Note: Descriptions are shown in the official language in which they were submitted.
WO 92t1 1844 PCr/USsl/09771
~f ~9~76
STABlLIZATlON OF PROTEIMS BY CATIONIC BIOPOLYMERS
Background of the InYention
This inven~ion is in the field of delivery systems forpharmaceutical agents and is especially rela~ed to methods for the
stabili~ation of proteins using cationic polymers.
Sustained release devices have been developed over the past
several years based on a broad range of technologies, directed to the
delivery of a wide selection of pharmaceutical agents. The physical
formats for such devices include use of microparticles, slabs or sirnilar
macroscopic systems designed for implantation, gels and emulsions, and
other preparations conceived to preserve the active agent in the delivery
system for an extended period of time.
The mechanism of release-from matrix-type sustained release
devices is generally understood to occur by hindered diffusion of the active
agent through the carrier matrix, or by erosion of the matl~x over tune
resulting in the liberation of the incorporated active agent. These
processes are not mutually exclusive, and both mechanisms may be
simultaneously active in the case of a given system.
In recent years sustained release devices have been used for the
delivery of protein pharmaceutical agents, primaril;y as a result of the
availability of recombinant proteins which have been developed for
therapeutic applications in a wide variety of pathological conditions.
~evelopment of such systems creates greater challenges to overcome than
in the ca~se of low molecular weight drugs and pharmaceutically active
substances, since prote~ns inherently have only marginal conformational
stability, and can frequently be susceptible to conditions or process~s
which result in inaclivaiion or desla~ration. ;n contrast to the degradation
or deterioration of low molecular weight pharmaceuticals, the struc~ral
alterations in proteins leading to inactivation need not involve changes ~n
the covalent saucture of the protein, but can be entirely the consequence
:
WO 92/11844 pcr/uss1/o977l
..
-2-
3 7 ~
of disruption of an ex~ensive system of noncovalent interactions which are
responsible for the preservation of the nativ~ three dimensional structure of
the protein. This is the basis for the greater lability of proteins.
Certain features of sustained release devices e xacerbate the
poten~l for the inactivation of protein active agents. These include the
fact that large amounts of solid protein are introduced into the delivery
system (either as pure preparations or n~ixed with additives and
excipients), and that the physical attributes of the delivery systems
themselves may present interfaces which promote denaturation. Hydrating
the solid protein under physiological conditions in vivo results in formation
of a protein gel or a highly concentrated solution of the protein. Under
these circumstances it is quite possible for the protein to become
aggregated or denatured due to interacttons with neighboring molecules or
upon exposure to the interface with the delivery system.
In order to overcome these potential problems, prote~lls h~ve been
formulated with excipients intended to stabili~e the protein in the milieu of
the pharmaceutical product. It has long been known that a variety of low
molecular weight compounds have the effect of preserving the activity of
proteins and enzymes in solution. These include simple salts, as described
by P. H. von Hippel and K.-Y. Wong, "Neutral Salts: the Generality of
Their Effects on the Stability of Macromolecular Conformations", Science
145, 577-580 (1964), buffer salts and polyhydroxylated compounds such as
glycerol, mannitol, sucrose and polyethylene glycols, K. Gekko and S. N.
Timasheff, "Mechanism of Protein Stabilization by Glycerol: Preferen~al
Hydra~on in Glycerol-Water Mixtures", Biochemistrv 20, 4667~676
~1981); K. Geldco and T. Morikawa, "Pleferential Hydration of Bovine
Serum Albumin in Polyhydric Alcohol-Water Mix~res", J. Biochem. 90,
39-50 (1981~; and J. C. Lee and L. L. Y. Lee, "Preferential Solvent
Interactions lietween Proteins and Polyethylene Glycols", J. Biol. Chem.
WO 92/l 1844 Pcr/uss1/o977
'5 --3--
2~937~
256, 625-631 (1981). Certain biocompa~ble polymers have also been
applied ~or this purpose, such as various polysaccharides and synthetic
polymers includmg polyvinylpyrrolidone, for example. Even benign
detergents such as polyoxyethylene sorbitan monooleate (Tween 80TM) have
been included to preserve bioactivity in pharmaceutical formulations. Use
of these materials has been irnplemented over many years, for example,
with soluble preparations of vaccines and insulin, long before recombinant
protein pharrnaceutical agents became available.
