Note: Descriptions are shown in the official language in which they were submitted.
'~O 93/08166 2 1 1 ~ PCT/US92/07314
BENZO-ISOQUINOLINE DERIVATIVES AND ANAL0GS AND
THEIR USE IN THERAPEUTICS
The present invention relates to novel compositions of matter. More particularly, the
present invention relates to new isoguinoline and pyridine derivatives, processes for preparing these
S compounds, and pharmaceutical compositions containing these compounds. The present invention
further relates to the use of these compounds in the treatment of certain psychiatric disorders, such
as anxiety, depression, sexual dysfunction schizophrenia and parkinsonism.
BACKGROUND OF THE INVENTION
Psychiat~ic diseases are thought to be due to dysfunctions in monoaminergic neuronal
10 systems, particularly those involving serotonin (5-HT,) and dopamine (DA).
Serotonin (5 HT~2
Anxiety is associated with increased activity in 5-HT systems. In animals where 5-HT hæ
been depleted, benzodiazepine anxiolytics are not active in anti-anxiety assays that they otherwise
are effeclive in. Serotonin neurons have autoreceptors that, when activated by agonists, depress
15 firing ra~es of 5-HT cells. These receptors are of the S-HT,A subtype. Because they depress 5-HT
neuronal activity, it can be expected that 5 HTIA agonists will be an effective anxiolytic. Clinical1y,
S-HTIA agonists have demonstrated anxioly~ic properties. The drug buspirone is the only currently
available marketed 5-HT,~ agonist having anxiolytic activity. This compound antagonizes dopamine
receptors at the same dose it stimulates S-HT,~ receptors. A similar drug, gepirone, also has
20 dopamine antagonist properties. These dopamine antagonist properties reduce the clinical utility
of these compounds because long term treatment with dopamine antagonists can p~duce tardive
dyskinesia.
Depression is a psychiatric condition thought to be associated with decreased 5-HT release.
~; Most anti-depressants potentiate the effects of 5-HT by blocking the termination of activity through
25 re-uptake into nenre terminals. Since some 5-HT", receptors are activated postsynaptically by 5-HT,
5-HT~ agonists may also be anti-depressants. Since the postsynaptic 5~ receptor may be less
sensitive than the autoreceptor, high doses of 5~ agonists, panicularly ve~ effective ones (i.e.~
those causing greater stimulation of the 5-HT,~ receptor~ a parameter referred to as "efficacy")~ can
be expected to be effective anti-depressants. Gepirone has already been demonstrated to have
30 ameliorative effccts on some depressive end points in some patients.
Serotonin is also involved in the regulation of feeding and sexual behavlor and in
cardiovascular regulation. Thus~ 5~HT~ agonists may be useful in treating overealing and sexual
dysfunction. These compounds have been shown to alter feeding and sexual behavior in animals.
5-HT,~ agonists are also known to depress sympathetic nerve discharge and thus lower blood
35 pressure. Thus, they may be useful in treating hypenension~ congestive heart failure by reducing
cardiovascular afterload and hean attack by removing sympathetic drive to the heart.
2 ~ 7 a
W O 93/08166 PC~r/US92/07314 "
-2-
DoDamine(DA)
Schizophrenia is thought to be due to hyperactivity in DA systems. Thus, currently
available anti-psychotics are DA antagonists. Dopamine autoreceptors depress DA neuron firing
rates, DA synthesis and release. Thus DA autoreceptor agonists can also be expected to be anti-
5 psychotics. DA agonists are also useful for ~:a~ng Parkinsonism, a disease caused by degenerationof DA neurons, and hyperprolactinemia, since DA agonists depress prolactin release.
Dopamine autoreceptor antagonists are a new clæs of drug that increæe releæe of DA by
releæing the DA neuron from autoreceptor control. Thus, these drugs can be expected to be useful
in conditions treatable with amphetamine and other similar stimulants which directly rclease DA.
10 Dopamine autoreceptor agonists are expected to be much milder stimulants than amphetamines
because they simply increase the release æsociated with the normal DA activity, by releasing the
cell from autoreceptor control, rather than directly releasing DA. Thus, DA autoreceptor antagonists
can be expected to be useful in treating overeating, attention deficit disorders, psychiatric, cognitive
and motor retardation in demented and elderly patients, and in treating nausea and dizziness.
Drugs acting on central DA transmission are clinically effective in treating a variety of
central nervous system disorders such as parkinsonism, schizophrenia, and manic-depressive illness.
In parkinsonism, for example, the nigro-neostriatal hypofunction can be restored by an increase in
postsynaptic DA receptor stimulation. In schizophrenia, the condition can be normalized by
achieving a decrease in postsynaptic DA receptor stimulation. Classical anti-psychotic agents
20 directly block the postsynaptic DA receptor. The same effect can be achieved by inhibition of in-
traneuronal presynaptic events essential for the maintenance of adequate neurotransmission, transporl
mechanism and transmitter synthesis.
In recent years a large body of pharmacological, biochemical and electrophysical evidence
hac provided considerable support in favor of the existence of a specific population of central
25 autoregulatory DA receptors located in the dopaminergic neulon it~elf. These receptors are part of
a homeostatic mechanism that modulates nerve i~pulse flow and transmitter synthesis and regulates
the amount of DA released from the nerve endings.
Direct DA receptor agonists, like apomorphine, are able to activate the DA autoreceptors
as well as the p~st synaptic DA receptors. The effects of autoreceptor stimulation appear to
30 predominate when apomorphine is administcred at low doses, whereas at higher doses the
attenuation of DA transmission is outweighed by the enhancement of postsynapdc receptor
stimulation. The anti-psychotic and anti-dyskinetic effects in man of low doses of apomorphine are
likely due to the autoreceptor-stimulator properties of this DA receptor agonist. This body of
Icnowledge indicates DA receptor stimulants with a high selectivity for central nervous DA
35 autoreceptors would be valuable in treating psychiatric disorders. The compounds of the present
invention have a variety of effects at S-HTI~ and DA receptors, and offer a variety of utilities
2 ~ 2 ~
`;1VO 93/08166 PCr/lJS92/07314
-3-
associated with those activities.
DESCRlPrION OF RELATED ART
The compounds of this invention may be described as having tricyclic moieties, or rings.
These rings, have been labeled A, B, C, in Formula 1, below, in the summary of invention section.
S The tricyclic moieties may be designated by the number of atoms in each ring. Thus, a 6-5-7 ring
system would have 6 atoms in the A ling, S atoms in the B ring, and 7 atoms in the C ring.
The compounds of this invention have a 6-6-6 ring system. One of the atoms in the C -
r ng is nitrogelL A 6-6-6 ring system where the A ring is arom~tic that contains a 6-atom C ring
which contains nitrogen might also be called a benzo-quinoline structure. Included below are
10 documents thal represent the h~6 system, "SET A," in addition to other systems. These other
systems are grouped and listed below for the sake of completeness. The first two references cited
in "SET A" are considered most relevant to ~is disclosure.
Some documents cited below describe compounds that belong in more than one group, but
gencrally the following groups are observed: The first group, SET A, contains 6-6-6 ring systems
15 or benzo-quinolone structures; the second gr~up, SET B, contains 6-5-6 ring systems or indeno-
pyridine structures; the third group, SET C, contains 6-6-5 ring systems or benzo-indolines; the
fourth group, SET D, contains 6-5-5 ring systems or indeno-pyrroles, and the fifth group SET E,
contains 6-7-5 ring systems or benzo-cyclohepta-pyridines.
These documents are described below:
SET A (6-6-6 ring systems)
European Patent Application, EP 410,535-A, application published 30 January 1991~
Derwent No. 91-030926/05, discloses octahydrobenzisoquinoline derivatives as antipsychotic agents.
Published PCI application, international publication number WO 90/06927, PCI`IUS89/05512,
intemational publication date, 28 une 1990, DeJwent No. 9~224487129, discloses
25 hexahydroindeno[l,2-c]pynoles, hexahydroindenopyrodines and hexahydrobenz[e]isoindoles as
selective S-HT receptor agents; United States Patent, US 4,341,786, issued 27 July 1982, discloses
octahydrobenzo[flquinoline compounds. Great Britain Pate~ GB 1,596,170, published 19 August
1981, Den~ent No. 55370A/31 discloses hexahydro-benz[flisoquinolines~ Great Britain Patent
Application, GB 2,126,58~A, application published 28 March 1984, Derwent No. 84-077373/13,
30 discloses octahydrobenzlflisoquinolines as psychotropic and analgesic agents. Great Britain Patent,
GB 2138-815-A, published 27 April 1983, Derwent No. 84-271,830 discloses benzo[flquinolines.
German Patent, DE 2,801,576, DenNent No. 55370/31~ discloses hexahydrobenz[f~isoquinolines.
SET B (~-5-6 ring systems)
Soviet Union Patent. SU 327193, Derwent No. 63503T-B~ discloses isoquinolines; and
35 Gelman Patent, DE 2.501.930. Derwent No. 58024X/31, discloses hexahydro-indeno wridines as
antidepressants.
2 0
WO 93/08166 PCr/US92/07314 "
SET C (6-6-5 ring systems)
Published PCT application, WO91/13872, published 19 September 1991, intemationalapplication No. PCT/US91/00117 discloses heterocyclic indole compounds. Published PCT
application WO91/11435, published 8 August ~ intemational application No. PCI`/US91/00018
S discloses benz(e)indole heterocyclic compounds. Published PCT application WO91/00856,
published 24 January 1991, intemational application No. PCI`/US90/03551 discloses carbocyclic-2-
amino tetralin derivatives. United States Patent, US 4,618,683, issued 21 Octnber 1986, Derwent
No. 86-298,374/45 discloses tetrahydro-benzole~indolines. European Patent, EP 95-666-A,
published I June 1980, Der vent No. 83-840180, discloses tetra:hydro-benzo-isoindolines.
10 Netherland Patent, NL 7216762, published 8 December 1981, Derwent No. 36797U-B discloses
benzolf]isoindolines. Belgium, BE 827,087, published 26 March 1974, Denvent No. 67323W/41,
discloses benz~isoindolines.
SET D (6-5-5 nng systems)
European Patent 170,093-A, published 25 luly 1984, Der vent No. 86-036877/06 discloses
15 indeno-wrroles.
SET E ~6-7-5 ring systems)
U.S. Patent 4,024,265, May 17, 19M, discloses benzolS,6~ cycloheptal1,2-clpyridines and
their use for treating depreæion.
SUMMARY OF INVENTION
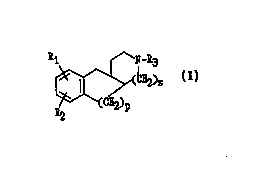
20The present invention encompasses: I) compounds of fonnula I below,
~s
(~)p FormuJa l
wherein p and s ar~ independent and may be either I or 2,
30 wherein Rl is -H, -Halo, -CN, -CO2H, -CO2Rl.,, -CONH2, -CONHR~.~, -CON(Rl.1)2, -SH, -SRl " -
SO2Rl l, -SO2NH2, -SO2NHR~," -SO2N(Rl,~)2, -ORl.l, -OSO2CF3, -OSO2Rl.l, -NH2, -NHRl ~, or -
N(R")2; wherein Rl.~ is -H, -(Cl-C~ alkyl), -(C,-C~, alkenyl), -(C3-CIo sycloalkyl), -(C6 aryl), -5
or 6 member heterocyclics, -(C,-C, alkyl)-(5 or 6 member heterocyclics), wherein R2 is -H, -Halo,
-CN, -CF3 -SH, or -SR2.,; wherein R2, = R, ~, wherein R3 is -H, -(C,-C8 alkyl), -(C,-C8 alkenyl), -
35 (C6 aryl), -(C3-Clo cycloalkyl), -(S or 6 member heterocyclics), -(C,-C8 alkyl)-5 or 6 member
heterocyclics), wherein R3., = R,, or a pharm~cologically acceptable salt thereof. 2. A
2it 8~?JQ
; ~W O 93/08166 PC~r/~S92/07314
-5-
pharmaceutical composition consisting essentially of a pharmaceutically acceptable carlier and an
effective amount of a compound of formula 1.
3. A method of treating central nervous system disorders, associated with serotonin and or
dopamine receptor activity comprising: administering an effective amount of a compound of
5 formula I to a patient in need thereof.
The compounds of this invention possess selective pharmacological properties and are useful
in treating central nervous system disorders including anti-depression symptoms, anxiolytic
symptoms, panic attacks, obsessive-compulsive disturbances, senile dementia, emotional disturbances
related to dementia disorders, and stimulation of sexual activity. Other central nervous system
10 disorders and conditions related to central dopamine transmission such as parkinsonism. schizophre-
nia, and manic-depressive illness may also be treated with the compounds of this invention. The
compounds of this invention are also useful to alle~/iate aggressive behavior, confusional delirious
states and impotence. In addition to tneir central nervous system pharmacological activities, the
compounds of this invention are also anti-diabetic, anti-obesity,anti-atherosclerotic, and anti-
15 hyper~ensive agents. Processes for preparation of these compounds, their pharmaceutical use andpharmaceutical preparations employing such compounds constitute funher aspects of the in
vention.
An object of the invention is to provide compounds for therapeutic use, especially
compounds having a therapeutic activity in the cent al nervous system. Another object is to provide
compounds having an effect on the 5-HTI~ receptor in mammals including man. A funher object
20 of this invention is to provide compounds having an effect on the subclass of dopamine receptors
known as the D2 receptor.
