Ihan the corresponding Arg-Gly-Asp-21mer. Ali, etaL, Peptides: Chemistry, StNcture and
Blolo~y, Proceedings ot the 11 th American Peptide Symposium, p.94-96, Rivier and Marshall
eds. ESCOM, Leiden (1990) describe moditications to the sequence Arg-Gly-Asp-Ser in a
platelet aggregation assay. Substitution of Lys tor Arg in this sequence as well as most other
1 0 substitutions grealiy deueased potency. Similariy, tor cyclic RGD peptides, moditication ol the
Arg residue (N,N-Et29Uan) produced a 10-fold bwer potency.
Garsky, et al., Proc Natl. Acad Sci. USA 86 4022-4026 (1989) describe a potent
platelet aggregation inhibitor trom the venom ot 1he saw-scaled viper Echis carinatus . These
arthors repon the inhibitor is a 49 amino acid protein containing ~he sequence -Arg24-Giy-Asp-
1 5 having an ICso.3.3x10-3M and Ihat replacing Arg24 with omithine (Orn) produces a mutant
inhibitor, (Om241Echistatin, having 3-toid bwer potency. Tetrapeptides containing the Om-
Gly-Asp sequence are also described hav~ng 10- to 50-toid bwer potency.
A search ot the Dayhon data base reveaied 2193 occurences ot the sequence KGD
(compare 2026 tor RGD). Ot proteins containing the KGD sequence, pbcental anticoaguiant
2 0 probin (PAP) anct two other members ot the lipoconin family, are reported to have
anticoa~ulant actlvay (Funakoshi, etal., Bbchemistry 26 5572-5578 (1987). However, these
protelns are not reponed to inhiba the tinal common pathway ot platelet activation by inhibaion
ot blndlng ot tibrinogen wah the GP llbllla receptor, rather anticoagulant activay is ascribed to
inhlbabn ot prothrombin activatbn as well as inhibltbn ot phospholipase A2 and subsequent
2 5 prostagland~n release.
Several synthetic cyclic peptldes contalnlng the lhioelher linkage have been
synthesked. Gero et al., ~hchem. Bbphys. Fles. Comm. (1984) 120, 840-845 describe a
pseudohexapeptide anabg ot somatostat~n where the ~roup ICH2-SI is substauted tor a
peptide bond. Slmilariy, i dwards el al., Bbchem. Bbphys. Fles. Comm. (1986) 136, 730-736
3 0 cornpare the bbbgbal activay ot linear and cycl~c enkephalin pseudbpeplide anabgs
contain~ng the thbmethylene ether linka~e. Other enkephalin related pseudopeptides and
macrocycles contalning the ICH2-SI substautbn tor peptides have been described, Spatola et
al., Bbpolymers (1986) 25, 229-244 and Spatola et aL,TetNhedron (1988) 44, 821-833.
None ot these reterences discbse a peptide containing the amino acid seouence
3 5 KGD havlng hbh platelet aggregatbn inhibitbn activit,v. None ot these reterences discbse a
peptide having high speciticity tor the GP llbllla receptor relative to other integrin receptors.
F~nally, none ot the reterences describe a small cyclic peptide stable to ring opening
containing eaher -Lys-Gly-Asi~ or -Om-Gly-Asp- having a higher inhibaion potency tor
"'..
' ` ' ' ' ` ` '
' ~
WO 92/07870 PCI~/US91/07809
5 2~23~
~ibrinogen/GP ~b~l~a ELISA than for vitronectin/vitronectin receptor ELISA or tor
libronectin/fibronectin receptor ELISA.
Accordingly, R is an object of this invention to produce a peptide having high platelet
aggre~ation inhibition activity. It is a lurther object o~ this invention to provide peptWes having
5 high specHicity lor the GP llbllla receptor. It is still a ~urther object to produce small cyclic
peptides that are stable to ring opening having the above described properties. It is still a
tunher object o~ this invention to provicie a platelet aggregatbn inhibitor exhibiting diminished
in vivo side ettects such increased bleeding times and optionally to provide such inhibRors
wRh increased litetime
1 0 These and other objects of this invention will be apparenn trom consideration ot the
Inventbn as a whole.
Summary ot the Inventlon
The objects ot this invention are accomplished by providing a polypeptide containing
the sequence Xaa-Gly-Asp having high speciticRy ~or the GP llbllla receptor relative to the other
1 5 integrin receptors, where Xaa is OmRhine (OM) or Lysine (Lys). Preterably the peptWe
contains the sequence Lys-Gly-Asp and connains 1ewer than about 345 amino acid resWues.
Also pretereably the peptide is cyclic, having trom 5-10 amino acids in the cycle. Most
preferabiy, the cyclic peptide has 5 amino acics lomning the ring ot the cycle. More preferably,
the ring ot the cyclic peptide contains trom aboun 17 to about 18 atoms, most preterabiy 18
2 0 atoms. A partiwlarly preterred compound ot the instant invennion is a polypeptide having the
stnucture
Xaal -Xaa2-Gly-Asp-Xaa3-R
Z
wh~rein
2 5 Xaa1 Is a D or L a amlno acid llnked to Z through the amino group; Xaa2 Is Om or Lys;
Xaa3 is a D or L a-amino acid linked through the side chain to Z;
Z Is an amide bond, disuitide, COCH2S, COCH2SO, or
COCH(C6Hs)S; and
3 û R is OH or NH2.
Also preterably, the ring will r,ontain a D amino acid most preterably linked to the Lys ot
the tripeptide sequence.
The inven~ion in its broad aspects relates to peptide derivatives whbh are usetul as
inhibitors ot platelet tunr,tion mediated by the GP llb Illa receptor and tor the prevention ot
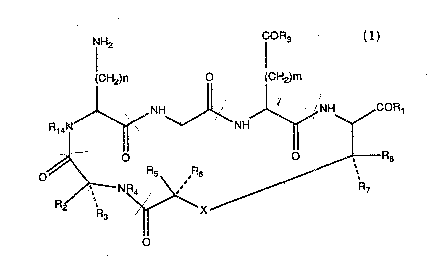
3 5 thrombus torrnation. Preterred compounds ot this inventbn are represented by Formula 1:
.
' : . ~ ' . ' ' . ' ' ` .
i, .. ..
. . ~ ,
'.
' .
r. . . ; , : ' :
WO 92/07870 PC~/US91/07809
NH2 CORg
(CH2)n 0 (CH2)m
R,4N~ J~N ~NH~COR,
0~ R5 ,R6 ~R8
2 F~J ~ X ~ R~
O
wherein
R1 and Rg are the same or difterent and are selected ~rom
S hydroxy,
C1 Cg alkoxy,
C3-C12 aikenoxy,
C6-C12 arybxy,
di-C1-C8 alkylamino-C1-Cg-alkoxy,
1 0 acylam~no-C1-Cg-alkoxy selected 1rom the group acetylaminoethoxy,
nicotinoylaminoethoxy, and succinamidoe~hoxy.
pivaloyloxyethoxy,
C6-C1 2 aryl-C1 Cg-alkoxy where the aryl group is unsubstituted or substituted
with one or more ol the groups nitro, hab (F, Cl, Br, 1), C1-C4-alkoxy, and
1 S amlno,
hydroxy-C2-Cg alkoxy,
dlhydroxy-C3-Cg-alkoxy, and
NR1oR11 wherein R10 and R11 are the same or di1terent and are hydrogen, C1-Cg-alkyl, C3-
Cg-aikenyl, C6-C1 2-aryl where the aryl group is unsubstituted or substituted with one
2 0 or more ol the groups nitro, hab (F, Cl, Br, 1), C1-C4-alkoxy, and amino, C6-C12 aryl-
C1-Cg-alkyl where the aryl group is unsubstituted or substituted by one or more ot the
groups nitro, hab (F, Cl, Br,l), C1-C4-alkoxy, and amino;
R2, R3, Rs, R6, R7, R8 are the same or ditferent and are selected ~rom
hydrogen,
2 5 C6-C1 2 aryl where the aryl group is unsubstituted or substituted by one or more
o1 the groups nitro, hydroxy, hab (F, Cl, Br, 1), C1-c8 alkyl, hab-C1-
C8 alkyl, C1-Cg-alkoxy, amino, phenyloxy, phenyl, acetamido,
benzamido. di-C1-Cg alkylamino, C1-Cg aikylamino, C6-C12 aroyl.
C1-Cg alkanoyl, and hydroxy-C1-C8 alkyl,
- - .
, . . . :.
: -, ~ . ;
, . , : ' ' : . . . '': , , . ` , ; : ': .- .
~. ,,, . , .. . ., - ~.
.. :,.~ ' : :' . :.
WO 92~07870 PCr/US91/07809 .
~ u 7 2 ~ 9 2 3 ~ ~
. .
C1-C12 alkyl either substitu!ed or unsubstituted, branched or straight chain
where the substituents are selected trom hab (F, Cl, Br, 1),
C1 Cg alkoxy,
C6-C12 aryloxy where the aryl group is unsubstituted or substauted by
S one or more ol the groups nitro, hydroxy, hab (F, Cl, Br, 1),
C1-Cg alkyl, C1-Cg-alkoxy, amino, phenyhxy, acetamido,
benzamido, di-C1-Cg alkylamino, C1-c8 alkylamino, C6-C12
aroyl, and C1-Cg alkanoyl,
isothioureido,
1 0 C3-C7 cycbalkyl,
ure do,
ammo,
C1-Cg alkybmino,
di-C1-Cg aikylamino,
1 5 hydroxy,
amlno-C2-Cg alkyithb,
amino-C2-Cg alkoxy,
acetamicb,
benzamido wherein the phenyl ring is unsubstituted or substituted by one
2 û or more of the groups nitro, hydroxy, hab (F, Cl, Br, 1), C1-c8
alkyl, C1-Cg-alkoxy, amino, phenybxy, acetamicb, benzamicb,
di-C1-Cg alkybmino, C1-Cg alkylamino, C6 C12 aroyl, and
C1-Cg alkanoyl,
C6-C1 2 arylamino wherein Ihe aryl group Is unsubstituted or subsli1uted
2 5 by one or more ol the cJroups nitro, hydroxy, haio, C1-Cg alkyl,
C1-Cg-alkoxy, amlno, phenybxy, acetamicb, benzamido, di-
C1-Cg aikybmino, C1-Cg alkybmino, C6-C12 aroyl, and C1-
C8 alkanoyl,
guanidino,
3 0 phlhallmicb,
mercapto,
C1-C8 alkynhb,
C6-C12 aryithb,
carboxy,
3 S caricoxamide,
carbo-C1-Cg alkoxy,
C6-C1 2 aryl wherein the aryl group is unsubstituted or substituted by
one or more o1 the groups nitro, hydroxy, halo, C1-Cg alkyl,
C1-Cg-alkoxy, amino, phenybxy, acetamido. benzam~do, di- . -
. . . . . . , ......... -.: . ,, , ::
: . - . : : ." . , :
WO 92/07870 PCr/US91/07809
2092315 8 ~ ~
C1-Cg alkylamino, C1-Cg alkylamino, hydroxy-C1-C8 alkyl,
C6-C12 aroyl, and C1-Cg alkanoyl, and
aromatic heterocycle wherein the heterocyclic groups have 5-10 ring
atoms and contain up to two O, N, or S heteroatoms;
5 R2 and R3, Rs and R6, or R7 and R8 may optbnally and independently be pined together to
torm a carbocyclic or heterocyclic ring ot trom tour to seven atoms where the
heteroaloms are selected lrom O, S or NR12 where R12 is selected trom
hydrogen, C1-Cg-alkyl, C3-Cg-alkenyl, C6-C12-aryl~ C6-C12-aryl-C1-
Cg-alkyl, C1-Cg alkar~oyl, and C6-C12 aroyl,
1 0 R4 is selected trom
hydrogen,
C1-Cg alkyl,
C3-C10 cycloalkyl,
C6-C1 2 aryl, and
1 5 C6-C12aryl-C1-Cg-alkyl;
R2 or R3 may be optionally pined with R4 to lorm a piperidine, pyrrolidine or thiazolidine ring;
R14 is selected trom
hydrogen, C1-Cg-alkyl, C3-Cg-alkenyl, C6-C12-aryl~ and C6-C12 aryl-C1-Cg- aikyl;
X is seiected from
anOorSatom,
an S atom bearing one or two O atoms,
NR13 wherein B13 is hydrogen, C1-Cg-alkyl, C3-Cg-alkenyl, C6-C12-aryl,
C6-C12 aryi-C1-C8-alkyl~ C1 Cg aikanoyi, and C6-C12 aroyl, and
C6-C12 aryl,
2 5 C1-Cg alkanoyl,
(CH2)k where k Is an Integer Irom 0 to 5;
n 18 an Integer lrom 1 to 6;
m 18 an Integer ~rom 0 to 4; and
pharrnaceutically acceptable sans thereot.
