Note: Descriptions are shown in the official language in which they were submitted.
-1- 20'9 32 2 1
N-sulfonylindoline derivatives carrying an amide functional group,
their preparation, and the pharmaceutical compositions in which
they are present.
The present invention relates to N-sulfonyl-
indoline derivatives, carrying an amide-functional group,
their preparation and the pharmaceutical compositions in
which they are present.
US Patent 3,838,167 describes some N-sulfonyl-
indole derivatives corresponding to the for~ula:
(R 2 n' ~ ~ RCOR 4
S~2R"3
in which
- R"~ is hydrogen, an alkyl or a substituted or unsub-
stituted phenyl;
- Rnz is a halogen, an alkyl, an alkoxy, a nitro or
trifluoromethyl;
~ Rn3 is an alkyl, a phenyl or an alkylphenyl;
- R , is an alkyl, a substituted or unsubstituted phenyl,
an alkoxy or a phenoxy;
lS - n' = O, 1 or 2.
These compounds 1 are synthesis inte~ediates for
the preparation of indole derivatives active on the
central nervous system, of formula:
H
in which R is an alkyl, a substituted or unsubstituted
phenyl or a hydroxyl.
The indoline derivatives according to the present
invention have an affinity for the vasopressin and
ocytocin receptors.
Vasopressin is a hormone known for its antidiuretic
effect and its effect in the regulation of the arterial
A
., ,~
- 2 - 2 ~
pressure. It stimulates several types of receptors: Vl,
V2, V1ar V1b and thus exerts cardiovascular, central,
hepatic, antidiuretic, emetic and aggregating effects, as
well as proliferative and mitotic effects, especially on
the vascular and hepatic tissues. Vasopressin receptor
antagonists can affect the regulation of the central or
peripheral circulation, especially the coronary, renal
and gastric circulations, as well as the metabolism of
water and the release of adrenocorticotrophic hormone
(ACTH). The vasopressin receptors, like those of
ocytocin, are also found on the smooth muscle of the
uterus. Ocytocin has a peptide structure similar to that
of vasopressin. Its receptors are also found on the
myoepithelial cells of the m~m~ry gland and in the
central nervous system. (Presse Médicale, 1987, 16 (10),
481-485, J. Lab. Clin. Med., 1989, 114 (6), 617-632 and
Pharmacol. Rev., 1991, 43 (1), 73-108).
Thus the compounds according to the invention are
useful especially in the treatment of complaints of the
central nervous system, the cardiovascular system and the
gastric sphere in humans and animals.
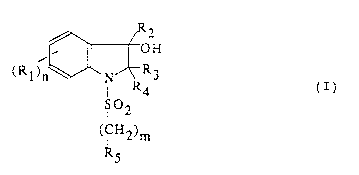
The present invention relates to compounds of
formula:
( R 1 ) n~ N~R 3
S~2 (I)
(CH2)m
R5
in which
- R1 is a halogen atom, a C1-C4 alkyl, a hydroxyl, a C1-C4
alkoxy, a benzyloxy group, a cyano group, a trifluoro-
methyl group, a nitro group or an amino group;
- Rz is a C1-C6 alkyl, a C3-c7 cycloalkyl, a C5-c7 cyclo-
alkene or a phenyl which is unsubstituted or mono-
substituted or polysubstituted by a C1-C4 alkyl, a
2 0 9 3 2 2
-- 3 --
C1-C4 alkoxy, a halogen, a trifluoromethyl group or an
amino group, or R2 is a nitrophenyl which is
unsubstituted or monosubstituted by a trifluoromethyl
~ group or monosubstituted or polysubstituted by a Cl-C4
alkyl or a halogen;
- R3 is a hydrogen atom;
- R4 is a carbamoyl group of formula CONR6R~;
- R5 is a Cl-C, alkyl; a 1-naphthyl; a 2-naphthyl; a 5-
dimethylamino-l-naphthyl; a phenyl which is un-
substituted or substituted by one or more substituent~s
selected from a halogen atom, a C1-C, alkyl, a tri-
fluoromethyl group, an amino group which is free or
substituted by one or 2 C~-C, alkyls, a hydroxyl, a
Cl-C~ alkoxy, a C2-C, alkenoxy, a C1-C, alkylthio, a
trifluoromethoxy group, a benzyloxy group, a cyano
group, a carboxyl group, a Cl-C~ alkoxycarbonyl group,
a carbamoyl group which is free or substituted by one
or two C~-C~ alkyls or a C1-C~ alkylamido group, or R5
is a nitrophenyl which is unsubstituted or monosub-
stituted by a trifluoromethyl group or a C2-C~ alkenoxy
or mono- or polysubstituted by a halogen, a C1-C,
alkyl, a Cl-C, alkoxy, a C~-C, alkythio, a trifluoro-
methoxy group or a benzyloxy group;
- R6 is a C1-C6 alkyl or R6 is identica1to R~;
- R7 is a 4-piperidyl group or a 3-azetidinyl group, the
said groups being substituted or unsubstituted on the
nitrogen by a C1-C~ alkyl, by a benzyloxycarbonyl or by
a C1-C, alkoxycarbonyl; a group (CH2)r which is itself
substituted by a 2-, 3- or 4-pyridyl group, by a
hydroxyl group or by an amino group which is free or
substituted by one or two C1-C, alkyls, a carboxyl
group, a C1-C~ alkoxycarbonyl group, a benzyloxy-
carbonyl group or a carbamoyl group which is free or
substituted by one or 2 C1-C, alkyls;
- or ~ and R7 together, with the nitrogen atom to which
they are connected, form a heterocycle selected from:
~ morpholine,
~ thiomorpholine,
~'
~ 2 ~
thiazolidine or 2,2-dimethylthiazolidine, un-
substituted or substituted by Ra/
piperazine, unsubstituted or substituted at the 4-
position by a group R"8,
~ an unsaturated, 5-membered ring containing a single
nitrogen atom and substituted by R8 or a saturated,
3-, 4-, 5-, 6- or 7-membered ring containing a
single nitrogen atom and substituted by R8 and Rg;
- R8 is R'8 or a group (CH2) r which is itself substituted
by a hydroxyl or by an amino which is free or sub-
stituted by one or two C1-C~ alkyls;
~ R'8 is a group (CH2)q which is itself substituted by a
carboxyl group, a Cl-C4 alkoxycarbonyl group, a
benzyloxycarbonyl group, a carbamoyl group which is
free or substituted by a hydroxyl or by one or 2 C1-C4
alkyls or an aminocarbothioyl group which is free or
substituted by one or 2 Cl-C4 alkyls;
- R"8 is R'8 or a group (CH2)2NH~ which is free or sub-
stituted by one or two Cl-c4 alkyls;
- Rg is hydrogen, a halogen, a group (CH2)rOR1o~ a group
(CH2)rNRIlRl2, a group (CH2)~NR11R'11 or an azido group;
- R1o is hydrogen, a Cl-C4 alkyl, a mesyl or a tosyl;
- R11, R'11 and R12 are each a hydrogen or a C1-C4 alkyl or
R11 is hydrogen and Rl2 is a benzyloxycarbonyl or a C1-C4
alkoxycarbonyl;
- n is 0, 1 or 2;
- m is 0, 1 or 2;
- q is 0, 1, 2 or 3;
- r is 0, 1, 2 or 3, with the limitation that r is not
zero when R8 or Rg is at the alpha-position of the
intracyclic amide nitrogen;
- s is O or 1;
as well as their possible salts.
The salts of the compounds of formula (I) according
to the present invention comprise those with inorganic or
organic acids which make possible a suitable separation
or crystallization of the compounds of formula (I), such
- 5 -
as picric acid, oxalic acid or an optically active acid,
for example a mandelic acid or a camphosulfonic acid, and
those which form pharmaceutically acceptable salts such
as the hydrochloride, the hydrogensulfate, the di-
hydrogenphosphate, the methanesulfonate, the maleate, thefumarate or the 2-naphthalenesulfonate.
The salts of the compounds of formula (I) also
comprise the salts with organic or inorganic bases, for
example the salts of alkali or alkaline-earth metals such
as the salts of sodium, potassium or calcium, the salts
of sodium and potassium being preferred, or with an
amine, such as trometamol, or even the salts of arginine
or lysine or of any pharmaceutically acceptable amine.
The compounds (I) exhibit cis-trans isomerism around
lS the 2,3 bond of the indoline. The different isomers form
an integral part of the invention.
By conver.tion, the compounds (I) in which R2 and ~4
are on the same side of the ring are called the cis
isomers.
By convention, the compounds (I) in which Rz and R4
are on opposite sides of the ring are called the trans
isomers. R2
~ OH
(Rl)n ~ N ~- 4
S~2 (I)
(CH2)m
R5
- cis isomer
(R l~N J~
S~2 (I)
(CH2)m
R5
trans isomer
- 6 - ~ ~ ~ 32 2 1
Moreover, the compounds according to the invention
have 2 asymmetric carbon atoms or more when R4 contains
one 1 or 2 asymmetric carbons. The optical isomers of the
compounds (I) form part of the invention.
In the present description and in the claims which
follow, halogen is understood as meaning a fluorine,
chlorine, bromine or iodine atom; alkyl group is under-
stood as meaning linear or branched hydrocarbon groups.
Preferred compounds (I) according to the invention
are those in which at least one of the following condi-
tions is satisfied:
- Rl is a chlorine or bromine atom or a methoxy group and
n = 1;
- R2 is a chlorophenyl, a methoxyphenyl or a cyclohexyl;
- R4 is a group CONR6R7 in which R6 and R~ or NR6R7 have
one of the following definitions;
~ NR6R7 is a pyrrolidino group which is substituted at
the 2-position by a group (CH2)q which is itself
substituted by a carboxyl or carbamoyl group with
q = 0, 1, 2 or 3.
~ NR6R7 is a piperidino group which is substituted at
the 4-position by an amino group, a C1-C, alkylamino
or a C1-C4 dialkylamino,
~ NR6R7 is a thiazolidino group which is substituted by
a group (CH2)q which is itself substituted by a
~ carboxyl or carbamoyl group with q = O, 1, 2 or 3.
~ NR6R7 is a pyrrolidino group which is substituted at
the 2-position by a group (CH2)q which is itself
substituted by a carboxyl or carbamoyl group with
q = 0, 1, 2 or 3 and which is substituted at the
4-position by an amino group, a C1-C, alkylamino or
a C1-C4 dialkylamino;
- R6 is a Cl-C, alkyl and R7 is a group (CH2) r which is
itself substituted by a carboxyl group or a carbamoyl
group with r = 1, 2 or 3;
- R5 is a phenyl substituted at the 3- and 4-position or
at the 2- and 4-position by a methoxy group, or R5 is
a phenyl substituted at the 4-position by a methyl
A
J i } ~
-- 7
group;
- m = 0-
The compounds (I) which are in the form of the cis
isomers are particularly preferred.
The following abbreviations are used in the
description and the examples.
DCM: dichloromethane
AcOEt: ethyl acetate
MeOH: methanol
EtOH: ethanol
Ether: ethyl ether
DMF: dimethylformamide
THF: tetrahydrofuran
TEA: triethylamine
DMSO: dimethyl sulfoxide
DIPEA: diisopropylethylamine
DCC: N,N'-dicyclohexylcarbodiimide
DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene
TBD: 1,5,7-triazabicyclo[4.4.0]dec-5-ene
DBN: 1,5-diazabicyclo[4.3.0]non-5-ene
DMAP: 4-dimethylaminopyridine
DMPU: 1,3-dimethyl-2-oxohexahydropyrimidinone
TMEDA: tetramethylethylenediamine
LDA: lithium diisopropylamide
HMPA: hexamethylphosphoramide
HOBT: 1-hydroxybenzotriazole hydrate
BOP: ben~otriazolyloxytrisdimethylaminophosphonium
hexafluorophosphate
TFA: trifluoroacetic acid
Lawesson's reagent: 2,4-bis(4-methoxyphenyl)-1,3-
dithia-2,4-diphosphetane-2,4-disulfide
M.p.: melting point
Saline solution: water saturated with sodium
chloride
Dry ice: solid carbon dioxide
TLC: thin layer chromatography
HPLC: high performance liquid chromatography
NMR: nuclear magnetic resonance
8 ~ ~ ~L ~
s: singlet
m: multiplet
bs: broad singlet
d: doublet
Hydrochloric water: dilute hydrochloric acid, about
lN
80% NaH: dispersion of sodium hydride in mineral oil
(Janssen Chemica)
Me: methyl
Et: ethyl
iPr: isopropyl, Pr: propyl
iPentyl: isopentyl
iBu: isobutyl
tBu:tert-butyl, Bu: butyl
Bz: benzyl
Ph: phenyl
RT: room temperature
The present invention further relates to the process
for preparing the compounds (I).
This process is characterized in that it camprises:
a) reacting a 2-aminophenone derivative of formula:
/ ~ CO-R2 (II)
(Rl)n~ NH2
in which R1, R2 and n have the meanings indicated above
for I, with a sulfonyl derivative of formula:
Hal-SO2-(CH2)m~Rs (III)
in which
- Hal is a halogen, preferably chlorine or bromine,
- m and R5 have the meanings indicated above for (I);
b) treating the resulting compound of formula:
2~g3
/ ~ O-R~
(R1)n ~ NH
S~2 tIV)
( ICH2)m
R5
with a halogenated deri~ative of formula:
Hal'-CH2COA (V)
in which
S Hal' is a halogen, preferably bromine, and A repre-
sents either the group NRbR7 or the group OR in which R is
a tert-butyl or a benzyl;
c) deprotecting the resulting ester of formula:
/ COR2
/ 11
(Rl)n~N-CH2COOR
S~2 (VI)'
( ICH2)m
R5
under suitable conditions, if applicable, when A is
OR;
d) treating, if applicable, the resulting acid from
Step c) of formula: / ~ COR2
/ I
(R1)n ~ I N-CH2COOH
S~2 ~VI)"
15(1CH2)m
or its acid chloride of formula:
/~CO R2
/ 11
(R1)n ~ N-CH2COC1 (VI)"'
SO2
( ICH2)m
R5
~ ,f~ i;.
with a compound HNR6R7 according to suitable amide
coupling techniques;
e) cyclizing the resulting compound from Step b) or
from Step d) of formula:
~COR2
(R1)n ~ ~-cH2coNR6R7
~~2 (VI)
(CH2)m
R5
in a basic medium in order to prepare the compound
(I) according to the invention;
f) separating, if appropriate, the cis and trans
isomers of the compound (I) and, if appropriate, separat-
ing the enantiomers. -
The 2-aminophenone derivatives (II) are known or
prepared by known methods, such as those described by
A.K. Singh et al., Synth. Commun. 1986, 16 (4), 485 and
G.N. Walker, J. Org. Chem., 1962, 27, 1929. The 2-amino-
2~-trifluoromethylbenzophenones and the other trifluoro-
methylated derivatives are prepared according to US
Patent 3,341,S92.
2,4-dimethoxybenzenesulfonyl chloride is prepared
according to J. Am. Chem. Soc., 1952, 74, 2008.
The sulfonyl derivatives of formula (III) are known
or prepared by known methods. Thus, for example, 4-
dimethylaminobenzenesulfonyl chloride is prepared accord-
ing to C.N. Sukenik et al., J. Am. Chem. Soc., 1977, 99,
851-858; p-benzyloxybenzenesulfonyl chloride is prepared
according to European patent application EP 229,566.
The alkoxybenzenesulfonyl chloride is prepared from
the sodium alkoxybenzenesulfonate, which is itself
prepared by reacting an alkyl halide with sodium hydroxy-
benzenesulfonate.
