Note: Descriptions are shown in the official language in which they were submitted.
W092/22662 2 ~10 9 9 7 PCT/CA92/002~1
M~OD~ ~OR ~ B B~N~E~I8 OF
~NOF~CO8YL~BD O~IG~8ACCE~RID~8 ~E~MIN~TING IN
DI-N AC~TY~LaCTO~A~I~Y~ 8TR~CT~R~S
BA~G~OUND OF T~ INVENTIO~
1. Field of the Invention
This invention is directed to methods for the
preparation of monofucosylated and sialylated
- d~-~ivati~es of the compound ~Gal~1-4)~GlcNAc-
~:: 5~3)~Gal(1-4~GIcNAc-OR. In particular, the
me~hod~ of this invention provide for a multi-step
: synthesis wherein selectiva monofucosylation is
ac omplished on tha 3-hydroxy group on only one of
the GlcNAc units found in the ~Gal(1-4)~GlcNAc-
(1 33~Gal(l-4)~GlcNAc-OR compound. In these
methods, monofucosylation is achieved by the use of
an ~ 3)fucosyltransferase. This invention is also
directed to compounds prepared by the herein
d~scxi~ed methods.
2. References.
The following references are cited in this
application as superscript numbers at the relevant
portion o~ the application:
la. Feizi, TIBS, 16:84-86 (1991~
20lb. Springer et al., Nature, 349:196-197 ~1991)
cEver et al., Thromobosi~ and Haemostasis,
66~80-87 (1991)
W092/22662 ~ PCT/CA92/00251
999 ~
2. SabQsan et al~, J. Amer. Chem. Soc., 108: 2068-
2080 (1986)
3, Toone et al., Tetrahedron 45:5365-5422 (1989)
4. Palcic et al., Carbohydr. Res., 190:1-11 (1989)
5. Walz et al., Sci nce, 250: 1132-1135 (1990~
6. Phillips et al., Science, 250:1130-1132 (1~90)
7. Tiem~yer et alO, Proc. Natl. Acad. Sci. USA,
88~ 8 1142 (1991)
8a. Holmes et al., J. Biol. Chem., 261:3737-3743
(1986)
8b, Holmes et al., Arch. Biochem. Biophys.,
: 274:633-647 (1989)
8co Basu et al., Indian J. Biochem. Biophys.,
~5 112-118 (lg88)
8d. Hanis~h et alO ~ 178:23_28 (1988)
;~ 9. Fukuda et al., J. ~iol. Chem., 261:237602383
(1986~ -
10 . Nudelman et a} ., J . Biol . Chem., 263 : 13942-
13951 (1988)
~ 11 . Howard et al ., J . Biol . Chem., 262 : 16830-16837
(I987)
12 . Johnson et al., Biochem. Soc. Trans. p. 3~6
( 1987 )
: 13. Foster et al., J. Biol. Chem., 266:3526-3531
~lg91~
1~. Smith et al., J. Biol. Chem., 262:12040-12047
(1987)
15 . Paulson at al ., J. Biol . Chem., 253: 5617-5624
(1~78)
16. Ichika~7a et al., J. Amer. Chem. Soc. 113:4698-
470~ (1991)
17. Nilsson et al., J. Carbohydr. Chem., 2o l-19
(1990)
18 . Gokhale et al ., Can. J . Chem., 68 : 1063-1071
~199D)
W092/22662 2 1 1 ~ 9 ~ i PCT/CA92/00~51
19. Mazid et al., U.S. Patent Appl. Serial No.
07/336,932 entitled: "Process for the
Separation and Purification of Sialyl
Transferases", filed: April 12th, 198~
20. Weinstein et al., J. Biol. Chem., 257:13835-
138~4 (1~2)
21. Unv~rzagt et al., J. Amer. Chem. Soc.,
112:9308-9309 (1990)
22. Ippolito et al., U.S. Patent Appl. Serial No.
07/714,161 entitled: "Methods for the Enzymatic
Synthesis of AlpAa-Sialylated Oligosaccharide
Glycosides~', filed June 10, 1991
23. Sialic Acids in "Cell Biology ~onographs"
Schauer, Editor, 10:6 (1982)
~4. Reuter et al., ~lycoconjug~e J., 5:133-135
(1988)
25. Weinstein et al., J. Biol. Chem., 2S7:13835-
13844 ~1982)
26. Paulson t al., J. Biol. Chem., 2~2:2356-2362
(1977)
~7. Paulsen, Agnew. Chem. Int. Ed. Eng., 25:155-173
: (1982)
2~.. Schmid , Agnew. Chem. Int. Ed. Eng., 25:212-235
(1986)
29. Fugedi et al., ~lycocon;. J., 4~97 108 ~lg87)
30. Alais ~t al., Carbohydr. Res., ~07~ 31 (1~90)
~ 31. Smith and Ziola, Immunology, 5~:245 (1986)
: 32. Sleytr et al., Arch. Microbiol., 146:19 (19B6)
33. Lowe et al., ell, 63:475-485 (1990~
39 34. Macher t al. Glycobiology 1(6):577-584 (1991)
35. Lowe et al., J. Biol. Chem., 266:17467-17477
(1991)
36. Piller et al., J. Biol. Chem., 258 (1983
12293-12~99.
5 37. Hosimi et al., Japan J. Med. Sci. Biol. 42
(1989) 77-82.
WQ9V22662 PCT/CA92/00251
9~
~ __ 4 __
38. Yates et al., Carbohydr. Res., 120 (1983~ 251-
268.
39. Hosomi et al., J. BiochPm., 95 (1987) 1655-
165g .
40~ ~ielenski et al., FEBS Lett. 158 (1983) 164-
168.
41. Hosimi et al., Japan J. Med. Sci. Biol~ 38
~1985) 1-8.
42. Van den Eijnden et al., J. Biol. Chem., 258
1~ (1983) 3435-3437,
43. Van den Eijnden et al., J. Biol. Chem., 263
(1988) 1246I-12471.
44. Basu et al., J. Biol. Chem., ~9 (19.84) 12~57-
12562.
45. Hosomi et al., Jpn. S. Vet. Sci. 51 ~1989) 1-~.
46. Holmes et al., J. Biol. Chem. 262 (1~87) 15649-
: 15658.
~ 47. Palcic et al., Glycobiology, l:205-209 (1991)
; 4~. Palcic et al~, Carbohydr. Res., ~2:315-324
2~ (19~7)-
4g. Wong et al., 3. Am. Chem. Soc., 113:8137-3145
( 1991)
50. Ekberg et al., Carbohydr. Res. 110:55-67 (1982)'~
51. Dahmen et al., Carbohydr. Res. 118:292-301
~1983)
52. Rana et al., Carbohydr. Res. 91:149-157 (1981)
53. Amvam-Zollo et al., Carbohy~r. Res. 150:199-212
(1986)
54. Paulsen et al., Carbohydr. Res. 104:195-219
(1982)
55. Chexnyak et al., Carbohydr. Res. ~28:269-2X2
(1984)
56. Fernandez-Santana et al., J. Carbohydr. Chem.
8:531-537 (1989)
57. Lee et al., Carbohydr. Res., 37:193 et seq.
(1974)
W092~22662 21 1 0 9 ~ 7 PCT/CA92/002~1
58. ~atcliffe et al., U.S. Paten~ Application
Serial No. 07~278,106, filed November 30, 1988.
59. Jiang et al., "Chemical Synthesis of GDP-
Fucose", U.S. Pa~ent Application Serial No.
07/848,223 filed March 9, 1992.
60. Wainstein et al., J. Biol. Chem., Vol. 2~7, No.
22, pp. 13835-13844 t9182).
61. Lemieux et al.~ Can. J. Chem., 5~:631-6~3
(1980).
62. Ichikawa et al~, Anal. Biochem., 202:215-238
(1992).
63. Schenkman et al., Cell, 65:1117-1125 (1991).
64. Thiem et al., A~gsw. Chem. Int. Ed.,
30(11):1~03-1505 (1991).
15 65. Venot et al., U.S. Patent Application Serial
No. 07/771,~59, filed October 2, lg91.
66. Lemieux et al., U.5. Pa~ent No. 4,137,401,
January 30, 1979.
67~ Ma~sumoko et al., ~nal. Biochem., 116 (1981
1~3-110.
: 68. Kukowska-~atallo et al., Genes and Developm~nt,
4:12~8-1303 (19~0).
69. ~umas et al., Bioorg. Med. Letters, 1:425-428
( 1~91) .
70. Prieel~ et al., J. Biol. Chem., 25Ç:10456~104~3
~ 81).
71. Eppenberg2r-Castori et al., Glyccconj~ J.
~: S:101-114 (1~89~.
72. Nunez, et al., Can. J. Chem., 59:2086-2095
, 3~ (~981~
73. Gokhale et al., Can. J. Chem., 68:1063-1071
( 19gO)
74. Schmidt, et al., Liebigs Ann. Chem., 121-12
( 1991)
75. Veeneman, et al., Tetrahedron Lett~, 32:6175-
~178 (1991)
,, . ., . . ~ .. . ., . . , . .. ., . . ., . . .. , .. . .. , . , i, .. . .
W092/22662 PCT/CA92/00251
9~
----6
All publications and patent applications
mentioned in this specification are indicative of
the level of skill of those skilled in the art to
which this inv~ntion pertains. All publications and
patent applications are herein incorporated by
reference in their entirety to the same extent as if
each individual publication or patent appliration
was specifically and individually indicated to be
incorporatPd by reference in its entirety.
3. State o~ the Art.
The art teache~ that specific oligosaccharides
such as sialylated and fucosyla~ed structures are
in~olved as ligands in cell adhesion phenomena.
Similarly, oligosaccharide glycosides relating t~
b}vod group determinant structures have been found
to impart immunosuppressive and tolerogenic
properties to mammals when th~ mammals were
previously challenged with an antigen. See Ippolito
et al.22, which application is ineorporated herein ~y
r~ference in its entirety. In this regard,
oligo~accharide glycosides relating to blood group
determinant strueture include the compsund
~Gal(1-4)~GlcN~c(1-3)~Gal(1-4)~GloNAc-OR depicted in
Figure 1 of thi~ application as compound la.
Ippolito et al. 22 fur~her discloses that blood
group determinant oligosaccharide glycosides having
a ialic acid group ~or an analogue thereof) at the
non-reducing sugar terminus of ~he oligosaccharide
glycosides and which are also monofurosylat2d
possess immunosuppressive and tolerogenic properties
(e.g., sialyl LewisX -- Compound III in FIG. 12 of
Ippolito et al. 2Z),
In view o~ the above, we desired to prepare a
monofucosylated derivative of compound la having a
sialic acid group (or an analogue thereof~ at the
non-reducing sugar terminus o~ this compound wherein
- 2 1 ~ U '! ~ ~
the fucosyl group was pendant to the 3-hydroxy of
only one of the GlcNAc groups.
A synthetic appro~ch employing enæymatic
sialylation and fucosylation steps is particularly
appropriate in order to provide ~n efficient route
for the preparation of sialylated and monofucosyl-
ated derivatives of compound la. In this regard,
since the work of Sabesan et al.,Z sialyltrans-
ferases, mostly the BG~ 4)BGlcNAc ~(2-6)- and the
: 10 BGal(1-3/4)~GlcNAc ~(2-3)-sialyltransferases
from rat liver and the BGal(1-3j~GalNAc
~(2-3)sialyltransferase from porcine submaxillary
gland have often been used for synthetic purposes.3
The former two sialyltransferases are useful in
sialylating a terminal B~al(1-4)~GlcNAc- group in an
: oligosaccharide glyccside. ~he latter
: sialyltransferase which has a wide acceptor
specificity, i5 useful in sialylating a terminal
BGal(1-3~GlcNAc- group in oligosaccharide
glycosides based on the LewisC (Type I) backbone.
In this regard, Palcic et al. 4 employs such
: sialyltransferases in the sequential enzymatic
sialyation and fucosylation to prepare sialyl Lewisa
derivatives. However, because of the low affinity
of the ~Gal(1-3)~GalNAc ~2-3)sialyltransferase for
the Type II backbone, the synthesis of sialylated
N acetyllactosaminyl structures, such as those
pres~nt in the sialyl LewisX,s sialyl dimeric LewisX6
o~ the corresponding internally monofucosylated
derivative7, by use of this sialyltransferase is
much more di~ficult. 4
By using fucosyltransferases of various
specificities, the biosynthetic pathway leading to
sialyl LewisX and the sialylated dimeric Lewi~X
struc~ures has been shown to proceed by the
sequential sialylation followed by fucosylation of
SI~BSTITUTE SH~ET
2 1 1 0 9 9 ~
-- 7/1 --
the Type II precursors~ 8a-d A similar process
"extension, sialylation, fucosylation" has also been
proposed9 to lead to internally fucosylated repetitive
Type II terminal structures, such as:
~Neu5Ac(2-3~BGal(1-4)BGlcNAc(1-3)BGal(1-4)-
SUBS~T~ S~tElET
W092~2662 ~ 9 9~ PCT/CA92/n02~1
[~Fuc(1-3)]BGlcN~c-.9 The identification of the new
terminal structure BGal(1-4)BGlcNAc(1-3)BGal(1-4)-
[~uc(1-3)]B~lcNAc-, defined by the antibody ACFH-
181, led to a proposed new biosynthetic pathway such
as 'lelongation followed by selective internal
fucosylation". While patterns of initial internal
monofucosylation of di-N-~cetyllactosaminyl
glycolipids have been observed for fucosyltrans-
ferases present in LECII Chinese Hamster Ovary
lQ mutant~ and in human colonic adenocarcinoma Colo 205
cells8b, these fucosyltransferases are not readily
av ilable and/or do not selectively lead to
monofucosylated structures. Similarly, while
fucosyltransferases possess.ing the specificity
requir~d for the synthesis of the internally
fucosylated structure ~Neu5Ac~-3)~Gal(1-4~GlcNA~-
~Gal(l-4)c~uc(l-3)]GlcNAc have been identified34 and
in one ca~e a recombinant enzyme35 has been
identified, their availability is also limited.
~oreover, other ~(1-3)fucosyltransferases do not
transfer L-fucose onto N-acetylglucosamine moieties
found in acceptors possessing a terminal aNeu5Ac(2-
3)BGal(1-4)~GlcN~c- sequence .12~13~34 It has already
been no~ed that "the order of addition of ~ 3)
fucose in N-acetyllactosaminyl sequ~nces of
glycoconjugates will then depend upon the particular
3)fucosyl-transferase present"l1.
