Note: Descriptions are shown in the official language in which they were submitted.
~11)3~&~
The invention relates to organo-functional polysiloxanes
with one or more functional or non-functional siloxane units,
which have the applicational and techn;cal advantages of a
macroscopic spherical shape and, unlike organosiloxanamine
copolycondensates already described (DE 39 25 359, DE 39 25
360, P 38 37 416, P 38 37 418), do not contain components of
the NR3 (with R = R'-SiO3/2) type.
Processes for the preparation of the new products in
particle sizes which are ideal for the application being
considered and with the currently appropriate physical
properties and applications for these novel materials are
described.
An lmch~red polymeric organosiloxane powder or organosiloxane
gels, which are obtainable by precipitation with a base such
as e.g. a lonia, are known and these are mechanically crushed
after hardening and are available as particulate materials.
Use of the corresponding polysiloxanes, e.g. in stirred
reactors, is connected with a considerable amount of friction
and associated technical problems. Accessibility of organic
functions on and in the polysiloxane structure is very poor
due to unfavorable, or a lack of, porosity.
Spherical organosiloxanes or silica gels are also known, with
particle sizes, however, in the region of a few micrometers.
(T. Kawaguchi, K. Ono; J. Non-Cryst. Solids 1990, 121, 383-
388, P. Espinard, J.E. Mark, A. Guyot; Polym. Bull. (Berlin)
1990, 24, 173-179, Jap. Kokai Tokkyo Koho / 02225328 A 2.
2103~3~3
I
In this case, the fundamental methods of preparation are
based on precipitation of siloxanes. Mainly due to process
restrictions, larger spherical particles could not be
S produced using this method. As a standard feature, the
particles size achieved is in the range from 1 to at most 10
micrometers.
Known (but not previously published) are methods for the
preparation of metal-cont~ininq organosiloxanamine
copolycondensates in the form of spherical particles with a
diameter of 0.01 to 3.0 mm (DE-PS 41 lo 705). In the case
of these products, the organosilanamine fulfills the task of
a subsequent stabilizing siloxane component, and also of a
catalyst for the hydrolysis and polycondensation reaction.
The invention provides shaped organosiloxane polycondensates,
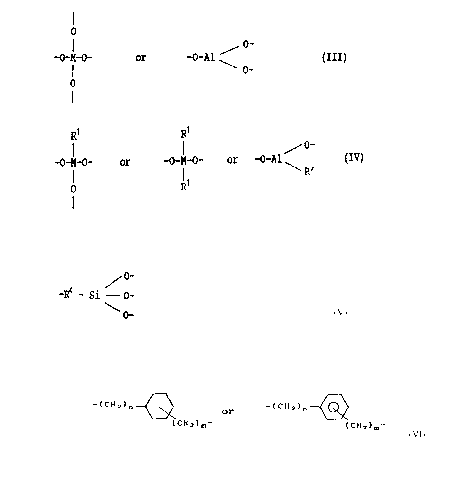
consisting of units of the formula.