The compounds of formula I where R3 equals H are novel and useful as intermediates in
the preparation of the compounds of formula 1.
DETAILED DESCRIPI ION OF THE INVENTION
The compounds of this invention are identified in two ways: by the descriptive name and
reference to labeled structures contained in appropriate charts. In app~priate situadons, the proper
stereochemistry is also represented in the charts.
ln this document the parenthetical term (Cn-Cm) is inclusive such that a compound of (Cl-
C~) would include compounds of one to 8 carbons and their isomeric forms. The vsuious carbon
....
30 moieties are defined as follows: Alkyl refers to an aliphatic hydrocarbon radical and includes
branched or unbranched forms such as methyl, ethyl, n-p~pyl, isopropyl, n-butyl, isobutyl, sec-
butyl, t-butyl, n-pentyl, isopentyl, n-hexyl, isohexyl, n-heptyl, isoheptyl~ and n-octyl.
Alkoxy as represented by -ORI when R~ is (Cl-C~) alkyl refers to an alkyl radical which is
attached to the remainder of the molecule by oxygen and includes branched or unbranched fonns
35 such as methoxy~ ethoxy~ n-propoxy~ isop~poxy, n-butoxy~ isobutoxy~ sec-butoxy~ t-butoxy~ n-
pentoxy~ isopentoxy, n-hexoxy~ isohexoxy~ n-heptoxy~ isoheptoxy~ and n-octoxy.
S~ 18~2~
WO 93/08166 PCr/US92/07314
-6-
Alkenyl refers to a radical of an aliphatic unsaturated hydrocarbon having at least one
double bond and includes both branched and unbranched forms such as ethenyl, I-methyl-l-e~henyl,
I-propenyl, 2-propenyl, I-butenyl, 2-butenyl, 3-butenyl, 2-methyl-1-butenyl, l-pentenyl, allyl, 3-
pentenyl, 4-pentenyl, I-methyl~pentenyl, 3-methyl-1-pentenyl, 3-methyl-allyl, I-hexenyl, 2-
hexenyl, 3-hexenyl, 4hexenyl, I-methyl~-hexenyl, 3-me~hyl-1-hexenyl, 3-methyl-2-hexenyl, 1-
heptenyl, 2-heptenyl, 3-heptenyl, 4-heptenyl, l-methyl~heptenyl, 3-methyl-1-heptenyl, 3-methyl-2-
heptenyl, I-octenyl, 2-octenyl, or 3-octenyl.
(C3-CI~)cycloalkyl refers to a radical of a saturated cyclic hydrocarbon which includes alkyl-
substituted cycloalkyl, such as cyclopropyl, 2-methylcyclopropyl, 2,2-dimethylcyclopropyl, 2,3
diethylcyclopropyl, 2-butylcyclopropyl, cyclobutyl, 2-methylcyclobutyl, 3-propylcyclobutyl,
cydopentyl, 2,2-dimethylcyclopentyl, cyclohexyl, cydoheptyl, or cyclooctyl.
Examples of aryl include phenyl, naphthyl, (o-, m-, p-)tolyl, (o-, m-, p-)ethylphenyl, 2-ethyl-
tolyl, 4-ethyl-o-tolyl, 5-e2hyl-m-tolyl, (o-, m-, or p-)propylphenyl, 2-propyl-(v-, m-, or p-)-tolyl, 4-
isopropyl-2,6-xylyl, 3-propyl~ethylphenyl, (2,3,4, 2,3,6-, or 2,4,5-)trimethylphenyl, (o-, m-, or p-
)fluorophenyl, (o-, m-, or p-trifluoromethyl)phenyl, ~fluoro-2~5-xylyl, (2,4-, 2,5-, 2,6-, 3,4-, or 3,5-
)difluo~ophenyl, (o-, m-, or p-)chlorophenyl, 2-chloro ~tolyl, (3-, ~, 5- or 6-)chloro~-tolyl, ~
chloro-2-propylphenyl, 2-isopropyl~chlorophenyl, 4-chloro-3-fluorophenyl, (3- or 4-)chloro-2-
fluorophenyl, (o-, m-, or p-,)trifluorophenyl, (o-, m-, p-)ethoxyphenyl, (~ or 5-)chloro-2-methoxy-
phenyl, and 2,4-dichloro(5- or 6-)methylphenyl.
Examples of heterocyclics include: (2-, 3-, or 4-)pyridyl, imidazolyl, indolyl, NU'-formyl-
indolyl~ NU'-C2-C~alkyl-C(O)-indolyl, [1,2,41-triazolyl, (2-, 4-, S-)pyrimidinyl, (2-, 3-)thienyl,
piperidinyl, pyrryl, pyrrolinyl, pyrrolidinyl, pyrazolyl, pyrazolinyl, pyrazolidinyl, imidazolinyl,
imidazolidinyl, pyrazinyl, piperazinyl, pyridazinyl, oxazolyl, oxazolidinyl, isoxazolyl, isoxazolidinyl,
morpholinyl, thiazolyl, thiazolidinyl, isothiazolyl, isothiazolidinyl, quinolinyl, isoquinolinyl,
benzimidazolyl, benzothiazolyl, benzoxazolyl, furyl, thienyl, and benzothienyl.
Each of these moieties may be substituted as appropriate.
Halo is halogen (fluoro, chloro, bromo or iodo) or trifluoromethyl.
LAH is lithilun aluminum hydride
LDA is !itluum diisopropylamide
THF is tet~hydrofuran
9-BBN-H or 9-BBN is 9-Borabicyclo[3.3.11nonane dimer
It will be apparent to those skilled in the art that compounds of this invention may contain
chiral centers. The scope of this invention includes all enantiomeric or diastereomeric forms of
formula I compounds either in pure form or as mixtures of enantiomers or diastereomers. The
compounds of forrnula I contain two asymmetric carbon atoms in the aliphatic ring moiety~
including the ring carbon atoms adjacent to the nitrogen atom. The therapeutic properties of the
2118~2~
`~0 93/08166 PCr/US92/0731
-7-
compounds may to a greater or lesser degree depend on the stereochemistry of a particular
compound.
Both organic and inorganic acids can be employed to form non-toxic pharmaceutically
acceptable acid addition salts of the compounds of this invention. ]llustrative acids are sulfuric,
5 nitric, phosphoric, hydrochloric, citric, acetic, lactic, tartaric, palmoic, methanesulfonic,
ethanedisulfonic, sulfamic, succinic, cyclohexylsulfamic, fumaric, maleic, and benzoic acid. These
salts are readily prepared by methods known in the art.
In clinical practice the compounds of the present invention will normally be administered
orally, rectally, or by injection, in the form of phalmaceutical preparations comprising the activc
10 ingredient either as a free bæe or æ a pharmaceutically acceptable non-toxic, acid addition salt.
such as the hydrochloride, lactate, acetate, mesylate, methanesulfonate, or sulfamate salt, in
association with a pharmaceutically acceptable carrier. The use and administration to a patient to
be treated in the clinic would be leadily apparent to a physician or phamlacist of ordinary skill in
the art.
I5 In therapeutical treatment the suitable daily doæs of the compounds of the invention are
I - 2000 mgApatient for oral application, and 0.001 - 20 mg/kg for intramuscular, intravenous or
subcutaneous application. The precise dosage will be apparent to an ordinarily skilled physician
or pharmacologist taking into account factors such as the age, weight, sex, and medical condidon
of the patient being treated. Also relevant is the potency of the particular compound. The potency
of a compound may be suggested by the standard tests described below.
These compounds are particularly effective anxiolytic and antidepressant agents. Central
nervous system disorders and conditions related to central dopamine transmission such as
parkinsonism, schizophrenia, and manic-depressive illness may also be treated with the compounds
of this invendon. Other uses for these compounds include panic attacks, obsessive-compulsive
disturbances, and senile dementia, particularly the emotional disturbances seen in dementia
disorders. In addition, central 5-HT receptor activation are believed to be involved in mediating
sexual disorder. These compounds would be useful to stimulate sexual activity and to alleviate
impotence. The compounds of this invention are also useful to alleviate aggressive behavior and
confusional delirious states.
The compounds of this invention can be made in accordance with the processes illustrated
in CHARTS A, B and C. The reactions and properties are divided into two parts. PART 1~
immediately below, contains compounds related to CHARTS A and B. The reactions of compounds
of CHARTS A and B are followed by the biological data for the compounds of CHARTS A and
B followed by detailed preparation steps. The compound claims that correspond to PART 1, the
compounds of CHARTS A and B are claims 14 - 24. Part Il contains the COMPOIJNDS that
correspond to CHART C. the reactions. the corresponding biological data and the detailed
~11892~
WO 93/08166 PCr/US92/07314
-8-
preparations. The compound claims that correspond to PART Il are claims 2 - 13.
PART I
Reactions of CHART A
Step I, the s~ting ketone, Al, is refluxed wi~ the sodium salt of triethyl phosphonoacetate
to give the ester, A~, along with some of its exo double bond isomer. The undesired isomer is
removed by treating the isomeric mixture wi~ LDA followed by quenching with acetic acid. Step
2, the ester, A2, is reduced to the alcohol, A3, with LAH. Step 3, the alcohol, A3, is treated with
silylchloride to protect the alcohol as the t-butyldimethylsilyl ether A4.
Step 4, the silyl ether, A4, is refluxed with 9-E~BN-H in TH~ and then treated with a
mixture of bromo ethylacetate and potassium 2,6-di-J-butylphenoxide to give the ester, A5. Step
5, the silyl gr~up is removed with aqueous acid to afford the alcohol, A6. Step 6, A6 is reduced
with lithium borohydride to afford the diol, A7. Step 7, the diol is converted into the dibromide,
A8, with NBS and triphenylphosphine. Step 8, the dibromide is heated with optically pure o~-
methylbenzylamine to afford a mixture of diastereomeric a~epines, A9, which are separated by
chromatography. The diastereomers are separately carried on to the final optically pure enantiomers
using the following procedures. Step 9, optically active azepine, A9, is t~eated with t-butyllithium
followed by trimethylsilylisocyanate to afford the carboxamido azepine, A10. Step 10,
hydrogenation over palladium hydroxide afforded All. Step 11, the hydrogenated azepine was
alkylated with bromopropane to afford A12. Step 12, the carboxamido group is replaced with a
cyano group, A13.
Structures are shown in CHART A.
Reactions of CHART B
Step 1, indanone, B1, is treated with pyrrolidine to convert it into the eneamine, B2. Step
2, the eneamine is alkylated with ethyl bromoacetate to afford the ca~boxylate, B3. Step 3, the
2~5 carboxylate undergoes a Reformatsky reaction with ethyl bromoacetate to afford the diester, B4.
Step 4, the diester is reduced with triethylsilane in trifluoroacetic acid to obtain the reduced diester,
B5. Step 5, the reduced diester is further reduced with LAH to afford the diol, B6. Step 6, the diol
is converted into the di-p-toluenesulfonate, A7. Step 7, the di-p-toluenesulfonate is converted into
the azepines B8 and B9 (trans and cis- fused rings, respectively).
Structures are shown in CHART B.
The compounds of this invention have high oral potency and a long duration of action.
This exceptionally good bioavailability combined with a long period of activity are beneficial to
effective clinical treatment. The preferred compounds of PART I are not only active but have
increased bioavailability as shown by metabolism studies. See PART I - TABLES I-IV, below.
35 One preferred compound is (-)-trans-l~carboxamido-5alO~dihydro-3N-n-propyl-6H-indeno~1,2-
d~azepine. compound number 3, below.
21~ .g~Q
`~0 g3/08166 P~r/US92/07314
g
The compounds of this invention are useful both as intermediates to produce other
compounds and they are useful to treat central nervous system disorders. The utility of the
compounds of this invention to treat central nervous system disorders is shown in behavioral,
physiological and biochemical tests. These tests are described below.
S CNS Receptor Binding Assay (PART 1 - TABLE 1): The Central Nervous System Binding
Assay measures the percent inhibition from experiments employing test compounds at I microMolar
concentration competing with valious radioligands for binding to whole brain membranes,
membranes prepared from specific brain structures, or membranes prepared from cel1 lines
expressing cloned receptors. When percent inhibition is equal to 10û the test compound binds as
10 well as the standard compound. The standard compound for the 5-HT,A receptor is 8-OH-DPAT.
The standard compo~nd for the dopamine Dl receptor is SCH 23390.
5-HT and DA Cell Flring (PART I - TABLE Il): Glass microelectrodes filled with
pontamine sky blue in 2M NaCI were used for extracellular recordings from Sprague-Dawley rats
anesthetized with chloral hydrate (400 mg~lcg i.p.). Drugs were injected intravenous and five
15 neurons were located in the dorsal raphe nucleus according to Aghajaruan et al., J. Pharmacol. Exp.
Ther. 137:178 (1970). DA neurons werc located in the substantia nigra pars compacta (SNPC) and
identified according to Bunney et al., l. Pharmacol. Exp. Ther. 185:560 (1973). For S HTIA agonist
effects, the ED50 is the dose required to depress dorsal raphe neuron firing by 50% of the maximal
depression obtainable. Lum and Piercey, Eur. J. Pharmacol. 149:9-15 (1988). For DA agonist
20 effects, the ED50 is the dose required to depress SNPC neuron firing by 50% for full agonists, or
for partial agonists, the dose required to depress SNPC neuron firing by 50% of the maximal
depression atlainable. Piercey and Hoffmann, l. Pharmacol. Exp. Ther. 243:391, (1987). For DA
antagonists, the ED50 is the dose reguired to reverse the depression of SNPC neuron firing caused
by a DA agonist (usually either 1-3 mgJkg amphetamine, or 100 ugQcg apomorphine). For partial
DA agonists, the antagonist ED50 is the dose required to reverse the agonist-induced depression of
SNPC neuron firing by 50% of the maximum reversal. Piercey and Hoffmarm, J. Pharmacol. Exp.
