Note: Descriptions are shown in the official language in which they were submitted.
wo 93/234lg 211 3 5 4 7 PCI`/DK93/001~i8
Purine derivatives
The present invention reiates to a method for treating ischaemia, epilepsy
and pain, to compounds for use in such a method and to pharmaceutical
compositior~s containing the said compounds.
10 Backaround of the Invention
Adenosine can be:considered to be a hormone which has been shown to
have a number of significant effects on the mammalian central neNous
system (CNS)~lAnnual Reports in Medicinal Chemistry, 1988, 23, 39-48;
15 ~ Irltemational Revlew of Neurobiology (Sm~thies, J.R. and Bradley, R.J.,
eds.) Academic~Press lnc., 1985, 27, 6~139], espe~ially under con~itions of
neuronal stress ~where :the compound appears to act as an endogenc)us
. ~
neuroprotectant;(Progress in Neurobiology, 1988, 31, 85-108, Trends in
Pharmacological ~Sciences, 1988, 9,:1 93-194). For exarnple, the concentra-
20~ tlorl o~f ade~osine: has` been demonstrated to rise greatly in certain brainregior~s foliowin~ :epileptic sekures or conditions of neuronal ischaemia/a^
noxia: ~Brain Research ;1990, 516, 24~256).
; :It has been established~for some years~now that centrally acting adenosine
25 ~ ~receptor agonists~;or~compounds which increase extraeellular adenosine
levels can exhibit what is termed neuromodulator activity. Such substances
., .
influence the release of neurotransmitters in regions of the central nervous
system (Annual Rev~ew of Neuroscience, 1985, 8, 103-124; Tr~nds in
Neurosciences, 1984,:164-168), with particular inhibitory effects on the
30 release of the excitatory amino acid glutamic acid (g7utamate~ (Nature,
1985, 316, 148-150, Journal of Neurochemistry, 1992, 58, 1683-1689).
::: :
:: : There are several CNS ailments for which this aden~sine receptor mediated
:~ .
418 ` ~-. - PCr/DK93~00158
neuromodulator activity could be of clear therapeutic benefit. Exampies of
these would include the trea~ment of convulsive disorders (European
Journal of Pharmacology, 1991, 195, 261-265; Journal of Pharmacology and
Experimental Therapeutics, 1982, 220, 70-76), prevention of neurodegenera-
5 tion under conditions of brain anoxia/ischaemia (N~uro~cience Letters,
1987, 83, 287-293; Neurosciencc, 1989, 30, 451-462; Pharmacology of
Cerebral Ischaemia 1990, (Kriegelstein, J. and Oberpichler, H., Eds.,
Wissenscha~tliche Verlagsgesellschaft mbH: Stut~gart, 1990, pp 439^448) or
the use of a purinergic agent in the treatment of pain (European ~)ournal of
Pharmacology, 1989,162, 36~369; Neuroscience Letters, 1991 ,121 ,
267-270).
Adenosine receptors represent a subclass ~P1) ~ the group of purine
; ~ ~ nucleotide and nucleoside receptors known as purinoreceptors. This
15 - subclass has been further classified into ~NO distinct receptor types whichhave become known~as A1 and A2. Extensive research has been carried
out in a quest to identify selective ligands at these sites lsee, for example,
Comprehensive Medicinal Chemistry, Volume 3, (Hansch, C., Sammes, P.G.
and Taylor, J.B., Eds., P~rgamon Press PLC: 1990, pp 601-642)]. Selective
~n; ~ 20 ligands exist for A1 and A2 adenosine receptors and the structure-activity
relationships of the various reference ligands have been reviewed (E~ioche-
:: mical Pharmacology, 1986, 35, 2467-2481) together with their therapeutic
potential ~Journ~l ~of Medicinal ~Chemistry, 1992, 35, 407-422~. Among the
known adenosine reccptor agonists most selective for the A1 receptor over
25 the A2 receptor are the examples where the adenine nucleus is substituted
with a cycloalkyl group on the amino function, for example N-cyclopentyla-
~: denosine and N-cyclohaxyladenosine (Journal of Medicinal Chemistry, 1985,
28, 1383-1384) or 2-chloro-N-cyclopentyladenosine (Naunyn-Schmiede-
berg's Aroh. Pharmacol. 1988, 337, 687-689).
However, these ligands are found to possess undesirable effects as to
~ r WO 93~2341B 2 1 1 3 5 4 7 PCr/DK93~00158
influence upon the cardiovascular system, rendering them unsuitable for the
treatment of CNS disorders such as cerebral ischaernia, epilepsy and pain.
GB 1,143,150 (equivalent to USP 3,551,409) and GB 1,123,245 disclose a
5 nurnber of adenosine derivatives, having interesting cardiac and circulatory
actions.
In EP 322,242A, a new use, as "agents to reduce plasma ~ree fatty acid
concentration or reducing heart rate and condition" is claimed for the
10 compounds listed below as well as physiolo~ically acceptable salts and
solvates thereof:
N-[~1S, ~-2-hydroxycyclopentylJadenosine
N-[(1R, ~)-2-hydroxycyclopentyl]adenosine
15 N l~-~hydroxycyclohexyl]-2-methyladenosine
N-[(~)-4-hydroxycyclohexyl]adenosine
N~ 2-hydr~xycyclopentyl]adenosine
N-~ttrans)-3-hydroxycyclohexyl]adenosine
-[~B-hydroxy-2-methylcyclopentyl~adenosine and
20 N-[~)-2-hydroxycyclohexyl]adenosine
Description o~the Invention
:: : : : `
It has now been discovered that a selected group of adenosine derivatives,
25 some of which are olaimed in GB 1,143,1~0, has potential therapeutic utility
for treating central nervous system ailments such as cerebral ischaemia,
:::
epilepsy and pain~in h~mans. They have a clear CNS effect in reievant
animal models at the same time as having a superior side-effect profile with
respect to cardiovascular properties. In addition, the compounds have utility
30 within myocardial isc~aemia. Specifically, the following compounds possess
therapeutic utility within the above-mentioned CNS indications:
WO 93~23418 PCI/DK93/00158
2 11 3 ~7 - 4 -
2-Chloro-N-(1-phenoxy-2-propyl)adenosine
2-Chloro-N-l(R)-1 -phenoxy-2-propyl]adenosine
2-Chloro-N-[(S)-1 -phenoxy-2-propyi~adenosine
;: 2-Chloro-N-(2-phenoxyethyl)adenosine
: ~ 5 2-Chloro-N-[(R)-1-phenyl-2-propyl]adenosine
2-Chloro-N-(1 -phenyl-3-butyl)adenosine
N-(1 -Phenoxy-2-propy!)adenosine
2-Amino-N-(1-phenoxy-2-propyl)adenosine
-[(1S, trans)-2-Hydroxycyclopentyl~adenosine
N-[(1R, trans)-2-Hydroxycycbpentylladenosine
2-Chloro-N-(cis-2-pheno)tycyclopentyl)adenosine
trans-2-Chloro-N-(2-phenoxycyclopentyl)adenosine
2-Chloro-N-t(R)-1-hydroxy-2-propyl]adenosine
2-Chloro-N-[(R)-1-phenylthio-2-propyl]adenosine
15~ 2-Chloro-N-[(R)~ (4 fluorophenoxy)-2-propyl]adenosine
2-Chloro-N-[(R)-2-phenoxy-1-propyl?adenosine
2-Chloro-N-[2-(phenylmethoxy)ethyl]adenosine
2-Fluoro-N-[(R)-1-pheno~*-2-propyl]adenosine
2-Methoxy-N-[(R)-1-phenoxy-2-propyl]adenosine
20 ~ N-(2-Methoxyeth~l)adenosme
: 2-Chloro-N-[(2-methoxyphenyl)methyl]adenosine
2-Chloro-N-[(R)-3-methyl-1 pheno~y-2-blutyl]adenosine
;2-Chloro-N-[(R)-~ (2-~2-propyloxy)phenoxy)-2-propyl~adenosine
: 2-Ghloro-N-l(R)-1-phenylsulphoriyl-2-propyl~adenosine
: :~ 25 N-l(2-methylphenyl)methyl~adenosine
2-Methyl-N-~(R)-1-phenoxy-2-propyl]adenosine
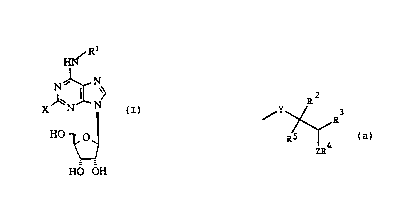
Accordingly, the present invention relates to adenosine analogues of
WO 93/23418 2 ~ I 3 5 4 7 pcr/DK93~ 5g
~ormula I
X ~ N
HO ~
:
HO OH
:: 1û wher~in X is hydrog~n, amino, halogen, hydroxy, lower alkoxy or lower alkyl
and
:
R' is 2
y ~R
5 ~
wherein Y is methylene or a valence bond,
2~and Rs is H or lower, straight or branched alkyl, ..
F~3 is H or lower alkyl, or
2Q ~; ~ R2 and ~R3 can:together~ form a cyclobutylj cyclopentyl, cyclohexyl or phenyl
Z is oxygen7 methylene,~s~Jlphur, sulphonyl or a valence bond,
R4 is H, lower alk~l, aralkyl a mono or bicyclic aromatic system optionally
substi~uted with halogén, hydroxy, haloal~l, alkyl, alkoxy, aryloxy, acyloxy
25 or alkylmercapto radi~als,
- or a pharmaceutically acceptable sa!t thereof as these ~ompounds have
:: : : : been found useful:intre~tment of a number of CNS-re!ated ailments, such as cerebral ischaemia, epilepsy and pain.
:: 30 Further, the compounds of formula (I) are found to be useful agents, for
lowering plasma free fatty acid (FFA) levels, as cardiovascular agents and
: :
WO 93/23418 PCI`/DK93/00158
21 1 35~ 7 - 6 -
also have application to myocardial ischaemia.