X - R1 (I)
and/or the formula
R2 _ y - R3 (II)
and of the formula
-O-M-O- or - o - Al = (III)
O _
o
~1~3~3
as well as optionally in addition of units of the formula
R1 R1
~ 0-
S -0-M-0- or -O-M-0- or -O-Al (IV)
I I - R'
O R1
in which the ratios of
(I) to (III) are in the range from 95 to S to 5 to
95 mol-~, preferably from 50 to 50 to 10 to 90 mol-~,
or
lS (II) to (III) or the sum of (I) plus (II) to (III) are 100
to 0 to S to 95 mol-%, preferably from 90 to 10 to 10 to
90 mol-%,
and
with ratios of the sum of (I), (II) and (III) to (IV) of
100 to 0 to 50 to 50 mol-%,
wherein R1 to R3 are identical or different and represent a
group of the formula
-R4 - Si - 0- (V)
O--
R4 being bonded directly to the group X or Y and
representing a linear or branched, fully saturated or
unsaturated alkylene group with 1 to 10 carbon atoms, a
cycloalkylene group with 5 to 8 carbon atoms, a phenylene
group or a unit of the general formula
- ( CH 2 ) n--~ or -( CH 2 ) n ~
( C112 ) m ~ ( CH 2 ) m~
in which n is a number from 1 to 6 and gives the number of
methylene groups adjacent to X or Y and m is a number from
21 03653
~ 0 to 6, wherein M is a Si, Ti or Zr atom and R' is a linear
or branched alkyl group with 1 to 5 carbon atoms or a
- phenyl group and X in formula (I) represents
5 -H, -Cl, -Br, ~ CN, -SCN, -N3, -OR' ', -SH, -COOH,
P (C6Hs) 2 ~ NH2, --N (CH3) 2~ --N ( C2Hs ) 2, --NH--~CH2) 2--NHz,
--NH--(CH2) 2--NH--(CH2) 2--NH2, --NH--C ( S )--NR2 ' ', --NH-C (O)--NR2 ' ',
--NR ' '--C ( S )--NR2 ' ', --O--C ( O )--C ( CH3 ) =CH2, --CH=CH2,
CH2--CH=CH2 ~ --CHz--CH2--CH=CH2 ~
~
and Y in formula (II) represents
15 =N-H, = N-CH3, ~ N--C2HS, -S- ~ --S2 ~ S3
S4 , =P--( C6Hs ); --NH--C ( S )--NH--, N--C ( S )--NR2 ' "
R' ' R' '
2 0 N--C ( S)--N \ , -NH--C (O)--NH--, N-C (O) -NR2 ' '
wherein R' ' is H or a linear or branched alkyl group with 1
to 5 carbon atoms, in the form of spherical particles with
25 a diameter of 0.01 to 2 . 5 mm, preferably 0. 05 to 1. 5 mm, a
specific surface area of 0.01 to 1000 m2/g, particularly 50
to 800 m2/g, a specific pore volume of 0.01 to 5 . 0 ml/g and
a bulk density of 90 to 1000 g/l, particularly 100 to
800 g/l.
Solid, shaped and well-defined products are obtained within
the claimed ranges. There are no problems with regard to
the relevant morphological, physical properties, i. e. the
porosity, or chemical stability.
~1~36~;~
_ 5
In a particular embodiment, the polycondensates are present
as random polycondensates, block polycondensates or mixed
polycondensates. Preferably, R1, R2 and R3 are defined as
5 / O-
-(CH2)3-Si ~ o-
O--
The suitable chemical composition of the polycondensates
according to the invention depends mainly on their intended
use. Depending on the desired application, a suitable
density of functional groups is selected by varying the
proportion of components of the formulas (I) and (II) and
of components with the formulas (III) and (IV), which serve
to cross-link the polysiloxane matrix and also to produce
suitable physical properties, without impairing the
intended mode of action by means of the organo-functional
groups which are incorporated.
The following compounds, which are in principle known, may
be used successfully, for instance, as monomeric units for
the shaped organosiloxane polycondensates :
Cl-CH2CH2cH2si(Oc2Hs)3
Ncs-cH2cH2cH2si(oc2Hs)3
NC-CH2CH2Ch2si(OcH3)3
CH2=CHsi(OcH3) 3
C6Hssi(oc2Hs)3
S[CH2CH2CH2Si(OcH3) 3 ] 2
HNt(CH2)10si(oc2Hs)3]2
Si(Oc2Hs)4
Ti(OC3H7)4
(Hsc2o)2si(cH3) 2
2103~53
. .
As can also be seen from the examples, the particle size
distribution, specific surface area, bulk density and thus
also the porosity can be set selectively within wide limits.
Preferred ranges are: diameter of particles: 0.05 to
1.5 mm; specific surface area: 50 to 800 m2/g; and bulk
density 100 to 800 g/l.
In general, random polycondensates are produced but it is
also possible, using selective precondensation, to obtain
block polycondensates, or mixed ploycondesates.