Ther., 243:391 (1987).
Sympathetic Nerve Discharge (SND) (PART I - TABLE III): SND ED50 (m~lkR): The
i.v. mg~lcg dose causing a 509'o depression in SND in chloralose anesthetized cats. Max. Decr. SND
qo Control: The maximum inhibition of sympathetic activity observed in the dose range tested
(0.001-1.0 mg/kg i.v.). 9bBP at SND ED50: The blood pressure of the chloralose anesthetized cats
in percent control at the dose causing 50% depression in SND. Max Decr. BP % Control: The
maximum reduction in blood pressure as percent of the control blood pressure in the same animals
observed in the dose range tested (0.001-1.0 mglkg i.v.).
Metabolism (PART I - TABLE IV): Intrinsic clearance of 3 concentrations of compound
(2, 5 and 15 ug/ml) foDowing a 60 minute incubation at 37C in the presence of a suspension of
;` 2 ~J
WO 93/OB166 PCr/US92/07314 ~
-10-
freshly prepared rat hepatocytes (5.0 million cells/ml). Aliquots of each incubate were withdrawn
during the incubation and analyzed for parent compound using HPLC methodology. lntrinsic
clearance is expressed as ml/minlS million cells. Metabolic stability relative to the control, ( ~ 1-
formyl-6,7,8,9-tetrahydro-N-di-n-propyl-8-amino-3H-benzle]indole, is determined.S Hypothermia (PART I - TABLE IV)- Starting with a dose of 30 mg/lcg, four mice are
injected subcutaneously with test compound. Twenty minutes latert the number of animals whose
body temperature has decreased by 2C or more are counted. If all four animals reach criteria. the
drug is considered "active," and subsequent readings are taken at 60 and 120 minutes after
administration of the drug. The time for the last statistically signific3nt drug effect on mean body
10 temperature is indicated u~ minutes. For all "active" compounds, doses are lowered by 0.5 Iog
intervals until a dose which does not lower body temperature by 2C in any animal is found.
Potency is given as mglkg ED50 (dose requi~ed to depress temperature in two of four mice) as
measured by Spealman-Karber statistics.
The compounds below have been subjected to one or more of the biological tests described
15 above. PART I - TABLES I - IV use the arbitrary numbers æsigned below.
Com~ound Number Name
cis-7-chloro-10-methoxy-5a,10b-dihydro-3N-n-propyl-GH-indenoll,2-dlazepine,
see CHART B stn~cture B9.
2 rrans-7-chloro-10-methoxy-5a,10b-dihydro-3N-n-propyl-6H-indenoll,2-
dlazepine, see CHART B structure B9.
3 (-~trans-lO carboxamido-5a910b-dihydro-3N-n-propyl-6H-indeno~ dlazepine,
see CHART A structure A12.
4 (+)-trans-10-carboxamido-5a,10b-dihydro-3N-n-propyl-6H-indenol1,2-
d)azepine, see CHART A st~ucnlre A12.
(-)-trans-7-carboxamido-5a,10b-dihydro-3N-n-propyl-6H-indenoll,2-dlazepine,
see CHART A stn~cture A12.
6 (I)-trans-7-carboxamido-5a,10b-dihydro-3N n-propyl-6H-indeno[1,2-dlazepine,
see CHART A structure A12.
7 (-)-trans~7~cyano-5a,10b-dihydro-3N-n~propyl-6H~indenoll,2-dlazepine, see
CHART A stlucnlre Al3.
8 (+)-trans-7~cyano-5a,10b-dihydro-3N-n-propyl-6H-indenol1,2-d]azepine~ see
CHART A structure A13.
2118~
WO 93/08166 PCr/US92/07314
PART I - TABLE I
BIOLOGICAL DATA - CNS Receptor Binding
5-HTIA Dopamine - Dl Dopamine - D2
Compound No. % inhibition % inhibition % inhibition
99 57 74
2 ----
3 93 13 45
4 21 0 2
24 10 24
6 -18 50 31
7 17 11 22
8 15 7 3s
1 5 ~ ~ _ .. ~ _
PART I - TABLE Il
BIOLOGICAL DATA - 5HT and DA Cell Firing
S-HTIA Piri;ng DA Firing
Compound No.(mglkg) (m~g)
2 -- 0.065
_ _ _ _
PART I - TABLE III
BIOLOGICAL DATA - Sympathetic Nelve Discharge (SND)
SNDED50 Max. Decr. SND %BP at Max. Decr. BP BP
Compound No. (mg/kg) % contsol SNDED50 % control
3 1.0 1.0 66.0 66.0
PART I - TABLE IV
BIOLOGICAL DATA - Metabolism and Hypothermia
Metabolic Stability Hypolhermia Duration
Compound No. (control is 1.0) (mg/kg) (minutes)
2 -- 13 120
3 0.95 0.97 (subcutaneous) --
3 1.3 (oral)
4 0.39 --
.~
Wo 93/08166 2 1 1 Q ~1 2 ~ PCr/US92/07~14
-12-
Without further elaboration, it is believed that one skilled in the art can, using the preceding
description, practice the present invention to its fullest extent. The following detailed examples
describe how to prepare the various compounds and/or perform the various processes of the
invention and are to be construed as merely illustrative, and not limitations of the preceding
S disclosure in any way whatsoever. Those skilled in the art will promptly recognize appropriate
variations from the procedures both as to reactants and as to reaction conditions and techniques.
PREPARATIONS AND EXAMPLES FROM CHART A
STEP 1
Preparation Al(a): 7-bromo~4chloro-1-carboethoxymethyl-3H-indene, see CHART A,
S'l~P 1, compound A2.
Tnethyl phosphonoacetate t34.2 ml) and sodium hydride (7.92 g of 50 % slurry in oil)
are placed sequentially in a flask with T~ (300 ml). After hydrGgen evolution ceases, the solution
is cooled to 0C and a THF (50 ml) solution of 7-bromo~chloroindanone is added. The solution
15 is brought to reflux and kept the~e for 18 hours. The solution is then cooled and partitioned into
ether and water. The ether layer is washed with water (2X) and dried over anhydrous sodium
sulfate and the solvent removed in vacuo. NMR shows a 3:1 ratio of the elldo olefin to the exo.
This mixture is added to a T~ solution of lithium diisopropylamide (1 eq.) at -78C. After five
minutes ~cetic acid (1 eq.) is added in THF (10 rnl) and the solution is allowed to warm to 25C
20 where it is partitioned between water and ether. The ether layer is washed with 2N aqueous
hydrochloric acid followed by water, saturated aqueous sodium bicarbonaee, and bnne. The ether
solution is dried over anhydrous sodium sulfate and the solvent removed in vacuo to afford 31 g
cf the title compound after chromatography with ethyl acetate/hexane ~2:98).
Preparation A1(b): 4bromo-1-carboethoxymethyl-3H-indene, see CHART A, STEP 1,
compound A2.
Use preparation A1(a), only start with ~brom~l-indanone.
STEP 2
. .
Preparation A2(a), 7-bromo-4chloro-1-(hydroxyeth~2-yl)-3H-indene, see CHART A,
STEP 2, compound A3.
A solution of 7-bromo~chloro-1-carboethoxymethyl-3H-indene (25.61 g, 81.2 mmol) in
diethylether ( 150 ml) is added over a 10 minute period to a suspension of lithium aluminum hydride
(10.0 g, 0.263 mol) in die~ylether (250 ml) with stirring at 0DC. The cold bath is removed, and
35 the mixture is stirred for 15 minutes. The mixture again is cooled to 0DC, and water (10 ml), 159h
NaOH (10 ml), and water ~30 ml) are added in succession. The cold bath is removed, and the
211~92~ `
WO 93/0816S PCr/~S92/07314
mixture is stirred for 10 minutes. The mixture is filtered, and the precipitate is washed well with
diethylether. The filtrate is dried (MgS04), and the solvent is removed under vacuum to leave the
title compound as a yellow solid (21.67 g, 98%).
.
S Preparation A2(b): 4-bromo-1-(hydroxyeth-2-yl)-3H-indene, see CHART A, SI'EP 2,
compound A3.
Use preparation A2(a), only start wi~h 4-bromo-1-carboethoxymethyl-3H-indene.
STEP ~
P~epa~ionA3(a): 1-(t-butyldimethylsilyloxyeth-2-yl)~7-bromo-4-chloro-3H-indene,see
CHART A, STEP 3, compound A4.
In this procedure 7-Bromo~chloro-l-(hydroxyeth-2-yl)-3H-indene (28.23 g, 103.2 mmol)
and imidazole (lSA g, 0.226 mol) are dissolved in dimethylformamide (200 ml), cooled to 0C, and
t-butyldimethylsilyl chloride (1~.08 g, 0.113 mol) is added. The mixture is stirred at room
15 temperature for 12 hours, and partitioned between diethylether and water. The ether solution is
wæhed several times wi~ water and once wilh brine. The solution is dried (Na2SO4~, and the
solvent is removed under vacuum to leave an amber oil (3g.94 g). Purification by flæh
chromatogra~hy ~230400 mesh silica gel; 10% toluene in hexane) gives the title compound as an
amber oil (37.26 g).
PreparationA3(b): 1-(t-butyldimethylsilyloxyeth-2-yl)-4-bromo-3H-indene,seeCHARTA, STEP 3, compound A4.
Vse Preparation A3(a) only start with 4-bromo-1-(hydroxyeth-2-yl)-3H-indene.
Sl'EP 4
PreparationA4(a): trans-7-bromo-1-(t-butyldimethylsilyloxyeth-2-yl)-2-carboethoxyme-
thyl-4-chloro-2,3-dihydro-3H-indene, see CHART A, STEP 4, compound AS.
Add 9-Borabicyclo[3.3~1~nonane (9-BBN-H) dimer (11.0 g, 0.045 mol, 1.05 eq.) to a
solution of l-(t-butyldimethylsilyloxyeth-2-yl)-7-bromo 4 chloro-3H-indene (33.28 g~ 85.8 mmol)
30 in dry tetrahydrofuran. Heat the mixture at reflux under argon in an oil bath maintained at 100C
for 41 hours and cooled in ice. A solution of 2,6-di-t-butylphenol (23.02 g, 0.112 mol) in dry
tetrahydrofuran (200 ml) is cooled to 0C, and a solution of potassium t-butoxide in tetrahydrofuran
(1.0 M, 90.1 ml, 9QI mmol) is added over 10 minutes. The mixture is stirred at room temperature
for 15 minutes and again cooled in ice. The solution is added to the borane solution above via
35 needlestock. Ethyl bromoacetate (15.1 g. 90.2 mmol) is added dropwise, and the mixture is stirred
at O~C for 30 minutes, room temper~ture for 2 hours, and at reflux for lS minutes. The mixture
2li~.~2a
WO 93/08166 PCr/US92/07314
-14-
is diluted with diethylether and washed with water, 10% sodium carbonate solution, and brine. The
solution is dried (MgSO4), and the solvent is removed under vacuwn to leave an oil. Purification
by flash chromatography (230400 mesh silica gel; pure hexane to 2% ethyl acetate in hexane) gives
the title compound as an oil (30.2 g, 74% yield).
s
PreparationA4(b): t7ans-4bromo-l~(t-butyldimethylsilyloxyeth-2-yl)-2-carboethoxyme-
thyl-2,3~dihydro-3H-indene, see CHART A, STEP 4, compound A5.
Use procedure A4(a) with the following modifications. 1-(t-Butyldimethylsilyloxyeth-2-yl)-
4-bromo-3H-indene ~38.8 g, 109.9 mmol) and 9-BBN-H (dimer) (14 g, 115.4 mmol) are dissolved
in T~ (10û ml) under a nitrogen atmosphere. This solution is refluxed for 48 hours and then
cooled. A slurry of potassium 2,6-di-t-butylphenoxide in THF is prepared by dissolving 2,6-di-t-
butylphenol (29.5 g) in THF (50 ml) and adding a 1.0 M TH~ solution of potassium t-butoxide
(115.4 ml). The slurry is stirred for 10 minutes and then cooled to 0C. The solution of the
organoborane is then added. Ethyl bromoacetate (12.8 ml) is added over a 3 minute period. The
slurry is stirred at 0C for 1 hour and then allowed to stir at 25C for I hour. The flask is cooled
to 0C and quenched with water and then partitioned between water and ether. The ether layer is
washed with water (2X) and bnne, then d~ied over sodium sulfate and the solvent removed in
vacuo. The resulting oil is placed on flash silica gel (6 cm X 45 cm) and eluted with ether/hexane
(0.5:99.5). This is raised to 5:95 to elute off the product which is obtained as an oil (18.7 g~ 38%
yield).
STEP S
Preparation A5(a): ~ans-7-bromo-2-carboethoxymethyl-4-chloro-2,3-dihydro-1-
(hydroxyeth-2-yl)-3H-indene, see CHART A, SIEP 5, compound A6.