The invention also relates to methods of preparing the above mentioned
compounds. These methods comprise:
Method A
A compound of formula (I) may be prepared by reacting a substance of
0 formu!a (~ wherein L represents a leaving group such as a halogen a~orn
(e.g. a chlorine or bromine atom) or a trimethylsilyloxy group, p', p2 and P3
are the same or dfflerent and represent hydrogen or a protecting group
such as benzoyl-, p-toluoyl-, lower alkanoyl- (e.g. acetyl-), a substituted silyl
group (e.g. a trimethyisilyl or t-butyldimethylsilyl group) or in the case of P3,
15: : a triarylmethyl group, or in the case of p1 and p2, a 2',3'-0-(1-meth~ethyli-
~; : dene function, with a substituted amine of general formula (Ill)
General ~rocess (A)
RI
X~XN H N/ ~m) X ~N
~ ~ p3 0 ~J _ p3 0 ~
: 25 p2 0 O PI ~2 0
~RI
~: 1 ~N
X N N
: 30
HO ~; (I)
- HO OH
Wo 93/23418 21 13 5 4 7 PCI/DK93/00158 ~
giving the compound of formula (IV~ as the reaction product. in cases
where p1, p2 and P3 are not hydrogen an additional step wil~ be required to
remove protecting groups from (IV); in cases where the groups p1, p2 and
P3 are for exampie acetyl or benzoyl, suitable conditions for deprotection
include the use of methanolic ammonia, an alkali metal carbonate in metha-
nol, or an alkali rnetal alkoxide in the corresponding alcohol. Where the
protecting groups are for example alkylsilicon or arylsilicon derivatives,
suitable methods for deprotection include, for example, treatment with tetra-
alkylammonium fluorides or aqueous hydrolysis in the presence of acid or
base. Where the pl and p2 groups comprise a 2',3'-0-(1-methyl)ethylidene
function or P3 comprises triarylmethyl, suitable conditions for deprotection
include, for example, hydrolysis with aqueous mineral acid.
Method B
~:
A compound of formula (I) wherein X represents -NH2, 0-alkyl or hydroxy,
may be prepared by r~acting a substance of general formula (V)
$N ~ Xl~N
p30 o ~ p30~ o
~ (V)~ ~ (IV)
. 25 p20 OPI p2 0
~: : .
E~
~ N>
HO ~ (I)
HO OH
WO 93~23418 . PCI`/I~Kg3/001~;8
2113~7
~where L is a leaving group as defined in method (A)3 with a nucleophile,
for example, ammonia or with an anion te.g. C, 6-alkoxide) to afford the
~: product (IV). In cases where p, p2 and P3 are hydrogen, compound (1) can
be obtained directly. However, in cases where pl, p2 and P3 are not hydro-
5 gen an additional step will be involved to remove protecting groups from
(IV); examples of ~onditions for removal of protecting groups are given in
process (A). In some reactions involving (~ with the anion Cl 6-alkoxide,
where p, p2 and/or P3 are for example acetyl- or benzoyl-, partial or full
deprotection may take place. In cases where only partial deprotection has
10 taken place, deprotection can be completed under conditions described in
method (A). ~ ~ ~
~: :
Accordingly, the present~invention provides a method for treating cerebral
i schaemia, epilepsy~and~ pain in hi~man or non-human animals, which
15~ ~ ~ method comprises ~administering an effective, non-toxic amount of a com-pound of formula~ ~I or a~ pharmaceuticatly acceptable salt thereof, to human
or~non-human animals~suffering from cerebral ischaemia, epilepsy or pain.
The~present invention~also provides the use of a compound of formula I or
a pharmaceutically ~acceptable salt thereof in the preparation of a medica-
20~ ~ ment~for use in~the treatment of cerebral ischaemia, epilepsy or pain.
T he~ present ir~vention further provides a pharmaceutical composition for use
in~the treatment ~of~ cerebral ischaemia, epilepsy or pain which comprises an
effective amount of a~ compound of formula I of a pharmaceutically accep-
~: 25 table salt thereof and a pharmaceutically acceptable carrier. Such composi-
tions may be prepared in the manner as described below.
;~
Various salts of compounds of formula ~1) can be prepared which can be
considered physiologically acceptable. These include addition salts derived
from inorganic or organic acids, for example, acetates, fumarates, glutara-
tes, glutaconates, lactates, maleates, methanesulphonates, phosphates,
WO 93/23418 211 3 5 4 7 PCI`/DK93/00158
salicylates, succinates, sulphates, sulphamates, tartrates and paratoluene-
sulphonates. In some cases, solvates of either the free nucleosides or the
acid addition salts can be isolated and these soivates may, for example, be
hydrates or alcoholates.
; ~ 5
- The compounds of the imention, together with a conventional adjuvant,
carrier, or diluent, and ff desired in the form of a pharmaceutically-accep-
table acid additlon sait thereof, may be placed into the form of pharmaceuti-
cal cornpositions and unit dosages thereof, and in such form may be
10~ employed as solids,~ such as tablets of filled capsules, or liquids, such assolutions, suspensions,~emulsions, elixirs, or capsules filled with the same,
all for oral use, in the~form of suppositories for rectal administration; or in
the~ form of sterll~e~ inJectabb solutions for parenteral use (including sub-
cutaneous.administration;;and infusion). Such pharmaceutical compositions
; 15 and unit dosage~ forms~thereof may comprise conventional ingredient^s in
conventional proportions, with or without additional active compounds or
pnnaples, and~such unit dosage forms may contain any suitable effective
amount of the~adenoslne~receptor agonist commensurate with the intended
r ~ daily~dosage range to ~be employed.
The ~compounds~;of~ s~invention can thus be used for the formulation of
pharmaceutical preparations, e.g. for oral and parenteral administration to
mammals including~ hùn~ans, ;in accordance with conventional methods of
galenic pharrnacy. ~Conventionàl excipients are such pharmaceutically
25 acceptable organic ~or inorgamc carrier substances suitable for parenteral orenteral application which do not deleteriously react with the active com-
pounds.
Examples of such carriers are water, salt solutions, alcohols, polyethyiene
30 glycols, polyhyroxyethoxylated castor oil, gelatine, lactose, amylose, magne-
~,
sium stearate, talc, silicic acid, fatty acid monoglycerides and diglyceridesl
: : ,
WO 93/23~18 PCI~/DK93/OOlS8
2113547 - 10-
pentaerythritol fatty acid esters, hydroxymethylcellulose and polyvinylpyrroli-
done.
The pharmaceutical preparations can be sterilized and mixed, if desired,
5 with auxiliary agents, emulsifiers, salt for influencing osmotic pressure,
buffers and/or colouring substances and the like, which do not deleteriously
react with the active compounds.
For parenteral application, particularly suitable are injectable solutions or
10 suspensions, preferably aqueous solutions with the active compound
dissolved in polyhydroxylated castor oil.
Ampoules are convenien~ unit dosage forms.
15 Tablets, dragees, or capsul0s having talc and/or a carbohydrate car;ier or
binder or the like, the ~carrier preferably being lactose and/or corn starch
and/or potato starcl~,~ are particularly suitable for oral application. A syrup,elixir or the like can be used in cases where a sweetened vehicle can be
emp!oyed.
20 ~
Generally,~the compounds ot~this invention are dispensed in unit form
comprising 0.05-100 mg in a pharmaceutically acceptable carrier per unit
dosage. The dosage~ of the compounds according to this invention is
0.1-300 mg/day, preferably 1~100 mglday, when administered to patients,
:~
25 e.g. humans, as a drug.
, .
;
A typical~tablet which rnay be prepared by conventional tabletting tech-
niques contains:
30 Active compound 5.0 mg
Lactosum 67.8 mg Ph.Eur.
wo 93/234l8 21 1 3 5 4 7 PCI`/DK93/00158
Avicel~ 31.4 mg
Amberlite~slRP 88 1.0 mg
Magnesii stearas 025 my Ph.Eur.
Owing to activity against pain or convulsive disorders and prevention of
neurodegeneration under conditions of anoxia/ischaemia the compounds of
the invention are extremely useful in the treatment of related symptoms in
mammals, when administered in an amount effective for agonist activity of
compounds of the invention. The compounds of the invention may accor-
10 dingly be administered to a subject, e.g., a living animal body, including ahuman, in need of an adenosine receptor agonist, and ~ desired in the form
of a pharmac~utically-acceptable acid addition salt thereof (such as the
hydrobromide, hydrochloride, or sulfate, in any event prepared in the usual
or conventional manner, e.g., evaporation to dryness of the free base in
15 solution together with~the~aeid), ordinarily conc~rrently, simultanously, or
together with a pharmaceutically-acceptable carrier or diluent, especially
and preferably in the form of a pharmaceutical composition thereof, whether
by oral, rectal, or~parenteral (including subcutaneous) route, in an effective
amount of adenosine receptor agonist, and in any event an amount which
2~ is effective for the treatment of anoxia, traumatic injury, ischemia, migraine
or other pain symptoms, epilepsy, or neurodegene~ ative diseases owing to
their adenosine rec~ptor agonist activity. Suitable dosage ranges are 1-200
milligrams daily, 1~100~milligrams daily, and especially 30-70 milligrams
daily, depending as usual upon the exact mode of administration, form in
25 which administered, the indication toward which the administration is
directed, the subject in volved and the body weight of the subject involved,
and the preference and experience of the physician or veterinarian
in charge.
30 The preparation of compounds of the invention is further illustrated in the
following examples:
WO 93/23418 - PCI~ Kg3/001~8
21135ll7
- 12 -
Hereinafter, TLC is thin layer chromatography, THF is tetrahydrofuran, TFA
is trifluoracetic acid and mp is melting point. Where melting points are
given, these are uncorrected. The structures of the compounds are con- ;
firmed by assignment of NMR spectra (from which representa~ive peaks are
quoted) and by microanalysis where appropriate. Compounds used as
starting materials are either known compounds or compounds which can
be prepared by methods known per se. Column chromatography was
carried out on Merck silica gei 60 (Art 9385). HPLC was carried out on a
Waters or Merck chromatograph with a multiwave~ength detector and a
~;~ 10 reversed phase C18 column (250 x 4 mm, 5,um, 100A; eluent flow rate 1
mV min at 3SC). Retention hmes are given in minutes.