For technical reasons, and also because of the ready
availability of the corresponding starting silanes, a C3
spacer group is preferred between the silicon atom and the
organic functional group.
The invention also provides processes for preparing the
polycondensates according to the invention. A process for
the preparation of shaped random organosiloxane
ploycondensates according to the invention is characterized
in that components of the general formulas (VI) to (VIII)
X-R5 (VI), R~-Y-R~ (VII~, M(OR8)2~R'a2 or
Al(OR~23R'ot (VIII)
corresponding to the stoichoimetric composition of the
polysiloxane being prepared wherein R5 to R7 are identical or
different and each represents a group of the general formula
(IX)
-R~-si(oR9)3 (IX~
.
6a
X, Y, R , M and R4 are each defined as in the formulas (I) to
(V) and R8 and R9 represent-a linear or branched alkyl group
with 1 to s carbon atoms, are dissolved in a solvent which is
predominantly water-miscible but dissolves the silane
components, and amount of water which is at least sufficient
for complete hydrolysis and condensation as well as
optionally hydrolysis and condensation catalyst from the
group HCl, H3PO4, CH3COOH, NH3, NR3'''wherein R''' represents
an alkyl group which contains 1 to 6 carbon atoms, as the
pure substance or in aqueous solution, is added to the
solution with stirring, then the reaction mixture is allowed
to gel with further stirring at a specific temperature in the
range from room temperature to 200~C, and at the start of
gelling or up to one hour afterwards 10 to 2000%, preferably
50 to 500% by weight, with reference to the total amount of
silane components used, of a predominantly water-immiscible
solvent, but one which dissolves and dilutes the (being)
gelled reaction mixture, is added, homogenized and
immediately or within a time interval of up to 3 hours later,
optionally increasing the originally fixed temperature 10 to
2000% by weight, preferably 50 to 500% by weight, with
reference to the total amount of silane components used, of
water is added, the siloxane-containing organic phase is
dispersed in the liquid two-phase system and the solid which
is formed after hardening of the droplets in the shape of
spheres is separated from the liquid phase after a sufficient
reaction time, at room temperature to 250~C, optionally
purified by extraction, optionally dried at room temperature
to 250~C, optionally under a protective gas or under vacuum,
and then optionally annealed and/or classified. Methanol,
ethanol, n- or i- propanol, n- or i- butanol or n-pentanol,
alone or in a mixture, are the preferred solvents for
hydrolysis.
211~3~g~ .
- Preferably a linear or branched alcohol with 4 to 12 carbon
atoms, toluene, xylene isomers (separately or in a mixture)
or tert.-butyl-methyl-ether is added to the (being) gelled
reaction mixture.
Some or all of the amount of water-insoluble solvent being
added at or after the start of gelling may be used from the
begi nn i ng of the process in addition to the solvent used at
that point.
One or more of the silanes of the formulas (VI) to (VIII) may
not be introduced to the mix from the beginning, but may be
introduced later, during or shortly after gelling, optionally
lS in the predominantly water-insoluble solvent being added.
Also, of the silane components, combined or each separately,
may be pre-condensed and then added to the reaction mixture.
A process for after-treating the shaped but not dried
organopolysiloxane condensates obtained in accordance with
the above processes is characterized in that the solid
obtained is subjected to a thermal treatment for 1 hour to
one week at 50 to 300~C, preferably 100 to 200~C, in the
liquid phase in the presence of at least the component water
or else in the mother liquor, wherein the excess pressure
corresponds to the sum of the partial pressures of the
components used.
Preferably the after-treatment is performed in the presence
of an acid or basic catalyst, preferably in the presence of
ammonia.
In principle, the corresponding halide or phenoxy compounds
may also be used as starting materials for the process
instead of alkoxysilyl compounds, but their use does not
~103653
6c
offer any advantaqes and may, e.g. in the case of the
chlorides, cause difficulties as a result of hydrochloric
acid being released during hydrolysis.