Except for the detergents, the mechanism by which these
substances exert their stabilizing effect has become evident in recent years
as a result of thorough investigation. It has been shown that stabilization
occurs as a result of a general thersnodynamic phenomenon prevalent in
these ternary systems, wherein the cosolute (for example, the polyol) is
preferentially excluded from the domain of the protein, and the protein is
preferentially hydrated. As a rcsult, the protein is stabilized by
enhancement of the hydrophobic interactions which are generally thought
to confer stability on the native tertiary structure of the protein, as
compared with the protein in the absence of the cosolute.
Use of these excipients may be associated with certain
disadvantages. For example, the thermodynamic effects require high
concentrations of the cosolute in order to be effective. Under certain
conditions, high concentrations of polysaccharides may even lead to phase
separation of the protein. Alternatively, low molecular weight excipients
have high solubilities and high diffusion coefficients, so that they are
depleted from the delivery device considerably more rapidly than the
active agent. The beneficial effects of the excipient are therefore transient,
OCCDg only in the ini~al stages of the dura~ion of ~e release of ~e
protein. This condi~on leaves the protein pharmaceu~dcal still wi~in the
.
wo 92/l 184q PCI/US91/09771
-4-
~i9937~
sustained release device, prone to inactivation due to interrnolecular
aggregahon and interaction with ~e surface of the device.
It is therefore an object of the present invention to enhance the
amount of release and stability of proteins incorporated into polymeric
matrices for controlled drug delivery.
It is a further object of the present invention to provide a method
and compositions that can be used with a variety of compounds to enhance
stability, with minimum effort and expense.
I~ is another object of the present invention to provide a method
and compositions that can be used as biodegra~able, biocompatible depots
for controlled drug delivery.
Summary of the Invention
A method is described for the incorporation of biologically active
agen~s, especially protein ph~maceutical agen~, in ~ fonn of specific
noncovalent complexes with polycationic reagents, into sustained release
systems, where the polycation stabilizes the protein against inactivation
while it resides in the delivery device, and retards release of the protein
due to the added effects of dissociation of the complex according to the
law of mass action. The end result is the release of the active agent with
retention of biological activity, with a high cumulative yield, over a
sustained period of time.
In a second embodiment of this method and compositions, the
polycation-protein complex itself serves as a depot for release of the
prote-in active agent, rather than a polymeric ma~c. ~ the most prefe~ed
embodirnent, the complexing polyelectrolyte is both biocompatible and
biodegradable.
', , " ~ , ..
~ . .; .
wo 92/11844 PCr/US91/09771
-5- ~9~76
Examples are provided demonstrating complex forma~on (for
example, between erythropoiehn and chitosan) and enhanced stabili~y and
release from polymeric devices of proteins (such as Factor Vl~.
Brief Description oP the Drawil}gs
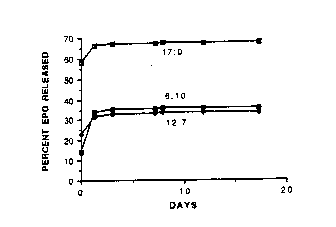
Figure 1 is the percent ery~ropoietin (EPO) released from
poly(DL-Iactide-co-glycolide) (50.50) microspheres in 50 mM sodium
phosphate pH 7.3 at 37C, for EPO:chitosan ratios, expressed as percents
of total solids, of 12:7, 6:10 and 17:0 over time (days).
Figure 2 is a graph of the cumulative units of Factor vm activity
released per mg of poly(lactic acid) microspheres, containing either
poly(arginine) (2 mg/ml) complexed with Factor VIII at 30% loading, over
time (days) or Factor vm in NaCl-CaCl2-glycine buffer.
Figure 3 is a graph of the percent cumulative release over time
(days) for bov~ne serum albumin (BSA):sucrose (5:5) ~ight squa~es);
BSA:protamine (5:5) (~iangles); and BSA (dark squares), all at 10% by
weight loading.
Detailed Description of the Invention
The majority of the prior art processes and phenomena relating to
stabilit~ and release of compounds from polymeric matrices is based on
general physical chemical principles, except for the process of erosion of
sustained release systems, which involves actual che nical degradation of
the matr~x. The method and compositions described herein, in contrast,
are based on a reversible chemical ~nteraction between the compound to be
released and a stabilizing compound.
