Note: Descriptions are shown in the official language in which they were submitted.
Wo93/10742 CA 2 1 1 7 3 04 PCI`/DK92/0034X
- 1 -
Chemical Compounds, their Preparation and Use
.
Much evidence has accumulated to suggest that neuroleptics exert their
antipsychotic action by blocking dopamine (DA) receptors in the brain. In
recent years, it has become clear that sorne neuroleptics (e.g. clozapine~
show an at,vpical profile: the compounds are nct only beneficial in treating
patients, who respond poorly to classical neuroleptic therapy, but the
compounds are also relatively devoid of extrapyrimidal side effects (EPS)
commonly seen with classical neuroleptics (Ereshefsky et al., Clin.Pharm 8,
691-709, 1989). In this respect it has been speculated that atypical neuro-
leptics are working mainly by blocking socalled A10 mesolimbic DA
systems (areas which are thought to be affected in psychosis~, while the
side effects of claxsical neuroleptics are produced by blockade of DA
receptors in the motor areas of the brain (A9 DA system (Gudelsky, Psy-
chopharmacology (Berl) 99: S13-S17, 1989)). The antipsychotic effect of
clozapine and related compounds might be due to its blockade of not only
DA-receptors (D-1, D-2, D-3, D-4) but also 5HT-receptor subtypes (5HT2-,
5HT3-, 5HT~c-, 5HT,A-), NA-~1-receptors, histamine and possibly other
receptors.
Furthermore, 5HT2-blockade may also be important (Meltzer, Schizphr. Bull.
17: 263-~7, 1991) to counteract the socalled negative symptoms of psycho-
5is ~delusions and social withdrawal) which are otherwise difficult to treat
with conventional neuroleptics.
~; Compounds reducing 5-HT neurotransmission have been suggested to be
useful for the treatment of various neurological and psychiatric diseases.
:
wo 93/10742 C A 2 1 1 7 3 0 4 - 2 - PCI'/DK92/00348
5HT2-antagonists, such as naftidrofuryl (Brain Res. 1989, 494(2) 387-90), are
described to exhibit a protective effect on ischemic neuronal damage in the
gerbil. Ritanserin, which is a potent and selective 5HT2-antagonist, has been
shown to have anxiolytic-antidepressant a~ivities in humans (Barone et al.,
Drug Clin. Pharm., 20 770 (1986)). Furthermore serotonergic mechanisms
are described to be involved as active factors, or inducing processes, in the
organization of sleep (Neuropharmacology, 19, 163 (1980)).
The piperidine derivative ketanserine, which is a 5HT2-antagonist with we,ak
~,-blocking properties has been shown to be useful for treatment of various
cardiovascular disorders.
Other similar piperidine derivatives are described in German Patent
1930818, EP 368388, EP 377528, EP 184258 and EP 402644.
This invention relates to piperidine derivatives, methods for making them
and pharmaceutical compositions containing them.
The compounds of this invention demonstrate high affinity for various
receptor subt,vpes including the 5HT2-, the NA-lx1-, the dopamine D,- and
D2- receptors or a combination of these. This invention relates to the use of
said compounds as rnedicaments useful for treating CNS-system, cardi~
vascular system and gastrointestinal disorders,~ such as treatment of
anxiety, sleep disorders, depression, psychosis, schizophrenia, migraine,
ischemic neuronal damage, asthma, hypertension, urticaria, analgesia and
emesis. ~ I
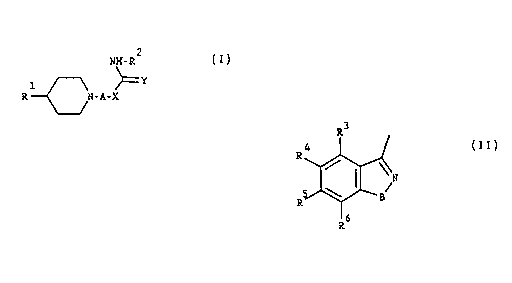
The present invention provides piperidine derivatives of formula l:
NH- R
' R{~N-A-X (I)
.
WO 93/10742 CA 2 1 1 7 3 04 PCr/DlC92/0034X
- 3 -
wherein A represents a straight or branched saturated hydrocarbon chain
containing from 2 to 6 carbon atoms;
R' is
R3
wherein R3, Ri, Rs and R6 independentiy are hydrogen, halogen or C1 6-
alkyl;
15 B is -O- or -NH-;
X is -O- or -NH-
Y is =O, =S or =NZ
wherein Z is hydrogen, C, 6-alkyl or -CN;
R' is selected frorn the group consisting of
~7
/D or 9
:;~ 30
wherein R7? R8, R9 and R' independently are hydrogen, Cl.6-alkyl, halogen,
WO93/10742 CA2 ] 1 7304 PCl/DK92/0034X
- 4 -
C1~-alkoxy or perhalomethyl;
-D- represents a 5- or 6-membered heterocycle containing one or more N-,
O- or S-atoms, or a pharmaceutically acceptable salt thereof.
The purified reaction product may be converted into a physiologically
acceptable salt. Such salts include acid addition salts formed with inorganic
or organic acids, for example hydrochlorides, hydrobromides, sulphates,
nitrates, oxalates, phosphates, tartrates, citrates, fumarates, maleates,
10 succinates, and sulphonates e.g. mesylates. If desirable, selected salts may
be subjected to further purification by recrystallization.
The invention includes within its scope all optical isomers of compounds of
the general formula I and their mixtures including racemic mixtures thereof.
