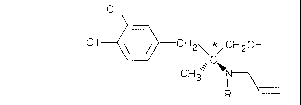Note: Descriptions are shown in the official language in which they were submitted.
- 1 - 20955~9
NEW ENANTIOMERS DERIVED FROM
(S)-2-AMINO-2-(3,4-DICHLOROBENZYL)-l-PROPANOL
The present invention relates to new compounds
derived from (S)-2-amino-2-(3,4-dichlorobenzyl)-1-pro-
panol, to a process for preparing them and to theirtherapeutic application.
In the European patent published under
No. 0,237,366, a description is given of amino alcohols
of formula:
i, I
_
''` I ~ C H 2 ~ !, H 2 OH
R~ R
in which: l-i2
Rl is lower alkyl,
R2 is H or lower alkyl,
R3 is H, lower alkyl, lower alkenyl, phenyl(lower alkyl)
or lower cycloalkylalkyl having from 3 to 6 carbon atoms
in the ring, or R2 and R3, together with the nitrogen atom
to which they are attached, form a saturated five- to
seven-membered heterocycle which can comprise, as a
second hetero atom not directly attached to the nitrogen
atom, an oxygen, a sulfur or a nitrogen, it being poss-
ible for this latter nitrogen hetero atom to bear a Cl to
C4 alkyl substituent, the racemic or optically active
forms of these amino alcohols and their addition salts
with acids.
The products of this invention show both psycho-
tropic and analgesic properties which are especially
suitable for the treatment of psychopathological and
neuropathological disorders, as well as for painful
syndromes of diverse etiologies.
In point of fact, it has now been found that, in
the set of particular compounds covered by the general
formula of the European invention No. 0,237,366, enantio-
0 9
-- 2
mers of (S) configuration which are new, since they were
not synthesized in this patent, possess unexpected
antihistaminic and antl-allergic properties which are
especially suitable for the treatment of symptomatologies
S caused by various allergens, and in particular mediated
by histamine release.
The present invention relates, by way of new
products, to the enantiomers of (S) configuration,
according to the rule established by Cahn-Ingold-Prelog,
which correspond to the formula I
C I
C1 ~`\\ ,~\; CH2\,~,CH20H
C! 13 ~N ,
( I ) R
in which R is hydrogen or a methyl radical, to their
addition salts with non-toxic organic or inorganic acids
and to their use as an anti-allergic, and in particular
antihistaminic, medicinal product.
The addition salts are those obtained with
therapeutically acceptable acids, among which there may
be mentioned, as examples, acetic, benzenesulfonic,
camphorsulfonic, citric, ethanesulfonic, fumaric, hydro-
bromic, hydrochloric, lactic, maleic, malic, methane-
sulfonic, nitric, pamoic, phosphoric, salicylic, stearic,
succinic, sulfuric and tartaric acids.
The preparation of the salts is carried out by
methods familiar to a person skilled in the art, such as
the addition reaction of the base of the product (I) with
one of the acids, in solution in a water-miscible solvent
such as ethanol or acetone or a water-immiscible solvent
such as chloroform or dichloromethane, and then to
separate the salt formed by concentration and/or cooling
of the reaction medium, or alternatively by precipitation
of the salt with a solvent in which it is insoluble, such
as, for example, diethyl ether.
The invention also relates to a process for
preparing the compounds of the invention (I), which
2~3~a ~9
-- 3
consists either in resolving thelr corresponding
racemates which are described in European Patent
No. 0,237,366, or in carrying out the chemical synthesis
from precursor enantiomers of (S) configuration.
The process employing resolution of the racemic
compounds consists in using an optically active acid to
obtain, with the racemic compound treated, either two
diastereoisomeric addition salts or two optically active
esters, which are separated and Erom which the enantiomer
of (S) configuration which is the subject of the inven-
tion and which is also termed "eutomer" in the text, and
moreover the antipode of (R) configuration which is
considered to be inactive and designated "distomer" in
the text, are generated by a suitable treatment. It is
also possible to use a method of direct resolution of the
racemic compound by high performance liquid chroma-
tography (HPLC). More specifically, these preparations
consist in the following:
a) as regards the process of resolution by means of
diastereoisomeric salts, this consists in reacting the
racemic compound, comprising the mixture of eutomer and
distomer in equal parts, in a suitable solvent with the
optically active acid to form two diastereoisomeric
salts, which are separated by their difference in solubi-
lity in the solvent used. Under these conditions, thediastereoisomer of lower solubility precipitates and is
isolated by filtration.
The separated salts obtained may be recrystal-
lized in selective solvents to obtain a satisfactory
optical purity, or else treated, generally in a basic
medium, to regenerate the respective enantiomeric amino
alcohols. The latter can then be purified by recrystal-
lization, or alternatively subjected to a second step of
purification by a fresh formation of diastereoisomeric
salts, this optionally being done with an optically
active acid different from that used before.
The enantiomers of acids commonly used for the
preparation of diastereoisomeric salts according to this
process are, to mention non-limiting examples, those of
_ 4 _ 209~9
~-phenylglycine, ~-phenylalanine and their N-carboxylated
derivatives, of malic acid, mandelic acid, tartaric acid
and their esterification derivatives with acids, or
alternatively camphanic acid, 3-bromo-8-camphorsulfonic
acid and its positional isomers, and ~-methoxy-~-tri-
fluoromethylphenylacetic acid.