Hydrolysis of the starting materials and optional cross-
linking agent must be performed in a solvent which is
predominantly water-miscible but which dissolves the
starting materials. Preferably therefore, alcohols are used
which correspond to the alkoxy grouping in the monomeric
precursor of the starting material or to the metal atoms in
the optionally used cross-linking agent. The following are
particularly suitable: methanol, ethanol, n- and i-propanol,
n- and i-butanol or n-pentanol. Mixtures of such alcohols
may also be used as the solvent for hydrolysis.
2103~
Instead of alcohols, other polar solvents which are
predominantly water-miscible may also be used, but it has
been shown that this is not as sensible from a technical
point of view due to the solvent mixture which is produced
with the hydrolytically eliminated alcohol.
Preferably the hydrolysis is performed with an excess of
water as compared with the stoichiometrically required
amount. The amount of water required for hydrolysis depends
on the rate of hydrolysis of each organosilane or cross-
linking agent used, in such a way that hydrolysis takes
place more rapidly with increasing amounts of water. An
upper limit can be set, however, by the occurrence of
demixing and the formation of a two-phase system.
Basically, hydrolysis in homogeneous solution is preferred.
On the basis of the two aspects mentioned, in practice
somewhat less water, with respect to the weight, is used
than organosilanes plus cross-linking agent.
The duration of hydrolysis depends on the tendency to
hydrolyse of the starting material and/or cross-linking
agent and on the temperature. The readiness to hydrolyse
and thus the rate of hydrolysis depends in particular on
the type of alkoxy groups adjacent to the silicon or
titanium, zirconium or aluminium atoms, wherein methoxy
groups are hydrolysed the most rapidly and there is a
slowing down with increasing chain length of the
hydrocarbon group. In addition, the duration of the total
hydrolysis and polycondensation procedure also depends on
the basicity of the organosilane. Hydrolysis and
polycondensation may be accelerated by the addition of
bases, preferably ammonia, or of inorganic or organic
acids, or else by the usual condensation catalysts, such as
dibutyltin diacetate.
2::10~653
Basically, all Br0nsted acids and bases may also be
considered as catalysts. Preventing precipitation of
siloxanes causes many technical difficulties when
performing the reaction and selecting the type and
concentration of catalyst. Surprisingly, it was possible to
prepare spherical products although the acid or base
catalysed hydrolysis of organosilanes is known and is used
in many different ways to prepare unshaped polysiloxanes
with undefined physical properties.
The requirement of keeping the starting material, which is
cross-linked with water and dissolved in solvent, at a
certain temperature while still being stirred results in
the rate of polycondensation, which is signalled by
gelling, being temperature dependent.
The temperature to be applied during hydrolysis or the
gelling phase is established empirically for individual
cases. It should be noted here that a fluid, gel-like
material which contains no solids is retained for the
subsequent process step, the so-called shaping phase.
The shaping phase, accompanied by the transfer of the
coherent fluid, gel-like mass (in which the condensation
reaction continues further) into separate spherical
particles, starts with the addition to the (being) gelled
reaction mixture of a predominantly water-insoluble
solvent, but one which dissolves the reaction mixture
adequately, in the designated amount.
Suitable solvents are e.g. linear or branched alcohols with
4 to 18 carbon atoms or phenols, linear or branched
symmetric or asymmetric dialkyl ethers and di- or tri-
ethers (such as ethyleneglycol-dimethyl ether), chlorinated
or fluorinated hydrocarbons, aromatic compounds or mixtures
of aromatic compounds substituted with one or more alkyl
21~3~;3
- g
groups, such as e.g. toluene or xylene, symmetric and
asymmetric ketones which are predominantly immiscible with
water.
Preferably, however, a linear or branched alcohol with 4 to
12 carbon atoms, toluene or o-, m- or p-xylene, separately
or as a mixture, is added to the (being) gelled reaction
mixture.