~he single fucosylation at the internal N-
acetylglucosamine unit of the ~(2-6~sialyl di-N-
: 30 acetyllactosaminyl sequence leading to the terminal
structure ~Neu5Ac~2-6)BGal(l-4~GlcNAc(1-3)~
~Gal(1-4)t~Fuc(1-3)]BGlcNAc-& (alsG proposed for a
sialylfucopentaose from human milk14j is in agreement
with the proposed mutually exclusive glycosylation
pattern of the BGal(1-4)BGlcN~c a(2 6)sialyltrans-
~erase and the BGal(1-3/4)~GlcNAc a~1-3/4)fucosyl-
", ,, ,, ; - .
, , ~
W092/22662 2 1 10 ~ 7 PCT/CA92/0U2~1
transferase in the synthesis of asparaginyl linked
oligosaccharides in glycoproteins.15
In view of the above, processes which would
enzymatically prepare sialylated and monofucosylated
derivatives of compound la without the need o
employ a fucosyltransferase specific for
monofucosylation would be particularly desirableO
The presant in~ention is based, in part, on the
discovery of synthetic pathways which utili7.es
enzymatic fucosylation and sialylation steps and
which r~sult in th~ selective formation of
monofucosylated derivatives of compound la without
the need to employ a fucosyltransferase which is
specific for monofucosylation on either of the
GlcNAc units o f compound l~.
8~ARY OF TE~ INV$NTIO~
Thi~ invention is directed, in part, to the
disco~ery that fucosylation onts the 3-hydroxyl
~roup of the GlcNAc saccharide in a ~Gal~l-4~GlcNAc
disaccharide via an ~ 3~fucosyltransferase (e.g.,
: ~Gal(l-3/4)~GlcNAc ~ 3/4) fucosyltransferase) is
. dependent on the presence of a 6-hydroxyl group on
the ~al saccharide and when this hydroxyl group is
blocked by a r~movable blocking group, fucosylation
on the neighboxing GlcNAc group is pravented. In
this aspect, tha methods of ~his invention employs
this characteris ic of ~ 3)fucosyltransferases to
provide for a means to selectively monofucosylate
c~mpound la which are used advantageously to prepare
compounds Sa and 12.
In another method aspect, the present invention
is directed to the disco~ery of enzymatic methods
and chemical/enzyma~ic methods to prepare the
compound aNeu5Ac(2-3)BGal(l-4)BGlcNAc(l-3)BGal(l-4)-
t~Fuc(1-3)]~GlcNAc-OR.
W092/22662 PCT/CA92~00251
~hu
s, in one of its method aspects, the present
invention is directed to a method for preparation of
a compound of the formula I:
OH OH OH
I ~ NHAc OH ~
7 ~ ~o~O~--O-R
OH HO OH NHAc
S wherein R is an aglycon group having at lea~t one
carbon atom, Y is L-fucose and Z is sialic acid or
an analogue of sialic acid, which method comprises
the ~ollowing steps:
(a~ preparing a compound of the ~ormula II
OH O-X OH
I ~ NHAC OH
110S~5~~~~ -~,0-R
OH OH HO OH NHAc
wher~in R is as defi~ed above and X is a removable
blocking group;
(b~ fucosylating the compound prepared in (a)
above with an ~(l 3)~ucosyltr~nsferase so as to form
a mono ucosylated derivative of the formula III:
OH C~X o~ -
I ~ NHAc OH ~
HO~ ~ ~ O ~ ~ O-R 11i
OH C)H HO QH Y NHAc
wherein X, Y and R are as defin~d above;
(c) removing the removable blocking group from
the compound formed in (b) above; and
(d) æialylating the compound formed in (c)
above with sialic acid or an analogue of sialic acid
using an ~12 3)sialyltransferase so as tD ~orm the
c~mpound of ~o~ula I.
In regard to the a~ove, the sialylation of the
oligosacch~ride glycoside so as to form an
SUBS~ITUTE SHEET
ISA/~P
W092/22662 21 1 0 9 9 7 PCT/CA92/00251
~(2-3)sialyl residue at the non-reducing sugar
terminus of ~he oligosaccharide glycoside is
necessarily after removing ~he blocking group
because sialylation with an ~(2-3)sialyltransferase
requires ~he presence of a free hydroxyl group at
the 6-position of ~he terminal galactose residue on
the oligosaccharide glycoside.
In another of its m~thod aspects, the present
invention is directed to a me~hod for preparation of
a compound of the formula IV:
~ ~ OH
1 ~ NHAc OH ~
Z O~O~o~O~O-R l~/
- OH OH HO OH NHAc
wherein R is an aglycon group having at least one
carbon at~m, Y~ is ~-~ucose and Z is sialie a id or
an analogue of siali~ a~id, which method c~mprises
the foll4wing steps:
(a) preparing a compound of the formula V
OH OH OH
I ~' NHAc ol~ ~
HO~"O~O~O~,~,,O-R V
OH OH HO O X' NHAc
wherein R is a~ defined above and X' is a removable
blocking group,
(b) sialyl~ting the compound formed in (a)
above with sialic acid or an analogue of sialic acid
usi~g an ~(2-3)sialyltransferase; and
(c) fucosylating the compound prepared in ~a)
a~ove wi~h an ~ 3)fucosyltransferase so as to form
a monofucosylated deriva~i~e of the formula VI:
OH OH OH
I ~ NHAc OH ~
ZO~OY'~O~O~_O-F~ Vl
OH OH HO OX' NHAc
SUBSTITUTE SHEET
ISAJEP
wog2r22662 ~9~ PCT/CA92/~0251
-- 12 -
wh~rein X', Y' and R are as defined abova;
(d~ removing th2 removable blo~king group from
the co~pound formed in (c) above so as to form a
compound of formula IV with the proviso that X' is a
~locking group o~her than sialic acid.
In regard to the above, the sialylation of the
oligosaccharide glycoside so as to form an
~(2-3)sialyl residue at the non-reducing sugar
termi~us o~ the oligosaccharide glycoside is
necessarily befor~ tha fucosylatisn step because
sialylation wi~h an ~(2-3~sialyltransferase will not
proce~d when Y' is L-fucose.
Preferred removable blocking groups for use in
the above described methods include sialic acid
groups and benzyl groups and any other group that
can be introduc~d either enzymatically or chemically
on the pre~uxsor leading to II or ~ and later
sel~ctiv~ly enzymatically or chemically r~mo~ed in
mild conditions compatible with the nature of the
product. In co~pound V, the blocking group X' i~;
not ~;ialic acid because this compound would be
difficult to syn~hesize.
In one of its co~position aspects, the present
i~Yention is ~irec~ed to a compound o~ the formula ~'
YII:
OH OH ~H
1 ~ NHAc OH
ZO ~ O ~ O ~ O ~ O-R
OH OH HO O H NHAc
wherein R i5 an aglycon having at least l carbon
atom~ Y and Y' are selected from the group
consisting of hydrogen and L-fucosyl wi~h the
provi50 that one of Y and Y', but not bo~h, is
hydrogen, and Z is sialic acid or a sialic acid
analogue.
S~JBSTIT~JTE SHEET
..
W092/22662 PCT/CA92/002~1
~09n7
l3 __
In a preferred embodiment, the aglycon moiety,
R, is selected from the group consisting of -(A)-Z'
wherein A represents a bond, an alkylene group of
from 2 to 10 carbon atoms, and a moiety of the form
~ (CH2-CR2G) n- wherein n is an integer equal to 1 to
5; R2 is elected from the group consisting of
hydrogen, methyl, or e~hyl; and G is selected from
the group con~i~ting of hydrogen, halogen, oxygen,
sulphur, nitrogen, phenyl and phenyl substituted
with 1 to 3 substituents selected from the group
co~sisting of amine, hydroxyl, halo, alkyl of from 1
to 4 carbon atoms ~nd alkoxy of from 1 to 4 carbon
a~oms; and Z' is selected from the group con isting
of hydrogen, methyl, phenyl, nitrophenyl and, when G
is not oxygen, sulphur or nitrogen and ~ is not a
bond, then Z' is al50 selected from the grsup
consisting of -O~, -SH, -NH2, -NHR3~ -N(R3)2,
; -C(O)OH, -C(O~OR3, -C(O)NH-N~2, -C(O)NH2, -C(O)NHR3,
C(O3N(R3~2, and -OR4 wherein each R3 is
ind~pendently alkyl of from 1 to 4 carbon atoms and
R4 is an alkenyl group of from 3 to 10 carbon atoms
with the proviso that when A is a bond then Z' is
. not hydrogen.
Preferably, the aglycon group is a hydrophobic
group. Mo~t prefera~ly, the aglycon moiety is a
hydrophobic group selected from the group consisting
of ~(CH2)gCOOCH3 and -~CH2~5OCH2CH=CH2 and
- ( C~2) 8CH20H -
The monosialylated and monofucosylated
compounds of this inventisn are particu~arly useful
in modulating a cell-mediated immune inflammatory
response. Accordingly, in another of its
composition a5pects, the present invention is
directed to a pharmaceutical compositi9n suitable
~or administration to a mammal (e.g., human) which
comprices a pharmaceutically inert carrier and an
effective amount of ~he compound of Formula I or IV
W092/22662 PCT/CA92/002~1
~,, h~ ~ 9 9 ---- 14
to modulate a cell-mediated immune response in said
mammal.
In another of its method aspects, the present
invention i~ directed to a mPthod for modulating a
cell-mediated immune r~sponse in a ma~mal which
m~thod comprises administering to said manmal an
amount of a compound of Formula I or IV eff ctive in
modulating said immune response.
BRIEF DE8CRIPTIO~ OF T~E D~A~ING~
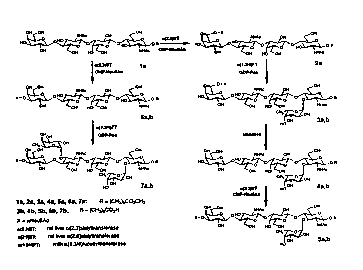
~OFIG. 1 illustrates the synthetic pathway
leading to Sialyl dimeric LewisX and internally
monofucosylated derivatives thereof. In FIG. 1, the
nomenclature for compound la is ~Gal(1-4)~GlcNAc(1-
3)~al~1-4)~GlcNAc OR, sometimes called di-N-
15 - acetyllactosaminyl tetrasaccharide. Similarly, the
hexasaccharide moiety present in rompounds 5~ and 5b
. in FI G . 1 is sometimes called VIM-2 epitope sr CD-
655 and 7~ and 7b are called sialyl dimeric LewisX.
FIG. 2 illustrates the synthetic pathway
leading to th~ externally monofucosylated
dQrivatives of the sialyl di-N-acetyllactosaminyl
hapt~n.
FIGURE 3 illus~rates an enzymatic pathway
leading to monofucosylated and monosialylated
compounds of Formula I.
,
FIGURE 4 illustrates an alternative chemical
synthesis of trisaccharide lg which can than be used
as per Figure 3 to prepare monofucosylated and
monosialylated compounds of Formula I.
W092/22662 PCT/CA92/00251
21 ~ 0997
-- 15 --
FI~URE 5 illustrates that the enzymatic pathway
set forth in Figure 3 can be used to extend the
structure of the hexasaccharides of Formula I.
DE~AI~D D~C~IPTION OF ~E~ INY$~TIO~
The present invention is directed, in part, to
the disc~ery that selective monofucosylation of
compound la (i.e.~ ~Gal~l 4)~GlcNAc(1-3)~Gal-
(1-4)~GlcNAc-OR), can be achieved by appropriately
blocking the 6-hydroxy group on the galacto~e unit
adjacent to ~lcNAc unit (in the non-reducing sugar
direction) so as to prevent fucosylation .of the
GlcNAc unitO
The present invention is also directed, in
part, to the discovery that the preparation o~
compounds of Formula I can be achieved by a complete
enzyma~ic process or by a chsmo/enzymatic processO
In eith r case, the synthetic steps employed in
~he synthesis of the monofucosylated de~iYatives are
critical to produce ~he monofucosylated deri~ati~es.
The prss nt inv4ntion is still further directed
. to the discoYery that the compounds of Formula I and
II are use~ul for ~n vivo modulation of a cell
m~diated immune response in mammals, including
humans.
~owever, prior to discussing this invention in
~urther detail, the following terms will first be
defined.
I
A. Definitions
As used herein, the following terms have the
de~initions gi~en b~low:
The term "cell-mediated immune response to an
antigen in a mammal" refers to those mammalian
immun~ responses which are mediated by cell-eell
in~eractions. Included within this term are cell
W092/22662 9~ - - 16 -- PCT/CA92/00251
mediated inflammatory responses to an antigen such
as DTH responses as well as cell-mediated
inflammatory responses arisi~g from myocardial
infarction, virus-induced pneumonia, shock and
sequelae (e.g., mul~iple organ failure~, adult
respiratory distress syndrome, psoriasis, art~ritis,
and ~he like. Preferably, the cell-mediated immune
response is a leucocyte-mediated response.
The term "N-acetyllac~osamine" or "LacNAc"
refers to ~he di~accharide ~Gal(1~4)~GlcN~c which is
represent~d by the for~ula:
HO OH
- I ~ NHAc
HO ~O~
OH . OH
The t~r~ "di-N-acetyllactosaminyl struc~ures"
:~eans that one N-acetyllactosamine unit i~
glycosidically linked in a ~ linkage to the 3-OH of
the ~Gal of the second unit attached to the aglycon.
The term "sialic acid" means all of the ~
naturally occurring structures of sialic acid
including (N-acetylated) 5-amino-3,~-dideoxy D-
glycero-D-yalacto-nonulopyranosylonic acid
~eu5Ac") and th~ natur~lly occurring analogue~ of
Neu5Ac, including N-glycolyl neuraminic acid
(Neu5Gc~ and 9-0-acetyl neuraminic acid (Neu5,9A~),
which are compatible with the selected
sialyltrans~erase. A complete list o~ naturally
occurring sialic acids known to date are provided by
Schauer~,
Naturally occurring sialic acids which are
recognized by a particular sialyltransferase so as
~o bind to the enzyme and are then a~ailable for
SUBSmlJTE SI IEET
ISJ~/EP
W092/22662 21~ ~ ~ g 7 PCT/CA92/00251
-- 17 --
transfer to an appropriate acceptor oligosaccharide
structure are said to be compatible with the
sialyltransferase and are sometimes referred to
herein as a "compatible naturally occurring sialic
acidl'.
The term l'analogues of sialic acid" refers to
analogues of naturally occurring structures of
sialic acid including those wherein the sialic acid
unit has been chemicalIy modified so as to introduce
and/or remove one or more functionalities from such
structures. For example, such modification can
result in the removal of an -OH functionality, the
introduction of an amine functionality, the
introduction of a halo functionality, and the like.