The enantiomers of these acids are commercially
available and their use is well known to a person skilled
in the art, thereby making them suitable for use in the
resolution of racemates comprising the eutomers of
formula (I) by preparation and then separation of dia-
stereoisomeric salts, or for the preparation of dia-
stereoisomers for purification of a product previously
enriched with respect to a eutomer (I) which is the
subject of the invention.
The preparation of the diastereoisomeric salts
consists in reacting, per mole of amino alcohol to be
separated or purified, from 0.25 to 2.00 mol of the
enantiomer of an acid in solution in a solvent or a
mixture of solvents which are preferably completely or
partially miscible with water, and which are usually
chosen from alcohols, ketones, ethers of low molecular
weight or alternatively acetonitrile.
Preferably, the reaction is performed in solution
in ethanol, methanol or acetone, by reacting from 0.5 to
1.25 mol of the enantiomer of an acid per mole of product
to be treated, at a temperature between 20C and the
boiling point of the solvent employed, this being gene-
rally preferred. Salification is complete after a period
of between 5 minutes and 3 hours. After this, the
reaction mixture is left, first at room temperature and
then, where appropriate, at about 0C, in order to
crystallize the less soluble diastereoisomer, which
crystallization may be promoted by seeding with a few
crystals of the expected salt if it has been prepared
already. On completion of crystallization, or at the
point considered expedient for obtaining the desired
optical purity, the crystallized diastereoisomer is
separated by filtration. The two phases each containing
s 2~95~09
a more or less purified diastereoisomer are treated
separately, either for purification of the diast~reo-
isomer, or for liberation of the enantiomeric amino
alcohol from its salt, this being carried out by an
alkaline treatment in an aqueous medium followed by
filtration of the enantiomer liberated, or alternatively
extraction with a water-immiscible organic solvent.
b) alternatively, the resolution of the racemic compound
or of the mixture comprising the eutomer (I) consists of
an esterification with an optically active carboxylic
acid to obtain a mixture of diastereoisomeric esters,
which are separated and then saponified to obtain the
separate enantiomers of the amino alcohol, including the
eutomer (I) and the corresponding distomeric antipode.
According to this process, the racemic or par-
tially enriched compound is subjected to an esterifi-
cation reaction with an optically pure carboxylic acid or
one of its derivatives such as a halide, an anhydride or
an ester. The (+) and (-) enantiomers of ~-methoxy-~-
trifluoromethylphenylacetic acid and their derivatives,
for example, are used. The reaction is carried out in an
inert solvent which may be chosen from toluene, tetra-
hydrofuran or alternatively dichloromethane or chloro-
form, and optionally in the presence of an organic or
inorganic base whose role is to catalyze the reaction
when the optically active acid derivative employed is an
ester, or alternatively to neutralize the acidic by-
products formed by the reaction. The catalysts are
favorably chosen from sodium and its alcoholates such as
sodium methylate, which is preferred, or alternatively
from metal alcoholates such as those of aluminum. The
bases used for the neutralization can be inorganic, such
as alkali metal or alkaline earth metal hydroxides or
else the salts of these metals, among which sodium
carbonate is preferred, or alternatively organic, for
example amines such as trialkylamines, N,N-dialkyl-
anilines or aromatic amines. Among these amines, tri-
ethylamine and pyridine are preferred.
The diastereoisomers obtained after reaction are
209~50~
-- 6
separated and purified by methods familiar to a person
skilled in the art, such as fractional crystallization or
a chromatographic separation method, and they are then
hydrolyzed under the action of a strong base such as an
alkali metal hydroxide and usually in an aqueous-
alcoholic medium so as to obtain the separated and
purified enantiomers of amino alcohols, and more
especially, in the context of the invention, the eutomer
of (S) configuration of formula (I).
c) the resolution of the racemic compound or the puri-
fication of a mixture enriched with respect to a eutomer
(I) by high performance liquid chromatography is carried
out by separation of the enantiomers on a column contain-
ing as stationary phase an acid glycoprotein, termed
~I-AGP, immobilized on a porous silica filler the spheres
of which have an average diameter of 5 ~m - CHIRAL-AGP
(R) columns - (Company Chrom Tech).
The mobile phase used for selective elution is a
mixture of pH 6 buffer solution (0.01M KH2PO4 solution
adjusted to pH 6 with N KOH) and a water-miscible polar
solvent which is chosen from aliphatic alcohols compris-
ing up to three carbon atoms and acetonitrile. The latter
solvent and isopropanol are preferred; they are mixed
with the buffer solution in the proportion of 0.1 to
25% (v/v) to elute the enantiomers from the stationary
phase. The elution is carried out under conditions suited
to the good separation of the isomers, namely a tempera-
ture of between 10 and 40C and a flow rate compatible
with the resistance of the material used but which can
reach the point of producing a pressure of the order of
107 Pa.
However, the preferred methods for preparing the
enantiomers of (S) configuration (I) are those employing
precursor enantiomers of (S) configuration according to
the rule established by Cahn-Ingold-Prelog.
The preferred process consists in reacting an
allyl halide with a precursor amino alcohol of (S)
configuration, of formula:
-7- 209~a9
,, I
i
'( H2` ~ ,0
CH ~ ~NH
( I I ) R
An alternative process consists in reducing, with
a complex hydride derived from boron or from aluminum,
the carbonyl function of a precursor amino ester of (S)
configuration, of formula
C I ~ CH2\ ~COO - R
CH3~ ~N~
( I I I ) R
in which R is hydrogen or methyl and Rl is hydrogen or a
lower alkyl radical such as methyl or ethyl.