The halogenated derivatives of formula (V) are known
or prepared by known methods, such as those described by
A.I. Vogel: A Text Book of Practical Organic Chemistry:
Longman, 3rd ed. l9S6, p. 383, or G. Kirchner et al.,
J. Am. Chem. Soc., 1985, 107, 24, 7072.
Step a) of the process is carried out in pyridine by
heating at a temperature between room temperature and the
boiling point of the solvent for a period of tLme of
between a few hours and a few days. If appropriate, the
reaction can be carried out in the presence of dLmethyl-
aminopyridine, which is used in a catalytic or stoichio-
metric amount.
Step b) of the process is carried out between the
sulfonamide of formula (IV) and an excess of the
halogenated derivative of formula (V), in a solvent such
as dimethylformamide or dimethyl sulfoxide, under an
inert atmosphere, at a temperature of between 0~C and
room temperature, for a time of between a few hours and
24 hours, in the presence of sodium hydride.
When the group -NR6R7 contains a second amine group,
that is to say whe-- R6 and/or R7 are ,ubstituted by an
amino group, it is possible to choose to use a
halogenated derivative (V) of formula Hal'-CH2-CO2R in
which R is a tert-butyl or a benzyl, in order to prepare
the intermediates of formula (VI)' and then (VI)". In
this case, Step c) for the formation of the acid of
formula (VI)" is carried out either by the action of
hydrogen in the presence of a catalyst such as palladium
on charcoal when R is benzyl, or in acid medium, when R
is tert-butyl, for example in the presence of TFA or in
the presence of hydrobromic acid in acetic acid or even
in the presence of ZnBr2 in DCM.
Step d) is then carried out under the conventional
conditions for amide coupling, for example in the
presence of BOP or HOBT and DCC.
The compounds HNR6R7 are known or prepared by known
methods. By way of example, the stereospecific synthesis
of (R)- and (S)-2-pyrrolidinylacetic acids is carried out
according to H. Rueger et al. in Heterocycles, 1982, 19
(9), 1677 from a proline derivative of suitable con-
figuration. The preparation of methyl N-Boc-3,4-dehydro-
~-prolinate is carried out according to J.R. Dormoy,
Synthesis, 1982, 753. The preparation of optically pure
derivati~es of pipecolic acid is described, for example,
in Tetrahedron, 1992, 48 (3) 431-442 and Tetrahedron,
1991, 47 (24) 4039-4062.
The preparation of the derivatives of aziridine-
carboxylic acid is carried out according to K. Nakajima
et al. in Bull. Chem. Soc. Jap., 1978, 51 (5), 1577.
Step e) of the process is closely related to an
aldolization reaction: the methylene group in the ~-
position of the amide is deprotonated and the carbonyl
group of the phenone then acts like an internal electro-
phile, resulting in cyclization with the appearance of
two asymmetric carbons (C ).
The reaction can be illustrated by the following
15 scheme: 1l 0
C--R2 '
S~2 H
(CH2)m
R5 OH
(R;~S O'~ H2
(CH2)m
R5
The principles of the aldol addition reaction have
been reviewed by C.H. Heathcock in Asymmetric Synthesis,
vol. 3: Stereodifferentiating addition reactions, part B,
111-112; Academic Press, 1984, edited by J.D. Morrison.
It is known that the aldol reaction of achiral amide
anions gives rise to the formation of 2 racemic
diastereoisomers of ~-hydroxyamides in a ratio which
depends largely on the experimental conditions used. The
following may be mentioned among these conditions: the
nature of the inorganic or organic base used, the nature
- 13 - 2093221
of the cations or counterions, the possible presence of
additives in the reaction medium, the solvent, the
reaction temperature and the structure of the compound
undergoing this reaction.
When the groups R6 and R7 do not contain a group
which is hydrolysable in alkaline medium, it is possible
to use sodium hydroxide in water, in the presence of a
cosolvent, with or without the addition of a phase
transfer catalyst; it is also possible to use a
quaternary ammonium hydroxide, for example benzyltri-
methylammonium hydroxide in methanol.
In order to carry out this aldolization reaction, it
is also possible to use organic bases, for example:
- guanidines such as 1,5,7-triazabicyclo[4.4.0]dec-5-
ene,
- amidines such as 1,8-diazabicyclo[5.4.0]undec-5-ene or
1,5-diazabicyclo[4.3.0]non-5-ene,
in a solvent or a mixture of solvents selected for
example from benzene, THF, dichloromethane, methanol and
dimethylformamide; the reaction is carried out under an
inert atmosphere at between -10~C and 110~C; the amount
of base used is at least stoichiometric; the reaction can
also be carried out without a sol~ent, at the temperature
of the bath.
Preferentially, Step e) of the process according to
the invention is carried out in the presence of 1,8-
diazabicyclo[5.4.0]undec-5-ene (DBU) in a solvent such as
dichloromethane or methanol, at a temperature of between
-10~C and the reflux temperature of the solvent.
It is also possible to use an alcoholate of a
primary, secondary or tertiary alcohol with lithium,
sodium, potassium, calcium or magnesium.
The alcoholate is used in a catalytic or stoichio-
metric amount in an anhydrous solvent, for example an
alcohol (if appropriate in the presence of a cosolvent
such as THF), or else in a stoichiometric amount in THF,
DMF or DMSO, if appropriate in the presence of crown
ethers, for example dicyclonexyl-18-crown-6; the reaction
14 - ~ Y
is carried out at between -15~C and 80~C.
The use of an amide of the type RR~NLi or RR~NMg~r,
in which R and R' are monovalent radicals, as a
deprotonating agent is a method of forming enolates of
amides, which are intermediates in the aldolization
reaction; this method has recently been reviewed by R.E.
Ireland et al., J. Org. Chem., 1991, 56, 650. The reac-
tion solvent can be benzene, hexane or THF used in
anhydrous form under an inert atmosphere. Adjuvants such
as LiF, LiCl, LiBr, LiI, LiBu, TMEDA, DMPU, HMPA or a
crown ether can be added. (M. Murakate et al., J. Chem.
Soc. Commun., lg90, 16571. By way of example, there may
be mentioned the use of lithium diisopropylamide at
between -78~C and -30~C in anhydrous THF under an inert
atmosphere or in THF in the presence of additives such
as, for example, tetramethylenediamine, D~PU or HMPA.
Examples of other known amides which can be used are
lithium cyclohexylamide and lithium 2,2,6,6-tetramethyl-
cyclohexylamide. It is also possible to prepare other
amides by reacting the requisite amount of butyllithium
in hexane with linear or cyclic secondary amines, the
reaction taking place in one of the sol~ents mentioned
above.
Finally, various pu~lications describe amides of
optically active secondary amines: L. Duhamel et al.,
Bull. Soc. Chim. France, 1984, II, 421; J.R. Whitesell et
al., J. Org. Chem., 1980, 45, 755; M. Murakata et al., J.
Chem. Soc. Chem. Commun., 1990, 1657; M. Yamaguchi,
Tetrahedron Lett., 1986, 27 (8), 959; P.J. Cox and N.S.
Simpkins, Tetrahedron: Asymmetry, 1991, 2 (1), 1.
The silylamides of lithium, sodium or potassium
constitute another group of bases which can be used,
among which there may be mentioned: (Me3Si)2NLi,
(Me2PhSi)2NLi, (Et3Si)2NLi, (Me3Si)2N~, (Me3Si)2NNa.
It is also possible to use mixed amides as described
by Y. Yamamoto, Tetrahedron, 1990, 46, 4563, for example
the lithium salt of N-(trimethylsilyl)benzylamine or an
analog in which the benzylamine is replaced with a chiral
- 15 - ~0~322 L
primary amine such as (R)- or (s)-~-methylbenzylamine.
When the compound of formula (I) to be prepared has
2 asymmetric carbon atoms, the use of chiral amides or
alcoholates in Step e) makes possible an enantiomeric
enrichment of each of the cis or trans stereoisomers. The
proportion of each of the enantiomers is then determined
by measurement on a chiral high performance liquid
chromatography column.
When the compound of formula (I) to be prepared has
3 or 4 asymmetric carbon atoms, the cyclization Step c)
can be accompanied by a diastereoisomeric enrichment and
the use of a suitable chiral base makes it possible to
modify this diastereoisomeric enrichment.
In Step f), the cis and trans geometric isomers of
the compound (I) formed are extracted by conventional
methods and separated by chromatography or fractional
crystallization.
If appropriate, the optical isomers of each of the
cis and trans isomers are separated, for example by
preparative chromatography on a chiral column followed,
if appropriate, by a fractional crystallization or by
formation of an optically active salt in the presence of
a suitably selected chiral acid or base.
Thus, when the compound according to the invention
has 2 asymmetric carbon atoms, the enantiomers can be
separated by chiral HPLC.
When the compound according to the invention has 3
or 4 asymmetric carbon atoms, the diastereoisomers can be
separated by using chromatographic methods and fractional
crystallization methods.
Several methods can be used to differentiate and
characterize the cis isomer and the trans isomer of a
compound (I). When R3 is hydrogen, a comparative analysis
is performed by high field NMR (250 MHz), coupled for
example with the study of the Overhauser effect (N.O.E.)
between, for example, the proton of the indoline (R3 = H)
and the proton of the hydroxyl.
- 16 -
The IR spectra of the cis isomer and the trans
isomer in solution in DCM are different. The cis isomer
most commonly has a strong, fine and symmetrical absorp-
tion band at around 3550-3520 cm~1, due to the hydroxyl
vibration, whereas the trans isomer has no resolved
vibration band in this region.
By means of the data collected, it has been found
that the cis isomer is generally the more mobile in TLC
on an aluminum oxide plate (60F254 neutral, Type E,
Merck), eluting with DCM containing variable proportions
of AcOEt. Similarly, in chromatography on an alumina
column (aluminum oxide 90, particle size 0.063-0.200 mm),
the cis isomer is most commonly eluted first, the eluent
being DCM containing variable proportions of AcOEt or
MeOH.
Thus the cis or trans isomerism of a compound (I)
according to the invention can most often be determined
by an analytical method. It is also possible to utilize
the analogy between similar compounds or between com-
pounds prepared from one another.
The absolute configuration of some compounds accord-
ing to the invention was determined by an X-ray analysis.
By deduction therefrom, taking into account the value of
the optical rotation, it is also possible to know the
absolute configuration of other compounds obtained in an
analogous fashion.
A compound (I) in which R1 is an amino group and/or
a compound in which R5 is a phenyl group which is sub-
stituted by an amino can be prepared by the conversion
of a compound (VI), obtained in Step b), in which R1 is a
nitro group and/or R5 is a phenyl group which is sub-
stituted by a nitro, the other substituents having the
meanings desired for (I), by catalytic hydrogenation, for
example in the presence of palladium on charcoal, or
rhodium on alumina or Raney nickel.
The compounds (I) in which the substituents R6 and/or
R7 or the group NR6R7 contain a C1-C4 alkoxycarbonyl group
make it possible to obtain, by hydrolysis of the ester,
- 17 _
the compounds (I) in which R6 and/or R7 or the group NR6R7
contain a carboxyl group, the other substituents of (I)
being unchanged. Furthermore, the compounds in which R6
and/or R7 or NR~jR7 contain a carboxyl group make it
possible to obtain, by a conventional amide coupling
reaction, the compounds (I) in which R6 and/or R7 or the
group NR6R7 contain a carbamoyl group which is free or
substituted by one or two C1-C4 alkyls, the othe- sub-
stituents being identical.
Finally, the compounds (I) in which R6 and/or R7 or
the group NR6R7 contain a carbamoyl group make it possible
to obtain, by a Hofmann rearrangement, the compounds (I)
in which R6 and/or R7 or the group NR6R7 contain an amino
group, the other substituents being identical (J. Org.
Chem., 1979, 44 (10), 1746).
Thus, according to the present invention, the
process for preparing compounds (I) in which R6 and/or R7
or the group NR6R7 contain an amino group which is free or
substituted by one or two C1-C4 alkyls can have two
variants:
i) Step b) of the process is carried out by treating
the compound (IV) obtained in Step a) with a halogenated
derivative (V) of formula Hal'-CH2CONR6R7 in which R6
and/or R7 or the group NR6R7 contain a precursor group of
the amine, for example a carboxyester, a carboxyl or a
carbamoyl; the cyclization Step e) is then carried out
and the precursor group of the amine is then converted
into the amine, for example the carboxyester group of~the
compound (I) thus obtained is hydrolyzed into a carboxyl
group, which is then converted into a carbamoyl group and
then into an amino group by the Hofmann rearrangement.
ii) Step b) is carried out by treating the compound
(IV) obtained in Step a) with an halogenated derivative
(V) of formula Hal'-CH2COOR in which R is a benzyl or a
tert-butyl; the ester of the compound (VI)' thus obtained
is deprotected by a suitable treatment, according to Step
c); a coupling is then carried out with the compound
HNR6R7 in which the amino group of R6 and/or R7 is, if
k ~ ~ ~
- 18 -
appropriate, protected; the compound (VI) thus obtained
is then cyclized according to Step e); and, if appro-
priate, the compound (I) in which the amino group is free
is prepared by deprotectic~ of the amine.
The compounds (I) in which the groups R6 and/or R7 or
the group NR6R7 contain a benzyloxycarbonyl or alkoxy-
carbonyl group as substituent of an amine group make it
possible to obtain the compounds (I) in which the amine
group is free, the other substituents being identical.
The compounds of formula (VI), useful as
intermediates for the preparation of compounds (I)
according to the invention, are novel and form part of
the invention. Likewise, the compounds (VI)' and (VI)"
are novel and form part of the invention.