In the preferred embodiment, the bido~cally ac~ve agent is a
protein or peptide (including nahlral, recombinant, synthetic, high and low
molecular weight proteins or peptides). It could also be a nucleic acid, a
'
wo 92/11844 PCr/US91/09771
-
-6- 2 ~ ~ ~ 3 7 6
polysaccharide, a carbohydrate or derivatives thereof, a low molecular
weight organic molecule or pharmacological agent. Complex formation
between proteins and biological polycations can be used for proteins whose
isoelectric point (pI) is acidic or neutral, as well as any protein having
acidic side chains clustered together on the surface of the protein when it
is in its native, active confo~nation. Proteins with acidic or neutral pI
values have a preponderance of acidic over basic side chains in ~heir
structures. These are the groups which are available for interaction with
the polycation, primarily by electrostatic interactions. The polycation has
the capability of binding several molecules of protein per molecule of
polycation. If the pxotein is also polyvalent in binding sites for the
polycation, the complex will likely aggregate or precipitate, in analogy ~o
the antigen-antibody precipitin reaction. If the protein is monovalent for
the polycation the complex will remain soluble, presumably as a complex
compAsed of many prot~ mole~:ules bound to each polyca~ion molecule.
The complexed protein is stabilized relative to the case of the absence of
the polycation, both in a~ueous solution or suspension, and when
incoIporated into sustained release devices.
The polycation must be biocompatible and, preferably,
biodegradable. A variety of polycations can be used. Simple polyamino
acids such as poly~ysine) or poly(arginine) are useful materials. Their
molecular weights should be 4,000 daltons or greater, preferably about
50,000 or greater. Protamine is another useful polycation. Chitosan is
useful primar~y for acidic proteins, since it precipitates at pH values
greater than about 6.5. Other biological polycations are also applicable for
the purposes of this inven~on.
The weight ra~o of protein to polycation can be in the range
l:1000 (when the protein has a very high biological ac~vity per un~t
weight, so that the overall dosing requirement is low) to 20:1 (in the
wo 92/11844 pcr/ussl/09771
1<'~.~
-7-
209~37~
converse situation). The preferred range for the weight ratio will be l:100
to 10:1. The pH at which the complex is fonned will affect the process.
The overall state of charge of the protein will be a ~nction of pH, since
proteins are polyampholytes. The pH must be one at which the protein
retains full biological activity, which is a property unique to each protein.
The pH may also affect the charge on the polycation in certain cases, or,
as with chitosan, actually affect its solubility. Of course, once in~oduced
in vivo, release devices incorporating these complexes will experience pH
values approxima~ng physiological pH.
The fabrication of sustained release systems containing protein-
polycation complexes differs little from the processes currently used for
incorporating protein formulations. Liquid formulations can be employed
in the manufacture of sustained release microspheres in conventianal
solvent evaporation procedures. Solid formulations, typically prepared as
lyophilized solids from the li~uid, can also be used. In particul~, solid
preparations of protein-polycation formulations can be micronized, i.e.,
fragmented to produce particles in the size ~nge from less ~an I
micrometer to about 5 micrometers, using the procedures outlined by
Gombotz, et al., in U.S. Serial No. 07/345,684 filed May 1, 1989, the
teachings of which are .incorporated herein, summarized as follows.
The biologically active molecule is first dissolved in a solvent that
can be lyophilized to forrn a solution having a concentration ranging from
approxirnately 0.1 to 25% (w/v). The solvent rnay be pure water or can
be buffered to a parLicular pH or ionic strength. The solvent may also be
organic. The solution may contain the biologically active molecule alone,
n~ix~res of two or more Iypes of biologically active molecules alone,
mixtures of biologically active molecules and stabilizers, or any
combination thereof. ~ order to reduce the particle size of these
preparations to the greatest extent, the composition should be suspended in
. . , -
W0 92/11844 PCr/US91/09771
~ .
2~37~
a medium ~n which not only the solvent but also the buffer s:alts are
vola~le under conditions of lyophilization. Examples of buffers removed
by lyophilization include ammonium bicarbonate and other vola~le
ammonium salts.