Certain analog~es of sialic acid are known in
the art and i~clude, by way of example, 9-azido-
Neu5Ac~ 9-amino-Neu5Ac, 9-deoxy-Neu5Ac, 9-fluoro-
Neu5Ac, ~-bromo-~eu5Ac, 8-deoxy-Neu5Ac, 8 epi-
Neu5Ac, 7-d~oxy-Neu5Ac, 7-epi-Neu5Ac, 7,8-bis-epi-
Neu5Ac, 4-0-methyl-~eu5Ac, 4-N-acetyl-Neu5Ac, 4,7-
di-d~oxy-Neu5Ac, 4-oxo-N u5A., 3-hydroxy-Neu5Ac,
3-fluor~-Neu5Ac acid as well as the 6-thio analogues
of Neu5Ac. The nomenclature employed herein in
descri~ing analogues of sialic acid is as sek forth
by Reut~r et al.24
Insofar as sialyltransferases are d~signed to
transer or donate compatible naturally occurring
sialic acids, analogues of Neu5Ac ar2 sometimes
r~ferred to herein as "artificial donors" whereas
the compatible naturally occurring sialic acids are
sometimes referred to herein as the "natural
donors".
The term "sialyltransferase" refers to those
enzymes which tran~fer a compatible naturally
occurring sialic acid, activated as its cykidine
monophosphate ( CMP) derivative, to the terminal
WO92J22662 PCT/CA92/002~1
? ~99 1 -- 18 -- "`
galactose group prese~t on natural acceptor
structures comprising those terminating in
~Gal(1-4)~GlcNAc and ~Gal(1-3)~GlcNAc di~accharides
and include enzymes produced from microorganisms
g~netical~y modified so as to incorporate and
expre~ all or part of the sîalyltransferase gene
obtained from ano~her source, including mammalian
~ources~
Such sialyltransfer ses comprise those that
havs been ide~tified in the literature as leading to
the follo~ing structures:
~rNeu5Ac ~ 2 -3 ) BGal ( 1-3 / 4 ) BGlcNAc-
~Neu5Ac ( 2--6 ) BGal ( 1-4 ) BGlcNAc-25 26
lS Analogues of sialic acid which are rec:ognized
by a particular ~ialyltransf erase so as to bind to
the enzyDIe and are then available $or trans~er to an
appropriate acc:eptor oligosacch~ride structure are
said to be l:o~patible with the sialyltransferase and
are someti~es referred to herein as a "compatible
analogue o~ sialic acid". Because the transfer
reaction employs a sialy}transferase, it goes
without saying that an analogue o~ sialic acid
employed in such a reaction must be a compatible
analogue o* sialic acid.
CMP-nucleotide deri~ati~e of Neu5Ac refers to
~he compound:
NH2
OH
H ~ ~} ' f J ~ ¢,~,\
CMP Neu5Ac
HO o~l
SlJBSmUTE S}lEF~
- ISAJEP
W092J22662 PCT/CA92/00251
2 l i 0 9 ~ 1
---- 19 ----
CMP-der.ivatives of analogues of sialic acid refer to
those compounds having struc~ures similar to that
above with the exception that ~he Neu5Ac residue is
replaced with an analogue of sialic acid.
The term "~ 3)fuco~yltransferase" refers to
any fucosyltransferase which transfers L-fucose and
compatible analogues of ~-fucose from GDP-fucose to
the 3 hydroxy position of GlcNAc in a LacNAc group
(~Gal(1-4)~GlcNAc) in an oligosaccha-ride glycoside
. 10 and which does not discriminate between ~Gal(1
4)~GlcNAc groups in the oligo-s~ccharide glycoside.
The particular ~ 3)fucosyl-transferase e~ployed is
compatible with the intended reaction. That is to
say that the selected ~ 3) fucosyltransferase will
~ind to the oligosaccharide glycoside e~ployed and
transfer L-fucose to the 3 hydroxy position of
GlcN~c in a ~Gal(1-4~GlcNAc group of the
oligosaccharide glycoside. Suitable fuco~yltrans-
ferases include the known ~Gal(}~3/4)~GlcNAc
~0 ~(1~3/4)f~cosyltransferase which is readily obtained
f human mil~4~70~72 and the ~Gal(1~4)~GlcNAc
: ~(1~3)fucosy}transferase which is also found in
human serum and is co-rec~vered with the
~Gal(1~3/4~GlcNAc a(l~3/4~fucosyltransferase. A
2S recombinant form of ~al(1~3/4)~GlcNAc
3/4)~ucosyltransferase is al~o available~69.
Compatibl~ analogues of L-fucose refer to
naturally ocourring and synthetic analogues of
fucose including those where the fucose unit has
been chemically modified so as to introduce and/or
xemove one or more functionalit~es from this
structure. For example, such modification can
result in the removal of an -OH functionality, the
introduction of an amine functionality, the
introduckion o~ a halo functionality, and the like.
Certain compatible analogues of fucose are
known in the art and include, by way of example,
W092/22662 99~ PCT/CA92~0025i
-- 20 --
3-deoxy-fu~ose, ~rabinose, and the like .18
The term "removable blocking group" refer~ to
any group which when bound to the 6-hydroxyl of the
galactose unit in a ~Gal(1-4)~GlcNAc group prevents
fucosylation of the 3-hydroxyl of the GlcNAc by an
a~1-3)fucosyltransferase and which group can be
removed by conventional chemical or enzymatic steps
to reestab}ish the 6-hydroxyl on the galactose unit.
The particular removable blocking group employed is
not critical and preferred removable blocking groups
include Neu5Ac and benzyl substituents and any other
group that can be introduced either enzymatically or
chemically on the precursor leading to II or V and
later selectively enzymatically or chemically
removed in mild con~itions compatible with the
nature of the product. One such additional
contemplated blocking group is ~-galactose which can
be removed enzymatically with an ~-galactosidase.
The term "removable protecting group" refers to
any group which when bound to one or more hydroxyl
groups o~ the galactose, N-acetylglucosamine, etc.
which pr~vent reactions from occurring at these
hydroxyl groups and which protecting group can be
removed by conventional chemical or enzymatic steps
to r~establish ~he hydroxyl group. The particular
re~ovable protecting group employed is not critical
and preferred remo~able hydroxyl pro~ecting groups
: include conventional substituents such as benzyl,
acetyl, chloroacetyl, benzylidine, t butyl-
diphenylsilyl and any other group that can be
introduced either enzymatically or chemically onto a
hydroxyl functionality and later selectively removed
sither by enzymatic or chemical methods in mild
conditions compatible with the nature of the
product. One such additional contemplated
protecting group is a ~-galactose which can be
rem~ed enzymatically with an ~-galactosidase.
W092~22662 PCT~CA92/002~1
2 1 1 ~) 9 9 i
. -- 2l --
The term "pharmaceutically acceptable salts"
includes the pharmaceutically acceptable addition
sal s of the compounds o~ Formula I derive~ from a
~ariety of organic and inorganic counter salt well
known in the art and include, by way of example
only, sodium, potassium, calcium, magnesium,
ammonium, te~ralkylammonium, and the like.
The term "aglycon" refer to the R substituent
on the hexasacch~ride glyco ides of formula I and
IV. In general, R is an aglycon having at least l
carbon atom. In a preferred embodiment, the aglycon
moiety, R, is selected from *he group consisting of
~(A)-Z' wherein A represents a bond, an alkylene
: group of from 2 to lO carbon atoms, and a moiety of
15 the form ~ (~H2-CR2G) n~ wherein n is an integer equal
to l to 5; R2 is selec~ed ~rom the group consisting
of hydrogen, methyl, or e~hyl, and G is selected
~rom the group con isting of hydrogen, halogen,
o~ygen, sulphur, nitrogen, phenyl and phenyl
substituted with 1 to 3 substituents select~d from
the group consisting of amine, hydroxyl, halo, alkyl
of from 1 to 4 carbon atoms and alkoxy of from 1 to
4 carbon atoms; and Z' is selected from the group
consisting of hydrogen, methyl, phenyl, nitrop~enyl,
and, wh~n G is not oxygen, sulphur or nitrogen and A
is n~t a bond, then Z~ is also selected from the
group consisting of -OHJ -SH, -NH2, -NHR3, -N~R3)2,
-C(O3O~, -C(O)OR3, -C(O)NH-NH2, -C(O)NH2, C(O)NHR3,
-C(O~N~3)z~ and -OR4 wherein each R3 is
independently alkyl of from l to 4 carbon atoms and
R4 is an alkenyl group of from 3 to lO car~on atoms
with the proviso that when A is a bond, Z' is not
hydrogen.
In those cases where the aglycon is one which
permits linkage of hexasaccharide glycoside I and/or
IV to a carrier~ then the aglycon is praferably
selected from the group consisting of -(A)-Z'~
W092/22662 ~Q g~1 PCT/CA92/00251
-- 22 --
wherein A is selected from the group consisting of
an alkylene group of from 2 to lO carbon atoms and a
moiety of the form -(CH2-CR~G) n~ wherein n is an
integer equal to 1 to 5; R~ is selected from the
group consisting of hydrogen, methyl, or ethyl; and
G is selected from-thè group consisting of hydrogen,
oxygen, sulphur, nitrogen, phenyl and phenyl
substituted with l to 3 substituents selected from
th~ group consisting of amine, hydroxyl, halo, alkyl
of from 1 to 4 carbon atoms and alkoxy of from 1 to
4 carbon atoms; and Z'' is selected from the group
consisting of hydrogen and, when G is not oxygen,
sulphur or nitrogen, then Z'' is also selected from
the group consisting o~ -OH, -SH, -NH2, -NHR6,
-C(O)OH, -C(O)OR6, -C(O)N~NH2, and -OR7 wherein each
R6 is independently alkyl of from l to 4 carbon
atoms and R7 is an alkenyl group of from 3 to lO
~ carbon atoms with the proviso that when A is a bond,
- Z is not hydrogen. In such cases, the -(A)-Z''
group defines a group capable of being linked ~o a
carrier or is capable of being derivatized to a
group which is capable of being linked to a carrier.
The choice of an appropriate carrier may be useful
in enhancing immunogenic properties.
Numerous aglycons are known in the art. For
example, a linking arm comprising a para-nitrophenyl
group (i.e., -YR = -OC6H4pNO2) has been disclosed by
Ekberg et al.50 At the appropriate time durin~
synthesis, the nitro group is reduced to an amino
group which can be protected as N-trifluoro-
~cetamids. Prior to coupling to a support, the
trifluoroacetamido group is removed thereby
unmasking the amino group.
A linking arm containing sulfur is disclosed by
Dahmen et al.51. Specifically, the lin3cing arm is
derived from a 2-bromoethyl group which, in a
substitution reaction with thio-nucleophiles, has
WO~/22662 PCT/CA92/00251
2110~9~
- 23 --
been shown to lead to linking arms possessing a
variety of terminal ~unctionàl groups such as
-OCH2CH2SCH2C02CH3 and -OCH2CH2SC6H4 PNH2
Rana et al. 52 discloses a 6-trifluoroacetamido-
hexyl linking arm (-O-(CH236-NHCOCF3) in which the
trifluoroacatamido protecting group can b~ removed
unmasking the primary amino group us~d for coupling.
Other exemplifica~ion of known linking arms
include the 7-methoxycarbonyl-3,~,dioxaheptyl
~0 linking arm53 (-OCH2-CH2)2OCH2CO2CH3; the
2-(4-methoxycarbonylbutancarboxamido~ethyl54
t OCH2CH2NHC(O~(CH2)4CO2CH3) the allyl linking arm55
~OCH CH=CH2) which, by radical co-polymerization wi h
an appropriate monomer, leads to co-polymers; other
allyl lin}cing arms56 [-O (CH2CH20~ 2CH2CH=CH2] .
Additionally, allyl linking arms can be derivatized
in the presence of 2 aminoethanethiolS7 to provide
for a linking arm -OCH2CH2CH2SCH2CH2NH2.
Additionally, as shown by Ratcliffe et al.5~, R
group can ba an additional saccharide or an
oligosaccharide containing a linking arm at the
reducing sugar terminus.
The carrier is generally a small molecular
weight, non-immunogenic or antigenic carrier
including the linking to a flusrescent label, a
rad~oactive label, biotin, or a photolabile linking
arm or a moiety to be targetted.
In either case, the aglycon moiety is
preferably a hydrophobic ~roup and more preferably a
hydrophobic moiety selected from the group
consisting of -(CH2) 8COOCH3 and -(CH2)5OCH2CH=CH2-
In particular, the use of a hydrophobic group and
most especially, a -(CH2) 8COOCH3 sr -(CH2~5QCH2CH=CH2
group may pro~ide for some enhancement in thP
kineti s of sialic acid transfer ~ia a
sialyltrans~era~e.
W092~22662 9g~ PCT/CA92/002~i
-- 24 --
As is apparent, hexasaccharide glycosides I and
IV descr~bed above are differsnt from
oligosaccharides and glycocQnjugates because the
aglycon moiety (R) is not hydrogen, a protein, or a
S lipid capable of forming a micelle or other large
aggregate structure.
B. Synthesis and MethodoloqY
Rl. Preparation of Starting Materials
Tetr.saccharide glycosides II and V are readily
prepared ~ither ~y complete chemical synthesis or a
chemical/~nzymatic synthesis as described below.
Specifically, tetrasaccharide glycoside II and V can
be prepared by chemically coupling the indiYidual.
saccharide units. Such coupling can r~adily be
~5 prepared using a convergent synthesis, i.e.,
approp~iate saccharide units are linked together to
: form two disaccharides which are then linked
~ogekher to form a tetrasaccharide. Alternatively,
th~ ynthesis of tetrasaccharide glycosides can be
conducted in a sequential synthesi~ starting with
the saccharide unit at the reducing sugar terminus
and sequential~y adding another sac~:haride unit
until ~e~rasaccharide glycosid2s II and V are
prepared.
I~ either c:ase, the first step of the synthe is
involves the addition of the aglycon moiety at the
anomeric carbon atom of the reducing sugar unit.
