Note: Descriptions are shown in the official language in which they were submitted.
2 ~
C~cloalkY1- and heterocYclyl-substituted imidazolyl-
pro~enoic acid derivatives
The invention relates to cycloalkyl- and heterocyclyl-
substituted imidazolylpropenoic acid derivatives, a
process for their preparation and their use in
pharmaceuticals, in particular as antihypertensives and
anti-arteriosclerosis agents.
It is known that renin, a proteolytic enzyme, cleaves off
the decapeptide angiotensin I in vivo from angio-
tensinogen, the decapeptide being in turn broken down inthe lung, the kidneys or other tissues to give the
antihypertensive octapeptide angiotensin II. The various
effects of angiotensin II, such as, for example, vaso-
constriction, Na+ retention in the kidney, release of
aldosterone in the adrenal gland and increase in tonus of
the sympathetic nervous system have a synergistic effect
in terms of an increase in bloo~ pressure.
Moreover, angiotensin II has the property to promote
growth and multiplication of cells, such as, for example,
heart muscle cells and smooth muscle cells, these cells
showing increased growth and proliferation in various
disease states (e.g. hypertension, arteriosclerosis and
cardiac inRufficiency).
A possible method of intervention in the renin-angio-
Le A 28 876 - 1 -
2 ~
tensin system (RAS) is, apart from inhibition of renin
activity, inhibition of the activity of the angiotensin-
converting enzyme (ACE) and blocking of angioten~in TI
receptors.
Imidazolyl derivati~es exhibiting a blocking effect on
the angiotensin II receptor are disclosed in Publications
EP 403,159 A2 and EP 425,211 Al.
The present invention relates to cycloalkyl- and hetero-
cyclyl-substituted imidazolylpropenoic acid derivatives
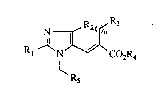
of the general formula (I)
N R2(~
Rl ~ ~ CO2R4 (I)
~ R5
in ~hich
R1 represents straight-chain or branched alkyl or
alkenyl each having up to 8 carbon atoms, which are
unsub~tituted or substituted by cycloalkyl of 3 to
6 carbon atoms, or
represents cycloalkyl of 3 to 8 carbon atoms,
R2 represents hydrogen, halogen, hydro~yl, nitro,
cyano, trifluoromethyl, trifluoromethoxy or penta-
fluoroethyl, or
Le A 28 876 - 2 -
2 ~
represents straight-chain or branched alkyl of up to
6 carbon atoms, or
represents aryl of 6 to 10 carbon atoms,
R3 represents cycloalkyl of 3 to 8 carbon atoms or
heterocyclyl of 3 to 8 carbon atoms and of up to 3
hetero atoms from the series consisting of S, O or
-N-A,
in which
A denote~ hydrogen or straight-chain or branched
alkyl of up to 6 carbon atoms,
n represents the number 0, 1, 2, 3 or 4,
R4 represents hydrogen, straight-chain or branched
alXyl of up to 8 carbon atoms, or represents phenyl,
R5 represents a radical of the formula
R7 ~ R7 ~ R7 ~ R~
R6 R6 R6
in which
R6 denotes hydrogen, halogen, cyano, nitro,
Le A 28 876 - 3 -
2~g~
trifluoromethyl, hydroxyl, trifluoromethoxy,
straight-chain or branched alkyl or alkoxy each
having up to 6 carbon atoms,
R7 denotes carboxyl, straight-chain or branched
alkoxycarbonyl of up to 6 carbon atoms or
tetrazolyl which may be Cl-C4-al~ylated,
and salts thereof.
The compounds according to the in~ention can also be
present in the form of their salts. In general, salts
with organic or inorganic bases or acids may be mentioned
here.
In thP context of the present invention, physiologically
safe salts are preferred. Physiologically safe salts of
cycloalkyl- and heterocycloalkyl-substituted imidazolyl-
propenoic acid derivatives can be salts of the substances
according to the invention with mineral acids, carboxylic
acids or sulphonic acids. Particular pr ferencP is given,
for example, to salts with hydrochloric acid, hydrobromic
acid, ~ulphuric acid, phosphoric acid, methanesulphonic
acid, ethanesulphonic acid, toluenesulphonic acid,
benzenesulphonic acid, naphthalenedisulphonic acid,
acetic acid, propionic acidl lactic acid, tartaric acid,
citric acid, fumaric acid, maleic acid or benzoic acid.
Physiologically safe salts can also be metal salts or
ammonium salts of the compound~ according to the
Le A 28 876 - 4 -
3 ~ ~
i~vention carrying a free carboxyl group. Particular pre-
ference is given, for example, to sodium salts, pota6si~m
salts, magnesium salts or calcium salt~, and to ammonium
salts derived from ammonia or organic amines, such as,
for example, ethylamine, di- or triethylamine, di- or
triethanolamine,dicyclohexylamine,dimethylaminoethanol,
arginine, lysine or ethylenediamine.
The compounds according to the inven~ion can be present
in ~tereoisomeric form~ whose relationship is either that
of mirror to mirror image (enantiomers) or is not that of
image to mirror image (diastereomers). The invention
relates to both enantiomers and diastereomers or to their
respective mixtures. The racemic forms, ~ust like the
diastereomers, can be resolved in a known manner into the
stereoisomerically uniform components ~cf. E.L. Eliel,
Stereochemistry of Carbon Compounds, McGraw Hill, 1962]
Heterocycloalkyl in general represents a 5- to 6-membered
ring having up to 2 hetero atoms from the series con-
sisting of ~, S, O or an NH group or N-Cl-C6-alkyl group,
such as, for example, dithiolanyl, pyranyl or piperidyl.
Dithiolanyl or pyranyl are preferred.