The soluhon is then atomized into a low temperature liquified gas
using any one of several devices, such as ultrasonic nozzles, pressure
nozzles, pneumatic nozzles and rotary nozzles. The liquified gas can be
liquid argon (-185.6C~, liquid nitrogen (-195.8C), liquid oxygen (-
182.9C) or any other gas that results in the immediate freezing of the
atomized particles into frozen particles. Oxygen is not preferred for
proteins since it is explosive and may also cause oxidation of the proteirl.
The liquified gas is removed by evaporation at a temperature at
which the solvent remains frozen, leaving behind frozen particles. The
frozen solvent is removed from the particles by Iyophilization to yield
porous par~c!es. These particles can ~ary in dia;ncter dcpcnding on the
technique used for their aton~zation, but generally range from
approximately 10 to 50 micrometers.
These protein particles can be incorporated into biodegradable
polymer microspheres using the processes taught by Gombotz, et al., U.S.
Serial No. 07/346,143 filed May 1, 1989, the teachings of which are
incorporated herein, or other more conventional techniques. Polymers that
can be used to forrn the microspheres include bioerodible polymers such as
poly(lactic acid), poly~actic-co-glycolic acid), poly(caprolactone),
polycarbonates, polyan~ides, polyanhydrides, polyamino acids, polyortho
esters, polyacetals, polycyanoacrylates and degradable polyurethanes,and
non-erodible polymers such as polyacrylates, ethylene-viny} ace~te and
other acyl substituted cellulose acetates and derivatives thereof, non-
erodible polyurethanes, polystyrenes, polyvinyl chloride, polyvinyl
'' ' .: .' ' ' . :, '
wo 92/l ~844 P~r/uss1tos77l
~: g
~99376
fluoride, poly(v~yl imidazole), chlorosulphonated polyolefins, and
polyethylene oxide.
The method of Gombotz, et al., is summarLzed as follows.
Polymer and agent to be encapsulated in solution or dispersion are
atomi~ed using an ultrasonic device into a liquified gas which overlays a
bed of frozen non-solvent. The microspheres are immediately frozen by
the Iiquified gas. The solvent is slowly removed from these spheres as
they thaw and sink onto and then into very cold non-solvent which extracts
the solvent as it and the spheres thaw, leaving microspheres conta~ning the
encapsulated agent. The liquified gas can be liquid argon (-185.6C),
liquid nitrogen (-195.8C), liquid oxygen (-182.9C) or any other gas that
results in the imrnediate freezing of the atomized par~cles into ~rozen
spheres.
The product microspheres have been shown to exhibit sustained release in
~irr~ and in ~vo uith a broad variety of proteins and e~zym~s.
The loadings of the active formulation of the protein-polycation
complex in such sustained release systems can be from S to 50% (w/w),
preferably in the range 10~0%.
Release of the protein active agent from microspheres containing
protein-polycation complexes can occur according to one of seve~al
mechanisms. First, dissociation of the protein from the complex would
occur only in situ in the domain of the sustained delivery system. The
free protein diffuses out of the device, while the polycation relT~ns
behind. The polgcation presumably is still bound in a network of the
protein-polycation complex (in the case of proteins tt-at are polyvalent for
~e polycation), or bound to other protein molecules (1n the case of
pro~eins that are rnonovalent for ~e polycation). In either case, it is likely
~at the diffusion coefflcient of the polycaticn molecule is much lower than
that of the free pro~ein, so that it remains within the device. Second, ~e
. .
:
Wo 92/11844 PCI/US91/09771
-1~
~i399~76
protein-polycation complex, to the extent that it is soluble, diffi~ses out of
the sustained de~ivery device into the release sink. Ilt then undergoes
dissociation to release the protein active agent into the medium. Third,
free ~i.e., uncomplexed) molecules of protein and po]Lycation leave the
sustained release device independently and possibly s~imultaneously. They
remain uncomplexed to the extent permi~ed by the law of mass ac~on. In
reality, it is l~sely that a combination of these effects is operative.
It has been discovered that proteins can form complexes with
biological polycations in Yitro; in many cases turbidity or formation of a
precipitate actuallLy occurs. This observation has led to the use of such
complexes as depots or reservoirs for stabilization of the protein active
agent and for incorporation into sustained release systems. In this
embodiment of this method and compositions, the polycation-protein
complex itself serves as a depot for release of the protein active agent,
rather than a polymeric maerix.