This is generally accomplished by using an
appropria~ely protect2d form of the reducing sugar
and then selectively modifying this sugar at its
an~meric center so as to introduce a leaving group
comprising halides, trichloroacetimidate,
thioglycoside, etc~ The sugar donor is then re~cted
under catalytic conditions (e~g., a soluble silver
salt su~h as ~ilver trifluoromethanesulfonate, a
W092/22662 PCT/CA92/002~1
21~ 0~3.4'1
-- 25 --
Lewis acid such as boron trifluoride etherate or
trimethylsilyltrifluoromethanesulfonate, or
thioglycoside promoters such as methyl
trifluoromethanesulfonate or dimethyl(mPthylthio~-
sulfonium trifluoromethanesulfonate) with an aglyconor an appropriate form of a carbohydrate acceptor
which po~sess one free hydroxyl group at the
position where the glycosidic linkage is to be
established. SPe, for example, Paulsen27, Schmidt28,
and FhgPd~ et al. 29~ the disclosures of each of these
references ar~ incorporated herein by reference in
their entirety. A large variety of aglycon moieties
: are known in the art and can be attached with the
proper eonfiguration to the anomeric center of the
reducing ~mit.
: Appropriate use of compatible protecting
groups, well known in the ar~ of carbohydrate
synthesis, will allow the further attachment of the
other saccharide units. Each of the steps required
to fsrm tetrasaccharide glycosides II and V is well
known in the art. For example, the synthesis of
compound l~ (FIG. l), i.e., the synthesi of a
protected form o~ a N-acetyllactosaminyl glycoside ~.
acceptor and its gly~osidation by an appropriate
form of a N-acetyllactosaminyl donor are w~ll known
in th~ art30.
The synthesis of saccharide precursors having
rem~able blocking groupts) is well known in the art
and the remoYable blocking group can be introduced
at an appropriate stag~ during synthesis of
tetrasaccharides II or V. For example, the
selecti~e opening of a 4',6'-O-benzylidene of a
glycoside and/or ~ thioglycoside of an appropriate
form of a lactosamine disaccharidP will pr~vide the
corresponding 6'-O-benzyl derivative. The protected
~orm o~ the 6'-O-benzyl thioglycoside wili be used
as a donor in a glycosidation reaction leading to
W092~22~62 ~ PCT/CA92/002~1
~ - 26 --
compound II, aft2r deprotection. The appropriate
form of the 6'-O-benzyl lactosaminyl glycoside will
be used as acceptor in a glycosidation reaction
leading ~o tetrasacchari~e Y aftar deprotaction.
When ~h~ removable blocking group i5 sialic
acid, then this group can ~e readily introduced into
tetrasaccharide VIII
OH OH OH
I ~ NHAc OH ~
Ho~"O~O~O~R \J ¦ ¦ ¦
OH HO O H NHAc
or into disaccharide IX
- S:)H OH
I ~ NHAc
HOS~O~OR IX
OH OH
~y use of a ~(2-6)sialyltransferase as described
- below. ~q
.
B~. Enzymatic sialylation.
As noted abo~e, deblocked (i.e., the removable
blocking group X or X~ is remo~ed3 p~ntasaccharide
glycoside III or tetrasaccharide glycoside V
(~'oligosaccharide ~lycoside~) is sialylate~ by
co~tacting the appropria~e oligosaccharide glycoside
with an ~t2-3)sialyltransferase and a compatible
CMP-derivative of a sialic acid or an analsgue
thereof under conditions wherein the sialic acid or
the analogue ~hereof is transferred to the non-
xeducing sugar tel~inus of ~he oligosaccharide
glycoside. Suitable condi~ions, known in the art,
SUBSTITUTE SHEET
ISA/EP
WO 92l22662 2 1 1 ~ 9 ~ i P~r/cA92/002~1
27 ----
include the addition of the appropriate
sialyltransferase to a mixture of the
oligosacc:haride glycoside and of the CMP-derivative
of the sialic acid in an appropriate buffer such as
0.1 M sodium cacodylate in appropriate conditions of
pH and temperature such as at a pH of 6 . 5 t:o 7 . 5 and
a temperature between 2 5 and 4 5 D C, pref erably 3 5-
40C for 12 hc~urs to 4 days. The resulting
sialylated oligosaccharide glycoside can be isolated
and purif ied using conventional methodology
comprising HPLC, gel-, reverse phase-, ion
exchange, or adsorption chromatography.
In this regard, when an analogue of sialic acid
is transferred to the oligosaccharide ~lycoside by
lS the sialyltransferase, the analogue is sometimes
referred tn as an artif icial donor and the
oligosac:charide glycoside is sometimes referred to
as an artif icial acceptor . Sialylation methods
employing an ~rtif icial donor and an . rtif icial
2û ac eptor are dPscri~ed by Venot et al .. , U. S . Patent
Application Serial No. 07/771, 007 filed Octob2r 2,
: 1992 which application is inccrporated herein by
- reference in its entire~y. Simil~rly, sialylation
methods ~mploying an artificial donor and an
arkificial accep~or are descri~ed by Ippolito et
al. ,22 which applications are also incorporated
herein by referencs in their entirety.
The enzymatic transfer of analogues of sialic
acid require the prior synthesis ~i.e., activation)
of their nucleot~de (CMP) derivatives. Activation
o~ the analogues o~ sialic acid is usually done by
using the ~nzyme CMP-sialic acid synthase which is
r~adily available and the literature provides
examples of the activation of various analogues of
sialic acid,
The present invention is based, in part, on the
discovery that, in Figure 1, sialylation of
W092/22662 ~99~ PCT/CA92/002~1
-- 28 --
deblocked pentasaccharide glycoside III so as to
form an ~ 3)sialyl residue at the non-reducing
sugar terminus of this oligosaccharide glyeoside is
necessarily after removing the removable blocking
group b cause sialylation with an ~2-3)sialy1-
transferase requires the presence of a free hydroxyl
group at the 6-position of the terminal galactose
r~sidue on the deprotected pentasaccharide glycoside
III.
This invention is based, in part, on the
further discovery that, in Figures 2, the
sialylation of the tetrasaccharide glycoside V so as
to form an ~(2-3)sialyl residue at the non-reducing
sugar terminus of this oligosaccharide glycoside is
necessarily before the fucosylation step because
sialyla$ion with an ~(2-3)sialyltransferase will not
proceed if there is an ~ fucose linked (1-3) to the
neighboring N-acetylglucosamine.
B3. Enzymatic fucosylation.
As noted bove, tetrasaccharide glycoside II or
pentasaccharide glycoside derived by sialylating
tetrasaccharide glycoside Y or trisaccharide lg
~'~oligosaccharide glycoside") are fucosylated by
contacting the appropriate oligosaccharide glycoside
with an ~ 3)fucosyltrancferase and a compatible
~DP-derivative of ~-fucose or an analogue of L-
fucose under conditions wherein the fucose is
transferred onto the 3-hydroxy group of one of the
GlcNAc moieties o~ the oligosaccharide glycosid
Suitable conditions, known in the art, inçlude the
addition of he ~ 3)fucosyltransferase to a
mixture of the oligosaccharide glycoside and of the
GDP-derivative of the L-fucose in a appropriate
buffer such as O~l M sodium cacodylate in
appropriate conditions of pH and temperature such as
at a pH of 6.5 to 7.5 and a temperature between 25
W092~22662 ` PCT/CA92/002~1
2 ~ g 7
-- 29 --
and 45~C, preferably 35 to 40~C for 12 hours to 4
days. The resulting ~ucosylàted oligo~accharide
glycoside can be isolated and purified using
conven~ional methodolo~y comprising HPLC, gel-,
reverse phase-, ion exchange-, or adsorption
chromatography.
As noted above, enzymatic fucosylation requires
the prior synthesis o~ ~DP-fucose. Preferably, GDP
fucose is prepared in the methods described by Jiang
e~ al.59
B4. Removal of the Remo~able Blocking Group
: ~he synthesis of both h~xasaccharides I and IV,
as per Figures 1-2, both require the removal of a
r~moYable blocking group. In general, the
appropriate oligosaccharide glycoside is treated
under conditions su~ficient to effect xemoval of the
blocking group. The specific conditions depend on
the blocking group employed and are well known in
the art. For ~xample, when a benzyl blocking group
is employed, this group is readily removed by
hydrogenation t~chniques known in ths art.
Similarly, when the blocking group is sialic acid,
it is removed in the manner depicted in the Examples
~et forth herein b~l~w.
Regarding Figures 1-2, Figure 1 illustrates the
synthesis of hexasaccharide glycoside I ~compound 5
and 5b) and hep~asaccharidP glycoside (compound 7
and 7b). Thus, the tetrasaccharide la wa~
transformed into 2~ by using the BGal~1-4)~GlcNAc
~(2-6) sialyltransferase from rat liver (Figure 1~.
It has been shown that a similar synthasis can be
achie~ed on gram scale. 16 P~ntasaccharide 2a was
then selectively ~ucosylated by thQ ~Gal(1 3)~GlcNAc
a(l-3/4)fucosyltransferase ~(}-3/4~FT] from human
milk4, giving the hexasaccharides 3a,b.
Quantit~tive desialylation of 3a,b by a suitable
W092~2266~ rl PCT/CA92/00251
3~.--
immobilized sialidase (e.g., a sialidase fromClostridium Perfringens) led to the fucosylatPd
derivatives a~b, the 1H-n.m.r. of which were in
agreement with ~hat of a synthetic material.17 The
free acid form 4b could be transformed into the
methyl ester 4~ by action of diazomethane in
methanol. Desialylation of a glycolipid possessing
of the same terminal hexasacrharide sequence has
already b~en mentioned10. Finally, sialylation of 4a
by the ~Gal(1-3/4)~1cNAc ~(2-3)sialyltransferase
from rat liver proYided the hexasaccharides 5a.b.
The 8-methoxycarbonyloctyl glycoside of the
starting tetrasaccharide la and trisaccharide 9 was
used wi h the intention of taking advantage of the
hydrophobi properties of the aglycone for
separation purposes and with the aim of possible
coupling of the products ~o carriers.4 In fact,
partial hydrolysis of the methyl ester could not
really be avoided, and this side reaction became
important in some cas~s (e.g., when the transferase
such as milk fucosyltransferase was not highly
purified). As a result, compounds 3a, ~a, 5a, 6a
and 7~ were isolated as the methyl ester and/or the
free a id forms (3~, 4b, Sb, 7b) of the aglycone
which were identified by l~-n.m.r. Howaver, the
free acid form of ~b can xeadily be reconverted back
to the methyl ester by treating the acid in dry
methanol with diazomethane.
Finally, although conversion of la into 2
appeared almost c~mplete, some losses occurred
during reco~ery of this derivative as it is not very
tightly retained on the hydrophobic C18 ~ilica gel.
Figure 1 al~o illustrates that heptasaccharide
7b was obtained by sequential sialylation of la by
the ~al(1-3~4)BGlcNAc ~(2-3)sialyltransferase~
followed by difucosylation of the intermediate 6a by
the ~al(1-3/4)~GlcNAc a(l-3/4)fucosyltransferase
W092~22662 21 1 0 9 ~ 7 PCT/CA92/002~1
-- 31 --
from human milk. In the conditions used, only the
difucosylated product was obtained~
Fi~ure 2 illustrates the synthesis of
hexasaccharide IV (compound l2).
Alternative synthPses for hexasaccharide I are
~et forth belo~ and generally involve a
chemical/enzymatis approach. O~e approach is a
totally enzymatic method which utilizes different
glycosyltransferases. This procedure is set forth
in Figure 3. Specifically, in this approach,
galartose is enzymatically transferred onto
GlcNAc-OR to form ~Gal(l-4)~GlcN~c-OR (LacNAc-OR).
SuitablP enzymes include the GlcNAc Bl-4
galactosyltransferase which transfers galactose from
uridine 5 (galactopyranosyl)-diphosphate ~UDP-Gal)
to the 4-position of GlcNAcB-OR, where R can be an
aglycon~ or a saccharide. This transferase is a
commercial and versatile enzyme and a~cepts some
modifications in the sugar portion of th~ donor47 and
in the acceptor4~49.
: N-acetylglucosamine is then transferred to th
3-position of the terminal ~-galactose of LacNAc-OR
(N-acetyllactosamine-O~ -- ~Gal~l-4)~Glc~Ac-OR) to
produce the ~GlcNAc(l-3)~Gal(l-4)~GlcNAc-OR
trisaccharide structure ~compound ~9~. Transferases
which transfer N2ce~ylglucosamine fr~m uridine 5 -
(N-acetylglucosamine~-diphosphate (U-DP~GlcNA~ ts
the 3 po~i~ion o~ the terminal B-ga~actose of a ND
acetyllactosamine moiety) are present in a variety
of ~ources such as human serum36-4~, human urine41,
Novikof~ tumor cell ascites fluid42~43, mouse ~-
lymphom~ cells44, human milX~s and human colonic
ad~nocarcinoma cells46.
The acceptor sp cificity of the transferases
3~ obtained, particularly from human serum36~3B and from
Novikoff tumor cell ascites fluid43, has been well
W092/22662 99 ¦ -- 32 -- PCT/CA92/002~1
characterized using synthetic oligosaccharides. This
enzyme re~uires a terminal ~Gal~l-4)~Glc(NAc~-OR
unit, where R can be an aglycone or a sacch~ride
moiety and Glc(NAc) ca-n be either GlcNAc or Glc.
The enzyme does not transfer to the structure
~Gal(1-4)~Fuc~1-3)}~Glc.~Ac (LewisX) in which tAe
fucose is attached to the penultimate GlcNAc~43.
As a result, in this process, enzymatic formation of
the ~GlcNAc(l-3)~Gal(l-4)~GlcNAc-OR should precede
fucosylation. In addition, this enzyme, in
combination with the GlcNAc B~l-4) galactosyl-
trans~erase could catalyze the synthesis of
oligQm~rs of N acetyllactosamine43.
~nzymatic transfer of fucose to the acceptor ~9
by the milk Gal(1-3~4)GlcNAc ~ 3/4)fucosyl-
transferase specifically occurs on the internal
GlcN~c leading to compound 20. The backbone of the
tetrasaccharide ~0 is then extended by transfer of a
galactose residue leading to compound 2l by the
bovine GlcNAc B~-4 g~lactosyltrans~erase. N~u5Ac is
~hen transferred to compound 21 (also sh~wn in
Fi~ure 1 a~ co~pounds ~,b) in the last step by the
r~t liver Gal(Bl-3/4)~lcNAc ~2-3 sialyltransferase60
pr~viding hexasaccharide 22 (also shown in Fi~ure 1
~5 as compounds 5~,b).
As a result, the last three ~teps of this
~ynthetic pathway, (l) fucosylation, (2) ~xtension
and (3) sialylation, differ from the propo~ed normal
biosynthetic pathway which sequentially proceeds
follo~ing the sequence: (1) extension, (2)
sialylation, and finally (3) fu~osylation.