As regards the (S) configuration of the compounds
of the invention and the precursors (II) and (III), this
was assigned by analogy to that of a precursor which is
common to them, methyl (S)-(-)-3-(3,4-dichlorophenyl)-
2-methylalaninate of formula (IV)
C I
C I j , CH2~ ~,COO- CH3
C
CH3 NH2
( IV)
the enantioselective synthesis of which was carried out
according to the process described by U.Schollkopf
(Synthesis, p. 969, 1981), and which consists in alkylat-
ing (2R,5SR)-(-)-2,5-dihydro-3,6-dimethoxy-2-isopropyl-5-
methylpyrazine (Merck product - ref. 818314) with 3,4-di-
chlorobenzyl chloride, and then in carrying out an acidhydrolysis of the (2R,5S)-5-(3,4-dichlorobenzyl)-2,5-di-
2 0 ~
-- 8
hydro-3,6-dimethoxy-2-isopropy:L-5-methylpyrazine to
obtain the methyl ester of formula (IV) which, on
N-alkylation with an allyl halide, yields the precursor
(III) in which R is hydrogen, or on reduction with an
organometallic hydride yields the precursor (II) in which
R is hydrogen or methyl, to obtain a eutomer (I).
The process for preparing the compounds (I) of
the invention which is especially preferred consists in:
- reacting an allyl halide with an amino alcohol (II) in
which R is hydrogen, to prepare a eutomer (I) of the
invention in which R represents hydrogen,
- and then carrying out a methylation of this eutomer (I)
with formaldehyde and formic acid to obtain the eutomer
(I) of the invention in which R represents a methyl
radical.
The implementation of this process consists in:
- reacting (S)-(-)-2-amino-2-(3,4-dichlorobenzyl)-1-pro-
panol, a compound described in Example lC of the European
Patent No. 0,237,366, with an allyl halide such as
chloride or iodide or, preferably, allyl bromide, in an
inert solvent such as toluene or acetonitrile. Option-
ally, a basic agent, inorganic such as sodium carbonate
or else organic such as triethylamine, is added to the
reaction medium in order to promote the reaction. In
practice, from 0.5 to 1.5 mol of allyl halide is used per
mole of amino alcohol employed, the reaction being
carried out in solution in 2 to 3 liters of the chosen
solvent. Depending on the solvent and the halide used, a
satisfactory result is obtained after a reaction period
of between 1 and 24 hours for temperature conditions of
between 20 and 110C. The preferred conditions, using
allyl bromide, are from 2 to 5 hours for a reaction
temperature of between 40 and 100C, the amino alcohol
(I) obtained being isolated and purified by familiar
methods, in particular by fractional crystallization, and
then by carrying out the N-methylation of this compound
(I) obtained as described above.
To this end, a reductive methylation reaction is
carried out with an aqueous formaldehyde solution and
9 2~3~3~33
formic acid according to the Eschweiler-Clarke process.
Per mole of amino alcohol (I) to be N-methylated,
a quantity of aqueous solution corresponding to 1.2 to
3.5 mol of pure formaldehyde and from 2 to 5 mol of pure
formic acid is employed.
In practice, the formaldehyde is added first to
the product, and then, after an exothermic reaction,
addition of the formic acid is performed and the reduc-
tion reaction is carried out by heating the mixture for
1 to 2 hours at 90-100C.
After treatments, the eutomer (I) in which R is
methyl is purified by conventional, in particular chro-
matographic, methods.
As regards the preparation of (S)-(-)-2-amino-
2-(3,4-dichlorobenzyl)-1-propanol, the state of the art
is taught in French Patent No. 2,378,746, Example 2-d,
for the synthesis of levorotatory 1-3-(3,4-dichloro-
phenyl)-2-methylalanine in 5 steps from methyl 2-iso-
cyanopropionate and 3,4-dichlorobenzyl bromide, and
furthermore, in European Patent No. 0,237,366, Example
l-C, where the reduction of this acid with the borane-
dimethyl sulfide complex is taught.
Advantageously, compared to this preparation
which comprises six steps in all, the preferred process
for the purposes of the invention, which is faster and
more economical, enables (S)-(-)-2-amino-2-(3,4-dichloro-
benzyl)-l-propanol to be prepared in four synthesis steps
from 3,4-dichlorophenylacetone. Essentially, the process
consists in reacting the ketone by the Bucherer-Berg
reaction with potassium cyanide and ammonium carbonate to
obtain (+/-)-5-(3,4-dichlorobenzyl)-5-methylhydantoin,
which is subjected in an alkaline aqueous-acetone medium
to a stereospecific salification with (R)-(+)-~-methyl-
benzylamine so as to obtain the addition salt thereof
with the (S)-(-) enantiomer of the hydantoin, which,
being insoluble, is filtered off, and from which (S)-(-)-
5-(3,4-dichlorobenzyl)-5-methylhydantoin is obtained in
a state of optical purity greater than 98% by treatment
in an acid solution.
203~
-- 10 --
The (S) enantiomer of this hydantoin is hydroly-
zed in a conventional manner in an alkaline aqueous
medium to obtain (S)-(-)-2-(3,4-dichlorobenzyl)alanine,
which is reduced as described in Example 1 of European
Patent No. 0,237,366 with the borane-dimethyl sulfide
complex to obtain (S)-(-)-2-amino-2-(3,4-dichlorobenzyl)-
1-propanol.
These preferred preparation processes are illus-
trated by the preparation and the examples described
below.