Specific compounds within the scope of the present invention include the
following, or pharmaceutically acceptable salts thereof:
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[3-(6-indazolylcarbamoyloxy)propyl]-
20 piperidine,
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[3-(~-indæolylcarbamoyloxy)propyl]-
piperidine,
4-(6-Fluoro-1,2-benzisoxæol-3-yl)-1-[2-(5-indæolylcarbamoyloxy)ethyl]-
piperidine, ~ ~
; 4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-[2-(6-indæolylcarbamoyloxy)ethyl]-
piperidine,
4-(6-Fluoro~1 ,2-benzisoxazol-3-yl)-1-[2-(6-(1-methylindazolyl)carbamoyloxy)-
~; ~ ethyl]piperidine,
WO 93/10742 C A 2 1 1 7 3 0 4 PCI/DK92/0034X
- 5 -
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1-[2-(5-(1 -methylindazolyl)carbamoyloxy)-
ethyl]piperidine,
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[3-(6-indolylcarbamoyloxy)propyl3-
5 piperidine,
4-(6-Fluoro-1,2-benzisoxazol 3-yl)-1-[2-(6-indolylcarbamoyloxy)ethyl]piperi-
dine,
4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-[3-(5-indolylcarbamoyloxy)propyl]-
piperidine,
4-(~Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[2-(5-indolyicarbamoyloxy)ethyl]piperi-
dine,
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1-[2-(5-(1 -methylindolyl)carbamoyloxy)-
ethyl]piperidine,
4-(~Fluoro-1 ,2-benzisoxazol-3-yl)-1-[3-(5-(1 -methylindolyl)carbamoyloxy)pro-
20 pyllpiperidine,
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1-[3-(6-(1 -methylindolyl)carbamoyioxy)pro-
pyl]piperidine,
4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-[2-(6-(1-methylindolyl)carbamoyloxy)-
ethyl~piperidine,
1 -[3-(6-Benzoxazolylcarbamoyloxy)propyl]-4-(6-fluoro-1 ,2-benzisoxazol-3-yl)-
plpendlne,
1-[2-(6-Benzoxazolylcarbamoyloxy)ethyl]-4-(6-fluoro-1,2-benzisoxazol-3-
- yl)piperidine,
WO 93/10742 C A 2 1 ~ 7 3 0 4 - 6 - PCI/DK92/00348
1 -[3-(5-Benzoxæolylcarbamoyloxy)propyl]-4-(6-fluoro-1 ,2-benzisoxæol-3-
yl)piperidine,
1 -[2-(5-Benzoxazolylcarbamoyloxy)ethyl]-4-(6-fluoro-1 ,2-benzisoxazol-3-
5 yl)piperidine,
1 -[2-(6-Benzothiæolylcarbamoyloxy)ethyl]-4-(6-fluoro-1 ,2-benzisoxazol-3-
yl)piperidine,
1-[2-(6-Benzothiæolylthiocarbamoyloxy)ethyl]-4-(6-fluoro-1,2-benzisoxæol-3-
yl)piperidine,
1 -[3-(6-Benzothiæolylthiocarbamoyloxy)propyl]-4-(6-fluoro-1 ,2-benzisoxazol-
3-yl)piperidine,
~(~FIuoro-1,2-benzisoxæol-3-yl)-1-[3-(6-(2-methylbenzothiæolyl)carbamoyl-
oxy)propyl]piperidine,
;`~
: 4-(6-Fluoro-1,2-benzisoxæol-3-yl)-1-[2-(6-(2-methylbenzothiæolyl)carbamoyl-
2Q oxy)ethyl]piperidine,
1-[3-(5-Benzothiazolylcarbamoyloxy)propyl]~(~fluoro-1,2-benzisoxazol-3-
yl)piperidine,
1-[2-(5-Benzothiazolylcarbamoyloxy)ethyl]-4-(6-fluoro-1,2-benzisoxazol-3-
yl3pipéridine, ~ i
1-[3-(5-Benzothiazolylthiocarbamoyloxy)propyl]-4-(6-fluoro-1,2-benzisoxæol-
`~ 3-yl)piperidine,
i
1-[2-(~Benzothiazolylthiocarbamoyloxy)ethyl]-4-(~fluoro-1,2 benzisoxæol-3-
yl)piperidine,
; ~
:
~:
WO93J10742 CA 2 1 1 7304 PCI/DK92/00348
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[2-(3,4,5-trimethoxyphenylcarbamoyl-
oxy)ethyl] piperidine,
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[2-(3,4,5-trimethoxyphenylthiocarbamoyl-
S oxy)ethyl~piperidine,
4-~6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[3-(3,4,~-trimethoxyphenylthiocarbamoyl-
oxy)propyilpiperidine,
1-[3-(3,4-Dimethoxyphenylcarbamoyloxy)propyl]-4-(6-fluoro-1,2-benzisoxa-
zol-3-yl)piperidine,
1 -[2-(3,4-Dimethoxyphenylcarbamoyloxy)ethyl]-4-(6-fluoro-1 ,2-benzisoxæol-
3-yl)piperidine,
1 -l2-(3,4-Dimethoxyphenylthiocarbamoyloxy)ethyl] -4-(~fluoro-1 ,2-benz-
isoxæol-3-yl)piperidine,
1 -[3-(3,4-Dimethoxyphenylthiocarbamoyloxy)propyl] -4-(6-fluoro-1 ,2-benz-
20 isoxæol 3-yl)piperidine,
1 -12-(3-Chloro-4-methoxyphenyicarbamoyloxy)ethyl]-4-(6-fluoro-1 ,2-benz-
isoxazol-3-yl)piperidine,
1-[2-(3-Chloro-4-methoxyphenylthiocarbamoyloxy)ethyl]-4~(~fluoro-1,2-
benzisoxazol-3-yl)piperidine,
1 -[3-(3-Chloro-4-methoxyphenylthiocarbamoyloxy)propyl]-4-(6-fluoro-1 ,2-
benzisoxæol-3-yl)piperidine.
` The invention also relates to methods of preparing the above mentioned
compounds. These methods include reacting a compound of formula ll
WO 93/10742 C Q 2 1 1 7 3 0 4 - 8 - PCI/DK92/0034X
Y= C= N-R2 (Il)
wherein Y and R2 have the meanings set forth above, with a compound of
formula lll
R ~CN - A - XH
wherein A, X and R1 have the meanings set forth above to form a com-
pound of forr~ula !.
15 For instance an isocyanate or isothiocyanate of 3,4,~trimethoxybenzene,
prepared by refluxing 3,4,5-trimethoxyaniline and phosgene or thiophos-
gene respectively in toluene, may be reacted with the desired piperidine
alkylamine or piperidine alkylhydroxy intermediate to obtain the desired urea
or carbamate of formula 1.
Cornpounds of formula 1, wherein X is -NH- and Y is =NZ wherein Z has the
meanings set forth above are prepared by standard procedures as
described in e.g. H.J. Petersen et al., J.Med.Chem. (1978) 21, 77~781, and
R. Lee Webb et al., J. Heterocyclic Chem. 24, 275 (1987).
The procedure includes rea¢ting a compound of formula IV
~, 1 ~
R--( N-A-NH
\ ( V)
WO 93/10742 PCl`/DK92/00348
CA 2 1 1 73U4 9
wherein A and R' have the meanings set forth above, with a compound of
formula V
N
H3CS-C-NH-R2 (V)
10 wherein R2 and Z have the meanings set forth above, or
reactiny a compound of formula Vl
~ N~ C_NR 2 (Vl)
, .
20 prepared by standard procedures, from a compound of formula Vll
NH-R
1 r~ ~CW
R--~, N - A - NH
2.5 ~ ~.
wherein A, R1 and R2 have the meanings set forth above and W is O or S.
; ~ with NH2-Z, wherein Z has the meaning set forth above, to form a com-
30 pound of formula 1, or
reacting a compound of formula lll, wherein X is -NH- and A and R1 have
:~
WO 93/10742 C A 2 ~ 1 7 3 0 4 pCI'/DK92/0034X
- 10-
the meanings set forth above, with a compound of formula Vlll
~, CN~ N
W~o~NH-R ~Vill)
prepared by the method described in R. Lee Webb and C.S. Labaw, J.,
Heterocyclic Chem. 19, 1205 (1982) from R2-NH2 and N-cyanodiphenoxy-
imidocarbonate.
Compounds of formula lll, wherein R', A and X have the meanings set forth
above, have been prepared b,~ alkylating the known piperidine IX (J.T.Strup-
czewski et ai., J.Med.Chem., 28, 761-769 (1985))
{~ (IX)
wherein R' has the meaning set forth above, using standard procedures.
The compounds of the present invention were tested for binding to various
CNS receptor subtypes as well as for analgesic activity in vitro in mice.
Detaileq conditions forl the in vitro and in vivo assays are described below:
:
IN-VITRO inhibition of DOPAMINE D2 receptor bindinq.