This addition of a solvent causes a dilution effect after
homogenisation with the reaction mixture and thus causes a
definite slowing down in the condensation reaction being
accompanied by an increase in viscosity.
Assessment of the amount of this solvent used in the
shaping phase depends in particular on what particle size
is being sought for each shaped organosiloxane compound. A
rule of thumb which may be applied is that less has to be
used for coarse particles (spheres with a larger diameter)
and more for fine particles ~spheres with a smaller
diameter).
In addition, the intensity with which the viscous
-homogeneous mixture consisting of reaction mixture and
predominantly water-insoluble solvent is dispersed in the
extra water added as dispersion agent in the shaping phase
also has a large effect on the particle size. The formation
of a finer particle range is regularly encouraged by
vigorous stirring. One of the known dispersion-aiding
agents, such as long-chain carboxylic acids or their salts
or polyalkylene glycols may be used in the normal
concentrations to stabilise the aqueous dispersion of the
organic phase (now containing siloxane).
According to one variant of the process according to the
invention, some or even the whole amount of the
2103~3
- 10
predominantly water-insoluble solvent being added at or
after the start of gelling is used in the hydrolysis step
alongside the solvent used there. If only some is added,
the residue is added after the start of gelling.
s
In the extreme case, addition of the whole amount, the
dispersion agent water may be added at or after the start
of gelling. This variant is preferred when the organosilane
and optional cross-linking agent mixture used exhibits an
extraordinarily high tendency towards hydrolysis and
polycondensation.
The preferred temperature at which dispersion of the
siloxane-containing organic phase in the aqueous phase is
lS performed and spherical solids are formed from the
dispersed phase, is generally the reflux temperature of the
whole mixture. Basically, however, the same temperatures as
thosé used in the gelling steps may be applied. The total
duration of the dispersion step and after-reaction is
generally 0.5 to 10 hours.
Both gelling and shaping may be performed at atmospheric
pressure or at an excess pressure which corresponds to the
sum of the partial pressures of the components of the
reaction mixture at the particular temperature being
applied.
When preparing the shaped, cross-linked or non-cross-linked
organosiloxanes according to the invention, this also being
independent of the type of alkoxy group, it may so happen
that one or more components in the mixture to be gelled has
a different hydrolysis and polycondensation behaviour. In
this case one version of the process according to the
invention provides for the cross-linking agent(s) and/or
the organo-functional silane not to be subjected to the
gelling process together, but to be gelled separately
2103~5~
11
first, to homogenise them with the predominantly water-
insoluble solvent and only then to add the cross-linking
agent(s) or organosilane to the homogeneous mixture.
However, the solvent and the silane component which is
still missing may also be added simultaneously to the
gelled mix.
Separation of the spherical shaped moist product from the
liquid dispersion agent may be performed using the usual
measures such as decanting, filtering or centrifuging.
The liquid phase may also be removed from the reactor, the
solids remaining behind being treated once or several times
with a low-boiling extraction agent, preferably in a low-
boiling alcohol, in order to facilitate subsequent drying
of the shaped material by at least partially exchanging the
mostly ~elatively high-boiling solvent from the shaping
phase for the low-boiling extraction agent.
Drying may be performed basically at room temperature to
250~C, optionally under a protective gas or under vacuum.
The dried, shaped solids may be annealed at temperatures of
150 to 300~C to harden and stabilise them.
The dried or annealed product may be classified into
various particle size fractions in the usual devices. One
or more of the working-up measures of extracting, drying,
annealing and classifying may be omitted, depending on the
circumstances. Classification may be performed with the
liquid-moist, dried or annealed product.
In order to compensate for different hydrolysis and
polycondensation behaviour by the monomeric components in a
random, optionally cross-linked, copolycondensate, the
2103653
12
monomeric components with the formulas (V) and (VIII) could
be initially pre-condensed.
A particularly important embodiment of the process
according to the invention provides for subjecting the
still solvent- and water-moist or -wet spherical material
to a thermal treatment for 1 hour to one week at
temperatures of 50 - 300~C, preferably 100 - 200~C, wherein
excess pressure may be applied if so required.