Alt~rnatively, as shown in Figure 4, compound
19 can be made in a totally synthetic sch~me
starting ~rom known precursors. Specifically,
compound l9 can be obtained by total chemical
synthesis following known procedures by which a
large variety of R group can be introduced. As a
W092~22662 2 1 1 0 3 9 ~ pcT/cAs2/oa2~l
-- 33 --
result, R can be an aglycQne or a saccharide moiety
itself a~tached to an aglycone. A wide variety of
glycosylation methods are available in order to
synthesize ~-glycosides. The present synthesis is
derived from the synthesis described by Alais et
al 3o
In addition~ glycosid~s s can be used,instead
of glycosyltransfera~es, for the synthesis of
glycosides in appropriate conditions. The main
characteristics of the use of both types of enzymes
have been reviewed by Ichikawa et al.~2 . The B-
galactosidases, N-acetylhexsaminidase or
sialidases~ could be used to synthesize some of
the saccharides.
In these synthe~is, the 2-N-phthalimido
protecting group is used in order to prefer2bly lead
- to the B-glycosides during glycosylation reactions.
Thus the blocked disaccharide glycosyl donor 113 iS
used in a glycosylation reactisn of the desired
alcohol ROR catalyzed by trimethylsilyltrifluoro-
methane~ulfonate to lead to the glycosidP 12~ Mild
de-0-acetylation provided 13. Disaccharide 13 is
reacted with acetone in the presence of an acid
catalyst, such.as p toluene sulfonic ~cid at 60C,
leading to a mixture of the 3,4- and of the 4,6-
isopropyliden d~rivative 14 and 15 which are
separated. The 3,4-isopropylidene derivative 14 is
totally acetylat~d in pyridin~ with acetic a~hydride
and the isopropylidene group hydrolyzed in a mixture
of acetic acid and water at 90-C providing ~6.
Glycosylation of diol 16 by the donor ?7 catalyzed
by trimethyl~ilyl~rifluoromethanesulfonate
preferably led to trisaccharide 1~ which was
deprotected using conventional procedures leading to
19.
As set ~orth above, L-fucose is then
tr~nsferred from GDP-fucose to the trisaccharide
WOg2~22662 PCT/CA92/0025l
~,9~l
acceptor 19 by using the Gal(B1-3/4) GlcNAc ~1-3/4
fucosyltransferase from human milk. Another
appropriate transferase from o~her source can also
be us~d. As evidenced ~y lH-n.m.r., only one
fucosyl unit is introduced. Furthermore, the 1H-
n.m.r. data, in particular the position of the
signal provided by H-5 of the ~Fuc is characteristic
of the presence of the ~al(Bl-4)~Fuc~1-3)]GlcNAc
` unit6l. The transformation is quantitative.
Galactose is then transferred from UDP-Gal to
tetrasaccharid~ 20 by the commercial bovine milk
GlcNAe ~1-4 galartosyltransferase. The
pentasaccharide obtained is identical to the same
compound obtained earlier by using a different
route65. The transformation is quantitative.
In a final step, Neu5Ac is trans erred to
pentasaccharide 21 by using the Gal(Bl-3/4)Glc~Ac
a(2-3) ialyltransferase from rat liver~5. Another
appropriate transferase from o~her sources can also
be used. This step is performed according to the
~rlier report and provides the same product as
de-~cribed in Venot et al. 65 .
As is apparent, more extended structures can be
obtain d from the pentasaccharide 21 ~s indica.ted in
Figure 5. For tha~ purpose, GlcNAc can b~
transferred to pentasaccharide 21 by the Gal(B~-
4~GlcNAc Bl-3 N-acetylglucosaminyltransferase.
Pentasaccharide 21 should be an acceptor for this
~ransferase since the ~-fucosyl residue is not
linked to the penultimate GlcNAc moiety. Further
sequential transfer of galactose and of Neu5Ac by
the appropriate glycosyltransferas~ will lead to
octasaccharides 26.
C. ~
H~xasaccharîde glycosides I and IV are
effective in suppressing mammalian cell-mediated
W092/22662 2 ~ i ~ 9 9 7 PCTJCA92/oO~Sl
-- 35 --
immune responses. Without being limited to any
theory, it is believed that these compounds affect
the cell mediated immune response in a number of
ways. Speci~ically, th~se compounds can inhibit the
ability of the immune response to become educated
about a ~pecific antigen when ~he compound is
administered simultaneously with the first expo~ure
of the immune system to the antigen. Also,
hexasaccharide glyccside. I and IV can inhi~it the
effector phase of a cell-mediated immune response
(eg., the inflammatory component of a DTH response)
when administered after second or later exposures of
the immune system to the same antigen.
Additionally, hexasaccharide glycosides ~ and IV
can induce tolerance to antigens when administered
at the time of second or later exposures of the
immune system to the antigen.
The suppression of the inf lammator~ component
of the immun~ response by hexasaccharide glycosides
I and IV is believed to require the initiation of a
secondary immune response ~i.e., a response to a
second exposure to anti~en). Hexasaccharide
glycoside I or IY is generally administered to the
pati~nt at least about O . 5 hours a~ter an
inflammatory episode, preferably, at least about 1
hour after, and most preferably, at least about 5
hours after an inflammatory episod~ or exacerbation.
Hexasaccharide glycosides I and I~ are
effectlve in suppr~ssing cell-modiated immune
responses to an antigen (eg. the inf lammatory
component of a DTH response) when admini~tered at a
dosage range of from about 0.5 mg to about 50 mg/kg
of body weight, and preferably from about 0.5 to
about 5 mg/kg of body w~ight. The specific dose
3S employed is regulated by the particular cell-
mediated immune response being tr~ated as well as by
the judgment of the attending clinician depending
W092~2~662 ~ PCT/CA~2/002~1
~ ~ -- 36 --
upon factors such as the severity of the adverse
immune resp~nse, the age and general condition of
the patient, and thP like. Hexasaccharide
glycosides I or IV is generally administered
parenterally, such as intranasally,
intrapulmonarily, transdermally and intravenously,
although other forms of administration are
contempl~ted. Preferably, the suppression of a
cel~-mediated immune response, eg. the inflammatory
component of a DTH response, is reduced by at least
about 1~% as opposed to control measured 24 hours
after administration of the challenge to the mammal
and 19 hours after administration of hexasaccharide
glycosides I or IV.
In addition to providing suppression of the
inflam~.atory component of the cell-mediated immune
respons~ to an antigen, administration of he
hexasaacharid glycosides I or IV also imparts a
tolerance to additional challenges from the same
20 antig~n. In this regard, re-challenge by the same
antigen weeks after administration of hexasaccharide
glyco~ides I or IV results in a significantly
reduced immune re ponse. f.
Ad~ini tration of hexasaccharide glycosides I
or IV simult2neously with first exposure to an
antigen imparts suppression of a cell-mediated
i~mune respon e to the antigen and tol.rance to
future challenges with that antigen. In this regard
the term "r~ducing sensitization'~ means that the
hexasaccharide glycosides I or IV, when administered
to a mammal in an effective amount along with a
su~icient amount of antigen to induce an immune
r~ponse, reduces the ability of the immuna system
o~ the mammal to become educated and thus sansitized
to the antigen administered at ~he same t ime as
hexasaccharide glycosides I cr IV. An "effective
amount'1 of this compound is that amount whiçh will
W092/~2662 ? ll o~ q~ PCT~CA92/002
-- 37 -
reduc~ sensitiza~ion (immunological education) of a
mammal, including humans, to an antigen administered
simultaneou~ly as determined by a reduction in a
cell-mediated response to the antigen such as DTH
responses as tested by the footpad challenge test.
Preferably the reduction in sensitization wîll be at
least about 2~% and more preferably at least about
30% or more~ Generally, hexasaccharide glycosides I
or IV are effective in reducing sensitization when
administer~d at a dosage range of from about 0.5 mg
to about 50 mg/kg of body weight, and preferably
from about 0.5 mg to about 5 mg/kg of body wei~ht.
The specific dose employed is regulated by the
sensitization being treated as well as the judgement
of the attending clinician depending upon the age
and general condition of the patient and the like.
"Simultaneous" administration of the compound with
; the antigen with regard ~o inhibiting sensitization
m~3ans that the compound is administered once or
2 0 continuously throughout a period of time within 3
hours o~ the administration of an antigen, mor
pre~erably the compound i5 administered within 1
hour of the antigen. _.
Th~ methods of this invention are generaLly
aehieved by use o~ a pharmaceutical composition
sultable for use in the parenteral administration of
an a~ec*ive amount of hexasaccharide glyco~ides I
or IV. These ompositions comprise a
pharmaceutically inert carrier such as water,
buffered saline, etc. and an effective amount of
hexasaccharide glycosides I or IV as to provide the
a~ove-noted dosage of the oligosaccharide glycoside
when administered to a patient. It is contemplated
that suitable pharmaceutical compositions can
additionally contain optional components such as an
adjuvant, a preservative, etc.
W092/22662 ~I PCT/CA92/002~1
99
-- 38 --
It is also contemplated that other suitable
pharmaceutical compositions can include oral
compositions, transdermal compositions or bandages
etc., which are well known in the art.
Hexasaccharide glycosides I and IV containing
an analogue of si~lic acid are also useful in
preparing artificial antigens which can cross-reart
with antigenic determinants having a similar
oligosaccharide stru~-ture as in hexasaccharide
glycosides I and IV but which contain a naturally
occurring sialic acid. Methods for the preparation
of artificial antigens and their uses are set forth
in U.S. Serial No. 07/771,Q07 filed concurrently
with this applic tion as attorney docket number
005~24-002 and entitled "~ETHO~S FOR THE ENZYMATIC
SYNTHESIS OF ALPHA-~IALYLATED OLIGOSACCHARIDE
GLYCOSID~S" which application is incorporated herein
by reference in its entirety.
The following examples are of4ered to
illu~trate the present in~ention and are not to be
construed in any manner as limiting it.
In these examples as w~ll as in the
application, all sugars disclosed are in their D
form except for fucGse which is in its L form.
2~ ~ In these examples, unless otherwise defined
below, the abbreviations employed have their
g~nerally accepted meaning:
C~P-Neu5Ac= cytidine-5'-monophospho-N-
acetylneuraminic acid
~TH = delayed-type hyper~ensitiYity
Fuc T = fucosyl transferase
Gal T = galactosyl transferase
GDP-Fuc = gu~nosine 5'-diphospho-L-fucos
ST = sialyl transferase
U = Units
UDP-Gal - uridine-5'-diphospho-D-galactose
2110997
-- 39 ~-
AG 1 x 8TH (formate form) = ion exchange resin AG 1 x
8 (formate form) available from Bio-
Rad Laboratories, Richmond, CA
Dowex 50W X 8TM (H~ form) = ion exchange resin Dowex
50W X 8 (H~ form) available from Dow
Chemical, Midland~ MI
IRC-50TM resin (H~ form) = ion exchange resin IRC-50
(H~ form) available from Rohm & Haas,
Philadelphia, PA
Commercially available components are listed by
manufactur~r and where appropriate, ~he order
number. Som~ of the recited manufacturers are as
follows:
Iatron = Iatron Laboratories, Tokyo, Japan
~erck = E. Merck AG, Darmstadt, Germany
Mi~lipore = Millipore Cor~., 8edford, M~.
Waters ~ Waters Associates, Inc., Milford, M~.
E~MP~ES
The following examples illustrate the
preparation of Compounds 5a and Sb which preparation~
is illustrated in FIG. 1. The synthethic pat-hway
u~ilized the following general m~thods:
&eneral Methods: All organic sol~ents usPd wer~ re-
distilled reagent grade. Pre-coated silica gel
plate~ (60-F254, ~. Merc~, Darmstadt) were run in
65:35:S, 65:35:8 and/or 60:40:10 mixtures of CHCl3,
C~3OH, and 0.2% CaCl2 solution, and detection was by
charring after spraying with a 5% solution of
sulphuric acid (H2SO4) in ethanol. Sep-Pa~TM C~
cartridges (Waters Associates, Milford, ~A3 were
conditioned as indicated by the supplier.
Iatrobeads (6RS-8060) were from Iatron Laboratories,
SUB~,TIT~ ~T~ ~
W092/22662 PCT~CA92/00251
211~9~7
-- 40 --
Tokyo, Japan and the ~G 50W X 8 ion exchange resin
was purchas~d from BioRad, Richmond, California.
CMP-Neu5Ac was purchased from Sigma Chemi~al Company
(St. Louis, Missouri) and GDP-fucose was obtained by
~hemical synthesis. 59 ~Gal(1-4)~GlcNAc(1-3)~Gal-
(1-4~GlcNAcoOR was obtained by following the
procedures of Alais et al30 with the appropriate
substitution of the aglycon. Evaporation of organic
solvents was done at 20-25C using a rotory
e~aporator connected to a water aspirator. 1H-
n.m.r. spectra have been run on at 300 and 500 ~Hz
using internal acetone (~=2.225) as reference and
samples were freeze dried twice from D2O and
dissolved in 99.99% D2O. The spectra of compounds
obtained as 8-m~thoxycarbonyloctyl glycosides all
show a singlet at ~=3.686 (CO2CH3) and a txiplet at
~=2.387 (7.5Hz, CH2CO2). The spec~ra of compounds
obtained as the 8-carboxyoctyl glycosides diff~r
from the respective 8 methoxy-carbonyloctyl
glycosides by the absenc~ of the singlet due to
CO2CH3 and the presence of a triplet at ~=2O314 ~t,
7.5Hz3 for CH2CO~H.
In exampl~s 1 to 6 below, preparati~e
sialylation was conducted as follows:
The rat liver BGal ( 1-3/4 ) BGlcNAc ~ ~ 2-~ ~ sialyl-
kransferas~ (EC 2.4.99.5) was purified by ffinity
chr~matography according to the procedure of ~azid,
et al. 19 but using a matrix obtained by covalently
linking the hapten B~al(1-3)BGlcNAcO(CH2)8CO
activated as in its N-succinimidyl ester to
epichlorohydrin activated Sepharose. 67
The ~Gal(1-4) ~GlcNAc ~(2-6)sialyltransferase
contained in the flow-through of the above affinity-
column, was further chromatographed on CDP-
hexanslamine Sepharose as reported.Z
The enz~matic sialylations were carried out at
37-C in a plastic tube using a sodium cacodylate
W092~226S2 2 ~10 9 9 7 PCT/CA92/002~1
-- 4l --
buffer (50 mM, pH 6.~) containing Triton CF-54
(0.5%), BSA (l mg/mL) and calf intestine alkaline
phosphatase.~1 The final reaction mix$ures were
diluted with H20 and applied onto C1~ Sep-Pak
cartridges as reported. 4 After washing with H20, the
products were eluted with CH3QH and the solvents
evaporated. The residue was dissolved in a 65:35:5
mixture of CHCl3, C~30H and H20 and applied on a
small column of Iatrobeads (O.200 to 0.500 g).