The present invention thus also relates to the
compounds of formula:
/~COR2
(R1)n ~ N-CH2 COA' 6
SO2
(CH2)m
R5
in which
A' is a group selected from: NR6R7, OH, OtBu, OBz;
- R1 is a halogen atom, a C1-C4 alkyl, a hydroxyl, a
C1-C4 alkoxy, a benzyloxy group, a cyano group, a
trifluoromethyl group, a nitro group or an ~amino
group;
- R2 is a Cl-C6 alkyl, a C3-c7 cycloalkyl, a C5-C7 cyclo-
alkene or a phenyl which is unsubstituted or mono-
substituted or polysubstituted by C1-C4 alkyl, a C1-C4
alkoxy, a halogen, a trifluoromethyl group or an amino
group, or R2 is a nitrophenyl which is unsubstituted or
monosubstituted by a trifluoromethyl group or mono-
substituted or polysubstituted by a C1-C4 alkyl, a Cl-C4
alkoxy or a halogen;
1 9 - 2 ~ ~¢ ~
- R5 is a Cl-c4 alkyl; a 1-naphthyl; a 2-naphthyl; a
5-dimethylamino-1-naphthyl; a phenyl which is unsub
stituted or substituted by one or more substituents
selected from a halogen atom, a C1-C4 alkyl, a tri-
fluoromethyl group, an amino group which is free or
substituted by one or 2 C1-C4 alkyls, a hydroxyl, a
C1-C4 alkoxy, a C2-C4 alkenoxy, a C1-C4 alkylthio, a
trifluoromethoxy group, a benzyloxy group, a cyano
group, a carboxyl group, a C1-C4 alkoxycarbonyl group,
a carbamoyl group which is free or substituted by one
or two C1-C4 alkyls or a C1-C4 alkylamido group, or R5
is a nitrophenyl which is unsubstituted or mono-
substituted by a trifluoromethyl group or a C2-C4
alkenoxy or mono- or polysubstituted by a halogen, a
C1-C4 alkyl, a C1-C4 alkoxy, a Cl-C4 alkylthio, a tri-
fluoromethoxy group or a benzyloxy group;
- R6 is a C1-C6 alkyl or R6 is similar to R7;
- R7 is a 4-piperidyl group or a 3-azetidinyl group, the
said groups being substituted or unsubstituted on the
nitrogen by a Cl-C4 alkyl, by a benzyloxycarbonyl or by
a C1-C4 alkoxycarbonyl; a group (CH2)r which is itself
substituted by a 2-, 3- or 4-pyridyl group, by a
hydroxyl group or by an amino group which is free or
substituted by one or two C1-C4 alkyls, a carboxyl
group, a Cl-C4 alkoxycarbonyl group, a benzyloxy-
carbonyl group or a carbamoyl group which is free or
substituted by one or 2 Cl-c4 alkyls;
- or R6 and R7 together, with the nitrogen atom to which
they are connected, form a heterocycle selected from:
~ morpholine,
~ thiomorpholine,
~ thiazolidine or 2,2-dimethylthiazolidine,
unsubstituted or substituted by R8,
~ piperazine, unsubstituted or substituted at the
4-position by a group R 81
~ an unsaturated, 5-membered ring containing a single
nitrogen atom and substituted by R8 or
- 20 - ~3~
a saturated, 3-, 4-, 5-, 6- or 7-membered ring con-
taining a single nitrogen atom and substituted by R8
and Rg;
- R8 is R'B or a group (CH2)r which is itself substituted
S by a hydroxyl or by an amino which is free or sub-
stituted by one or two Cl-C~ alkyls;
~ R'8 is a group (CH2)q which is itself substituted by a
carboxyl group, a Cl-C4 alkoxycarbonyl group, a
benzyloxycarbonyl group, a carbamoyl group which is
free or substituted by a hydroxyl or by one or 2 Cl-C4
alkyls or an aminocarbothioyl group which is free or
substituted by one or 2 C1_C4 alkyls;
~ R"8 is R'8 or a group (CH2)2NH2 which is free or sub-
stituted by one or two C1_C4 alkyls;
- Rg is hydrogen, a halogen, a group (CH2)rORlo~ a group
(CH2)rNRllRl2, a group (CH2)sCONRllR'll or an azido group;
~ R10 is hydrogen, a C1_C4 alkyl, a mesyl or a tosyl;
- Rll, R'11 and Rl2 are each a hydrogen or a Cl-C4 alkyl or
Rll is hydrogen and Rl2 is a benzyloxycarbonyl or a C1-C4
alkoxycarbonyl;
- n is 0, 1 or 2;
- m is 0, 1 or 2;
- q is 0, 1, 2 or 3;
- r is 0, 1, 2 or 3, with the limitation that r is not
zero when R8 or Rg is at the alpha-position of the
intracyclic amide nitrogen;
- s is 0 or 1;
According to another aspect of the present inven-
tion, the compounds (I) according to the invention in
which either R, or the group NR6R7 contains a carboxylgroup are useful for the preparation of analogous
decarboxylated compounds.
2. 2 ~
According to this aspect, the invention relates to
the use of the compounds of formula (I)'
I ~N~ 3 (I)~
CONRvIRVlI
1 2
(CH2)m
RS
in which Rl~R2,R3, Rs, m and n have the meanings
indicated above for co~oun~ of foLmula (I)
-RYI is a Cl-C6 alkyl,
-RVII is a group (CH2)rCOOH with r ~ i, 2 or 3,
- or RVI and RVII together, with the nitrogen atom to
which they are connected, constitute a heterocycle
selected from :
. thiazolidine or 2,2-dimethylthiazolidine,
substituted by a (CH2)qCOOH group
. piperazine substituted at the 4-position by a
(CH2)qCOOH group,
; an unsaturated, 5-membered ring containing a single
nitrogen atom and substituted by a (CH2)qCOOH group
. a saturated, 3-, 4-, 5-, 6- or 7-membered ring
containing a single nitrogen atom and substituted by
a (CH2)qCOOH group,
with q = O, 1, 2 or 3
for the preparation of a compound of formula (I)"
having the same configuration around the 2,3 bond of
the indoline as the starting material
1~
- 22 -
CO ' ' (I)
(CH2)m
R5
in which
- R1, R2, R3, R5, m and n are as defined above,
- R'VI is a Cl-C6 alkyl,
- R'VII is a group (CH2)rH~
- or R'VI and R'VII together, with the nitrogen atom
to which they are connected, constitute a
heterocycle selected from :
lS . thiazolidine or 2,2-dimethylthiazolidine,
substituted by a (CH2)qH group,
. piperazine substituted at the 4-position by ,a
(CH2)qH group,
. an unsaturated, 5-membered ring containing a single
nitrogen atom and substituted by a (CH2)qH group or
~ a saturated, 3-, 4-, 5-, 6- or 7-membered ring
containing a single nitrogen atom and substituted by
a (CH2)qH group.
The free-radical decarboxylation reaction is
carried out according to D. H. R. Barton et al. in J.
Chem. Soc; Chem. Commun.; 1984, 1298.
The affinity of the compounds according to the
invention for the vasopressin receptors was determined in
vitro using the method described in J. Biol. Chem., 1985,
260 t5), 2844-2851. This method consists in studying the
displacement of tritiated vasopressin bound to the Vl
sites of rat liver membranes. The 50% inhibitory con-
centrations (IC50) of the compounds according to theinvention for the binding of tritiated vasopressin are
A
3 ~ ~ ~
~ 23
low, ranging up to 109M.
Furthermore, the inhibition of the platelet
aggregation induced by vasopressin was measured on a
human platelet rich plasma (human PRP) using the method
described in Thrombosis Res., 1987, 45, 7-16. The com-
pounds according to the invention inhibit the aggregation
induced by 50 to 100 nM concentrations of vasopressin
with low ID50 values (inhibitory doses) which range up to
10-9M. These results show the antagonistic activity of the
compounds according to the invention towards the V
receptors.
The affinity of the compounds (I) according to the
invention for the V2 receptors was measured by a method
adapted from P. Crause et al., Molecular and Cellular
Endocrinology, 1982, 28, 529-541.
The compounds according to the invention of cis
configuration around the 2,3 bond of the indoline have a
marked selectivity for the V1 receptors.
The affinity of the compounds (I) according to the
invention for the ocytocin receptors was determined in
vitro by the displacement of tritiated ocytocin bound to
the receptors of a membrane preparation of gestating she-rat
glands. The IC50 values of the compounds according to the
invention are low, of between 105M and 10-8M.
The compounds according to the invention are active
after administration by various routes, especially
orally.
No sign of toxicity is observed with these compounds
at the pharmacologically active doses.
Thus the compounds according to the invention can be
used in the treatment or prevention of various
vasopressin-dependent complaints, especially cardio-
vascular complaints such as hypertension, cardiac
insufficiency, thrombosis or coronary vasospasm, in
3S particular in smokers; complaints of the central nervous
system, for example cerebral edemas, psychotic states,
appetite disorders or memory disorders; complaints of the
renal system, such as renal vasospasm or necrosis of the
- 24 -
renal cortex; and complaints of the gastric system, for
example ulcers or else the syndrome of inappropriate
secretion of antidiuretic hormone (SIADH).
The compounds according to the invention can also be
used as antiemetics, especially in motion sickness, and
as antiproliferative agents, for example in cancer or
atherosclerosis.
In woman, the compounds according to the invention
can also be used for the treatment of dysmenorrhea or
premature labor.
The present invention further relates to pharma-
ceutical compositions containing an effective dose of a
compound according to the invention, or of a pharma-
ceutically acceptable salt, and suitable excipients. Said
excipients are chosen according to the pharmaceutical
form and the desired mode of administration.
In the pharmaceutical compositions of the present
invention for oral, sublingual, subcutaneous, intra-
muscular, intravenous, topical, intratracheal,
intranasal, transdermal or rectal administration, the
active principles of formula I above, or their possible
salts, can be administered to animals and humans in unit
forms of administration, mixed with conventional pharma-
ceutical carriers, for the prophylaxis or treatment of
the above disorders or diseases. Appropriate unit forms
of administration include forms for oral administration,
such as tablets, gelatin capsules, powders, granules and
solutions or suspensions to be taken orally, forms for
sublingual, buccal, intratracheal or intranasal admini-
stration, forms for subcutaneous, intramuscular or
intravenous administration and forms for rectal admini-
stration. For topical application, the compounds accord-
ing to the invention can be used in creams, ointments or
lotions.
To obtain the desired prophylactic or therapeutic
effect, the dose of active principle can vary between
0.01 and 50 mg per kg of body weight and per day.
- 25 -
Each unit dose can contain from 0.5 to 1000 mg,
preferably from 1 to 500 mg, of active ingredients in
combination with a pharmaceutical carrier. This unit dose
can be administered 1 to 5 times per day so as to
5administer a daily dosage of 0.5 to 5000 mg, preferably
1 to 2500 mg.
If a solid composition in the form of tablets is
prepared, the main active ingredient is mixed with a
pharmaceutical vehicle such as gelatin, starch, lactose,
magnesium stearate, talc, gum arabic or the like. The
tablets can be coated with sucrose, a cellulose
derivative or other appropriate substances or they can
also be treated so as to have a prolonged or delayed
activity and so as to release a predetermined amount of
active principle continuously.
A preparation in the form of gelatin capsules is
obtained by mixing the active ingredient with a diluent
and pouring the resulting mixture into soft or hard
gelatin capsules.
20A preparation in the form of a syrup or elixir or
for administration in the form of drops can contain the
active ingredient in combination with a sweetener, which
is preferably calorie-free, and methylparaben and
propylparaben as antiseptics, as well as with a flavoring
and an appropriate color.
Water-dispersible granules or powders can contain
the active ingredient mixed with dispersants or wetting
agents or with suspending agents such as polyvinyl-
pyrrolidone, as well as with sweeteners or taste
correctors.
Rectal administration is effected using
suppositories which are prepared with binders melting at
the rectal temperature, for example cocoa butter or
polyethylene glycols.
35Parenteral administration is effected using aqueous
suspensions, isotonic saline solutions or sterile and
injectable solutions which contain pharmacologically
compatible dispersants and/or wetting agents, for example
- 26 -
propylene glycol or butylene glycol.
The active principle can also be formulated as
microcapsules, if appropriate with one or more carriers
or additives.
Apart from the products of formula I above or one of
the pharmaceutically acceptable salts, the compositions
of the present invention can contain other active
principles which may be useful in the treatment of the
disorders or diseases indicated above.
Thus the present invention relates to pharmaceutical
compositions containing a plurality of active principles
in association, one of which is a compound according to
the invention.
The following examples illustrate the invention.
The compounds are characterized by their melting
point (M.p.~C) (or their boiling point B.p.) and/or their
NMR spectrum recorded at 200 MHz in DMSO, and/or their
optical rotation (~D) measured at 25~C (except when
otherwise indicated).
The measured value of the optical rotation is
dependent on the amount of residual solvent present in
the prepared product.
Except when otherwise indicated, the designation
"cis isomer' or "trans isomer" signifies that the
isolated compound is a mixture of enantiomers, either of
cis configuration or of trans configuration.
The optical purity of the compounds is studied by
high performance liquid chromatography (HPLC).
EXAMPLE 1
N-methyl-N-methoxycarbonylmethyl-5-bromo-3-(2-
fluorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
2-indolinecarboxamide, cis isomer.
A) Methyl N-bromoacetylsarcosinate.
This compound is prepared according to T.D. Harris
et al. in J. Heterocyclic Chem., 1981, 18, 423.
B) 5-Bromo-2-(3,4-dimethoxyphenylsulfonamido)-2'-fluoro-
benzophenone.
2~39~221
- 27 -
20 g of 2-amino-5-bromo-21-fluorobenzophenone are
heated at 85~C for 48 hours in 120 ml of dry pyridine in
the presence of 20 g of 3,4-dimethoxyphenylsulfonyl
chloride. The mixture is cooled, poured into ice-cold
water, the solid is filtered off, the solid is extracted
with AcOEt, the organic phase is washed with water, a
solution of hydrochloric acid (lN), water and then saline
water. After drying over magnesium sulfate and evaporat-
ing the solvent under vacuum, a solid is obtained which
is recrystallized from DCM/isopropyl ether.
m = 28 g
M.p. = 125-128~C.
C) 5-Bromo-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-(N'-
methyl-N~-(methoxycarbonylmethyl)carbamylmethyl)]amino-
2'-fluorobenzophenone.
3.5 g of the compound prepared in Step B are dis-
solved in anhydrous DMF at 0~C under argon and 250 mg of
80% sodium hydride are added; after 15 minutes, 4.85 g
of the compound prepared in Step A are added and the
mixture is left stirring at RT for 12 hours. The reaction
mixture is poured into water, the solid is filtered off
and then the solid is dissolved in AcOEt, the organic
phase is washed with water and then with saline water and
the solvent is evaporated under vacuum. The oil obtained
is filtered on silica by eluting with a DCM/AcOEt
(85/15 ; v/v) mixture. It is recrystallized from a
DCM/isopropyl ether/MeOH mixture.
m = 3.2 g
M.p. = 136-137 C.
D) N-methyl-N-methoxycarbonylmethyl-5-bromo-3-(2-f luoro-
phenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-
indolinecarboxamide, cis isomer.
3.2 g of product obtained in the preceding step are
dissolved in DCM (3 ml), 750 mg of DBU are added and the
mixture is left stirring at RT for 24 hours. The reaction
mixture is poured onto a silica column; by eluting with
DCM/AcOEt (90/10 ; v/v), a product is obtained which is
the mixture of the two isomers (cis and trans) of the
;' ''~ ~ " 7'4 ~
.h i
- 28 -
expected compound. This product is triturated in a
hexane/isopropyl ether mixture and the solid obtained is
filtered. The filtration liquors are chromatographed on
an alumina column which was preequilibrated in a
S DCM/AcOEt (70/30 ; v/v) mixture. The least polar compound
is eluted with a DCM/AcOEt (60/40 ; v/v) mixture and is
then recrystallized from a DCM/hexane/isopropyl ether
mixture.
M.p. = 95~C with evolution of gas.
EXAMPLES 2 and 3
2-[(4-Benzyloxycarbonyl)-l-piperazinyl]carbonyl-5-
chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxyindoline, cis isomer and trans isomer.
A) 1-Bromacetyl-4-(benzyloxycarbonyl)piperazine.
A mixture of 22 g of 4-benzyloxycarbonylpiperazine
and lO.1 g of triethylamine in 200 ml of ether is cooled
to 0~C. 20.2 g of bromoacetyl bromide in 100 ml of ether
are added over 30 minutes and the mixture is left to
return to RT. After 4 hours, the reaction mixture is
washed with water, dried, concentrated and then chroma-
tographed on silica. The mixture DCM/AcOEt (95/5 ; v/v)
elutes the expected compound which is recrystallized from
DCM/isopropyl ether.
m = 9 g
M.p. = 100-101~C.
B) 2',5-Dichloro-2-(3,4-dimethoxyphenylsulfonamido)benzo-
phenone.
5.6 g of 2-amino-2',5-dichlorobenzophenone and 5 g
cf 3,4-dimethoxyphenylsulfonyl chloride are heated in
pyridine at 100~C overnight. The pyridine is evaporated
to dryness, water is added and extraction is carried out
with ethyl acetate cont~i n ing a small amount of DCM.
After washing more than once with water and drying over
sodium sulfate, the extract is evaporated under vacuum
and 7.7 g of the expected product are recrystallized in
a DCM/AcOEt mixture.
M.p. = 164~C.
~ 29 -
C) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-(4-
benzyloxycarbonyl-l-piperazinylcarbonylmethyl)]amino-
benzophenone.