Acetic acid (180 ml) is added to a solution of mans-7-bromo-1-(t-butyldimethylsilyloxyeth-
2-yl)-2-carboethoxymethyl~chloro-2,3-dihydro-3H-indene (29.2 g, 61.3 mmol) in tetrahydrofuran
(60 ml). Water (60 ml) is added, and the mixture is stirred at room temperature for 18 hours. The
solvent is removed under vacuum, and the oil is dissolved in diethylether, washed twice with 109h
aqueous sodium carbonate followed by brine, and the solution is dried overmagnesium sulfate. The
solvent is removed under vacuum leaves a yellow oil (24.5 g). Purification by flash
chromatography (230-400 mesh silica gel, 20% ethyl acetate in hexane) gives the title compound
as a colorless oil (18.4 g, 60% yield from the olefin).
Prepara~ionAS(b): nans~bromo-2-carboetho~ymethyl-2,3-dihydro-l-(hydroxyeth-2-yl)~
3H-indene, see CHART A~ SrEP 5, compound A7.
Use prepa~ation AS(a) with the following modifications. Trans4-bromo- I-(t-
3 ` ~
WO 93~08166 PCr/USg2/07314
-15-
butyldimethylsilyloxyeth-2-yl)-2-carboethoxymethyl-2,3-dihydro-3H-indene (46 g) is dissolved in
acetic acid/IHF/water (3:1:1, 250 ml) and stirred for 16 hours. The solvent is removed in vacuo
to obtain 22.3 g of pure oil.
STEP 6
Preparation A6(a): bans-7-bromo-4-chloro-2,3-dihydro-1,2-di(hydroxyeth-2-yl)-3H-indene, see CHART A, STEP 6, compound A7.
Lithium borohydride (2.0 M in tetrahydrofuran, 45 ml, 90 mmol) is added to a solution of
trans-7-bromo-2-carboethoxymethyl~chloro-2,3-dihydro- 1 -(hydroxyeth-2-yl)-3H-indene ( 16.07 g,
0.0444 mol) in diethylether (400 ml) at room temperature. The mixture is stined for 3 hours,
allowed to stand at 0C for 155 hours, and ~efluxed for 30 minutes. The mixture is cooled in ice,
and water (100 ml) is slowly added. When the reaction subsided, 10% hydrochloric acid (100 ml)
is slowly added, and the mixture is stirred for 30 minutes. The layers are separated, and the
aqueous layer is extracted with diethylether. The combined ether solution is washed with water,
saturated aqueous sodium bicarbonate, and brine. The solution is dFied over magnesium sulfate,
and the solvent removed under vacuum to leave the title compound as a cololless oil (15.0 g,
100%).
Preparation A6(b): ~ans-4bromo-273-dihydro-1,2-di(hydroxyeth-2-yl)-3H-indene, see
CHART A, STEP 6, compound A7.
Use preparation A6(a) only start with trans~bromo-2-carboethoxymethyl-2,3-dihydro-1-
(hydroxyeth-2-yl)-3H-indene.
STEP 7
Prepara~on A7(a): Irans-7-bromo-4chloro-2~dihydro-1,2-di(bromoeth-2-yl)-3H-indene,
see CHART A, STEP 7, compound A8.
ln this procedwe, srans-7-bromo~chloro-z~3-dihydro-l~2-di(hydroxyeth-2-yl) 3H-indene
( 19.7 g) is dissolved in methylene chloride (300 ml) and TH~ (500 ml) with triphenylphosphine (38
g) and cooled to 0C. N-Bromosuccinimide (25 g) is added in portions. The solution is stirred for
20 minutes and the solvent removed in vucuo~ The residue is dissolved in ethyl acetate (200 ml)
and silica gel (300 g) is added. With swirling. hexane (800 ml) is added. The slurry is filtered and
the solvent removed in vacuo. The residue is eluted do~,vn a flash silica gel column (4 cm X 25
cm) with ethyl acetatel hexane (10:90). Solvent removal in vacuo afforded the dibromide in 89%
yield.
Prepalation A7(b): ~rans-4bromo-1,2-di(bromoeth-2-yl)-2,3-dihydro-3H-indene~ see
Wo 93/~ 3 ~ 2 ~) PCl/US92/07314
-16-
CHART A, STEP 7, compound A8.
Use preparation A7(a) only start with trans-4bromo-2,3-dihydro-1,2-di(hydroxyeth-2-yl)-
3H-indene (19.7 g, 69.1 mmol). Title compound is afforded in 89% yield (25.~ g of an oil).
STEP 8
Preparation A8(a): preparation and separation of the diastereomers of A9, ~ans 10-bromo-
7-chloro-3N-[(S)-methylbenzyl]-6H-Sa,lOb-dih~droindeno[l,2-d]azepine, see CHART A, STEP
8, compound A9.
A mixture of tran3-7-bromo~chloro-2,3-dihydro-1,2-di(bromoeth-2-yl)-3H-indene (19.31
g, 0.0307 mol), ~S)-(-)-a-methylbenzylamine (3.91 g, 0.0323 mol), and potassium car'oonate (12.73
g, Q0921 mol) in acetonitrile (200 ml) is stirred at reflux on the steambath for 48 hours. The
solvent is removed under vacuum, and the residue îs partitioned between water and diçthylether.
The aqueous layer is extracted with diethylether, and the com~ined organics ale washed with brine
and dried (MgSO4). The solvent is removed under ~acuum to leave an orange oil (14.22 g).
Purification by flash chromatography (230 400 mesh silica gel, 5-30% ethyl acetate in hexane~ give
the desired amine as a mixture of diastereomers (7.05 g). Fu~er purification by mediwn pressure
liquid chromatography in a Michell-Miller column (230 400 mesh silica gel, 2-2.5% ethyl acetate
in hexane) gives hVO diastereomers. Mastereomer I ~higher Rf)(2.88 g). Diastereomer 2 (lower
Rf) (2.52 g~.
Preparation A8(b): preparation and separation of the diastereomers of A9, ~rans-7-bromo
3N-I~S)-methylben~yl]-6H-5a,l0b-dihydroindenol~ d]aæpine, see CHART A, STEP 8.
compound A9.
Use procedure A8(a) only start with trans-4-bromo-1,2-di(bromoeth-2-yl~2,3-dihydro-3H-
indene (25.3 g). The reaction is refluxed for 48 hours to afford compound. The diastereomers are
separated with flash silica gel ch~matography (Scm X 30cm) eluting with ether/hexane (1:99). The
higher Rf fraction is isolated æ a crystalline solid (m.p. 117) by crystallizing from hexaneltoluene
(7.2 g) la]25589 = -8.45 (c = 0.355, chloroform). The lower Rf diastereomer remains an oil (8.75
g)- 1]25589 = +5.82 (c = 1.545, chloroform). The remainder is a mixture of starting material and
diastereomeric products which are resubjected to the reaction conditions.
STEP 9
Preparation A9(a)(1-2): preparation of two diastereomers of A10, see CHART A~ STEP
9, compound A10. follow the appropriate paragraph below.
Preparation A9(a)(1): preparation of Irans-lO-carboxamido-7-chloro-5a.10b-dihydro-3N-
- `~1VO 93/08166 2 1 1 8 ~ 2 0 PCr/US92/07314
-17-
[(S)-methylbenzyl]-6H-indenol1~2-d~azepine from diastereomer 1. A solution of ~rans- 10-bromo-
7-chloro-3N-l(S)-methylbenzyl]-6H-Sa,lOb-dihydroindeno[1,2-d]azepine (diastereomer 1; 2.88 g,
7.12 mmol) in dry, argon degassed tetrahydrofuran (30 ml) is cooled to -78C, and t-butyllithium
(1.7 M in pentane, 8.4 ml, 14.3 mmol) is added over 15 seconds. The mixture is stirred at -78C
S for S minutes and added via needle stock to a solution of trimethylsilylisocyanate (2.42 g of 85%,
17.9 mmol) in dry tetrahydrofuran (10 ml) and dioxane (15 ml) at -78C. The cold bath is removed
and the mixture is allowed to warm to room temperature. Water is added, the mixture is stirred for
15 minutes, and the volatiles were removed under vacuum. The residue is shaken with diethylether
and water, the precipitate is filtered, washed with diethylether, and dried under vacuum at 60C to
10 give a solid (1.86 g). A sample (Q20 g) is crystaUized from ethanol to give the title compound as
a colorless solid (0.15 g, m.p. 231-232C).
- Preparation A9(a)(2): p~eparation of trans-1O-carboxamido-7~hloro-5a,l0b-dihydro-3N-
l(S)-methylbenzyll-6H-indenoll,2-d~azepine from diætereomer 2, see CHART A, STEP 9,
15 compound A10.
The amide is prepared in a manner similar to that for diastereomer 1, using procedure
A9(a)(1), except stalting with diastereomer2 of trans-10-bromo-7-chloro 3N-l(S~methylbenzyll-6H-
5a,10~dihydroindeno[1,2-d]azepine. The product is a colorless solid. A sample may be
crystallized from acetonitrile (m.p. 192-193C).
Preparation A9(b)(1-2): preparation of two diastereomers of A10, see CHART A, STEP
9, compound A10, follow the appropriate paragraph below.
Prepa~ion A9(b)(1), prepa~ation of (+)-trans-7-carboxamido-Sa,lOb-dihydro-3N-[(S)-
25 methylbenzyll-6H-indenol1,2-dlazepine.
Use preparation A9(a)(1) only start wi~ (+)-~rans-7-bromo-3N-[(S)-methylbenzyll-6H-
Sa,lOb-dihydroindenol 1,2-d]azepine (7.26 g). This produces 5.57 g of ~e isomer as solid. [ocl~S589
= +29.11 (c = 0.56 methanol).
....
30 Prepa~ion A9(b)(2): (-)-bns-7 carboxamido-5a,10b-dihydro-3N-I(S)-methylbenzyll-
6H-indenol1,2-dlaupine.
- Use preparation A9(a)(1) only start with (-~trans-7-bromo-3N-[(S)-methylbenzyll-6H-
5a,10b dihydroindeno[t,2-d]azepine (8.75 g). Afforded 6.7 g of the title compound as a solid.
Ia]25589 = -25.97~ (c = 1.525 methanol).
3S
Preparation A9(c): trans-10-aminosulfonyl-7-chloro-5a,l0b-dihydro-3N-l(S)-
WO g3/08166 2 1 1 ~ ~ 2 t~ P~/US92/0~31~ ~ ;
-18-
methylbenzyl]-6H-indenol1,2-d]azepine, CHART A, STEP 9, compound A10.
The diastereomeric title sulfonamide is prepared from the diastereomerof ~rans-10-bromo-7-
chloro-3N-l(S)-methylbenzyl]-6H-Sa,lO~dihydroindeno[1,2-dlazepine, see procedure A8(a), by
performing a metal-halogen exchange with t-butyllithium, as in procedure A9(a)(1), followed by
conversion of the aryllithium to the sulfonamide according to the reference: S. L. Graham, et al.,
J. Med. Chem., 32:2548 (1989).
STEP 10
Preparation AlO(a)(1-2), Examples 1-2: preparation of two diastereomers of All, see
CHART A, STEP 10, compound All, follow the appropriate paragraph below.
P~leparation AlO(a)(l), Example- 1: (+)-~rans-1O carboxamido-5a,10b-dihydro-6H-
indenoll,2-dlazepine.
A mixture of (+~rans-10-carboxamido-Sa,lO~dihydro-3N-[(S)-methylbenzyl]-6H-
indeno[l,2-d]azepine (1.70 g, 4.61 mmol), 20% palladium hydroxide on carbon (0.65 g), 12 N
hydrochloric acid (0.4 ml) in water (15 ml), and absolute ethanol (75 ml) is hydrogcnated in a Parr
appa~anls for 16 hours with an initial hydrogen pressure of 50 psi. The mixture is filteled through
H diatomaceous earlh and washed well with ethanol. The combined filtrate is evaporated leaving a
foam (1.37 g).
Procedure AlO(a)(2), Example 2: (-)-tr~ns-10-carboxamido-5a,10b-dihydro-6H-
` indenoll,2-dlaupine.
Use the same procedure as AlO(a)~l), except substituting (-~trans-10 calboxamido-Sa,lOb-
dihydro-3N-[(S)-methylbenzyll-6H-indenol1,2-d]azepine in place of the equivalent (+) isomer.
Preparation AlO(b), Examples 34, preparation of two diastereomers of Al l, see CHART
A, STEP 10, compound Al l, from the appropriate paragraph below.
Preparation AlO(b)(l), Example 3: (+)-trans-7-carbo~amido 5a,10b-dihydro~6H-
30 indenoll,2-dlazepine.
Use procedure AlO(a)(l) only start with (+)-trans-7-carboxamido-5a,10b-dihydro-3N-[(S)-
methylbenzyl]-6H-indeno[1,2-dlazepine (5.5 g). The isomer is produced as a white solid, m.p.
222C decomp. ~al255~9 = +28.68 (c = 0.68 methanol).
Prepa~ion AlO(b)(2), Example 4: (-)-trans-7-carbo~amido-5a,l0b-dihydro-6H-
indeno[l,2-dlazepine.
JI~ '
VO 93/08166 PCr/US92/07314
_19_
UsepreparationA10(a)(1)onlystartwith~-)-trans-7-carboxamid~Sa,lOb-dihydro-3N-[(S)-
methylbenzyl]-6H-indeno[1,2-d]azepine. The title compound is produced æ a white solid, m.p.
223C, decomp. [ak55B9 = -29.43 (c = 0.72 methanol).
S Preparation A10(c): ~ans~ aminosulfonyl-Sa,lOb-dihydro-6H-indeno[l,2-dlazepine,
see CHART A, ST~P lQ compound All.