Preference is given to compounds of the general formula
(I)
in which
R1 represents straight-chain or branched alkyl or
Le A 28 876 - 5 -
2~92g~g
- alkenyl each having up to 6 carbon atoms, which are
unsubstituted or substituted by cyclopropyl, cyclo-
butyl, cyclopentyl or cyclohexyl, or
represents cyclopropyl, cyclopentyl or cyclohexyl,
R2 represents hydrogen, fluorine, chlorine, bromine,
iodine, trifluoromethyl, trifluoromethoxy, penta-
fluoroethyl or straight-chain or branched alkyl of
up to 6 carbon atoms,
R3 represents cyclopropyl, cyclopentyl, cyclohexyl,
cycloheptyl, dithiolanyl or pyranyl,
n repre~ents the number O, 1, 2 or 3,
R4 represents hydrogen or straight-chain or branched
alkyl of up to 6 carbon atoms,
R5 represents a radical of the formula
lS ~ R7 ~ R7 ~ R7 or ~ R~
in which
R6 denotes hydrogen, fluorine, chlorine, bromine,
trifluoromethyl, trifluoromethoxy or straight-
chain or branched alkyl of up to 4 carbon
atoms,
Le A 28 876 - 6 -
~21~
R7 denotes carboxyl, straight-chain or branched
alkoxycarbonyl of up to 3 carbon atoms, or
denotes tetrazolyl,
and salts thereof.
Particular preference is given to compounds of the
general formula (I)
in which
Rl represents straight-chain or branched alkyl or
alkenyl each having up to 4 carbon atoms, or
represents cyclopropyl,
R2 represents hydrogen, fluorine, chlorine, bromine,
iodine, trifluoromethyl, pentafluoroethyl, ~ri-
fluoromethoxy or straight-chain or branched alkyl
of up to 4 carbon atoms,
R3 represents cyclopropyl, cyclopentyl or cyclohexyl,
n represents the number O, 1 or 2,
R4 represents hydrogen or straight-chain or branched
alkyl of up to 4 carbon atoms,
R5 represents a radical of the formula
Le A 28 876 - 7 -
~92~
R~ ~ R7 ~ R7 or
6 ' R6 R6 R6
in which
R6 denotes hydrogen, fluorine, chlorine or
bromine,
R7 denotes carboxyl, straight-chain or branched
alkoxycarbonyl of up to 3 carbon atoms, or
denotes tetrazolyl,
and salts thereof.
Furthermore, a process for the preparation of the com-
pounds according to the invention of the general formula(I) has been found, which process is characterised in
that
aldehydes of the general formula (II)
N R2
~II)
~ Rs H
in which
La A 28 876 - 8 -
2~92~
R1, RZ and R5 have the abovementioned meaning,
are first converted by reaction with compounds of the
general formula ~III)
R3-(CH2)n-c02-T (III)
in which
R3 and n have the abovementioned meaning~
and
T represents straight-chain or branched alkyl of up to
8 carbon atoms, or denotes phenyl,
in inert solvents in the presence of a base into the
compounds of the general formula (IV)
N ~ R2(~n 3 (IV)
R~ N ~ CO2T
Rs
in which
R1, R2, R3, n and ~ have the abovementioned meaning,
Le A 28 876 - 9 -
2~3~23~
the free hydroxyl function is then blocked by introducing
a protective group, and, in a last step, an elimination
reaction in inert ~olvents is carried out in the presence
of a base,
and, in the case of the acids (R4/R7 = H), the esters are
hydrolysed,
and, if desired, the substituPnt~ R1 and R2 are varied by
cu3tomary methods, for example by hydrogenation or
alkylation,
and, in the case where R7 represents alkylated tetrazolyl,
the -NH function i alkylated.
The process according to the invention can be illustrated
by the following reaction scheme by way of example:
Le A 28 876 - 10 -
~2~g
N ~CI
H3C-~H2C)3 ~ N ~ ~ LDA
3~ ~CH2-CO2CH, THF
CO2CH3
H3C (H2c)3 l~CO~CH3 CH2C12 ~3C-(H2C)3 ~;~CC~21CH3
OH DMAP ~ ¦ OAc
~CO2CH3 \~CO2cH3
Cl ~CDH1~
DE3U H3c-(H2c~3 J~C2CH3
Toluol
~CO;!CH3
A hydroxyl-prot-ecting group in the context of ~he above-
mentioned definition is in general a protective group
from ~he series consisting of benzylo~ycarbonyl, methane-
sulphonyl, toluenesulphonyl, 2-nitrobenzyl, 4-nitro-
bonzyl, 2-nitrobenzylo~ycarbonyl, 4-nitrobenzyloxy-
carbonyl, tert.-butoxycarbonyl, allyloxycarbonyl,
~e A 28 876 - 11 -
2 ~
4-methoxycarbonyl, acetyl, trichloroacetyl, 2,2,2-tri-
chloroethoxycarbonyl, 2,4-dimethoxy~enzyloxycarbonyl,
2-(methylthiomethoxy)ethoxycarbonyl, benzoyl, 4-methyl-
benzoyl, 4-nitrobenzoyl, 4-fluorobenzoyl, 4-chlorobenzoyl
or 4-methoxybenzoyl. Preference is given to acetyl,
methanesulphonyl and toluenesulphonyl.
Suitable solvents for the process are conventional
organic solvents which remain unchanged under the reac-
tion conditions. These preferably include ethers, such as
diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl
ether, or hydrocarbons, such as benzene, toluene, xylene,
hexane, cyclohexane or petroleum fractions, or halogena-
ted hydrocarbons, such as dichloromethane, trichloro-
methane, carbon tetrachloride, dichloroethylene, tri-
chloroethylene or chlorobenzene, or ethyl acetate,triethylamine, pyridine, dimethyl sulphoxide, dimethyl-
formamide, hexamethylphosphoric triamide, acetonitrile,
acetone or nitromethane. It is also possible to use
mixtures of the solvents mentioned. For the various
steps, tetrahydrofuran, methylene chloride and toluene
are preferred.