This treatment under "vaporising" or digesting conditions
also predominantly serves to improve the mechanical
strength and porosity of the shaped material and may also
be performed in the dispersion which is obtained last in
the preparation process, which contains a liquid and the
solid product phase, or in water on its own.
The previously described embodiment of an after-treatment
of the shaped, but not dried, organosiloxane
copolycondensate which is obtained thus comprises
subjecting the solid produced in the form of spheres, in
the presence of at least the component water or of the
liquid phase which was present last in the preparation
process as a vapour or a liquid, to a thermal treatment for
1 hour to one week at temperatures of 50 - 300~C,
preferably 100 - 200~C, optionally under excess pressure.
The presence of an acid, basic or metal-containing catalyst
may be of advantage here. A particularly advantageous
embodiment provides for the use of ammonia.
The novel, shaped organosiloxane copolycondensates are
characterised in particular by using the quantitative
hydrolysis yields, by elemental analyses and by the
determination of the individual functional groups.
210~3
_ 13
Purely optically, there is no difference between the
copolycondensates obtained by the different methods of
preparation. Depending on preliminary treatment, the
spherically shaped copolycondensates according to the
invention have a particle diameter of 0.01 to 2.5,
preferably 0.05 to 1.5 mm, a specific surface area of 0.01
to 1000, preferably 150 to 800 m2/g, a specific pore volume
of 0.01 to 5.0 ml/g and a bulk density of 50 to 1000 g/l,
preferably 100 to 800 g/l. The adjustable pore diameters
lo -are in the range 0.01 to more than 1000 nm.
Specific control of synthesis permits the preparation of
products in the most technically applicable spherical shape
and with the desired physical and morphological properties.
The spherical polycondensates may be used, optionally after
further additional chemical modification, as active
substance carriers in general or else as carriers for the
preparation of noble metal catalysts.
A further use of all the copolycondensates according to the
invention is use for the adsorptive bonding of gaseous
organic compounds and/or water vapour, preferably of
organic solvents.
It is in particular the pore volume, pore diameter, and
surface properties which are critical for this adsorptive
action.
These factors may be affected on the one hand by the
methods of preparation and after-treatment according to the
invention and on the other hand also by the chemical
composition, e.g. by the incorporation of hydrophobic
cross-linking groups in the polysiloxane structure.
Recovery of the adsorbed organic compounds or water is
~103~S3
14
readily achieved by raising the temperature and/or by
flushing out with warm air.
In the following, the invention is explained in more detail
by using working examples.
Example 1
383.8 g of Si(oc2H5)4 are introduced into a 3 l double-
walled glass vessel together with S00 ml of ethanol and
100 ml of 1-octanol and heated to 80~C with stirring. 125
ml of water (pH = 4.0) are added, the mix is cooled to 60~C
and 0.1 ml of tributylamine is added. The mix itself is
maintained at a temperature of 60~C with slow stirring.
After 20 minutes the mix gels, i.e. the viscosity increases
noticeably. The rate of stirring is immediately increased
(600 rpm) and 116.2 g of NC-CH2CH2CH2-Si(oCH3)3, dissolved in
400 ml of octanol, are added. 1500 ml of water (50~C) are
added to the homogeneous solution after 10 min. and the
organic solution is dispersed in the water. The emulsion
which is present is heated and ~oiled under reflux for 2
hours. After cooling the mix, the solid which is produced
is filtered off under suction and extracted three times
with ethanol. The product is dried at 150~C for 24 hours
under N2. After classifying the solid, 177 g (9S.9~ of
theory) of product are obtained in the form of a solid with
spherical particles in the particle size range from 0.1 to
0.6 mm (of which 65% is in the range from 0.2 to 0.4 mm)
and with the composition NC-(CH2)3-Sio3/2.3Sio2.
