Note: Descriptions are shown in the official language in which they were submitted.
-` 2~ ~ ~3~
The present invention relates to 3-(6-quinolyl-
methyl)-41f-imidazol-4-one deri~atives, to th~ir prepara-
tion and to their application in therapy.
The compounds of the invention ccr~2spond to the
formula (I)
N~Z~ 11 ( I )
in which
Rl repre~ents an unbranched or branched (C2-C5)alkyl
group,
R2 and R3 represent, each independently of one another,
either a hydrogen atom, or an unbranched or branched (Cl-
C5)al~yl group, or a (CM2)n-aryl ~roup where n = O to 3,
or R2 and R3 with the imidazole ring can form a spiro-
cyclo(c3-ca)alkyl group.
The preferred compounds o~ the invention are compounds having
formula (I) wherein,
R~ represents an unbranched or branched l~-C5)alkyl group,
and R3 represent, each independently of on~ another, an
unbranched or branchad (C~-C5~alkyl group,
or ~ and R3 with the i~idazole ring can form a 6pirocyclo
(C~-C~)alkyl group.
Among the~ the compounds of choice are those having formula
(I) ~herein,
Rl represents a butyl group,
, ... ,. ,," . ... ~ - , .r ~; ~
~ --`` 21113~.~
R2 and R3 represent, each independently of one another, an
unbranched or branched (Cl-C5) alkyl group,
or R2 and R3 with the imidazol,e ring can form a spirocyclo
(C3-C8)alkyl group.
The compound~ of the invention ma~ ~e presented
in frea form or in the form of pharmaceut.cally accep-
table organic or inorganic salts.
According to the invention, th~ co~.~cunds of
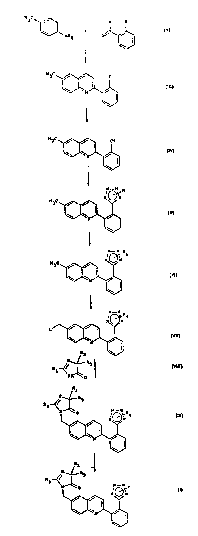
formula (I) may be synthesized according to Sc~eme 1.
4-~ethylbenzenamine (para-toluidine) is r acted
at the re~luxing temperature with a benzaldehyde of for-
mula ~II), in which X represents a bromine or iodine
atom, in the pre~ence of a catalyst such as 4-methylben-
zenesulfonic acid (para-toluenesulfonic acid), in solu-
tion in benzene. After cooling, propiolic acid is aàded
and the mixture is heated to the refluxing temperature so
as to obtain the compound of formula (III). Next, a
mixture of the compound of formula (III) and cuprous
cyanide is heated in a solvent cuch as pyridine so as to
obta~n 2-(6-methyl-2-quinolyl)benzonitrile (IV), which is
reacted with an organometallic a~ide such as trimethyltin
azide, or a metal azide such a~ sodium azide, so as to
obtain a compound over which a stream of gaseous hydro-
chloric acid i~ passed in order to obtain the quinoline
of formula (V). The first reaction is performed in a
solvent ~uch as xylene at the refluxing temperature; ~he
second reaction is perfor~ed in a solvent such as a
toluene/tetrahyd,oluran mixture at room temperature.
The tetrazole group of the quinoline of formula
(V) i8 then protected with a protective group R4, where R~
. ,.... . . ~ .. ~ . . ~ . . .:: .
2 1 1 :~ 3 3 ~
.
Sc heme
",
I
.
:
~ ~ .. ..
-~ .
: I .: .:
~ . I . -: :. ~
p_ " :
~ L
' ~
: :` 2~ .2
represents a group of formula CR,R6R7 in which Rs~ R6 and
R7 aro, each independsntly of one another, a (Cl-Cz)alkyl
group or an aryl ~roup; in this step, the compound (V) is
reacted with a protective agent such a~, for example,
trityl chloride at room temperature in a solvent such as
dichloromethane, in the presence of a base such as N-
methylmorpholina or triethylamine, and a compound of
formula (VI) in which R5, R6 and R7 are as defined above
is obtained. Next, the methyl group at position 6 of the
quinoline of formula (VI~ is functionalized by introduc-
ing a leaving group L, for example a bromo radical, into
it, and a compound of formula (VII) where R5, R6 and R~
are as defined a~ove is obtained; the reaction is per-
form~d at the refluxing temperature in a solvent such as
carbon tetrachloride, in 'he presence of an initiator
~uch as benzo~il peroxide or ~ azobisisobuty.onitrile,
at the refluxing temperature. Finally, the compound of
formula (VII) is condensed, in dLmethylforma~ide at a
temperature of 0C to 50C, in the presence of a base
such ~s potassium hydroxide or potassium carbonate, with
an imidazolone of formula (VIII) in which ~, R2 and R3 -~
are as defined above~ and a compound of formula ~IX) is
obtained, which compound is sub~ected to deprotection.