The bases which can be used for the process according to
the inven~ion are in general inorganic or organic bases.
These preferably include alkali metal hydroxides, such
as, for example, sodium hydroxide or potassium hydroxide,
alkaline earth metal hydroxides, such as, for example,
barium hydroxide, alkali metal carbonates, such as sodium
carbonate or potassium carbonate, alkaline earth metal
Le A 28 876 - 12 -
carbonates, such as calcium carbonate, or alXali metal
alkoxides or alkaline earth metal alkoxides, such as
sodium methoxide or potassium methoxide, sodium ethoxide
or potassium ethoxide or potassium tert.-butoxide, or
S organic amines (trialkyl(Cl-C6)amines), such as triethyl-
amine, or heterocycles, such as 1,4-diazabicyclo[2.2.2]-
octane (DABCO), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU),
pyridine, diaminopyridine, methylpiperidine or morpho-
line. The bases used can also be alkali metals, such as
sodium, or hydrides thereof, such a~ sodium hydride.
Preference i3 given to lithium diisopropylamide and DBU.
The base is generally used in an amount of 0.05 mol to
10 mol, preferably 1 mol to 2 mol, relative to 1 mol of
the compound of ~he formula (III).
The process according to the invention is in general
carried out in a temperature range from -lOODC to +100C,
preferably at -78~C.
The process according to the invention is in general
carried out at atmospheric pressure. However, it is also
possible to carry out the process at superatmospheric
pre~sure or a~ reduced pre~sure (for example in a range
from 0.5 to 5 bar).
~he protective group is in general introduced in one of
the solvents listed above and a base, preferably in
methylene chloride together with dLmethylaminopyridine.
Le A 28 876 - 13 -
~0~2~
Blocking in general taXes place in a temperature range
from 0C to +60C, preferably at room temperature at
atmospheric pressure.
Elimination is in general carried out in one of the
solvents listed above, preferably in toluene, and in the
presence of one of the bases listed, preferably DBU.
Elimination in general takes place in a temperature range
from +30C to ~130C, preferably at +50C to +100C, and
atmo pheric pressure.
Suitable bases are the inorganic bases customary for
hydrolysis. These preferably include alkali metal
hydroxides or alkaline earth metal hydroxides, such as,
for example, sodium hydroxide, potassium hydroxide or
barium hydroxide, or alkali metal carbonates, such as
sodium carbonate or potassium carbonate or sodium
bicarbonate or alkali metal alkoxides, such as sodium
methoxide, sodium ethoxide, potassium methoxide,
potassium ethoxide or potassium tert.-butoxide.
Particularly preferably, sodium hydroxide or potassium
hydroxide are used.
Suitable solvents for hydrolysis are water or the organic
solvents customa~y ~or hydrolysis. ThesP preferably
include alcohols, such as methanol, ethanol, prop nol,
isopropanol or butanol, or ethers, such as te~rahydro-
furan or dioxane, or dimethylformamide, or dimethyl-
sulphoxide. Particularly preferably, alcohols, such as
-
Le A 2~ 876 - 14 -
2V~ 8~
methanol, ethanol, propanol or isopropanol, ar~ used. It
is also possible to use mixtures of the ~olvents men-
tioned.
Hydrolysis can also take place with acids, such as, for
example, trifluoroacetic acid, acetic acid, hydrochloric
acid, hydrobromic acid, methanesulphonic acid, sulphuric
acid or perchloric acid, preferably with trifluoroacetic
acid.
Hydrolysis is in general carried out in a temperature
range from 0C to +100C, preferably from +20C to +80C.
In general, hydrolysis is carried out at atmospheric
pressure. However, it is also possible to work at reduced
pressure or at superatmospheric pressure (for example
from 0.5 to 5 bar).
When hydrolysis is carried out, the base is in general
used in an amount of 1 to 3 mol, preferably 1 to 1.5 mol,
relative to 1 mol of the ester. Particularly preferably,
molar amounts of the reactants are used.
When the reaction is carried out, the carboxylates of the
compounds according to the in~ntion are formed in the
first step as intermediates, which can be isolated. The
acids according to the invention are obtained by ~reat-
ment of the carboxylates with the customary inorganic
acids. These preferably include mineral acids, such as,
2S for example, hydrochloric acid, hydrobromic acid,
Le A 28 876 - 15 -
2 ~
sulphuric acid or phosphoric acid. When the carboxylic
acids are prepared, i~ has proven advantageou~ to acidify
the basic reaction mixture of the hydrolysis in a second
step without isolating the carboxylates. The acids can
then be isalated in the usual manner. In the case of
basic heterocycles, the treatment of the solutions of the
carboxylates with the acids listed above also makes it
possible to obtain the salts of the heterocycles with the
inorganic acids.
Alkylation is in general carried out using alkylating
agents, such as, for example, (Cl-C6)alkyl halides,
~ulphonic esters or substituted or unsubstituted (C1-C6)-
dialkyl or (Cl-C6)-dialylsulphonates, preferably methyl iodide or dimethyl sulphate.
Alkylation in general takes place in one of the ~olvents
listed above, preferably in dimethylformamide, in a
temperature range from 0C to +70C, preferably from 0C
to +30C, and a~mospheric pressure.
Hydrogenation in general proceeds by customary methods in
one o~ the sol~ents listed above, preferably in methanol,
using hydrogen and Pd/C in the presence of sodium
acetate.
In general, hydrogenation is carried out at eleva~ed
pressure ~about 2 to 3 bar). ~owever, it is also possible
to work at reduced pressure or at atmospheric pressure.
Le A 28 876 - 16 -
Hydrogenation is in general carried out in a temperature
range from 0C to +40C, preferably at room temperature.