3 0 As used herein and unless speci~ied otherwise: alkyl, alkenyl and alkynyl denote
straight and branched hydrocarbon chains having single, double and triple bonds,respectiveiy; C6-C12 aryl groups denote unsubstituted aromatb ring or tused rings such as,
lor example, phenyl or naphthyl; hetero denotes the heteroatoms O, N, or S; aromatic
heterocyclb groups have 5-10 ring atoms and contain up 1O tour heteroatoms; halogen or hab
3 5 denote F, Cl Br, or I atoms, alkoxy denotes an alkyl group anached to O.
Examples ot C1-Cg all~yl or C2-Cg alkenyl groups include methyl, ethyl, propyl,
isopropyl, butyl, t-butyl, pentyl, isopentyl, hexyl, vinyl, allyl, butenyl and the like; examples o~
C3-C1o-cycbalkyl groups include cyclopropyl, cyclopenlyl, cycbhexyl, and the like; aromatic
t
':
: ` ~
' ` ~ `:
': ' . . ~ , ':
,- . ~' ' ' ` ~ ,' "'I
WC) 92~07870 ~ 1~ 9 2 31 ~ PCI~/US91/07809
heterocyclic groups include but are not limited to pyridyl, thienyl, furyl, indolyl, benzthienyl,
imWazolyl, thiazolyl, quinolinyl and isoquinolinyl.
The present inventbn includes a method of making the compounds of Forrnula 1.
The present invention also includes a method for reducing platelet aggregation in a
5 mammal. Thls method involves administering a therapeutkally effective arnount of the
compounds o~ the present Invention alone or in combination with a phanmacoiogically
acceptable carrier This general method may aiso be applied to treat a mammal having an
increased propensity ~or thrombus formation.
Additionally, the present invention is directed to compositions o~ matter ~or reducing
1 0 platelet aggregation in a mammal; treating a mammal having an increased propensity for -
thrombus ~om ation; or inhibiting binding of a ligand to GP llb Illa in a mammal; wherein each of
these compositbns contains as an active ingredient one or more of the cyclic peptides defined
in Fom~ula 1.
Detalled Descrlptlon o~ the Inventlon
1 5 The instant invention is the resuit of the surprising discovery that KGD containing
polypeptide are potent inhibitors of platelet aggregation and in the Fg/GP llbllla ELISA. The
most potent KGD-containing inhibitors are small cyclk peptides and thus these peptides are
pre~ened.
n has also been discovered that the instant KGD peptides are weak inhibitors of the
2 0 fibronectin (Fn)/fibronectin receptor (FnR) interaction and o~ the vnronectin (Vn) hitronectin
receptor VnR interaction. In comparing inhibitbn ot varbus KGD-containing poiypeptides n
has been ~ound that inhibitbn potency, as measured by ICso, is 1rom 10-500 told higher (i.e. a
bwer ICso) in the Fg/GPllbllla ELISA than in the Vn/VnR ELISA. Thus, the instantpolypeptides exhibit high specHkny ~or the GPllbllla receptor. It is contemplated that this high
2 5 specHblty wlll reduce the number and severity o~ side effects ~or these antithrombotic
compounds. It 18 believed these compounds will exhiblt high platelet aggregatbn ~nhibnbn
without substantlally Increaslng bleeding ~ime in a mammal.
Polypeptides o~ this invention can be made by chemkal synthesis or by emphying
recombinant technobgy. These methods are known in the art. Chemical synthesis, especialiy
3 0 solld phase synthesis, is pre~ered tor short (eg. iess than 50 residues) poiypeptides or lhose
containlng unnatural or unusual amino acids such as; D-Tyr, Omnhine, amino adipk acid, and
the like. Recombinant procedures are pre~ered ~or bnger polypeptides or tor mutant or variant
peptides containing the KGD sequence.
When recombinant procedures are selected, a synthetic gene may be constnucted de novo or
3 5 a natural gene may be mutan~gized by, ~or example, casette mutagen~s~s. Set ~orth bebw are
exemplary general recombinant procedures.
General i~ecomblna1~ Procedures. From a purified protein and its amino acid
sequence, a KGD-containing protein may be produced using recombinant DNA techniques.
These techniques contemplate, in simplified form, taking the gene, enher natural or synthetic,
., ' - .
. - , . . . . .
. , ' ` ' ' .' ' ' ' '' .
. --.
,,
. . . .
'. ',
WO 92/07870 PCI/US91/07809
2~923i~ 1 0
tor the protein; inserting it into an appropriate vector; inserting the vector into an appropriate
host cell; culturing the host cell to cause expression ot the gene; and purifying the protein
produced thereby.
Somewhat more particularly, the DNA sequence encoding a KGD-containing protein is
5 cloned and manlpulated so that a may be expressed in a convenient host. DNA encoding
parent polypeptides can be obtained trom a genomic library, from cDNA derived ~rom mRNA
trom cells expressing the protein, or by synthetkally constnucting the DNA sequence
~Sambrook, J., Frasch, E.F., and Maniatis, T., (1989), Moleevlar Cbning (2d ed.), Cold
Springs Harbor Laboratory, N.Y.).
1 0 The parent DNA is then inserted into an appropriate plasmid or vector which is used to
transtorm a host cell. In general, plasmid vectors containing replication and control
sequences which are derived from species compatible wah the host cell are used in
connection wah those hosts. The vector ordinarily carries a replication sae, as well as
sequences which encode proteins that are capable ot providing phenotypic selection in
1 5 transtormed cells.
For example, F. ~j may be trans~ormed us~ng pBR322, a plasmid derived trom an F
mli species (Mandel, M. et al. (1970) J. MoL Bbl. 53, 154). Plasmid pBR322 contains genes
tor ampicillin and tetracycline resistance, and thus provides easy means ~or selection. Other
vectors include dfflerent ~eatures such as different promoters, which are otten important in
2 0 expressbn. For example, plasmids pKK223-3, pDR720, and pPL-lambda represent
expressbn veaors with the tac, trp, or PL promoters that are currently available (Phamlacia
Biotechnology) .
A preterred vector is pB0475. This vector contains origins o1 replicatbn lor phage and
.~.9Qli which albw it to be shuttled bstween such hosts, thereby ~acilitating both mutagenesis
25 and expression (Cunnir~ham, B., etaL t1989), &ience243, 1330-1336; Wells, J. and
Cunnlngham, B., co-pending applicatbn WO 90/04788, published 3 May 1990. Other
pre1erred vectors are pRlT5 and pR1T2T ~Phammacia Bbtechnobgy). These vectors contain
appropria1e promolers lolbwed by the Z dbmain ol protein A, albwing genes inserted into the
veaors to be expressed as tusbn pro~eins. Funher discussbn ol 1hese veaors may be
3 0 ~ound below.
Other preterred veaors can be constn~cted using standard techniciues by combining
1he relevant 1raits ot the vectors described above. Relevant ~raas include lhe promoter,1he
ribosome binding sae, the decorsin or omatin gene or gene 1usbn (lhe Z dbmain ot protein A
and decorsin or omatin and as linker), the antibbtic resistance markers, and the appropriate t
3 5 origins ot replicatbn.
The host cell may be prokaryotic or eukaryotic. Prokaryotes are preterred tor cbning
and expressing DNA sequences to produce parent polypeptides, segment substitutedpolypeptides, residue-substauted polypeptides and polypeptide variants. For example, ~.
9Qli K12 strain 294 (ATCC No.31446) may be used as F. ~QIi B,
: , . . ; , .
- . . , : - , . ~ .
, . , ~: : . ;.:
: ` . . , . , ' . ', ,.;
., : .. .. . . . . . .
WO 92/07870 PCT/US91/07809
2~2315
QL X1776 (ATCC No. 31537), and F ~j c600 and c600hfl, ~,~ W3110 (F-, gamma-.
prototrophic /ATCC No. 27325), bacilli such as Bacillus subtilis, and other enterobacteriaceae
such as Salmonella typhimurium or Serratia marcesans, and various pseudomonas species.
The prelerred prokaryo~e is F. ~QIi W3110 (ATCC 27325). When expressed by prokaryotes
the polypeptides typically contain an N-terminal methbnine or a tommyl methionine and are not
glycosylated. In the case ot tusion proteins, the N-temminal methbnine or tormyl methionine
resides on the amino terminus ot the ~usion protein or the sbnal sequence of the fusion
protein. These examples are, ot course, ~nnended to be iltustraUve rather than limning.
In addnion to prokaryotes, eukaryotic organisms, such as yeast cultures, or cells
1 0 derived trom multicellular organisms may be used. In principle, any such cell culture is
workable. However, interest has been greatest in vertebrate cells, and propagation ot
vertebrate cells in culture (tissue culture) has become a reproducible procedure ( rSsue
Culture, Academic Press, Knuse and Patterson, ednors (1973)). Examples ot such useful
host cell lines are VERO and HeLa cells, Chinese Hamster Ovary (CHO) cell lines, W138, 293,
1 S BHK, COS-7 and MDCK cell lines.
Gene Fuslons A variatbn on the above procedures contemplates the use of gene
fusions, wherein the gene encoding the desired protein is associated, in the vector, with a
gene encoding another protein or a gragment of another protein. This results in the desired
protein - here, a KGD-containing protein - being produced by the host cell as a tusion with
2 0 another protein. The ~other~ protein is Onen a protein or peptide which can be secreted by
the cell, making it possible to isolate and purify the desired protein 1rom the culture medium
and eliminatin3 the necessity ol destroying the host cells which arises when the desired
proteln remains inside the cell. AitemaUvely, the 1usbn protein can be expressedIntracellularly. It is useful to use tusion proteins that are highly expressed.