The compounds of formula ~IX)
R2
N ~ / ~ R4 (IX)
/J~3 '
'"",,. ~", ~,~ ", " " ,~ ", .: i:' ' ' " '''; ' ' ' '' ~ ` ' ~` '~ '' ''` ' ~ '
~ `` 2 ~
in which R~, R2, R3 and R~ are as defined abo-.-e, are new
and form part of the invention.
The 3tar~ing compounds are commerciall~ available
or described in the literature, or may be prepared
accor~ing to methods which are described therein or which
are known to those skilled in the art.
Thus, the imidazolones of formula (VIII) may be
synthe~ized according to the method described by Jacquier
et al ~Bull. Soc. Chim. France, 1971, 3, 1040-1051~, by
condensation o~ an alXyl imi~ate and an amino ac~ d ester
such as, ~or example, methyl l-amino-l-cyclopropylcar-
boxylate.
Methyl l-amino-l-cyclopropylcarboxylate is
prepared according to the method described by
Valdyanathan (J. Orq. Chem., 1989, 54, 1810-1815).
- The compounds of formula (VII) are prepared
according to the method described in the European Patent
Application published under ~oO 0540500 by the Applicant
The ~xamples which follow illustrate the
: invention.
The microanalyses and the IR and NMR spectra
confirm the structure of the compounds obtained.
!
Example 1
: 5-butyl-6-~t2-t2~ tetraæol-5-yl~phenyl~-6-quinolyl3me- ::
thyl~-4,6-diazaspiro~2.4]hept-4-en-7-one
.
1.1. 6-bromomethyl-2-[2-t2-((triphenylmethyl)2~-tetrazol-
5-yl]phenyl~quinoline
1.1.1. 2-(2-bromophenyl)-6-methylquinoline . .
50 ~ (270 mmol~ of 2-bromcbenzaldehyd~ are heate~
2 ~ 3 ~ ~
to the refluxing temperature, in a round-bottomed flask
surmounted by a Dean and Stark apparatus, wi.h 29.5 g
(276 mmol) of para-toluidine and 0.5 g of pa-a-toluene-
sulfonic acid dissolved in one liter of benzene. When the
removal of water is complete (approximately 5 ml), 8.3 ml
(135 mmol) of propiolic acid are added to the reaction
medium which has been cooled beforehand to about 50~C. A
substantial evolution of C02 iS noted, and the mixture is
brought to reflux for 3 hours. The reaction is monitored
by thin-layer chromatography in a mixture of dichloro-
methane and hexane (70:30). Under these experLmental
conditions, it has been necessary to add a 20 % excess of
propio}ic acid followed by 1 hour of reflux in order to
complete the reaction. The solvent is evaporated off
under reduced pressure ànd the residue is purified by
chromatography on a silica col~n, elutin~ with a mixture
:
of dichloromethane and hexane ~70:30).
~ 22 g of ths expected derivati~e are recovered in
;~ the form of a crys~allized compound.
` M = 22 g
;~ ~H NMR (200 MHz, CDC13): ~ 2.55 (s, 3~), 7.25-7.70 (~,
7H), 8.02-8.15 (m, 2
In the same manner, 2-(2-iodophenyl)-6-methyl-
. : quinoline is prepared from 2-iodobenzaldehyde.
M.p. = 77-77.5C
1.1.2. 2-(6-methyi-2-quinolyl)benzonitrile
A mixture containing 15 g (50 m~.ol~ of the
2~ fl ?
.
compound obtained above in 1.1.1 and 5 g (56 mmol) of
cuprous cyanide is heated to 160C for 12 hours, under
argon, in 60 ml of pyr.idine. The reaction is monitored by
thin-layer chromatography in dichloromethane. The
pyridine is evaporated off under reduced pressure and the
residue is taken up with dichloromethane. The organic
phase is washed several tLmes with aqueous ammonia
solution un~il the aqueous phase is colorless. After a
final wash wi~h water, the oryanic phase is dried over
magnesium sulfate and the solvent is evaporated off. The
residue is taken up with petroleum e~her.