The compound~ of the general formula (I~) are in part
known ~R5 = phenyl, see, for example, EP 425,211) or can
be prepared by reac~ing
compounds of the general formula (V)
J~o (V)
Rl N
H
in which
R1 and R2 have the abovementioned meaning,
with compounds of the general formula (VI)
M (VI)
L Rs
in which
R5 has the abovementioned meaning
and
Le A 28 876 - 17 -
~ ~3 ~
M represents halogen, preferably bromine,
in one of the solvents listed above, preferably dimethyl-
formamide, in the presence of one of the bases listed
above, preferably potassium carbonate or potassium tert.-
butoxide.
The compounds of the general formulae (V) and (VI) are
known.
The compounds of the general formula (III) are known or
can be pxepared by customary methods by esterification of
the purchasable acids [see, for example, MSD Book 2,
1593 D].
The compounds of the general formula (IV) are new as
specific representatives of their class and can be
prepared, for example, by the abovementioned process.
The preparation processes given above are solely given
for illustration. The preparation of the compounds
according to the invention of the general formula (I) is
not limited to these processes, an~ modification of these
processe~ being analogously applicable ~o the prepara-
tion.
The cycloalkyl- and heterocyclyl-sub~tituted imidazolyl-
propenoic acid derivatives exhibit an ~nforeseeable,
useful pharmacological action spectrum.
Le A 28 876 - 18 -
2 ~
The compounds according to the invention show a specific
A II antagonistic action, since they competitively
inhibit the binding of angiotensin II to the receptors.
They suppress the vasoconstricting and aldosterone
secretion-stimulating effects of angiotensin II.
Noreover, they inhibit the proliferation of smooth muscle
cells .
Accordingly, they can be used in pharmaceu icals for the
treatment of arterial hypertension and arteriosclerosis.
Moreover, they can be used for the treatment of coronary
heart diseases, cardiac insufficiency, disorders in
cerebral performance, ischemic brain diseases, peripheral
circulatory disorders, functional disorders of kidney and
adrenal gland, diseases of the respiratory tract of
bronchospactic and vascular origin, sodium retention and
oedemas.
Testing for inhibition of contraction induced by aaonists
Ra~bits of both sexes are anaesthetized by a blow to the
back of the neck and exsanguinated or, depending on the
case, narcotised with nembutal (about 60-80 mg/kg i.v.)
and sacrificed by opening the thorax. The thorax aorta
i~ removed, freed of any adhering connective tissue,
divided into annular segments of l.5 mm in width and
transferred individually under an initial load of about
3.5 g to organ baths of lO ml containing a Rrebs-
Henseleit nutrient solution which is temperature
1e A 28 876 - l9 -
2 ~ 8 $
controlled at 37C, gassed with Carbogen and has *he
following composition: 119 mmol~l of NaCl; 2.5 mmol/l of
CaCl2 x 2H20; 1.2 mmol/l of RHzP04; 10 mmol/l of glucose;
4.8 mmol/l of KCl; 1.4 mmol/l of MgS04 x 7H20 and
25 mmol/l of NaHC03.
The contractions are measured isometrically by Statham
UC2 cells via bridging amplifiers (ifd Mulheim or DSM
Aalen) and digitalized by means of A/D converters (system
570, Keithley Munich~ and evaluated. Agonist dosage-
response curves ~DRC) are recorded hourly. With each DRC,3 or 4 individual concentrations are applied to the baths
at 4 min intervals. Completion of the DRC and subsequent
washing cycles (16 times about 5 sec/min each with the
abovementioned nutrient solution) are followed by resting
or incubation phase of 28 minutes during which the
contractions usually again reach the initial value.
The maximum of the, in the usual case, 3rd DRC is used as
reference value for evaluating the test substance to be
tested in further runs, the test substance being applied
to the baths at the beginning of the incubation time in
a dose increasing in each of the following DRCs. Each
aorta ring is stimulated all day with always the same
agonists.
Le A 28 ~76 - 20 -
2318g-74~0
Aqonists and ~heir standard concentr~tions Application
volume per sinqle dose ~= 100 ~
~Cl 22.7;32.7;42.7;52.7 mmol/l
1-~oradrenalin 3x10-3;3x10-8;3x10-7;3x10-5 g/ml
Serotonin 10~;10';10 6;10 5 g/ml
B-HT 920 10-7 1o~6 10-5 g/ml
Methoxamine 10-7;106;105 g/ml
Angiotensin II 3x109;10~;3x108;10-7 g/ml
For calculating the IC50 ~concentration at which the
substance to be tested causes 50~ inhibition), the effect
observed in each case at the 3rd (= submaximum) agonist
concentration is-used as the basis.
Example No. IC50[nM]
7 8
8 18
9 llO0
.
The compounds according to the invention cause dose-
dependent inhibition of the angiotensin II-induced
contraction of the isolated rabbit aorta. The contraction
induced by potassium depolarisation or other agonists was
not inhibited or only slightly inhi~ited at high con-
centrations.
Measurements of_the blood pre~Cure of rats infused with
anaiotensin II
Mals Wistar rats (Moellegaard, CopenhagQn, Denmark) of
body weight 300-350 g are anaesthetized with thiopental
(100 mg/kg i.p.~. After ~racheotomy, a ca~heter is
inserted into the femoral artery for measuring the blood
pre~sure and a catheter i8 inserted into ~he femoral
veins for angiotensin II infusion and a catheter is
- 21 -
~ ~3 ~
231g9-74
lnserted for administration of the substance. After
admini~tration of the ganglion blocker pentolinium
(5 mg/kg i.v.), angiotensin II infusion (0.3 ~g/kgJmin)
is started. As soon a~ the blood pressure values have
reached a stable plateau, the test substances are
administered either intravenously or orally as a suspen-
sion or solution in 0.5% tylose. The changes in blood
pressure under the effect of this substance are given in
~xample No. ¦ Dose ¦ mmHG
7 ¦ 0.01 mg/kg i.v. -27
7 0.01 mg/k~_p_o. -19
Determination of the antihypertensive efficiency in
conscious hvpertensive rats
The oral antihypertensive efficiency of the compounds
according to the invention was te3ted in alert rats
having surgically induced unilateral kidney arterial
stenosis. To this endl the right kidney artery was
narrowed using a silver clip of 0.18 mm clearance. In
this form of hypertension, the plasma renin activity is
increased during the first six weeks after the inter-
vention. The arterial blood pressure of these animals was
measured non-invasively at defined intervals after
administration of the substance using a ~tail cuff~'. The
substanc~s were applied intragastrally (I~orally~
suspended in a tylose suspension, via a gastric tube in
various do~es. The compounds according to the invention
lower the arterial blood pre sure of hypertensive rats in
clinically significant dosage.