,
EXAMPLE 1 (Method A)
15~ ~ 2-Chloro-N-(1-phenoxv-2-Dropyl)adenosine
: ~ ~
title~compound was prepared by reacting 1-phenoxy-2-propylamine
(16.62 9, 0.11 mol)~ with~ 9-~2~3~5-tri-o-acetyl-B-D-ribofuranosyl)-2~6-dichlor
9H-purine~ (24.6 9,;~55;~mmol) in dioxan (250 ml) in the presence of triet-
20 ~ hylamine p.23 g ~71.5 ~mmo!) followed by deprotection of the product USih9
a~solution of sodlum~(O.15 9, 6.5 mmol) in methanol (250 ml~. The reaction
mi~*ure~ w~ neutralized wnh citric acid, and treated with a mixture of ethyl
acétate ~300 ml) and water (200~ml).~ The ethyl acetate phase was separa-
ted,~ dried ~MgS()4)~and ~evaporated before being purKied by flash ehroma-
tography on silica gel, el~*ing initially with dichloromethane, and later with amKture of dichloromethane and ethanol (9:1). This provided the title
2-chloro-N-(1-phenoxy-~-propyl)adenosine (18.2 9, 76%) (a mixture of
diastereoisomers) as an amorphous foam, lH NMR (DMSO-d6)~ 1.31 (3H,
d, -CH3), 3.53 - 3.59 (1 H, m, H-5'~,), 3.64 - 3.71 (l H, m, H-5'b), 3.95 (1 H, q,
H-4'), 4.06 4.20 (3H, 2 m, H-3' and -CH2-), 4.54 (1 H, m, H-2'), 4.65 (1 H, m,
-CHGH3~, 5.07 ~1 H, t, 5'-OH), 5.21, 5.50 ~2H, 2d, 2'- and 3'-OH), 5~84 (1 H, d,
': ~
:
WO 93J23418 2113 5 4 7 PCI-/DK93/OOlS8
- 13-
H-1'), 6.87 - 7.00 (3H, m, Ar-tl), 7.23 - 7.32 ~2H, t, Ar-H), 8.31 - 8.45 (2H, m,
H-8 and N-H).
The corresponding maleate salt was pr~pared by dissolving the above
2-chloro-N-(1wphenoxy-2-propyl)adenosine (t.7 9, 3.9 mmol) in THF (10 ml),
adding diethyl ether (60 ml) followed by maleic acid (0.45 9, 3.9 mmol). The
residue on evaporation was treated with diethyl ether ~50 ml) whereupon
the maleate sal~ precipitated and was collected by filtration (1.15 g), m.p.
1 02-1 04C.
1 0
C23H26CIN507:requires G, 50.0; H, 4.7; N, 12.7. Found: C, 50.3; H, 4.9; N,
12.7%.
15 ~ E)(AMPLE 2 (Method A)
2-Chloro-N-~(R~-~enoxy-2-Propyl~adenosine
(R)-N-(tert-Butoxycarbonyl)-2-amino-1-propanol
(R)-2-Amino-1-propanol (15.û 9, 200 mmol) was dissolved in 1N sodium
hydr~xide (198 ml) and THF (85 ml) was introduced. The reaction mixture
was cooled to 0C~ and a solution of di-tert-butyl dicarbonate ~52.4 9, 240
:mmol) in THF (230 ml) was added dropwise over 30 min. The reaction
mixture was stored at 4C for 72 h., allowed to reach room temperature and
filtered. The fiitrate~ was evaporated to remove THF and the aqueous phase
was extracted with eth~l acetate (2 x 200 ml). T~e combined extracts were
drled (MgS04), evaporated and the crude product was dissolved in dich-
~; loromethane (100 ml) and extracted into water (5 x 200 ml). The combined
: 30 aqueous extracts were evaporated in vacuo. The resultant oil crystallised
whiist standing at room temperature to provide the required alcohol
WO 93/23418 PCI`/DK93/OOlS8
211~5~7
- 14-
~15.359, 44%), mp 59 - 61C, 1H NMR ~ûMSO-d~ (3H, d, -CHCH3),
1.45 (9H, s, butyl-CH3), 3.50 (1H, dd, -CH2a-), 3.65 (1H, dd, -CH2b-), 3.70-
3.80 (1H, m, CH).
5 (R)-N-(tert-Buto)~ycarbonyl)-1-phenoxy-2-propylamine
(R)-N-~tert-butoxycarbonylj-2-amino-1-propanol (10.0 g, 57 mmol), triphe-
nylphosphine (22.5 9, 8ff mmol) and phenol (5.4 9, 57 mmol) was dissolved
in toluene (200 ml). Diethyl azodicarboxylate (14.9 9, 86 mmol) in toluene
; (100 ml) was slowly~ added keeping the temperature below 35C (Mitsuno-
bu, O., Synthesis,~1981, 1; Manhas, M.S.; Hoffman, W.H.; Lal, B.; Bose,
A.K, J. Chem.~ Soc. Perkin Trans l, 1974, 461). The resulting yellow solution
was stirred for 16 h at room temperature before being washed with 1N
hydrochloric acid~ ~3~;x 100 ml). The organic phase was drie~ (MgSO4),
15~ evaporated in vacuo, ~and the residual oil was purified ~y flash chromato-
graphy~elutmg with~heptane/ethyl a~etate (4/t) giving the desired product
(8.0 g,~59%)~ 'H~NMR~(DMSO-d6)~ 1.10 (3H, d, -CH3), 1.38 (9H, s, butyl-
CH3)~,~3.70 - 3.90~(3H,~m,~-CH-CH2-j, 6.85 - 6.95 (3H, m, Ar-H), 7.2~ ~2H, t,
Ar-H).
(R)-1-Phenoxy-2-propylamine
(R)-N-(tert-Butoxycarbony!)-1-phenoxy-2-propylamine (8.0 g, 33 mmol) was
dissolved in ethyl~acetate ~ ml). A solution of hydrochloric acid ~9) in
ethyl acetate (6N, 1 00ml) was added dropwise at room temperature. Tl~e
reaction mixture was stirred at room temperature for 2Qh during which time
; a heavy precipitate was~forrned. The reaction mixture was concentrated to
half the originai volume before the product was collected by filtration and
dried in vacuo ~to;provide the tnle compound as a white solid hydrochloride
(4,3 9, 69%) m.p. 186 -189C. 'H NMR (DMSO-d6)~ 1.31 (3H, d, -CH3), 3.51
- 3.60 (1H, m, -GH-), 4.05 (1H, ddi -CH2a-~, 4.12 (1H, dd, -CH2b-), 6.95 -
WO 93t23418 211 3 S 4 7 PCJ/DK93/00158
- 15-
7.00 (3H, m, Ar-H), 7.32 ~2H, t, Ar-H).
2-Chloro-N- [~R) 1 -phenoxy-2-propyl]adenosine
(R)-1-phenoxy-2-propylamine (4.3 9, 23 mmol) was reacted with 9-~2,3,5-tri-
O-benzoyl-~-D^ribofuranosyl)-2,~dichloro-9H-purine (11.2 g, 18 mmol) in
dioxan ~150 ml) in the presence of diisopropylethylamine (5.3 g, 41 mmol).
The reaction rnixture was stirred at room temperature for 18 h, heated at
5ûC for 4h, and stirred at room temperature for 60h before being filtered
and evaporated. The produc~ (a~ter purifieation by flash chromatography)
~; ~ was debenzoylated with methanoli~ ammonia to provide the title
2-chloro-N-[(R)-1-phenoxy-2-propyl~adenosine (af~er column
chromatography):~as a foam (4.2 9, 64%), 'H NMR (DMSO-d6)~ 1 31 (3H,
d, -CH3), 3.52-~3.59 (1H, m, H-5~a)~ 3.63-3.72 (1H, m, H-5'b), 3.92 - 3.99
and 4.10 - 4.21 (4H, 2 m, H-3', H-4' and -CH2-), 4.52 (lH, dd, H-2'), 4.65
(1 H, mj -CH3C_-), 5 07 (1 H, t, 5'-OH), 5.22, 5.49 (2H, 2d, 2' and 3'-OH),
5.84 (1H, d, H-1'), 6.88 - 7.02 (3H, m, Ar-H)t 7.24 - 7.33 ~2H, dd, Ar-H), 8.32
8.45 (2H, s & m,~ H-8 ~and N-H).
20~ C19H22ClNsO5 requires C, 52.4; H, 5.1; N, 16.1. Found: C, 52.0; H, 5.2; N,
8/ ~ ;
EXAMPLE 3 (Method A!
2~i 2-Chloro-N-~(S)-1-Phenox-~-2-propvlladenosine
2-Chloro-N-~(5)-1-phen~xy-2-propyl]adenosine was prepared by the pro-
cedure described for Example 2, except that (S)-2-amino-1-propanol was
used in the first step, providing the opposite diastereoisomer to Example 2.
30 The nucleoside was obtained as a hemihydrate:
C19H22ClN5OsØ5 H2O requires ~, 51.8; H, 5.2; N, 1~.9. Found: C, 51.8; H,
.
.
WO 93/23418 . PCI`/DKg3/001~8
21135~7
- 16-
5.3; N, 15.6%.
EXAMPLE 4 ~Method A
5 2-Chloro-N-~2-~henox~ethvl)adenosine
: .
The title compound was prepared by reacting 2-phenoxy~thylamine
hydrochloride (QBO g, 4.6 m mol) with 9-(2~3~5-tri-o-benzoyl-B-D-ribofuran
osyl)-2,6-dichlorQ-9H-purine (2.0 g, 3.2 mmol) in dioxan (25 ml) in the pre-
sence of triethylamine~ ~1.0 9, 9.6 mmol) followed by deprotection of the
: purffled product using methanolic ammonia to provide the title nuoleoside
(0.75 ~, 60%) (following flash chromatography) as an amorphous foam, 1H
NMR (DMSO-d6)~ 3.52 - 3.59 (1H, m, H~5~a)~ 3.64 - 3.71 (1H, m, H-5'~), 3.82
(2H, q, -t~H2~ 3.96 (1H, q, H-4'), 4.14 ~1H, m, H-3'), 4.52 (1H, q, H-2'~, 5.12
15: : ~(1H,~t,~5'-OH), 5.25,~5.54~(2H, 2d, 2'- and 3'-OH), 5.85 (1H, d, H-1'), ~.92 -
7.02 (3H, m,: Ar H) 7 26: 7.34 (2H, t, Ar-H) 8.~46 (1 H, m, H-8), 8.56 (1 H, br
C,8H20CIN505Ø75~20 requires C, 49.7; H, ~.0; N, 16.1. Found: C, 49.7; H,
20~ S.O; N, 15.7%.~
: EXAMPLE 5 ~Method A)
2-Chlo!o-N-~(R)-1-Phenvl-2-propvlladenosine
:: The titlc compound~was:prepared by reactlng L-amphetamin (0.49 9, 3.6
mmol) with 9-(2~3~tri-o-benzoyl-B-D-ribofuranosyl)-2~6-dichloro-9H-purine
~: ~ (1.9 9, 3.0 mmol3: in~ dioxan (2~ ml) in the presence of diisopropylethylamine
(0.~8 9, 4.5 mmol) followed by deprotection of the purified product using
~,
methanolic ammonia. Evaporation of the reaction mixture provided a gum-
: :;: ~:
WO 93/23418 211 3 ~ 4 7 PCI/DK93/00158
my residue which crystallized on addition of dichloromethane (10 ml;, to
provide the title compound (0.26 9, 38%) as a solid, m.p. 132.5 - 135.5C. A
further sample of the title compound (0.26 9) was obtained by flash Ghro-
matography of the mother liquors. 1H NMR ~DMSO-d6)~ 1.22 (3H, d, -~3),
2.67 - 2.79 & 2.92 - 3.03 (2H, 2m, -CH2-), 3.51 - 3.58 (1 H, m, H~5~a)~ 3.62 -
:~ ~ 3.68 (1H, m, H-5'~,), 3.94 (1H, q, H-4'), 4.12 (1H, m, H-3'), 4.41 - 4.54 (2H,
m, H-2' and CH3CH-), 5.06 (1H, t, S'-OH), 5.22, 5.49 (2H, 2d, 2' and 3'-OH),
~;: 5.82 (1H, d, H-1'), 7.10 - 7.33 ~5H, m, Ar-H), 8.28 - 8.44 ~2H, m, H-8 and N-
H).