M = 9.6 g ~.p. 5 157 C Yld - 78 %
1.1.3. 6-methyl-2-~2-(lH-tetrazol-5-yl)phenyl]quinoline
hydrochloride :~
9.6 g (3~.3 mmol) of nitrile obtained above in ~`
1.1.2 and 14.96 g (72.7 mmol) of trLmethyltin azide are
introduced into 110 ml of xylene. This mixture is brought
to reflux for 15 hour~. After cooling, the solid is
filtered off and sus~ended in 115 ml of toluene and 7 ml
of tetrahydrofuran. The mixture is cooled in an ice bath
and subjected to a stream of hydrogen chloride gas
bubbled through for 2 hours. The insoluble fraction is
recovered by filtration and then washed with toluene and
with water.
M = 13 g
2 ~ 3 ~ ~f 1'' i
' .
1.1.4. 6-m~thyl-2-[2-[l(or 2)-~triphenylmethyl)-l(or 2)~-
tetrazol-5-yl]phenyl~quinoline
80.5 g (219 mmol) of the compound obta n~d above
in 1.1. 3, 60 ml ~54~ mmol) of N-methyLmorpholin2 and
73.1 g ~262 m~ol) of trityl chloride are added at room~-
temperature to one liter of dichloromethane. The solution
is stirred overnight and taken up with water, and the
organic phase i washed twice with water and then dried.
The solvent is evaporated off and the residue is crystal-
lized in a minimum amount of ether.
M = 119 g ~.p. = 176-177C Yld = 87 % ~ - :
1.1.5. 6-bromomethyl-2-[2-~l(or 2~-(triphenylmethyl) ol (or
2)~-tetrazol-5-yl]phenyl]quinoline
10 g (189 mmol) of the compound obtained above in
1.1.4 are added to 300 ml of carbon tetrachloride, and
the mixture ls brought to about 60C until dissolution is
complete. At this temperature, 3.7 g (20.8 mmol) of N-
bromosuccinLmide and 60 mg ~0.37 mmol) of ~ azobisiso-
butyro~itrile are added in a single portion. The mixture
is brousht to the refluxing temperature for 2 to 3 hours
until the N-bromosuccinimide has disappeared. 100 ml of
water and 300 ~il of dichloromethane are added to the
cooled mixture. The organic phase is washed several ti~es
~ith water and then dried. The solvent i~ evaporated off
and the residue is ground in diisopropyl ether. A 90 %
pure product is obtained, which product will be used as ~ :
it is.
- -~ 2 ~
M = 10.3 g
1.2. 5-butyl-4~6-diazaspiro[2.4]hept-4-en-7-one hydrochloride
A mixture of 15.6 g (121 mmol) of ethyl pen-
tanimidate and 13.5 g (117.4 mmol) of methyl l-amino-l-
cyclopropylcarboxylate is heated to the refluxing temper-
ature for 8 hours in lS0 ml of xylene to which 20 drops
of acetic acid are added. The solv~nt is evaporated off,
the residue is taken ~p with ether and the mixture is
acidified with 12 N hydrochloric acid. A precipitate is ~`
obtained and is filtered off.
H NMR, 200 M~z(CDCl3) d [sic] 0.95 tt, 3H), 1.4S (m, 2H)
1.6-2.0 (unresolved complex, 4H~, 2.20 (m, 2H), 3.0
(t, 2H). `
1.3. 5 butyl-6-tt2-~2-tl(or 2~-(triphenylmethyl)-l(or 2)~- :
tetrazol-5-yl~phenyl];6-quinolyl]methyl~-4,6-diaza spiro[2.4]
hept-4-~n-7-on~
1 g (7.24 ~mol) of potassium carbonate and 1.42 g
(1.87 mmol) of the 80 % pure compound obtained
above in step 1.1 and 6 ml of dimethylformamide are added
to 0.35 g (1.73 mmol~ of the hydrochlori~e obtained in ~ :
step 1.2. The mixture is left stirring at room temper-
ature overnight. The reaction medium is taken up with
water and extracted with dichloromethane. The organic
phase is recovered, the sclven~ is evaporated off and the
residue is dried over magnesium sulfate. The residue is -
purified ~y chromatography on a column of silica sel,
"
eluting with a toluene/ethyl acetate (80;20) mixture.