Moreover, the compounds according to the invention cause
-` ~V~2~
23lss-74sa
concentration-dependent specific binding of radioactive
angiotensin II.
Exam le No. Dose mmHG
P _ ,
7 10 mg/kg P ~ ~ -2
9 10 mq/kg p . o . . - :
Interaction of the compound~ accordina to the invention
with the anaiotensin II receptor in membrane fractions of
5 the (bovine) adrenal cortex
Bovine adrenal cortices (AC), freshly r~moved and care-
fully freed of marrow and capsule, are comminuted in
~ucrose solution (0.32 ~) by means of an Ultra-Turrax
(Janke ~ Kunkel, Staufen i.B.) to give a coarse membrane
homogenate, which was then partly purified in two centri-
fugation ~teps to give membrane fractions. The tests
regarding receptor binding are carried out with partially
purified membrane fractions of bovine AC using
radioactive angiotensin II in an a~say volume of 0.25 ml,
which specifically contains the partially purified
membrane~ (50-80 ~g), 3H-angiotensin II (3-5 nM), test
buffer ~olution (50 mM tris, pH 7.2, 5 mM ~gCl2, 0.25%
BSA) and the substan~es ~o b~ tested. After an incubation
time of 60 min at room temperature, the unbound radio-
activity of the samples is separated off by means ofmoistened glass fibre filters (~hatman GF/C), and the
bound radioactivi~y is measured by spec~rophotometry in
a scintillation cocktail after washing the protein with
ice-cold buffer solution (50 m~ trisJHCl, pH 7.4, 5% P~G
6000j. Analy~is of the crude data was carried out using
computer programB to give ~i and IC5~ values ~Ri: IC53
v21ues corrected for radioactivity u~ed; IC5~ values:
concentration at which he substance to be tested causes
2 ~
2~1g9-7480
50~ inhibition of the total binding of radioligands3.
.
.
Example No. Ki [nM]
. 7 11
__ _ 51
: 9 1100 :
Test of the inhibition of the proliferation of smooth
mu~cle cells by_the compounds according to the invention
In order to determine the antiproliferative effect of the
compound~, smooth muscle cell3 are us2d which are
obtained from rat aortas by the media explant method tR.
Ro~s, J. Cell. Biol. 5Q, 172, 1971~. The cell~ are
inoculated in suitable culture dishes, usually 96-hole
plates, and cultured in medium 199 containing 7.5~ FCS
and 7.5~ NCS, 2 mN L~glutamine and 15 mM HEPES, pH 7.4 in
5~ CO2 at 37C for 2-3 days. The cells are then
synshronised for 2-3 days by serum withdrawal and then
growth-stimulated u~ing 3erum or other factors. Test
compound~ axe added sLmultaneously. After 16-20 hours,
1 ~Ci3~-thymidine is added, and, after another 4 hoursr
the extent of incorporation of this sub~tance in the TCA-
precipitable DNA of the cell~ i3 determined.
To deterrnine the halfmaximal inhibition of thymidine incorporation (ICso) caused
by addition of 10 % FCS, the compounds were sequentially diluted in the range of2 0 lO~M to 1~9M.
-- 24 --
2~2~
23189-74
The new active compound can be converted in a known
manner into the customary formulations, such as tablets,
sugar-coated tablets, pills, granules, aerosols, syrups,
emulsions, suspensions and solutions using inert, non-
toxic, pharmaceutical~y suitable carriers ox solvent~. In
these, the therapeutically active compound should in each
case be present in a concentration of about 0.5 to 90% by
weight of the total mixture, i.e. in amounts sufficient
for achieving the dosage range given.
The invention also extends to a commercial package
containing a compound of the invention, together with instruc-
tions for its use for treatment of arterial hypertension and
arteriosclerosis.
- 24a -
Formulations are prepared, for example, by extending the
active compounds with solid and/or carriers, if appro-
priate with the use of emulsifiers and/or dispersants, it
being possible, for example in the case where water is
used as a diluent, to use, if desired, organic solvents
as solubilisers.
Application takes place in the usual m~nner, preferably
orally cr parenterally, in particular perlingually or
intravenously.
In the case of parenteral application, it is possible to
use solutions of the active compounds obtained by using
suitable liquid caxriers.
In general, it has proven advantageous to administer
amounts of about 0.001 to 1 mg/kg, preferably about 0.01
to 0.5 mg/kg of body weight, in the case of intravenous
application/ in order to obtain efficient results, while
the dosage in oral application is about 0.01 to 20 mg/kg,
preferably 0.1 to 10 mg/kg, of body weight.
Never~heless, it may be necessary in some cases to
deviate from the amounts mentioned a~ a function of the
body weight or the type of application me~hod, the
individual behaviour towards the medicament, its type of
formulation and the time or interval at which administra-
tion takes place. Thus, in some cases, it may be suffi-
cient to do with less than the abovementioned minimumamount, while in other cases the upper limit mentioned
Le A 28 876 - 25 -
2 ~
must be exceeded. In the case of application of larger
amounts, it may be advisable to spread it over the day in
several individual doses.