Method description
Principle:
wo 93/l0742 pcr/DK92/oo34x
~A 2 l l 7304 - 11
Radioactive-labelled ligand 3H-Spiroperidol is incubated with isolated cell-
membrane fragmen~s at 37C for a given period of time. Following complet-
ed incubation, the incubate is filtered through GF/B filters which are rinsed
following filtration to remove unspecifically adhered radioactivity. As
opposed to low-molecular compounds, membrane fragments are not rinsed
through the filters, the radioactivity bound to the filters is indicative of theamount of ligand bound specifically as well as nonspeclfically to the mem-
branes.
Rssue PreParatis:~n:
The procedure is perFormed in ice bath. Polytron kinematica is rinsed with
milli-Q-H2O before and after use. Male Wistar r~ts, 150-200 9 are decapitat-
ed, striatum is removed quickly and weighed (approx. 50 mg). Striatum is
transferred to a centrifuging vial containing 10 ml ice-cold D2 buffer. Homo-
genization is performed applying polytron kinematica (homogenker) setting
6 for 20 sec. The homogenizer is rinsed with 10 ml D2 buffer in another
centrifuging vial. The 10 ml rinsing buffer is added to the tissue vial. Centri-fugation at 18,000 rpm for 10 min. at 4C. Final pellet is transferred to 1,000
x vol. of same buffer. (Ex. 50 mg striatum in 50 ml D2 buffer). Can be
stored at 0C for at least 4 hours. Note that the tissue must be monoge-
neous (uniform) before use. If not, brief homogenization is performed.
Assav:
2,500 ~I tissue (homogeneous)
25 ,ul 3H-Spiroperidol (0.05 nM)
25 ~LI test substance/H2O/blind (Domperidone 0.2 ,I~M)
Incubation for 20 mir~. at 37C - 10 min. on ice bath.
.
10 ml ice-cold 0.9% NaCI is added to the tubes and filtered through GF/B
WO 93/10742 PCI/DK92/0034X
~CQ;~ 1 1 7 3`()~ - 12 -
filters (use gloves). This procedure is repeated. The filters are placed in
counting vials and 4 ml opti-flour is added (perform in fume cupboard, use
gloves). Counting is performed at window 0-19 of the beta-counter
(Pachard). Note that receptor box and lid are rinsed thoroughly in H2O after
S use to avoid contamination. Further, the analytical site is cleaned carefully
every day after use.
Test substances:
10 Dissolved in H2O, EtOH, MeOH or DMSO and further diluted in H2O. The
D2 binding will stand concentrations of up to approx. 20% of these solvents
without affecting the binding. Most stock solutions are stable at 4C,
attention is, however, paid to any precipitation, change in colour etc. Test-
substance dilutions are aiways made fresh everv dav. When weighing out
15 test substances, it is attempted to weigh out approx. 1 mg of substance.
Less than 0.8 mg must never be weighed out and only infrequently more
than 2 mg ffor economy reasons), dependent, however, on conc./assay.
Results:
The test value is given as IC50 indicating the concentration inhibiting specificbinding by 50%.
IN-VITRO inhibition of ~1-receptor bind~g,
Method description
P~indDle:
30 Radioactive-labelled ligand 3H-Prazosin is incubated with isolated cell-
rnembrane fragments àt 25C for a given period of time. Following complet-
- ed incubation, the incubate is filtered through GF/B filters, which are rinsed
Wo 93/10742 C A 2 1 1 7 3 0 4 pcr/DK92/oo348
- 13-
following filtration to remove unspecifically adhered radioactivity. As op-
posed to low-molecular compounds, membrane fragments are not rinsed
through the filters, the radioactivity bound to the filters indicates the amountof ligand bound specifically as well as nonspecifically to the membranes.
rlssue preParation:
The procedure is performed in ice bath. Polytron kinematica is rinsed with
milli-Q-H2O before and after use. Male Wistar rats, 150-200 9 are decapitat-
10 ed, cortex is removed quickly and weighed (approx. 500 mg). Cortex istransferred to a centrifuging vial containing 10 ml ice-cold D2 buffer. Homo-
genization applying polytron kinematica (homogenizer) setting 6 for 20 sec.
The homogenizer is rins2d with 10 ml D2 buffer in another centrifuging vial.
The 10 ml rinsing buffer is added to the tissue vial. Centrifugation at 18,000
rpm for 12 min. at 4C. This is repeated once. Final pellet is added to 400 x
vol. of same buffer. (ex. 500 mg cortex in 200 ml D2 buffer). Can be stored
for 30 min. at 0C.
Assay:
2,000 ,ul tissue
25 ~l 3H-Prazosin (0.5 nM)
25 ~I test substance/H20/blind Phentolamine (10 ~LM)
25 Incubation for 30 min. at 25C.
10 ml of ice-cold 0.9% NaCI is added to the tubes and filtered through GF/B
filters (use gloves). This procedure is repeated. Filters are placed in count-
ing vials and 4 ml opti-flour is added (perform in fume cupboard, use
30 gloves). Counting is performed at window ~19 of the beta-counter
(Pachard). Note that~receptor box and cover are rinsed thoroughly in H2O
- after use to avoid contamination. Further, the analytical site is cleaned
WO 93flO742 C A 2 1 1 7 3 0 4 PCr/DK92/0034~
- 14 -
carefully every day after use.
Test substances:
5 Dissolved in H2O, EtOH, MeOH or DMSO and further diluted in H2O. The
binding will stand concentrations of up to approx. 5% of $hese solvents
withoLn affecting the binding. Most stock solutions are stable at 4C.
Attention is, however, paid to any precipitation, change in colour etc. Test-
substance dilutions are alwavs made fresh everv dav. When weighing out
10 test substances, it is attempted to weigh out approx. 1 mg of substance.
Less than 0.8 mg must never be weighed out and only infrequently more
than 2 mg (for economy reasons), dependent, however, on conc./assay.
Results:
The test value is given as IC50 indicating the concentration inhibiting specificbinding by 50%.
IN-VITRO inhibition of DOPAMINE D1 receptor bindinq,
Method description
Principle:
Radioactive-labelled li~and 3H-SCH 23390 is incubated with isolated cell-
membrane fragments in incubation buffer at 30C for a given penod of time.
Following completed incubation, the incubate is filtered through GF/B filters,
which are rinsed following filtration to remove unspecifically adhered ra-
dioactivity. As opposed to low-molecular compounds, membrane fragments
are not rinsed through the filters, the radioactivity bound to the filters
indicates the amount of ligand bound specfflcally as well as nonspec~lcally
to the membranes.
WO 93/10742 C A 2 1 1 7 3 0 4 PCI/DK92/00348
rlssue preparation:
Male Wistar rats, 150-200 g are decapitated. Striatum is removed quickly,
weighed (approx. 50 mg) and carefully homogenized in 100 x vol. of buffer I
5 applying glass/teflon homogenizer 10 up/down strokes. Ex.: 50 mg striatum
is homogenized in 5,000 ~I buffer 1. The homogenate is centrifuged at
18,000 rpm for 20 min. at 4~, and the supernate is decanted. This step is
performed three times, and each time the pellet is resuspended and
homogenized in 100 x vol. of buffer 1. Following the third centrifugation, t~he
10 pellet is suspended in 100 x vol. of resuspension buffer and homogenized.