2.3 g of the benzophenone prepared in Step B are
placed in 10 ml of DMF and treated with 200 mg of 80%
sodium hydride in oil. After 30 minutes, 5.3 g of the
compound prepared in Step A are added and the mixture is
stirred for 60 hours at RT. The mixture is poured into
water, the precipitate is filtered, taken up in DCM,
dried and then concentrated and chromatographed on
silica. The DCM/AcOEt (90/lO ; v/v) mixture elutes the
expected product which crystallizes from a DCM/isopropyl
ether mixture.
m = 2 g
M.p. = 173-175~C.
D) 2-[(4-Benzyloxycarbonyl)-1-piperazinyl]carbonyl-5-
chloro-3-(2-cAlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxyindoline, cis isomer and trans isomer.
1 g of the compound obtained in the preceding step
is suspended in 20 ml of methanol and 20 ml of THF and is
treated with 75 mg of sodium methylate. After 2 hours,
the mixture is neutralized by the addition of a small
amount of dry ice, is concentrated to dryness and then
taken up in water; the mixture is then extracted with
DCM, the extract is dried and concentrated. The crude
product is chromatographed on alumina, and the DCM/AcOEt
(80/20; v/v) mixture elutes the 2 isomers successively.
The least polar isomer is recrystallized from a
DCM/hexane mixture. This compound is the cis isomer.
m = 262 mg
M.p. = 169-179~C.
The most polar isomer is recrystallized from the
DCM/isopropyl ether mixture.
m = 200 mg
M.p. = 209-211~C.
EXAMPLE 4
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-(1-piperazinylcarbonyl)indoline,
_ 30 ~ 322~
cis isomer.
200 mg of the cis isomer prepared in the preceding
example are dissolved in 10 ml of ethanol and 5 ml of THF
and are hydrogenolyzed at RT in the presence of 10~ Pd/C.
After 30 minutes, the mixture is filtered on Celite~, the
filtration liquors are concentrated and then chroma-
tographed on silica. The MeO~/DCM (10/90 ; v/v) mixture
elutes the expected product which is recrystallized from
a DCM/isopropyl ether mixture.
m = 110 mg
M.p. 230-233 C.
EXAMPLES S and 6
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-morpholinocarbonylindoline, cis
isomer and trans isomer.
A) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
(morpholinocarbonylmethyl)]aminobenzophenone.
g of 2',5-dichloro-2-(3,4-dimethoxyphenyl-
sulfonamido)benzophenone are treated with 350 mg of 80
sodium hydride in 30 ml of DMF at RT for 20 minutes.
4.5 g of morpholinebromoacetamide are added and then the
mixture is stirred at RT for 48 hours. The mixture is
poured into water, the precipitate is filtered, it is
dissolved in DCM, the solution is dried and concentrated.
The product formed is recrystallized from a DCM/isopropyl
ether mixture. 5.4 g are obtained.
M.p. = 173-176~C.
B) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-morpholinocarbonylindoline, cis
isomer.
1 g of the product obtained in the preceding step is
dissolved in the methanol (10 ml) and THF (20 ml) mixture
and is treated with 92 mg of sodium methylate at RT for
1 hour. The mixture is neutralized with dry ice, the
solvents are partly evaporated, the mixture is taken up
in water, extracted with DCM and the extract is dried,
concentrated and chromatographed on alumina. The
DCM/AcOEt (70/30; v/v) mixture elutes the least polar
~ 31 - ~g3~
isomer which is recrystallized from a DCM/isopropyl ether
mixture.
m = 215 mg: cis isomer
M.p. = 260-264~C.
S C) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-morpholinocarbonylindoline, trans
isomer.
By the chromatography of the preceding step, a more
polar product is collected by eluting with the AcOEt/MeOH
(90/10) ; v/v) mixture. After recrystallizing from a
DCM/isopropyl ether mixture, there is obtained:
m = 513 mg: trans isomer
M.p. = 240-241~C.
EXAMPLE 7
N-Methyl-N-carboxymethyl-5-bromo-3-(2-fluorophenyl)-
1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-indoline-
carboxamide. cis isomer.
200 mg of the compound prepared in Example 1 are
dissolved in 3 ml of MeOH and 1 ml of water cont~ining
13 mg of sodium hydroxide. After stirring for 24 hours at
RT, one drop of concentrated sodium hydroxide solution is
added to bring the reaction to an end and then, after 15
minutes, the mixture is acidified to pH 3 by addition of
a potassium hydrogensulfate solution. Water is added, the
mixture is extracted with AcOEt and the extract is washed
with water and dried over magnesium sulfate and the
solvent is evaporated under vacuum. The product obtained
is recrystallized from DCM/isopropyl ether.
M.p. = 206-208 C.
EXAMPLES 8 and 9
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-(4-ethylcarboxylatepiperidino-
carbonyl)indoline, cis isomer, trans isomer.
A) Ethyl N-bromoacetyl-4-piperidinecarboxylate.
This product is prepared from ethyl 4-piperidine-
carboxylate, which is commercially available.
2 ~ ~
- 32 -
B) 2~5-Dichloro-2-[N-(3~4-dimethoxyphenylsulfonyl]-N-(4-
ethylcarboxylatepiperidinocarbonylmethyl)]aminobenzo-
phenone.
8 g of 2',5-dichloro-2-(3,4-dimethoxyphenylsulfon-
amido)benzophenone are dissolved in 100 ml of DMF and
then 541 mg of sodium hydride are added. After stirring
for 30 minutes, 9.5 g of the compound of Step A are added
and the mixture is left stirring for 18 hours at RT. The
mixture is concentrated under vacuum, taken up in water,
extracted with ethyl acetate and the extract is dried and
concentrated. The oil obtained is chromatographed on
silica, eluting with the AcOEt/DCM/hexane
(40/10/50 ; v/v/v) mixture. The expected product
crystallizes from ether.
m = 3.5 g
M.p. = 128~C.
C) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-(4-ethylcarboxylatepiperidino-
carbonyl)indoline, cis isomer, trans isomer.
A mixture containing 3.4 g of the compound prepared
in the preceding step and 869 mg of DBU in 10 ml of
chloroform is brought to 60~C for 18 hours. The reaction
mixture is then filtered on an alumina column, eluting
with a DCM/AcOEt (90/10; v/v) mixture in order to obtain
the cis isomer.
m = 700 mg
M.p. = 110~C.
Pure ethyl acetate elutes the trans isomer.
m = 610 mg
M.p. = 187~C.
EXAMPLES 10 and 11
N-methyl-N-(2-pyridylethyl)-5-chloro-3-(2-chloro-
phenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-
indolinecarboxamide, cis isomer, trans isomer.
A) N-[2-(2-chlorophenylcarbonyl)-5-chlorophenyl]-N-(3,4-
dimethoxyphenylsulfonyl)glycine acid.
a) 2',5-Dichloro-2-(3,4-dimethoxyphenylsulfonamido)-
benzophenone.
- 33 - ~93~
This compound is prepared in Example 2-3, Step B.
b) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-
N-benzyloxycarbonylmethyl]aminobenzophenone.
172 g of the product prepared previously are
dissolved in 800 ml of DCM and cooled to 0~C. 11.7 g of
80% sodium hydride is added progressively under nitrogen
and then, after 30 minutes, 256 g of benzyl bromoacetate
are added and the mixture is left stirring for 24 hours
at RT. The DMF is evaporated, the residue is taken up in
water, extracted with DCM, and the extract dried and
concentrated. The expected product crystallizes from
isopropyl ether and is then recrystallized from a
DCM/isopropyl ether mixture.
m = 136.5 g
M.p. = 102-104~C.
c) N-[2-(2-Chlorophenylcarbonyl)-5-chlorophenyl]-N-
(3,4-dimethoxyphenylsulfonyl)glycine acid.
50 g of the benzyl ester obtained previously are
dissolved in 500 ml of AcOEt and 2.5 g of 5$ Pd/C are
added under nitrogen. The solution is vigorously stirred
and a stream of hydrogen is passed in for 5 hours. At the
end of the hydrogenation, the product crystallizes. The
mixture is filtered on Celite~, the cake is washed
copiously with hot DCM and then the organic phase is
concentrated. The expected product crystallizes and is
then recrystallized from the DCM/isopropyl ether mixture.
m - 33.7 g
M.p. = 177-178~C.
B) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
(N'-(2-(2-pyridyl)ethyl)-N'-methyl)carbamoylmethyl]amino-
benzophenone.
2 g of the acid prepared in Step A are placed in
30 ml of DCM and 1.13 g of 2-(2-methylaminoethyl)-
pyridine, then 844 mg of triethylamine and finally 1.92 g
of BOP are added and then the mixture is left stirring
for 18 hours at RT. The mixture is taken up with water,
the organic phase is separated, washed with a sodium
carbonate solution, dried and concentrated. After
~ 34 - ~3~
chromatography on silica, the expected product is
collected by eluting with the DCM/MeOH (95/5); v/v)
mixture.
m = 2 g
M.p. = 150~C.
C) N-methyl-N-(2-pyridylethyl)-5-chloro-3-(2-chloro-
phenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-
indolinecarboxamide.
A mixture containing 1.7 g of the product obtained
in the preceding step and 442 mg of DBU in DCM is heated
at 55~C for 18 hours. The reaction mixture is chroma-
tographed on alumina. The AcOEt/DCM (40/60); v/v) mixture
elutes the cis isomer:
m = 410 mg
M.p. = 191 C.
Pure AcOEt elutes the trans isomer:
m = 790 mg
M.p. = 154~C.
EXAMPLE 12
2-(4-Carboxypiperidinocarbonyl)-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, cis isomer.
500 mg of the cis isomer prepared in Example 9 are
placed in 5 ml of methanol in the presence of 48 mg of
sodium hydroxide in 1 ml of water. After stirring for 18
hours, the mixture is poured into water, acidified with
dilute hydrochloric acid, then extracted with DCM and the
extract dried and concentrated. The solid obtained is
?urified by chromatography on silica, eluting with the
DCM/MeOH (95/5; v/v) mixture and the product obtained is
then crystallized from a DCM/isopropyl ether mixture.
m = 250 mg
M.p. = 150 C.
EXAMPLES 13 and 14
N-Methyl-N-(l-methyl-4-Piperidyl)-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
2-indolinecar~oxamide, cis isomer and trans isomer.
- 3S ~ 9 3 ~ 2 ~
A) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
(N'-methyl-N~-(methyl-4-piperidyl)carbamoylmethyl]amino-
benzophenone.
2 g of the acid prepared in Example 10-11, Step A in
50 ml of DCM are mixed with 650 mg of 4-methylamino-1-
methylpiperidine in the presence of 1.90 g of BOP. After
stirring for 2 hours at RT, the organic phase is washed
with carbonated water, dried and concentrated. The
residue is then chromatographed on silica, eluting with
the DCM/MeOH (90/10 ; v/v) mixture. 1.2 g of the expected
product are obtained.
M.p. = 165-166~C.
B) N-Methyl-N-(methyl-4-piperidyl)-5-chloro-3-(2-
chlorophenyl)-l-(3~4-dimethoxyphenylsulfonyl)-3-hydr
2-indolinecarboxamide, cis isomer and trans isomer.
650 mg of the product obtained in the preceding step
are treated overnight with 100 mg of sodium methylate in
5 ml of methanol. Dry ice is added, the solvent is
evaporated, the residue is taken up in carbonated water,
extracted with DCM and the extract dried and concentrated
and then chromatographed on silica. The methanol/DCM
(5/95 ; v/v) mixture elutes the 2 isomers successively.
Each is then recrystallized from a DCM/isopropyl ether
mixture.
The trans isomer is the least polar under these
conditions,
m = 205 mg
M.p. = 181~C.
Cis isomer: m = 150 mg
M.p. = 97~C: contains 0.25 M of isopropyl ether.
EXAMPLES 15 and 16
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[4-methyl-1-piperazinylcarbonyl]-
indoline, cis isomer and trans isomer.
A) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((4-methyl-1-piperazinyl)carbamoylmethyl))aminobenzo-
phenone.
~ 36 - ~3~1
This compound is obtained by the action of N-methyl-
piperazine on the acid prepared in Example 10-11 Step A.
M.p. = 165-167~C.
B) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[4-methyl-1-piperazinylcarbonyl]-
indoline, cis isomer and trans isomer.
The compound of the preceding step is cyclized by
proceeding as in Example 12-13. The 2 isomers formed are
separated by chromatography on alumina. The DCM/AcOEt
(75/25; v/v) mixture elutes the least polar product: the
cis isomer, which is recrystallized from a DCM/isopropyl
ether mixture.
M.p. = 120~C: contains 0.25 M of isopropyl ether.
The DCM/MeOH mixture elutes the most polar compound,
the trans isomer which is then recrystallized from-
methanol.
M.p. = 189~C.
EXAMPLES 17 and 18
N-Isopropyl-N-methoxycarbonylethyl-5-chloro-3-(2-
chlorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
2-indolinecarboxamide, cis isomer and trans isomer.
A) N-Isopropyl-N-(methoxycarbonylethyl)bromoacetamide.
90 g of isopropylamine are added dropwise to 130 g
of a solution, cooled to -10~C, of methyl acrylate in
300 ml of methanol. After 72 hours at RT, the mixture is
evaporated and the residue is then distilled. The oil
obtained (168.3 g) is methyl 3-(N-isopropyl)-aminopro-
pionate.
B.p. = 73-78~C at 15 mm Hg.
29 g of the compound obtained in 100 ml DCM are
mixed with 20.2 g of bromoacetyl bromide in 100 ml of DCM
at 0 C. After 12 hours at RT, the solvent is evaporated,
the residue is taken up in water, extracted with ethyl
acetate and the extract dried and concentrated. The oil
obtained is used as it is in the following step.
B) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
(N'-isopropyl-N'-methoxycarbonylethyl)carbamoylmethyl]-
aminobenzophenone.
- 37 -
This compound is obtained by following the usual
procedure, by reacting the product prepared in Step A
with 2',5-dichloro-2-(3,4-dimethoxyphenylsulfonamido)-
benzophenone in the presence of sodium hydride.
M.p. = 135-137 C (recrystallization: DCM/isopropyl
ether).
C) N-Isopropyl-N-methoxycarbonylethyl-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
2-indolinecarboxamide, cis isomer and trans isomer.
This product is obtained by cyclizing the compound
prepared in Step B, in the presence of DBU. The cis
isomer is separated by chromatography on alumina, eluting
with a DCM/AcOEt (g0/10; v/v) mixture. The product is
then crystallized from an AcOEt/hexane mixture.
M.p. = 153-155~C.
The trans isomer is obtained by eluting the alumina
column with ethyl acetate. The product is then
recrystallized from a methanol/isopropyl ether mixture.
M.p. = 182-185 C.
EXAMPLES 19 and 20
N-Methyl-N-methoxycarbonylmethyl-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
2-indolinecarboxamide, cis isomer and trans isomer.
The 2 isomers of this compound are prepared accord-
ing to the procedure described in Example 1. They areseparated by chromatography on alumina. The DCM/AcOEt
(80/20 ; v/v) mixture elutes the cis isomer. This
crystallizes from a DCM/isopropyl ether mixture in the
form of a white powder containing 0.25 mole of isopropyl
ether. It is converted to a foam by heating in vacuum.
The NMR spectrum of the cis isomer (Example 19) is
given in Figure 1.
The trans isomer is eluted with pure AcOEt. It is
recrystallized from DCM/isopropyl ether.
M.p. = 176-178~C.
The NMR spectrum of the trans isomer (Example 20) is
given in Figure 2.
- 38 - ~ ~ 3~ ~
EXAMPLES 21 and 22
N-Methyl-N-carboxymethyl-5-chloro-3-(2-chloro-
phenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-
indolinecarboxamide, cis isomer and trans isomer.
These compounds are each prepared from the compounds
described in Examples 19 and 20 according to the pro-
cedure described in Example 8.
Cis isomer: M.p. = 220-222 C after recrystallizing
from a DCM/isopropyl ether/MeOH mixture.