1.4. S-butyl-6-~[2-t2-(lH-tetra201-S-yl)phenyl]-6-quinol-
yl]methyl]-4,6-diazaspiro[2.4~hept-4-en-7-one
.500 mg of the compound obtained in step 1.3,
dissolved in 20 ml of an acetic acid/methanol ~gO:10)
mixture, are heated for 5 hours to the refluxing tempera-
ture. ~he solvents are evaporated off and a gum is
obtained, which gwm is purlfied by chromatography on a
column of ~ilica gel, eluting with an ethyl acetate~meth-
anol/acetic acid (100:5:0.5) mi~ture. The product is
crystallized in water.
NMR d tsic] 0.77 (t, 3H), 1.17-1.4 (m, 2~), 1.~2-1.75
(m, 6~), 2.5 (t, 2H), 4.~8 (s, 2~), 7.5-8 (m, 8~), 8.3
(d, 1~), 16.2-16.6 (~nresolved complex, 1~
1.5. 5-butyl-6-tt2-~2~ tetrazol-5~yl)phenyl}6-quinolyl]
methyl`]-4,6-diazaspiro[2.4]hept-4-an-7-one, dihydrochloride
Compound obtained in ~tep 1.3 is dissolved in a ~ini~al
volumo of ether and 2 eguivalents of an aqueou~ ~ N ~olution
of hydrochloric a~d are added. The reaction m~xture i8 lef~
6tirring at room temperature overnight. The chlorhydride
cri~tallized in th~ reaction mixture, is filtered and washed
with ether.
Melting point = 12~ ~C (daco~po6ition)
2~ J
' 11
E~cample 2
2-butyl-3-[[2~[2-(l~f-teLrasol-5-yl)phenyl]-6-quinolylJme-
thyl]-1,3-diazaspiro[4.4]non-1-en-4-one
2.10 2-butyl-3-~[2-t2-~l(or 2)-(triphenylmethyl)-l(or 2)~-
tetrazol-5-yl]phenyl]-6-~uinolyl]methyl]-1,3-diaza~pi
ro~4.4~non-1-en-4-one
2.1.1. 2-butyl-1,3-diazaspirot4.4]no~-1-en-4-~one chlorhydride
Title compound is prepared as described in step 1.2 from
ethyl l-amino-l-cyclopentylcarboxylate.
2.1.2. 2-butyl-3-[[2-~2-Cl(or 2)-(triphenylmethyl)-l(or 2)~
tetrazol-5-yl]phenyl]-6-quinolyl~ethyl]-1,3-d~azaspirot4.4
non-l-en 4-one
1 g (7.24 mmol) of potassiu~ carbonate and 1.25 9
(1.64 ~ol) of the 80 ~ pure [~ic] compound Gbtained in
step 1.1 are added to 0.35 g (1.52 mmol)of compound
obtained in ~tep 2.1.2 dissolved in 6 ml
of -dimethylformamide. The reaction mixture is left
stirring at room temperature overnight. It is taXen.up
with water and extracted with toluene. The organic phase
is recovered, the solvent is evaporated off and ~he ~ :
organic phase [sic] is dried over magnesium sulfate. The
residue is purified by chromatography on a column of
silica gel, eluting with a toluenefethyl acetate (85:15)
mix~ure. :~
0.8 g of the expected product is oDtained.
~"` 2~3~
12
2.2. 2-bu;yl-3-~[2-[2~ -t~tra~ol-5-yl)p~.enyl]-6-
quinolyl]methyl]-1,3-diazaspiro[4.~]non-1-en-4-one
20 ml of a m2thanol/acetic acid (~0:10) ~ x~ure
containing 0.8 g of tha co~pcund obtained above in s~ep
1.3 are heated to the refluxing temperature for 6 hours.
The solven~s are removed under reduced pressure and the
residue is crystallized in ether.
0.3 g of product is obtained.
NMR d Isic] 0.8 (t, 3H), 1.17-1.37 (m, 2H), 1.38-1.62 (m,
2H~, 1.62-2 (m, ~H), 2.4 (t, 2H), 4.95 (s, 2H), 7.55 ~d,
2H), 7.6-8 (m, 6H), 3.5(d, lH), 16.2-16.6(unresolved
co~plex, lH).
amPle ~
2-butyl-3-~2-t2-(1~-tetrazol-5-yl)phenyl~-6-quinolyl]
methyl]-1,3-diaPaspirot4,5]dec-1-en-4-on~
3.~. 2-butyl-3-lt2-[2-[l(ou ~)-(triphenyl~athyl)-l(ou 2)H-t~-
trazol-5-yl)phenyl~6-quinolyl]methyl]-1,3-diazaspirot4,5~ :~
dec-1-en-4-one ~ -
3.1.1. 2-~utyl-1,3-diaz~spirot4,5]d~c-1-sn-4-one
A ~ixture of 11,3 g (8,8 mmol) of ethyl pe~tanimidate and of
11,56 g (6,7 mmol) of ethyl 1-amino-1-cyclopentyl~arboxylat~
in 100 ml o~ xylen containing 0,6 ~l o~ ac~tic acid i8 heat~d
to refluxing temperature. Then reaction mixture i~ allowed to
ool, residue is eliminated by filtrat~on and ~olvent ~:
evaporated under reduced pressure.Titl~ compound cri6t~11ized
in ether.
