Note: Descriptions are shown in the official language in which they were submitted.
~92~7~3
HOECHST ARTIENGESEL~SCHhF~ HOE 92/F 080 Dr.WN~St
Description
Carboxylic acid e~ters of 2-amino-7~1,3-dihydroxy
2-propoxy)methyl]purine, their preparation and their use
It has already been found (s~e ~P 452 6803 that subv
stitu~ed purin~s of the formula
R4
I 1 ~ ,J,
N ~ N ¦ R5
possess antiviral acti~ity~
It has now emerged that certain carboxylic acid esters of
substituted purines have exceptional antiviral activity
and, in addition, exhibit particularly marked bioavail-
ability following oral administration.
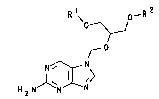
Accordingly, the invention relates to compounds of the
formula 1
Rl, o--R2
0~
~ Nr (1)
H N ~`N N
in which the radicals R1 and/or R2, independently of each
other, are acyl radicals of the formula
-~(=o)-R3 (2),
where
R3 i~ (cl-c3)-alkyl~ and one of the radicals Rl and R,2 can
also be hydrogen, and thei~ phy~iologically tolerated
-- 2
salts.
Particularly Lmportant compounds of the formula 1 a~
previously described are those in which
R3 is methyl, and one of the radical~ R1 and R2 can also
~e hydrogen.
The abovementioned compound of the formula 1 in which R3
is methyl is of very particular importance.
The compounds of the formula 1 according to the invention
can possess one or mors chiral cen~rs in the acyclic
~ide chain. Th~ compounds are as a r~le pre~ent as
racemates; preparation or i~olation of the pure enan-
tiomexs is po~ible. ~he invention therefore relates koth
to the pure enantiomers and to mixtures thereof, ~uch as,
for example, the corresponding rac~mate.
Salts of the compounds according to the invention which
are particularly suitable for t;herapeutic purpoæes are
salts of physiologically tolerated acids, ~uch as acetic
acid, lactic acid, malic acid, p-toluenesulfonic acid,
methanesulfonic acid, i~ethionic acid, hydrochloric acid,
sulfuric acid or phosphoric acicl.
The present invention furt~ermore relates to a process
for preparing compounds according to the invention,
wherein the compound of the formula 1'
H~ 0~~
r~
N~CN>
H2N l N N
is reacted with carboxylic acids of the formula 3
R4 - COOH (3)
7 ~
where R4 is (Cl-C3~-alkyl, in an organic solYent ~nd in
the pre~ence of ~ dehydrating agent and with the addition
of a base, to give compound~ of the formula 1, and the
monoesters and diesters are subsequently separated from
each other u~ing conventional methods.
The abovementioned compvund of the formula 1' can be
prepared, for example/ as described in EP 452 ~60.
Dicyclohexylcarbodiimide, for example, i~ a preferred
dehydrating agent for caxrying ou~ the abovemen~ioned
reaction. Suitabl~ bases are, for e~ample, N,N'-dim~thyl~
aminopyridine or its derivatives.
Examples of suitable organic solvents fo carrying out
the reaction are dimethylformamide, N-meth~1-2-pyrroli-
done, pyridine, dichloromethane, 1,2-dichloroe~hane or
tetrahydrofuran.
For carrying out the raaction, the compound of the
ormula 1~ is preferably dissolved sr ~uspended in the
organic solvent and, preferably at -10C to ~40~C, in
particular at room t~mperature, and with stirring, mixed
succe~si~ely with pre~erably at lea~t 2 equivalent~, in
particular 2.2-3 equ~valents, of th~ corresponding
~arboxylic acid, where appropriate di solved in the
corresponding solvent, with catalytic quantities, prefer-
ably 0.2-0.4 equivalents, of N,N-dLmethylaminopyrîdine,
and with preferably at least 2 equiv~lents~ in particular
3-5 equivalent~, of dicyclohexylcarb~diimid2, with the
mixture sub~equently being etirred at -10C to +50C,
preferably at rom room temperature to +40C, or
1-24 hours, preferably 6-10 hours. To complete the
r~ac~ion, a ~urther eguivalent of the corresponding
carboxylic acid, 0.2 equivalent of N,N-dimethylamino-
pyridine and 2 equival~nts of dicyclohexylcarbodiimide
are added, where appropriate, after this time and the
mixture is stirred for a further 5-24 hours, preferably
5 10 hours. After this; the precipitated dicyclohexylur~a
~ ~ 3 ~
~ 4 -
is filtered off and the product according to th~ inven-
tion is isolated. The i~olation is effected by chroma-
tography, for example, or by crystallization, preferably
- after co~centrating the filtrate - by chromato~raphy,
for e~ample on silica gel with, for ex~mple, dichloro~
methane/methanol 9/1.