Trans isomer: M.p. = 222-225 C after recrystallizing
from a DCM/isopropyl ether mixture.
EXAMPLES 23 and 24
N-Methyl-N-carbamoylmethyl-5-chloro-3-(2-chlorophenyl)-
1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-indoline-
carboxamide, cis isomer and trans isomer.
Each isomer is obtained from the corresponding
isomer of the acid prepared in Example 21-22.
605 mg of the trans isomer of the acid obtained in
the preceding example are dissolved in 10 ml of DCM, and
435 mg of BOP and 260 mg of DIPEA are added. After
5 minutes at RT, 6 ml of 20% aqueous ammonia are
added with vigorous stirring and the mixture is left
stirring for 4 hours. A sodium carbonate solution is
added and the mixture is then extracted with DCM. The
organic phase is washed successively with water, a sodium
hydrogensulfate solution, and water and is then dried
over magnesium sulfate. After evaporating, the residue is
chromatographed on silica gel and is eluted with an
AcOEt/MeOH (95/5; v/v) mixture. The product obtained is
crystallized twice from a DCM/EtOH mixture at 0 C.
M.p. = 236 C.
The NMR spectrum of the trans isomer (Example 23) is
given in Figure 3.
Using the same procedure, the cis isomer is
prepared.
The expected product crystallizes from DCM/isopropyl
ether. The micronized compound, dried in vacuum at 70~C
for 8 hours, contains 0.25 mole of isopropyl ether.
X~2.4~
39
The NMR spectrum of the cis isomer (Example 24) is
given in Figure 4.
EXAMPLES 25 and 26
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-1-(4-hydroxy-1-piperidyl)carbonyl-
indoline, trans isomer.
This compound is prepared from N-[2-(2-chlorophenyl-
carbonyl)-5-chlorophenyl]-N-(3,4-dimethoxyphenyl-
sulfonyl)glycine acid described in Example 11-12, Step A.
The process is then carried out as in Example 11-12
for the addition of 4-hydroxypiperidine, in the presence
of BOP and triethylamine. The product obtained is then
cyclized according to the usual method in the presence of
DBU. The 2 isomers are separated by chromatography on
alumina. The DCM/MeOH (99/1; v/v) mixture elutes the cis
isomer.
The product crystallizes from a DCM/hexane/MeOH
mixture and the solid obtained is then triturated in
DCM/hexane to provide an amorphous powder.
The cis isomer is characterized by its NMR spectrum
at 388~R.
1-1.8 ppm:m:4H:CH2 at positions 3 and 5 of the piperidine
2.8-3.65 ppm:m:5H:CH2 at positions 2 and 6 of the piper-
idine and CH at position 4
3.75 ppm:2s:6H:20CH3
4.15 ppm:d:lH:OH on piperidine
5.45 ppm:s:lH:CH (indoline)
6.1 ppm:s:lH:OH indoline
6.8-7.6 ppm:m:lOH:H aromatic
DMS0:2.4 ppm
DOH:2.75 ppm
The DCM/MeOH (97/3; v/v) mixture elutes the trans
isomer which is recrystallized from DCM/isopropyl ether.
M.p. = 232-234~C.
~Yhl~SES in the (L)-Proline series: Examples 27,
28, 29and 30.
2 ~
EXAMPLES 27 and 27a
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[(2S)-l2-methoxycarbonyl)pyrrol-
idinocarbonyl]indoline, (cis isomers: 2 compounds).
A) Methyl (L)-N-(bromoacetyl)prolinate.
g of triethylamine and 20 g of bromoacetyl
bromide in 30 ml of DCM are added simultaneously to a
solution of 16.7 g of methyl (L)-prolinate hydrochloride
- in 20 ml of DCM while maintaining the temperature at -5~C
and the mixture is then stirred at RT for 24 hours. Water
is added, and the mixture is washed with a solution of
KHS04, with water, with a sodium bicarbonate solution and
with water and is then dried over magnesium sulfate.
After evaporating, an oil is obtained which is dried
under vacuum. This oil, pure by TLC, is used as it is in
the following step.
B) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((2S)-(2-methoxycarbonyl)pyrrolidinocarbonylmethyl)]-
aminobenzophenone
4.66 g of 2',5-dichloro-2-(3,4-dimethoxyphenyl-
sulfonamido)benzophenone are dissolved in 40 ml of
anhydrous DMF under argon, at 0~C, 340 mg of 80% sodium
hydride are added and then, after 30 minutes, 6.5 g of
the compound obtained in Step A. After 4 days at RT, the
mixture is poured into water, extracted with AcOEt, the
extract washed with water, with saline water and then
dried over magnesium sulfate and evaporated under vacuum.
A solid containing a small amount of the starting
brominated derivative is eluted with a DCM/AcOEt (85/15;
v/v) mixture by chromatography on silica gel. A sample
is recrystallized from DCM/isopropyl ether.
m = 1.2 g
M.p. = 141-142~C
~D5 = -43.7~(C = l; MeOH/THF: 8/2; v/v)
Analysis Calculated C:54.81 H:4.44 N:4.41
Found 54.40 4-54 4-55
- 41 ~ 2 ~
C) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[(2s)-(2-methoxycarbonyl)pyrr
idinocarbonyl]indoline, (cis isomerism).
1.1 g of the compound obtained in the preceding step
are heated in 4 ml of methylene chloride for 24 hours
with one equivalent of DBU. HPLC analysis of an aliquot
shows the existence of the expected 4 isomers. After 24
hours, the reaction mixture is poured onto an alumina
column, pre-equilibrated in the DCM/AcOEt (90/10; v/v)
mixture and is eluted with the DCM/AcOEt (90/10; v/v to
70/30; v/v) mixture. 510 mg of a mixture of the 2 least
polar compounds are obtained in the ratio 4/1 (measured
by HPLC).
1~) Two successive crystallizations from DCM/iso-
propyl ether while cold provide the major compound.
m = 180 mg
~D5 = - 247~(c = 0.4; chloroform)
M.p. = 187-190~C.
2~) The crystallization mother liquors of the
preceding compound are chromatographed on alumina by
eluting with DCM/AcOEt (85/15; v/v). The preceding
compound is thus separated from the second, the latter is
dissolved in the m; nimum amount of DCM and is then pre-
cipitated by addition of the minimum amount of hexane.
~26 = +136~(c = 0.24; chloroform)
EXAMPLE 28
2-((2S)-2-Carboxypyrrolidinocarbonyl)-5-chloro-3-(2-
chlorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, cis isomer.
430 mg of the compound prepared in Example 27 are
dissolved in 6 ml of methanol, 41 mg of sodium hydroxide
in 1 ml of water are added and the mixture is stirred for
24 hours at RT. The mixture is acidified to pH 3 with a
few drops of a potassium hydrogensulfate solution and is
extracted with ethyl acetate. The extract is washed with
water and is then dried over magnesium sulfate. Chroma-
tography is carried out on a silica column prepared in a
DCM/pentane (80/20; v/v) mixture. The unreacted ester
- 42 _ ~3~
elutes the expected acid which is then recrystallized
from DCM/isopropyl ether.
M.p. = 232-234 C
~ Z6 = -2S4~(c = 0.3; chloroform).
EXAMPLES 29 and 29a
2-((2S)-2-Carbamoylpyrrolidinocarbonyl)-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, (cis isomers : compounds).
230 mg of the compound prepared in Example 28 are
dissolved in 5 ml of DCM, 50 mg of DIPEA and then 165 mg
of BOP are added and the mixture is left for 5 minutes at
RT. The mixture is cooled in an ice bath and a stream of
gaseous ammonia is then bubbled through for 1 minute and,
after 15 minutes, for a further 1 minute. Water and then
a large volume of ethyl acetate are added in order to
obtain two phases. The organic solution is washed with a
sodium carbonate solution, water, a potassium hydrogen-
sulfate solution, water and then saline water. After
drying, the residue is chromatographed on silica by
eluting with a DCM/MeOH (93/7; v/v) mixture. The product
obtained is triturated in a DCM/isopropyl ether/hexane
mixture. It contain~ 1/3 mole of isopropyl ether.
~26 = -189~(C = 0.23; chloroform).
The compound of Example 29 can be prepared according
to another procedure.
A) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((2S-2-carbamoylpyrrolidinocarbonylmethyl)]aminobenzo-
phenone.
33.9 g of the acid prepared in Example 10-11, Step
A are dissolved in 300 ml of chloroform. 15 g of thionyl
chloride are added and the mixture is brought to reflux
for 1 hour and a half. The mixture is evaporated to
dryness, the residue is then taken up in DCM and
evaporated again. The mixture is dissolved in 300 ml of
DCM, brought to 0~C and 10.5 g of (L)-prolinamide hydro-
chloride are added, and then 18 g of DIPEA in 20 ml of
DCM are slowly added without allowing the temperature of
the reaction mixture to exceed 3~C.
_ 43 -
After one night at
RT, the reaction mixture is washed with sodium
bicarbonate ( twice ) and then with potassium hydrogen-
sulfate ( twice ); the reaction mixture is dried andconcentrated. The crude product obtained is dissolved in
the minimum amount of DCM and added dropwise to isopropyl
ether (1.2 1) with stirring. After stirring for 2 hours,
the precipitate ootained is filtered and then dried under
vacuum for 6 hours at 60~C. 42 g are collected.
~D5 = -40.8O(c = 1.007; chloroform).
B) 2-((2S)-2-Car~amoylpyrrolidinocarbonyl)-5-chloro-3-(2-
chlorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-
hydroxyindoline, (cis isomers: 2 compounds).
5 g of the product prepare~ in the preceding step
are dissolved in 50 ml of methanol. The solution is
cooled to -10~C; 1.35 g of DBU is added and the mixture
is maintained for 60 hours at -10~C. A compound crystal-
lizes; it is filtered (cis compound 1). The crystal-
lization liquors are neutralized with ~HS04 and the
mixture is e~aporated to dryness It is taken up in
water, extracted twice with DCM, and the extracts are
dried and concentrated. The crude product obtained is
chromatographed on silica by eluting with an AcOEt/DCM
(28/72; v/v) mixture. A mixture is collected which is
dissolved in the minimum amount of methanol while hot;
the insoluble material is filtered off,the liquors are
then placed overnight at -4~C and the cis compound 2
crystallizes.
m = 1.25 g
~D5 = -196~(c = 0.351; chloroform).
The analysis of the NMR spectrum shows the presence
of one mole of MeOH per mole of product. The recrystal-
lization of the product from ethanol makes it possible to
remove the solvent in the crystals.
M.p. = 154-162~C
~D5 = - 204~(c = 0.3; chloroform)
~D5 = -131~(c = 0.27; chloroform/methanol: 8/2: v/v)
_44 - ~3~
This compound is identical, the solvent excepted, to
that prepared by the first procedure of the present
example.
The compound which crystallized in Step B) above,
called cis compound 1, is recrystallized from methanol.
M.p. = 190~C
~ D = +115~(c = 0.3; chloroform)
EXAMPLE 30
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[(2S)-2-(hydroxymethyl)pyrrolidino-
carbonyl]indoline, cis isomers.
A) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-(2-
(hydroxymethyl)pyrrolidinocarbonylmethyl)]aminobenzo-
phenone.
This compound is obtained by reacting (L)-prolinol
with the acid prepared in Example 10-11, Step A, by
following the usual procedure.
B) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[(2S)-2-(hydroxymethyl)pyrrolidino-
carbonyl]indoline, cis isomer.
1.5 g of the compound of the preceding step is
cyclized in the presence of 380 mg of DBU in 2 ml of DCM.
After 3 days at RT, 1 ml of DCM is added and the mixture
is then heated at 40~C overnight. The formation of 3
major compounds is observed by TLC on silica (eluent
AcOEt).
The least polar fraction is eluted by chromatography
on silica using DCM/AcOEt (60/40 to 80/20; v/v)-. A
chromatography on alumina is then carried out by eluting
with DCM/MeOH (99/1; v/v). The fraction obtained is
homogeneous by TLC. The product is recrystallized three
times from DCM/isopropyl ether. The expected product is
obtained with an HPLC purity greater than 99%.
m = 155 mg
- M.p. = 194-197 C
~25 = -195~ (c = 0.2; chloroform).
~3~
_ 45 -
~ Y~ SES in the (D)-Proline series: Example 31.
EXAMPLE 31
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[(2R)-2-(methoxycarbonyl)pyrrol-
idinocarbonyl]indoline, cis isomer.
A) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((2R)-2-(methoxycarbonyl)pyrrolidinocarbonylmethyl)]-
aminobenzophenone.
This compound is obtained from the acid prepared in
Example 10-11, Step A (3 g) to which are added 1.2 g of
methyl (D)-prolinate and 2.B g of BOP in 10 ml of DCM in
the presence of 1.15 g of triethylamine. The mixture is
left for 1 hour at RT and is then diluted with DCM, the
organic phase i~ washed with sodium carbonate and with
potassium hydrogensulfate, dried and concentrated. The
crude product is chromatographed on silica, eluting with
a DCM/AcOEt (95/5; v/v) mixture. The product obtained is
then recrystallized from a DCM/isopropyl ether mixture.
M.p. = 140-141~C.
~25 = +28.5~(C = 0.27; chloroform).
B) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxypheny.-
sulfonyl)-3-hydroxy-2-[(2R)-2-(methoxycarbonyl)pyrrol-
idinocarbonyl]indo'ine, cis isomer.
1.5 g of the preceding compound is brought to reflux
2S overnight in 5 ml of DCM in the presence of 360 mg of
DBU. The mixture is chromatographed on alumina. The
mixture DCM/AcOEt (95/5; v/v) elutes the least polar
fraction (m = 300 mg) which is recrystallized twice in
a DCM/isopropyl ether mixture.
M.p. = 186-188 C
~D5 = +245O(c = 0.4; chloroform).
This compound is the enantiomer obtained from (D)-
proline of that described in Example 27.
EXAMPLE 32 and 32a
N-Methyl-N-methoxycarbonylmethyl-5-chloro-3-(2-chloro-
phenyl)-l-(4-ethoxyphenylsulfonyl)-3-hydroxy-2-indoline
carboxamide, trans isomer and cis isomer.
_ 46 _ ~3~ 1
A) 2',5-Dichloro-2-[N-(4-ethoxyphenylsulfonyl)-N-(N'-
methyl-N'-(methoxycarbonylmethyl)Carbamoylmethyl~amino-
~enzophenone.
5.7 g of 2',5-dichloro-2-(4-ethoxyphenylsulfonamido)-
benzophenone are dissolved, under argon, in 40 ml of DMFand 400 mg of 80% sodium hydride are added at 0~C; after
15 minutes, 4.3 g of methyl N-(bromoacetyl)sarcosinate
are added. After 48 hours, the expected product is
extracted in the usual way and is then purified by
chromatography on silica by eluting with DCM/AcOEt
(90/10; v/v) and recrystallizing in a DCM/isopropyl ether
mixture.
M.p. = 158-160 C
B) N-Methyl-N-methoxycarbonylmethyl-5-chloro-3-(2-chloro-
phenyl)-1-(4-ethoxyphenylsulfonyl)-3-hydroxy-2-indoline-
carboxamide, trans isomer.
1 g of the compound obtained in the preceding step
is dissolved in 4 ml of DCM and treated for 90 minutes at
RT with 312 mg of TBD. A solution of potassium hydrogen-
sulfate is added, the DCM is evaporated under vacuum, the
mixture is extracted with AcOEt and the extract is washed
and dried over magnesium sulfate. The expected product is
obtained by chromatography on silica gel by eluting with
DCM/AcOEt (90/10; v/v).
2S m = 590 mg
M.p. - 168-171~C after recrystallizing from
DCM/hexane.