Use procedure lO(a)(l) only start with trans-10-aminosulfonyl-7-chloro-5a,10b-dihydro-
3N~[(S)-methylbenzyl]-6H-indeno[l,2-d]azepine, from preparation 9~c).
STEP 11
Prepas~tion All(a)(1-2), Examples S-6: prcparation of two diastereomers of A12, see
CHART A, Sl~P 11, compound A12, follow the appropriate paragraph below.
Prepar~tion All(a)(l), Example 5: (+)-trans-1~carboxan~ido-5a,10b-dihydro-3N-n-
propyl-6H-indeno[1,2-dlazepine.
A mixture of (+)-~rans- 10-carboxamido^5a,10hdihydro-6H-indenol l ,2-dlazepine
hydrochloride (1.37 g,4.61 mmol), l-bromopropane (1.76 g,14.3 mmol), potassium carbonate (1.91
g, 13.8 mmol), triethylamine (0.73 g, 7.2 mmol), and acetonitrile (SO ml) is stirred at reflux for 17
hours. The solvent is removed under vacuum, and the residue is partitioned between water and 1:1
20 THFldiethylether. The aqueous layer is extracted again with the same solvent, and the combined
extracts were washed with brine and dried (MgSO4). The solvent is removed under vacuum to
leave a solid (1.04 g). Crystallization from acetonitrile gives colorless crystals ~m.p. 198-199C).
la]D= +218.7 (c = 0.785, 25C, T~
Preparation All(a)(2), Example 6~ bans-10-carboxamido-Sa,lOb-dihydro-3N-n-
propyl-6H-indenoll,2-d]azepine.
Use the same preparation as above except substituting (-)-trans-10-carboxamido-Sa,10~
dihydro-6H-indeno~1,2-d~azepine hydrochloride for the equivalent (+) isomer (0.85 g, m.p. 198-
199C). 1OC~D = -222.3 (c = 0.865, 25C, THI:~.
Preparation All(b)(1-2), Examples 7-8: preparation of t~,vo diastereomers of A12, see
CHART A, STEP 11, compound A12, f~m the appropriate paragraph below.
P~paration Al l(b)(l). Example 7: (+)-bans-7-carboxamido-5a,10b-dihydro.3N-n-propyl-
35 6H-indeno~ d)azepine.
Use preparation Al l(a) only start wi~ (+)-trans-7-carbox~nido-Sa,lO~dihydro-6H~
WO 93/OB166 PCI /US92/07314 / '
-2~-
indeno[l,2-d~azepine. The isomer is isolated as a white solid which is converted into the fumaric
acid salt with one equal of acid in methanol/ether (m.p. 185C). [a]25589 - +26.47 (c = 0.665
methanol).
Preparation Al l(b)(2),Example 8~ rans-7-carboxan~ido-Sa,lOb-dihydro-3N-n-propyl-
6H-indenol1,2 dlazepine.
Use preparation All(a) only start with (-)-trans-7-carboxamido-Sa,lOb-dihydro-6H-
indeno[1,2-d~azepine. The isomer is isolated as a white solid which is converted into the fumaric
acid salt with one equal of acid in methanollether (m.p. 185~ a]2S589 = -23.84~ (c = 0.99
10 methanol).
PrepasationAll(c): ~ns~ aminosulfonyl-5a,l0b-dihydro-3N-n-propyl-6H-indenol1,2-
dlazepine, see CHART A, STEP 10, compound Al l.
Use procedure All(a) only start with trans-1~aminosulfonyl-Sa,10~dihydro-6H-indeno[1,2-
15 d~azepine, from procedure AlO(c~.
STEP 12
Preparation A12(a)(1-2): preparation of t vo diastereomers of A13, see CHART A, STEP
12, compound A13, follow the appropriate paragraph below.
PreparationA12(a)(1): (+)-trans-10-cyano-5a,10b-dihydro-3N-n-propyl-6H-indenoll,2-
dlazepine. (+)-trans-lO-calboxamido-5a,10~dihydro-3n-n-propyl-6H-indeno~1,2-d]azepine (0.33
g, 1.22 mmol) is dissolved in THF (lS mL) and t~iethylamine (1.0 mL, 7.42 mmol) then cooled to
O~C. Titanium (IV) Chloride (0.33 mL, 3.04 mmol) is added. After stirdng at 20C for 2 hours, it
25 is partitioned between methylene chloride and dilu~e aqueous sodium carbonate. The organic layer
is washed with water and brine, then dried over sodium sulfate. Product is dissolved in ether and
hydrochloric acid in ether is added. Product is recrystallized from methanol and ether.
Preparation A12(a)(2)~ rans~ cyano-Sa,lOb-dihydro-3N-n-propyl-6H-indenop,2-
d]azepine. (-)-trans-10-carboxamido-5a,10b-dihydro-3n-n-propyl-6H-indeno[1,2-d]Dzepine (0.28
. . .
30 g, 1.02 mmol) is dissolved in THP (15 mL) and triethylamine (0.87 mL, 6.24 mmol) then cooled
to 0C. Titanium (IV) Chloride (0.28 mL, 2.56 mmol) is added. After stirring at 20C for 2 hours,
it is partitioned between methylene chloride and dilute aqueous sodium carbonate. The organic
Iayer is washed with water and brine. The product is dissolved in ether and hydrochloric acid in
ether is added. Product is recrystallized from methanol and ether.
Prepa~tion A12(b)(1-2). Examples 9-10: preparation of two diastereomers of A13, see
21~8~2~
~VO 93/08166 PCr/US92/07314
-21 -
CHART A, STEP 12, compound A13, follow the appropriate paragraph below.
Preparation A12(b)(1), Example 9: ~+)-trans-7-cyano-5a,10b~dihydro-3N-n-propyl-6H-
indenoll,2-dlazepine.
The title compound is prepared from (+)-trans-7-carboxamido-Sa,lOhdihydro-3n-n-propyl-
6H-indeno[1,2-d]azepine (0.33 g, 1.22 mmol) is dissolved in T~ (lS mL) and triethylamine (1.0
mL, 7.42 mmol) then cooled to 0C. Titanium (IV) Chloride (0.33 mL, 3.04 mmol) is added. After
stirring at 20C for 2 hours, it is partitioned between methylene chloride and dilute aqueous sodium
carbonate. The organic Iayer is washed with water and brine, then dried over sodium sulfate to
yield brown oil (0.29 g). The oil is dissolved in ether and hydr~chloric acid in ether is added. The
tan solid is recrystallized from methanol and ether to give a gray solid (0.22 g, m.p. 252C).
Preparation A12(b)(2), Example 10: (-}~ans-7-cyano-5a,10b-dihydro-3N-n-prop~1-6H-
indenoll,2-dlazepine.
The title compound is prepared from (-)-trans-7-carboxamido-Sa,lOb-dihydro-3n-n-propyl-
6H-indenol 1,2-d]azepine (0.28 g, 1.02 mmol) is dissolved in THF (IS mL) and triethylamine (0.87
mL, 6.24 mmol) then cooled to 0C. Titanium aV) Chloride (0.28 mL, 2.56 mmol) is added. After
stirring at 20C for 2 hours, it is partitioned hetween methylene chloride and dilute agueous sodium
carbonate. The organic layer is washed with water and b~ine, then dried over sodium sulfate to
yield brown oil (0.23 g). The oil is dissolved in ether and hydrochloric acid in ether is added. The
tan solid is recrystallized from methanol and ether to give a gray solid (0.21 g, m.p. 252C).
Preparation A12(c): ~ans-7-aminosulfonyl-5a,10b-dihydr~3N-n-propyl-6H-indenoll,2-
dlazepine, see CHART A, STEP 12, compound A13.
The title compound is preparcd from t~ans-7-aminosulfony1-Sa,lO~dihyd~6H-indenol1,2-
2S d]azepine in a manner similar to that for the preparation of n ans- l~aminosulfonyl-Sa,lO~dihyd~
3N-n-propyl-6H-indeno[1,2-d]azepine, see preparation Al l(c).
PREPARATIONS AND EXAMPLES FROM CHART B
STEP I
Preparation Bl(a): 4Chloro-7-melhoxy-l-(N-pyrrolidinyl)indene. see CHART B, SrEP1, compound B2.
A solution of the ~chloro-7-methoxy- I-indanone t3S.OO g, 0.178 mol~ and pynolidine (76.0
g, 1.07 mol) in dry tetrahydrofuran (800 ml) is cooled to 7~C, and a solution of titanium
tetlachloride (18.6 g, 0.098 mol) in pentane (SO ml) is added over a period of 7 minutes. The
mixture is stirred at 10C for 30 minutes at room temperature for 3.5 hours. The mixture i~ filtered~
and the fil~e is evaporated under vacuwn to leave the title compound as a green oil (44.4 g).
.,.
WO 93/081q~ 3 9 ~ PCr/US92/07314
-22-
S~EP 2
Preparation B2(a): 2-(Carboethoxymethyl)-4-chloro-7-methoxy-1-indanone, see CHART
B, SrEP 2, compound B3.
S A solution of the 4-chloro-7-methoxy-1-(N-pyrrolidinyl)indene (44.4 g, 0.178 mol). ethyl
bromoacetate (32.7 g, 0.196 mol), and diisopropylethylamine (34.5 g, 0.267 mol) in acetonitrile
(150 ml~ is stirred at reflux in an oil bath maintained at 95C for 4 hours and at room temperature
for 15 hours. The mixture is titrated with water, and 10% hydrochloric acid is added to adjust the
pH to 4-5. The m~xture is heated on the steam bath for 15 minutes, cooled, and extracted with
diethylether and with 1:1 diethylether/tetrahydrofuralL The combined extracts are washed with 10%
hydrochloric acid (3 times), saturated sodium bicarbonate, and brine. The solution is dried
(MgSO4), and the solvent is removed under vacuum to leave an amber oil (41.3 g). Crystallization
from ethyl acetate/hexane gives title compound as yellow erystals (28.g g). The filtrate is purified
by flash chromatography (230400 mesh silica gel, 4:1 hexane/ethyl acetate) to give title compound
as a solid (4.70 g, total yield 68%).
STEP 3
Preparation B3(a): 1,2-Bis(carboethoxymethyl)-4-chloro-2-hydroxy-7-methoxyindane,
see CHART B, SI'EP 3, compound B4.
- 20 A few crystals of iodine were added to a mixture of 2-(ca~boethoxymcthyl~chloro-7-met-
hoxy-l-indanone (8.48 g, 30.0 mmol), ethyl bromoacetate (10.0 g, 60.0 mmol), and zinc dust (5.~9
g. 90.1 mmol) in diethylether (50 ml) and benzene (100 ml) at room temperature, and the mixture
is heated to reflux. After 30 minutes, no significant reaction occurred. A few more crystals of
iodine are added, and the reflux is continued. The reaction is started a~er 30 minutes, and the
2S mixture is refluxed for an additional 1.5 hours. The mixture is cooled, diluted with water, and
washed with 10% hydrochloric acid, saturated sodium bicarbonate, and brine. The solution is dried
(MgSO4), and the solvent is removed under vacuum to leave the compound, B4(a), as an oil (10.89
g)-
STEP 4
Preparation B4(a)~ Bis(carboethoxymethyl)-4-chloro-7 methoxyindane, see CHART
B, SI~P 4, compound BS.
Trifluoroacetic acid (~0 ml) is added to a mixture of 1,2-bis(carboethoxyme~hyl)~chloro-2-
hydroxy-7-methoxyindane (9.75 g, 0.026 mol) and triethylsilane (6.1 g, 0.052 mol) at room
35 temperature with stirring. The mixture is sdrred at ~oom temperature for 3 hours, and the solvent
is removed under vacuum. The material is dissolved in diethylether and washed with saturated
~ ~WO 93/08166 2 1 1 8 9 2 0 PCI/US92/07314
-23-
sodium bicarbonate (twice) and brine. The solvent is removed under vacuum to leave an oil (8.9
g). Purification by flæh chromatography (230400 mesh silica gel, 10-20% ethyl acetate in hexane)
gives the title compound æ a mixture of cis-trans isomers (1.57 g).
STEP 5
Preparation B5(a): 1,2-Bis(2-hydroxyethyl)-4chloro-7-methoxyindane, see CHARTB,
STEP 5, compound B6.
A solution of 1,2-bis(carboethoxymethyl)~chloro-7-methoxyindane (1.72 g, 4.85 mmol)
in diethylether (50 ml) is added to a suspension of lithium aluminum hydride (1.5 g, 39.5 mmol)
at room temperature. The mixture is stirred for 45 minutes, cooled in ice, and water (1.5 ml), 15%
sodium hydroxide (1.5 ml), and water (4.5 ml) were added in succession. The mixture is stirred
at room temperature for 15 minutes, and the precipitate is filtered and washed with tetrahydrofuran.
The filtrate is dried (MgSO~), and the solvent is removed under vacuum to leave the title compound
(1.34 g) as a mixture of cis-trans isomers.
STEP 6
Preparation B6(a)~ Bis(p-toluenesulfonyloxyeth-2-yl)-4chloro-7-methoxyindane, see
CHART B, STEP 6, compound B7.
A solution of the 1,2-bis(2-hydroxyethyl)~chloro-7-methoxyindane (1.57 g, 5.80 mmol)
is cooled in ice, and p-toluenesulfonyl chloride (3.32 g, 17.4 mmol) is added. The mixture is stirred
at 0C for I hour and allowed to stand at 0C for 20 hours. Water (12 ml) is added, and the
mixturc is stirred at room tcmperature for 45 minutes. The mixture is diluted with diethylether,
washed with 10% hydrochloric acid (3 times), sa~urated sodium Wcarbonate, and brine~ The
solution is dried (MgSO,), and the solvcnt is removed under vacuum to leave the title compound
25 as a yellow oil (2.9 g).