The mQnoesters can be obtained ~y monitoring the re~ction
by thin layer chromatography, terminating it prematurely,
and thereafter purifying as described above, for example
by chromatography~
The isolation of optically active compounds, which may be
necessary, is effected by state-of-the-art methods.
The invention furthermore relates to the use of the
compounds of the formula 1 according to the invention as
antiviral agents for the treatment or prophylaxis of
viral diseasesO
The compounds according to the invention are particularly
active against herpes simplex virus type 1, herpes
simplex virus type 2, human cytomegalovirus, murine
cytomegalovirus, varicella zoster virus, Epstein-Barr
virus and human herpes virus 6 ~HV-6).
Additionally, the present invention relates ~o pharma-
ceuticals containing at least one of the compounds
according to the invention.
The pharmaceuticals according to the in~ention may be
used preferably enterally (orally) but also parenterally
(intravenously), rectally or locally (topically~. They
may be administered in the form of solutions, powders
(tablets and capsules including microcapsules), ointments
(creams or gels) or ~uppositorieB. Suitable auxiliairy
substances for such formulation~ are the pharmaceu~ically
cu~tomary liquid or 601id filler~ and ex~enders/
solvents, emulsifiers, lubricant~, taste corrigents,
coloran~s andfor buffering ~ubstances. Dosage~ o~ 0.1-10,
preferably 0.2-8~ mg/kg of body weight are e~pedient~y
administered. They are expediently administered in dosage
units whi~h contain at least the effective daily quanti~y
of the compounds accordin~ to *he invention, e~g. 30-300,
preferably 50-250, mg.
Efficacy against systemic herpes simplex 1 virus (HSV-l)
infec~ion: specific pathogen-free NMRI mice, 15-18 ~ in
weight, were infected in~raperitoneally with ~SV-l and
subse~uently given oral therapy in the :Eorm of the
compounds according to the invention. The treatment was
first given 3 hours after infection and was then con
tinued twice daily for 4 days. ~he success of the treat~
ment was determined on the basis of the course of the
disease and the survival rate as compared with the
untxeated infection control. ~he latter received phy~io-
logical saline solution instead of the compounds accord-
ing to the invention. The period of observation was two
weeks. ~able 1 shows the resultl3 of this investigation.
Table 1 ~n~iviral efficacy against HSV-1 in the NMRI
mouse on oral administration
Ex~mple Dosage Average ~3urvival Survivors/
(~m~1/kg) time (day~) group ~ize
1 9 x 10 ~ 14 days 5/5
9 x 30 > 14 day~ 5/5
9 x 100 > 14 days 5/5
2 9 x 30 5t5
9 x 100 ~/5
3 g x 30 5/5
9 x 100 5/5
7 ~3
Continuation of Table 1
Example Dosage ~verage survival SurviYoxs/
(~moltkg) time ~days~ group ~ize
Formula 1, 9 x 10 10.5 i O.7 3~5
R1=~2=H 9 x 30 9.5 t 1.3 1/5
(- compound 9 x 100 5/5
of the
formula 1')
Control 9 x 10 7.8 t 1.1 1/5
Determination of oral bioavailabili~y:
Rhesus monkey~ were placed in indiYidual cages 24 hour6
before the start of the exp~riment. The feed was with-
drawn 12 hours before the start of the experiment~ while
unlimited ~uantities of drinking water were avaiIable
before and during the experiment. In each case two
~nimals received the compounds ac:cording to the invention
and one animal received the compound of the formula 1',
in each case at 105 ~mol/kg, administered in a volume of
~0 5 ml of tap water by means of a ~tomach tube. This dose
was rinsed down with a further ~0 ml of water.