C1gH22CtNsO4Ø5 H2O requires C, 53.2; H, 5.4; N, 16.3. Found: C, 53.3; H,
5.4; N, 16.3%.
15 ~ EXAMPLE 6 (Method A)
,
2-Chloro-N-(1-phenyl-3-butvl)adenosine
3-Arnino-1-phenylb~ane (0.67 9, 3.6 mmol~ was reacted with 9-~2,3,5-tri-
O-be~zoyl-~-D-ribofuranosyl)-2,6-dichloro-9H-purine (1.9 9, 3 mmol) in
dioxan (25 ml) in the presence of diisopropylethylamine ~0.58 g, 4.5 mmol).
The reaction mixture was stirred at room temperature for 18 h, filtered and
eYaporated. The product ~af~er purification by flash chromatography) was
debenzoylated with methanolic ammonia to provide the title 2-chloro-N-~1-p-
~: 25 henyl-3-butyl)adenosine (mixture of diastereoisomers) as a foam (0.72 9,
56%), 'H NMR (DMSO-d6~ 1.12 (3H, d, -CH3), 3.53- 3.61 (1H, m, H-5'~),
3.64 - 3.72 (1H, m, H-!i-b), 3.93- 3.99 (lH, m, H-4'~, 4.11 - 4.17 (1H, m, H-
3'), 4.22- 4.36 (1H, m, -CH2CH-), 4.53 (lH, dd, H-2'), 4.65 (1tl, m, -CH~CH-
), ~.10 (1H, t, 5'-OH), 5.25, 5.51 (2H, 2d, 2' and 3'-OH), 5.83 (1H, d, H-1'),
7.12 - 7.30 ~5H, m, Ar-H), 8.28- 8.37 (1H, m, N-H), 8.40 (1H, s, H-8).
WO 93/23418 PCI-/DK93/00158 ....
2il3547
- 18 -
C~oH24ClNsO4.H2O recluires Ct 53.2; H, 5.8; N, 15.5. Found: C, 53.9; H, 5.7;
N, 15.6%.
E)(AMPLE 7 (Method A~
N~ Phenoxv-2-propvl)adenosine
1-Phenoxy-2-propylamine (û.33 9, 2.18 mmol) was reacted with 6-chloro-
purine riboside (i.e. 9-B-D-ribofuranosyl-9H-purine) (0.5 9, 1.7 mmol) in
dioxan (30 ml) in the presence of diisopropylethylamine (0.28 9, 2.2 mmol~.
The reaction mixture was heated at reflux for 5 h, cooled and evaporated.
;~ The residue was purified by flash chromatography on silica gel to provicle
:~ the N~ Phenoxy-2-propyl)adenosine (a mixture of diastereoisomers) as a
foam (0.06 9, 7%)~, 1H NMR (DMSO-d3~ 1.32 (3H, d, -CH3), 3.52 - 3.60 (1H,
: 15: ~ m, ~H-5~a~ ~.64 - 3.71 (1 H, m, H-5'~, 3.93 - 3.99 and 4.12 - 4.20 (4H, 2m, m,
H~4', k' ~'; ad -CH2-), 4.62 (1H, q, H-2'), 4.68 - 4.82 (1H, m, -CHCH3), ~.40
1H, t, ;)i-oH)~ 5.20, 5.45 (2H, 2d, 2'- and 3'-OH), 5.90 (1H, d, H-1'), 6.88 -
6.98~ ~3H, m,~ Ar-H), 7~23 - 7.31 (2H, t, Ar-H), 7.85, 8.21, 8.38 (3H, 3s, H-2, H-
8~ and N-H).
: EXAMPLE 8 (Method A)
2-Amino-N-(1-phenoxv-2-~ropvliadenosine
1-Ph~noxy-2-propylamine (2.90 9, 19.2 mmol) and 9-(2,3,5-tri-O-acetyl-J3-D-
ribofuranosyl)-2-amino-6-chtoro-9H-purine (6.94 9, 16.2 mmol) were dissol-
ved in dioxan (50: mi? ar.d triethylamine (4.~ ml, 33.2 mmol) was introduced.
Aft~r stirring the reaction mixture for 18 h at room temperature, diisopropy-
lethylamine (2.08 9, 16.1 mmol) was added and the solution was heated a~
80C for 1û0 h. Following column column chromatography, a 1.3 9 sample
of the resultant 2',3',5'-tri-O-acety1-2-amino-N-(1-phenoxy-2-propyl~adenosi-
WO g3/234l8 2113 5 ~ 7 PCI-/DK93/00158
- 19-
ne was deprotected using saturated methanolic ammonia (50 ml). The reac-
tion mixture was evaporated, and the residue dissolved in in a mixture of
ethyl acetate (150 ml) and water (150 ml). The phases were separated and
the ethyl acetate phase was washed with watef (2 x 150 ml). The ethyl ace-
tate phase was then extracted with pH 2 diiute hydrochloric acid, and this
acidic aqueous phase was washed with ethyl acetate (2 x 100 mi), and
basffiled with sodium bicarbonate solution before extraction with ethyl aceta-
te (100 ml). The ethyl acetate phase was was dried (MgSO4) and eva-
porated to give the title compound (0.43 9, 33%) a mixture of diastereoiso-
mers as an amorphous foam, 1H NMR (DMSO-d6)~ 1.28 (3H, d, -CH3),
3.49 - 3.57 (1H, m, H-5',1), 3.61 - 3.68 (1H, m, H-5'b), 3.85 - 3.94 and 4.07 -
4.15 (4H, 2m, m, H~', H-3' and -CH2-), 4.50 ~1H, q, H-2'), 4.68 (1H, br, -
CHCH3), 5.11, 5.36~ (2H, 2d, 2'- and 3'-OH), 5.40 (1H, t, 5'-OH) 5.73 (1H, d,
H-1'), 5.83 (1H, br, -~H2), 6.88 - 6.96 (3H, m, Ar-H), 7.22 - 7.31 (2H, t, Ar-H),
15 ~ ~ 7.95 ~1H, s, H-8). ~ `;
ClgH24ClNsO5~0~75 H20 requires Cj 53.1; H, 6.0; N, 19.5. Found: C, 53.0; H,
6.0; N, 19.2%.
20~ EXAMPLES 9 and 10 (Method A)
N-~(1R. Trans)-2-hvdroxYcvcloPentvlladenosine and
N-r(1 S, Trans)-2-hvdroxvcvclopentvQadenosine
Trans-2-hydroxycyclopentylamine (0.35 g, 3.46 mmol) (prepared by reaction
of cyclopentene oxide with ammonia in a sealed vessel: see example 11)
was reacted with 6 Ghloropurine riboside (i.e. 9-s-D-ribofuranosyl-9H-purine)
(0.5 g, 1.7 mmol)~in dioxan (30 ml) in the presence of triethylamine (0.93 9,
9 mmol). The reaction mixture ~was heated at 100C for 70 h, cooled and
evaporated. The resultant residue was purified by flash chromatography
eluting with a mixture of ethyl acelate and methanol (19:1). The fractions
WO 93/23418 PCI/DK93/00158 ,~
S 4 1
- 20 -
found to contain the highest amounts of N-l(1 R, trans)-2-hydroxycyclopen-
tyl~adenosine following HPLC examination, were combined and evaporated
to a solid (0.17 9). Recrystallisation from methanol provided the pure N-
~(1 R, trans)-2-hydroxycyclopentyqadenosine (0.11 9, 1 8%) mp 233-235C.
1H NMR (DMSO-dd~ 1.43 - 2.12 (6H, 4m, -CH2CH2CH2-), 3.52 - 3.59 (1H,
mj H~5~a)~ 3.54- 3.71 (1H, m, H-5'b), 3.97 (1H, q, H-4'), 4.07 (1H, br, -
CHOH) 4.15 (1H, q, H-3'), 4.61 (1H, q, H-2'), 4.87, 5.21, 5.41 - 5.47 (4H, d
& 3m, OH groups), 5.89 (1H, d, H-1'), 7.75 (1H, br d, -NH), 8.21 and 8.37
(H-2 and H-8).:
::
~ ~ :
The mother liquors~from~the above recrystallisation were evaporated and
purffled by short path chromatography on silica gel (Art. 7729) and the
product recrystallised to provide N-l(1S, trans)-2-hydroxycyclopentyl]adeno-
sine (0.05 gj 4%), 'H NMR ~(DMSO-d6)~ 1.44 - 2.13 (6H, 4m, -CH2CH2CH2-),
5: 3.52 - 3.59 (1H, m, H-5'a), 3.54 - 3.71 (1H, m, H-5'b), 3.96 (1H, q, H-4'), 4.15
(1Hj q, H-3'), 4.60 (1H,~ q, H-2'), 5.20, 5.41 - 5.47 (3H, d & m, 2', 3' and 5'-OH),~5.88 (1H,~d, H-1'),~7.75:(1H, br d, -NH), 8.19 and 8.36 ~H-2 and H-8).