After washing with the same solvent mixture, the
products were eluted with a 65:35:8 and/or 65:40:10
mixtures of the same solvents. The appropriate
fractions (t.l.c.) were pooled, the solvents
evaporated in vacuo, the residue run through a smali
column of AG 50W X 8 (Na' form) in H20 and the
products recovered aft2r freeze drying in vacuo. In
all cases, th~ 8-methoxycarbonyloctyl glycosides
w~re eparated from the corresponding 8-carboxyoctyl
glycosides.
In exa~ples l to 6 below, preparatiYe
~ucosylation was conducted as follows:
The ~GlcN~c ~ 3/4)fucosyltranferaRe was
purified from human milk, as reported.4 The
~nzymatic reactions were carried out at 37~C in a
~ plastic tube using a sodium cacodylate buffer (lO0
mM, p~ 6.5), MnCl2 (lO mM), ATP (1.6 mM), NaN3 (1.6
mM~. The reaction products were isolated and
purifisd as indicated above.
~xample 1 -- Preparation of 8-Methoxycarbonyloctyl
(5-Acetamido-3,5-dideoxy-a-D~glycero-D-
galac$o-2-nonulopyranosylonic acid)-(2-6)-
O-B-D-galactopyranosyl-(l-4)-0-2-
acetamido-2-deoxy-glucopyranosyl-(l-3)-O-
B-D-galactopyranosyl-(l-4~-0-2-acetamido-
2-deoxy-glucopyranoside t2 ~
Compound 1 (6.5 mg), CMP-Neu5Ac (17 mg),
~Gal(1-4)BGlcNAc ~(2-6)sialyltransferase ~50 mU) and
W09~/22662 ~h~9~ PCT/~A92~0~51
- 42 --
alkaline phosphatase (lS U) were incubat~d for 48
hours in 2.5 mL of the above buffer. Isolation and
purification provided 2~ (3.0 mg).
~xample 2 -- Preparation of 8-Methoxycarbonyloctyl
(5-Acetamido-3,5-dideoxy-~-D-glycero-D-
galacto-2-nonulopyranosylonic acid)-(2-6)-
0-B-D-galacto-pyranosyl-(l-4)-0-2-
acetamido-2-deoxy-glucopyranosyl-~l-3)-0-
B-D-galactopyranosyl~ 4)-0-~ L-
. fucopyranosyl-(l-3)-0-]2-acetamido-2-
deoxy glucopyranoside (3a) and the 8-
carboxyoctyl glycoside (3b~
CompQund 2a (3.0 mg), ~DP-fucose (5 mg),
BGlcNAc ~(l-3/4~fusosyltransferase (lO mU) were
incubated for 68 hours in the buffer (1.3 mL).
Isolation and purification provided 3~ ~l.2 mg) and
3b (0.5 mg)-
~: Exampl~- 3 -- Preparation of 8-Methoxycarbonyloctyl
~: *-D-galac~opyranosyl-(l 4~-0-2-acetamido-
: 20 ~ 2-deoxy-B-D-glucopyranosyl~ 3)-0-B-D-
galactopyranosyl-(1-4)-O-f ~ fuco-
pyrano~yl-(l-3)-0-]2-acetamido-2-deoxy-B-
D-glucopyranoside l~) and the 8-
carboxyootyl glycoside l4bL
Compounds 3~ and 3b (1.7 mg) were incubated ~
: with Clostridium Perfringens neuraminidase
immobilized on agarose (Sigma Ch~mical ~ompany, 1 U)
i~ a buffer of sodium cacodylate (50 mM, pH 5~2, 2
mL) at 37-C. After 24 hours the mixture was diluted
with water ~lO mL) and filtered through Amicon PM-lO
membrane. The flow-through and washings were
lyophilized and the residue dissolved in water ~3
mL) and ~pplied to two Ct8 cartridge. Each cartridge
was washed with water (lO mL) prior to elution with
methanol t20 mL)~ After evaporation of the solvent,
the residue was chromatographed on Iatrobe~ds (210
mg) as indicated above giving (~a, O.B mg) and 4b
(O.7 mg~. 4b was dissolved in dry methanol and
W092/22662 PCT/CA92~0025l
-- 43 --
treated with diazomethane un~il t.l~c. indicated thecomple~e con~ersion in~o 4a.
Example 4 -- ~reparation of 8-Methoxycarbonyloctyl
~5-Acetamido-3,5-dideoxy ~-D-glycero-D-
galacto-2-nonulopyranosylonic acid)-(2-3)-
0-B-D-gala~topyranosyl-(1-4)-0-2-
acetamido 2-deoxy-B-D-glucopyranosyl-(1-
33-0-B-D-galactopyranosyl-(1-4~-0-t~
fucopyranosyl~ 3)-0-]2-acetamido-2-
deoxy-B-D-glucopyranoside t5~) and the 8-
carboxyoctyl glycoside ~5b)
Compound ~a (l.S mg), C~P-Neu5Ac (8 mg),
~Gal(1-3/4)BGlcNAc a~2-~sialyltransfPrase (17 mU),
alkaline phosphatase (5 U), were incubated for 40
hours in the sialylation buffer (1.5 mL). Isolation
and purification provided Sa (0.7 mg) and Sb (0.55
~ mg) .
Exa~ple S -- Preparation of ~-Methoxycarbonyloctyl
(5-Acetamido-3,5-dideoxy-~ D-glycero-D-
galacto-2-nonulopyranQsylonic acid) (2-3)-
0-B-D-gal~ctopyranosyl~ 4)-0-~-
.- acetamido-2-deoxy B-D-glucopyranosyl-(1-
3)-0-B-D galactopyranosyl (1-4)-0-2~
acetamido-2-deoxy-glucopyranoside(~).
Compound 1~ (S mg), CMP-~eu5Ac (15 mg), ~G~l(1
3/4)BGlcNAc ~ 3)~ialyltransferase ~46 mUI, and
alkaline phosphatase (15 U) were incubated in ~he
~ialylation buffer (2,5 mL) for 48 hours. Isolation
~nd purification of the product gave 6~ (2.5 mg).
~xample 6 -- Prepara~ion of 8-Meth~xycarb~nyloGtyl
(5-Acetamido-3,5-dideoxy-~ D-glyoero-D~
galacto-2-nonulopyranosylonic acid) (2 3)-
0-B-D galactopyranosyl-(1-4)-0-[~-L-
~ucopyranosyl~ 3)-0-3~-acetamido-2-
deoxy-glucopyranosyl-(1-3)~9-R-D-
galactopyranosyl-(1-4)-0-[~-L-
fucopyranosyl-(1-33-0-]2-acetamido-2
deoxy-glucopyranoside ~7~
.. , , ~ . . . . . . ... . .. . . .
PCr/CA92/00251
Comps:~und 6~ (2 . 5 mg), GDP-fucose (8 mg) and the
BGlcNAc ~(1~3/4)fucosyltransferase (19 mU) were
incubated in the enzymatic buffer (2.0 mL) for 4B h.
Isolation and purification of the product give 7b
~1.7 mg).
lH~NMR data for the compounds prepared in
Examp}es 1 to 6 above are set forth in the following
Table I:
'
~.
WO 92~22662~ ~ r PCr/CA92/00251
2 ~ 3 ~ !
---- 45
_ ~ ~ _ O ~ a ~ ~,0 -- 0O ~ _
~ ~.~0 O ~0 ~ ~. ~ ~ ~ I
~ 00 . , _ _
`~1 ~s ~ o ~_ ~ _ _
~ ~ ~0 v~ Xv~o~ 0~ ~ 2'`~ l
1- - - . _ . . _ I
~1 o ~o oo ~- o COV~ ~r _ _ S ~ I
~ ~ ~ ~ ô. ~o ~ ~ ~ ~ I
~ ........ . . . . . .
u~ ~1 ~}co ~ o ~_ ~s 8-~ ~o ~ ~o
E _ r _ ~ l
~ ~ ~ ~ S ~ ~ ~ ~ O ~ ~ I
o ~ e~ ~ 1~ oo~ ~ ~ ~` 5~
:1 1 1 ---- . .
~1 ~ s~ ~_ s ~ ~ u~ ._
: ' ~ ~ ~ ~u~ ~ r~ U~
~ - -- - ~ -
V~ ô G r ~ ~ O~ ~ X
_ _~ ~ _~ _U~D ~ r-~ _ _
_ _ __ . ___ __ ~c
1~ ~ 1 ~ o Y vl Z e L:~= o
S~S~ITUTE S~T
W092/22662 PCT/CA9~/002~i
- 46 --
B. KINETIC DATA
Experiments were conduc~ed to determine the
relative rates of transfer of fucosylation onto the
3-hydroxy of the GlcNAc in disaccharide glycosides
~Gal~1-4)~GlcNAc-OR having different substit~ents at
the 6-position of the galactose. Fucosyltransferase
assays were conducted with ~(1-3/4)fucosyl-
transferasQ in a manner similar to that described in
the art4 and gave the following results:
Substituent at
the 6-position Relati~e Rate
of~qalactose R . of Transfer
-OH -(CH2)~co2cH3 lOO
H -(CH2) 2 ~ CH2 ) 2C02CH3
- 15 The remaining low xelative rate of transfer
obtained on the 6'-deoxy deriYative may be due to a
small amount of ~Gal ~1-2)fucosyltransferase which
was present in the preparation of the
~ 3~4)fucosyltransferase.
The above res~lts indicate that the presence of
a hydroxyl group at the 6-position of galactose is
necessary for efficient fucosylation of 3-hydroxy of-~
: the ~lcNAc in disaccharide glycoside~ ~Gal(l-
4)~1cNAc-~R using ~(1-3/4~fucosyltransferase.
The following Examples 7 to 8 illustrate
alternative methods for preparing for compounds of
Formula I.
i
In th~se examples, during the chemical
synthesis, unless otherwise specially indicated, khe
work up generally included extraction with
dichloromethane followed by the normal sequential
washings of the organic phase with water, a dilute
solution of sodium carbonate and water. The organic
W092/22662 2110 9 9 7 PC~/CA92/002~1
.
-- 47 --
solvent were then dried over magnesium sulfate, the
solid filtered and the solvent evaporated in vacuo
as indicated.
Evaporation of organic solvents was done at 20-
25C using a rotory evaporator connected to a water
aspirator. lff-n.m.r. spectra were run at 300 MHz
using internal acetone (~=2.225) as reference and
samples were freeze dried twice from D20 and
di~solved in g9.99% D20~ The spectra of compounds
obtained as 8-mathoxycarbonyloctyl all show a
singlet at ~=3.686 (C02C_3) and a triplet at ~=2.387
(7.5Hz, CH2C02). The spectra of compounds obtained
as the 8-carboxyoctyl glycosides differ from the
respective 8-methoxycarbonyloctyl glycosides by the
absence of the singlet due to C02CH3 and the presence
of a triplet at ~=2.314 (t, 7.5Hz) for CH2CO~H,
- Prs~arative Enzvmatic Sialy~lation
The rat liver ~Galt1-3/4)BGlcNAc ~(2-3)sialyl
transferase (EC 2.4.99.5) was purified by affinity
chromatography according to the procedure of Mazid~
et al.19 but using a matrix obtained by covalently
linking the hapten ~Gal(1-3)BGlcNAcO(CH2)8C0
acti~ated as in its N-succinimidyl ester to
epichlorohydrin activated Sepharo-~e. 67
2~ ~he enzymatic sialylations were carried out at 37C
in a plastic tube using a sodium cacodylate buff r
(50 mM, pH 6.5) containing Triton CF-54 (0.5%), BSA
:, (1 mg/mL) and calf intes ine alkaline phosphatase. 69
The final reaction mixtures were diluted with ~2
and applied onto C18 Sep-Pa~ cartridges a~ reported.4
A~ter washing with H20, the products were eluted
with CH30H and ~he solvents evapsrated. The residue
wa~ dissolved in a 65:35:5 mixture of CHC13, CH30H
and H20 and applied on a small column of Iatrobeads
~0.200 to 0.500 g). After washing with the same
W092/22662 ~ - 48 -- PCT/CA92/00251
solvent mixture, the produc~s were eluted with a
65:35:8 and~or 65:40:10 mixtures of th~ same
solv~nt The appropriat~ fractions (t.l.c.) were
pooled, the solvPnts evaporated in vacuo, the
residue run through a small column of AG 50W X 8
(Na~ form) in H20 and the products recovered after
freeze dxying in vacuo. In all cases, the 8-
methoxy-carbonyloctyl glycosides were separated from
the corresponding 8-carboxyoctyl glycosides.
Pre~arative EnzYmatic FucosYlation
Enzymatic Conditions
The BGlcNAc ~ 3/4)fucosyltransferase (EC 2.4.1.6~)
was purified from human milk, as reported.4 Th~
enzymatic reactions were carried out at 37C in a
plastic ~ube using ~ sodium cacodylate buffer (lOO
mM, pH 6.5), MnCl2 (lO mM), ATP (1.6 mM), NaN3 (1.6
mM). The r~action products were isolatad and
purified as indicated above.
ynthesis of GDP-Fucose
A. Preparation of Bis(tetra-n-butylammonium) ,~
hydroaen Phos~hate
Tetra-n-butylammonium hydroxide (40% a~. w/w,
about lSOg) was added dropwise to a solution of
phosphoric acid (85% aq, w/w, 18g, 0.155 mmol) in
wat~r ( 150 mL) until ~he pH reached 7 . Wa~er was
then eYaporated in vacuo to give a syrup which was
co-~vaporated with dry aceto-nitrile (2 x 400 mL)
~ollowed by dry toluene (2 x 400 mL). The resultin~
white solid (75g) was dried in vacuo and stored ~ver
phosphorus pentoxide under vacuum until used.
W092~22662 21 i O 9 9 7 PCT/CA92/00251
-- 49 --
B. Pre~aration of ~-L-Fucopyranosvl-l-~hosPhate
A solution of bis(tetra-n-butylammonium~
hydrogen phosphate (58g, 127.8 ~mol) in dry
acetonitrile (300 mL) was stirred at room
temperature under ni~rogen in the presence of
molecular sieves (4b, 20g) for about one hour. A
solution of tri-0-acetyl fucosyl-l-bromide (freshly
prepared from 31g, 93 mmol of L-fucose tetraacetate
in the manner of Nunez et al.n) in dry toluene (100
mL) was added dropwi~e in about 0.5 hour to the
above solution, cooled at 0C. After one more hour
at 0C, the mixture was brought to room temperature
and stirred for 3 hour. Tlc (1:1 toluene:ethyl
acetate) indicated a main spot on the base line and
several faster moving smaller spots.