Startinq compounds
Example I
Methyl 4-bromomethylbenzoate
Br
~ co2CH3
45 g (0.3 mol) of methyl p-toluate and 53.4 g (0.3 mol)
of N-bromosuccinimide are heated to boiling for 1 hour
together with 1.97 g of azobisi~obutyronitrile in 375 ml
of carbon tetrachloride, the mixture is then cooled to
0C, and the succinimide is filtered off. The residue is
chromatographed on silica gel 60 using ethyl acetate/
petroleum ether (1:10).
Yield: 55.7 g (81% of theory~
RF = 0.58 (ethyl acetate/petroleum ether = 1:10)
Le A 28 876 - 26 -
Example II
Methyl 4-bromomethyl-3-chlorobenzoate
Br
C~ CO2CH3
Analogously to the procedure of Example I, the title
compound is prepared from 9.0 g (48.8 mmol) of methyl
3-chloro-p-toluate.
Yield: 7.3 g (57~ of theory)
RF = 0.62 (ethyl acetate/petroleum ether = 1:5)
Example III
Methyl 5-bromomethylthiophene-2-carboxylate
Br
~ co2CH3
Analogously to the procedure of Example I, the title
compound is prepared from 20 g (0.13 mol) of methyl
5-methylthiophene-2-carboxylate.
Yield: 22 g (73~ of theory)
Le A 28 876 - 2? -
2 ~ 9 ~ ç~
RF = ~.43 (dichloromethane/methanol = 10:1)
Example IV
Methyl 4-[(2-n-butyl-4-chloro-5-formylimidazol-1-yl)-
methyl]benzoate
N ~ Cl
N ~
~`
CO2CH3
20 g (0.107 mol) of 2-n-butyl-4-chloro-5-formylimidazol
(prepared according to EP 324,377) ar~ stirred under
inert gas in DME with 13 g (0.107 mol) of potassium tert.-
butoxide at 25C for 30 minutes, 36 g (0.157 mol) of the
compound from Example I in DMF are then added dropwise,
and the mixture is stirred at 25C for 20 hours. After
concentrating, the crude product is chromatographed on
silica gel 60 using ethyl acetate/petroleum ether (1s23.
Yield: 25.5 g (69% of theory)
RF = 0-7 (ethyl acetate/petroleum ether = 1:2)
Le A 28 876 - 28 -
2~2~8~
Example V
Methyl 4-[(2-n-butyl-4-chloro-5-formylimidazol-1-yl)-
methyl]-3-chlorobenzo~.te
N ~ Cl
N ~
Il I
Cl~--CO2CH3
Analogously to the procedure of Example IV, the title
compound is prepared from 3.16 g (12 mmol) of the com-
pound from Example II.
Yield: 2.3 g (78% of theory)
RF = 0.38 (ethyl acetate~petroleum ether = 1:5)
Le A 28 876 - 29 -
2~2~
Exam~le VI
Methyl 5-[t2-n-butyl-4-chloro-S-formylimidazol-1-yl)-
methyl]thiophene-2-carboxylate
~0
~S
~ CO2CH3
14.4 g (77 mmol) of 2-n-butyl-4-chloro-5-formylimidazol,
20 g (85 mmol) of the compound from Example III and
21.3 g (154 mmol) of potassium carbonate are stirred in
300 ml of DM~ at 25C for 20 hours. After concentrating,
the residue is taken up in 300 ml of dichloromethane and
250 ml of water, the organic layer is separated off,
dried over sodium sulphate and concentrated. The product
thus obtained i~ chromatographed on silica gel 60 using
ethyl acetate/petroleum ether (1:3).
Yield: 14.9 g (57% of theory)
RF = O . 54 (ethyl acetate/petroleum ether - 1:3)
Le A 28 876 - 30 -
209~s~
Example VII
Methyl 4-[(2-n-butyl-5~formylimidazol-1-yl)methyl]-
benzoate
N ~
N ~
Il I
\~--CO2CH3
20 g (63 mmol) of the compound from Example IV in 300 ml
of methanol are hydrogenated at 25C in the presence of
2 g of 5% palladium on activated carbon and 8.6 g
(63 mmol) of sodium acetate trihydrate at a hydrogen
pressure of about 3 bar for 3 hours. The solution is then filtered off from the catalyst
and concentrated, and the residue is chromatographed on silica gel using ethyl
acetate/petroleum ether (3:1).
Yield: 13.4 g (75% of theory)
RF = O.48 (ethyl acetate/petroleum ether = 3:1
Le A 28 876 - 31 -
2 ~3 ~
Example VIII
Methyl 4-~t2-n-butyl-5-formylimidazol-1-yl~methyl]-
3-chl~robenzoate
N ~
N ~ 0
Cl ~ C02CH3
Analogou~ly to the procedure of Example VII, the title
compound is prepared from 3.1 g (8.4 mmol) of the com-
pound from Example II.
Yield: 2 g ~71% of theory)
RF = 0-54 (ethyl acetate/petroleum ether = 5:1)
Le A 28 876 - 32 -
~Q~2~
Example IX7
Ethyl 5-[(2-n-butyl-4-chloro-5-formylimidazol-1-yl)-
methyl]furan-2-carboxylate
N ---r'Cl
H3C ~ N ~ O
~0
~ co2C2H5
Analogously to the procedure of Example VI, the title
S compound was prepared from 18.7 g (0.1 mol) of 2-n-butyl-
4-chloro-5-formylimidazol and 24.1 g (0.103 mol) of ethyl
5-bromomethylfuran-2-carboxylate.
Yield: 22.8 g (65% of theory)
RF = 0.38 (ethyl acetate/petroleum ether = 1:2
L~ A 28 876 - 33
Exa~ple X:
Ethyl 5-t(2-n-butyl-5-formylimidazol-1-yl)methyl~furan-
2-carboxylate
N ~
H3C--~N ~fSSO
~0
~ co2C2H5
Analogously to the procedure of Example VII, the title
compound was prepared from 14.51 g (42 mmol) of the
compound from Example IX.