EXAMPLE 11 (Method A)
20 ~ : ~
2-Chloro-N-(cis-2-~henoxycvclopentvl)adenosine
trans-N-(tert-BLnyloxycarbonyl)-2-hydroxycyclopentylamine
This compound~was ;prepared as a mixture of enantiomers by reaction of
cyclopentene epoxide (8.0 9, 95.1 mmol) with a 25% aqueous ammonia
,
: ~ solution ~35 ml) in~ a sealed glass vessel at 110C for 1.5 h. The reaction
mixture was cooled ~an~ evaporated to half its original volume before 1 N
sodium hydroxide- solution (95 ml) and THF (100 ml) were introduced at
0C. A solution of; dl-tert-butyl dicarbonate (21.8 g, 99.6 mmol) in THF ~50
ml) was added dropwise and the reaction mixture stirred at room tempera-
ture for 18 h. The phases were separated and the aqueous phase was
WO 93~2341X 2 1 1 3 5 ~ 7 Pcr/DK93/oo1~8
- 21 -
washed with ethyl acetate (100 ml). The organic phases were combined
and washed with saturated brine (100 ml), dried (MgSO4) and evapora~ed.
The solid residue was recrystallised from a 10:1 mixture of heptane and
ethyl acetate (55 ml) to provide an analytical sample of trans-N-(tert-bLty-
loxycarbonyl)~2-hydroxycyclopentylamine (4.06 g, 21%), mp 103 - 105C.
:~ C~0H19NO3 requires C, 59.7; H, 9.5; N, 7Ø Found: C, 59.6; H, 9.8; N, 7.0%.
The above trans-N-(tert-butyloxycarbonyl)-2-hydroxycyclopentylamirle was
: : 10 converted into cis-2-phenoxy-cyclopentyl-amine by the seq~lence of reac-
, ~
tions described in Example 2 ~i.e. phenyl ether formation by the Mitsunobu
procedure resulting in inversion at the 2-position, fol5Owed by acidic hydroly-
sis of the BOC- group using TFA3.
: ~cis-2-Phenoxyc~clopentylamine (0.75g, 4.23 mmol) was combined with
9-(2~3~tri-Q-benzoyl-B-D-ribofuranosyl)-2~6-dichloro-9H-purine (2.95 g, 4.7
mmol~ and tri~thylamine (0.64 9, 6.3 mmo!) in dioxan (30 ml) and stirred for
50 h. The reaction mixture was filtered, evap~rated and the
residue dissolved in ethyl acetate and washed with water (2 x 50 ml). The
20~ organic phase was :dried (MgSO4), evaporated and the residue coevapora-
ted to give cis-2',3',5'-tr:O-benzoyl-2-chloro-N-(2-phenoxycyclopentyl)ade-
nosirle (3.1 g, 90%) as an amorphous foam, which was deprotected using
saturated methanolic ammonia (50 ml). After 70 h at room temperature the
reaction mixture was evaporated and the residue purified by flash chroma-
tography on silica gel, eluting with a mixture of dichloromethane and rnetha-
nol (19:1). The title cis-2-chloro-N-(2-phenoxy~yclopentyl)adenosine (0.925
9, 53%~ was oMained~ as an amorphous~foam (a 1:1 mixture of diastereol-
sorners), 1H NMR (DMSO-d6)~ 1.58 - 2.15 (6H, 3m, -Ctl2CH2CH2-), 3.51 -
3.60 (1H, m, H-5'~,), 3.62 - 3.71 (lH, m, H-5'b), 3-95 (1 H, br q, H-4'), 4.12
(1H, br q, H-3'), 4.46 - 4.62 ~2H, m, H-2' and -CH), 4.82 - 4.89 (1H, m, -CH),
5.07 (1 H, br t, 5'-OH), 5.22, 5.49 (2H, 2d, 2'- and 3'-OH), 5.83 ~1 H, d, H-1'),
:
WO 93~23418 pcr/DK93/ool5g
2~135~
- 22 .
:
6.80 - 6.91 (3H, m, Ar-H), 7.15 - 7.24 ~2H, t, Ar-H), 7.99, 8.22 (lH, d & m, N-
H), 8.41, 8.45 (1H, 2s, H-8).
/
C21H24ClNsOsØ5 H20 requires C, 53.6; H, 5.4; N, 14.9. Found: C, 53.5; H,
5.3; N, 14.7%.
:
~ : ,
EXAMPLE 12 (Method A)
1 0 Trans-2-chloro-N-(2-Phenoxycvclopentvl)adenosine
-N-(tert-buty~oxycarbonyl)-2-hydroxycyclopentylamine
Trans-N-(tert-butyloxycarbonyl)-2-hydro~yclopentylamine (24.7 9, 123
5~ mmol) (prepared~as;described in Example 11) was dissolved in THF (500
ml); and~ ~nitrobenzoic acid (20.51 9, 123 mmol) ;was added, followed by
triphenyiphosphine; ~48.28 ~g, ~184 mmo!). A solution of diethylæodicarboxy-
late~(æO6 9,~184 mmol1 1n THF (250 ml) ~was introduced dropwise. The
reaction mixture~was stirred for 18 h at room temperature, evaporated and
20~ purffled by~flash~chromatography~eluting with a mixture of cycohexane and
ethyl acetate~(4~ to~ provide the intermediate ~nitrobenzoy! ester as a solid
(2,5.5 g),~TLG P~ 0.52 ~[Si~: cyclohexane/ ethyl acetate (1:1)]. This ester was
j suspended~in~a~;mixture;~of ~a mixture of methanol (180 ml) and 25% aque-
QUS ammonia solution ~20 ml) and the mixture was stirred at room tempera-
25 ture for 70 h before~ evaporation to a residue. Purification by flash chromato-
graphy eluting with a~mixture of cycohexane and ethyl acetate ~4:1) provi-
ded ~ractions contair~ing~the;title compound which crystallised on evapora-
tion to~afford cis-N-pert-butyloxycarbonyl)-2-hydroxycyclopentylamine as a
solid (11.0 g, 44/O),~mp 64 - 65C.
~ ~
, ~ ~
This cls-N-(tert-butyloxycarbonyl)-2-hydroxycyclopentylamine was converted
WO 93/23418 2113 5 9 7 PCI`/DK93/00158
- 23 -
into trans-2-phenoxycyclopentylamine hydrochloride by Mitsunobu phenyl
ether formation and deprotection - the methods described in Example 2.
9-(2~3~5-Tri-o-benzoyl-B-D-ribofuranosyl)-2~6-dichloro-9H-purine (3.0 9, 4.7
rnmol) was dissolved in dioxan ~30 ml) and trans-2-phenoxycyclopentyiami-
ne hydrochloride (0.95 g, 4.4 mmol) was added followed by triethylamine
(0.64 9, 6.3 mmol). The rea~tion mixture was stirred at room temperature
for 72 h and purified by flash chromatography on silica gel to provide a
foam to which saturated methanolic ammonia (10û ml) was added. After 16
h at room temperature, ~he reaction mixture was evaporated and purffiled by
~; flash chromatography to provide the title trans-2-chloro-N-(2-phenoxycyclo-
pentyl)adenosine (0.70 9, 35%) as an amorphous foam ~a mixture of dia-
stereoisomers), 1H NMR (DMSO-d6)~ 1.56 - 2.30 (6H, 3m, -CH2CH2CH2-),
3.52- 3.60 (1~1, m, H-5'a)~ 3.63- 3.71 (1H, m, H-5'b), 3.96 (1H, q, H-4'), 4.13
(jH, q, H-3'), 4.50- 4.61 (2H, m, H-2' and -CH), 4.82 - 4.89 (1H, m, -CH),
5.08 ~1H, t, 5'-OH), 5.23, 5.49 ~2tl, 2d, 2'- and 3'-OH), 5.83 (1H, d, H-1'),
6.90, 7.07 and 7.25 (SH, t,d,t, Ar-H), 8.43 (1H, s, H-8~, 8.60 (1H, d, N-H).
H24ClNsO~; 0.5 H2O requires C, 53.6; H, 5.4; N, 14.9. Found: C, 53.4; H,
~; 20 5.5; N, 14.5%.
EXAMPLE 13 (Method A)
2-Chloro-N-~fR)-1-hvdro)~v-2-propylladenosine
R)-2-Amino-1-propanol ~0.23 9, 3.0 mmol), 9-(2,3,5-~ri-O-benzoyl-~-D-ribo-
furanosyl)-2,6-dichlQro-9H-purine (1.7 9, 2.7 mmol) and triethylamine (0.30
~: ~ g, 3.0 mmol) were dissolved in dioxan (20 ml) and stirred for 200 h at room
temperature. Following purification by column chromatography, the resultant
2',3',5'-tri-O-benzoyl-2-chloro-N-[(R)-1-hydroxy-2-propyl]adenosine was de-
: ~:
WO 93/23418 PCI'/DK93/0015X
21135~7
- 24 -
protected using methanolic ammonia to provide the title 2-chloro-N-[tR)-1-
hydroxy-2-propyl]adenosine as an amorphous foam (0.5 9, 54%3, 'H NMR
(DMSO-d6)~ 1.17 (3H, d, -CH3), 3.35 - 3.72 (4H, m, H~~a~ H-5'b and -t:~H2-),
3.96 (1H, q, tl-4'), 4.14 (1H, m, H-3'), 4.52 (1H, dd, H-2'), 5.08 (lH, t, 5'-
OH), 5.22, 5.49 (2H, 2d, 2' and 3'-OH), 5.83 (1H, d, H-1'), 8.0 (1H, d, N-H)
8.40 (1H, s, H-8).
Cl3H,7ClNsO5Ø75 H20 requires C, 41.9; H, 5.0; N, 18.8. Found: C, 42.1; H,
5.2; N, 15.8%.
EXAMPLE 14 (Method A)
2-Chloro-N-r(R)-1-~henvlthio-2-Propvlladenosine
; (R)-N-tertbutyloxycarbony!-1-phenylthi~2-propylamine
Thiophenol (1.5 g, 14 mmol) was dissolved in dry THF (100 ml) and a 60%
oil dispersion of sodium hydride (0.30 g, 14 mmol) was added in portions
under nitrogen. After stirring for 15 min. at room temperature, the mesylate
,, , :
20 ester of N-te*-butoxycarbonyl-2-hydroxypropylamine (3.2 g, 14 mmol) was
::
added in three portiorls and the reaction mixture was heated at 70C for 18
h. After coollng, water~(30 ml) w~s added, the~aqueous phase was separa-
ted and washed with dichloromethane ~50 ml). The cornbined organic ph~-
ses were dried (MgSO4) and evaporated to give (R)-N-tertbutyloxycarbonyl-
: ~ 25 1-phenylthio-2-propylamine as a fawn oil (3.2 g, 85%), TLC R~ 0.64 ~SiO2:
heptane/ ethyl acetate~ 1)].