The mixture was filtered over a pad of Celite
~- (which was ~urther washed with acetonitrile) and the
solvents evaporated in vacuo to give a red syrup.
This material was di~solved in water (400 mL) and
ex~racted with ethyl acetate ~250 m~, twice). The
a~uesus layer was then evaporated in vacuo leaving a
y~llowish syrup to which a solution of ammonium
hydroxide (25% aq., 200 mL) was added. The mixture
was stirred at room temperature for 3 hours af.ter
which tlc (65:35:8 chloroform:methanol:water~
indicated a ba~eline spot. The solvent was
evaporated in vacuo to give a yellowish syrup which
waæ diluted with water (400 mL). The pH of this
solution was checked and brought to 7, if necessary,
by addition of a small am~unt of hydrochloric acid.
The solution was slowly absorbed onto a co}umn of
ion exchange resin Dowex 2 X 8 ~200-400 mesh, 5 x 45
cm, bicarbonate form which had been prepared by
sequential washing of the resin with methanol (800
mL), water (1200 mL), ammonium bicar~onate (1 M,
1600 mL) and water (1200 mL)~. Water (1000 mL) was
WO 92/22662 r PCT/CA92/OOt51
~ -- 50 --
then run thr~ugh the column followed by a solution
of ammonium bicarbonate (0O5-M, 2.3 mL/minutP,
overnight). The eluate was collected in fractions
. (15 mL) and the product detected by charring after
spotting on a tlc plate. Fractions 20 to 57 were
pooled and evaporated in vacuo leaving a white solid
which was further co-evaporated with wa~er 53 x 300
mL~ and freeze drying of the last 50 mL and then
drying of the residue with a vacuum pump to give ~-
L-fucopyransyl-l-phosphate (9.5g, 40%) as a 12:1
mixture of ~ and ~ anomers containing some ammonium
acetate identified by a singlet at ~=1.940 in the
1X-n.m.r. spectrum. This product was slowly run
through a column of Dowex 5 X 8 resin (100 200 mesh,
kriethylammonium form~ and eluted with water to
provide the bis triethylammonium salt of ~-L-
fucopyransyl-l-phosphate as a sticky gum aft~r
freeze drying of the eluata. 1H-n.m.r~
~ 4-840 (dd~ J1,2 = 31,P = 7~5 Hz, H-1), 3.82 (q, lH,
J5 6 6.5 ~Z, H-5), 3~750 ~dd, lH, J34 3.5, J45 1.0
Hz, H-43, 3.679 (dd, 1~, J23 lO.Q Hz, H-3), 3.S20
(dd, lH, H-2), 1.940 ~s, acetate~ 2~ (d, H-6),
I~t~gral of the signals at 3.20 (q, J 7.4 Hz, NC 2)
and 1.280 and 1.260 (~CH2CH3 and H-6) indicates that ~~
the product is the bis-triethyl-ammonium salt which
may loose s~me triethylamine upon extensive drying.
13C-n.m.x- ~:9B-3 (d, Jc 1P 3-4 Hz, C-l~, 72-8 (d~
JC,2P 7.5 Hz, C-2), 16.4~C-6): 31P-nmr ~: +2 S(s~
~-L-fucopyransyl-1-phosphate appears to slowly
degrade upon prolonged storage (1+ days) in water at
22C and, accordingly, the material should not be
left, handled or stored as an aqueous solu ion at
22C or higher temperatures. In the present ca~e~
this material was kept a~ -18C and dried in vacuo
over pho~phorus pentoxide prior to being used in the
next step.
.. , , .. ., .. . . , , , . . ~ , . .. ... , .... . . . . . ~ . .. ....
W092~22662 2 ~ 1 ~ 3 9 7 PCT/CA92/00251
C. Preparation of Guanosine 5'-(~-1-fucopy-
ranosyl)-diphosphate
Guanosine 5'-(~-1-fucopyranosyl)-diphosphate
was prepared from ~-L-fucopyranosyl-l-phosphate
using two different art recognized procedures as set
forth below:
PRO~D ~ $~
~ -L-fucopyranosyl-l-phosphate and guanosine 5'
mono-pho~phomorpholidate (4-morpholine-N,N'-di-
cyclohexyl-carboxamidine salt, available from Si~ma,
St. Louis, ~issouri, "GMP-morpholidate"3 were
reacted as described in a recent modification74~ sf
Nunez's original procedure~. Accordingly, tri-n-
octyl~min (0.800g, available from Aldrich Chemical
Company, Milwaukee, Wisconsin) was added to a
mix$ure of ~ fucopyranosyl-l-phosphate (triethyl-
ammonium salt, l.OOg, about 2.20 mmol) in d~y
pyridine ~}0 mL) under nitroyen the ~olvent removed
in vacuo. The process was repeated three times with
care to allow only:dry air to enter the fla~k. G~P
morpholidate (2.4g, about 3.30 mmol) wa~ dissolved
in ~ 1:1 mix~ure of dry dimethylformamide and ~
W ridine (10 mL). The solvents were evaporated in
: vacuo and the proc~d~re repeated three times as
a~ove. The residue was dissolved in the same
mix~ure of solvents (20 mL) and the solution added
to the reaction flask accompanied by crushed
molecular sieves (2g, 4A~. The mixture was stirred
at room temperature under nitrogen~ Tlc ~3:5:2 25~ .
3~ aq~ ammonium hydroxide, isopropanol and water)
showed spots corresponding to th~ starting G~P-
mo~pholidate (Rf-0.8, U.V.), guanosin~ 5'~
fucspyranosyl)-diphosphate (Rf-0.5, U.V. and
charring), followed by the tailing spot of the
star$ing fucos~ phosphate (Rf~0.44, charring).
W092/22662 ~ PCT/CA92~002$l
-- 52 --
Additional U.V. active minor spots were also
present. After stirring for 4 days at room
temperature, the yellowish mixture was co-evaporated
in vacuo with toluene and the yellowish residue
further dried overnight a~ the vacuum pump leaving a
thick residue (2.~3g). Water ~10 mL) was then added
into the flask to give a yellow cloudy solution
which was added on top of a column of ~G 50W-X12
(from Biorad) resin (100-200 mesh, 25 x 1.5 cm, Na~
form~. The product eluted with water after the void
volume. The fractiQns which were active, bo~h by
U.V. and charring after spotting on a tlc plate,
were recovered and the solution freeze-dried
overnight in vacuo providing a crude material
(1.96g~.
This residue was dissolved in water (10 mL
overall) and slowly absorbed onto a column of
hydrop~obic Cl8 silica gel (Waters, 2.5 x 30 cm)
which had ~een conditioned by washing with water,
methanol and water (250 mL each~. Water was then
run through the column (O.4 mL/min) and th~ eluate
collected in fraction~ tO.8 mL) which were checked
by tlc t3:5:2 25% aq. ammonium hydroxide,
i~opropanol and water3. ~-L-fucopyranosyl-l-
p~o phate, (R~-0.54, charring) was eluted in
fractions 29 to 45. A product showing a strongly
U.V. a tiY2 spot (Rf-0.51) eluted mainly in
fractions 46 to 650 Other minor U.V. active spots
~f higher or lower Rf were observed. Fractions 59
to 86, which contained guanosine 5'~
fucopyranosyl)-diphosphate (Rf-0.62), also showed a
narrow u.v, active spot (Rf-0.57). Fractions 59 to
86 were pooled and freeze-dried o~ernight providing
O.353g of material enriched in guanosine 5~
3~ fucopyranosyl)-diphosphate. 1H-n.m.r. indicated
that this material was ontaminated by a small
W09~22662 21~ 0 9 ~ 7 PCT/CA92/002~1
-- 53 --
amount of impurities giving signals at ~ - 4.12 and
_ 5.05.
Fractions 29 to 45 and 47 to 57 w~re separately
pooled and freeze-dried providing recovered ~-L-
fuco-pyranosyl-l phosphate (0.264g and 0.223g,
re~pectively, in which the second fraction contains
some impurities). Occasionally, pooling of
appropriate fractions pro~id~d some amount of
guanosine 5'~ fucopyranosyl)-diphosphate in good
purity (l~n.m.r.). Genarally, all the material
~nriched in guanosine 5'-(~ fuco-pyranosyl)-
diphosphate was dissolved in a minimum amount of
water and run on the same column which ~ad been
regenerated by washing wi~h large amoun~s of
methanol followed by water. The fractions
containing the purified guanosine 5'~
fucopyranosyl3-diphosphate (tlc) were pooled and
freezed dried in vacuo leaYing a whîte fluffy
material ~187 mg, 16%). 1H-n.m.r. was identical to
the pr~vîously reported data~.
ROC~D~R~ ~2
~-L-fucopyranosyl-l-pho~phate and guanvsine
S'~monophosphomorpholidate (4~morpholine-N,N'-di-
cyclohexyl-carboxamidine salt -- "GMP-morpholidate")
w2ra reacted in dry pyridine as indicated in the
original proceduren. Accordingly, the ~-L-
fucopyranosyl-l-phosphate (triethyl-ammonium alt,
O.528g, about 1.18 mmol) was dissolved in dry
pyridine (20 mL) and the solvent remov~d in vacuo.
The process was repeated three times with care to
allow only dry air to enter the ~lask~ GMP-
morpholid te (1.2g, 1.65 mmol~ and pyridine (20 mL)
were added into the reaction flask, the solvent
evaporated in vacuo and the process repeated three
times ~s above. Pyridine (20 mL~ was added to the
W092/22662 ~ PCT/CA92./002~l
9~ ~
-- 54 -
final residue and the heterogeneous mixture was
stirred for 3 to 4 days at room temperature under
nitrogen. An insoluble mass.was formed which had to
be occasionally broken down by sonication.
The reaction was followed by ~lc and worked up
as indicated in the first procedure ~o provide the
GDP-fucose ~120 mg, 16%).
Preparative Enzvmatic GalactosYlation
The bovine milk BGlcNAc B(1-4~ galactosyltransferase
(EC 2.4.1.22, specific acti~ity 6.~ units/mg of
protein) and UDP-Gal were obtained from Sigma. The
enzymatic reactions were carried out at 37~ in a
plastic tube using a sodium cacodylate buffer (100
mM, pH 7.5) containing 20 mM manganese di~hloride.
The reaction products were purified as indicated
above in the case of the preparative sialylation.
In some cases, depending upon the enzymatic
preparation, it may happen that the terminal methyl
est~r of the aglycone is hydrolyzed. As a resultO
the final products may possibly ~e isolated as
...
saccharides posssssing the aglycone terminated by a
me~yl ester of a free acid group. These two
c~harides are separated during the step of the
~hromatography on Iatrobeads as indicated above.
: 25 The two forms of the aglycone of the saccharide are
identi~ied by 1H-n.m.r.
Example 7 - Synthesis of 8-Methoxycarbonylo tyl
(2,3,4,6-tetra-0-acetyl-~-D-
- galactopyranosyl~ 4)-0-3~6-di-0-
acetyl-2-deoxy-2-phthalimido-B-D-
glucopyranoside (Compound 19)
W092/22662 21~ 3 9 ! PCT/CA92/00251
-- ~5 --
A. Synthesis of Compound 12
A solution of trimethylsilyltrifluoro-
methanesulfonate (0.504 mL, 2.6 mmol) in
dichloromethane (4 mL) was added to the mixture of
the disaccharide donor 113~ (2.0 g, 2.6 mmol),
drierite (~.0 g, crushed) and 8-methoxycarbonyl-
octanol (1,9 g, 10.0 mmol) in dichloromethane (30
mL) at ~C. ~fter stirring for 0.5 h at 4C, the
mixture was slowly warmed up to room temperature for
1 ho After cooling to 4C, a second portion of the
catalyst (0.250 mL, 1.3 mmol) in dichloromethane (2
mL) was added. After slowly warming up and stirring
at room temperature for 1 h, the reaction was
stopped by addition of triethylamine. After
filtration, the crude product recovered after the
~sual work up was dried in ~acuo, and acetylated in
a 2:1 mixture of pyridine and acetic anhydrid~.
After addition of methanol, the mixtu~e was worked
up as usual, and the solvents co-evaporated with an
excess of toluene. The residue was chromatographed
on silica gel using a 2Ol mixture of toluene and
ethyl acetate providing compound 12 (1.40 g, 60%).
H-n.m.r.(CDCl3): ~ 7.90-7.70(m, 4H, aromatics~
5~75(dd, 1~, J2,3 10.5 J3,~ 9.5Hz, H-3~ 5.34(m, 2H,
incl. H-l and H-4 ), 5.15(dd, lH, J1 2 8.0, Jz 3,
10.5Hz, H-2'), 4.97(dd, lH, H-3 ), 3.67(s, 3H,
CO2CH33, 2.25(t, 2H, J 7.5Hz, CX2CO2), 2.1~-1.94(6s,
18H, 6 OAc), 1.45(m, 4H) and 1.08(m, 8H):
methylenes.
B. Synthesis of Compound 5 -- 8-Methoxy~
rarbonylo~tyl (2,6-di-O-acetyl-3,4-O-
isopropylidene-B-D-galactopyranosyl) (1-4)-O--
(3,6-di-O-acetyl-2-deoxy-2-phthalimido-B D-
glucopyranoside (~5)
A lM solution of sodium methoxide in methanol
~0.200 mL) was ~dded to a solution of compound 12
WO92l22662 PCT/CA92/00251
'?J ~ 9~
-- ~6 -- -
~1.40 g, 1.65 mmol) in methanol (40 mL) cooled at
4C. After 1.5 h at 4 D C, the solution was deionized
using IRC-50 resin ~H+ form~. The resin was
filtered, the solvent evapora~ed and the product
dried in vacuo (1.0 g, 94%).
A solution of the a~ove material (0.776 g, 1.2
mmol) and paratoluene sulfonic acid monohydrate (60
mg) in dry acQtone (60 mL) was refluxed for 3 h.
A~ter neutralization with triethylamine, the solvent
was e~porated and the residue chromatographed on
silica gel using a 100:1 mixture of ethyl acetate
and methanol providing compound 1~ (0.575 g, 70%);
1H n~m.r. (CD30D, DOH at 4.80~: 7.80-7.60(m, 4H,
aromatics~, 5.10(d, lH! J1 2 8.0Hz, H-l), 4.38~m, 2H,
H-l and H-3), 3.70(s, 3H, CO2CH3), 2.31~t, J 7.5Hz,
CH2CO2), 1.65-1.00[m, incl. 1.57 and 1.45 (2s,
C (C~3) 2J~ Further elution provided the 4,6-
isopropylidene derivative (0.200 g, 24%).