2 S The use ot gene lusions, though not essential, can facilitate the expressbn ot
helerologous prolelns In ,~QII as well as the subsequent purilkatbn ot those gene products
~Harris, T. J. R. ~1983) In Genetic Engineering, ~lliamson, R., Ed., Academic, London, Vol. 4,
p. 127: Uhlen, M., Moks, To. ~1989) Me~hods Enzyrno/. (in press)). Protein A tusions are
onen used because the binding of protein A, or more specitically the Z domain of protein A, to
3 0 IgG provides an ~affinity handle~ for the puriticatbn ol the fused protein. it has also been
shown that many heterologous protelns are degraded when expressed dlrectiy in E,~QIj, but
are stable when expressed as tusbn proteins (Mars~on, F. A. O., (1986) Biochem 1 240, 1).
A KGD-containing protein expressed a ~usbn protein may be properly folded or
require folding to obtain the native stnucture. The properiy folded fusion protein may be
3 S active and useful as a GP llb Illa antagonist and Inhibitor ot platelet aggregatbn. More
preferred wouid be the correctly foided~ native~ protein that is obtained from the fusion
protein by methods known in the art. Fusion proteins can be cleaved using dhemicals, such
as cyanogen bromide, whkh cleaves at a methionine, or hydroxyiamine, whkh cleaves
between an asn and gly. Using standard recombinant DNA methodology, the nudeotide
. .
.. . . , : ~ . . . '' ~
WO 92/07870 PCr/US9~/07809
2~92315 12 ~,.
base pairs encoding these amino acids may be inserted just prbr to the 5 end of ~h~ KGD-
containing protein gene.
Aitemative~, one can empby proteolytic cleavage of fusbn proteins, which has been
recently reviewed (Carter, P. ~1990) in Ptotein Putification: Ftom MolecularMeehanisms to '.
5 L~tge-&ale Ptocesses, Ladisch, M. R., Willson, R. C., Painton, C. C., and Builder, S. E.,
eds., American Chemical Society Symposium Series No. 427, Ch 13,181-193).
Proteases such Factor Xa, thrombin, subtilisin and mutants, and a number of other
have been successfully used to cleave fusion proteins. Typically, a peptide linker ~hat is
amenable to cleavage by the protease used is inserted between the other protein (e.g., the
1 0 Z domain o~ protein A) and the KGD^contaning protein ot interest. Using recombinant DNA
methocblogy, the nucleotide base pairs encoding the linker are inserted between ~he genes
or gene tragments coding for the other proteins. Proteolytic cleavage o~ the partially puritied
tusion protein containing the correct linker can then be carried out on either the native fusbn
protein, or the reduced or denatured tusbn protein.
1 5 The protein may or may not be properly lolded when expressed as a fusbn protein.
Also, the specitic peptide linker containing the cieavage site may or may not be accessible to
the protease. These factors detemmine whether the tusion protein must be denatured and
re~olded, and it so, whether these procedures are employed before or atter cleavage.
When denaturing and refoiding are needed, typkally the protein is treated with a2 0 chaotrope, such a guanidine HCI, and is then treated with a redox buflier, containing, for
example, reduced and oxidized dRhiothreitol or glutathione at the appropriate ratios, pH, and
temperature, such that the protein ot interest is refoided to its native stnucture.
C~L_~es When peptldes are not prepared
uslng recombinant DNA technobgy, they are preferabiy prepared using solid-phase
2 5 synthes~s, such as that 6enerally described by Merr~eid, J. Am. Chem. Soc. (1963) 85, 2149.
anhouoh o~her er.-ulvalent chemhal syntheses known ~n Ihe art are employabie as previous~y
mentbned. Solid-phase synthesis is initialed lrom the C-terminus of the peptide by coupling a
protected ~-amino acid to a suitabb resin. Such a starting rnaterial can be prepared by
anachlng an c~-amino-protected amino acid by an ester linkage to a chbromethylated resin or a
3 0 hydroxymethyl resin, or by an amide bond to a BHA resin or MBHA resin. The preparatbn o~
the hydroxymethyl resin is described by Bodansky etal., Chem. Ind. (Loncbn) (1966)
3B,1597-1598. Chbromethylated resins are commercially ava~iable trom BbRad Laboratories,
Richmond, CA and Iro m Lab. Systems, Inc. The preparatbn ot such a resin is descriced by
Stewart et aL, ~SOIKf Phase Peptide Synthesis~ (Freeman & Co., San Francisco 1969),
3 5 Chapter 1, pp.1-6. BHA and MBHA resin suppOfts are commercially avaaable and are generally
used only when the desired polypeptide being synthesked has an unsubstituted amide at the
C-terminus.
The amino acids are coupled to the peptide chain using techniques well known in the
art for the formatbn of peptide bonds. One method involves converting the amino acid to a
, ... , , ............ .~, ~ . : , ... . . - , : .. .
: , , . . ~ - : . , ; . ~ ., . ............................ :~, .
.. . - , . . . ..
- . : .
WO 92/07870 PCI`/US91/07809
,~ 13 2~92315 s
derivative that will render the carboxyl group more susceptible to reaction with the ~ree N-
terminal amino group o~ the pep~ide ~ragment. For example, lhe amino acid can be converted
to a mixed anhydride by reaction o~ a protected amino acid with ethykhbro~ommate, phenyl
chlorotormate, sec-butyl chloro~ormate, isobutyl chbrotommate, pivabyl chlorWe or like acid
5 ehbrides. Alterna~vely, the amino acid can be converted to an active ester such as a 2,4,5-
trlehlorophenyl esler, a pentachbrophenyl ester, a pentatluorophenyl ester, a p-nitrophenyl
ester, a N-hydroxysuccinlmide ester, or an ester tormed 1rom 1-hydroxybenzotriazole.
Another coupling method involves use of a suUable coupling agent such as N,N -
dicycbhexykarbodiimide or N,N -diisopropyl~arbodiimide. Other appropriate coupling
1 0 agents, apparent to those skilled in the art, are discbsed in E. Gross & J. Meienho~er, The
Peptictes: Analysis, Structure, Biology, Vol. 1: Major Methods of Peptide Bond Formation
(Academic Press, New York, 1979).
It should be recognized that the a-amino group of each amino acid employed in the
peptide synthesis must be protected during the coupling reaction to prevent side reactions
1 5 involving there active a-amino function. It should also be recognized that cerlain amino acids
contain reactive side-chain 1unctional groups (e.g. sulfhydryl, amino, carboxyl, and hydroxyl)
and that such functbnal groups must also be protected with suitable proterting groups to
prevent a chemkal reaabn from occurring at that site during both the initial and subsequent
coupling steps. Suitable protecting groups, known in the art, are described in E. Gross & J.
2 0 Meienho~er, The Peptid~s: Analysis, StnJcture, Biobgy, Vol.3: Protectbn o~ Functional
Groups in Peptide Synthesis (Academic Press, NewYork, 1981).
In the seleclbn o~ a partkular side-chain protecting group to be used in synthesizing
the peptides, the ~olbwing general nules are tolbwed. An -amino protecting group (a) must
render the -amino tunction inert under lhe conditbns employed in the coupling reaction, (b)
2 5 must be readlly removable after the coupllng reactbn under conditbns lhat will not remove
sb~-ehaln protèetlng groups and will not aner 1he structure o~ the peptide lragment, and (c)
must sllmlnate the possibllity ot racemizatbn upon activatbn immediately prbr to coupling. A
slde-ehaln proteetlng group (a) must render the side chain tunctional group inert under the
conditbns empbyed in the coupling reactbn, (b) must be stable under the conditbns
3 0 empbyed In removing the -amino proteeting group, and (e) must be readily removable upon
completbn ot the desired amino acid peptide under reaetion condabns that wlll not alter the
stnueture ot the peptide chain.
It will be apparent to those skilled in the art that the proteeting groups known to be
usetul tor peptide synthesis will vary in reactivity with the agents empbyed ~or their removal.
3 5 For example, cortain proteeting groups sueh as tfiphenylmethyl and 2-(p-
biphenylyl)isopropybxyearbonyl are very bbile and can be eleaved under mib acid conditbns.
Other proteeting groups, such as t-butybxycarbonyl (BOC), t-amybxycarbonyl. adamantyl-
oxyearbonyl, and p-methoxybenzybxycarbonyl are less labile and require moderately strong
acids, such as tritluoroaoetic, hydrochhric, or boron trilluoride in aoetk acid, tor their removal.
; `. ' '' : ' ............ -. . ' ' .... . ' , ':
. '. : ~ ' '': ', :~' ~. . '
::: . : .' '
WO 92/07870 PCr/US91/07809
20923~i 14 ~ I
Slill other protecting groups, such as benzybxycarbonyl (CBZ or Z), habbenzybxycarbonyl, p-
nitrobenzybxycarbonyl cycbalkybxycarbonyl, and isopropyloxycarbonyl, are even less labile
and require stronger acids, such as hydro~qen tluoride, hydrogen bromide, or boron
trHluoroacetate in trHluoroacetic acid, 10r their removal. Among the ciasses ot usetul amino ~i
acld protecting groups are included:
~1) lor an -amino group, (a) aromatic urethane-type protecting groups, such as
lluorenylmethybxycarbonyl (FMOC) CBZ, and substituted CBZ, such as. e.g., p-
chbrobenzybxycarbonyl, p-6-n~trobenzybxycari~onyl, p-bromobenzyloxycarbonyl,
and p-methoxybenzybxycarbonyl, o-chlorobenzybxycarbonyl, 2,4-
1 0 dichbrobenzybxycarbonyl, 2,6-dichbrobenzybxycarbonyl, and the like; (b) aliphatic
urethane-type protecting groups, such as BOC, t-amybxycarbonyl.
isopropybxycarbonyl, 2-(p-biphenylyl)-isopropybxycarbonyl, allyloxycarbonyl and the
like; (c) cycloalkyl urethane-type protect~ng groups, such as cycbpentybxycarbonyl,
adamantybxycarbonyl, and cycbhexyloxycarbonyl; and d) allyloxycarbonyl. The
1 5 preterred -am~no protecting groups are BOC or FMOC.
(2) lor the side chain amino group present in Lys, protectbn maybe by any ot thegroups mentioned above in (1) such as BOC, p-chlorobenzyloxycarbonyl. etc.
(3) lor the guanid~no group ol Arg, protect~on may be by nitro, tosyl, CBZ,
adamantyloxycarbonyl, 2,2,5,7,8-pentamethykhroman-6-sultonyl or 2,3,6-trimethyl~-
2 0 methoxyphenylsuiionyl, or BOC.
(4) lor the hydroxyl group ot Ser, Thr, or Tyr, protection maybe, tor example, by
C1-C4 alkyl, such as t-butyl; benzyl (BZL); substltuted BZL, such as p-methoxybenzyl, t
p-nitrobenzyl, p-chlorobenzyl, o-chbrobenzyl, and 2,6-dichbrobenzyl.
(5) lor the carboxyl group ol Asp or Glu, protectbn may be, tor example, by
2 5 ester~ncatbn us~n3 groups such as BZL, t-butyl, cycbhexyl, cycbpentyl. and the l~ke.
(6) lor the imidazob nltrogen ol His, the tosyl moiety is suitably empbyed.