The tissue is now ready for use. The tissue is stable at 0C for 8 hours.
Assay:
15 600 ,ul incubation buffer
100 ~LI 3H-SCH 23390 (0.2 nM)
100 ,ul tissue
200 ,ul test substance/H20/blind (cis-flupentixol 2 ~M)
20 Incubation for 60 min. at 30C.
10 ml of ice-cold 0.9% NaCI is added to the tubes. Filtration is performed
through GF/B filters (use gloves). This procedure is repeated. Filters are
placed in counting vials and 4 ml opti-flour is added (perform in fume
25 cupboard, use gloves) and counting is performed at window ~19 of the
beta-counter (Pachard). Note that receptor box and lid are rinsed thorough-
ly in H2O after use to avoid contamination. Further, the analytical site is
cleaned carefully every day after use.
;
30 Test substances:
Dissolved in H2O, EtOH, MeOH or DMSO and further diluted in H2O. The
WO 93/10742 C A 21 1 7 3 0 4 PCI'JDK92/0034g
- 16-
D1 binding will stand concentrations of up to approx. 20% of these solvents
without affecting the binding. Most stock solutions are stable at 4C.
Attention should, however, be paid to any precipitation, change in colour
etc. Test-substance di!utions are alwavs made fresh everv dav. When
5 weighing out test substances, it is attempted to weigh out approx. 1 mg of
substance. Less than 0.~ mg must never be weighed out and only in-
frequently more than 2 mg (~or economy reasons), dependent, however, on
conc./assay.
1 0 Results:
The test value is given as IC50 indicating the concentration inhibiting specificbinding by 50%.
15 IN VITRO inhibition of 5Hr2-reoeptor bindinq
Method descri~tion
Princip!e:
Radioactive-labelled ligand 3H-Ketanserine is incubated with isolated cell
membrane fragments at 37C for a given period of time. Following comptet-
ed incubation, the incubate is filtered through GF/B filters, which are rinsed
following filtration to remove unspecifically adhered radioactivity. As op-
25 posed to low-molecular compounds, membrane fragments are not rinsed
through the filters, the radioactivity bound to the filters indicates the amountof ligand bound specifically as well as nonspecifically to the membranes.
rlssue preparation:
The preparation is made in ice bath. Polytron kinematica is rinsed with milli-
Q-H2O before and after use. Male Wistar rats, 15~200 9 are decapitated.
WO 93/10742 PCI/DK92/0034X
CA2 1 1 7304
Frontal cortex is removed quickly and weighed (approx. 200 mg). Frontal
cortex is added to centrifuging vial containing 10 ml ice-cold D2 buffer.
Homogenization applying polytron kinematica (homogenizer) setting 6 for
20 sec. The homogenizer is rinsed with 10 ml D2 buffer in another centrifug-
5 ing vial. The 10 ml rinsing buffer is added to the tissue vial. Centrifuged at18,000 rpm for ~ 0 min. at 4C. Final pellet is transferred to 125 x vol. of
same buffer. (Ex 200 mg in 25 ml D2 buffer). Can be stored for approx. 30
min~ at 0C.
1 0 Assay:
1250 ~LI tissue
25 ,ul 3H-Ketanserine (0.4 nM)
25 ,ul test substance/H20/blind cyproheptadine (2 ~M)
Incubation for 15 min. at 37C.
10 ml ice-cold 0.9% NaCI is added to the tubes. Filtration is performed
through GF/B filters (use gloves). This procedure is repeated. The filters are
20 placed in counting vials and 4 ml opti-flour is added (prepare in fume
cupboard, use gloves). Counting at window 0-19 of the beta-counter
(Pachard). Note that receptor box and lid are rinsed thoroughly in H2O after
use to avoid contamination. Further, the analytical site is cleaned carefully
every day.
Test substances:~ ~
; Dissolved in H2O, EtOH, MeOH or DMSO and further diluted in H2O. The
5HT2 binding will stand concentrations of up to approx. 5% of these sol-
30 vents without affecting the binding. Most stock solutions are stable at 4C.
Attention should, however, be paid to any precipitation, change in colour
etc. Test-substance dilutions are always made fresh eve day. When
WO 93/10742 C A 2 1 1 7 3 0 4 PCI/DK92/0034X
- 18-
weighing s)ut test substances, it is attempted to weigh out approx. 1 mg of
substance. Less than 0.8 mg must never be weighed out and only in-
frequently more than 2 mg (for economy reasons), dependent, however, on
conc./assay.
The test value is given as ICso i.e. the concentration inhibiting specific
binding by 50%.
Antagonism of acetic add-induced
writhin~s in mice
Principle:
In mice i.p. injection of acetic acid induces a writhing syndrome which is
antagonized by analgesics (Siegmund et al., 1957; Eckhardt et al., 1957).
Me~od:
Acetic acid 0.5 per cent is injected i.p. (0.15 ml/10 9 body weight) to 6 mice
(NMRI, either sex weighing 20-25 9) pretreated with physiological saline
(controls) and to 6 mice pretreated with test substance. In the controls
acetic acid induces a syndrome characterized by contraction of abdomen,
25 turning of trunk and extension of hind limbs. Saline and test substances are
administered s.c. 30 min. before acetic acid. The number of writhings is
counted 5-15 min. after injection of acetic acid.
Resutts:
Initially, the dose of test substance is equivalent to 5-10 per cent of LDso~ If- this dose decreases writhings, 3-5 dose levels are tested. The activity is
WO93/10742 CA2 1 1 7304 PCl/DK92/0034X
- 19-
.
expressed as per cent protection:
100 avera~e writhings of treated group x 100
average writhings ot daily control groups
The effect of active substances is evaluated by a dose response curve, log
dose on the abscissa, and per cent protection on the ordinate. The potency
is expressed as the dose (E~D50 in mg/kg) giving 50 per cent protection
against writhings.
Specificitv of test:
Analgesics and various other drugs inhibit acetic acid-induced writhings in
mice. This test is used as a screening test for analgesics. Additional results
15 from other screening tests are required to exclude activc anti-writhing
substances without analgesic effect.
References:
20 Eckhardt, E. et al.
Ethiology of chemically induced writhing in mouse and rat. Proc. Soc. exp.
Biol. 98, 186-188, 1958.
Siegmund, E. et al.
25 A method for evaluating both non-narcotic and narcotic analgesics. Proc.
Soc. exp. Biol. 95, 729-731,1957.
The compounds of this invention typically binds to NA-~l, 5HT2-, DA-D1-,
` and DA-D2-receptors, with IC50 values in the order of 0.1 nM to 1 ,~LM.