C) N-Methyl-N-methoxycar~onylmethyl-5-chloro-3-(2-chloro-
phenyl)-1-(4-ethoxyphenylsulfonyl)-3-hydroxy-2-indoline-
car~oxamide, cis isomer.
2.96 g of the compound obtained in Step A are
suspended in 20 ml of methanol and 10 ml of THF; 100 mg
of sodium methylate are added and the mixture is then
left for 7 hours in the refrigerator. Water is added, the
mixture neutralized with a potassium hydrogensulfate
solution and a part of the methanol is evaporated under
vacuum. After extracting with AcOEt, the residue is
chromatographed on alumina and is then eluted with a
_47 _ ~ Xx~
DCM/AcOEt (80/20; v/v) mixture. 850 mg of the expected
product are obtained which are recrystallized from a
DCM/isopropyl ether mixture.
The NMR spectrum is given in Figure 6.
By using methods similar to those described above,
intermediate compounds (VI) for the synthesis of com-
pounds (I) according to the invention were prepared.
The compounds (VI) prepared are described in Table
1 below.
The compounds (I) prepared are described in Table 2
below.
R' N CH2-CON\
5~2 7
R, 5 ~J¦
3 ~
_ 48
Table 1
R'l R~5 R'2 N~R6 M.p. (~C) or IR
\R7 Solvent
Br- 3,4-CH30 F- ICH3 82-83
-N-CH2c~2cH2c6H5 DcM/isopropyl ether
Cl- 3,4-CH30 Cl- $H3 164-166
-N-CH2CH2C02CH3 DCM/iSopropyl ether
Cl- 3,4-CH30 Cl- ,CH3 ~ 128
-N-CH2CH2N-~ DCM/isopropyl ether
Cl- 3,4-CH30 Cl- ~ 105
-N-CH2CH2C02cH3 DCM/iSopropyl ether
Cl- 2,4-CH30 Cl- N~ N-C02CH2C6HS 142-143
\--J MeOH
CH30- 3,4-CH3 Cl -N-CH2CH2C02CH3 IR (1)
Cl- 3,4-CH30 Cl- IPcn~l 85
-N CH2CH2C02CH3 Isopropyl ether/DCM
E~
Br- 3,4-CH30 Cl- -N-cH2cH2co2cH3 IR (2)
Cl- 3,4-CH30 Cl- -N 199
~ DCM/isopropyl ether
Cl- 3,4-CH30 Cl- ~ 135
N-CH2C02cH3 Isopropyl ether/
DCM/AcOH
Cl- 3,4-CH30 Cl- ~ 113
N CH2CH2-C02CH3 DCM/isopropyl ether
_ 49 -
Cl- 3,4-CH30 Cl- ~t 160
-N-CH2-C02CH3 isopropyl ether
Cl- 3 ,4-CH30 Cl- N~ NH-CO2-tBu 197-198
Cl- 3,4-CH30 ~C02CH2c6H5
IR (1) (DCM) 1740 cm~1 fine
1680 cm~1 broad
IR (2) (DCM) 1735 cm~1 fine
1660-1680 cm~1 split
(3) This compound is characterized by its optical
rotation:
~D5 = -36.8~(c = 0.44; chloroform).
Table 2
lRl2
R 1 ~ OH / C \
I CON~ 6
For each compound of formula (I) which has the
substituents R'1, R~5, R~2 and NR6R7 of the table below,
the cis isomer is shown and then the trans isomer,
except when otherwise indicated.
~a~3~2:~
- 50
Example ~'1 R'5 Rl2 ~, R6 M.p. (~C) or NMR
\R7 Solvent
33 Br- 3,4-CH30 F- IfH3 ~ 87-95
34 -N-CH2C02CH2C6H5 N2rR
100 - 103
36 Cl- 3,4-CH30 Cl- ,CH3 154-157
N-CH2CH2C02CH3 DcM/isopropyl ether
37 cis Cl- 3,4-CH30 CL- ICH3 140-144
-N-cH2cH2co2H DCM/isopropyl ether
.
38 Cl- 3,4-CH30 Cl- ,CH3 ~ 222-225
mixture -N-CH2CH2N-~ DCM/isopropyl ether
39 Cl- 3,4-CH30 Cl- ~ N~R
-'~-CH2CH2C02CH3 N~.
2 0 41 Cl - 3,4- CH30 Cl - E~ 166
cis -N-cH2cH2co2H DCM/isopropyl ether
42 119
43 Cl - 3,4 - CH30 Cl - -N3 /CH3AcOEt/isopropylether
2 5 228
MeOH
44 Cl- 3,4-CH30 Cl-~H2C6H5CH 179
cis -N-CH2CH2N~CH3 DCM/isopropyl ether
109
Cl- 3,4-CH30 Cl- -N N-CX2co2E~ DCM/isopropyl ether
46 196
DCM/isopropyl ether
47 ' ~ 134 (iPr)20
48 Cl- 2,4-CH30 Cl- -N ~.~-CO~C~2C6~5 195
' DCM/isopropyl ether
~ 1 _
49 Cl- 3,4-CH30 Cl- Et 236
cis CH2cH2coNH2isopropyl ether
154
Sl CH3 3,4-CH3 Cl- Et isopropyl ether
-N-c~2cH2co2cH3 87
isopropyl ether
52 Cl- 2,4-CH30 Cl- ~N NH 194
\ MeOH/isopropyl ether
cis
53 Cl- 3,4-CH30 Cl- iPentyl 195
cis N-CH2CH2C02CH3 isopropyl ether/DCM
54 138-140
Br- 2,4-CH30 Cl- I isopropyl ether
-N-cH2cH2co2cH3 140
isopropyl ether
56 _~ 242-245
57 Cl- 3,4-CH30 Cl--N S DCM/isopropyl ether
\J 225
MeOH/isopropyl ether
Cl- 2,4-CH30 Cl- 228
58 -N~}N(CH3)2MeOH/isopropyl ether
58 a 221
DCM/MeOH
131-134
59 Cl- 3 4-CH30 Cl-~L DCM/isopropyl ether/
-N-cH2cH2cH2co2cH3
59a hexane
121
3 5 I DCM/etherfhexane
- 52 _ ~ ~ ~ 32 2 1
162- 166
Cl - 3,4-CH30 Cl- iBu DCM/isopropyl ether
61 N-cH2cH2co2cH3 130
DCM/isopropylether/
hexane
62 Cl- 3,4-CH30 Cl- Pr 176
63 -N-CH2cO2cH3isopropyl ether/DCM
64 Cl - 3,4 - CH30 Cl - 148
~ isopropyl ether/DCM
-N-CH2CH2C02CH3 122
lS isopropyl ether/DCM
66 Cl- 3,4-CH30 Cl-
67 N-CH2 C02CH3 168
isopropyl ether
68 Cl- 3,4-CH30 Cl- iBu 179-182
cis N-(CH2)2C~2H DCM/isopropyl ether
69 Cl- 3,4-CH30 Cl- ~ 139
cis N-CH2C~2HDCM/isopropyl ether
Cl- 3,4-CH30 Cl- ~ 130
cis N-CH2C~2Hisopropyl ether
71 Cl- 3,4-CH30 Cl- ~ 136
cis N-(CH2)3C~2H DCM/iSopropyl etherl
72 Cl- 3,4-CH30 Cl- ~ 135
cis N-CH2-C~NH2 DCM/isopropyl ether
_ 53~
73 C1- 3,4-CH30 Cl- 197
N3NHco2c(cH3)3MeoH/isopropyl ether
74 ' 211
MeOH
Cl- 3,4-CH30 C1- N3NH2
cis
76 Cl- 3,4-CH30 Cl- ~ Fumarate
cis N ~ NH2 152-156
DCM/isopropyl ether
76a Cl- 3,4-CH30 Cl- ~ 137
N isopropylether/MeOH/
76b ~ C02CH2C6H5 hexane
183-185
hexane
20 - Example 34
analysis: calculated C:55.54 H: 4.24 N:3.93
found55.72 4.573.83
NI~R spectra at 200 MHz (DI~SO: 2.5 ppm)
- Example 34: Figure S
25 - Example 38
0.7-1.1 ppm: m:6H:2CH3(Et)
2-4 ppm:m:l7H:2CH2(Et), 2C_2-N, N-CH3, 20CH3
5.2-5.7 ppm:3s:1H:H (indoline)
6.2-8.2 ppm:m:llH:OH + aromatics
30 - Example 39
0.3-1.2 ppm:m:3H:CH3 (Et)
1.5-4.3 ppm:m:15H:CH7-CO, CH2 (Et), CH2-N, 20CH3, CO2CH3
5.2-5.6 ppm:3s:lH:H (indoline)
6.2-8.2 ppm:m:llH:OH + aromatics
35 - Example 40
0.8-1.1 ppm:m:3H:CH3 (Et)
2.2-3.9 ppm:m:lSH:CH2CO, CH2(Et), CH2N, C02CH3, 20CH3
5.3-5.7 ppm:2s:1H:H (indoline)
~32~ 1
6.6-8.2 ppm:m:llH:OH + aromatics
- Example 63
0.4-1 ppm: split t:3H:CH2-CHz-CH3
5 ppm:m:2H:CH2-CHz-CH3
2.5-4.4 ppm:m:13H:CH2-CH2-CH3, NCH2COOCH3, 20CH3
5.2-5.8 ppm; bs :lH:H (indoline)
6.5-8.3 ppm:m:llH:OH + aromatics
- Example 66
O to l.S ppm:m:3H:CH2-CH3
2.3-5.8 ppm:m:14H:CH2-CH3, NCH2COOC~, 20CH3, H (indoline)
6.1-8.3 ppm:m:llH:OH + aromatics
- Example 75
1.95 ppm:bs:2H:NH2
2.7 to 5.3 ppm:m:l2H:20CH3, 2NCH2, H(indoline), C HNH2
6 to 8.3 ppm:m:llH:OH + aromatics
- Example 76a
~D5 = +102 (c = 0.35; chloroform)
- Example 76b
~25 = -158 (c = 0.2; chloroform).
Some compounds according to the invention described
in Table 2 are useful in the preparation of other com-
pounds according to the invention. For example, compound
41 was obtained from compound 39 by treatment in basic
medium in methanol MeOH/H20. Compound 49 was prepared from
compound 41 by treatment with aqueous ammonia in the
presence of DIPEA and BOP.
EXAMPLE 77
N-Ethyl-N-(2-aminoethyl)-5-chloro-3-(2-chlorophenyl3-1-
(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-indoline-
carboxamide, (cis iscmer).
500 mg of compound 49 are dissolved in 10 ml ofacetonitrile and 10 ml of water and 252 mg of pyridine
and 380 mg of bis(trifluoroacetoxy)iodobenzene are added.
After stirring for 2 hours, the mixture is taken up in a
soiution of hydrochloric acid, extracted with ether,
alkalized with dilute sodium hydroxide solution,
extracted with DCM and the extract is dried and con-
centrated. An oil is obtained and the expected product
2 ~3 9 ~
then crystallizes from ether.
m = 150 mg
M.p. = 164~C
EXAMPLE 78
N-Ethyl-N-[(lS)-1-(ethoxycarbonyl)ethyl]-5-chloro-3-(2-
chlorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
2-indolinecarboxamide, (cis isomer).
A) N-[2-(2-Chlorophenylcarbonyl)-5-chlorophenyl]-N-(3,4-
dimethoxyphenylsulfonyl)glycine acid chloride.
A mixture containing 11 g of the acid prepared in
Example 10-11, Step A) and 5 g of thionyl chloride in
10 ml of chloroform is heated for 1 hour at 60~C. The
mixture is left to return to RT, concentrated under
vacuum and the residue taken up in DCM ( twice ). A
yellow oil is obtained which is used as it is in the
following step.
IR: 1800 cml (C=O)
B) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
(N'-ethyl-N'-((lS)-1-(ethoxycarbonylethyl)ethoxy-
carbamoylmethyl)]aminobenzophenone.
The preparation of this compound was carried outaccording to J. Org. Chem., 1985, 50, 945-950.
5.15 g of (L)-Boc(N-Et)AlaOEt is treated with 10 ml
of TFA at 0~C in order to remove the Boc group. The
mixture is concentrated under ~acuum, taken up in 20 ml
of DCM, cooled to -78~C and 2 equivalents of TEA and the
acid chloride prepared in the preceding step, dissolved
in DCM, are added. After 18 hours at RT, the mixture is
extracted with DCM, the extract is washed with water and
then chromatographed on silica by eluting with a
DCM/AcOEt (90/10; v/v) mixture. The expected product
crystallizes from isopropyl ether.
M.p. = 112~C
m = 8 g
C) N-Ethyl-N-~(lS)-1-(ethoxycarbonyl)ethyl]-5-chloro-3-
(2-chlorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-
hydroxy-2-indolinecarboxamide, (cis isomer~.
- 56 - ~ ~93~2~
The compound obtained in the preceding step is
stirred at RT for 18 hours in 10 ml of THF and 20 ml of
ethanol, in the presence of 1.46 g of DBU. The mixture is
concentrated under vacuum, the residue is taken up in
S DCM, washed with water, concentrated and the product
chromatographed on alumina by eluting with AcOEt/DCM
( 10/90; v/v~ .
N~
0-0.9 ppm: split d:3H:CH-C~
0.9-1.7 ppm:m:6H:2CH3 (ethyl)
2.6 to S.8 ppm:m:12H:20C~, NCH2, OCH2, NCH, coca
6.1 to 8.3 ppm:m:llH:OH - 10H aromatics
EXAMPLES 79 and 80
N,N-Di[2-(methoxycarbonyl)ethyl]-5-chloro-3-(2-chloro-
phenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-
indolinecarboxamide, cis isomer and trans isomer.
A) N,N-Di[2-(methoxycarbonyl)ethyl]benzylamine.
Preparation according to J. Am. Chem. Soc., 1950,
72, 3298.
107 g of benzylamine in 200 ml of ethanol are cooled
in an ice bath and 172.2 g of methyl acr,late in 250 ml
of ethanol are slowly added. After 13 days at RT, the
solvent is evaporated under vacuum and a part of the oily
residue is then distilled.
B.p. = 135-140 C at 0.6 mm Hg
m = 30 g
IR: 1730 cm~1
B) N,N-12-dimethoxycarbonyl)ethyllamine.
27.9 g of the amine obtained in the ?receding step,
placed in 500 ml of methanol, are mixed with 3 g of 5%
palladium on charcoal and are treated under hydrogen
pressure for 1 hour. The mixture is filtered on Celite~,
rinsed with methanol and the solvent evaporated under
vacuum; the residual oil is used as it is in the follow-
ing step.
C) N,N-Di[2-(methoxycarbonyl)ethyl]bromoacetamide.
A mixture containing 14.3 g of the amine prepared in
the preceding step, 100 ml of DCM and 10.6 ml of TEA is
~ 57 ~ ~ 9 3 2 2~
cooled in an ice bath; 15.3 g of bromoacetyl bromide are
added dropwise and the mixture is then left stirring for
48 hours at RT. The mixture is extracted with DCM, the
extract is washed with water and then a chromatography is
carried out on silica by eluting with a DCM/MeOH
(97/3 ; v/v) mixture. The expected product is obtained in
the form of an oil.
m = 15.9 g
IR: 1650 cml and 1730 cm1.
D) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
[N',N -di(2-(methoxycarbonyl)ethyl)carbamoylmethyl]]-
aminobenzo phenone .