Elemental analysis : % C % H % N ~ Si
Theory: 15.9 2.0 4.6 37.4
Found: 14 2.3 3.2 35.7
Bulk density: 683 g/l (anhydrous)
- 21~3~3
Example 2
After extraction with ethanol, the product prepared in the
same way as in example 1 is first subjected to a
hydrothermal treatment at 150~C in 5% aqueous ammonia
solution (24 h) and then dried as in example 1. A solid is
obtained as in example 1, but with a bulk density of
405 g/l.
Example 3
138.8 g of StCH2CH2CH2Si(OCH3)3]2 and 161.2 g of Si(oC2H5)4
are initially introduced into a 3 1 double-walled glass
vessel together with 300 ml of ethanol and 120 ml of 1-
octanol and heated to 75~C with stirring. 49 g of NH3
solution (25 % by weight in water) and 55 ml of distilled
water are added and the mix is cooled to 70~C. After 5
minutes the mix gels, the rate of stirring is immediately
increased (600 rpm) and 240 ml of octanol are added. 900 ml
of water (50~C) are immediately added to the homogeneous
solution and the organic phase is dispersed in the water.
The emulsion which is present is heated and boiled under
reflux for 2 hours. The mix is filtered under suction, 5%
strength NH3 solution is added to the isolated solid and
stirred in a laboratory autoclave at 150~C for 24 h. After
cooling the mix, the solid which is produced is filtered
off under suction and extracted three times with ethanol,
with stirring.
The product is dried under N2 for 4 h at 60~C, for 4 h at
sooc, for 4 h at 120~C and finally for 12 h at 150~C. After
classifying the solid, 101 g of product in the form of a
solid with spherical particles in the particle size range
from 0.3 to 0.8 mm and the composition
S[(CH2)3-siO3/~i2sio2 are obtained.
~1~3~53
16
Elemental analysis: % C % H % S % Si
Theory: 21.2 3.6 9.4 32.9
Found: 23 4.1 9.9 30.7
Bulk density: 164 g/l (anhydrous)
Example 4
62.4 g of CH3CH2CH2Si(OCH3)3 and 237.6 g of Si(oc2Hs) 4 are
initially introduced into a 3 1 double-walled glass vessel
together with 300 ml of ethanol and heated to 80~C with
stirring. 71 g of HCl solution (37% by weight in water~ and
90 ml of distilled water are added stepwise, the mix is
boiled under reflux for 2 h and then cooled to 70~C. After
15 minutes the mix gels, the rate of stirring is
immediately increased (600 rpm) and after 1 minute 300 ml
of octanol are added. After another 1 minute 900 ml of
water (50~C) are added to the homogeneous solution and the
organic phase is dispersed in the water. The emulsion which
is present is heated and boiled under reflux for 2 h.
The mix is filtered under suction, 5% strength NH3 solution
is added to the isolated solid and stirred in a laboratory
autoclave at 150~C for 24 h.
After cooling the mix, the solid which is produced is
filtered off under suction and extracted three times with
ethanol, with stirring.
The product is dried under N2 for 4 h at 60OC, for 4 h at
90~C, for 4 h at 120~C and finally for 12 h at 150~C. After
classification of the solid, 92 g of product, in the form
of a solid with spherical particles in the particle size
- 210~6~3
17
range from 0.1 to 0.8 mm and the composition CH2CH2CH2-
sio3,2.3sio2 are obtained.
Elemental analysis: % C % H % Si
Theory: 13.1 2.6 40.8
Found: 13.0 2.8 39.7
Bulk density: 240 g/l (anhydrous)
Exam~le 5
60-6 g of NCS-CH2CH2CH2si(OC2H5) 3 and 239.5 g of Si(oC2H5) 4 are
initially introduced into a 3 l double-walled glass vessel
together with 300 ml of ethanol and heated to 80~C with
stirring. 71 g of HCl solution (37~ by weight in water) and
45 ml of distilled water are added stepwise, the mix is
boiled under reflux for 40 minutes, then cooled to 70~C.