Melting point = 124-125 C.
3.1.2. 2-butyl-3-[[2-t2-[1(or 2)-~triphenylmethyl)-l~or 2)~
tetrazol-5-yl)phenyl]-6-quinolyl~methyl~-1,3-diazaspiro
[4,5]dec-1-en-4-one
1,64 g (7,88 mmol) of compound obtained in step 3.1.1, 3 g
(21,7 mol) of potassium carbonate and 5 g (7,47 mmol) of the
-
2~3~
13
90 % pure compound obtained in ~tep 1.1 are added to 25 ml of
dimethylformamide. The reaction mixture is le~t stirring at
room temperature overnight and is poured into 100 ml o~
water. A~ter filtration, t~e residue is purified by chromato-
graphy on a column of silica gel eluting with a toluen/ethyl
acetate (80:20) mixture.
2,3 g of the expected product is obtained in th~ form of a
302. 2-butyl-3-~2-~2-(1~-tetrazol-5-yl)pheny~]-6-quinolyl]
methyl]-1,3-diazaspiro~4,5]dec-1-en-4-one
20 ml of methanol/acetic acid (90:10) mixture conta~ning
0,59 g (0,8 mmol) of the compound obtained above in step 3.1
are heating to the refluxing temperature for 5 hour3. The
solvents are removed under vacuum and the residue i8 cris-
tallized in ether.
0,2 g of product are obtained.
Melting point = 122-123 C
N~R : d 0,8(t,3H), 1,17-1,37(~2H), 1,37-1,88 (unresolved
complex, 12H), 2,4(t,2H), 4,9(~,2H), 7,5~d,2H), 7,6-B(m,6~),
8,35(d,lH)
The ta~le which follo~s illustrates the
struct~res and physical properties of a faw compounds
according to the invention.
.
r~
..:
1~ .
O a ~ N V a ~ u) r ~ I
~ D 5~ 1` o t~ ~0
æka: ~ ~ ,, ~,,
~\ /
_ _ _ '~
i ~ ~ _ Y _
_
11 1 1 1 ~ ~
. ~.
-- ~
¦ r ~. ~ r 8
~xOO
= n~`n ~ ,=, ~ ~r n
7 ~` ~_ ~ ~
~ ~ N ~ ~1 0 N ~
~a ~~ ` ''Ot`~
_ t~ , , , .,: :~
O N N ~: :
~ 1 ~ ~ ~
~ t~ ~ ~
~ ~ q ~ o ','~
P: ~ C~ ~ .C -~C 5
~; _ _ ; U O U O
:
The compound~ of the invention were subjected to
pharmacological studies which demonstrated their
angiotensin II-antagonist properties.
Test of bindinq of r3Hlangiotensin II to ra~bit adrenal
cortex.
Male Fauve de Bourgogne rabbits weighing 2 to
3 kg are used. After sacrifice by cervical dislocation,
the adrenal ~lands are excised and the cortex is
dissec~ed on a culture dish cooled in ice. It i~ placed
in 10 ml of an ic~-cold 10 mN tris(hydroxymethyl)amino-
methane buffer solution containing 0.33 ~ sucrose and 1
mM ethylsnediami~etetraacetic acid and whose pH has ~een
adjusted to 7.4 with hydrochloric acid. The tissue is
homogenized using an electrical Potte_ apparatus with 13
to-and-fro movements of the plunger at a speed of
1200 rpm. The volume o. the preparation is adjusted to
25 ml with Tris-sucrose ~uffer before centrifuging for
15 min at 1075 x g. Th~ su2ernatant is ret~ined. The
pellet is homogenized again, after resuspension in 10 ml ~ .
of Tris-sucrose buffer, ~y transfer to the electrical
Potter, and then centrifuged under the conditions
described above. The supernatant obtained is added to the
: . first, and they are centrifuqed for 30 min at 47,800 x ~.