This ~R)-N-(tert-butoxycarbonyl)-1-phenylthio-2-propylamine was oonverted
into ~R)-1-phenylthio-2-propylamine hydrochloride by acidic hydrolysis using
~; 30 the method described in Example 2.
WO 93/23418 ~ 2113 5 4 7 PCI`/DK93/00158
- 25 -
(R)-1-Phenylthio-2-propylamine (0.4 g, 1.96 mmol) was reacted with
9-(2~3~5-tri-o-benzoyl-B-D-ribofuranosyl)-2~6-dichloro-9H-purine (1.2 9, 1.9
mmol) in dioxan ~15 ml) in the presence of triethylamine (0.4 9, 4 mmol).
The reaction mixture was stirred at room temperature for 72 h, heated at
50C for 24 h, cooled, filtered and evaporated. The product (a~ter purifica-
tion by flash chromatography) was debenzoylated with methanolic ammonia
to provide the title 2-chloro-N-[(R)-1-pheny~hio-2-propyl]adenosine (after co-
lumn chromatography) æ~a foam (0.47 9, 52%), 1H NMR (DMSO-d6)~ 1.34
(3H, d, -CH3), 3.01 (lH,~dd,~;-C-H), 3.52- 3.60 (lH, m, H-5'.), 3.62-3.72 (1H,
m, H-S'b), 3.95 (1H,~q,~H4'), 4.13 ~1H, m, H-3'), 4.30 - 4.45 (1H, m, -C-H),
4.53 (1 H, m, H-2'), 5.09, 5.22, 5.50 (3H, 3 br, 2', 3'and 5'-OH), 5.84 (1 H, d,H-1'), 7.19 ~1H,~t, Ar-H), 7.30 ~2H, t, Ar-H), 7.45 (2H, d, Ar-H), 8.29 - 8.45
(2H, s & m, H-8 and N-H).
15~ ~ CtgHæClNsO4S requires C, 50.5; H, 4.9; N, 15.5. Found: C, 50.6; H, 5.1; N,
EXAMPLE 15 (Method A~
R)-2-Chloro-N-r1-(~fluoror henoxv)-2-Propvlladenosine
(R)-1-(4-fluorophenoxy)-2-propylamine (0.29 9, 1.4 mmol) (prepared from 4-
fluorophenol by the~method desrcribed in example 2) was reacted with
9-(2',3',5'-tri-O-benzoyl-s-D-ribofuranosyl~-2,6-dichloro-9H-purine ~0.89 9, 1.4- ~, 25 ~ mmol3 in dioxan (30 ml) in the presence of triethylamine (0.42 9, 3 mmol~.
The reaction mixture~was~stirred at room temperature for t8 h, and heated
at 60C for 4 h. The reaaion mixture was filtered and evaporated to a resi-
due which was purdied by~flash chromatography. The resultant 2',3',5'-tri-O-
benzoyl-2 chloro-N-[(R)-1-(4-fluorophenoxy)-2-propyl]adenosine was depro-
tected using methanolic ammonia to provide the title 2-chloro-N-l(R)-1-(4-
fluorophenoxy)-2-propyl]adenosine ~0.21 9, 40%) (after column chromato-
:,: : ~
~,
WO 93/23418 PCI/DK93/00158,
~113547
- 26 -
graphy), mp 172-173C; 1H NMR (DMSO-d~)~ 1.29 (3H, d, -CH3), 3.52-
3.60 (1 H, m, H-5'~), 3.64 - 3.72 (1 H, m, H-5'b), 3.92 - 4.00 (2H, rn, H-4' and -
C-H), 4.0S - 4.20 ~2H, m, H-3' and -G-H), 4.53 ~1H, m, H-2'), 4.65 (1H, m, -
CH3CH-), 5.08, 5.24, 5.50 (3H, 3 br, 2', 3' and 5'-OH), ~.86 (1 H, d, H-1'),
6.8~- 7.15 (4H, 2 m, Ar-H), 8.30 - 8.46 (2H, m, H-8 and N-H).
C1gH2,ClFN5O5 requires C, 49.8; H, 4.7; N, 15.3. Found: C, 49.4; H, 4.7; N,
14.9%.
~MPLE 16 (Method A)
2-Chloro-N-~(R)-2-phenoxv-1 -propv~adenosine
(R)-2-Phenoxy-1-propylamine~ ~0.6 9, 2.9 mmol) (prepared by the method
described in example 2) was reacted with 9-(2,3,~tri-O-benzoyl-s-D-;ibo-
furanosyl)-2,~dichloro-9H purine ~1.5 9, 2.4 mmol) in dioxan (20 ml) in the
presence of triethylamine ~(0.5 9, 5.3 mmol). The reaction mixture was stirred
at room temperature ~for 72 h before being fil~red and evaporated. The
produc~, following purifica~ion by flash chromatography, was treated with
~ saturated methanolic ammonia ~30 ml) for 18 h and evaporated to provide a
~:: :; solid residue. This solid~ was washed thoroughly with dichloromethane to
provide the tit,e 2-chloro~-N-[(R)^2-phenoxy-1-propyl]adenosine ~0.7 9, 65%),
mp 1:75-177C, lH NMR ~DMSO-d6)~ 1.39 (3H, d, -CH3), 3.56 (1H, ABX,
: H-51a), 3.68 (1H, m,: H-5'b~, 3.33 - 3.40 ~1 H, m, -C-H), 3.83 - 3.92 (1 H, m, -C-
H), 3.96 (1H, q, H-4'~, 4.14 (1H, m, H-3'~, 4.53 ~1H, dd, H-2'), 4.70 (1H, ~, -
C-H), ~.08, 5.34, 5.50 (3H, 3 br, 2', 3' and S'-OH), 5.85 (1 H, d, H-1'), 6.9û
(1H, t, Ar-H), 7.11 (21~, a, Ar-H), 7.28 (2H, t, Ar-H), 8.45 (1H, s, H-8), 8.63
(1 H, t, N-H).
C19HzClN5O5 req~ires C, 52.4; H, 5.1; N, 16.1. Found: C, 52.5; H, 5.1; N,
15.9%.
: :~
.
5~.~ WO 93/23~1~ 21 1 3 5 4 7 PCl'lDK93/00158
EXAMPLE 17 (Method A)
2-Chloro-N-r2-(phenvlmethoxv)ethvl~adenosine
~e titl~ compound was prepared by reacting 2-(phenylmethoxy)ethylamine
hydrochloride (0.51 9, 2.7 mmol) with 9-(2,3,~tri-0-benzoyl-B-D-ribofuran-
osyl)-2,~dichloro-9H-purine (1.43 9, 2.25 mmol), followed by debenzoyla-
tion of the purified product using methanolic ammonia to provide the title 2-
chloro-N-[2c~phenyimethoxy)ethyl]adenosine (0.38 9, 44%) (after column
chromatography) as a solid, mp 115 -124C, 'H NMR (DMSO-d6)~ 3.~0-
3.58: (1H, m, H-5'~), 3.60 - 3.70 (4H, m, H-5'b, -CH2- and -CH-), 3.95 (1 H, q,
: H-4'), 4.04 - 4.16 (2H, m, H-3' and -CH-), 4.52 (1tl, br s, H-2' and -Ct~2-),
~: ~
5.07 (1 H, t, 5'-OH), :5.21 j 5.50 (2H, 2d, 2'-and 3'-OH), 5.84 (1 H, d, H-1'), 7.22
7.36: (5H, m Ar-Hj, :8.25 - 8.40 (2H, m, H-8 and N-H).
1 5
: ~ .
C19H22ClN50s. 0.1 H20 requires C, 52.1; H, ~.1; N, 16Ø Found: C, 51.8;
H, 5.3;~ N, 15.6%. ~ ~ :
EXAMPLE 18 (Method A~
2-Fluoro-N-~(R~-1 -Dhenoxy-2-Dropvlladenosine
9-(2,3,~Tri-Q-acetyl-s-D-ribofuranosyl~-6-chloro-2-fluoro-9H-purine (1.03 g,
2.38 mmol) P~T Publication No. WO 93/08206, (R)-1-phenoxy-2-propylami-
2~ ~ ne (0.36 9, 2.33 mrnol) and triethylamine (0.29 g, 0.28 mmol) in dioxan ~20
; :: :: ml) were stirred at room temperature for 18 h. The reaction rnixture was
fllltered and evaporated to~a residue which was purified by flash chroma~o-
graphy. The resuttant;2',3',5'-tri-0-acetyl-2-fluoro-N-[(R)-1-phenoxy-2-pro-
pyl~adenosine was~ deprotected using methanolic ammonia to provide the
t itle 2-fluoro-N-l(R)-1-phenoxy-2-propyl]adenosine (0.28 9, 23%) (after co-
Iumn chromatography), mp 148-1~0C; 'H NMR (DMSO-d6)~ 1.33 (3H, d, -
:
WO 93/23418 PCr/DK~3/00158
2113547
- ~8 -
3.59 (1 H, m, H-51a), 3.63 - 3.71 (1 H, m, H-5'b~, 3.92 - 3.99 ~2H, m, H-4' and -
C-H), 4.10 - 4.1S (2H, m, H-3' and -C-H), 4.51 (1H, q, H-2'), 4.61 (1H, m, -
~H3CH-), 5.06 (1 H, t, 5'-OH), 5.22, 5.48 ~2H, 2d, 2' and 3'-OH), 5.82 (1 H, d,
H-1'), 6.89 - 6.97 (3H, m, Ar-H), 7.~5 - 7.30 (2H, t, Ar-H), 8.39 (1 H, s, H-8),8.49 (1H, d, N-H).
:: C1gHzFN5O5 requires~C, 54.4; H, 5.8; N, 16.7. Found: C, 54.7; H, 5.5; N,
~ 1 6.4%.