One anlmal received 42 ~mol/kg of the compound of the
formula 1', a~ a concentration of 10 mg/ml in phy~io-
logical sodium chloride solution, injected into a vein in
the upper arm.
Blood samples were removed from the femoral veins of all
the anLmals at variou~ tim~ interval~ up to 24 hours
after administration of the subst2nces.
The blood was kept at 4C until it had ~oagulated and was
then centrifuged. ~he serum thus obtained was stored at
-20C until the fiamples were analyzed. 200 ~l of serum
were introduced into a reaction vessel and diluted with
100 ml of water. After addition of 30 ~l of 50~ streng~h
7 '~ ~
trichloroacetic acid, th~ mixture was homogenized in a
~Vor ex Mixer and subsequently centrifuged for
3 minutes. 50 ~1 of the ~upernatant 501ution were ex
amined hy ~PLC.
Chromatographic ~eparationo column ~Nucleosil Cl~,
125 x 4.6 mm; mobile phase: phosphoric acid, 0.01 molar;
flow rate: 1.0 ml/min; detection: fluorescence, ex.:
310 nm, em.: 360 nm; retention time: 3.5 min (compound of
the formula 1, where R1 is = R2 = Hr i.e. compound of the
formula 1').
The calculation was effected by evaluating the area undPr
the data ~AUD). The metabolic avail~bility of the com-
pound of the formula 1~, ollowing oral administration of
the compound of Example 1, wa~ on average 37.80% (34.47%
and 41.16~. Following administration ~f the compound of
Example 2, a bioavailabilit~ of the compound of the
formula 1' of on average 20.11% (23.73% and 16.50%) was
achieved (the said compounds are metabolized in the body
to compounds of the formula 1~).
By contrast, the bioavailability of the compound of the
formula 1~, following oral administration, is on average
only 15%.
Determination of oral bioa~ailahility ~mou~e):
The following values for the bioavailabilitie of the
~5 compounds of the formula 1~ were obtained in mice follow-
ing oral administration of the compounds according to the
invention:
~a~7~
Compound Method of Dose AUD(o >OO~ Bioavail-
administration[mg/kg] C(~gxmin)/ml] ability ~YO]
Formul a 1 ' ; .v. 25 13 .39 100
p.o. 1~0 9.23 17.2
Example 1 i.v. 25 13.21 98.6
p.o. 100 27.32 51.1
~
Example 2 p.o. 100 36.03 67.3
.
Example 3 p.o. 100 38.40 71.7
The present invention is illustrated in more detail by
the following examples and by the con ent of ths patent
claims.
Examples
1~ Compound of the formula 1 where R1 = R2 z acetyl:
1.2 g (5 mmol) of 2 amino-7-[(1,3-dihydroxy-2-propoxy~-
methyl]purine (compound of the formllla 1 where Rl = R2 =
hydrogen; prepared as described in European Patent
Application EP 452 6~0 A~ are dissolved in S0 ml of
anhydrous dimethylformamide. 0.~3 ml ~11 mmol) of acetic
acid, 0.2 g ~1.6 mmol) of N,N-dimethylaminopyridine ~nd
3.96 g (19 mmol) of N,~'-dicyclohexylcarbodiimide are
added successi~ely and the mixture is stirxed at room
temperature for 6 hour~. After this time, a further
O.32 ml (5.5 mmol~ of acetic acid, 0.1 g (O.8 mmol) of
N,N-dim~thylaminopyridine and 1.98 g (9.5 ~mol) of
N,N~-dicyclohexylcarbodiimide ar~ added and the mixture
is stirred at room temperature ~or a further 17 hours.
The precipitated N,N'-dicyclohexylurea is filtered off,
the filtrate is concentxated in vacuo and the re~idue is
chromatographed on silica gel with dichloromethane/-
methanol 9/1 as the eluent mixture. 1.51 g (93.5% of
r~
- 9 -
theory)of2 amino-7-[(1~3-bi~-acetoxy-2-propoxy~me hyl]-
purine are obtained as ~olorle6s cry~tal~ with a melting
point of 158C.
lH-NMR (270 MHz, d6-DMSO) ~ tppm~s 8.65 (~lH), 8.43
5 (8rlH)/ 6-27 (s,2EI), 5.68 (6,,2H), 4.15--3086 ~m,5H3, 1.83
( s , 6H ) .