The requirements for a polycation-protein complex to serve as a
reservoir for the sustained release of the protein as the ac~àve agent in a
pharmaceutical formulation can be surnmarized as follows. First, the
assoc~ation constant for the formation of the complex should be relatively
high, a proper~y which may be achieved by virtue of cooperativity in the
process of forming the complex. A consequence of having a high
association constant is that the concentration of free protein will remain
relatively low. Under such conditions, when the release mechanism is
govemed by diffllsion, the rate of release can be dirninished because the
flux is p~roportional to ~e concentration gradient established between the
inner and outer phases. With a low concentration of protein es~ablished in
~e inner phase, the rate of diffusion will be low. Second, the
concentration of polycation should be relatively low, so that the ac~ve
agent is the prevalent component by weight in the formuhtion, if so
.
.
.. ,- , .~ ' ~
WO 92/~ 1844 PCr/US~l/0977l
7 6
desired. This is readily achievable because the high association constant
ensures that most or all of ~e polycation par~cipates in complex
formation.
Third, the molecular weight of the polycation should be relatively
high, so that its diffusion coefficient will be low. In this way the active
agent will be preferentially depleted firom the ma~ix or depot prior to the
polycation.
The present inven~on will be further understood by the following
non-limiting examples.
Example 1: Formation of a complex between bovine serwn
albumin and chitosan.
1 g of chito~san was dissolved in 100 ml of 1 æ acetic acid. The
pH of the resulting solution was 3Ø The solu~on was titrated with
sodium hydroxide to pH of 6.0, avoiding precipitation and gel formation
by ~u~ c.' ito~n. This is termed neu~ralized chitosan.
12.0 mg of bovine serum albumin (BSA) was dissolved in 1.0 ml
5 mM ammonium bicarbonate. 20 microliter aliquots of neutralized
chitosan were added to the BSA, as well as to a buffer blank. A thick
cloudy precipitate formed with the BSA, which was more profound and
extensive than that observed with buffer alone. The latter is ascribed to
pH-induced precipitation of chitosan. Centrifugation was used to
determine whether precipitation occurred in the liquid supernatant with
successive additions of chitosan. Generation of incremental turbidity
ended at about the point where 200 microliters of the chitosan solution had
been added to the BSA, coIIesponding to 2.0 mg chitosan.
The equivalence point was reached at a weight ratio of
BSA:chitosan of about 5:1.
.
wo 92/1~844 PCr/US91/09771
f
-12-
3 7 6
Example 2: Fo~nation of a complex between bo~ne
hemoglobin and chitosan.
10.2 mg of bovine hemoglobin (EIb) was dissolved in 1.0 ml
deionized water. Up ~o 40 microliters of neutralized chitosan was added
in portions. With the first additions a dark agglome~ate formed,
corresponding to partial depletion of color from the solution. Further
addition of chitosan did not lead to a quantitative precipitation of ~e Hb.
Example 3: Preparation of and in vitro ~elease from PLGA
microspheres containing the erythropoietin~hitosan
complex.
Chitosan acetate at pH S WélS used to dissolve recombmant human
erythropoietin (EPO) with varying ratios of chitosan:EPO. These
formulations were micron~zed according to the method set forth in
Gombotz et al. (U. S. Serial No. 07/345,684) and incorporated into
copoly~l),L-lactide, glycolide) (50:50, Boe}lringer-Ingelhe~m R~3 503)
using the procedures of Gombotz et al. (U. S. Serial No. 07/345,143).
The final loading ratios, in weight percentages of the final microsphere
preparation, were 6% EPO: 10.3% chitosan, 12% EPO: 6.8% chitosan,
and 17% EPO alone.
These microspheres were subjected to in v~tro release stu~ies at
37C, using the follo~1ving release buffer: 50 n~ sodium phosphate, 0.9%
MaCI, 2% (w/v) ovalbun~n, pH 7.2. The release results are shown in
Figure 1. It is evident that, as compared to the abænce of chitosan,
incorpoIation of the polycation profoundly reduces the burst effect upon
~e release of EPO from the microspheres.
Example 4: Prepar~tion and in vi~o release of P~ A microspheres
containing the Factor Vm-poly(arginine) complex.