STEP 7
Preparation B7(a), Examples 11-12: preparation of the diastereomers, cis and Irans-7-
chloro~ metho~y-Sa,lOb-dihydro-3N-n-propyl-6H-indenoll,2-dlazepine, see CHART B, S~EP
.~
30 7, compound B8. A mixture of 1,2-bis~-tolucnesulfonyloxyeth-2-yl~chloro-7-methoxyindane
(2.9 g, 5.0 mmol), n-propylamine (0.33 g, 5~6 mmol), and potassium carbonate (2.07 g, IS.0 mmol)
in acetonitrile is sdrred at reflux in an oil bath maintained at 90C for 3 hours. n-P~pylamine (0.72
g, 12.2 mmol) is added. and the reflux contDued for 19 hours. The mixture is diluted with
diethylether and washed with water, 10% sodium carbonate, and brine. The solution is dried
35 (MgSO4), and the solvent is removed undervacuum to leave an oil (1.45 g). Purification by gravity
cl~matography (7~230 mesh siUca gel, 0.7% isopropylamine and 10% ethyl acet~te in hexane)
WO 93/08166 ~ 2 -~ - PCr/l,'S92/07314
-24-
gives two compounds. The faster moving compound (0.70 g) is dissolved in diethylether and
acidified with excess e~hereal hydrochloric acid. The precipitate is centrifuged. washed with
diethylether, and crystallized fivm methanoVdiethylether to give the trans-compound as a colorless
solid (0.69 g, m.p. 195-196C). The slower moving compound (O.S5 g) is converted to the
S hydrochloride salt in a similar manner giving the cis-compound as an off-white solid (0.47 g, m.p.
245-246C).
WO 93/08166 2 1 1 8 ~ 2 (1 PCr/US9~/07314
CE~ART A page 1
R2 A 1
step 1 1
Rl rC ~2CII3
R~
A 2
Step 2 1 :~
o~
R
~ .
R2 A 3
Step 3
211 ~?~13
WO 93/08166 PCI~/US92/07314
-26-
C~9RT A page 2
Step 3
Xl ~Si-~e2t-Butyl
~ '
R2
~4
Step4
2t-B3tyl
~"c'~C l2CII3
R2 A S
Step 5
Rl ~O~o
~G c~ 2~3
R2 A 6
Step 6
;~N0 93/08166 2 i l ~ ~ 2 Q Pcr/US92/07314
-27-
CHART A page 3
Step 6
R
R
Step ~ 1 A 7
R
~~'~EIr
R~
A 8
Step 8
C~3
~2
A 9
Step 9
W~ 93/08166 2 ~ 2 n PCI/lJS92/0731
-28-
G~[ART A pa~e 4
Step 9 ¦
c~
Rl~
2 A 10
Step 10
R2 A 11
Seep 11
N~'
Rl~J
R2 A 12
Step 12
2 ~
;~0 93/08166 PCI`/US92/07314
-29-
CIIART A page 5
Step 12
R ,
R,.
A 13
WO 93/08166 211 g 3 2 ~ PCI`/US92~07314
-30-
CEART B pa8e 1
R~
step
R
B 2
Ste~ 2
I~d 0~2C33
B 3
Ste~ 3
, W093/08166 2~ 20 PCI/US92/07314
C~ART B page 2
Step3
Rl~ o_ C~2C~3
R~--b-~2C~3
Ste,o 4 1 3 4
Rl ~C~ 2C~3
~g~ 3
B S
Step 5
0
Rl ~
~~
R2 B 6
. Step 6
~3~
WO 93/08166 ~ 2 {~ pcr~us92/o7314
-32-
CHART B page 3
Step6
OSO2Tolue~e
Rl ~ :
~~~OSO2Tol~
Step 7
Rl f N~
R2 B 8
`~O 93/08166 211 ~ ~ 2 ~ PCl'/US92/07314
-33-
PART 11
Reactions of CHART C
Step 1, carboxylic acid, Cl, is reduced with borane/dimethylsulfide to give the benzylic
alcohol, C2. Step 2, the benzylic alcohol, C2, is converted into the benzylic bromide, C3, with
S NBS and triphenylphosphine. Step 3, the benzylic bromide, C3 is alkylated with the enolate of
- ethyl acetate to give C4. Step 4, C4 is reduced to the alcohol, C5, with LAH. Step 5, the alcohol,
C5, is oxidized under Swem conditions to afford the aldehyde, C6.
Step 6, the atdehyde, C6, is treated with vinylmagnesium bromide to afford the allylic
alcohol, C7. Step 7, the atlylic alcohol, C7, is treated with triethylorthoacetate to cause a Claisen
10 rearrangement to give the ester, C8. Step 8, the ester, C8, is saponified with sodium hydroxide in
aqueous methanol to afford the acid, C9. Step 9, the acid, C9, underwent a Curtus rearrangement
with the aid of diphenylphosphorylazide to afford the carbamate, C10.
Step 10, the carbamate, C10, is cycliz'ed to the 1,2,3,4,4a,5,6,10b-
transoctahydrobenzo[f~isoquinoline, Cll, using paJaformaldehyde and borontrifluoride-etherate.
15 Step 11, the carbamate, Cl 1, is saponified by refluxing potassium hydroxide in aqueous ethanol to
afford the amine C12. Step 12, the amine, C12, is resolved into pure enantiomers by crystaDization
with opticatly active di-~toluoyltartaric acid in methanol. From this point, step 12, each enantiomer
is independently converted through the subsequent steps to afford opticatly pure products.
Step 13, the amine, now the pure enantiomer, C13, is converted into the propionamide, C14,
20 with propionylchloride. Step 14, the propionamide, C14, is reduced with LAH to give C15.
Altematively, from Step 13, the amine, the pure enantiomer, C13, can be converted directly into
C15 by atkylation with bromopropane. Step 15, C15 is converted into the carboxamide, C16, by
subsequent treatment with t-butyllithium and trimethylsilylisocyanate. The hydro-debrominated
anatog, C17, was isolated as a side product. The hydro-debrominated analog, C17, can be
25 synthesized in high yield by treatment with t-butyllithium followed by a water quench.
Step 16, the carboxamide, C16, is dehydra~ed to the nitrile, C20, with the aid of Burgess'
reagent. Step 17, the carboxamide, C16, is hydrogenated to remove the chlorine using palladi-
um/carbon, affording C18. Step 18, the compound, C18. is dehydrated using Burgess' reagent to
afford the nitrile, C19.
30 Structures are shown in CHART C.
The compounds of this invention have high oral potency and a long duration of action.
This exceptionally good bioavailability combined with a long period of activity are beneficial to
effective clinical treatment. The preferred compounds of PART II are not only active but have
incre~sed bioavailability as shown by metabolism studies. See PART 11- TABLES I-II, below.
35 The preferred compounds from the list below and PART 11 - TABLES I and Il are compounds
numbered 1, 2~ 7~ and 11.
WO 93/08166 2 1 ~ ~ ~ 2 ~ PCI`/US92~07314
-34-
The compounds of this invention are useful both as intermediates to produce other
compounds and they are useful to treat central nervous system disorders. The utility of the
compounds of this invention to treat central nervous system disorders is shown in behavioral.
physiological and biochemical tests. These tests are described below.
CNS Receptor Binding Assay (PART Il - TABLE 1): The Central Nervous System Binding
Assay measures the percent inhibition from experiments employing test compounds at 1 microMolar
concentration competing with various radioligands for binding to whole brain membranes,
membranes prepared from specific brain structures, or membranes prepared from cell lines
expressing cloned receptors. When percent inhibition is equal to lO0 the test compound binds as
well as the standard compound. The standard compound for the dopamine Dl receptor is SCH
23390.
Metabolism (PART n - TABLE Il):
HeDatocvte. Intrinsic clea~nce of 3 concentrations of compound ~2, 5 and 15 ug/ml)
following a 60 rninute incubation at 3rC in the presence of a suspension of freshly prepared rat
hepatocytes (5.0 million cellslml). Aliquots of each incubate were withdrawn during the incubation
and analyzed for parent compound using HPLC methodology. lntrinsic clearance is expressed as
ml/min/5 million cells. Metabolic stability relative to the control, ( ~) l-fonnyl-6,7,8,9-tetrahydro-N-
di-n-propyl-8-amino-3H-benzle]indole, is determined.
Microsome. Micromes prepared from Sprague Dawley rat livers were incubated at 37 C
with 5 or 12.5 micromolar substrate. The metabolic half-lives of the test compounds were
determined from the HPLC from plots of LN(peak area) vs time. These were ratioed with the
corresponding half-life of 5-(dipropylamino)-5,~dihydro~H-imidazo(4,5,l-y~quinoline-2(1H)~ne,
(-), monohydrobromide hydrate to give relative metabolic half lives.
The compounds below have been subjected to one or more of the biological tests described
above. PART n - TABLES I -11 use the numbers assigned below.
WO 93/08166 213~ ~ ~ ? PCl/US92J07314
Compound Number Name
1. (-)-10-Bromo-7-chloro-1,2,3,4,4a,5,6,10b-trans-octahydro-3N-
propylbenzo[f~isoquinoline, See CHART C STEP 14 compound C15, EXAMPLE 1.
2. (+)-10-Bromo-7-chloro-1,2,3,4,4a,5,6,10b-trans~octahydro-3N-
5 propylbenzolfJisoquinoline, See CHART C STEP 14 compound C15, EXAMPLE 2.
3. (-)-7-Chloro-10-carboxamido-1,2,3,4,4a,5,6,10b-trans-octahydro-3N-
propylbenzolt~;soquinoline, See CHART C, STEP 15, compound C16, EXAMPLE 3.
4. ~+)-7-Chloro-10-carboxamido-1,2,3,4~4a,5,6,10b-~rans-octahydro-3N-
prnpylbenzolf~isoquinoline, See CHART C, STEP 15, compound C16, EXAMPLE 4.
5. (-)-7-Chloro-1,2,3,4,4a,5,6,10b-trans-octahydro-3N-propylbenzo[fpsoquinoline,compound C17, from prepara~ion C15, EXAM~LE 5.
6. (+)-7-Chloro-1,2,3,4,4a,5,6,10b-t~ans-octahydro-3N-propylbenzolfJisoquinoline,
compound C17, from preparation C15, EXAMPLE 6.
7. (-)-7-Chloro-10-cyano-1,2,3,4,4a,5,6,10b-trans-octahydro-3N-
15 propylbenzolf~isoquinoline. see CHART C, STEP 16, cornpound C20 EXAMPLE 7.
8. (+)-7-Chloro-10-cyano-1,2,3,4,4a,5,6,10b-trans-octahydro-3N-
propylbenzo[fJisoquinoline. see CHART C, STEP 16, compound C20, EXAMPLE 8.
~ . (-)-10-Carboxamido-1 ,2,3,4,4a,5,6,1 Ob-octahydro-trans-3N-
propylbenzol~isoquinoline, see CHART C, STEP C17, compound C18. EXAMPLE 9.
10. (+)-10-Carboxamido-1,2,3,4,4a,5,fi,10b-octahydro-tr~ns-3N-
propylbenzo[f~isoquinoline, see CHART C, STEP 17, compound C18, EXAMPLE 10.
I 1. (-)-lO Cyano-1,2,3,4,4a,5,6,10b-octahydro-tr~ns-3N-propylbenzo[~isoquinoline.
see CHART C. STEP 18. compound Cl9. EXAMPLE 11.
12. ~+)~ Cyano-1,~,3,4,4a,~,6,10b-octahydro-trans-3N-propylbenzo[flisoquirloline,
see CHART C, STEP 18, compound Cl9, EXAMPLE 12.
2~?
WO 93/08166 PCI/US~2~07314
-36-
PART Il - TABLE I
BIOLOGICAL DATA - CNS Receptor Binding
Dopamine - D1 I)opamine - D2
Compound No % inhibition % inhibition
92.09 97.06
2 16.00 81.~4
3 14.54 ~3.42
4 3.18 27.37
25.08 89.87
6 40.95 95.92
7 83.17 100.00
8 9.40 24.67
9 24.77 2.90
10 48.35 4.87
11 36.66 100.0~
12 15.14 90.18
_
. . . ~
PART 11 - TABLE 11
BIOLOGICAL DATA - Metabolism ~hepatocyte and microsome)
Hepatocyte Microsome
Compound No. (control is 1.0) ~control is 1.0)
. ~ _
. 0.120 --
2 0.140 --
3 ----
4 ----
, 0.120 0.20
6 0.040
7 0.~30 0.91
8 0.200 -
9 0.500 1.30
1.610 --
1l 0,130 0.48
12 0.070 0.28
~ ..... --............ .. _ .
.
Without fur~er elaboration, it is believed that one skilled in the art can, using the preceding
description~ practice the present invention to its fullest extent. The following detaiied examples
describe how to prepare the vanous compounds and/or pçrform the various processes of the
50 invention and are to be construed as merely illustrative, and not limitations of the preceding
WO 93/08166 2 ~ 2 0 PCI`/US92/07314
-37-
disclosure in any way whatsoever. Those skilled in the art will promptly recognize appropriate
variations from the procedures both as to reactants and as to reac~on conditions and techniques.
PREPARATIONS AND EXAMPLES FROM CHART C
S STEP 1
Preparation Cl, Bromo-2-chlorobenzyl alcohol, see CHART C, STEP 1, compound C2.