:
~: 10 ample 19 (Method B)
2-Methoxy-N-[~R)-1 -phenoxv-2-propvlladenosine
2-Methoxy-N-[(R)-1-phenoxy-2-propyl]adenosine was prepared by reacting
15 ~: 2-chloro-~ R)1-phenoxy-2-propyl)adenosine ~Example 2) (0.30 9, 0.69
mmo!) with a mixture of sodium hydroxide (0.32 g, 8.0 mmol) and methanol
: (15 ml) in a ~sealed~ vessei ~ 80 - 90C for 4h. The cooled reaction mixture
was:neutraiised with concentrated hydrochloric acid and evaporated to
dryness. Water ~30 ml) was added and the mixture was extracted with dich-
20 ~ ~ ~ loromethane (2~x 30 ml). The combined extracts were dried (MgSO4) and:coe~aporated with :dichloromethane ~30: ml), giving the title con~pound as a
foam (0.19 9, 60%), ~H NMR (DMSO-d6)~ 1.32 (3H, d, -CHC~3), 3.5~ (1H,
m, H-5'~,), 3.65 (1 H, m,~ H5'b), 3.72 (3H, sj -CH3), 3.91 - 3.99 and 4.10 - 4.20
(4H, 2 m, H-3',~H-4' and -CH2-), 4.51 (1Hj dd, H-2'), 4.67 (1H, m, -CHCH3),
~, 25 5.84 ~1 H,~ d, H-1'~, ;6.89 - 6.98 (3H, m, Ar-H), 7.26 (2H, dd, Ar-H) 8.12 (1-H,
br, -NH), 8.46 (~ H, s, H-8).
:: :
:~
... WO 93/23418 2113 5 4 7 pcr/DK~3/ool58
- 29 -
EXAMPLE 20 (Method A)
N-(2-Methoxyethvl)adenosine
The title compound was prepared by the procedure described in example 7
by reacting 2-methoxyethylamine hydrochloride (0.27 9, 3.6 rnmol) with 6-c-
hloropurine riboside (i.e. 9-B-D-ribofuranosyl-6-chloro-9H-purine) (1.0 9, 3.5
mmol) in dioxan (30 ml) at room temperature for 72 h with triethylamine
(1.04 ml, 7.5 mmol) present. The reaction mix~ure was filtered and evapora-
ted and the resultant residue was recrystallised from methanol ~tOO ml) to
` provide the title compound (0.80 g, 82%) as a solid, mp 151 -152C, 'H
NMR (DMSO-d6)~ 3.26 (3H, s, -CH3), 3.50- 3.58 (3H, m, H-5'~, and -CH2-),
3.60 - 3.70 (3H, m, H-5'b~and -CH2-), 3.96 (1H, q, 11-4'), 4.14 (1H, dd, H^3'),
4.60 (1 H, dd, H-2') 5.20, 5.45 ~2H, 2d, 2'-and 3'-OH), 5.~2 ~1H, t, 5'~0H)
5.87 ~1 H, d, H-1'), 7.80 (1 H, br s, -NH) 8.22, ~.35 (2H, 2s, H-2 and H-8).
C13H1gN505 requires C, 48.0; H, 5.9; N, 21.5. Found: C, 47.8; H, ~.9; N,
21.3~/o. ~:
20~ EXAMPLE 21 ~ethod A)
2-_hloro-N-~(2-methoxvphenyl~ethv!ladenosine
The title c~mpound~was prepared by reacting (2-methoxyphen~l)methylami-
~, 25 ne (û.55 9, 4.0 mmolj with 9-(2,3,~tri-0-benzoyl-~-D-ribofuranosyl)-2,6-dichl-
oro-9H-purine (1.01 g, 1.6 mmol), followed by d~benzoylation of the purified
product using meth~nollc ammonia~to provide the title 2-chloro-N-~(2-met-
hoxyphenyl)methyl]adenosine (0.31 9, 45%) (after column chromato~raphy~
as a solid; mp 116 119~C, tH NMR (DMSO-d6)~ 3.51 - 3.60 (lH, m, H-5ta),
3Q 3.61 - 3.7û (1H, m, H-5'b)~ 3.95 (1H, q, H-4'), 4.13 (1H, m, H-3'), 4.52 (1H, q,
H-2'), 5.06 (1 H, t, 5'-OH), 5.22, 5.50 (2H, 2d, ~'-and 3'-OH~, 5.85 (1 H, d,
WO 93/23418 PCI`/DK93/001~i8 ~
211 35~7
- 30-
H-1'), 6.83 - 7.25 ~4H, 2t, 2d, Ar-H~, 8.43 (1H, s, H-8), 8.72 (1H, t, N-H).
EXAMPLE 22 (Method A)
5 ,-Chloro-N-~(R)-3-methvl-1-phenoxv-2-butylladenosine
(R)-3-methyl-1-phenoxy-2-butylamine (0.6 9, 2.8 mmol) was reacted with
9-(2~3~5-tri-o-benzoyl-B-D-ribofuranosyl)-2~dichloro-9H^purine (1.4 g, 2.2
mmol) in dioxan (20 ml) in the presence of triethylamine ~0.5 9, 5.0 mmol).
10 The reaction mix~ure was stirred at room temperature for 40 h before being
filtered and evaporated. The product (after purfflcation by flash chromato-
graphy) was debenzoylated with methanolic arnmonia to provide the pro-
duct (after column chromatography) as a foam which solid fied on Goeva
: ~ :
poration with dichloromethane. 2-Chloro-N-l(R)-3-methyl-1-phenoxy-2-bu-
15~ tyi3adenosine (0.46 9, ;44%)~ was o~tained as a white solid, mp 95 - 1 00C,
1H~ NMR (DMSO-d6)6 0.95,~0.98 (6H, 2d, 2 x -CH3), 2.10 (1H, m, -CH(~H3)2),
3.53 - 3.60 (1 H,~ m, H-5'~), 3.63 -3.71 (1 H, m, H-5'b), 3.95 (1 H, q, H-4'),
4.07 - 4.23 (3H, m,~ H-3' and -CH2-), 4.96 (1 H, m, -C-H), 4.56 ~1 H, q, H-2'),
5.08 (1 H, t, 5'-OH), 5.23, 5.49 (2H, 2d, 2' and 3'-OH), 5.84 ~1 H, d, H-1'),
20~ 6.87 -~6.97 (3H, m, Ar-H), 7.24 - 7.31 (2H, dd, Ar-H), 8.36 (1 H, d, -N-H), 8.40
(1H,~s, H-8)- ~
C2lH26ClNsOs.O.S~H20 requires C, 53.3; H, 5.8; N, 14.8. Found: C, 53.4; H,
5.7; N, 14.8%.
~; EXAMPLE 23 ~Method A)
2-Chloro-N-~tR)-1-(2-(2-propvloxv3phenoxy)-2-p,ropvlladenosine
(R)-1-(2-(2-Propyloxy)phenoxy)-2-propylamine (prepared from 2-(2-propy-
loxy)phenol by the procedure described in example 2) 10.54 9, 2.2 mmol)
::
WO 93~2341X 21 1 3 5 ~ 7 pcr/DK93~oo1s8
- 31 -
was reacted with 9-(2,3,5-tri-O-acetyl-~-D-ribofuranosyl)-2,6-dichloro-9Hpuri-
ne (2.0 9, 4.5 mmol) in dioxan ~30 ml) in the presence of triethylamine (2.19
g, 22 mmol). The reaction mixture was stirred at room ~emperature for 18 h
before being filtered and evaporated. The product ~after purification by flash
5 chromatography) was debenzoylated with methanolic ammonia to provide
the 2-chloro-N-~(R)-1-(2-(2-propyloxy)phenoxy)-2-propyl]adenosine (a~ter co-
lumn chromatography) as a foam (0.47 9, 39%), 1H NMR (DMSO-d6)~ 1.04,
1 .CN6 (6H, 2d, 2 x -CH3), 1.31 (3H, d, -CH3), 3.53 - 3.60 (1 H, m, H-5'~), 3.64 -
3.71 (1H, m, H-5'bJ, 3-95 (1H, q, H-4'), 3.98 - 4.15 (3H, 2m, H-3' and -C H2-),
4.35 (1H, p, -C-Hj, 4.51 (~H, q, H-2'), 4.72 (1H, m, -C-H), 5.08 (1H, t, 5'-
OH), 5.22, 5.48 (2H, 2d, 2' and 3'-OH), 5.85 (1H, d, H-1'), 6.82 - 7.08 (5H,
,
m, Ar-H), 8.32 (l H, d, -N-H), 8.41 (1 H, s, H-8).
CæH28ClN506.1.0 H20 requires Cj 51.6; H, 5.9; N, 13.7. Found: C, 52.0; H,
15~ ;58 N 133%
EXAMPLE Z4 (Method A)
20 ~ 2-Chloro-N-r(Rj-1-phenvlsulphonvl-2-propylladenosine
Phenylsulphonyl-2-propylamine (0.4 9, 1.7 mmol) was reacted with
~(2,3,5-tri-O-benzoyl-~-D-ribofuranosyl)-2,6-dichloro-9H-purine (1.7 9, 1.0
mmol) in dioxan (20;m~) in~the~presence of triethylamine (0.4 g, 4.3 mmol).
25 The reaction mixture was stirred at room temperature for 48 h, heated at
~ I
60C for 4 h, cooled, filtered and~evaporated. The produ~t (after purification
by~ flash chromatogrPphy)~was deber~oylated with methanolic ammonia to
provide the title 2-chloro-N-~(R)-1 -phenylsulphonyl-2-propyl] adenosine (after
column chromatography~as a foam (0.2 g, 24%), 1H NMR (DMSO-d6)~ 1.24
~ . ~
30 ~ (3H, d, -CH3), 3.45 (1H, dd, -C-H), 3.53 - 3.61 (2H, m, H-5'~ and -C-H),
3.64 - 3.71 ~1H, m, H-5'b), 3.86 ~1H, dd, -C-H), 3.97 (1H, q, H-4'), 4.14 (1H,
,
:: ~
WO 93/23418 P~/DK93/00158
21135~7
^ 32-
m, H-3'), 4.53 (1 H, m, H-2'), 5.09 (1H, t, 5'-OH~, 5.23, 5.50 ~2H, 2 d, 2' and
3'-OH), 5.83 (1H, d, H-1'), 7.45 - 7.82 (5H, m, Ar-H), 8.21 (1H, s, -N-H), 8.38
(1 H, s, I 1-8).