(7) lor the phenollc hydroxyl group ol Tyr, a protecting group such as
tetrahydropyranyl, tert-butyl, trityl, BZL, chlorobenzyl, 4-bromobenzyl, and 2,6-
dichbrobenzyl are suitably empbyed. The prelerred protecting group is 2,6-
3 0 dichlorobenzyl.
(8) lor the side chain amino group ol Asn or Gln, xanthyl (Xan) is prelerably
employed.
(9) lor Met, the amino acid is prelerably lelt unprotected.
(10) lor the thio group ol Cys, p-methoxybenzyl is typically empbyed.
3 5 The C-terminal amino acid, e.g., Lys, ~s protected at the N-amino position by an
appropriately selected protecting group, in the case ol Lys, BOC. The BOC-Lys-OH can be
lirst coupled to Ihe benzyhydrylamine or chbrome~hylated resin according to the procedure
set lorth in Horiki et al.. Chemistry Letters, (1978)165-168 or using isopropylcarbodiimide at
about 25C tor 2 hours with stirring. Following the coupling ot the BOC-protected amino acid
.: ~ . - . , -...
... . : .......... . : : , . .
. ~ ~ . . :. , .
WO 92/07870 PCr/US91~07809
~: 15 ~923~ :
to the resin support, the ~-amino protecting group is removed, as by using trifluoroacetic acid
(TFA) in methylene chioride or TFA alone. The deprotectbn is carried out at a temperature
between about 0C and room temp0rature. Other standard cleaving reagents, such as HCI in
dbxane, and conditions lor removal ol specinc a-amino protecting groups are described in
5 Schroder & Lubke, supra, Chapter 1, pp. 72-75.
Atter removal ol the cl-amino protecting group, the remaining ct-amino and side-chain
protected amino acids are coupled step within the desired order. As an alternative to adding
each amino acid separately in the synthesis, some may be coupled to one another prior to
addition to the solid-phase synthesizer. The selectbn of an appropriate coupling reagent is
1 0 within the skill o~ the art. Particulariy suitabie as a coupling reagent is N,N -dicycbhexyl
carbodiimide or diisopropylcarbodiimWe.
Each protected amino acid or amino acid ser.juence is introduced into the solid-phase
reactor in excess, and the coupling is suitabiy carried out in a medium ol dimethyifommamWe
(DMF) or CH2CI2 or mixtures thereot. if incomplete coupling occurs, the coupling procedure is
1 5 repeated belore removal ot the N-amino protecting group prbr to the coupling ol the next
amlno acid. The success ot the coupling reactbn at each stage o1 the synthesis may be
rnonitored. A pre~erred method ot monitoring the synthesis is by Ihe ninhydrin reaction, as
described by Kaiser et aL. AnaL Biochem, (1970) ~4, 595. The coupling reactions can be
performed automatically using well known methods, for example, a Bbsearch 9500 Peptide
2 0 Synthesizer.
Upon completbn of the desired peptide sequence, the protected peptide must be
cleaved trom the resin wpport, and all protecting groups must be rernoved. The cleavage
reactlon and removal ot the protecting groups is witably accomplished simultaneously or
stepwise. When the resin support is a chbro-methylated polystyrene resin, lhe bond
2 5 anchoring the pept~de to the resln Is an ester linkage ~ormed between the ~ree carboxyl group
0t Iha C-lerrnlnal r0sidue and one ot the many chbromethyl groups present on the resin
matrix. it wlll be appreciated Ihat Ihe anchoring bond can be cleaved by reagents thal are
known lo be capable ot breaking an esler linkage and ot penetraling the resin matrix. One
especialy convenient method is by Irealment with liquid anhydrous hydrogen tluoride. This
3 0 reagent not oniy will deave the peptide trom the resin but also will remove all protecting
groups. Hence, use ot 1his reagent will directly atford the lully deprolected peptide. When the
chbrc melhylated resln is used hydrogen tluoride Ireatment results in Ihe 10rmatbn ol the tree
peptide acids. When the benzhydrylamine resin is used, hydrogen ~luoride treatment results
directty in the tree peplide amines. Reaction with hydrogen tluoride in Ihe presence ot anisole
3 5 and dlmelhylsullide al O C lor one hour will simultaneously remove the side-chain protecting
groups and release the peplide 1rom Ihe resin.
When it is desired lo cleave the peplide without removing protecting groups, theprotected peptide-resin can undergo methanolysis to yield the protected peptide in which the
C-terminal carboxyl group is methylated. The methyl ester is then hydroiyzed under mild
, .- , ':.
.
.. . : , -
' ' . ' -,' ' '
- ` . .~ ~ -
.
WO 92/07870 2 0 9 ~ 31~ 1 6 PCl/US91/07809
alkaline conditbns to give the tree C-terminal carboxyl group. The protecting groups on the
peptide chain then are removed by treatment with a strong acid such as Ibuid hydrogen
lluoride. A particularly useful technique ~or methanolysis is that ot Moore et aL. Pep~des, Proc.
F~th Amer. Pept. Symp., M. Goodman and J. Meienhoter Eds. (John vViley N.Y. 1977) p.
518-521 in which the pro~ected peptide-resin is treated with methanol and potassium cyanide
In the presence ot crown ether.
Another method tor cleaving the protected peptide trom the resin when the
chbromethylated resin is empbyed is by ammonoiysis or by treatment with hydrazine. it
desired the resuiting C-terminal amide or hydrazide can be hydrolyzed to lhe tree C-temminal
1 0 carboxyl moiety and the protecting groups can be removed conventionally.
It will also be recognized that the protecting group present on the N-terminal rl-amino
group may be removed preterentially ei~her betore or atter the protected peptide is cleaved
trom the support.
Puritkation ot the polypeptides ot the invention is typically achieved using
1 5 conventional procedures such as preparative HPLC (including reversed phase HPLC) or other
known chromatographb technbues such as gel permeation ion exchange partition
chromatography attinity chromotography (including monocbnal antibody columns) or countercurrent distribution.
Polypeptide chains are poiymerized by crosslinking monomer chains with
2 0 poiytunctbnal crosslinking agents including compound 1 either directly or indirectly through
multitunctbnal polymers. Ordinarily two substantialiy identical polypeptides are crosslinked at
their C or N temmini using a bitunctional crosslinking agent. The agent is used to c~sslink the
terminal amino and/oi~ carboxyl groups. Generally both terminal carboxyl groups or both
terminal amino groups are crosslinked to one another anhough by selectbn ot the appropriate
2 5 crosslinking a~ent the alpha amino ol one polypeptide is crosslinked to Ihe terminal carboxyl
~roup ol the other polypeptide. Prelerably the poiypeptides are substituted at their C-termini
with cystelne. Under conditions well known ~n the art a d~sult~de bond can be ~ormed between
the termlnal cysteines thereby crosslinking the polypeptide chains. For example disuHide
brW~es are conveniently lommed by metal-cataiyzed ox~daUon o~ the Iree cyste~nes or by
3 0 nucleophllic substautbn ot a suitabiy modilied cysteine residue. Selectbn ol the crosslinking
agent will depend upon the Wentities ol there active side chains ol the amino acWs present in
the polypeptWes. For example dlsullide crosslinking would not be prelerred it cysteine was
present In the polypeptide at additbnal sites other than the C-terminus. Also within the scope
hereol are peptides crosslinked with methylene bridges.
3 5 Suitable crosslinking sites on the peptWes aside lrom the N-temminal amino and C-
terminal carboxyl groups include epsibn amino groups lound on Iysine residues as well as
amino imlno carboxyl sullhydryl and hydroxyl groups bcated on the side chains ot intemal
residues ol the peptides or residues introduced into tlanking sequences. Crosslinking
through extemally added crosslinking agents is suitably achieved e.g. using any ot a number
.
, . , ~ ., , - .: ,. , :. . ~ : .
, . : . ~ ~ .,.,.; .,: ' ,. . . : .
WO 92/07870 PCI'/US91107809
- 17 ~0923~ l
ot reagents tamiliar to those skilled in the art, tor example, via carbodiimide treatment of the
polypeptide. Other examples of suitable multifunctional (ordinarily bifunctional) crosslinking
agents include 1,1-bis(diazoacetyl)-2-phenylethane; glutaraidehyde; N-hydroxysuccinimide
esters (Bragg and Hou, Atch. Bhchem. Biophvs. (1975) 167, 311-321; Anjaneyla and Staros,
Int. J. Pep. Pro. ~s. (1987) ~0,117-124), such as esterswith4-azidosalicylic acid;
homobifunctbnal imidoesters including disuccinimidyl esters such as 3,3-dithiobis
(succinimidyl-propionate) and dimethyladipimidate dihydrochloride (Zahn, Agnew. Chem.
(1955) 67, 561-572; Golden and Harrison, Biochemistr~(1982) 21, 3862-3866); bifunctional
maleimides such as bis-N-maleimido-1,8-octane; disuccinimidyl suberate (Novick e~ al., J. Biol.
1 0 Chem. (1987) 262, 8483-8487), ~j~(suHosuccinimidyl) suberate (Lee and Conrad, J.
Immunol. (1985) 134, 518-525); heterobifunctional crosslinking reagents (Lomants and
Fairbanks, Arch. Biochem. Biophys. (1976)167, 311-321; Anjaneyula and Staros, ~L2~;
Partis e~ aL, J. Pro.Chem. (1983) 2, 263-277; Weitman et al., BioTechniqves, (1983)1, 148-
152; Yoshtake e~ al., J. Biochem. (1982) 92, 1423 1424), including those with an N-
1 5 hydroxysuccinimide moiety at one end and a maleimido group on the other end; succinimidyl
4-(N-malelmidomethyl) cycbhexane - 1 - carboxylate (SMCC) (Mahan et aL AnaL Biochem.
(1987)162, 163-170); suHo-SMCC (Hashida et aL, J. Applied Biochem. (1984) 6,56-63); m-
maleimidobenzoyl-N-hydroxysuccinimide ester (MBS); suHo-MBS; succinimidyl 4-(~
maleimidophenyl) butyrate (SMPB); suHo-SMPB; N-succinimidyl(4-bcbacetyl)aminoben ~oate
2 0 (SIAB); suHo-SlAB; 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochbride (EDC); and
N-hydroxysuHosuccinimide. Crosslinking agents such as methyl-3-I(p-azido-phenyl)dithb]
propioimidate yield photoactivatable intermediates which are capable o~ tomming crosslinks in
the presence ot light. It necessary, sensitive residues such as the side chains ot the diargininyl
group are protected during crosslinking and the protecting groups removed thereatter.
2 5 Polymers capable o~ muitiple crossl~nk~ng serve as ~nd~rect crossl~nk~ng agents. i-or
,example, cyanogen bromiaie activaled carbohydrates and Ihe systems descfibed ~n U.S.
pa~ents 3,959,080; 3,969,287; 3,691,016; 4,195,128; 4,247,6~,2; 4,229,537; 4,055,635
and 4,330,440 are suitably moditied tor crossllnking the peplides herein. Crosslinking to
amino groups ot the peptides is accomplished by known chemistries based upon cyanuric
3 0 chbride, carbonyl diimidazole, aidehyde reactive groups (PEG alkoxide plus diethyl acatal ot J
bromoacetaidehyde; PEG plus DMSO and acetk anhydride, or PEG chbride pius the
phenoxide ot 4-hydroxybenzaldehyde). Also usetul are succinimidyl active esters, activated
dithiocarbonate PEG, and 2,4,5-trichlorophenyl-chbrotorrnate- or p-nitrophenyl-
chbro10rmate-activated PEG. Carboxyl groups are derivatked by coup0ng PEG-amine using
carbodiimide. Ordinarily, however, the crosslinking agent is not a multitunctional poiyrner but
~nstead ~s a srnall molecule being bss than about 500 in MW.