Preparation: (S)-(-)-2-amino-2-(3,4-dichlorobenzyl)-
1-propanol [(II); R = H]
- Stage 1: (+/-)-5-(3,4-dichlorobenzyl)-5-methylhydantoin
160.0 g (0.79 mol) of 3,4-dichlorophenylacetone
in 700 ml of ethanol are introduced into a reactor, and
the mixture is heated to 40C to obtain dissolution.
53.0 g (0.81 mol) of potassium cyanide, 149.0 g of
ammonium sesquicarbonate and 700 ml of water are then
added.
The mixture is heated with stirring to a tempera-
ture of between 65 and 70C for 15 hours. The suspension
is cooled with stirring to 10-15C and left for 16 hours
at this temperature. The insoluble matter is filtered off
and washed with water and then with diisopropyl ether
before being dried under vacuum at 50C to constant
weight.
Weight obtained 155.0 g Yld 72% M.p. 240C
- Stage 2: (S)-(-)-5-(3,4-dichlorobenzyl)-5-methyl-
hydantoin
100 ml of demineralized water are mixed with
150 ml of acetone in a reactor set up in the reflux
position, and 3.7 g (0.091 mol) of sodium hydroxide
pellets are then added.
50.0 g (0.183 mol) of the racemic hydantoin
35 obtained in the preceding stage and 22.2 g (0.183 mol) of
(R)-(+)-~-methylbenzylamine are then added successively.
With stirring, the mixture is brought to reflux
until the reactants have dissolved. 100 ml of water are
then introduced while heating the mixture gradually to
- 11- 2o9!~.rjQ~
70C, and the solution is thereafter cooled slowly with
stirring.
Crystallization starts to be seen at about 60C,
and the mixture is then maintained for 1 h 30 min at 50C
S in order to complete the crystallization and thereafter
cooled to approximately 20C for 30 minutes.
The crystalline precipitate is filtered off and
washed with twice 100 ml of a 2:1 (vol/vol) acetone/water
mixture.
The insoluble matter is suspended without further
treatment in 250 ml of water, and the mixture is acidi-
fied to pH 1 by the gradual addition of concentrated
hydrochloric acid solution (d = 1.18).
The insoluble matter is filtered off and washed
with water. The optical purity of the product, determined
by HPLC on an aliquot fraction, is 98.6~.
The wet product is recrystallized under reflux in
200 ml of a 1:1 (vol/vol) water/acetone mixture.
After cooling to 15C for two hours, the
insoluble matter is filtered off, washed with an
acetone/water mixture and then dried under vacuum at
50C
18.79 g of S-(-)-5-(3,4-dichlorobenzyl)-5-methyl-
hydantoin are obtained.
Yield = 75~ Optical purity = 100% (HPLC)
M.p. 276C [~]Do = 28.4 (c = 1, EtOH)
- Stage 3: (S)-(-)-2-(3,4-dichlorobenzyl)alanine
4.7 g (115 mmol) of sodium hydroxide pellets are
dissolved in 25 ml of water in an autoclave. 7.0 g
(25.6 mmol) of the (S)-(-)-hydantoin prepared in Stage 2
above are added. After the apparatus is hermetically
sealed, the mixture is heated with stirring for 8 hours
to 140-145C under a pressure of 3 to 4 bars.
After cooling to 20-25C, the chestnut-brown
solution which is obtained is acidified with stirring
with concentrated hydrochloric acid solution to
pH 5.8-6.2. After cooling to 10C for 30 minutes, the
insoluble matter is filtered off, washed with water and
then with toluene and dried under vacuum at 80C.
2 Q 9 ~ i Q ~
- 12 -
Weight = 5.5 g Yld = 86%
[~]20 = _ 6~9O (c = 1.5, CH30H)
- Stage 4: (S)-(-)-2-amino-2-(3,4-dichlorobenzyl)-1-pro-
panol
62.5 ml of tetrahydrofuran (THF) and then 2.48 g
(10 mmol) of (S)-(-)-2-(3,4-dichlorobenzyl)-alanine are
introduced into a reactor protected from moisture and
under a nitrogen atmosphere. 3.80 g (50 mmol) of borane-
dimethyl sulfide complex (BMS) are added dropwise, in the
course of 30 minutes and at a temperature of 20C to the
suspension obtained. Stirring is maintained for 15
minutes at room temperature and the mixture is then
heated to reflux for 4 hours 30 minutes. After cooling to
5C, 7.5 ml of methanol are introduced gradually without
exceeding 20C, followed, in an identical manner, by 7.5
ml of N sodium hydroxide solution. The suspension ob-
tained is left overnight, and the insoluble matter is
filtered off and removed. The filtrate, evaporated under
vacuum and on a water bath, gives a white residue which
is taken up with 50 ml of water. The mixture is acidified
to pH 1 by adding concentrated hydrochloric acid (d =
1.18).
The solution obtained is extracted with twice
15 ml of ether. The ether phases are discarded, and the
acid phase is alkalinized in the cold state to pH 12 by
adding concentrated sodium hydroxide solution (d = 1.38)
and then saturated with sodium chloride.
The alkaline mixture is extracted with 3 times 20
ml of ether, and the combined ether phases are washed
with saturated sodium chloride solution and then dried.
After evaporation of the ether, the amino alcohol
is obtained in the form of a white and amorphous solid
residue.