S-Bromo-2-chlorobenzoic acid (107.9 g, 4S8.2 mmol) in THF (1000 mL) is cooled to 0C.
Borane-methyl sulfide complex (1.3 eq) is added. After 25 minutes at room temperature the
solution is refluxed for 30 minutes. Water, 300 mL, is added at 0C, then 2N hydrochloric acid is
10 added. After stirring 45 minutes ~ room temperature, additional acid is added and the mixture
extracted with ether/methylene chloride. The organic layer is washed successively with more
aqueous acid, water, saturated aqueous sodium bicarbonate and brine. The organic layer is then
dned over sodiurn sulfate, filtered, and stripped of solvent to yield a white solid, 100.9 g (99%),
mp 90.0-92.0C.
STEP 2
Preparation C2, 5-Bromo-2-chloroben~yl bromide, see CHART C, STEP 2, compound
C3.
5^Bromo-2-chlorobenzyl alcohol (106.6 g, 481.4 mmol) in methylene chloride (1000 mL)
is cooled to 0C. N-Bromo-succinimide (1.2 eq) is added over 60 minutes and the ice bath is
20 removed. After 2 hours, methanol (8 mL) is added. The solution is poured into 40~0 ether in
hexane (2100 mL); silica gel (800 mL, 230~00 mesh) is added. The mixture is f;ltered through
another 200 mL of silica gel and the silica gel plug is rinsed with 40% ether in hexane (1050 mL~.
Solvents are removed and the residue is flash chromatographed on a silica gel column (24 x 7 cm),
followed by elution with methylene chloride/ethyl acetate/hexane ~2:10:88) to yield a red solid,
25 108.9 g (79%), mp 72.5-74.5C.
STEP 3
Preparation C3, ethyl 3-(5-bromo-2-chlorophenyl)propiw~ate, see CHART C, STEP 3,compound C4. `
S-Bromo-2-chlorobenzyl bromide (108.9 g, 382.9 mmol) in TH~ (475 mL) is added to ethyl
....
30 acetate (60.0 mL, 612.7 mmol) in lithium diisopropylamide (1.5 eq in TH~, 650 mL) at -78C.
After sdr ing for 2 hours, glacial acetic acid (I equivalent) is added. The mixture is pardtioned
between ether and 2N hydrochloric acid. The organic layer is washed with water, saturated aqueous
sodium bicarbon~te, and brine. lt is dried over sodium sulfate, filtered, and the solvents removed
under vacuum. The residual oil is flash cl~omatographed on a 20x7 cm silica gel column. The
35 silica gel column is eluted with 7.5% ethyl acetate in hexane to yield 87.2 g (78%) orange oil.
STEP 4
2 ~ t ~ ~ 2 s)
WO 93/08166 PCr/US92/07314 ''
-3g-
Preparation C4, 3-(5-Bromo-2-chlorophenyl)propanol, see CHART C, STEP 4, compound
CS.
Ethyl 3-(S-bromo-2-chlorophenyl)propionate (87.2 g, 299.1 mmol) in ether (300 mL) is
added to lithium aluminum hydride (12.0 g, 299.1 mmol) in ether (300 mL) at 0C. After 30
S minutes, water (10 mL), sodium hydroxide (10 mL, 15% aqueous), and water (30 mL) are added
successively and stirred at room temperature for lS minutes. The slurry is filtered and the filtrate
is d~ied over sodium sulfate to yield a pale orange oil (68.9 g, 92%).
STEP S
P~paration C5, 3-(5-Bromo-2-chlorophenyl)propionaldehyde, see CHART C, STEP S,
compound C6.
Dimethyl sulfoxide (43.1 mL, 0.61 M) in methylene chloride (300 mL) is cooled to -78C.
Oxalyl chloride (26.5 mL, 0.30 M) is added. After 30 minutes, 3-(S-bromo-2-chlorophenyl)-
propanol (68.9 g, 0.28 M~ in methylene chloride (250 mL) is added. After 20 minutes, tAethyl-
amine (192 mL, 1.38 M) is added. The slurry is warmed to room temperature. Water is added and
the mixture is extracted with ether. The organic layer is successively wæhed with 2N hydrochloric
acid, water, saturated aqueous sodium bicarbonate, and brine. It is dried over sodium sulfate and
yields 68.3 g (100%) pale oil.
STEP 6
Preparation C6, 5-(S-Bromo-2-chbrophenyl)penten-3-ol, see CHART C, SlEP 6
compound C7.
Vinylmagnesium bromide (339 mmol) in THF (900 mL) is cooled to 0C. 3-(5-Bromo-2-
chlorophenyl)propionaldehyde (68.3 g, 276 mmol) in TH~ (500 mL) is added and the solution
stirred at room temperature for 2.5 hours. Saturated aqueous ammonium chloride is added, then
2N hydrochloric acid is added. The mixture is extracted with ether. The ether layer is washed with
water then with brine. The washed ether layer is dried over sodium sulfate and yielded 71.6 g
(g4%) yellow oil.
STEP 7
Preparation C7(a). ethyl 7-(5-bromo-2-chlorophenyl)-~rans-hept-~enoate, see CHART
C, STEP 7, compound C8.
S-(S-Bromo-2-chlorophenyl)penten-3-ol (71.6 g~ 260 mmol~, triethylorthoacetate (164 mL
885 mmol)~ and propionic acid (1~1 mL, 099 mmol) are combined and heated at tS0C for 2 hours
with removal of ethanol. The solution is cooled to 0C and 2N hydrochloric acid is added. The
mixture is extracted with ether. The ether layer is washed with water then with brine. The either
layer is then dried over sodium sulfate and yields 84.12 ~ (94%) of yellow oil.
STEP 8
Preparation C8~ 7-(5-Bromo-2-chlorophenyl~-~ans-hept-~enoic acid~ see CHART C.
``VO 93/08166 2 1 1 ~ ~ 2 ~ PC,/US92~073l4
-39-
STEP 8 compound C9.
Ethyl 7-(5-bromo-2-chlorophenyl)-trans-hept4-enoate (~4.1 g, 243 mmol), potassium
hydroxide (47 g, 730 mmol), methanol (400 mL), and water (50 mL) are refluxed for 2.5 hours.
The solvents are removed under vacuum and the residue is partitioned between ether and water.
S The aqueous layer is acidified with hydrochloric acid and extracted with ether. The ether layer is
washed with waler and brine and is dried over sodium sulfate. It yields 57.8 g (7~%) yellow solid.
A portion is rec~ystallized from ethyl acetate, mp 53.5-55.0C.
.STEP 9
Prepa~ation C9, 6-(5-Bromo-2-chlorophenyl)-~rans-hex-3-enyl ethyl carbamate, seeCHART C, STEP 9, compound C10.
7-(5-Bromo-2-chlor~phenyl)-trans-hept~enoic acid (57.8 g, 182 mmol), 1,4-dioxane (600
mL), diphenylphosphoryl azide (39.2 mL, 182 mmol),and triethylamine (25.4 ml,, 182 mmol) are
stirred at room temperature for I hour, then refluxed 30 minutes. Nitrogen evolves. Ethanol (æ.l
mL, 364 mmol) is added and refluxing continues for 2 hours. The solution is cooled to O~C and
sodium hydroxide (1.1 equivalent) and water are added. After 30 minutes, the mixh~re is partitioned
between ether and water. The e~er layer is washed successively with water, 2N hyd~chloric acid,
water, saturated aqueous sodium bicarbonate and brine. The solution is dried over sodium sulfate.
The yellow oil is flash ch~matogra~hed on a 16 x 7 cm silica gel column and eluted with 5%
followed by 10% ethyl acetate in hexane to yield 37.2 g (57%) yellow oil.
STEP 10
Preparation C10, 10-Bromo-3N-carboethoxy-7-chloro-1,2,3,4,4a,5,6,10b-trans-
octahydrobenzo[f]isoquinoline, see CHART C, STEP 10, compound Cll.
6-(5-Bromo-2-chlorophenyl)-~rans-hex-3-enyl ethyl carbamate (46.4 g, 129 mmol) is
dissolved in methylene chloride ~250 mL). Paraformaldehyde (4.3 g, 135 mmol) is added and then
boron trifluoride etherate (17.4 mL, 142 mmol). The slurry is wasmed to 30C and after 5 minutes
it is poured onto sodium carbonate and ice. The slurry is extracted with ether. The ether layer is
washed with brine and then dried over sodium sulfate. This produces a thick orange oil which is
flash chromatographed on a 19 x 7 cm silica gel column and eluted with 2% acetone in hexane to
yield 41.6 g (83%) of pale oil.
STEP I I
Preparation Cl l, 10-Bromo-7-chloro-l,2,3,4,4a,5,6,10b-t~uns-octahydrobenzolf]-
isoquinoline, See CHART C. STEP 11, compound C12.
l~Bromo-3N-calboethoxy-7-chloro 1,2,3,4,4a.5,6,10~trans-octahydrobenzn[~isoquinoline
(41.1 g. 110 mmol) is refluxed with potassium hydroxide (28.5 g~ 441 mmol), methanol (200 mL).
and-water (20 mL) for 65 hours. The solvents are removed under vacuum and the residue is
partitioned between water and ether/methylene chloride. The organic layer is washed with water
Wo 93~08~ ) 2 ~ PCr/US92/07314 ~ ~
-40-
twice and then with brine. The organic layer is dried over sodium sulfate to yield 30.9 g (93%
crude) of pale oil.
STEP 12
Preparation C12, enantiomeric separation of 10-bromo-7-chloro-1,2,3,4,4a,5,6,10b-trans-
S octahydrobenzo[f)isoquinoline, see CHART C, STEP 12, compound C13.
A hot methanol solution (650 mL) of 10-bromo-7-chloro- 1,2,3,4,4a,5,6,10b-~rans-
octahydrobenzo[flisoquinoline (30.9 g, 103 mmol) is combined with di-p-toluoyl-D-tartaric acid
(22.5 g, 56.5 mmol). Upon cooling, the resultant white solid is filtered. The filtrate is dried of
solvent under vacuum, then pa~titioned between aqueous sodium hydroxide and ether. The ether
layer is washed with water and brine, then dried over sodium sulfate to yield a pale oil. The above
procedure is repeated five more times alternating between the D and the L folms of ~e acid. Thc
combined ~salts are then converted to free base and converted back to the v-sa1ts as described
above. A second cycle yielded the free base as a white waxy solid, 9.0 g (29%). ~a]~5589=
-212.9 (c= 0.68, MeOH).
NOTATION COMMENT
Steps 13 - 19, below, show how ~e enantiomers, "~a)" and "(b)" are independentlyconvelted to give optically pure products. Enantiomer~ for aminosulfonyl deriva~ives are labeled
with preparation numbers that include the letters "(c)" and "(d)."
STEP 13
Pleparation C13(a), (-)-10-Bromo-7-chloro-1,2,3,4,4a,~,6,10b-trans-octahydro-3N-propionylbenzo[t~isoquinoline, See Chart C, STEP 13, compound Ct4.
(-)-1 0-Bromo-7-chloro- 1 ,2,3,4,4a,S,6,10b-tr~ans-octahydro benzolf~isoquinoline ( 1.75 g, 5.82
mmol), triethylamine (0.97 mL, 6.98 mmol), and methylcne chloride (12 mL) are combined and
cooled to 0C. Propionyl chloride (0.57 mL, 6.40 mmol) is added. Af~r I hour water is added and
the mixture is extracted with ether. The ether layer is washed with 2N hydrochloric acid, water,
saturated aqueous sodium bicarbonate, and brine. The ether layer is dried over sodium sulfate to
yield 2.17 g (100%) thick oil.
Preparation C13(b), (+)-10-Bromo-7-chloro-1,2,3,4,4a,5,6,10b-trans-octahydro-3N-propionylbenzolfJisoquinoline, See Chart C, STEP 13, compound C14.
Use the same procedure as above, for C13(a), only start with (+)-10-Bromo-7-chloro
1,2,3,4,4a,S,6,10~t~ans-octahydro benzolflisoquinoline.
STEP 14
Preparation C14(a), EXAMPLE 1, (-)-10-Bromo-7-chloro-1,2,3,4,4a,S,6,10b-trans-
octahydro-3N-propylbenzolf]isoquinoline, See CHART C, ST~P 14. compound CIS.
(-)-1~Bromo-7-chloro-1.2,3,4,4aS,6,10~rans-oc~ahydro 3N-propionylbenzo[flisoquinoline
(2.17 g, 6.08 mmol) in ether (6 mL) is added to lithium aluminum hydride (0.23 g, S.82 mmol) in
~ WO 93/08166 2 1 1 8 ~ 2 0 PCr/US9~/07314
-41 -
ether (6 mL) at O~C. The mixture is refluxed for I hour, then water (0.20 mL), sodium hydroxide
(0.20 mL, 15% aqueous), and water (0.60 mL) are se~guentially added. The mixture is filtered and
the fil~ate is dried over sodium sulfate tO yield 1.88 g thick oil. The oil is flash chromatographed
on a 2.2 x 15 cm silica gel column and eluted with 15% ethyl acetate in hexane to yield 1.77 g
5 (89%) of an oil. la]25589= -176.55~ (c=1.70 CHC13). The oil is dissolYed in ether and is acidified
with hydrochloric acid in ether. The resulting white solid is recrystallized from methanol and ether
to yield a white solid, m.p. 277C (decomposition).