14.3 g of 2',5-dichloro-2-(3,4-dimethoxyphenyl-
sulfonamido)benzophenone are placed in 180 ml of DMF and
1.1 g of sodium hydride are added in portions. After
stirring for 1 hour at RT, the mixture is cooled in an
ice bath and 14.3 g of the product prepared in the
preceding step are added and the mixture is left stirring
for 72 hours at RT. The mixture is extracted with DCM,
the extract is washed with water and then chromatographed
on silica by eluting with a DCM/AcOEt (93/7; v/v)
mixture.
m = 28.4 g
M.p. = 130~C.
E) N,N-Di[2-(methoxycarbonyl)ethyl]-5-chloro-3-(2-chloro-
phenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-
indolinecar~oxamide, cis isomer.
12 g of the compound prepared in the preceding~step
and 0.930 g of sodium methylate in 150 ml of methanol are
mixed at 0~C and the mixture is then left stirring
overnight at RT. The reaction mixture i~ neutralized by
addition of 5~ KHS04 and the solvent is then evaporated
under vacuum. The residue is chromatographed on alumina
by eluting with a DCM/AcOEt (8/2; v/v) mixture. 2.4 g of
the expected product are recovered which are crystallized
from methanol.
M.p. = 175~C.
z ii
- 58 -
F) N,N-Di[2-(methoxycarbonyl)ethyl]-5-chloro-3-(2-chloro-
phenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-2-
indolinecarboxamide, trans isomer.
The chromatography of the preceding step is con-
tinued and elution is carried out with a DCM/MeOH
(9.5/0.5; v/v) mixture. 1.82 g of the trans isomer is
obtained which crystallizes from isopropyl ether.
M.p. = 85~C.
EXAMPLES 81, 82 and 83
2-((2R)-2-Carbamoylthiazolidinocarbonyl)-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, (cis isomer: 2 compounds and trans
isomer).
A) (L)-4-Thiazolidinecarboxamide.
This compound is prepared according to J. Med.
Chem., 1981, 24, 692.
B) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((2R)-2-carbamoylthiazolidinocarbonylmethyl)]-amino-
benzophenone.
This compound is obtained by the usual methods from
the acid prepared in Example 10-11, Step A).
M.p. = 125 C after crystallizing from ether.
C) 2-((2R)-2-Carbamoylthiazolidinocarbonyl)-5-chloro-3-
(2-chlorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-
hydroxyindoline.
4.3 g of the product obtained in Step B) are
cyclized in 90 ml of MeOH at RT in the presence of 1 g of
DBU. The mixture is concentrated, the residue is taken up
;n water and DCM, the layers are separated, the organic
layer is washed with RHSO~ and then dried and concen-
trated. The residue is chromatographed on alumina by
eluting with DCM/MeOH (97/3; v/v). The compound is
obtained in the cis form (mixture of 2 diastereoisomers):
1.5 g, and then in the trans form (mixture of 2
diastereoisomers): m = l g.
a) The cis fraction is crystallized from MeOH/DCM in
order to obtain cis compound 1.
~ ~ ~3 3 ~
5 9
M.p. = 176~C after crystallizing from isopropyl
ether.
~ D = +57 (c = 0.1; chloroform).
b) The crystallization liquors of the preceding
product are chromatographed on silica by eluting with
AcOEt/DCM (30/70; v/v). The cis compound 2 obtained is
recrystallized from ether.
M.p. = 205~C
~D = -185~(c = 0.3; chloroform).
c) The trans fraction (mixture of 2 diastereo-
isomers) is recrystallized from isopropyl ether.
M.p. = 170~C
EXAMPLES 84, 85, 86 and 86a
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[(2S)-2-(N,N-dimethylthio-
carbamoyl)pyrrolidinocarbonyl]indoline, cis isomerism
(2 compounds), (trans isomerism: 2 compounds).
A) (L)-(N'-Boc)-N,N-Dimethylprolinethioamide.
This compound is prepared according to J. Med.
Chem., 1989, 2178.
2.36 g of (N'-Boc)-N,N-dimethylprolinamide are
heated in anhydrous toluene under argon at 80~C for 4
hours in the presence of 2.3 g of Lawesson's reagent.
After 24 hours, the solvent is evaporated and isopropanol
is added. The precipitate formed is separated, the
isopropanol is evaporated and the residue is chroma-
tographed on silica by eluting with hexane/AcOEt (30/70;
v/v). The product obtained is recrystallized while-cold
from DCM/isopropyl ether (30/70 ; v/v).
M.p. = 62~C
B) 2',5-Di~hloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((2S)-2-(N',N'-dimethylthiocarbamoyl)pyrrolidino-
carbonylmethyl)]aminobenzophenone.
3 g of the product prepared in the preceding step
are dissolved in 10 ml of DCM and treated at 0~C for 2
hours with 10 ml of TFA. The mixture is evaporated to
dryness, then 20 ml of DCM and 6.1 g of the acid prepared
in Example 10-11, Step A) are added at 0~C and the
_ 60 -
mixture is neutralized with 3 g of DIPEA. 5.15 g of BOP
are dissolved in 30 ml of DCM and this solution is added
to the preceding solution at 0~C over 30 minutes; the pH
is maintained at neutral by the addition of DIPEA and the
mixture is left stirring for 3 hours at 0~C. After one
night at RT, the mixture is extracted in the usual way
and then chromatographed on silica by eluting with
DCM/AcOEt (85/15; v/v). The product obtained is
recrystallized from isopropyl ether.
M.p. = 182-185~C
~D = -72~ (c = 0.32; chloroform).
C) 5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxy-2-[(2S)-2-(N,N-dimethylthio-
carbamoyl)pyrrolidinocarbonyl]indoline (cis isomerism :
2 compounds, and trans isomer: 2 compounds).
3.8 g of the compound obtained in the preceding step
are dissolved in 15 ml of DCM and the mixture is heated
at reflux for 36 hours in the presence of 850 mg of DBU.
The different isomers formed are separated by successive
chromatographic runs on silica.
a) Using DCM/AcOEt (85/15; v/v), the expected
compound is eluted first in the form of a mixture of 2
cis diastereoisomers. The least soluble diastereoisomer
is crystallized twice from a DCM/isopropyl ether/
methanol mixture and is then recrystallized from the
m; nimll~ amount of DMF at 60~C followed by addition of
2 volumes of ethanol.
- M.p. = 270~C
~D - - 278O(c = 1; chloroform).
b) The crystallization liquors of the preceding
mixture are taken up in and the second cis diastereo-
isomer crystallizes from a DCM/isopropyl ether mixture.
M.p. = 249-251~C
~D = +42~(C = 0.22; chloroform).
c) The chromatography fractions eluted last, as well
as the crystallization mother liquors of fractions a) and
b), are combined, and are chromatographed again on silica
by eluting with hexane/AcOEt (20/80; v/v). Isolated first
h~9~
- 61_
is a fraction which is recrystallized 3 times from a
DCM/isopropyl ether mixture and an insoluble material is
removed on a paper between each recrystallization. The
trans isomer 1 is thus obtained.
M.p. = 191-193
~D = +74.5~(C = 0.2; chloroform).
d) The second fraction contains trans isomer 2 which
is recrystallized from a DCM/isopropyl ether mixture and
crystallizes with 1/3 mole of isopropyl ether.
M.p. = 170~C
~ D = -266~(C = 0.14; chloroform).
EXAMPLES 87, 88 and 89
2-((2S)-2-Carbamoylpyrrolidinocarbonyl)-5-chloro-3-
cyclohexyl-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, (cis isomers: 2 compounds, trans isomer).
A) 5-Chloro-2-[N-(3,4-dimethoxyphenylsulfonyl)amino]-
cyclohexylphenone.
A solution of 35.6 g of 2-amino-5-chlorocyclohexyl-
phenone and 39.5 g of 3,4-dimethoxyphenylsulfonyl
chloride in 340 ml of pyridine is left stirring for 24
hours at RT. The solvent is evaporated under vacuum and
the residue is then washed with water and with an acid
solution (0.5 N HCl). The expected product crystallizes
from ethanol.
M.p. = 135 C
m = 56.1 g.
B) 2-[(N-Benzyloxycarbonylmethyl-N-(3,4-dimethoxyphenyl-
sulfonyl))amino]-5-chlorocyclohexylphenone.
3.2 g of sodium hydride are added in portions to
52.6 g of the compound prepared in the preceding step in
520 ml of DMF and the mixture is left stirring for 1 hour
at RT. After cooling in the ice bath, 21 ml of benzyloxy-
carbonylmethyl bromide are added dropwise and the mixture
is left stirring for 24 hours at RT. The solvent is
evaporated under vacuum and the residue is taken up in
water. It is extracted with DCM and the extract is washed
with water; the product obtained is used as it is in the
following step.
~ h
- 62 -
C) N-(5-Chloro-2-(cyclohexylcarbonyl)phenyl)-N-(3,4-
dimethoxyphenylsulfonyl)glycine.
The compound obtained in the preceding step is
placed with 3.9 g of 5% palladium on charcoal in 700 ml
of acetic acid under hydrogen (1 atmosphere). At the end
of the reaction, the palladium is filtered on Celite~ and
rinsed with hot acetic acid; the solvent is evaporated
under vacuum and the residue is taken up in water. It is
extracted with DCM and the extract is washed with water
and then with a concentrated NaHCO3 solution. The residue
obtained is chromatographed on silica by eluting with a
DCM/MeOH (97/3; v/v) mixture. The expected product
crystallizes from ethanol.
M.p. = 160~C
m = 22.4 g.
D) 5-Chloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-((2S)-
2-carbamoylpyrrolidinocarbonylmethyl)]-aminocyclohexyl-
phenone.
A mixture containing 9.92 g of the acid prepared in
the preceding step, 3 g of (L)-prolinamide hydrochloride
and 3.5 ml of DIPEA in 75 ml of DCM is cooled to 0~C.
8.84 g of BOP in solution in DCM are added and the pH is
maintained at 7 by addition of DIPEA. The mixture is left
stirring for 24 hours at RT. The mixture is extracted
with DCM, and the extract washed with a saturated NaHCO3
solution, a saline solution, a 5% RHS04 solution and again
with a saline solution. The product is chromatographed on
silica by eluting with a DCM/MeOH (96/4; v/v) mixture.
The expected product solidifies in isopropyl ether.
M.p. = 110~C
m = 7.3 g
QD = -53.9~(C = l; chloroform)
E) 2-((2S)-2-Carbamoylpyrrolidinocarbonyl)-5-chloro-3-
cyclohexyl-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, cis isomer (2 compounds)~ trans isomer.
5.9 g of the compound prepared in the preceding stepand 1.67 g of DBU are placed in 60 ml of methanol with
stirring at 0~C for 48 hours. The solvent is evaporated
~ ~ 9 6~
- 63 _
under vacuum, water is added, the mixture is extracted
with DCM and the extract is then washed with a 5% KHS04
solution. The product is chromatographed on alumina by
eluting with DCM/MeOH (98/2; v/v).
a) The least polar fraction contains the 2 cis
isomers. This fraction is recrystallized from methanol.
The first compound thus obtained (cis 1) is pure by HPLC.
M.p. = 185~C.
By recrystallization of the mother liquors from
MeOH, a second compound is obtained (cis 2). HPLC purity:
75% (it contains 25% cis 1).
M.p. = 132~C.
b) The most polar fraction contains the trans isomer
in the form of an apparently single compound which is
obtained by recrystallizing from methanol.
M.p. = 240~C
~ D = -55.1~(C = 1; chloroform).
EXAMPLE 89a
2-((2S)-2-Carbamoylpyrrolidinocarbonyl)-5-chloro-3-
cyclohexyl-1-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, cis isomer (2 compounds)~ trans isomer.
By using a procedure similar to that described for
Examples 87, 88 and 89, an analogous compound in the (D)-
proline series is prepared.
The compound obtained after crystallizing from a
DCM/MeOH mixture has the trans configuration.
M.p. = 238~C
~D = +164~(c = 0.245; chloroform/methanol, 8.2, v/v).
The NMR spectrum of this compound and of that
described in Step E b) of the preceding example are
identical.
EXAMPLE 9O
5-Chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-2-[(2S)-2-(N-methylaminocarbonyl)pyrrolidino-
carbonyl]-3-hydroxyindoline, cis isomer.
920 mg of the compound prepared in Example 28 are
placed, with stirring, in 20 ml of DCM containing 371 mg
of BOP for 15 minutes, a stream of monomethylamine is
~ 64 -
bubbled through for 10 minutes and the stirring is
maintained for an additional 30 minutes. The mixture is
taken up in water, the layers separated, the organic
layer washed with potassium hydrogensulfate and with
S sodium carbonate, dried and concentrated. The residue is
chromatographed on silica by eluting with DCM/methanol
(97.5/2.5; v/v). The expected product is collected which
crystallizes from an isopropyl ether/DCM mixture.
m = 750 mg
M.p. = 158 C
~D = -216~(c = 0.3; chloroform).
By operating as in the previously described examples
(Examples 27 to 31 and 90) and by using derivatives of
(L)-proline (except when otherwise indicated), other
intermediate compounds (VI) for the synthesis of the
compounds (I) according to the invention were prepared.
The compounds (VI) prepared are described in Table
3 below.
The Compounds (I) prepared are described in Table 4
below.
Table 3
Cl ~1 -CH2-CO-N,
SO2
R'5~1
~ 65 -
R~5 N' R6 M.p.(~C) ~25
R/7 l (chloroform)
3,4-CH30 H ~ +45
- c = 1.015
CONH2
3,4-CH30 -N ~ 161-163 -70.8
COOCH3 DCM/isopropyl ether c 0.48
/~
2,4-CH30 -N 145-148 -17.5
H~ ~ c ~ 3.36
COOCH3
3,4-CH30 H~ 10
COOCH2 ~ DCM/isopropyl ether
3,4-CH30 H,.. ~_J 148
CH2COOCH3 isopropyl ether/MeOH
2 0 Table 4
C1
R' 1 ~ OH ,/~\
CON' 6
d'
R 5 ~JI
Example R l R~5 N~ R6 M.p.(~C) ~ 5
R7 (chloroform~
91 cis 1 Cl 3,4-CH30 ~ ~ 115 + 188
H MeOH c = 0.33
92 cis 2 CONH2 204 -114
Cl YeOH/DMF I c = O.31
2a~2~
~ 66 -
198-201
93 cis Cl 3,4-CH30H ~ DCM/isopropyl
ether
COOCH 1
~ ~ 205-207
94 cis Cl 2,4-CH30' ~ DCM/isopropyl
COOCH
3 ether
-N~ 221 -242
95 cis Cl 2,4-CH30 Hl~ ~DCM/isopropyl c ~ 0.254
~Q~ ether
96 cis Cl H~' ~ c 0 32
/~
H~ -214
CON(CH )
97 cis Cl 3,4-CH30 ~ 2 c = 0.32
~ 105-115 +174.6
98 cis 1 Cl 3,4-CH30Hl~ ~ DCM/isopropylc = 0.3
H2COOCH3ether
99 cis 2 175 -214.6
DCM/isopropyl c - 0.3
ether
100 trans 1 -155
c = 0.2
101 trans 2 177 +95.2
DCM/isopropyl c - 0.2
ether
102 cis 1 Cl 3,4-CH30 -~ ~ 135 -162
isopropyl ether
C H , C O O H
~ 67 -
~/ 1 145 -167
103 cis 1 Cl 3,4-CH30 H~ CM/isopropyl c = 0.4
CH2 CONH2 ether
' /~
103a Cl 3,4-CH30 ,H~
cl s 1
cis 2 , CH2 CONH2
104 cis 1 Cl 3,4-CH30 -N/--¦ 210 -177.5
Hl, ~l ether c ~ 0.2
CH2 NH2
- 104a Cl 3,4-CH30
cis 1 H~
cis 2
CH2NH2
200 -195
105 CH30 3,4-CH30 -~ j EtOH c--0.2
H~;
106 CONH2 215 +127
MeOH c ~ 0.2
-63.3
107 CH30 3,4-CH30 (D) ~ 198 c O.117
2 5 H (CHCl3J~0H
CONH2 - ''
8~ ;v~v)
~ 274 -225
108 Cl 2,4-CH30 H~ DCM/MeOH c = O.372
3 0 CONH2 (CHCl3/~H
8/2;v/v)
/~
108a Cl 3,4-CH30 H~ -198.7
cis c - 0.24
CONHOH
- 68 -
: Example 92: Other measured optical rotation:
~D5 = - 39.5~(c = 0.17; CHCl3/MeOH: 8/2:v/v).