30 Furthermore the cornpounds are able to antagonize the acetic acid induced
writhing in mice with ED50-values typically in the order of 0.1 mg/kg to 100
- mg/kg~
`~ ~
WO 93/1074:2 C A 2 1 1 7 3 0 4 PCl`/DK92/0034X
- 20-
The compounds of the invention, together with a conventional adjuvant,
carrier, or diluent, and if desired a pharmaceutically acceptable acid addi-
tion salt thereof, may be placed into the form of pharmaceutical composi-
tions and unit dosages thereof, and in such form may be employed as
5 solids, such as tablets or filled capsules, or liquids, such as solutions, sus-
pensions, emulsions, elixirs, or capsules filled with the same, ali for oral use,
in the form of suppositories for rectal administration; or in the form of sterile
injectable solutions for parenteral (including subcutaneous) use. Such
pharmaceutical compositions and unit dosage forms thereof may comprise
10 conventional ingredients in conventional proportions, with or without additio-
nal active compounds or principles, and such unit dosage forms may
contain any suitable effective central nervous system ailment alleviating
amount of the active ingredient commensurate with the intended daily
dosage range to be employed. Tablets containing one (1) milligram of
15 active ingredient or, more broadly, one (1) to thirty (30) milligrams, per
tablet, are accordingly suitable representativé unit dosage forms.
The compounds of this invention can thus be used for the formulation of
pharmaceutical preparations, e.g. for oral and parenteral administration to
20 mammals including humans, in accordance with conventional methods of
galenic pharmacy.
Conventional excipients are such pharmaceutically acceptable organic or
inorganic carrier substances suitable for parenteral or oral application which
25 do not deleteriously react with the active compound.
Examples of such carriers are water, salt solutions, alcohols, polyethylene
glycois, polyhydroxyethoxylated castor oil, gelatine, lactose, amylose,
magnesium stearate, talc, silicic acid, fatty acid monoglycerides and digly-
30 cerides, pentaerythritol tatty acid esters, hydroxymethylcellulose and polyvi-
nylpyrrolidone.
wO 93t10742 pcr/DK92/oo34x
CA21 1 7304 21 -
The pharmaceutical preparations can be sterilized and mixed, if desired,
with auxiliary agents, such as lubricants, preservatives, stabilizers, wetting
agents, ernulsifiers, salt for influencing osmotic pressure, buffers and/or
coloring substances and the like, which do not deleteriously react with the
5 active compoun~s.
For parenteral application, particularly suitable are injectable solutions or
suspensions, preferably aqueous solutions with the active compound
dissolved in polyhydroxylated castor oil.
Ampoules are convenient unit dosage forms.
For oral application, particularly suitable are tablets, dragees, or capsules
having talc andtor a carbohydrate carrier or binder or the like, the carrier
15 preferably being lactose and/or corn starch and/or potato starch. A syrup,
elixir or like can be used when a sweetened vehicle can be employed.
Generally, as to broader ranges, the compound of the invention is dis-
pensed in unit dosage form comprising 0.05-100 mg in a pharmaceutically
acceptable carrier per unit dosage.
A typical tablet which may be prepared by conventional tabletting tech-
niques contains:
Active compound 1.0 mg
Lactosum 67.8 mg Ph.Eur.
Avicel~ 31.4 mg
Amberlite~ IRP 88 1.0 mg
Magnesii stearas 0.25 mg Ph.Eur.
`~:
30 The following examples illustrate the specific methods employed in produc-
tion of a representative number of compounds embraced by this invention.
~::
WO 93/10742 PCI/DK92/00348
CA21 1 730~
EXAMPLE 1
4-(6-Fluoro-1 ,2-benzisoxæol-3-yl)-1 -13-(3,4,5-trimethoxyphenylcarbamo-
- yloxy)propyl]piperidine, oxalate
A. 4-(6-Fluoro-1,2-benzisoxazol-3-yl)piperidine, hydrochloride (5.0 g, 20
rnmol), 3-bromopropanol (2.0 ml, 21.6 mmol) and potassium carbonate (6.5
g, 47 mmol) in 300 ml dry acetone were refluxed for 16 h. The mixture was
10 cooied to room temperature, filtered and concentrated in vaeuo. The
resulting compound was recrystallized from ethanoltwater to give 4.3 9 of
the desired compound. M.p. 139-141C.
B. A mixture of 3,4,5-trimethoxyaniline (365 mg; 2.0 mmol) in toluene
(20 ml) and phosgene (6 ml 20% in toluene; 12 mmol) was refluxed for 6 h.
The solvent was removed under reduced pressure to give crude 3,4,~
trimethoxyphenylisocyanate. To the crude product was added 3(4-(~fluor~
1,2-benzisoxazol-3-yl)-piperidino)propanol (420 rrg; 1.5 mmol) in DMF (10
ml). The mixture was stirred at 1 00C for 2 h and then at room temperature
for 16 h, whereupon it was taken up in ethyl acetate and waten The organic
phase was washed with water and saturated sodium chloride and con-
centrated in vacuo~ The resulting oil was taken up in acetone/ethanol (4:1,
V/V) and oxalic acid (150 mg~ in 2 ml acetone added to precipitate the
desired product. The product was washed wlth ice cold ethanol giving 550
mg of the title compound. M.p. 77-80C. MS (70 eV): m/z 487 (9%, M+), 287
(31), 233 (40), 209 (56), 194 (45), 140 (67), 96 (100).
EXAMPLE 2
4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-[3-(3,4-ethylenedioxyphenylcarbamo-
yloxy)propyl]piperidine, oxalate
: `
A mixture of 1,4-benzodioxan-6-amine (300 mg; 2.0 mmol) in toluene (20
WO 93/10742 PCT/DK92/00348
CA21 1 7304 23-
ml) and phosgene (10 ml 20% in toluene; 19 mmol) was refluxed for 6 h.
The solvent was removed under reduced pressure to give crude 3,4
ethylenedioxyphenylisocyanate. To the crude product was added 3-[4-(~
fluoro-1,2-benzisoxazol-3-yl)piperidino~propanol (420 mg; 1.5 mmol) in DMF
(10 ml). The mixture was stirred at 100C for 2 h and then at room tempera-
ture for 16 h, whereupon it was taken up in ethyl acetate and water. The
organic phase was washed with water and saturated sodium chloride and
concentrated in vacuo. The resulting oil was taken up in acetone/ethanol
(4:1, v/v) and oxalic acid (150 mg) in 2 ml acetone added to precipitate the
desired product. The product was washed with ice cold ethanol to give 600
mg of the title compound. M.p. 109-110C. MS (70 eV): m/z 455 (32%, M+),
278(23),233 (49), 177 (89), 140 (48), 121 (42), 96 (100).
EXAMPLE 3
1 -[3-(6-Benzothiazolylcarbamoyloxy)propyl]-4-(~fluoro-1,2-benzisoxæol-3-
yl)piperidine, oxalate
.. . . _ .... _ _ _ _
A mixture of 6-aminobenzothiæole (300 mg; 2 mmol) in toluene (20 ml) and
phosgene (10 ml 20% in toluene, 19 mmol) was refluxed for 6 h. The
solvent was removed under reduced pressure to give crude ~benzothiazo-
Iylisocyanate. Using the procedure of example 1 the crude 6-benzothiazoiyl-
isocyanate was combined with 3-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-piperi-
dino~propanol (410 mg; 1.5 mmol) in 10 ml DMF to give the 0.7 9 of the title
compound. M.p. 176-177C. MS (70 eV): m/z 454 (3%, M+), 27 (25), 233
(30j, 190 (17), 176 (55), 150 (37), 140 (53), 96 (100).