Weight 2.10 g M.p. 86C Yld = 90%
35 [~]20 = _ 1.8 (c = 5; CH30H [lacuna~
Example 1 - (S)-(-)-N-allyl-2-amino-2-(3 4-dichloro-
benzyl!-1-propanol [(I); R = H]
35.0 g (0.15 mol) of (S)-(-)-2-amino-2-(3,4-di-
chlorobenzyl-1-propanol, [~]20 = -1.8 (c = 5, methanol),
2 ~ a s
- 13 -
are dissolved in 350 ml of anhydrous acetonitrile in a
reactor protected from moisture.
12.9 ml (18.08 g - 0.15 mol) of allyl bromide are
added to the solution with stirring at 20-25C and in the
course of approximately 5 minutes. The solution is kept
stirring for one hour at the same temperature and then
heated gradually to 50-60C. This temperature is main-
tained for two and a half hours, during which period a
precipitate appears. The suspension is then cooled with
a bath of ice-cold water at 5C. The precipitate is then
filtered off, taken up with 800 ml of water, alkalinized
to pH 11 with 10 N sodium hydroxide solution and
extracted with 3 times 250 ml of diethyl ether.
The combined ether phases are extracted with
saturated NaCl solution and then dehydrated over Na2S04.
The ether is evaporated off under vacuum and on a water
bath. The crude solid residue is taken up for dissolution
in 500 ml of hexanes under reflux. After cooling, the
crystalline white precipitate of purified (S)-(-)-N-
20 allyl-2-amino-2-(3,4-dichlorobenzyl)-1-propanol is
filtered off and then brought to constant weight in an
oven at 40C under vacuum.
Weight: 25.4 g M.p. = 87-88C Yld 62%
[~]20 = -12 (c = 0.5, N HCl)5 Note: observed deviation not significant for:
(c = 5, C2H50H) and (c = 1, C2HsOH/N HCl)
- IH NMR (CDCl3 TMS): ~ (ppm) 1.00 (s, 3H); 1.00-2.90
(m, 2H exch. D20); 2.65 (s, 2H); 3.10-3.40 (m, 4H);
5.00-5.35 (m, 2H); 5.65-6.15 (m, lH); 6.90-7.45 (m,
3H).
Example 2 - (S)-(+)-N-allyl-2-methvlamino-2-(3.4-di-
chlorobenzyl)-l-propanol [(I); R = CH3]
13.5 g (49 mmol) of the (S)-N-allylaminopropanol
prepared in Example 1 above are mixed with 13.5 ml of 37%
w/v formaldehyde solution (equivalent to 5.0 g
133 mmol) in a reactor equipped with a powerful stirrer.
The mixture is warmed to about 40-50C to promote
homogenization with stirring, and then cooled to 15C.
2095~
- 14 -
9.3 ml of 100% formic acid (d = 1.22, equivalent to
11.35 g - 247 mmol) are then added dropwise. The clear
solution obtained is maintained for two and a half hours
with stirring on a boiling water bath. After cooling,
200 ml of water are added, and the mixture is acidified
to pH 1 by adding concentrated hydrochloric acid and
extracted with 3 times 100 ml of diethyl ether. The ether
phases are discarded, the acid phase is alkalinized to
pH 11 with 10 N sodium hydroxide solution without exceed-
ing 25C and the mixture is then extracted with 3 times150 ml of ether.
The combined ether phases are washed with NaCl
solution and then dried over Na2SO4. The ether is evapo-
rated off, and the crude residue of 14.0 g is purified
according to the so-called "Chromatoflash" chromato-
graphic technique on a column packed with silica.
Elution with a 95:5 (v/v) dichloromethane/-
methanol mixture enables purified (S)-(+)-N-allyl-2-
methylamino-2-(3,4-dichlorobenzyl)-1-propanol to be
obtained in an oily form, which crystallizes slowly to a
product of low melting point.
Weight = 11.5 g Yld = 81%
[~]20 = + 13 (C = 1, N HCl)
- IH NMR (CDCl3 - TMS): ~ (ppm) 0.95 (s, 3H); 2.30
(s, 3H); 2.70 (s, 2H); 2.95 (s; lH exch.D20); 3.0S-
3.45 (m, 4H); 5.00-S.3S (m, 2H); 5.60-6.0S (m, lH);
6.9S-7.40 (m, 3H)
- HPLC - Determination of the optical purity on
a "Chiral-AGP (R) - supplier Chromtech" column, eluting
with a mixture comprising O.OlM KH2PO4 adjusted to pH 6
and Isopropanol - (v/v)
result: % purity > 98
- Hydrochloride: the eutomer (I) obtained above
is dissolved in 150 ml of dichloromethane. While main-
3S taining a temperature below 2SC, an excess of etherealhydrogen chloride is added to the solution, and the
mixture is then left stirring for 1 hour while protected
from moisture.
The solvents are evaporated off under vacuum and
2 n ~ 3
- 15 -
on a water bath. The solid residue is dissolv~d in the
minimum quantity of methanol at 20-25C, and the purified
hydrochloride is obtained by precipitation after adding
anhydrous diethyl ether.
M.p. 171-172C
- Elemental analysis (Cl4H20Cl3NO = 324.69)
Calculated C 51.79 H 6.21 Cl 32.76 N 4.31 O 4.93
Found C 51.71 H 6.30 Cl 32.72 N 4.20 O 5.22
- IR (KBr): 3250, 2600, 1470, 1400, 1060, 1030, 950,
830 cm~l
The distomeric antipodes of the compounds pre-
sented in Examples 1 and 2 are, respectively:
- (R)-(+)-N-allyl-2-amino-2-(3,4-dichlorobenzyl)-1-
propanol
M.p. 890C [~]Do = + 11 (c = 0.5, N HCl)
- (R)-(-)-N-allyl-2-methylamino-2-(3,4-dichlorobenzyl)-1-
propanol
M.p. < 50C [~]20 = -14 (c = 1, N HCl)
They were prepared according to the procedure described
in the above examples, from (R)-(-)-2-amino-2-(3,4-di-
chlorobenzyl)-l-propanol described in Example 1 B of
European Patent No. 0,237,366.