The peptides ot this invention also may be contommationally stabilked by cyclkation.
The peptides ordinarily are cyclked by covalently bonding the N and C-temminal dornains ot
one peptide to the corresponding domain ot another peptide ot this invention so as to torm
_~.. . . .
, ~ . . . . . .
:.... . ~ - '~. . - ~
WO 92/07870 PCI-/US91/07809 1~
20~2315 18 ~
cyciooligomers containing two or more iterated peptide sequences, each intemal peptide
having substantially the same sequence. Furlher, cyclked peptides (whether cyciooligomers
or cylcomonomers) are crosslinked to tomm 1-3 cyciic stnuctures having trom 2 to 6 peptides
comprls0d therein. The peptides preterably are not covalently bonded through -amino and
maln chaln carboxyl groups (head to tail), but rather are aoss-linked through the side chains of
resldues iocated in the N and C-terminal domains. The linking sites thus generally will be
between the side chains ot the residues.
The cyclic structures of the present invention will have the general tormula:
A B
\/
1 0 wherein A and B represent the peptldes o~ this invention and are the same or diflerent. A anF
B are single peptides or head-to-tail poiymers ot two or more o~ such peptides. C represents
one or more bonds or crosslinking moieties.
Many suitable me~hods per se are known tor preparing mono-or poly-cyciized peptides
as aontemplated herein. Lys/Asp cyclization has been accomplished using N-Boc-amino
1 5 acids on solid-phase support with Fmoc/9-11uorenylmethyl (OFm) sWe-chain protection ~or
Lys/Asp; the process is r,ompleted by piperidine treatment folbwed by cyclization.
Glu and Lys side chains aiso have been crosslinked in preparing cyclk or bkycik
peptides: the peptide is synthesized by solid phase chemistry on a p-methylbenzhydrylamine
resin. The peptide is cleaved from the resin and deprotected. The cyclk peptide is tommed
2 0 usin~ diphenylphosphorylazide in diluted methyltommamide. For an altemative procedure, see
Schlller et aL, Peptide Proteln Res. (1985) 25, 171-177. See also U.S.Patent 4,547,489.
Dlsullide crosslinkad or cyclized peptides are generated by conventbnal methods.The method ol Pelton et ~1., (J. Med. Chem (1986) 29, 237û-2375) is suitable, except that a
greater proportbn ot cyclooligomers are produced by conducting 1here actbn in more
2 5 concentraled solu~bns lhan 1he dilu~e reaction mixture described by Pelton et al., lor the
productbn o~ cycbmonomers. The same chemis~ry is usetul tor synthesis ot dimers or
cyclooli~omers or cycbmonomers. Also usetul are thbmethylene bridges ( Tetrahedron
Leners (1984) 25, 2067-2068). See also Cody e~ al., 1 Med. Chem. ~1985) 28, 583.The desired cyclk or polymeric pep~ides are puritled by gel till-ation tolbwed by
3 û reversed-phase high pressure liquid chroma~ography or o~her conventional procedures. The
peptides are sterile tinered and tormulated into conventional pharmacobgically accep~able
vehlcles.
Seecltlc Chemlcal Synthe~lçp~ocedures The produc~s o~ Formula I and ~he
preterred substituen~s can be made by using one o1 ~he me~hods depkted bebw or by o~her
3 5 me~hods known in the art (see e.g., Spatola et al., Te~rahedron (1988) 44, 821 -833, and
reterences cited therein). The de1initions o~ the substituent groups are the same as tor
Fommula I except where noted~
WO 92/07870 2 ~ 9 2 31~ PCr/US91/07809
METHOD A
NHR~5 fORg
(CH2)n 0 (CH2)m~0 ~ 5 0
~HR~ 6 R7 R~r
R3 ll lll
7HR15 CORg
(CH2)n 0 (CH2)m
NH Jl~ 1 NH CO-Polymer Support
R1,N ~ ~ NH ~ ` ~
O ll 1. Cleave R16
~ ~XW R1~,X ~ R8 2. Cyclee
R R6
lV
NHR,5 fORs
(CH2)n 0 (CH2)m
R~N~NH JI~NH~NH~CO-PolymerSupport
O 1. Cleave 1rom resin
O~ ~ O 11 / ~ R8 2. Cleave R15
R~ NR~X
R Ro
V
A peptide derivatlve bound to a polymer support, depicted by intermedlate ll, may be
5 prepared by ser,iuenlial coupling ot individual amino aeid derivatives by standard technir.-ues.
(Merritieid, R. E~., J. Am. Chem. Soc. (1963) 85, 2149-2154; Stewart. J. M. and Young, J. D.,
SolAd Phase Peptide S~n~hesis (1984), Pieree Chemical Co., Rockiord, IL and additbnal
reterenees eited in the above publicatbns). When the te~rapeptide derivative ll is obtained,
the terminal amino group iS acylated with a suitabie carboxylic acid derivative lll. The acylation to
1 0 yieid IV may be accomplished uSing a number o~ standard m!hods which require activation of
the carboxylic acid group ot 111. For exarnple, activation rnay be obtained by the addition d an
WO 92/0~870 2 0 9 ~ 3 1 ~ PCI-/US91/07809
equimolar amount of dicyclohexylcarbodiimide or related carbodiimide reagent. It desired an
aWitive such as 1-hydroxybenztriazole or N-hydroxysuccinimide may be incorporated.
Altematively, the carboxyl group may be activated by conversbn to a hab derivative. For
example, the chbride may be obtained by treatment of the acid with thbnyl chbnde or oxalyl
5 chbrlde In a compatible solvent such as dichbromethane, toluene, or ethylene dkhloride it
desired. The substituent W is chosen such that it is readily displaceable by the group X.
Suitable substituents W are, 10r example, halo atoms such as bromine or bdine or activated
oxygen 1unctions such as methanesultonyloxy or p-toluensultonybxy and related sultonk acid
esters.
1 0 Cyclization to the resin bound intermediate V may be accomplished by selectively
exposing the nucleophilic group X by removal of R16 and albwing X to react such that it
dispbces group W with 1emlation of a new chemkal bond. For example, i~ X is a sultur or
oxygen atom and R16 is a triphenylmethyl group, then R16 may be selectively cleaved trom X
using a very dilute solution ot a strong acid such as tri~luoroacetic acid in a solvent compatible
1 5 with the polymer resin. Examples o~ resin corrlpatible solvents are dimethylacetamide,
dlmethyltormamide or dkhbromethane and the like.
The end result o1 the cleavage process is replacement of the R16 group with a
hydrogen atom. A~ter cleavage of R16, the resin bound peptide derivative V (R16 = H) is
albwed to react in a suitable solvent such as dimethylacetamide un!il cyclizatbn is complete. If
2 0 desired, a base such as N-methylmorpholine may be incorporated into the rear,tion. Other
protecting groups in the peptide molecule IV must be stable to the reaction conditions chosen
to 10mm V. For example, Rg may be a group which affords an ester such as methoxy, ethoxy,
benzyloxy, t-butybxy and the like or an amide or substituted amide. R1s may be a protecting
group such as t~n butybxycarbonyl. Final cleavage o1 ~he cyclized peptide product Irom the
2 5 polymer resin may be accomplished In a variety ol ways dependent upon the type ot resin
used and lhe chemical llnkage between the cyclized peptide and the resin. It, tor example, the
resln Is derived Irom a polymerked p-alkoxybenzyl alcohol derivative, then cleavage o~ the
peptide-resln linkage may be carried out using a strong acid such as tritluoroacetk acid. If
destred, additives such as phenol, anisole and ethanedithbl may be added to the reaction.
3 0 The groups Rg and R1s may be chosen, it desired, to also be cleavable concunently
with cleavage o1 the cyclized peptide Irom the polymer res~n. Examples ol such chemical
groups are Rg . t-butybxy, deavage o~ whbh yields Rg . OH and R1s t-butybxycarbonyl,
cleavage ot whhh at~ords R1s ~ H. The cnude product thus obtained may be turther purified
using dhromatographb or other methods of chernical purUkatbn to obtain 1.
3 5 Further defivatkation ot I may be carfied out ~ desired. For example, il X is S, treatment
o~ I with a stokhbmetrb amount ot an oxidizing agent such as 3-dhloroperoxybenzok acid or
similar agent will produoe the sultoxide derivative where X is SO. Use of an exoess arnount o~
oxidant will atlord the sullone derivative where X is S02.
METHOD B
. . . . . . . .
.: . ~. : - . ,,
WO 92/07870 . PCT/US91/07809
t';~' 2~2315
NHR,5 COR9
I
~CH2)n 0 (CH2)m
R~N~ V~NH~NH~cO-Polymersupport
~, OR X '~Ra Cleavage trom resin
Rs R6
lV
NHR,5 fORg
(CH2)n 0 (CH2)m
R~N ~ V~NH~NH~COR, Cyclize
0~ ~<W R~oX
R R6
Vl
Altematively, 1he linear peptide derivative IV, prepared as described above in Method
A, may be cieaved 1rom the resin prior to cyclka~bn to yield Vl. For example, it IV is
5 synthesked on a polystyrene resin the cleavage can be accomplished using aciuid hydrogen
tburicie. The groups Rg, R1s and R16 may, il desired, be cleaved concurrently under these
conditbns. I~ concurrent cleavage is des~red, lhen examples o~ suitable substituents Rg are t-
butybxy, benzybxy or cycbhexyloxy, R1s is t-butybxycarbonyl and R16 Is triphenylmethyl or
p n~thylbenzyl il X Is ellher O or S, or t butoxycarbonyl N X is NR1 3. Cleavage ot these groups
1 0 wouid result In Rg being OH and R1s and R16 be~ng hydrogen. The peptide derivative Vl may
then be cyclked h solutbn in the presence ot a weak base such as ammonium hydroxide.
The group W ~s as described ~n Method A. The rewlt~ng cnude I may then be puri~ed as
described above in Method A.
The purHled I may be lurther transtommed as described in Method A. Additbnally and H
1 5 desired, when X ~s NR13 and R13 ~s hydrogen, I may be acylated with, lor example, acetyl
chioride, aceUc anhydrlde or benzoyl chbride, methanesultonyl chbride or p-toluenesultonyl
chbride and the like.
Method C
Intermediate Vl may be prepared by the sequential coupling ot amho acid derivatives
2 0 in solutbn without the use ol polymer resin or other solid supports. The methods usetul tor
solutbn phase peptide synthesis are well documented in the chemical literature and are
,~ ,
- . .
.; , . . ..
~ , : . ' ' , !