EXAMPLE 4
30
1 -[3-(3,4-Ethylenedioxyphenylthiocarbamoyloxy)propyl]-4(~fluoro-1,2-
benzisoxazol-3-yl)piperidine, oxalate
. _
WO 93/10742 C A 2 1 1 7 3 04 PCr/DK92/0034X
- 24 -
To a mixture of 1,4-benzodioxan-6-amine (1~.1 g; 100 mmol) and triethyl-
amine (20.2 g; 200 mmol) in toluene (350 ml) was added dropwise over 10
min. thiophosgene (11.5 g; 100 mmol) in toluene (50 ml). The mixture was
stirred at 80C for 30 min., cooled to room temperature and filtered. The
5 filtrate was evaporated. The product was redissolved in toluene and con-
centrated in vacuo. The resulting oil was taken up in warm petroleum ether,
which was filtered. The filtrate was concentrated to a small volume, which
afforded 7.2 9 of 3,4-ethylenedioxyphenylisothiocyana~e.
Starting from 3,4-ethylenedioxyphenylisothiocyanate (390 mg; 2.0 mmol)
and 3-[4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidino]propanol (420 mg; 1.5
mmol~ using the procedure described in example 1 was prepared 550 mg
of the title compound: M.p. 101-104C. MS (70 eV): m/z 471 (0.5%, M+),
278 (61), 233 (58), 193 (100), 151 (17), 140 (60).
EXAMPLE 5
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -[2-(3,4,5-trimethoxyphenylcarbamoyl-
oxy)ethyl]piperidine, oxalate
A. 4-(6-Fluoro-1,2-benzisoxazol-3-yl)piperidine, hydrochloride (2.6 9, 10
mmol), 2-bromoethanol (1.3 ml, 15 mmol) and potassium carbonate (4.1 g,
30 mmol) in 25 ml dry acetone were refluxed for two hours and then stirred
at 60C for 16 h, whereupon extra 0.4 ml (5 mmol) 2-bromoethanol was
added. The mixture was then refluxed for 4 h, cooled to room temperature,
concentrated~ in vacuo and taken up in water and methylene chloride. The
,
organic phase was washed with water and saturated sodium chloride, dried
over MgSO4 and concentrated in vacuo. Purification by column chromato-
graphy (silica gel; methylene chloride:methanol:conc. ammonium hydroxide
(80:20:0.5, v/vlv)) gave 2.2 g of 2-[4(6-fluoro-1,2-benzisoxazol-3-yl)piperidi-
no]ethanol. M.p. 119-120C.
WO 93/10742 C A 2 1 1 7 3 04 PCl`/DK92/0034X
- 25 -
B. Starting from 3,4,5-trimethoxyphenylisocyanate (600 mg, 3 mmol)
and 2-[4-(6 fluoro-1,2-benzisoxazol-3-yl)-piperidino]ethanol (400 mg; 1.5
mmol) using the procedure described in example 1 was prepared 450 mg
of the title compound. M.p. 158-160C. MS (70 eV): m/z 473 (32%, M+), 246
(38), 233 (100), 209 (41).
EXAMPLE 6
1 -(3-(6-Benzthiazolylthiocarbamoyloxy)propyl)-4-(6-fluoro-1,2-benzisox~ol-3-
10 yl)piperidine, oxalate
.. . . .
Using the procedure described in example 4 starting from 3-[4-(6-fluoro-1,2-
benzisoxazol-3-yl)piperidino]propanol (280 mg, 1.0 mmol) and 6-benzothia-
zolylisothiocyanate (210 mg, 12 mmol), prepared from ~aminobenzothiaz~
le and thiophosgene, was prepared 280 mg of the tltle compound. M.p.
108-112C, MS (70 eV): m/z 470 (0.2%, M+), 278, (38), 233 (30), 192 (62),
150 (65), 140 (75), 96 (100).
E)CAMPLE 7
4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1 ^(3-(3,4-methylenedioxyphenylcarbamoyl-
oxy)propyJ)piperidine, oxalate
Using the procedure described in example 1 starting from 3-[4-(6-fluoro-1,2-
benzisoxazol-3-yl)piperidino]propanol (140 mg, 0.5 mmol) and 3,4methyl-
enedioxyphenylisocyanate (240 mg, 1.5 mmol), prepared from 3,4-methyl-
enedioxyaniline and phosgene, was prepared 210 mg of the title com-
pound. M.p. 133-136C. MS (70 eV): m/z 441 (20%, M+), 303 (15), 278 (16),
233 (53), 163 (52), 140 (37), 96 (100).
WO 93/10742 PCI/DK92/00348
CA211730~ -26-
EXAMPLE 8
1 -[2-(6-Benzothiazolylcarbamoyloxy)ethyl]-4-(6-fluoro-1,2-benzisoxæol-3-
yl)piperidine, oxalate
Using the procedure of example 3 crude ~benzothiazolylisocyanate, pre-
pared from 6-aminobenzothiazole (500 mg, 3.4 mmol), was combined with
2-[4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidino]ethanol (450 mg, 1.7 mmol) in
10 ml dry DMF to give 100 mg of the title compound. M.p. 13~134C. MS
(70 e\/): m/z 440 (1%, M+), 264 (23), 233 (69), 150 ~100).
E~CAMPLE 9
4-(~Fluoro-1,2-benzisoxazol-3-yl)-1 -[2-(3,4-methylenedioxyphenylcarbamoyl-
oxy)ethyl]piperidine, hydrochloride
.
Starting from 3,4-methylenedioxyphenylisocyanate (320 mg, 2 mmsl) and 2-
[4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidino]ethanol (270 mg, 1 mmol) in 5
ml dry DMF using the procedure described in example 1 was prepared 210
mg of the title compound as free base. This product was dissolved in 5 ml
ethanol/acetone (50%, v/v) and ethanolic hydrochloric acid added to
precipitate 180 mg of the desired product as white crystals. M.p. 22~229C.
MS (70 eV): m/z 428 (47%, M+), 246 (42), 233 (100), 208 (21), 190 (47), 163
(70).
E)(AMPLE 10
4-(6-Fluoro-1,2-benzisoxæol-3-yl)-1-[3-(phenylcarbamoyloxy)propyl]piperidi-
ne, hydrochloride
Phenylisocyanate (0.36 g, 3 mmol) and 3-[4-(~fluoro-1,2-benzisoxæol-3-
yl)piperidino]propanol (0.3 9, 1.1 mmol) was refluxed in toluene (25 ml) for
:
WO 93/10742 PCI/DI~92/0034X
CA2117304 27-
6 h. The mixture was cooled to room temperature and hydrochloric acid in
ether was added. The resulting precipitate was recrystallized from etha-
nol/ether and isopropanol/ether to give 180 mg of the title compound as
white crystals. M.p. 204.5-205.5C. MS (70 eV): mtz 397 (39%, M+), 278 (4),
259 (26), 233 (50), 178 (28), 96 (100).
EXAMPLE 1 1
N-Cyano-N'-(3,4-methylenedioxyphenyl)-N"-3-((6-fluoro-1 ,2-benzisoxazol-~
10 yl)piperidino)propyl)guanidine, oxalate
A mixture of N-cyanodiphenoxyimidocarbonate (1.2 g, 5 mmol), 3,4-methy-
lenedioxyaniline (0.7 9, 5 mmol) and 2-propanol (25 ml) was stirred at room
15 temperature for 16 h. The formed precipitate was taken up in methylene
chloride, treated with activated carbon. Evaporation of the solute and
trituration with ether gave 1.2 g of N-cyano-N'-3,4-methylenedioxyphenyl-O-
phenylisourea. M.p. 172-174C.