The low toxicity of the products of the invention
and the unexpected pharmacological properties which have
been discovered justify the value of the compounds in
preventive or curative treatments of allergic mani-
festations, and in particular of those caused by
histamine release.
The acute toxicity of the compounds (I) was
studied orally in male mice. To this end, the products
were administered in aqueous solution on the basis of
2 ml per 100 g of body weight. The animals were observed
during the three hours following administration, and then
daily for 14 days, after which they were sacrificed and
autopsied.
The LD50 values (lethal doses causing the death of
50% of the animals) are in the region of 1,000 mg/kg,
which may be perceived as a relatively low toxicity of
the products.
2~3~
- 16 -
The anti-allergic properties of the eutomers (I)
were demonstrated by their capacity to inhibit, in guinea
pigs sensitized to ovalbumin, an anaphylactic broncho-
constrictor effect produced by an aerosol containing this
same allergen.
The technique performed consists in previously
sensitizing guinea pigs weighing 250 to 300 g by two
intraperitoneal injections, separated by an interval of
24 hours, of 0.5 ml of a sterile suspension of physio-
logical saline (0.9~ NaCl) containing 20 ~g of ovalbumin(Grade V; Sigma) and 100 mg of aluminum hydroxide
Al(OH)3-
At the same time, unsensitized control animalsare treated with two injections of suspension not con-
taining ovalbumin.
The actual experiment is performed after a periodneeded for the sensitization of the animals, which is
between 14 and 21 days. It is generally arrived at after
18 days, and consists in first administering increasing
quantities of the test product in solution in water
orally to groups of 5 animals, on the basis of 0.5 to
1.0 ml per animal, corresponding to doses of 1 to
10 ~mol/kg of product.
The animals are then anesthetized 50 minutes
after this administration by an intraperitoneal injection
of a solution of urethane. They then undergo a surgical
preparation which consists in:
- placing a cannula in the left carotid in order to be
able to determine the blood pressure and heart rate in
continuous fashion,
- placing a cannula in the right jugular vein for the
intravenous administration of solutions,
- after an upper cervical tracheotomy, inserting a
polystyrene cannula (internal diameter 2.65 mm, length 9
to 11 mm) in order to ventilate the animal (Harvard
50-1728 pump; Kent - Great Britain) at a frequency of 60
insufflations per minute and on the basis of a volume of
1.2 ml/kg each.
After this operation, and stabilization of the
2095~9
- 17 -
intratracheal pressure and the b:Lood pressure, an aerosol
of ovalbumin is administered by means of an ultrasonic
nebulizer (Pulmosonic apparatus; model 2511). The appa-
ratus delivers 30 insufflations in 30 seconds via the
tracheal ventilation in a total volume of 30 ~l of an
ovalbumin suspension containing 5 mg/ml. After 60
minutes, the intratracheal pressure measured in mm of Hg
is determined for each animal of the test groups and
those of the control group. These measurements enable the
extent of the inhibition of bronchoconstriction produced
by the test product to be determined. The results are
expressed as the ED50 f the product, which is the effec-
tive dose, expressed in micromols per kg (~mol/kg)
capable of inhibiting 50% of the bronchoconstriction
caused by ovalbumin in the sensitized animal under the
conditions of the test.
Determined under these conditions, the inhibitory
ED50 of the hydrochloride of the eutomer (I) of Example 2
is 1.84 ~mol/kg).
Furthermore, the antihistaminic properties were
studied in guinea pigs by the bronchoconstriction test
performed according to the method of H. Konzett and
R. Rossler (Arch. Exp. Path. Pharmak. - Naunym
Schmiedeberg - 195, 71-74, 1940).
The products of the invention were studied in
this test after administration in solution or in suspen-
sion via the intravenous and intraduodenal routes, and
also in powder form via the intranasal route.
Irrespective of the administration route, the
preparation of the animals employed in the test consists
in first anesthetizing male guinea pigs weighing 350 to
400 g by intraperitoneal (i.p.) injection of a sterile
and isotonic solution containing 25% (w/v.) of ethyl
carbamate on the basis of 6.0 ml/kg, and in then fitting
the animals with a tracheal cannula to permit measurement
of the pulmonary pressure, a cannula in the right jugular
vein for the intravenous (i.v.) administration of solu-
tion, and also a cannula in the left carotid artery for
qualitative observation of the blood pressure.
209550~
- 18 -
The animals are then connected to a pump
(Harvard, ref. 50-1718) in order to maintain an arti-
ficial ventilation at a frequency of 60 insufflations per
minute; the intratracheal pressure is recorded by an
assembly comprising a transducer, an amplifier and a
recorder (Gould, respective refs.: PLOEZ, 13-4615-50 ar.d
8188-G4400-06).
Fitted in this way, the animals are left undis-
turbed for 10 minutes before the beginning of the actual
experiment, which is carried out in different ways
according to the administration route envisaged.
a) i.v. administration
In base form, the test products are dissolved in
an isotonic solution (0.9% NaCl) containing 10% (v/v) of
Transcutol (R). Dilutions are performed by adding
isotonic solution. In salt, in particular hydrochloride,
form, the solutions are prepared in the isotonic
solution.