Yield: 7.31 g (57% of theo~y)
RF = O.39 (ethyl acetate/petroleum ether = 1:1)
Le A 28 876 - 34 -
2f~2~
Example XI:
Ethyl 5-[(2-n-butyl-5-formylimidazol-1-yl)methyl]-
thiophene-2-carbo~ylate
N
H3C ~ ~
~ CO2CH3
Analogously to the procedure of Example VII, the title
compound was prepared from 6.5 g (19.1 mmol) of the
compound from Example VI.
Yield: 4 g (68% of theory)
RF = O . 21 (ethyl acetate/petroleum ether = 1:1)
Le A 28 876 - 35 -
Example XII:
Methyl 3-~2-n-butyl-1-{(2-ethoxycarbonylfuran-5-yl)-
methyl}-lH-Lmidazol-5-yl]-2-cyclopentylmethyl-3-hydroxy-
propionate
A
N ~ ~ ~
H3C ~ N ~ CO2CH3
~0
~ ~o2cH2cH3
5.16 ml (8.25 mmol) of a 1.6 N solu~ion of n-bu~yllithium
in n-hexane are injected at -78C under inert gas into a
solution of 0.89 g (8.75 mmol) of N,N-diisopropylamine in
10 ml of THF. The reaction solution is then warmed to 0C
for a short period, again cooled to -78C, and 1.17 g
(7.5 mmol) of methyl 3-cyclopentylpropionate in 5 ml of
THF are added. The mixture is stirred at 78C for 30
minutes, 1.52 g (5 ~mol) of the compound from Example X
in 15 ml of THF are added, and stirring at 78DC is
continued for 4S minutes. The mixture is then slowly
warmed to 25C, 20 ml of sat. ammonium chloride solution
are added, and the mixture is ex~racted three times with
50 ml each of ethyl acetate. The organic layer is dried
over sodium sulphate, concen~rated, and the residue is
purified on silica gel using ethyl acetate/petroleum
ether (2~1).
Le A 28 876 - 36 -
2 ~
Yield: 1.1 g (48% of theory)
RF = 0.09 (ethyl acetate/petroleum ether = 2:1, mixture of
diastereomers)
Example XIII:
Methyl 3-[2-n-butyl-l-{(4-methoxycarbonylphenyl)methyl}-
lH-imidazol-5-yl]-2-cyclopentylmethyl-3-hydroxypropionate
H3C ~ ~ CO~CH3
¦ OH
~ C2CH3
Analogously to the proceduxe of Example XII, the title
compound was prepared from 885 mg (8.75 mmol) of the
compound from Example VII and further reacted without
purification.
RF = 0.17 (sthyl acetate/petroleum ether = 4:1, mixture of
diastereomers)
Le A 28 876 ~ 37 -
2~92~8~
xample XIV:
Methyl 3-[2-n-butyl-1-{(2-methoxycarbonylthiophen-5-yl)-
methyl}-lH-imidazol-5-yl]-2-cyclopentylmethyl-3-hydroxy-
propionate
3 ~Y~CO2VCH3
~S
~ CO2CH3
Analogously to the procedure of Example XII, the title
compound was prepared from 1.53 g (5 mmol) of the com-
pound from Example XI.
Yield: 1.25 g (54% of theory)
Rp = 0.12 (ethyl acetate/petroleum ether = 2:1, mixture of
diastereomers)
Le A 28 876 - 38 -
Example XV:
3-Acetoxy-3-[2-n-butyl-1-{(2-ethoxycarbonylfuran-5-yl)-
methyl}-lH-imida~ol-5-yl]-2-cyclopentylmethylpropionate
H3C ~ ~ ~ CO2CH3
OA~
~0
~ CO2CH2CH3
2.45 g (5.36 mmol) of the compound from Example XII are
dissolved in 50 ml of dichloromethane, 264 mg ~2.16 mmol)
of N,N'-dimethylaminopyridine (DMAP) and 0.76 ml
(8.04 mmol) of acetic anhydride are added, and the
mixture i5 stirred at 25C for 16 hours. It is diluted
with ether, washed with water (1 x 40 ml) and sat. sodium
bicarbonate solution, the organic layer is dried over
sodium sulphate and concentrated. The crude product thus
obtained is chromatographed on silica gel using ethyl
acetate/petroleum ether (3.2).
Yield: 1.93 g (72% of theory)
RF = 0.65 (ethyl acetate/petroleum ether = 3:2)
Le A 28 876 - 39 -
2~92~
Example XVI:
Methyl 3-acetoxy-3-[2-n-butyl-1-{(4-methoxycarbonyl-
phenyl)methyl}-l~I~imidazol-5-yl] 2-cyclopentylmethyl-
propionate
H3C ~ ~ CO2CH3
I OAc
~ CO2CH3
Analoyously to the procedure of Example XV, the title
compound was prepared from the crude product from Example
XIII.
Yield: 1.7 g (66% of theory)
RF = 0.63 (ethyl acetate/petroleum ether = 4:1, mixture of
diastereomers)
~e A 28 876 - 40 -
~92~
Example XVII:
Methyl 3-acetoxy-3-~2-n-b-1tyl-1-{(2-methoxycarbonyl-
thiophen-5-yl)methyl}-lH-imidazol-5-yl]-2-cyclopentyl-
methylpropionate
N ~
H3C ~ ~ CO2CH3
OAc
~ COzCH3
Analogou~ly to the procedure of Example XV, the title
compound was prepared from 1.2 g (2.6 mmol) of the
compound from Example XIV.