1-(3-Aminopropyl)-4-(6-fluoro-1 ,2-benzisoxazol-3-yl)piperidine (420 mg, 1.1
mmol), N-cyano-N'-3,4-methylenedioxyphenyl-O-phenylisourea (320 mg, 1.2
mmol), 0.4 ml triethyl amine and 25 ml 2-propanol were stirred at room
temperature for 4 days. The mixture was concentrated in vacuo and then
taken up in water and methylene chloride. The organic phase was washed
with water and saturated sodium chloride, dried over magnesium sulphate
and concentrated in vacuo. The product was purified by column chroma-
tography (silica gel; ethyl acetate:methanol (4:1, v/v)),~ and then taken up in
2 ml dry acetone. Oxalic acid (50 mg) in 1 ml acetone was added precipi-
tating 90 mg of the desired product as white crystals. M.p. 112-115C. MS
~70 eV): m/z 464 (M~, 1%), 422 (4), 233 (10), 220 (32), 137 (100).
\
WO 93/10742 PCI/D~C92/0034X
`C~ ~ 1 .1 i3 04 28 -
E)(AMPLE 12
1-[3-(3,4-Methylenedioxyphenylcarbamoyloxy)propyl]-4-(6-fluoro-1 H-indæol-
3-yl)piperidine
A. To a mixture of 6-fluoro-3-(4-piperidinyl)-1 H-indazole (438 mg, 2
mmol) and dry potassium carbonate (1.1 9, 6 mmol) in 30 ml methyl
isobutyl ketone was added 3-bromo-1-propanol (276 mg, 2 mmol). The
mixture was refluxed for 48 h, cooled, filtered and concentrated in vacuo.,
The crude product was purified by chromatography on silica gel 60 eluting
with ethyl acetate:methanol (9:1, v/v). Concentration of the appropriate
fractions afforded 300 mg (53%) of 3-[1-(1-hydroxyprop-3-yl)-4-piperidinyl]-
6-fluoro-1 H-indazole as an oil.
H-NMR (DMSO-d6, ~): 1.62 (t, 2H), 1.7-2.1 (br., 8H)f 2.39 (t, 2H), 2.95 (br.,
3H), 3.48 (t, 2H), 6.90 (dt, 1H), 7.21 (dd, 1H), 7.78 (q, 1H), 12.70 (s, 1H).
B. A solution of 3-[1-(1-hydroxyprop-3-yl)-4-piperidinyl]-6-fluoro-1H-
indazole (230 mg, 0.83 mmol) in 5 ml dry DMF was added 3,4methylenedi-
oxyphenylisocyanate (291 mg, 1.65 mmol) in 3 ml dry DMF. The reaction
was heated to 100C for 2 h and 16 h at 80C. The reaction was cooled to
room temperature and added a mixture of 50 ml water and 200 ml ether,
filtered and separated. The ether phase was washed with water, brine and
dried with sodium sulphate and concentrated in vacuo. The crude product
was purified by chromatography on silica gel 60 eluting with ethyi
acetate:rnethanol (9:1, vh). Concentration of the appropriate fractions~
afforded 50 mg (11%) of the title compound as an amorphous solid.
'H-NMR (DMSO d6, ~): 1.71-1.92 (m, 6H), 2.1 (m, 2H), 2.42 (br., 2H), 2.98
(br.d, 3H), 4.11 (t, 2H), 5.95 (s, 2H), 6.82 (m, 2H), 6.91 (t, lH), 7.14 (s, 1H),
7.21 (dd, 1H), 7.80 (dt, jH), 9.5 (s, 1H), 12.68 (s, 1H).
WO 93/10742 C A 2 1 1 7 3 0 4 PCI/DK92/0034X
- 29 -
Analysis: Cz3H2sN4O4F, 0.75 H2O:
Calculated: C 60.85; H 5.88; N 12.34%
Found: C 60.69; H 5.76; N 12.31%
MS (70 eV): m/z 440 (M+, 1%), 277 (14), 232 (100), 218 (12), 189 (37), 163
(80), 70 (16).
EXAMPLE 13
1-[2-(3,4-Methylenedioxyphenylcarbamoyloxy)propyl]-4(6-fluoro-1 H^indazol-
3-yl)piperidine
A. A mixture of 6-fluoro-3-(4-piperidinyi)-11 t-indazole (500 mg, 2.3
mmol) and propylene oxide (1 g, 17 mmol) in 25 ml toluene was heated to
50C in an autoclave for 7 days. The cooled reaction was concentrated in
vacuo and purified by chromatography on silica gel 60 eluting with ethyl
acetate:methanol (4:1, v/v). Concentration of the appropriate fractions
afforded 350 mg (54.9%) of 3-[1-(2-hydroxyprop-1-yl)-4-piperidinyl]-~fluoro-
20 1 H-indazole as a foam.
1H-NMR (DMSO-d6, ~): 1.04 (d, 3H), 1.87 (m, 4H), 2.18 (m, 4H), 2.93 (br. d,
3H)t 3.78 (m, 1H), 4.25 (br., 1H), 6.91 (dt, 1H), 7.20 (ddl 1H), 7.78 (q, 1H),
12.58 (s, 1H).
B. Starting from 3-[1-~2-hydroxyprop-1-yl)-4-piperidinyl]-6-fluoro-1H-
indazole (300 mg, 1.1 mmol) and 3,4-methylenedioxyphenylisocyanate (380
mg, 2.2 mmol) using the procedure described in example 12A was pre-
pared 40 mg (9%) of the title compound as an amorphous solid.
1H-NMR (DMSO-d6, ~): 1.21 (d, 3H), 1.85 (m, 4H), 2.20 ~m, 2H), 2.4 (m, 1H),
2.55 (m, 1H), 3.0 (m, 2H), 3.1 (m, 1H), 4.9 (m, 1H), 5.98 (s, 2H), 6.8 (d, 1H),
6.89 (m,2H), 7.14 (s, 1H), 7.19 (dd, 1H), 7.73 (dt, 1H), 9.51 (s, lH), 12.68 (s,
WO 93/10742 PCI/DK92/0034f~
CA21 17304 -30-
1H).
MS (70 eV): m/z 441 (M++, 1.1%), 440 (M+, 1), 259 (29), 232 (100), 218 (7),
189 (48~, 163 (41), 137 (15).
EXAMPLE 14
1 -[6-(3,4-Methylenedioxyphenylcarbamoyloxy)hexyl]-4-(6-fluoro-1 ,2-benzis-
oxæol-3-yl)piperidine
A. A mixture of 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole
hydrochloride (1 g, 3.9 mmol), lithium carbonate (865 mg, 1.7 mmol) and 6-
chloro-1-hexanol (534 mg, 3.9 mmol) in 5 ml dry DMF was heated at 100C
for 48 h. The reaction was poured into water and the aqueous mixture was
extracted with ethyl acetate. The ethyl acetate phase was washed with
water, brine, dried with sodium sulphate and concentrated in vacuo. The
crude product was purffled by chromatography on silica gel 60 eluting with
ethyl acetate:methanol (9:1, v/v). Concentration of the appropriate fractions
afforded 310 mg (25.6%) of 3-11-(1-hydroxyhex-6-yl)-4-piperidinyl]-6-fluoro-
1,2-benzisoxazole as an oil.