`~:
EXAMPLE 2~ (Method A)
~l~LL2-methylphenvl)methvlladenosine
:
The title compound was prepared by reacting (2-methylphenyl)methylamine
(1.51 g, 12.5 mmol) with;6-chloropurine riboside (2.87 g, 10.0 mmol) in
dioxan (100 ml)~in th~ presence of diisopropylethylamine (1.94 9, 15.0
mmol). The reaction mixtur~ was heated at 60C for 6 h, cooled, filtered and
evaporated. The residue~ wa s pur~ied by flash chromatography, eluting initi-
ally with dichloromethane, and later increasing polarity to dichloromethane/
15~ ~ ethanol (9:1), to~provide the product (2.6 g, 70%) as a solid which wa^s
r ecrystallised from ~methanol to give N-[~2-methylphenyl)methyl]adenosine
as~ white~ crystals (1;.75~ g, 47%), mp 161.5 - 163.5C, lH NMP~ (DMSO-d6)~
2.35~(3H,~ S! -CH3),;3.53 -~3.6û (1H, m, H-5~ 3.65 - 3.72 (1H, m, H-5'b), 3.98
(1H, q, H-4'), 4.16 (1H, m, l~i-3'), 4.64 (1H, q, H-2'), 5.41 (1H, t, 5'-OH), 5.21,
5.48 ~2H, 2d, 2'-and 3:0H), 5.92 (l H, d, H-1'), 7.06 - 7.24 (4H, m, Ar-H),
8.20~ and~ 8.4û (3H, s ~and br s, H-2, H-8 and N-H).
EXAMPLE 26 (Method ~
2-Methvl-N~ 1-phenoxv-~-propviladenosine
(R)-1-phenoxy-2-propyla~nine (0.56 9, 3 mmol) was reacted with 9-(2,3,~tri-
O-acetyl-~-D-ribofuranosyl)-6-chloro-2-methyl-9H-purine ~û.43 gl 1 mmol)
, :
pfepared from 2-methylinosine (Journal of Organic Chemistry, 1967, 32,
3258 - 3?603 by standard acylation and chlorination steps] in dioxan (20 ml)
in the presence of triethylamine ~0.41 9, 4 mmol). The reaction mixture was
::
~ WO 93/23418 211 3 S ~ 7 PCI-/DK93/00158
heated at 50C for 70h, and at 90C for 3 h. before being filtered and eva-
porated. The product ~after purification by flash chromatography) was de-
benzoylated with methanolic ammonia to provide the titte 2-methyl-N-[(R)-
1-phenoxy-2-propyl]adenosine ~after column chromatography) as a foam
(0.21 9, 50%), lH NMR (DMS~d6)~ 1.30 (3H, d, -CH3), 2.43 (3H, s, -CH3),
3.53 - 3.60 (1 H, m, H-5'Q), 3.66 - 3.73 (1 H, m, H-5'b), 3.94 (1H, dd, -C-H),
3.99 (1H, q, H-4'), 4.12 - 4.22 ~2H, m, H-3'and -C-H), 4.54 ~lH, dd, H-2'),
4.76 (1H, m, -CH3CH-), 5.20, 5.52 (2H, 2d, 2' and 3'-OH), 5.73 (1 H, t, 5'-
OH), 5.87 (1H, d, H-1'), 6.90 - 7.31 (5 H, t, m, t, Ar-H), 7.74 (1 H, br d, N-H),
~; 10 8.28 ~1H, s, tl-8).
C2oH2sClN5O5Ø33 H2O requires C, 57.0; H, 6.1; N, 16.6. Found: C, 57.0; H,
6.2; N, t6.8%.
~, :
~,
i ,: :
. :
~:
::
,
.
WO 93/23418 PC~/DK93/00158
21135~7
- 34 -
Evaluation of the compounds.
.
Methods for assessing adenosine receptor binding in vitro have been revie-
5 wed lAdenosine Receptors, (Cooper, D.M~F. and Londos, C., eds.) Alan R.
Liss, inc., New York, 1988, 43-62].
Evaluation of these: compounds in established animal models has indicated
that the compounds according to the invention possess desirable central
10 nervous system properties. For example, they act as anticonvulsant agents,
are effective in ~animal ~models of pain, and show cerebroprotective effects in
laboratory test animals subjected to simulated cerebral ischaemia. In addi-
tion, the ~compounds may~ have efficacy as neuroprotec~ive agents in cases
of cerebral oedema:and traumatic head injury.
Evaluation of in vitro bindin~ to adenosine A1 and A2 receptors
The :affinity: of the known and novel compounds described in this invention
for:the adenosine~A1~receptor has been determined essentially as descri-
20 ~ ~: ; bed in the literature using [3H]-R-PiA as a radioligand (Naunyn-Schmied~-
berg's Archives~of Pharmacoiogy, 1980, 313, 179-187). Affinity ~or the A2
receptor was :measured~ using the radioligand [3H]-CGS 21680 (European
Journal of Pharmacology, 1~B9, 168, 243-24~), and the values for represen-
tative compounds~are~giverl; In table l below. In vitro receptor binding values
25 obtained for the reference standard adenosine agonists CPA [~-(cyclopen-
tyl)adenosine~ and _-PlA [~-(1-phenyl-2-propyl)adenosine]) are included for
comparison.
.
"~ WO 93/23418 PCl`/DK93/001~i8
2113S47
- 35-
Method descr~ption
~ ~ DMCM INDUCED SEIZURES IN MICE
: ~ S
In this moclel, seiz~;res are induced by i.p. (intraperitoneal) dosing of methyl6,7-dimethoxy-4-ethyl-~-carboline-3-carboxylate DMCM at 15 mg/kg.
DMCM is an inverse agonist t~ the benzodiazepine receptor, presumably
producing sekures by decreasing the po~ency of inhibition of the GABA
receptor/benzodiæepine receptor/chloride ionophore complex.
15 mg/kg of DMCM dissolved in 0.02 N HCI (1 mg/ml) is administared i.p. in
a:volume of 300:~1 to male NMRI mice weighing 20 + 2 9. This induces two
15~ ~different responses: a) some animals manifest a brief loss of righting;efle-
xes :or;take up~ an upright ~position in which they have a mild short clonus of
the~upper extremities, b) o~her animals man~est intense clonic and tonic
:;convulsions of all extremities often followed by death. DMCM is
adminlstered 30 :min~after an intraperitoneal injection of a tes~ compound.
20 ~ ~ L atency: time for the~ presence o~ intense clonic and tQnic convuisions and
death is noted until~1~5 min after administration of DMC::M. At least S doses
of each test compound are tested with 8 mice per dose.
An anticonvulsive ~EDW value is determined as the dose (mg/kg) protecting
~, 25 50/O of the animals against clonic eonvulsions; some representative values
are shown in table ll.:
The above method is a described in Petérsen, E.N., Eur. J. Pharmacol. 94,
117-124, 1983; P:etersen, E.N., Eur. J. Pharmacol. 195, 261-265, 1991.
3~)
WO 93/23418 PCI`/DK93/001!i8
2113~ ~7
- 36-
Blood pressure in anaesthetised rats
Test compounds are generally dissolved in DMSO and diluted in 5% chre-
mophore/saline before being dosed to nembutal anaesthetised 200 g fema-
5 le Sprague Dawley rats which have not been starved or fasted. The rats arebreathing spontaneously; blood pressure (BP) and heart rate (HR) is mea-
sured 5 minutes af er a bolus i.v. injection. Each measurement is repeated
twice. Results for representative compounds are shown in table ll.
10 Neuro~rotective effect: Gerbil BCAO ischemia model.
:: :
Transient ~lobal ischaemia was produced in Mongolian gerbils (60-70 9
males) anaasthetiæed~with~2% halothane in 70% nitrous oxide and 30% oxy-
`gen. The common carotid arteries were occluded for 5 min. and tt e animals
15 were allowed to recover ~for~ 4 days. The animals were reanaesthe~ized de-
capitated and the brains quickly removed and frozen in powdered dry ice.
Coronal sections ~(20 um)~ were taken through the brain at the level of the
hippocampus and stained with cresyl violet and hematoxylineosin. The brain
se~tions were rated for neuronal damage in~the hippocampus CA1 region
20~ u sing a scale from 0 (undamaged~ to 3 (total damage of CA1). The body
temperature of ail the~animais was maintained at 37C throughout the sur-
gery and the animals;~were placed in warmed boxes during the recovery
period. Each expenment consisted of a drug and a vehicle control group (n
= 10-15j. Test compounds were administered 30 min. after reperfusion.
2~
.
~: :
:
WO 93/23418 2 11 3 5 ~ 7 pcr/DK93/oo158
- 37 -
TABLE I
In V~ro evaluation of the compounds
5 ~denosine agonist A1 receptor A2 receptor Ratio
tested binding binding A21A1
, nM) (Kj, nM)
43 ~ ~ 57 ~7
10: (2) 18 ~: 318 18
(3) :~ 100 ~ 7413 74
(4)~ 166 3095 19
5): 4 ~ ~ 1 23 31
(6) ~ ~ 36 ~ 802 22
1 5~ 1 8: ~ 791 44
:: (8) ;~ 123 ~ ~ 2188 ~ : 18
(9) ~ 3.3 ~ 3270 g91
(10)~ : 3.1 ~ 1320 426
~ 397 11
: :20 ~(1 2) : 6 ~:: 383 64
3) ~ 7: :~ 2241 : : ~ 320
14) ~ :15 : 893 ~ 60
: (17) ~ 340~ m6 ~3
, ~ .
~: 25(18) 8 310 39
,, . . ~
(1 9) 88 2332 27
(20)~: ~77~ 2432: ~: 32
(26) ~ 69 ~ 1200 17
: -
:: :; CPA 1.6~ 173 108
`: 3()(E3)-plA 2.0 134 67
~;; :
: :
~,
WO 93/23418 PCr/DK93/00158 . .
21135~7
- 38-
TABLE ll
Pharrnaco~g_al evaluation of the comPounds
DMCM seizures % fall in BP
CompoundED50(mg/kg)i.p. 0.1 mg/kg i.v.
; (~xample No.): HR
.4 0
: 10 (2) : 3.9 8
~ ,
4) 1 3.3 0
: (6) 3.8 5
(8): 24.9~
10) ~ 0.1 ~ -
~11 ) 2.0 15
12~ 6.7~ ~ 25
::(14): : : ~99 15
5) ~13.6:: 16
(19~ ~ 6.7
(~S)~ ~;: 0.5 ~ ~ ; 4~
(26) 1.9 : ; 15
,~ ~: : : ::: ~ ::
25 ~ ~
"
,,:
~:` :
::
:~'
:: :
:: ::
:; :