2. Compound of the ormula 1 where R1 = R2 = butanoyl:
1.2 g (5 mmol) of 2-amino-7-[(1,3 dihydroxy 2-propoxy)-
methyl]purine (compound of the formula 1 where R1 = R2 =
hydrogen; prepared ~s described in ~uropean Patent
Application EP 452 680 A) are dissolved in 50 ml of
anhydrous dLmethylformamide. 1.0 ml (11 mmol) of butyric
acid, 0.2 g ~1.6 mmol~ of N,N-dLmethylaminopyridine ~nd
3.96 g (19 mmol) of N,N'-dicyclohexylcarbodiLmide are
added successively and the mixture i~ ~tirred at room
temperature for 6 hours. After thi~ time, a further
0.5 ml (5.5 mmol~ of butyric acid, 0.1 g (0.8 mmol) of
N,Ndimethylaminopyridine and 1.98 g (9.5 mmol) of
N,N'-dicyclohexylcarbodiimide are added and the mixture
is stirred at room temperature for a further 17 hours.
The precipitated N,N'-dicyclohe~lurea i~ filtered off,
the filtrate is concen~rated in vacuo and the residue is
chromatographed on siliea gel with dichloromethane/
methanol 9fl as the eluent mixture. 1.84 g (97.1 ~ of
theory) of 2~amino-7-[~1,3 bis-butyryloxy-2-propoxy)-
methyl]purine are obtained as colorless crystals with a
melting point o~ 1~3C.
H-NMR (270 MHz, d6-DMSO) ~ tppm]: 8.64 (~ ), 8.43
(s,lH), 6.27 (s,2H), 5.67 (s,2~), 4.18-3.79 (m,5H), 2.05
(t,4H), 1.42 (m,4H), 0.83 (t~6H).
3. Compound of the formula 1 where R1 = R2 = propanoyls
1.2 g (5 mmol) of 2-amino~7 [(1,3-dihydroxy 2~propoxy)-
methyl]purine (compound of the formula 1 where R1 = R2 =
hydrogen; prepared as described in European Patent
Application EP 452 680 A) are dissolved in 50 ml of
anhydrous dimethylfoxmamide. O.B2 ml (11 mmol) of
7 P~ ~
-- 10 --
propionic acid, 0.2 g (1.6 mmol) of N,N dime~hylamino-
pyridine and 3.96 ~ (19 mmol) of ~,N~ dicyclohexyl-
carbodiimide are added successively and the mixture i~
stirred at room temperature for 6 hour~. After this t~me,
a further 0.41 ml ~5.5 mmol) of propionic acid, 0.1 g
(O.8 mmol) of N,N-dimethylaminopyridine and 1.98 g
(9.5 mmol) of N,N'-dicyclohexylcarbodiLmide are added and
the mixture is ~tirred at room temperature for a further
17 hours. The precipitated N,Nr-dicyclohexylurea is
filtered off, the filtrate i5 concentrated in vacuo and
the residue is chromatographed on ~ilica ~el with
dichloromethane/methanol 9/1 as the eluent mixture.
1.53 5 (87.2% of theory) of 2-amino-7-~(1,3-bis-
propionyloxy-2-propoxy)methyl]purin~ are obtained a~
colorless crystal with a melting point of 143C.
H-NMR (270 MHz, d6-DMSO) ~ ~ppm]- 8.64 (s,lH), B.42
(s,lH), 6.2~ (s,2~), 5.67 (s,2H), 4.13 (m,2H), 4.05-3.89
(m,3H), 2.10 (q,4H), 0-92 (t,6H3.
4. Compound of the formula 1 where R~ = hydrogen and R2
= acetyl:
Colorless crystals, m.p.: 153C, lH-NMR (200 MHz, d6-DMS0)
[ppm]: 8-62 (s,lH)~ ~-40 (8,1H), 6.23 (s,2H), 5.63
(m,2H), 4.85 (t,lH3, 4.15-4.00 ~m,lH), 3.95-3.83 (m,lH),
3.72-3.60 (m,lH), 3.40 (m,2H), 1.73 (s,3H).