Yield: 1.23 g (94% of theory)
RF = 0.67 (ethyl acetate/petroleum ether = 2:1, mixture of
diastereomers)
Le A 28 876 - 41 -
2~2~
Preparation examples
Example 1
Methyl 3-[2-n-butyl-1-{(2-ethoxycarbonylfuran-5-yl)-
methyl}-lH-imidazol-5-yl]-2-cyclopentylmethyl-
5 2-propenoate
H3C ~ ~ CO2CH3
~0
~ CO2CH2CH3
1.9 g (3.78 mmol) of the compound from Example XV are
dissolved in 60 ml of toluene, 1.13 ml (7.56 mmol) of
1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) are added, and
the mixture is stirred at 80C for 6 hours. 1.13 ml
(7.56 mmol) of DBU are then added again, and the fl~sk
contents are stirred at 80C for 16 hours. After cooling,
the mixture is washed with sat. sodium chloride solution
(1 x 30 ml), the organic layer is dried over sodium
sulphate, filtered and concentrated. The residue is
chromatographed on silica gel using e~hyl ace~ate/
petroleum ether (1:2).
Yield: 1.16 g (69% of theory)
RF = O.68 (ethyl acetate/petroleum ether = 1:1)
Le A 2B 876 - 42 -
2~9~
Example 2:
Nethyl 3-~2-n-butyl-1-{(4-methoxycarbonylphenyl)methyl}-
lH-imidazol-5-yl]-2-cyclopentylmethyl-2-propenoate
H3C ~ ~ C02CH3
~ CO2CE~3
Analogously to the pxocedure of Example 1, the title
compound was prepared from 1.6 g (3.2 mmol) o the
compound from Example XVI.
Yield: 0.61 g (43~ of theory)
RF = O.63 ~ethyl acetate/petroleum ether = 2:1)
Le A 28 876 - 43 -
2~9~
Example 3:
Methyl 3- ~ 2-n-butyl 1- { ( 4-methoxycarbonylthiophen-5-yl ) -
methyl ~ - lH-imidazol -5 -yl ] -2 -cyclopentylmethyl-
2 -propenoate
H3C/ ~o2CH3
~S
~CO2CH3
Analogously to the procedure of Example 1, the title
compound was prepared from 1. 2 g ( ~ . 38 mmol ~ of the
compound from Example XYII.
Yield: 0.5 g (4796 of theory)
RF = O . 55 ( ethyl acetate/petroleum ether = 1: 1 )
Le A 28 876 - 44 -
2 ~
Example 4:
3-[2-n-butyl~ (4-carboxyphenyl)methyl}-lH-imidazol-
5-yl]-2-cyclopentylmethyl-~-propenoic acid
H3C
CO2H
A solution of 1 g of sodium hydroxide in 20 ml of methan-
ol/water (1:1) is added to a solution of 1.26 g(2.88 mmol) of the compound from ExamplP 2 in 30 ml of
methanol. The mixture is stirred at 50C for 16 hours,
then acidified with dil. hydrochloric acid, poured into
300 ml of watex and extracted three tLmes with 75 ml each
of dichloromethane and three times with 40 ml each of
ethyl acetate. The organic layers are dried over ~odium
sulphate, filtered and freed of solvent. The crude
product is purified on silica gel (toluene/ethyl acetate/
glacial acetic acid 10:30:0.2~toluene/methanol/glacial
acetic acid 35;5:0.2), giving 342 mg (29~ of theory) of
product.
RF = 0.25 (toluene/methanol~glacial acetic acid = 35:5:1)
Le A 28 876 - 45 -
-`` 2~2.~
xample 5:
3 - [ 2 - n - b u t y 1 - 1 - ~ ( 2 c a r b o x y t h i o p h e n -
5-yl)methyl}-lH-imida~ol-5-yl~-2-cyclopentylmethyl-
2-propenoic acid
N
H3C ~ ~ CO2H
~S
W`
co2~
Analogously to the procedure of Example 4, the title
compound was prepared from 500 mg (1.13 mmol) of the
compound from Example 3.
Yield: 101 mg (21% of theory)
RF = 0.13 (toluene/ethyl acetate/glacial acetic acid =
10:30:0.2)
Le A 28 876 - 46 -
~9~
Example 5:
3-[2-n-butyl-1-{(2-carboxyfuran-5-yl)methyl}-lH-imidazol-
5-yl]-2-cyclopentylmethyl-2-propenoic acid
H3C
~0
~ CO~H
Analogously to the procedure of Example 4, the title
compound wa~ prepared from 1.11 g (1.13 mmol) of ~he
compound from Example 1.
Yield: 380 mg (39% of theory)
RF = 0 . 07 ( toluene/ethyl acetate/glacial acetic acid =
10:30.1)
Le A 28 876 - 47 -
2~9~
Example 7:
3-[2~n-butyl-1-{(4-carboxyphenyl)methyl}-lH-imidazol-
5-yl]-2-cyclopentylmethyl-2-propenoic acid, disodium salt
H3C ~ ~ O2~Na+
ll l
\~~C02~Na~
340 mg (O.83 mmol~ of the compound from Example 4 are
dissolved in methanol/water, 1.66 ml of 1 N sodium
hydroxide solution are added, and the mixture is then
freed of the solvent. 376 mg (100% of theory) are
obtained.
Le A 28 876 - 48 -
Example 8:
3-[2-n-butyl-1-{(2-carboxythiophen-5-yl)methyl}-
lH-imidazol-5-yl]-2-cyclopentylmethyl-2-propenoic acid,
disodium salt
H3C ~ - C02~Na+
~S
~ C02~Na+
Analogously to the procedure of ~xample 7, the title
compound was prepared from 330 mg (O.79 mmol) of the
compound from Example 5.
Le A 28 876 - 49 -
2~
Example 9:
3-[2-n-butyl-1-{(2-carboxyfuran-5-yl)methyl}-lH-imidazol-
5-yl~-2-cyclopentylmethyl-2-propenoic acid, disodium ~alt
H3C~CO2~Na+
~0
l! ,1
~ ~ C02~Na~
Analogously to the procedure of Example 7, the title
compound was prepared from 376 mg (0.94 mmol) of the
compound from Example 6.
L~ A 28 876 - 50 -