The treatment of the animals consists of a series
of administrations of histamine dihydrochloride solution
on the basis of 100 ~mol/kg before and after treatment
with the solution of test product, which is carried out
according to the following time schedule:
t = - 20 minutes - histamine (100~mol/kg)
t = - 10 minutes - ditto controi
t = 0 - injection of the test product
t = + 10 minutes - histamine (100 ~mol/kg)
t = + 20 minutes - ditto
t = + 30 minutes - ditto
t = + 40 minutes - ditto
t = + 50 minutes - ditto
t = + 60 minutes - ditto
The results of the tests are presented in
Table 1.
b) i.d. administration
In base form, the test products are suspended in
a solution containing 1% w/v of carboxymethylcellulose
tCMC), and, in salt form, they are dissolved in an
isotonic solution before being administered via a cannula
2ns~ 3
-- 19 --
whose end is placed in the animal's duodenum.
The time schedule for the successive admini-
strations of histamine via the i.v. route and that for
the test solution via the i.d. route are identical to
that described above in a).
The results of these tests are presented in
Table 2.
c) intranasal-powder (In.pl adm:inistration
The test product is administered via a nasal
insufflation of a powder consisting of 20 mg of lactose
loaded with the appropriate quantity of the test product.
Before this insufflation, the artificial ventilation of
the animal is momentarily stopped in order to facilitate
the pulmonary penetration of the compound. The time
schedule for the administration of histamine via the i.v.
route and that of the test product via the In.P. route is
as follows:
t = - 15 minutes - histamine (100 ~mol/kg)
t = - 5 minutes - ditto control
t = 0 minutes - insufflation test product
t = 5 minutes - histamine (100 ~mol/kg)
The results of these tests are presented in
Table 3.
- Calculation and expression of the results:
For each administration route and for each time
of administration of histamine, the activity of the
products is expressed as the percentage change in ampli-
tude of bronchoconstriction, calculated relative to that
produced by the "control" administration of histamine
which precedes the administration of the test product.
The % change is calculated according to the formula:
Bc(tx) - Bo(tx)
% change = 100 - ( x 100)
Bc(to) - Bo(to)
in which:
Bc(tx) represents the amplitude of the bronchocon-
striction in mm at time x,
Bo(tx) the amplitude of the base-line respiration in mm
at time x,
Bc(to) and Bo(to) the corresponding amplitudes in mm on
2~ 3 ~
- 20 -
receipt of the "control" administration of histamine
which precedes that of the test product.
The inhibitory activity of the test products on
histamine-induced bronchoconstriction is expressed at
different times of the studies as their ED50, which is the
effective dose of product, expressed in ~mol/kg, capable
of inhibiting 50% of the histamine-induced bronchocons-
triction at the time in question.
Failing this expression, the results are ex-
pressed as a % change at the time in question and for a
given concentration of the test product. Under these
conditions, a positive percentage corresponds to a
potentiating effect on bronchoconstriction and, con-
versely, a negative percentage to an inhibitory effect.
Statistical analyses of the results were carried
out by the Student t test. A value of p < 0.05 is con-
sidered significant.
The compounds of the invention described in
Examples 1 and 2 were employed in these tests. In
addition, as a comparative product for the eutomer of
Example 1, the racemic (R,S) compound corresponding to
it, and which is described and prepared in Example 7 of
European Patent No. 0,237,366, was employed. As regards
the eutomer of Example 2 and its hydrochloride, the
hydrochloride of the racemic (R,S) compound which is
also the corresponding distomeric antipode of (R) con-
figuration prepared as described above, were employed
comparatively in these tests.
Fenspiride (INN) hydrochloride, known as an
antagonist of allergens which are active on the airways,
and Cromakalim (INN), which is a compound known to be a
bronchodilator, were also employed in these tests as
reference compounds.
- 21- 2~53~
U~ o
I
h .~
_ Il~ ~`1 rt O J~ a
ll U~ ~ rt ~t
J' 1~ ~+ rt I .0
_ I~ ~t
O (` ~ rt rt ~ rt
rt O O 11~ ~ D O .~
t0~1 + ~ Ul
r l O
O CO ~ ~ O C I
ll ~ ~u~om ~ ~ I
J' 1'`'''' '`J + I co rt C
_ t~ ~a I
O d' Cl~ ~ 1 . ~D rt l
ll U~ t~l 1` 11~ 00 CO ~I ~ ~
L~ 1~ I ~t + \D r
l l ~ I
00 0 ~ O ~t ~r a~ U~
CO r-~ a~ t CO ~ ~ a
~ ~ ~r o o ~ + ~r o
X~ ~ I I I .~ I
O~ 1~ 1` CO N t-~Il- rt
rt rt co ~~ ~ 1~ . ~ h I
Oll. . . . . ~D . ~ ~
O~ ~O rt ~t ~ + d' ~ -
_~ I_ l l O ' I
O ~ _ _ o a
1~1 rt _U~ __U~ ~ l S .IJ
~ ' 5
V ~ X
- 22 ~
o
O
--= . ~
O I~N ~O . I ~ a~
l ~ ~ ,o ~
o cor~ ,r~~ I ~ I
L~ L~ ¦ C O + - r
_ ~ I ~ I
o a~Ir) OD~ l l
ll ~O~ I ~ I
1~ ¦~ O + r I
o c~ a~ ~ . .,1 I
~ O O O + .r I
~ ~ Ln r~ ~D ~ l
X _r 1 r-l O + r c
r-1 r-l _ I ~1 + ~ .r1
o t~ ~) r
r1 _ ~ _ _ .C ~
r-l O _ _ ~ _ ~ ~r1
~: ~ o a
O _ :) O ~J D
r1 O L S S ~,1 ~,1 , a
s X ~ oal ~ ,I H U
,1 a ~, ~ s ~ ~
E ~ ,~ I I L~ ~ Xa~ 4
~ _ = ~ 6 o~o ~
~ ~ 9 ~
- 23 -
- test via the in.P. route (Results ED50 ~mol/kg)
Compound Config.ED50 at t - 5
Example 2 of the invention
- base (S) 1.28
5 ¦ - hydrochloride (S) 1.77
¦Cromakalim (INN) 1.88
Table 3: Inhibitory effect via the In.P. (intranasal-
~owder) route on bronchoconstriction caused bY
lOOnmol/k~ of histamine
The results of these tests demonstrate convin-
cingly the inhibitory activity of the compounds of the
invention with respect to ovalbumin- and histamine-
induced bronchoconstriction in guinea pigs.