H-NMR (CDCI3, ~): 1.39 lm, 4H), 1.58 (m, 4H), 2.10 (m, 6H), 2.41 (t, 2H),
3.09 (br.d, 3H), 3.65 (t, 2H), 7.04 (dt, 1H), 7.23 (dd, 1H), 7.75 (q, 1H).
MS (70 eVj: m/z 320 (M+, 28%), 233 (47), 190 (15), 182 (50), 96 (10G), 82
(100),~ 82,(21)i 5S ~23). /
B. Starting from 3-~1-(1-hydroxyhex-6-yl]-4-piperidinyl]-~fluoro-1,2-
benzisoxæole (270 mg, 0.84 mmol) and 3,4-methylenedioxyphenylisocya-
:
nate (297 mg, 1.69 mmol) using the procedure descrjbed in example 12B
was prepared 210 mg (51.8%) of the title compound as an amorphous
solid.
~'~
WO 93/10742 . PCr/DK92/0034X
C~21 1 7304 31
H-NMR (CDCI3, ~): 1.4 (m, 4H), 1.6 (m, 4H), 2.11 (m, 6H), 2.42 (t, 2H), 3.09
(br.d, 3H), 4.1~ (t, 2H), 5.95 (s, 2H), 6.70 (m, 2H), 7.05 (m, 2H), 7.24 (m,
2H), 7.72 (q, 1H).
MS (70 eV): m/z 483 (M~, 1.5%), 320 (22), 233 (45), 182 (32), 163 (100),
130 (7~), 96 (70), 77 (35).
EXAMPLE 15
4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-(3-(2-bromo-4,5-methylenedioxyphenyl-
carbamoyloxy)propyl)piperidine, hydrochloride
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -(3-(3,4-methylenedioxyphenylcarbamoyl-
oxy)propyl)piperidine (example 7) (220 mg, 0.5 mmol) was dissolved in 2 ml
glacial acetic acid and the mixture stirred at room temperature under N2.
Br2 ~0.25 ,ul, 0.5 mmol) dissolved in 1.0 ml glacial acetic acid was added.
Th~ mixture was stirred for 2 h whereupon aqueous K2C03 was added to
neutralize the solution, which was then extracted with ethyl acetate. The
organic phase was dried over magnesium sulphate and concentrated in
vacuo. The product was taken up in acetone and HCI in ether added to
aystallize the desired product as 40 mg white crystals. M.p. 21~218C. MS
(70 eV): m/z ~21 (17%, M+), 519 (17%, M+), 278 (20), 243 (35), 241 (34~,
233 (56), 96 (100).
E)CAMPLE 16
4-(6-Fluoro-1 ,2-benzisoxazol-3-yl)-1 -(3-(3,4-methylenedioxyphenylthiocarba-
moyloxy)propyl)piperidine, hydrochloride
; ~ 3,4-Methylenedioxyphenylisothiocyanate (360 mg, 2 mmol) and 3-[4-(6-
fluoro-1,2-benzisoxazol-3-yl)piperidino]propanol (4.20 mg, 1.5 mmol) in 5 ml
dry DMF were stirred at 100C for 2 h and then at 60C for 16 h. The
~`
i ~ ` ' ,,i :
WO 93/10742 PCl/DK92/0034X
CA2 1 1 7304 32 -
mixture was cooled to room temperature and taken up between water and
ether. The organic phase was washed with water and saturated sodium
chloride, dried over magnesium sulphate and concentrated in vacuo.
Purification of the product by columr) chromatography (silicagel; ethyl
5 acetate:methanol (4:1, v/v)) gave an oil, which was taken up in dry acetone.
HCI in ether was added to crystallize the desired product as 550 mg white
crystals. M.p. 181-185C. MS (70 eV): m/z 457 (1%, M+), 278 (55), 233 (56),
179 (100)-
EXAMPLE 17
4-(B-Fluoro-1,2-benzisoxazol-3-yl)-1 -(3-(2-methoxyphenyl)carbamoyloxy)-
propyl)piperidine, hydrochloride
2-Methoxyphenylisocyanate (270 mg, 2 mmol) and 3-[~(6-fluoro-1,2-benz-
isoxazol-3-yl)piperidino]propanol (280 mg, 1 mmol) was dissolved in 100 ml
dry toluene and refluxed for 16 h. 50 ml ethyl acetate was added to the
cooled mixt-Jre, which was then washed with water and saturated sodium
20 chloride. 2.5 ml (1.9 M) HCI in ethanol was added and the solution was
concentrated to about 30 ml crystallizing 310 mg of the desired product.
M.p. 174-175C. MS (70 eV): mlz 427 (30%, M~), 289 (19), 233 (33), 96
(1 00) .
EXAMPLE 18
4-(6-Fluoro-1,2-benzisoxazol-3-yl~-1 -(3-(3-chloro-4-methoxyphenyl3carbamo-
yloxy)propyl, hydrochloride
Starting from 3-chloro-4-methoxyphenylisocyanate, prepared from 3-chloro-
4-methoxyaniline (470 mg, 3 mmol) and phosgene (7.5 mmol) as described
in examp!e 1, and 3-~4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidino]propanol
(560 mg, 2 mmol) using the procedure of example 1, was prepared 500 mg
wo 93/1074Z C A 2 1 1 7 3 0 4 Pcr/DKg2~003-x
mg of the desired product. M.p. 212-21 5C.
EXAMPLE 19
1-[2-(3,4-Methylenedioxyphenylcarbamoyloxy)propyl]-4-(6-fluoro-1,2-benz-
isoxazol-3-yl)piperidine, oxalate
A. A mi~ture of 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazol (1.5 9, 6.8
mmol) and propylene oxide (2 9, 34.4 mmol) in 25 ml acetonitrile was
heated to 50C in an autoclave for 3 days. The cooled reaction was con-
centrated in vacuo and purified by chromatography on silica gel 60 eluting
with ethyl acetate:methanol (9:1, v/v). Concentration of the appropriate
fraction afforded 1.3 9 (68.4%) of 3-[1-(2-hydroxyprop-1-yl)-4-piperidinyl~-6-
fluoro-1,2-benzisoxazole. M.p. 45-47C. MS (70 eV): m/z 278 (M~, 18%), 233
(100), 190 (28), 109 (12), 96 (70), 82 (25), 68 (14), 55 (22).
B. Starting from 3-[1-(2-hydroxyprop-1 -yl)-4piperidinyl~-6-fluoro-1,2-
benzisoxæole (500 mg, 1.1 mmol) and 3,4-methylenedioxyphenylisocyanate
(500 mg, 2.2 mmol) using the procedure described in example 12B was
prepared 100 mg (17%) of the title compound. M.p. 163-164C. MS (70 eV):
m/z 441 (M~, 11%), 260 (32), 233 (100), 190 (25), 163 (17), 136 (33), 96
(58).
Analysis: C2sH26N3Fo9~ 0-5 H20
Calculated: C 55.55; H 5.03; N 7.77%
Found: C 55.74; H 4.91; N 7.39%