,
W O 92~07870 , . P ~ /US9~/07809
2 0 9 2 3 15 2 2 ~?
known to those skilled in the art (Houben-Weyl, Methoden der Organischen Chemie, 4th
Edn., Vol. 15, Georg Thieme Verlag, Stuttgart 1974). The attached substituents R1, Rg, R15
and R16 may be chosen such that they are transfommable concurrently or sequentially as
described in Methods A and B above. Cyclizatbn ot ol Vl wherein R16 is H under conditions
5 descrlbed above In Method B will provide compounds of Formula 1.
The starting materials required tor the processes described herein are known in the
literature or can be prepareci using known methods and known starting materials.Isomeric Products
In products ot Formula I carbon atoms bonded to tour nonidentical substituents are
1 0 asymmetric. Accordingly, the compounds may exist as diastereoisomers, enantiomers or
mixtures thereot. The syntheses described above may employ racemates, enantiomers or
diastereomers as starting materials or intermediates. Diastereomeric products resulting trom
such syntheses may be separated by chromatographic or crystallkation methods. Likewise,
enantiomeric product mixtures may be separated using the same techniques or by other
1 S methods known in the art. Each ol the asymmetric carbon atoms, when present in compounds
ot Fommula 1, may be in one ot two contiguratbns (R or S) and both are within the scope of the
present inventbn. The carbon atoms bearing the (CH2)n sidechain and the (CH2)m sidechain
are generally pretenred to have the S connguration. The carbon atom bearing the substituents
R2 and R3 is generally preferred to have a configuratbn corresponding to that of a D amino
2 0 acid. The contiguration may be assigned R or S depending on the chemical compositbn o~ R2
and R3.
The compounds described in this inventbn may be isolated as the ~ree acid or base or
convened to sans ot varbus inorgank antt organb acids anci bases. Such salts are within the
scope of this invention. Examples ot such salts Include ammonium, metal satts like sodium,
2 5 potssslum, cablum and ma;nes~um; sans with organic bases like dicycbhexylamine, N-methyl-
D-glwamlng 8nd the like; and Saits with amlno acids llke arginine or lysine. Salts with inorgank
anci orç anlc aclds may be llkewise prepared, tor example, using hydrochbrk, hyrirobromic,
suifurk, phosphoric, tritluoroacetb, methanesuifonk, malb, maleic, tumark anci the like. Non-
toxb and physbbgkally compatible salts are particularly usetul anhough other less desirable
3 0 sans may have use in the processes ot Isolatbn and purifkatbn.
A number ot methods are useful lor the preparation ot the salts described above anci
are known to those skilled in the art. For example, reactbn ot the tree acid or tree base fomm of
a compound ot Fommub I with one or more molar ec;uivabnts of the desired acid or base in a
soivent or solvent mixture in whbh the san is insoluble; or ~n a soivent like water after whkh the
3 5 soivent is removed by evaporatbn, distillation or ~reeze drying. Altematively, the tree acid or
base torm ot the product may be passed over an bn exchange res~n to torm the des~red san or
one san torm ot the product may be convened to another using the same general process.
The compounds described in the present invention inhibit the binding ot tibrinogen to
its receptor on platelets, GP llb Illa, anci thus prevent the aggregation ot platelets and the
:- . .:
:
. . -.
WO 92/07870 PCI`/US91/07809
!, . ~ 2 3 2 0 9 2 3 1 5
formation of platelet plugs, emboli and thrombii in the circulatory system in mammals.
Thrombosmbolic disorders have bsen shown to be dirsctly related to the susceptibilay of
bbod platelets to aggregate. Mammals exposed to medical procedures such as angioplasty
and thrombolytlc therapy are particularly susceptible to thrombus formatbn. The compounrJs
S o~ Ihe prBsent Invention can be used to inhibit thrombus tormation tolbwing angbplasty. They
may also bs used In combination with thrornbolytic agents such as tissus plasminogen
acllva~or and its derivaUves (US palents 4,752,603: 4,766,075: 4,m,043 EP 199,574; EP
0238,304; EP 228,862; EP 297,860; PCT W089/04368: PCT WO89100197), streptokinaseand its dsrivatives, or urokinase and its derivatives to prevent arterial reocclusbn folbwing
1 0 thrombolytic therapy. When ussd in combinatbn wah the above thrombolytk agents, the
compounds of the present invention may be administered prbr to, simultaneously wah~ or
subsequent to the antithrombolytic agent. Mammals exposed to renal dialysis, bbod
oxygenatbn, cardiac cathsterization and similar medical procsdures as well as mammals fmed
with certain prosthetic devices are also susceptible to thromboembolb disorders. Physblogk
1 5 conditbns, with or without known cause may also lead to thromboembolb disorders. Thus, the
compounds described herein are useful in treating thromboembolb disorders in mammals.
The compounds dewribed herein may also be used as adjuncts to anticoagulant therapy, for
exampls in combinatbn with aspirin, heparin or warfarin and other anticoagulant agents. The
applbatbn of the compounds described hsrein for these and relatsd disorders will be
2 0 apparent to those skilled in the art.
' ' " '""~ ,,"",~ ", " ~", ",,~ "; ~ ,. .
; . .~: . . ..
; ,.,.,, ..~
;
:
~ , .
WO 92/07870 . PCl`/US9]/07809
` 20~i~3~j 2
Platelet Inhlbltlon Assays
The evaluation of inhibaors of ~he fibrinogen-platele1 interaction is guided by in vi~ro
recep~or binding assays and in vi~ro pla~elet aggrega1ion inhibition assays.
In-varo bbloglcal activay ot the r,ompounds ol Fom~ula I was monaored using a
moditled tlbrlnogen-GP llb Illa ELISA based on lhe method ot Nachman and Leung (J. Clin.
Invest, (1982) 69, 263-269) which measures the inhibhion of fibrinogen binding to purNied
human platelet GP llb Illa receptor. Human fibrinogen was prepared by the method of Lipinska,
et aL (J. Lab. Clin. Med. (1974) 84, 509-516). Platebt GP llb Illa was prepared by the me~hod ot
Fitzgerald, et al. (AnaL Biochem. (1985) 151, 169-177.
1 0 Mbro~aer pla~es are coa~ed wah tibrinogen (10 ~ml) and ~hen blocked wah TACTS
buffer containing 0.5% bovine serum albumin (BSA). (TACTS buffer contains 20mM Tris.HCI,
pH 7.5, 0.02% sodium azide, 2 mM calcium chloride, 0.05% Tween 20, 150 mM sodiumchbride.) The pla~e is washed wah phospha~e buffered saline (PBS) containing 0.01% Tween
20 and ~he sample ~o be de~ermined aWed, tolbwed by addition ot solubilked GP llb Illa
1 5 recep~or (40 i~yml) In TACTS, 0.5% BSA. Atter incubation, the plate is washed and 1 ilg/ml ot
murine antl-platelet monoclonal antibody AP3 (P. J. Newman etal. Blood(1985) 65, 227-232) ~ .
is aWed. Atter ano~her wash a goa~ anti-mouse IgG conjuga~ed to horseradish peroxidase is
added. A tinal wash is perlormed and developing reagent butter (10 rng o-phenylenediamine
dihydrochloride, 0.0212/~ hydrogen peroxide, 0.22 mM cara~e~ 50 mM phospha~e, pH 5.0) is
2 0 added and ~hen ~ncuba~ed until cokor deveiops. The reacthn is s~opped wah 1 N suituric acid
and the absorbance at 492 nm is recorded.
In addnbn to the GP llb Illa ELISA assay, piatelet aggregathn assays may be pertomled
in human platelet rich piasma (PRP). Fitty millil-ners ot whole human blood (9 parts) is drawn on
3.6% sodlum citrate (1 part) trom a donor who has not taken aspirin or related medkatbns tor at
2 5 least two weeks. The blood Is centrituged at 160 x 9 tor 10 min at 22 C and then aliowed to
stand tor 5 mln aner whlch the PRP is decanted. Platelet poor plasma (PPP) is isolated ~rom
the remalnlng biood atter centraugathn at 2000 x 9 tor 25 min. The platelet count ot the PRP
was adjusted to ca. 300,000 per mkrolner wah PPP.
A 225 ~L aliciuot ot PRP plus 25 IlL ot either a dilution ol the test sample or a control
3 0 (PBS) Is Incubated tor 5 min in a Chrono-iog Whole Bbod Aggregometer at 25 C. An
aggregaling agern (collagen, 1 mglml; U46619, 100 ng/ml; or ADP, 8 ~N) b added and the
platelet aggregation recorded.
In the management ot thromboembolic disorders the compounds ot this inventhn maybe utilked in composithns such as tablets, capsules or elixers tor oral administratbn;
3 5 supposltofies tor rectal administration; sterile solutbns or suspensions 1Or injectable
admlnlstrathn, and the like. Animals in need o1 treatment using compounds o1 this inventbn
can be administered dosages that will provide optimal etticacy. The dose and method ot
administraUon will vary trom animal ~o animal and be dependen~ upon such ~acto~s as weigM,
, - - : . ,: . . .
., ,
WO 92/07870 . PCI/US91/07809
25 2~.9231~ ~
diet, concurrent medication and other factors which those skilled in the medkal arts will
recognize.
Dosage Formulatlons
Dosage formula~ions o~ the cyclic polypeptides o~ the present inventbn are prepared
ior storage or administralion by mixing the the cyclk polypeptide having the desired degree o1
purity with physhbgically acceptable carriers, excipienls, or stabilizers. Such materials are
non-toxb to the recipients at the dosages and concentratbns employed, and include buffers
such as phosphate, citrate, acetate and other organk acW salts; antbxidants such as ascorbic
acid; bw molecular weight ~less than about ten residues) peptides such as poiyarginine,
1 0 proteins, such as senum albumin, gelatin, or immunoglobulins; hydrophilk polymers such as
polyvinylpyrrolidinone; amino acids such as giycine, glutamic acid, aspartic acid, or arginine;
monosaccharides, disaccharides, and other carbohydrates including cellulose or its
derivatives, glucose, mannose, or dextrins; chelating agents such as EDTA; sugar alcohols
such as mannUol or sobitol; counterbns such as sodium and/or nonbnic suriactants such as
Tween, Pluronics or polyethyleneglycol.
Dosage lormulatbns ot the cyclic poiypeptides o1 the present inventhn ~o be used 10r
~herapeu~k adminis~rathn must be sterile. S~erility is readily accomplished by nltration through
s~erile nltrathn membranes such as 0.2 microne membranes. Cyclk polypeptide torrrlula~ions
ordinarily will be stored in Iyophilized 10rm or as an aqueous solutbn. The pH of the cyclk
2 0 poiypeptide prepara~bns typkaliy will be between 3 and 11, more pre1erabiy 1rom 5 to 9 and
most pre1erably from 7 ~o 8. 1~ will be unders~ood ~ha~ use o1 certain of ~he 10regoing
exeipients, earriers, or stabilizers will resuit in the 10rma~hn o1 eyclie polypep~ide sans. While
the pre1erred route of administrathn is by hypodermb injeetbn needle, other methods of
administra~ion are also antkipated such as suppositories, aerosols, oral dosage tormulatbns
2 5 and topbal 10rmulathns such as ointments, drr ps and dermal patehes.
Therapeutlc cyclic polypeptide tommulatbns generally are placed into a con~ainerhavlr~ a sterih aeeess port, for example, an intravenous soluthn bag or vial having a stopper
plereeable by hypodermle Injection needle.