In addition, they demonstrate the specificity of
the antihistaminic activity of the compounds of
Examples 1 and 2 (S) configuration which are the subject
of the invention.
Indeed, in comparison to their racemic (R,S)
analogs, the compounds of the invention prove ap-
proximately at least twice as active.
Moreover, via the i.v. or In. route, the com-
pounds of the invention show an activity equal to, if not
greater than, the reference compounds with which they are
compared.
More surprisingly, it is found that, as regards
the product of Example 2 of the invention, both via the
i.v. route and via the i.d. route, the corresponding
distomer of (R) configuration shows, perceptibly though
not significantly in statistical terms, a potentiating
effect on histamine activity.
These anti-allergic, and in particular antihista-
minic, properties in the bronchoconstriction test are
seen not to be obvious from the standpoint of the teach-
2095~a~
- 24 -
ing of the state of the art, and to be noteworthy from
the standpoint of the observed dissociation of activity
between the eutomers of formula (I) and their antipodes.
They justify the subject of the invention, which
relates, by way of new products, to the enantiomers tI)
of S configuration which are:
- (S)-(-)-N-allyl-2-amino-2-(3,4-dichlorobenzyl)-1-
propanol,
- S-(+)-N-allyl-2-methylamino-2-(3,4-dichlorobenzyl)-1-
propanol,
and their salts, to a process for preparing them and to
their use in the form of medicinal products intended for
preventive or curative treatments of allergic states, in
particular of those caused by histamine release.
In the form of suitable pharmaceutical com-
positions, the compounds of the invention, as a result of
their anti-allergic activity, are useful in the treatment
of asthmatic states, and in particular for inhibiting the
bronchoconstriction or bronchospasm states of allergic
asthma or of those resulting from acute or chronic
bronchitis. The antihistaminic activity of the compounds
justifies their application in the symptomatologies
caused by histamine release, such as, for example, in
allergies of the nasal and conjunctival mucosae, allergic
rhinitis and certain forms of edema, of dermatosis, of
pruritus or of eczema.
The low toxicity of the products permits the use
of daily dosages of up to 1,000 mg, administered orally,
in order to obtain the expected effects. They are
usually, however, from 50 to 500 mg per day, administered
orally, divided if necessary into several doses.
The products of the invention and their pharma-
ceutically acceptable salts are administered in the form
of compositions appropriate to the routes which are
suited to the nature and extent of the disorder to be
treated.
These compositions are, for example, tablets,
dragées, capsules, powders, suppositories, gels, suspen-
sions or alternatively solutions to be injected or
- 25 - 20~5a~
swallowed.
They are prepared by methods familiar to a person
skilled in the art. They comprise from 1 to 50% by weight
of active principle consisting of a compound (I) or one
of its salts, and from 99 to 50% by weight of a pharma-
ceutically sultable vehicle which is compatible with the
active principles and the physical form of the com-
position envisaged.
As non-limiting examples, the preparation of
tablets and of injectable isotonic solutions with the
compounds of the invention is presented.
Tablets
- Formula
Active substance (I) according to
Example 2 5 to 75 mg
Polyvinylpyrrolidone 2 mg
Carboxymethylstarch 8 mg
Magnesium stearate 3 mg
Lactose 60 to 76 mg
Monocrystalline cellulose 122 to 76 mg
per tablet weighing 200 mg.
- Manufacture
Dissolve the polyvinylpyrrolidone in the pro-
portion of 0.1 to 1.0% by weight in water, a low mole-
2S cular weight alcohol such as ethanol or an aqueous-
alcoholic mixture.
Separately, make an intimate mixture of the
active substance, lactose and half the quantity of
cellulose and of carboxymethylstarch, and wet this
mixture with the solution obtained above.
Granulate the paste, dry the granules and size
them on a sieve. Add the rema~nder of the components, mix
intimately and then tablet on the basis of 200 mg per
unit.
35 In~ectable isotonic solution
- Formula
Active substance (I), hydrochloride of
Example 2 10 mg
Sodium chloride 9 mg
2095~9
- 26 -
Distilled water q.s. 1.0 ml
- Manufacture
The solution is distributed in ampoules, which
are sterilized by the usual thermal means after sealing;
the solution can also be sterilized by filtration and
distributed in ampoules which are then sealed, these
operations being performed in a sterile atmosphere.