Human recombinant Factor vm was recons~tuted to 200 units/ml
in 0.2 M NaCl, 0.55 M glycine, 0.005 M CaCl2, 12 mg/ml hurnan serum
WO 92/11844 PCr/US91/09771
, .
-13-
2~37~
albumin. To this solution was added polyarginine [(Arg)81 at 2 mg/ml.
The solution was subjected to a change in composition of the bu~fer to O.l
M proline, 2.5 mM CaCl2, pH 7.35 by passing the reconstib~ted mixture
through a SephadexR G-25 column equilibrated with ~e proline - CaC12
solution. The product was then micronized according to the proccdure of
Gombotz, et al., in U.S. Serial No. 07/345,684, and incorporated ~nto
microspheres comprised of poly~actic acid) as the carrier matrix, at a
loading of the for nulated Factor vm preparation of 30% (w/w) using the
procedure described by Gombotz, et al. in U.S. Serial No. 07/345,143.
This preparation is referred to as "poly(arginine)" in Figure 2.
A similar microsphere preparation was made using human
recombinant Factor vm reconstituted to lO0 unitstml in 0.1 M NaCl,
0.275 M glycine, 0.0025 M CaC12, 6 mg/ml human serum album~n. This
preparation was similarly incorporated into PLA microspheres at 30%
loading. This prepa~a~on is re~erred to as "NaCl-Glyc~ne" ~n };igure 2.
The two rnicrosphere preparations were subjected to in vi~ro
release experiments at 37C, by immersLng approximately lO mg of
microspheres in l.0 rnl aliquots of a release buffer consisting of O.l M
NaCl, O.l M glycine, lO mM HEPES, 2.5 mM CaCl2, 2 mg/lT~l hurnan
serum albumin, pH 7.2 m a l.5 ml microfuge tube, and agitated gently.
Fresh aliquots of release medium were applied for each time point. The
activity was assayed using the CoatestR kit for Factor VIII produced by
Kabi Vitrum and distributed by Helena Laboratories, Inc. The color
resulting from release of p-nitrophenolate from a synthetic substrate, as
deternnned in microtiter plate format using a plate reader, and expressed
as the cumulative percent of inçolporated acti~ity released per mg of
microspheres, is given for the two preparations in Figure 2. It is evident
~at Factor VIII formuL~ted wi~ (Arg)n has led to markedly enhanced and
sustained release l~ne~cs compared to omission of (Arg)n.
wo 92/11844 PCT/US91/09771
~; ' `
-14-
~' ~3~7 ~
Example 5: Bo~ine Serum Albumin-Protamine Complex Release
from Copoiy(lactid~glycolide) Microspheres.
A globulin-free preparation of bovine semm albumin (BSA)
obtained from Sigma Chemical Co. was mixed 1:1 (w:w) with sucrose or
WIth protan~ine sulfate. The resulting solu~ons, as well as a solution of
BSA alone, were micronized according to Gombotz, et al., as described in
U.S. Serial No. 07/345,684. The protein-excipient formulations were
incorporated into microspheres of copoly(DL-lactide,glycolide) (50:50)
following the procedures of Gombotz, et al., U.S. Serial No. 07/345,143,
with total loadings of 10% by weight. These microspheres were placed in
20 rnM sodium phosphate, 0.15 M sodium chloride, 1.5 rnM sodium
azide, pH 7.S, at 37 C to measure in vitro release.
The cumulative release over 68 days is shown in Figure 3. The
surge in release that occurs between about days 20 and 28 is ascribed to
dcgradatioil Or the polymeT matrix, expos~ng fresh reservoirs Or prv~ein for
release to the medium. The results show that incorporation of protamine
sulfate gives enhanced release characteristics as compared to the
incorporation of an equa} arnount of sucrose. The extent of release in the
first hour, termed the burst, is diminished, and the steady, near-zero-
order release of protein is sustained for a longer duration. For BSA
without added excipients, the burst release is the lowest of the three cases
shown, but the degradation phase releases a large fraction of the protein
over a relatively short period of time; further release continues for the
remainder of the time period considered. Of the three preparations shown,
the incorporation of protan~ine sulfate leads to the most monotonic release
of rotein after ~e burst.
. . . . .
,