Therapeutkally effective dosages may be determined by either in vitro or in vivo3 0 methods~ One method of evaluating therapeuticaliy effective dbsages conslsts of taking the
eyelb poiypeptide cycb-S-aeetyl-Gly-Lys-Giy-Asp-Cys-OH and detemmining a 50/0 inhibitory
eoneentratbn (ICso) o~ inhibiting flbrinogen bind~ng to the GP llb Illa piatelet receptor.
Simibriy, in a platelet aggregatbn assay using the same eyeib peptide, the ICso is measured.
Based upon sueh in vitro assay teehniques, a therapeuticaliy effective dosage range may be
3 5 determined. For each partkular cyclie poiypeptide ot ~he present invention, individual
determlnations may be made to de~ermine ~he op~imal dosage required. The range of
therapeutically ef1eetive dosages will na~urally be influenced by lhe rou~e of adminis~ra~ion. For
injection by hypodermic needle it may be assumed ~he dosage is delivered into ~he body s
. ~
. . .
:
WO 92/07870 PCI`/US91/07809
2~231'~ 26 ~
fluids. For other routes of administration, the absorption efficiency must be individually
deterrnined tor each cyclic polypeptide by methods well known in pharmacology.
The range of therapeutic dosages is from about 0.001 nM to 1.0 mM, more preSerably
trom 0.1 nM to 100 llM, and most preferably Irom 1.0 nM to 50 iiM.
Typkal lormulation ol compounds ol Fommula I as pharmaceutical compositions are
dlseu8sed below.
About 0.5 to 500 mg ol a compound or mixture of compounds of Formula 1, as the Iree
aeld or base torm or as a pharmaceutkally acceptable salt, is compounded with a physiok~gkally
acceptable vehicle, carrier, excipient, binder, preservative, stabilizer, llavor, etc., as called lor
t 0 by accepted phamlaeeutical practke. The amount of active ingredient in these compositions is
such that a suitable dosage in the range indicated is obtained. . -
Typical adjuvants which may be incorporated into tablets, capsules and the like are a
binder such as acacia, com starch or gelatin: an excipient such as microcrystalline cellubse; a
disintegrat.ng agent like com starch or alginic acid; a lubricant such as magnesium stearate; a
1 5 sweetening agent such as sucrose or lactose; a tlavoring agent such as peppermint,
wintergreen or cherry. When the dosage lorm ~s a capsule, in addition to the above materials it
may also contain a liquid carrier such as a latty oil. Other materials ôl various types may be used
as coatings or as rnodiliers ol the physkal torm ot the dosage una. A synup or elixer may
contain the active compound, a sweetener such as sucrose, preservatives like propyl paraben,
2 0 a cobring agent and a llavoring agent such as cherry. Sterile composabns lor injectbn can be
lommulated aceording to conventbnal pharmaceutkal practke. For example, dissolution or
suspenslon ol the active compound in a vehicle such as water or naturally occurring vegetable
oil like sesame, peanut, or cottonseed oil or a synthetk tatty vehkle like elhyl oleate or the like
may be desired. Bulters, preservatives, antbxidants and the like can be incorporated
2 5 according to accepted pharrnaceutical practke.
EXAMPLES
In the loliowlng Examples, common ~ amino acicts may be described by the standard
Ihree lener amlno aeid eode when relerring ~o intermediates and linal products. By common -
amlno aeids Is meant those amino aeids incorporated Into protelns under mRNA directbn.
3 0 Standard abbreviatbns are listed in The Merck Index, 10th Edabn, pp Misc-2 - Misc-3. Unless
otherwise designated the eommon a-amino acids have the natural or ~L~- eordiguratbn at the
alpha ealbon atom. Il the eode is preeeded by a ~D~ this signilies the oppOsae enantbrner ot
the eommon c~-amino aeid. Moditied or unusual c~-amino aeids sueh as norteueine (Nle~ and
omahine (Orn) are desbnated as described in U.S. Patent and Trademark Office Of ~kial
3 5 Gazette 1114TMOG, May 15, 1990. Il the produet or intemlediate name is preceded by
eyelo~ this shall be taken to mean that the peptide has been eyelized, e.g. eompounds ol
Fomlula I or V.
Example 1
Bromoacetyl-Gly-Lys-Gly-Ase-Cys-OH
.: ' , . ,' ' . ~: ,
; . . . .. -
: . . .~ , . - . , .
- , ~ - . .
', ~ . . ~ .
:
WO 92/07870 PCI'/US91/07809 ,'
2 7 2 ~ 9 ~ 3 1 ~ .
The title compourld is prepared in prolected fomm by standard solid phase peptide ~.
synthesis on 2% cross-linked polystyrene resin (Merrilield resin). Treatment ol the resin bound
intemmediate with liquid hydrogen nuoride induces concomitant cleavage of the protecting
groups from the litle compound as well as cleavage ol Ihe peplide trom Ihe resin. The crude
5 peptlde Is purlfled by reverse phase high performance liquid chromatography (HPLC) using a
4.6 mm x 250 mm column contalning 10 micron 300 Angstrom pore ske C-18 packing. The
elullon of the column is wah an acetontrile/0.1% aqueous traluoroacetk acW gradient going
trom 0% - 40% acetonitrile linearly over 80 minutes. The tale compound elutes at 14 minutes.
Example 2
1 0 Cycb-S-acetyl-GIy-Lvs-Gly-As~Cys-OH
The compound prepared in Example 1 is disso~ed in deionked water (1 mg/ml) and
the pH of the solution is adjusted to 7.0~.5 with ammonium hydroxide. Alter stirring tor 4 hr at
ambient temperature the reaction solution is acidilied to pH 3.0 - 3.5 with trifluoroacetic acid
and then Iyophilized. The resulting cnude product is purified by HPLC using the conditions
1 5 described in Example 1. The desired title compound elutes alter 11 minutes.
Example 3
Cycb-S-acetyl-Gly-Lys-Gly-As~Cys-OH
Sromoacetyl-Gly-Lys~t-butyloxycarbonyl)-Gly-Asp(beta-t-butyl)-Cys(S-
triphenylmethyl)-O-(polymer resin) is prepared using standard solid phase peptide synthesis
2 0 utillzing fluorenylmethoxycarbonyl (FMOC) protecting group chemistry on a p-alkoxybenzyl
alcohol resin. Repeated treatment of the resin bound peptide wah a 1% soluUon oftrilluoroacetb acid in dbhbrome~hane results in cleavage ot ~he S-~riphenylme~hyl group as
ev~denced by ~he br~gh~ yelbw ot ~he solu~bn. Trea~ment ~s cont~nued un~l d~ss~pa~ion o~ the
yelbw cobr (ca. 1.5 L ol the cleavage solutbn Is required per gram of resin bound peptide.)
2 5 ~tter complete cleavaDe ol the S-triphenylmethyl group the resin bound peptide is washed
several llmes wlth a 5% solutbn of N-me~hylmorphollne in N N-dime~hylace~amide and then
shaken In pure N N-dimethylacetamide for 12 hr to complete the cyclizatbn. Treatment of the
cyclked resin bound peptide wah ~rifluoroaceUc acid containing (vh) 1% phenol 1% anisole
and 1% ethanedlthiol eflerts concomaant cleava~e o~ ~he remaining pro~ective groups and
3 0 cleavage of the deslred product from ~he resin. Purifica~bn of ~he cnude product as described
In Example 2 aflords the U~le compound ~dentical ~o ~ha~ described above.
Exampl- 4
Syrdh~i~ Other Comeounds of Fommula I
Using ~he me~hods described ~n Examples 1 and 2 ~he compounds l~s~ed ~n Table I
3 5 may be prepared. The compounds are depbted by Fomlub I wherein m ~ 1 and n 3 R1 and
Rg are OH R14 is hydrogen and X is S. Crude products are purified using HPLC as described
in Example 2.
,~ .
, .
' -
W0 92/07870 2 0 9 2 3 1 ~ 2 8 PCI'~US91/07809
The ~ollowing amino acid derivatives may be used in place of Boc-Gly ~or coupling to
the alpha-amine group ot Lys to obtain the substituents R2 and R3 shown bebw. When Boc-
Glycine is used, R2 and R3 are both hydrogen (H).
Amlno Acid Derivative R2 R3
- :
Boc-L-Ala methyl H
Boc-D-Ala H methyl
Boc-L-Val 2-propyl H
Boc-D-Val H 2-prowl
Boc-D-Thr H 1-hydroxy-1-ethyl
Boc-L-Thr 1-hydroxy-1-ethyl H
Boc-D-Asn H carboxamidomethyl
Boc-L-Asn carboKamidomethyl H
Boc-D-Gln H 2-carboxamidoethyl
Boc-L-Gln 2-carboxamidoethyl H
Boc-D-~0-2-bromobenzyloxy- H 4-hydroxybenzyl
carbonyl)Tyr
Boc-L-(0-2-bromobenzybxy- 4-hydroxybenzyl H
carbonyl)Tyr
Boc-D-Phe H benzyl
Boc-L-Phe benzyl H
Boc-D-Leu H 2-methyl-1-prowl
Boc-L-Leu 2-methyl-1-propyl H
Boc-D-Met H 2-methylthioethyl
Boc-L-Met 2-methylthbmethyl H
Boc-D-Nle H 1-butyl
Boc-L-Nle 1-butyl H ;
Boc-D-lle H 2-butyl
Boc-L-lle 2-butyl H
Boc-D-Asp(~-0-cycbhexyl) H carboxymelhyl
Boc-L-Asp(~-0-cycbhexyl) carboxymethyl H
Boc-D-Glu(~0-cycbhexyl) H 2-carboxye~hyl
Boc-L-Glu(y 0-cyclohexyl) 2-carboxyethyl H
Boc-D-(~benzyl)Ser H hydroxymethyl
Boc-L-(0-benzyl)Ser hydroxymethyl H
Boc-D-(Nim- H 4-imidazolylmethyl
benzybxymethyl)His
WO 92/0~870 . PCI'/US91/07809
-~ 29 2~231~ 1
Boc-L-(Ni'n- 4-imidazoiylmelhyl H
benzybxymethyl)His
Boc-D-Trp H 3-indolylme~hyl
Boc-L-Trp 3-indolylmethyl H
Boe-D-~Ne-2- H 4-amino-1-butyl
chborocarbobemzyl-oxy) Lys
Boc-L-~Ne-2- 4-amino-1-butyl H
ehbroearbobenzyl-oxy)Lys
Boe-D-~N~- H 3-amino-1-propyl
carbobenzyoxy)Orn
Boc-L-(N~- 3-amino-1-propyl H
carbobenzyoxy)Orn
Boe-D-(O-benzyl)Thr H 1-hydoxy-1-ethyl
Boe-L-(O-benzyl)Tht 1-hydoxy-1-ethyl H
Boe-L-Pro gives R2 ~ R4 CH2CH2CH2
Boe-D-Pro gives R3 I R4 ~ CH2CH2CH2
The substiluted bromoaeetb aeids listed bebw may be used in place ot bromoaceticacid in E-xample 1 and in cornbination with lhe arnino acid derivatives listed above to provide
5 the compounds shown in Table 1 wah variable substituents at Rs, R6. When bromoacetic acid
is used in eombination with the amino acid derivatives listed above Rs and R6 are hydrogen
(H)-
1-naphthyl--bromoacetic acid
2-naphthyl--bromoaeetie aeid
1 0 phenyl-a-bromoaeetb aeld
2-1rmurome~hylphenyl--bromoaeetk aeid
3-1rilluromelhyphenyl-rl-bromoaee~b aeid
4 lrUlurom~hylphenyl-r,~-bromoaeelb aeid
4-blphenyl--bromoaeetb aeld
1 S 2-bromopropionle aeld
2-bromobutyrie aeid
2-bromopentanole aeid
L-Pennlelllamine may be substituted tor L-eysteine in Example 1 to produee
eompounds in Table 1 where R7 and R8 are methyl.