After 215 minutes the mix gels, the rate of stirring is
immediately increased (600 rpm) and after 1 min. 300 ml of
octanol are added. After 5 minutes, 900 ml of water (50~C)
are added to the homogeneous solution and the organic phase
is dispersed in the water. The emulsion which is present is
heated and boiled for 2 h under reflux.
After working-up in the same way as in example 4, a shaped
polysiloxane with the composition NCS-CH2CH2CH2-Sio3~2.5sio2
was obtained.
ExamPle 6
57.8 g of CH2=CH2Si(OCH3) 3 and 242.2 g of Si(oc2Hs) 4 are
initially introduced into a 3 l double-walled glass vessel
together with 300 ml of ethanol and 120 ml of 1-octanol and
~1~36~
.. .
18
heated to 80~C with stirring. 75 ml of water (pH = 4.0) are
added, the mix is cooled to 70~C and 2.0 ml of
triethylamine are added. The mix itself is kept at a
temperature of 60~C with slow stirring. After 15 minutes,
the mix gels, the rate of stirring is immediately increased
(600 rpm) and 240 ml of octanol are added. 900 ml of water
(50~C) are immediately added to the homogeneous solution
and the organic phase is dispersed in the water. Further
working-up is performed in the same way as in example 1. A
shaped polysiloxane with the following composition was
obtained : CH2=CH2-Sio3~2.3Sio2.
ExamPle 7
81.9 g of C8H17Si(oCH3)3 and 218.2 g of Si(oc2Hs)4 were
reacted in precisely the same way as described in example 6
and a shaped polysiloxane of the composition CôH"Sio3~2.3Sio2
was obtained.
Exam~le 8
In the same way as in example 6, 73.1 g of
phenyltriethoxysilane and 226.9 g of polydiethyl silicate
40 (pre-condensed tetraethoxysilane, corresponding to 40%
sio2 content) were reacted and a product with the
composition C6HsSio3/Z~5sio2 was obtained.
Sieve analysis: 0.2 - 0.3 mm : 31%
0.3 - 0.6 mm : 59%
0.6 - 0.8 mm : 10%
BET surface area: 642 mZ/g
Mesopores (2-30 nm): 0.72 ml/g
Macropores: 0.84 ml/g
21~3~
19
Example g
In the same way as in example 6, 26.9 g of
propyltrimethoxysilane and 273.1 g of tetraethoxysilane
were reacted and a product with the composition
C3H~i3/2.8SiO2 was obtained.
BET surface area: 784 mZ/g
Mesopores (2-30 nm): 0.48 ml/g
10 Macropores: 1.24 ml/g
Bulk density: 390 g/l
Example 10
The polysiloxane obtained in example 9 was stirred with 5%
NH3 solution before drying for 24 h at 150~C.
BET surface area: 491 m2/g
Mesopores (2-30 nm): 1.81 ml/g
Macropores: 3.35 ml/g
Bulk density: 192 g/l
Example 11
In the same way as in example 6, 83.44 g of
chloropropyltriethoxysilane and 216.6 g of
tetraethoxysilane were reacted and a shaped polysiloxane
with the composition Cl-CH2CH2CH2SiO3/2.3SiO2 was obtained.
Chlorine content: 10.4% by wt. (Theory: 11.4% by wt.)
Spec. surface area: 649 m2/g
Micropores (< 2 nm): 0.42 ml/g
Mesopores (2-30 nm): 0.02 ml/g
35 Macropores: 0.75 ml/g
Bulk density: 545 g/l
- 2103653
Exam~le 12
In the same way as in example 6, but using 1 ml of
triethylamine, 50.9 g of HNtCH2CHzCH2Si(OC2H5)3]2 and 249.1 g
of tetraethoxysilane were reacted and a shaped polysiloxane
with the composition HNtCHzCHzCH2SiO3/2]2~l0SiO2 was obtained.
Spec. surface area: 112 m2/g
Mesopores (2-30 nm): 0.22 ml/g
Macropores: 4.47 ml/g
Bulk density: 167 g~l