: The pellets are finally taken up with 150 volum9s
(equivalent to 100 mg of tissue in 15 ml of buffer) of a
50 m~ Tris-HCl buffer solution containing 150 mM NACl,
5 mM ethylenediaminetetraacetic acid, 1.25 ~y/ml of
3 ~
bacitracin, 100 ~M phenylmethylsulfonyl fluoride and
0.2 ~ of ~ovine serum albumin (pH = 7.4 at 25C).
This suspension contains the microsomes of the
adrenal cortex, and will be used as it is in the studies
described below.
lO0 ~l aliquot fractions of suspension are
incubated in the presence of l3H]angio~ensin II (New
England Nuclear, specific actiYity 61 Ci/mmol~ in a final
volume of 1 ml of Tris-HCl buffer whose composition has
been descri~ed above. A~ter incubation for 30 minutes at
25C, the microsomes are recovered by filtration on
0.45 ~m Millipore HANP~ cellulose nitrate filters condi-
tioned be~orehand by immersion in a 1 % solution of
bovine serum albumin. The fil~ers are washed three t~mes
with 5 ml of ice-cold ~ris-~Cl buffer. The q~antity of
radioactivity bound to the tissues and retained on the
filters is measured by scintillation spect~ometry.
: Non-speci~ic binding of ~3H]angiotensin II is
measured ~y incubation in ~he presence of 1 ~m non-
radioacti~e angiotensin II. This non-specific bindins
represent~ 5 to lO ~ of the total .quantity of radio-
activity bound to the filter. The non-specific binding is
the difference between the total radioactivity collected
on the filter and the non-specific binding. The binding
of t3HI2ngiotensin is measured in the presence of dif-
ferent concentrations of ~est compounds, and the IC50, the
concentration of the test compound which inhibits 50 % of
the specific binding of ~3H~angiotensin II, is determined
"~
- 16 -
graphically.
The IC50 concen~rations of the compounds of the
invention are les~ than 50 nM.
Inhibition of the resPOnse to anqi~tensin II on rat
arterial blood pressure
Male rats (Sprague-Dawley, Charles Ri~er France)
weighing 250 to 280 g are used, the rats ~eing anes-
thetized with pentobarbital sodium (55 mg/kg i.p.) and
maintained under artificial respiration (Harvard~ Respir-
ator: respiration rate.70 ml per minute, air volume 12 ml
per 100 g o~ body weigh~ he animals are ~pithed- using
a metal rod introduced via the orbit of the right eye and
inserted over the length of the Yertebral column. The
left and right vagus ne~v~s are sectior,ed (~ilateral
vagotomy); the right carotid artery is ligated, the left
carotid artery ~eing catheterized in order to measure the
arterial blood pressure using a pressure cell (Stath~m~
type P23Db). A femoral vein is catheterized for the
purpose of administration of various compounds.
The changes in mean arterial blood pressure
induced by angictensin a~inistered intravenously at a
dose of 0.5 ~g~kg before the administration of the
compound~ of the in~ention, and those induced by angio-
tensin administered under the same condi.ions S minutes
after the intravenous administration of the co~pounds of
the invention or 30 minutes after the oral a~ministration
thereof, are measured. The compounds of the invention are
administered at doses ranging from O.01 to 100 mg/kg.
The ?ercentage inhibition of the control response
to angiotensin II is used in order to assess the
angiotensin II-anta~onist potential of the compounds of
the invention.
The compounds of the invention or their suitable
sal~s may be used for the treatment of various forms of
hypertensive pathologies and of cardiac, renal or pul-
monary insufficiencies, 25 well as for the treatment of
glaucoma.
The compounds of the invention or their suitable
salts ~ay also be u~ed in combination with other substan- :
ces having cardiovascular activity, such a3 diuretics, ~:
~-blockers, ~-~lockers, calcium antagonists or ~ ~:
angiotonsin I-converting enzyme inhibitors.
The compounds of the invention or their suitable
salts may be presented in all pharmaceutical forms suited
to the treatment, for oral, parenteral, intramuscular or
rectal administration: tablets, capsules including hard
gelatin capsules, sterile solutions or suspensions,
suppositories, and the liXe.
For the treatment of slaucoma, the compounds of
the invention may b~ present2d in the for~ of tahlets,
hard: gelatin capsules, injections or topical ocular
formulations.
The compounds of the invention may be adm~nis-
tered to patients in a quantity which can range from 1 to
1000 mg per day per patient, in one or more doses.
,; - , ,: :