2 0 TABLE I
Soleeted eompounds ot Fonnula I
R2 R3 R4 R5, R6 R7 R8
H (4) (4) H,H H H
, . .
' ~ ;
.
.
WO 92/07870 . PCI~/US91/07809
2~9231~ 30 ~ ~
H 1-hydroxy-1- H H, H H H ~ .
ethyl
H H H H, 1-napMhyl H H
H 1-hydroxy 1- H H, phenyl H H
ethyl
H H H H, 4-biphenyl H H
H 4-hydroxy- H H, H H H
benzyl
H H H H, phenyl H H
H H H H, H CH3 CH3
H 2-propyl H H, H H H
H methyl H H, H H H
.. ... . .
H carboxamido H H, H H H
methyl
H H H H, 2-trl- H H
tluoromelhyl
phenyl
H 41midazolyl- H H, H H H
methyl
H 2-methyl- H H, H H H
1-propyl ,
H 4-hydroxy- H H, phenyl H H
benzyl
H H H H, 4-biphenyl H H
WO 92/07870 . PCI`~US91/07809
~'~' 31 209231a r
H H H H, 1-naphthyl H H
H 2-me~hyl- H H, H H H
thioethyl
H hydroxy- H H,H H H
methyl
H 3-indolyl- H H, H H H
methyl
H carboxamidoe H H, H H H
thyl
H H H H, 4-tfi- H H
tluoromethylp
henyl
H H H H, 2-naphthyl H H
H H H H, propyl H H
H 3-amino-1- H H, H H H
propyl
H H H H,3-tfi- H H
11uoromethylp
henyl
H H H H, ethyl H H
H 2-butyl H H, H H H
4-1midazolyl- H H H, H H H
methyl
benzyl H H H, H H H
.. ~.... . . .
:- . . . - ' ' ~ - ,
~ ' ' - ,~ ' ' '"' ~ , ' ' '
, ., . ~
.. , . ~ . ~
WO 92/07870 . PCT~/US91/07809
32
H 1-hydroxy-1- H H, phenyl H H
ethyl
H 4-hydroxy- H H, H CH3 CH3
benzyl
. H 4-amirlo 1- H H,H H H
butyl
.
CH3 H H H, H H H
(5) H (5) H,H H H
,~ .
H H H H, 2-naphthyl H H
-
1-hydroxy-1- H H H, H H H
~thyl
H H H H, phenyl H H
H 2~arboxy-1- H H, H H H
ethyl
1 bulyl H H H,H H H
2 propyl H H H,H H H
2 butyl H H H,H H H
4~hydroxy H H H,H H H
bonzyl
H H H H, ethyl H H
4-amino-1- H H H, H H H
butyl
H H H H, 1-propyl H H
. .. ; . . . ~, , .,, , . , . ., ~ . ,
--
.
: - . ~ .. ~ , . ..
: ?. : ~ '
WO 92/07870 PCr/US91/07809
~ 33 2~9231~
hydroxy- H H H, H H H
methyl
H H H H, 2-tri- H H
~luoromethylp
henyl
H carboxy- H H, H H H
methyl
2 methyl- H H H,H H H
t thioethyl
3-indolyl- H H H, H H H
me~hyl
3-amino-1- H H H, H H H
propyl
carboxamido H H H, H H H
methyl
benzyl H H H, H H H
H H H H, 3-tri H H
tluoromethylp
henyl
2-methyl- H H H, H H H
l~propyl
H H H H,CH3 H H
H H H H, H H
pentatluoro-
phenyl
2-carbox H H H,H H H
amidoethyl
. : . .: . . ;
.: .` .
WO 92/07870 PCI/US91/07809 `
, 3 4 (~ I :
H H H H 4-tri- H H
tluoromethylp
henyl
2-carboxy- H H H H H H
ethyl
carboxy- H H H H H H
melhyl
: ' ' ' ' . '
Example 5
'. Cycb-S-acetyJ-~D-Tvr)-Lys-Gly-As~Cvs-NH2
Synthesis oS the title compound is accomplished using standard Boc-synthetic
protocols on a 4-methylbenzylhydrylamine resin to obtain ~irst a linear peptWe as described in
5 Example 1. Cleavage ol the linear peptide 1rom ~he resin with hydrogen tluoride tolbwed by
cyclkatbn as described in Example 2 affords the title cornpound after HPLC purification. Using
th's procedure the 'olbwing compounds are anabgously obtained.
Cycb-S-acetyl-(D-Ala)-Lys~ly-Asp-Cys-NH2
Cycb-S-acetyl-(D-Val)-Lys-Gly-Asp-Cys-NH2
1 0 Cycb-S acetyl-(D-Leu)-Lys-Giy-Asp-Cys-NH2
Cycb-S-acetyl-(~lle)-Lys-Giy-Asi~Cys-NH2
Cycio-~acetyl-(D-Phe) Lys-Gly-Asp-Cys-NH2
, Cycb-S-acetyl-(D-Pro) Lys-Gly-Asp-Cys-NH2 .,
Cycb-S-acetyl-Gly-Lys-GIY-Asp Cys-NH2
15 Ex~mpl~ 6
Cycb-S-ace~yl-Gly-Lys-Gly-Ase-Cys-OH sunoxitfe
The purifled product trom Example 2 is dissoived in water at a concentratbn ot 10 mg
por mL. The pH ot the solution is adjusted to 7. A 50% solutbn of hydrogen peroxide is
added to rrake a tinal concentratbn ot 3% hydrogen peroxide and the resuning reaction
2 0 mlxture Is stlrred ovemlght at room temperature. The solutbn is baded directly onto an
octadecylsilyl reverse phase chromatography column. The suHoxide isomers tormed in the
reactbn are eluted wah a linear grad~ent o~ acetonarile ~n 1% tr~nuoroacetb acld in water.
_.. ~. . .. .
. . ,, ,~ : . , ~ ; : :
:- :.; - ~ . .. .. ; . -,
-~ . .
;,; . ' ' '`'`; , ~ ' ' ~:
WO 92/07870 . PCI'/US91/07809
,~: 35
Example 7
Inhibitionof 1ibrinogen bi[ldi~ GP 11~
Microtiter plates are coated with fibrinogen (1o i-~ml) and then blocked with TACTS
buffer containing 0.5% i3SA. (TACTS bufler contains 20mM Tris.HCI, pH 7.5, 0.02% sodium
azide, 2 mM cakium chloride, 0.05% Tween 20,150 mM sodium chbride.) The plate is washed
with phospha~e buflered saline conta~ning 0.01% Tween 20 and a dilutbn d the sample to be
delemmlned added, loliowed by aWition o~ sohbilized llbllla receptor (40 i-~ml) in TACTS,
0.5% BSA. A~ter incubation, the plate is washed and murine monocbnal anti-platelet antibody
AP3 ~1 i~ml) added. A~ter another wash goat and anti-mouse IgG conjugated to horseradish
1 0 peroxidase is added. A nnal wash is periormed and developing reagent buffer (10 mg o-
phenylenediamine dihydrochbride, 0.0212% hydrogen peroxide, 0.22 mM citrate. 50 mM
phosphate, pH 5.0) is added and then incubated until cobr devebped. The reaction is
stopped with 1 N suHuric acid and the absorbance at 492 nm is recorded and the ICso values
determined.
1 5 Example 8
Inhibition of Vitronectin i3indin9 tP. Y3~i~e~
a. Coat 9~well microliter plates (Nunc Maxisorp) with human vitronectin (Telbs) made up
at 15 i-~ml in PBS. Use 50 i-lUwell. Incubate ovemight 4 C.
b. Remove coat solutbn. Wash plates one time with 200 il' ' assay buHer (50 mM Tfis,
2 0 100 mM NaCI,1mM CaC12,1mM MgC12,1mM MnCI2, p~-i 7.4) wXh the addabn of 3.5% BSA in
assay buHer. Add additbnal 15qii assay bufferlwell. Bbck plate ~or 1 hour at room
temperature.
c. Prepare test compounds in assay bu~fer. Prepare human varonectin receptor, purifled
at 50 ~ml concentratbn.
2 5 d. Acid 25 ~ll o~ test r;ompounds or buHer control into the plates. Add 25 lli d the
receptor solutbn ~nto the plates. Inwbate at room 1emperalure with shaking 1Or ~i hour.
e. Prepare antibody solutbn dufing Incubatbn. Comb~ne 4B12 mab (a i33 specffic
antlbody) at 1 :1650 wnh a rabbit F(ab )2 anti-mufine Fc-HRP conjugate (Pel-Freez)1 :7500 in
assay buHer.
3 0 ~. Decant plates. Wash 4 times ~150 ~Uwell) with PBS, .05% Tween 20. Add in 50 i-~i ot
the antibody solutbn per well. Incubate 1 hour at RT wffh shaking.
9. Prepare OPD (onhophenyhnediamine) sut~strate,10m~ OPD (SigmaJ Into 15 ml o~
phosphate-citrate. Then add 6 ~i o~ 30% H22 to the soiutbn. Do this 5 minutes be~ore use.
h. Wash plates with PBS/Tween20 4 times. Add 75 ~li ol the OPD soiutbn to each well.
3 5 Let reactbn proceed 20-30 minutes. Add 75 ~ ot 1 M ti2SO4 to stop reactbn. Read
absorbance at 492nm.
In view o~ the eHicacy o~ these cyclic polypeptides as inhibitors ot fibrinogen binding to
GP llb Illa, and the teasibility as demonstraled herein o~ producing these cyclic polypeptides,
the present invention may have application in the treatment o~ a brge group ot disorders r
, . . .
.
:~,
- ' ~ ' ` '
. . . ..
WO 92/07870 2 ~ 9 2 3 1 ~i 3 6 PCI~US91/07809
associated with, or characterked by, a hyperthrombotic state. Representative of such
disorders are genetk or acquired deticiencies of factors whkh normally prevent ahyperthrombotic state; medical procedures such as angioplasty and thrombolytk therapy;
mechankal obstnuctions to blood nOw~ such as tumor masses, prosthetk synthetk cardiac
5 valves, and extracorporeal perlusbn devkes; atherosclerosis; and coronary artery disease.
The present invention has of necessity been discussed herein by reterence to certain
specilb methods and materlals. It is to be understood that the discussion ot these specifk
melhods and materlals in no way constitutes any limitation on the scope ot the present
Inventbn, which extends to any and all alternative materials and rnethods suitable lor
1 0 accomplishing the objectives of the present invention.
._.. . . .
., .~ , . . . . .
~ - , - . .
. : ' ' ~ ' .' ' . ' .'.
,' '~Ai 1:
'
,. , ~ '. ,. '