The compound of Example 107 is the enantiomer of
that of Example 106.
The compound of Example 108a was prepared from the
compound of Example 28 by reacting with hydroxylamine
hydrochloride in DMF and by activating with the reagent
BOP in the presence of DIPEA.
Some compounds according to the invention, described
in Table 4 above, are useful for the preparation of other
compounds. Thus, the compound of Example 99 makes it
possible to obtain the compound of Example 101, then that
of Example 103 and finally that of Example 104.
EXAMPLE 109
2-((2S,4S)-4-Azido-2-(methoxycarbonyl)pyrrolidino-
carbonyl)-5-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxy-
phenylsulfonyl)-3-hydroxyindoline, cis isomer.
A) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((2S,4R)-4-hydroxy-2-(methoxycarbonyl)pyrrolidino-
carbonylmethyl)]aminobenzophenone.
15 g of the acid prepared in Example 10-11, Step A)
and 6.25 g of methyl (2S,4R)-4-hydroxyprolinate hydro-
chloride are heated to 0~C in 150 ml of DCM in the
presence of 7.4 g of DIPEA. A solution of 12.7 g of BOP
in 30 ml of DCM is added dropwise over 30 minutes and the
amount of DIPEA necessary to neutralize the solution is
added. After one night at RT, the mixture is extracted in
the usual way and chromatographed on silica by elution
with a DCM/AcOEt (60/40; v/v) mixture. The expected
product crystallizes from a DCM/ether/isopropyl ether
mixture.
M.p. = 128-131~C
~D = +8.5O(C = 0.38; chloroform).
B) 2',5-Dichloro-2-[N-(3,4-dimethoxyphenylsulfonyl)-N-
((2S,4R)-4-mesyloxy-2-(methoxycarbonyl)pyrrolidino-
carbonylmethyl)]aminobenzophenone.
2 g of the compound obtained in the preceding step
are dissolved at 0~C in 10 ml of DCM. 550 mg of
- 69- 2~932~
triethylamine and then 550 mg of methanesulfonyl chloride
are added and the mixture is left at 0~C for 20 hours.
Water is added and the organic layer is washed with 0.5 N
hydrochloric water, with water and then with a sodium
bicarbonate solution, dried over magnesium sulfate and
evaporated. The oil obtained is used as it is in the
following step.
C) 2-[N-((2S,4S)-4-azido-2-(methoxycarbonyl)pyrrolidino-
carbonylmethyl)-N-(3,4-dimethoxyphenylsulfonyl)]amino-
2',5-dichlorobenzophenone.
11 g of the product prepared in the preceding step
are heated in 60 ml of DMSO at 80-90~C in the presence
of 2.7 g of sodium azide for 18 hours. The mixture is
poured into water, extracted with ethyl acetate, the
organic layer washed with water, dried and chroma-
tographed on silica by eluting with a pentane/AcOEt
(50/50; v/v) mixture. An oil (10 g) is obtained.
~D = -25.5~(C = 0.39; chloroform, T = 26 C).
D) 2-((2S,4S)-4-Azido-2-(methoxycarbonyl)pyrrolidino-
carbonyl)-5-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxy-
phenylsulfonyl)-3-hydroxyindoline, cis isomer.
3.38 g of the product obtained in the preceding step
are cyclized under the usual conditions in the presence
of DBU. The expected product is obtained which is
recrystallized from DCM/isopropyl ether.
m = 755 mg
M.p. 200-202~C
- ~D - -176~(c = 0.21; chloroform, T = 26~C).
EXAMPLE 110
2-[(2S,4S)-4-(N-Benzyloxycarbonyl-N-methyl)amino-2-
(methoxycarbonyl)pyrrolidinocarbonyl]-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, cis isomer.
A) Methyl ester of (N-Boc)-4-hydroxyproline.
The starting material is the hydrochloride of the
methyl ester of (2S,4R)-4-hydroxyproline.
19 g of this compound are suspended in 100 ml of
THF, 22.9 g of (Boc)2O are added and then the mixture is
70 - ~ 32h ~
cooled to 0 C. 21.2 g of triethylamine in 25 ml of THF
are added dropwise and then the mixture is stirred for
12 hours at 0~C and 4 hours at 60~C. Water is added, the
mixture is extracted with ethyl acetate, the organic
layer is washed with water, with a potassium hydrogen-
sulfate solution (4 times), with water and then with
saline water. The solvent is evaporated and an oil
(21.6 g) is isolated which contains a small amount of
(Boc) 2~ .
B) Methyl ester of (2S,4R)-(N-Boc)-4-mesyloxyproline.
A solution of 22.9 g of the product prepared in the
preceding step in 250 ml of DCM is cooled to 0~C. 22.9 g
of mesyl chloride in 10 ml of DCM are added dropwise,
then 9.4 g of triethylamine in 100 ml of DCM are added
dropwise and the mixture is left to return to RT over-
night. The mixture is evaporated to dryness, water is
added, the mixture is extracted with AcOEt, the organic
layer is washed with water and saline water and dried
over magnesium sulfate. After a second evaporation, an
oil is obtained which is used as it is in the following
step.
C) Methyl ester of (2S,4S)-(N-Boc)-4-azidoproline.
This compound is prepared from that obtained in Step
B. 15.2 g of the methyl ester of (N-Boc)-4-mesyloxy-
proline are dissolved in 70 ml of DMSO and thesolution is heated at 90~C for 5 hours in the presence of
3.05 g of sodium azide. The mixture is cooled, water is
added, the mixture is extracted with AcOEt, the organic
layer is washed with water and saline water and dried
over MgSO4. The oil obtained is purified by chromatography
on silica by eluting with the AcOEt/hexane (40/60; v/v)
mixture.
~D = -37.8O(c = 3; chloroform)
lit. ~D = -36.6~(c = 2.8; chloroform) D.J. Abraham et
al., J. Med. Chem., 1983, 549, 26.
D) Methyl ester of (2S,4S)-(N-Boc)-4-aminoproline.
8.45 g of the compound obtained in Step C are
dissolved in 100 ml of methanol, 500 mg of 10% Pd/C are
- 71 - 2B~3221
added and the mixture is hydrogenated at 40~C for 18
hours. The catalyst is filtered off, half the methanol is
evaporated, 100 ml of 0.5 N HCl are added, the remainder
of the methanol is then evaporated and the unreacted
starting material is extracted with AcOEt. The aqueous
phase is treated with sodium carbonate and the fraction
containing the expected product (m = 4.35 g) is extracted
with AcOEt.
E ) Methyl ester of (2S,4S)-(N-Boc)-4-(N'-benzyloxy-
carbonylamino)proline.
The crude product obtained in the preceding step is
dissolved in 15 ml of ether and 15 ml of DCM at 0~C.
2.3 g of DIPEA and then 3.03 g of benzyl chloroformate
in 5 ml of DCM are added, over 70 minutes at 0~C. After
3 hours, the solvents are evaporated at RT under vacuum;
water and ethyl acetate are added and the organic phase
is washed successively with a potassium hydrogensulfate
solution (3 times), with water (3 times), with a sodium
carbonate solution (3 times), with water (3 times) and
with saline water. The product is chromatographed on
silica by eluting with hexane/AcOEt (40/60; v/v) mixture
in order to obtain the expected product.
~D = -16.4~(c = 0.3; chloroform).
F) Methyl ester of (2S,4S)-(N-Boc)-4-(N'-benzyloxy-
carbonyl-N'-methyl)aminoproline.
2 g of the compound obtained in the preceding step
are dissolved in 20 ml of DMF at 0~C, under argon, in the
presence of 2.25 g of methyl iodide. 170 mg of 80% sodium
hydride are added in portions and then the mixture is
stirred at 0~C for 90 minutes. The mixture is extracted
with water and ethyl acetate; the organic phase is washed
with water and then saline water. The product is chroma-
tographed on silica by eluting with a hexane/AcOEt
(50/50; v/v) mixture. 1.55 g of the expected product is
o~tained.
~3 = -38.8~(C = 0.38; chloroform).
~ 72 - ~ f~t~
G) 2',5-Dichloro-2-[(2S,4S)-N-(3,4-dimethoxyphenyl-
sulfonyl)-N-(4-(N'-benzyloxycarbonyl-N'-methyl)amino-2-
(methoxycarbonyl)pyrrolidinocarbonylmethyl)]-amino-
benzophenone.
This product is obtained by the usual methods.
~D = -22.4~(C = 0.37; chloroform).
H) 2-[(2S,4S)-4-(N-Benzyloxycarbonyl-N-methyl)amino-2-
(methoxycarbonyl)pyrrolidinocarbonyl]-5-chloro-3-(2-
chlorophenyl)-l-(3,4-dimethoxyphenylsulfonyl)-3-hydroxy-
indoline, cis isomer.
This product is obtained by cyclizing in the
presence of DBU according to the usual methods. The
crystals formed -are crystallized from DCM/isopropyl
ether.
M.p. = 129~C
~ D = -129~(c = 0.321; chloroform).
The isomeric p~rity by HPLC is 99~.
The compounds prepared in Examples 109 and 110 are
used to prepare the compounds according to the invention
described in Table 5 below:
Table 5
Cl~ OH~
S ~2 (2S)\~
COOCH,
~'OCH3
OCH3
Example Rg ~D t chloroform)
111 cis -NH2 -189.6
c = 0.4
112 cis -NHCOOCH2 ~ \ c - 0.24
113 cis -NHCH3 -152.6
c = 0.28
114 cis -191
-N(CH3)2 c = 0.19
The compound of Example 112 makes it possible to
successively prepare the compounds of Examples 115 and
116 described in Table 6 below and the compound of
Example 114 makes it possible to prepare the compound of
Example 116a.
Table 6
Cl ~ ~C
~~2 (2S ~
CONH2
OCH3
OCH3
- 74 -
Example Rg ~D(chloroform)
115 cis /~ 151
-NHCOOCH2 ~ \ c = 0.27
116 cis -NH2 -161.4
c = 0.26
116a cis -NtCH3)2
The compound of Example 116a can be prepared either
by conversion of the compound of Example 114, or from
(2S,4S)-(N-Boc)-4-(dimethylamino)prolinamide, the
preparation of which is carried out as follows:
1) Methyl (2S,4S)-(N-Boc)-4-aminoprolinate is
prepared from methyl (2S,4S)-4-azidoprolinate according
to T.R. Webl in J. Org. Chem., 1991, 56, 3009.
2) Methyl (2S,4S)-(N-Boc)-4-(dimethylamino)-
prolinate.
4 g of the compound prepared in 1) are dissolved in50 ml of acetonitrile, 12.8 ml of30~ formalin are added
and then, over 5 minutes, 3 g of sodium cyanoborohydride.
After the reactants have been in contact for 2 hours,
acetic acid is added to bring the solution to a pH of 6.
After 3 hours, the acetonitrile is evaporated, water,
potassium carbonate and solid sodium chloride are added
and the mixture is extracted with 4 volumes of ethyl
acetate. The organic phase is evaporated, the residue is
dissolved in 1 N hydrochloric acid and extraction is
carried out with AcOEt. Solid sodium carbonate and then
solid sodium chloride are added to the aqueous phase and
extraction is carried out with AcOEt. After evaporating,
the residue is chromatographed on silica gel by eluting
with a DCM/MeOH (95/5; v/v) mixture and an oil which
solidifies is isolated.
m = 2.1 g
IR (DCM): 1755 cm~1, 1695 cm1.
~ J~ s
3) 534 mg of the ester prepared in 2) are dissolved
in 4 ml of MeOH and are treated with sodium hydroxide
(116 mg) in 1 ml of water for 48 hours at RT. The mixture
is acidified with 0.5 N hydrochloric acid to a pH of 3.5
and is evaporated to dryness. An azeotropic drying of the
residue is carried out in the presence of benzene (5
times) and then the residue is dried under vacuum for 8
hours. 2 ml of DMF and 3 ml of DCM are then added and the
mixture is cooled to 0~C. 865 mg of BOP, and DIPEA, are
added to bring the reaction mixture to neutrality. After
minutes, a stream of gaseous ammonia is bubbled
through twice for 30 minutes. After 2 hours at RT, the
DCM is evaporated, carbonated water and sodium chloride
are added and the mixture is extracted with 4 volumes of
AcOEt. After evaporating, the residue is chromatographed
on silica. The mixture (DCM/MeOH/NH40H; 84.5/15/0.5;
v/v/v) elutes a solid (m = ~85~g) which is recrystal-
lized from a DCM/isopropyl ether mixture.
M.p. = 183-186~C.
~D5 = - 63.1~(c = 0.24; chloroform).
EXAMPLE 117
Decarboxylation of N-(2-carb~xyethyl)-N-ethyl-3-(2-
chlorophenyl)-5-chloro-l-(3~4-dimethoxyphenylsulfonyl)
3-hydroxy-2-indolinecarboxamide, cis isomer.
630 mg of the compound prepared in Example 41 are
placed in solution in 20 ml of THF under an argon
atmosphere and then 101 mg of N-methylmorpholine at -15~C
and 118 mg of isobutyl chloroformate are added. After
stirring for 5 minutes, 127 mg of N-hydroxy-2-
pyridinethione and 101 mg of TFA are added, the mixture
is held at -15~C with stirring for 15 minutes, 900 mg of
tert-butylmercaptan are then added and the mixture is
left to return to RT. The reaction mixture is then
irradiated for 1 hour 30 with a tungsten filament lamp
(150 watts). The mixture is concentrated, taken up in
water, extracted with DCM and the extract dried and
concentrated. The residue is chromatographed on silica by
eluting with DCM/AcOEt (95/5; v/v). The expected product
~3~
_76
is obtained.
m = 300 mg
M.p. = 215 C.
This compound is similar to that of Example 125
described in the European Patent Application EP 469984.
It has the cis configuration around the 2,3 bond of the
indoline as in the starting material.
EXAMPLE 118
Decarboxylation of 2-((2R)-2-(carboxymethyl)pyrrolidino-
carbonyl)-S-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxy-
phenylsulfonyl)-3-hydroxyindoline, cis isomer.
The operation is carried out as in the preceding
example from the compound described in Example 102.
The product obtained is recrystallized from a
DCM/isopropyl ether mixture.
M.p. = 215-220~C
QD = -214.S~ (c = 0.2; chloroform).
This compound is 2-((2S)-2-methylpyrrolidino-
carbonyl)-5-chloro-3-(2-chlorophenyl)-l-(3~4-dimeth
phenylsulfonyl)-3-hydroxyindoline, cis isomer~
EXAMPLE 119
Decarboxylation of 2-(2-carbox~yLlolidinocarbonyl)-
S-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxyphenyl-
sulfonyl)-3-hydroxyindoline, cis isomer.
The operation is carried out as in the preceding
example by using the compound prepared in Example 28 as
the starting material. The product obtained is recrystal-
lized from an isopropyl ether/DCM mixture.
M.p. = 263~C
QD = -201.5~ (c = 0.2; chloroform).
This compound is 2-pyrrolidinocarbonyl-S-chloro-3-
(2-chlorophenyl)-1-(3,4-dimethoxyphenylsulfonyl)-3-
hydroxyindoline, cis isomer.