Compo~nd 14 (0.515 g, 0.84 mmol) was ac~tylated
in a 2 :1 mixture of pyridine and acetic anhydride
~or 24 h at 22. ~fter addition 3f m~thanol and the
u~ual wnrk up, the solvents were co-evaporated with
an excess of toluene and the residue chromatographed
on silica gel.using a 100:3 mixture of chloroform
and methanol providing compound 1~ (0.646 g, 90%);
~]D ~ 13.3- (C, 1 chloroform); 1H-n.m.r.(CDCl3);
7.90-7~70(m, 4H, aromatics), 5-74(J~2 8.5, J23
10.5Hz, H-3), 5.34(d, lH J12 8.5Hz, H-l), 4.88(dd,
lH, J1 2 -J2 3 6.5Hz, H-2 ), 3.67(s, 3H, CO2CH~),
2.23(t, J 7.5Hz, CH2CO2), 2.14, 2.13, 2.10, 1.91(4s,
12H, 4 OAc~ 30-1.54~m, incl. 1.53 and 1.32(2s,
C(CH3)2] -
W092/22662 2 ~1~ 9 9 7 PCT/CA92/00251
- 57 --
C. Synthesis of Compound lS -- 8-Methoxycarbonyl-
octyl (2,6-di-0-acetyl-~ galactopyranosyl)-
(l-4)-0-3,6-di-0-acetyl-2-deoxy-2-phthalimido-
B-D-glucopyranoside
Compound 15 (0.575 g, 0.68 mmol) in 90% acetic
acid (l~ mL~ was heated at 80 for 2 h. Aft~r
dilution with dichloromethane, ~he solvent was
washed with water, a solution of sodium bicarbonate
and water. After drying over magnesium sulfate, the
solvents were evaporated in vacuo, and the residue
chromatographed on silica gel providing compound 16
(0.452 ~, 82%); [~]D +12.1 (C, 1.03 hloroform).
D~ Synthe is of Compound 8 -- 8-Methoxy-
car~onyloctyl (3,4,6-tri-0-acetyl-2-deo~y-2-
phthalimido~ glucopyranosyl)~ 3)-0-(2,6-
:~ di-0-acetyl B-D-galactopyranosyl)-(1-4~-Q-3,6-
di-O-acetyl-Z-deoxy-2-phthlamido-B-D-
glucopyranoside (l8)
Trimethylsilyltrifluoromethanesulfonate (0.036
mL, 0~060 mmol ) in methylene chloride (O.5 mL) was
added to a solution of ~ompound 16 (O.lO0 g, 0.123
mmol) in methylene chloride (5 mL). A solution of
the~ imidate 17 (0.102 g, 0.1~5 mmol) in met~ylene .~'
chloride ~4 mL) was slowly added to the above
soIutivn cooled at -70. The mixture was further
stirred at that temperature for 0.5 h. An
additisnal portion of the catalyst (0.018 mL, 0.030
mmol) in methylene chloride (O.5 mL~ was fur*her
added. After 0.5 h at -70, the reaction was
stopped by addition of triethylamine, and th
mixture worked up as usual. The recovered residue
was chromatographed on silica gel using a lO0:2
mixture o~ chloroform and methanol providing
compound l~ (0.120 g, 80%); 1H-n.m.r.(CDCl33: ~
7~95-7.60 (m, 8H, aromatics), 5.74(dd, lH, J2 3''
10.5 J3 4 9.0Hz, H-3 ), 5.61(dd, lH, J23 10.5,
WO92~2?662 PCT/C~92/00251
~ ~Q 9 -- 58 --
J3~ 8.5Hz, H-3~, 5.48(d, lH, J1..2.. 8.5Hz, H-l ),
5.27(d, lH, ~12 8.5Hz H-1),-5.14(dd, lH, J~.5
10.0Hz, H-4 ), 4.90(dd, lH, J1~2 8-0 J3.~ 10-0HZ~
H-2 ~, 3.68(s, C02CH3), .22(t, J 7.5Hz, CH2C02),
2.12(two), 2.10, 2.04, 1~86, 1.85, 1,56(6s, 21H, 7
OAc), 1.~0~m, 4H), and 1.20(m, 8H): methylenes.
E. Synthesis of Compound 13 -- 8-Methoxycarbonyl-
octyl (2-acetamido-2-deoxy-B-D-
gIucopyranosyl)-~1-3)-O-(B D-
galactopyrancsyl)-(1-4)-0-2-acetamido-2-
deoxy-B-D-glucopyranoside
Hydrazine acetate (1~27 g, 13~8 mmol) was added
~o compound 1~ (0.120 g, 0.098 mmol) in anhydrous
ethanol (15 mL). The mixture was refluxed for 18 h.
The solvent were then co-evaporated with an excess
of toluene. After drying in vacuo, the residue was
acetylated in a 2:1 mixture of pyridine and acetic
anhydride for 48 h. Af*er quenching the exc~ss of
acetic anhydride with some methanol, the reaction
mixture was worked up as usual. The recover~d
solvents were evaporated in vacuo and the residue
co-e~aporated with an excess of toluene. The
residue was chromatographed on silica g21 using a
}00:9 mixture of chloroform and methanol as eluant
provided the peracetylated trisaccharide
i~termediate. This material was de-O-acetylated in
anhydrous methanol ~5 mL) in the presence of 0.2
sodium methoxide in m2thanol (0.200 mL). After
overnight at 22C, de-ionization with Dowex 50 X 8
and filtration, the solvent was e~aporated in vacuo.
The reco~erPd product was chromatographed on ioGel
P-2 and eluted with a 1:1 mixtur~ of water and
ethanol whi~h provided the pure trisaccharide lS
~0-044 g, 60%); [~D -4-8 (C~ 0.48, water); lH-
n.m.r. (D2O): data provided in Table II.
W092~22662 2 ~ ~ Q ~ 3 ~ PCT/CA92/00251
.~
__ Sg __
Example 8 - Synthesis of 8-Me~hoxycarbonyloctyl ~5-
acetamido-3,5-didèoxy-~-D-glycero-D-
galacto-2-nonulopyranosylonic acid~-(2-3)-
0-(B-D-galactopyranosyl)-(1-4)-0-(2-
acetamido-2-deoxy-B-D-gluropyranosyl)~
3)-0-(B-D-galactopyranosyl)-(1-4)-0-[~-L-
fucopyranosyl-(1-3)-0]-2-acetamido-2-
d~oxy B-D-glucopyranoside ~22) (Compound
2~ he CD-65/VIM-2 Saccharide)
A. Synth~sis of Compound ~0 -- 8-~etho~y-
carbonyloctyl (2-acetamido-2-deoxy-B-D-
glucopyranosyl)-(1-3)-0-~B-D-
galactopyranosyl)-(1-4~-0-[~-L-
fucopyranosyl-(1-3)-Q]-2-acetamido-2-
d~oxy-B-D-glucopyranoside
Compound 19~ mg), GDP-fucose (33 mg) and the
~Glc~Ac ~ 3/4jfucosyltransferase (5~ mU) were
incubated for 72 hours in the buf~er (4 mL) as
indicat~d a~ove. Isolation and purification
~: 20 pr~vided the compo~nd 20 (14.0 mg). 1H-n.m.r. data
is included in Table II.
~:~ B. Synthesis of ~ompound 21 -- 8-Metho~ycarbonyl-
octyl (B-D-galactopyranosyl)~ 4)-0-(2-
acetamido-2-deoxy-B-D-glucopyranosyl)~
3)-0-(B-D-galactopyranosyl~-(1-4) 0~
fucopyranosyl~ 3)-0] 2-ac~tamido-2-
d~oxy-B-D-glucopyranoside
Compound 20 (14.0 mg), UDP-Gal ~25 mg), BGlcN~c
~tl-4~ galactosyltransferase (14.5 U, Si~ma) were
incu~ated for 48 hours in the buffer described above
~3.2 mL). Isolation and purification provided
: compound 21 (13.2 mg). lH-n.m.r. data is included
in Table I~.
C. Synthesis of Compound 22 -- 8-~ethoxycar~onyl-
octyl ~5-acetamido-3,5-dideoxy-~-D-glycero-D-
galacto-2-nonulopyranosylonic acid)-[2-3)-0
D galactopyr~nosyl)-(1-4) 0-(2-acetamido-2-
deoxy-B-D-glucopyranosyl)~ 3)-0-(B-D-galacto-
pyranosyl) (1-4)-0-[~-L-fucopyranosyl-(1 3)-0]-
2-acetamido-2-deoxy-B-D-glucopyranoside
Compound ~2 was synthesized from compound 21 as
lndicated above65.
WO 92~22662 PC~/CA92/OD2~1
---- 60 ----
99 ~
Table II: IH-n . m . r . 5tructural Parameters
_ ~ _. ~ ._ . ~
¦ Unit Hydro~en lg-,b 20~ 21~ 22
; _" . _ ~ . . . ,
¦ pGlcNAc 1 (d)4.50 (7.5) 4.52 P.5) 4.52 ~8.0) 4.53 ~8.0) l
I (A) _ _ ¦
¦ ~tGal ~B) t ~d) 4.45 ~8.0~ 4.4317.0) 4.4317.5) 4.4318.0)
4 (dJ4.15 ~3.0) 4.09 l3.5) 4.1013.2) 4.1013.0)
_. . . I
~BGIcNAc 1 (d)4.66 l8.5) 4.67 ~8.5) 4.7017.0 4.69 ~8.0)
I ~:)
. . . . . .
l ~Gal ~D) 1 ~d) 4.48 ~7.8) 4.4617.7)
E 3 ~d~
.. . ~ . . . Il
oFuc 1 Id~ 5.0914.0) 5.1 0 ~3.0 5.09 ~3.8)
Iq) 4.81 1~.5) 4.81 (6.~) 4.81 ~6.5J
6 ~d) 1.14 1.15 1.15 l
_ ... ... -- . . Il
l oNeu5Ac 3~,~ ldd) 2.76 ~4.5, 13.0) ¦I 3 ~t) t.~9 ~12.0)
_ ., . . . . . - 11
NHAc s 2.02, 2.01 2.02, 2.01 2.03, ~.02 2.02 Ithree) ¦~
CH,C0, t 2.38 (7.5) 2.381~.5) :Z.38 R.5) 2.38 ~7.5
02Ct~3 3 3.6~ 3.69 3.69 3.~9
_=~ ~===
g, ~0, 11 and 12 how multiplets around 1.4~-1.63
(4~i~ and 1. 30 (8H~: met.hylenes
b J in HZ
,. ~
interchangeable
i
SU~Sml.JTE 5HEET
IS~VEP
W092/22662 ~ 3 7 PCT/CA92~00251
.
-- 61 --
C. IMMUNOSUPPRESSIVE PROPERTIES
E~amples 9 and lO illustrate the
immunosuppressive properties of hexasaccharide
glycoside 5a.
Example 9 -- Inhibition of DTH Inflammatory Response
DTH inflammatory r~sponses were measured using
the mouse footpad swelling assay as described by Smith
and Ziola3l. Briefly, groups of Balb/c mice were
immunized with lO ~g of the Llll S-Layer protein, a
bacterial surface prokein32 from Clostridium
thermohydrosulfuricum Llll-Ç9 which has been shown to
induce a strong inflammatory DTH response. Seven days
later, each group of mice was footpad-challenged with
~g of L 111 S-~ayer protein. The resulting
înflammatory footpad swelling was measured with a
Mitutoyo Engineering micrometer 24 hours after
challenge.
To assess the effect of hexasaccharide glycoside
5~ on the inflammatory DTH response, groups of mice
received lOO ~g of this compound, injected in~o the
tail vein, 5 hours after challenge. Control groups
received 100 ~L of phosphate-bu~fered saline (PBS).
T~e results of this experiment are shown in Table II
below~ In this ta~Ie, smaller increases in footpad
swelling, as compared to control, evidence the fact
~hat the tested compo~nd poss sses immunosuppressive
properties in that it red~lces the degree o* footpad
~welling in response to an antigen.
TABLE II
COMPOOUND TESTED INCREASE IN FOOTPAD SWELLING (mm-l~
Control 3.3
Hexasaccharide Glycoside 5~ 1.5
The above results indicate tha~ mice injected
with hexasaccharide glycoside 5~ had less than 50% of
3~ the footpad swelling as compared to the control mice.
....
,'
,. :, , I , ., ,-" . .
`` 211U9~7
-- 62 --
Example 10 -- Persistence of Suppression of the DTH
Inflammatory Response at 11 Weeks After
Challenge
i. The identical groups of mice treated with
hexasaccharide glycoside Sa in Example 7 were re-
challenged with L111 S-Layer protein 11 weeks after
primary immunization. Mice treated with the PBS
control responded with the usual degree of footpad
swelling whereas mice treated with hexasaccharide
glycoside Sa sh~wed a reduction in footpad swelling of
about 40%, i.e., the mice treated with hexasaccharide
glycoside 5a exhibited only about 60% of the footpad
swelling exhibited in mice treated with PBS.
This anti-inflammatory effect of hexasaccharide
glycosides 5a, given 5 hours after the first challenge
(ona week after primary immunization), had somewhat
weakened eleven weeks after primary immunization but
ne~ertheless providPd for a significant reduction in
inflammation as compared to PBS tre~ted controls.
In additi:on to providing suppres~ion of cell-
medi~ted immune responses, the above data demonstrate
that treatment with a hexasaccharidP glycoside as per
this inv~ntion also imparts tolerance to additional
challenges from the same antigen.
2S Fxample 11 -- Effect Hexasaccharide Glycoside 5a has
on Endothelial-Leukocyte Adhesion Molecule-1
(E~AM-l) Dependent Cell Adhesion to Acti~ated
Vascular Endothelium
$his example examines whether hexasaccharide 5a
could inhibit ELAM-l dependent cell adhesion to
a~tivated vascular endothelium. Specifically, an ~n
~itro cell binding assay was preformed as de cribed by
Lowe et a}33. Briefly, human umbilical vein
endothelial cells (HI~JECs purchased from Cell Systems,
Seatt~e, WA, USA) were stimulated with TNF~ ~10 ng/ml)
to express ELAM-l . Human tumor cell lines, U937 or
HL60, whieh ha~e been shown to bind to HUVECs, in an
~;~BS~ T