Preparation C14(b), EXAMPLE 2, (+)-10-Bromo-7-chloro-1,2,3,4,4a,5,6,10b-~ans-
octahydro-3N-propylbenzo[f3isoquinoline, See CHART C, STEP 14, compound C15.
Use the same procedure as above, for C14(a), only start wlth (+)-1~Bromo-7-chloro-
1,2,3,4,4a,5,6,10b-trans-octahydro-3N-propionylbenzo[f3isoquinoline.
STEP 15
Preparation Cl5(a),EXAMPLE 3, (-)-7-Chloro-l~carboxamido-1,2,3,4,4a,5,6,10b-trans-
octahydro-3N-propylbenzo[f3isoquinoline, See CHART C, STEP 15, compound C16.
(-)-1~Bromo-7-chloro-1,2,3,4,4a,5,6,1Ob-trans-octahydro-3N-propylbeni~o[f3isoquinoline
(1.20 g, 3.50 mmol) in THF ~10 mL) is cooled to -78C and t-butyllithium (4.1 mL, 1.7 M in
pentane) is added. Trimethylsilylisocyanate (1.1 mL, ?.ûO mmol) is added. After 75 minutes at
room temperature, water is added and the mixture extracted wi~ ether. The ether layer is washed
with saturated a~ueous sodium bicarbonate and brine. The washed ether layer is dried over sodium
20 sulfate. The ctude product is flash chromatographed on a 2.2 x 15 cm silica gel column and eluted
with 5:1 methylene chloride:methanol. Product obtained is 0.86 g (80%) of a white solid, m.p.
220C (decomposition). [a]~5589 = -255.82 (c-0.455 CHCI3~. This solid is dissolved in isopropanol
and ether, therl hydrochloric acid in ether is added. The white solid is recrystallized from
isopropanol and ether, m.p. 165C (decomposition).
Preparation C15(b), EXAI~PLE 4, (+)-7-Chloro-l~carboxamido-1,2,3,4,4a,5,6,10b-trans-
octahydro-3N-propylbenzo[t~isoquinoline, See CHART C, STEP 15, compound C16.
Use the same procedure as above, for Cl5(a~, only start with (+)-10-bromo-7-chloro-
1 ,2,3.4,4a,5,6.10b-trans-octahydro-3N-propylbenzo[f]isoquinoline.
EXAMPLE 5, isolation of ~-~-7-Chloro-1,~,3,4,4a,5,6,10b-trans-octahydro-3N-propylbenz-
olflisoquinoline, compound C17, from preparation C15. This compound is isolated from (-)-7-
chloro- I O-carboxamido- 1 ,2,3,4,4a,5 ,6.1 Ob-trans-octahydro-3N-propylbenzolf]isoquinoline
chromatography using an initial elution of 20:1 methylene chloride:methanol to yield 0.30 g (1.14
mmol) pale paste. This is dissolved in methanol and ether and hydrochloric ~cid in ether is added.
35 The white solid is recrystallized from methanol and ether to yiçld 0.14 g white solid. m.p. 250C
(decomposition), lal2558~ -92.73 (c= 0.33 MeOH).
211~2~
WO 93/08166 PCr/lJS92/07314
-42-
EXAMPLE 6. isolation of (+)-7-Chloro-1,2,3,4,4a,5,6,10b-trans-octahydro-3N-
propylbenzolflisoquinoline, compound C17, from preparation C15. This compound is isolated
from (-)-7-ch1Or~-10-carboxamido-1,2,3,4,4a,5,6,10~trans-octahydro 3N-propylbenzo[flisoquinoline
using the same procedure as above.
Preparation C15(c) and Cl5(d) ~+) and (-)-7-chloro-10-aminosulfonyl-1,2,3,4,4a,5,6,10b-
trans-octahydro-3N-propylbenzolfJisoquinoline, See CHART C, STEP 15, compound C16. The
title sulfonamides are prepared from the enantiomers of 10-Bromo-7-chloro-1,2,3,4,4a,5,6,10b-trans-
octahydro-3N-propylbenzolflisoquinoline, compound C15, via metal-halogen exchange with t-
10 butyllithium, followed by conversion of the aryllithium to the sulfonamide according to thereference: S.L. Graham, ct al., J. Med. Chem., 32:2548 (1989).
STEP 16
Prepa~ation C16(a), EXAMPLE 7, (-)-7-Chloro-10-cyano-1,2,3,4,4a,5,6,10b-trans-
octahydro-3N-propylbenzo[l~isoquinoline. see CHART C, Sl'EP 16, compound C20.
(-)-7-Chloro-10-carboxamido-1,2,3,4,4a,5,6,10~trans-octahydro-3N-propylbenzolflisoqu-
inoline (0.45 g, 1.47 mmol) is dissolved in THF (7.5 mL) and methylene chloride (9.0 mL) and
Burgess' reagent (0.87 g, 3.67 mmol) are added. After 3 hours at room temperature it is poured
onto 10% aqueous sodium carbonate at 0C and extracted with ether. The ether layer is wæhed
with brine and dried over sodium sulfate. The amber pæte is flash chromatographed on a 1.2 x 16
20 cm silica gel column, eluted with 10:1 methylene chloridehnethanol to yield 0.41 g (97%) opaque
tan oil, la]255S9 -209.6, (c = 1.09 MeOH). This is dissolved in methanol and ether and
hydrochloric acid in ether is added. The white solid is recrystallized f~m methanol and ether, m.p.
275C (decomposition).
Prepa~ation C16(b), EXAMPLE 8, (+)-7-Chloro l0-cyano-1,2,3,4,4a,5,6,10b-~ns-
25 octahydro-3N-propylbenzolflisoquinoline. see CHART C, STEP 16, compound C20.
Use the same procedure æ above, for C16(a), only start with (+)-7-chloro-10-calboxamido-
1,2,3,4,4a,5,6,10~trans-octahydro-3N-propylbenzolflisoquinoline.
STEP 17
PrepD~ion C17(a), EXAMPLE 9, (-)-10 Carboxamido-1,2,3,4,4a,5,6,10b-octahydro-tr~ns-
30 3N-propylbenzotflisoquinoline, see CHART C, STEP C17, compound C18.
(-)-7-Chloro- 10-carboxamido- 1,2,3,4,4a,5,6,10b octahydro-trQns-3N-propylbenzotflisoqu-
inoline (I.Sg) is placed in a hydrogenation flask with palladium hydroxide/carbon (0.45g) and
ethanol (120 ml). This is hydrogenated under 50 p.s.i. for 12 hours then filtered through
diatomaceous earth. Solvent evaporation is followed by dissolving the residue in methylene
35 chloride and 10% aqueous sodium carbonate. The organic layer is separated and washed with water
and then brine. and dried over sodium sulfate. Solvent removal affords a white solid. 10~1255w = -
21~ 2~
WO 93/~8166 ` PCr/~S92/07314
43-
307.80 (c = 0.68 methanol). The amine is converted into the crystalline maleate salt with maleic
acid in methanollether (needles) []25S89 = -214.95 (c = 1.07 methanol), m~p. 240C (decomposi-
tion).
Preparation C17(b), EXAMPLE 10, (+)-10-Carboxamido-1,2,3,4,4a,5,6,10b-octahydro-
S trans-3N-propylbenzolflisoquinoline, see CHART C, STEP 17, compound C18. Use the same
procedure as above, for C17(a), only start with (+)-7-chlor~-10-casboxamido-1,2,3,4,4a,5,6,10b-
octahydro~trans-3N-propylbenzolf~isoquinoline.
Prepa~ion C17(c) and C17(d), (+) and (-) -(10)-aminosulfonyl-1,2,3,4,4a,5,6,10b-trans-
octahydro-3N-propylbenzo[flisoquinoline, see CHART C, SrEP 17, compound C18.
STEP 18
Preparation C18(a), EXAMPLE 11, (-)-10-Cyano-1,2,3,4,4a,S,6,10b-octahydto-~rans-3N-
propylbenzo[~isoguinoline, see CHART C, STEP 18, compound Cl9. (-)-10-Carboxamido-
1,2,3,4,4a,5,6,10hoctahydro-trans-3N-propylbenzo[flisoquinoline (300 mg, 1.1 mmol) is dissolved
in methylene chloride. Burgess' reagent (3 eq. 785 mg) is added and ~e solution is stirred for two
15 hours. The solution is poured into 10 % aqueous sodium carbonate and extracted. The organic
layer is washed with water and then brine. Drying over sodium sulfate and solvent removal affords
an oil [0~5589 = -202.64 (c = 1.89 chloroform). l'his is converted to the crystalline hydrochloride
salt witb etheral hydrogen chloride and methanol, m.p. 325C (decomposition).
Preparation C18(b), EXAMPLE 12, (+)-10-cyano-1~2,3,4,4a,S,6,10b-octahydro-~rans-3N-
20 propylbenzo[flisoquinoline, see CHART C, STEP 18, compound Cl9. Use the same procedureas above, for C18(a), only s~art with (+)-10-carboxamido-1,2,3,4,4a,5,6,10b-octahydro-Jrans-3N
-
propylbenzo[f~isoquinoline.
The follow~ng altemative procedures are provided fnr the synthesis of some compounds.25 The following synthesis steps begin in place of step 13 and are numbered 131, 132
etc., see page
S of CHART C.
STEP 131
Preparation C131, Example 13, (-)-10-Bromo-7-chloro-1,2,3,4,4a,~,6,10b-1rans-octahydro-
3N-benzylbenzolfJisoquinoline, see CHART C, STEP 131, compound C132~ Begin with staning
30 materials for preparation C13 from STEP 13 above. Benzyl ch1oride (5.4 mL~ 45.4 mmol) is added
to triethylamine ~6.6 mL, 47.6 mmol),
(-)-l~bromo^7-chloro 1,2,3,4,4aS,6,10b-~rans-octahydrobenzo[flisoquinoline (13.0 g~ 43.24 mmol)~
and dime~ylformamide (90 mL~ at 0C. After 30 minutes, the ice bath is removed and the slurry
stirred at room temperatur~ for two days. Water and e~her are added. The organic layer is washed
35 with water and brine and is dried with sodium sulfate to yield an amber oil.
[0~]25589--139.32 (c= 1.04~ MeOH).
211~ 2ii
WO 93/08166 PCI/US92/07314
Notation comment. In the compounds of this series the preparations followed by a lower
case (a) are where R = H, lower case (b) indicates R = CH3.
STEP 132
Preparation C132(a), Example 14, (-)-7-Chloro-10-sulfonamido-1,2,3,4,4a,5,6,10b-trans-
octahydro-3N-benzylbenzolfJisoquinoline, see CHART C, STEP 132, Compound C133, R = H.
tert-Butyllithium (8.0 mL, 1.7 M in pentane) is added to (-)-10-bromo-7-chloro-1,2,3,4,4a,5,6,10~
trans-octahydro-3N-berl2ylbenzo[flisoquinoline (2.58 g, 6.60 mmol) in THF (15 mL) at -78C.
After 10 minutes, sulfuryl chloride (0.8 mL, 9.90 mmol) is added in a quick dose and the cold bath
10 removed. After 1 hour, the solvents are removed under vacuum. THl~ (15 mL) is added and the
mixture cooled on an ice bath and ammonia sah~rated THF (15 mL) added in a quick dose to the
slurry, and the ice bath removed. Ammonium hdyroxide (10 mL) is then added. After 2 hours,
water and ether are added. The organic layer is washed wi~ water and brine and dried over sodium
sulfate to yield an ~nber foam, m.p. 97- 1~2C. [a]25589~ -158.2 (c= 0.56, MeOH).
Preparation Cl 32(b), Example 15, ~ 7-Chloro-10-methylsulfonamido-1,2,3,4,4a,~,6,10b-
trans-octahydro-3N-benzylbenzolf]isoquinoline, see CHART C, STEP 132, Compound C133, R
= CH3. tert-Butyllithium (9.3 mL, 1.7 M in pentane) is added to (-)-l~bromo-7-chloro-
1,2,3,4,4a,5.6,10~trans-octahydro-3N-benzylbenzo~f~isoquinoline (3.0 g, 7.68 mmol) in THF (7.5
20 mL) at -78C. After 10 mimltes, sulfuryl chloride (2.6 mL, 15.8 mmol) is added in a quick dose
and the cold bath removed. THF (7.5 mL) is added and after I hour, the solvents are removed
w~der vacuum. TH~ (15 mL) is added and methylamine saturated TH~; is added. After 50 minutes,
water and ethyl acetate are added. The organic layer is washed with water and brine and dned over
sodium sulfate to yield a yellow foam. The foam is flash chromatographed on a 19 x 2 cm silica
25 gel column and eluted with 10% me~ylene chloride and 25% ethyl acetate in hexane to yield a
yellow foam, m.p. 99C. [oc]25589= -148.5D (c= 0.60, MeOH).
STEP 133
Preparation 133(a), ~xample 16, (-)-10-Sulfonamido-1,2.3,4,4a,5,6,10b-~rans-
30 octahydrobenzolf]isoquinoline hydrochloride, see CHART C, STEP 133, Compound C134, R =
H. (-)-7-Chloro-l~sulfonamido-1,2,3.4,4a,5,6,10~trans-octahydro-3N-benzylbenzo-[f]isoquinoline
(0.20 g, 0.51 mmol), ethanol (50 mL), and palladium hydroxide, 20% on carbon (0.40 g) are shaken
under hydrogen (48 PSI) for 6 hours. The mixture is filtered through diatomaceous earth and
solvents removed wlder vacuum to yield a white solid. 0.18 g. Recrystallization from ether and
35 methanol yields a pink solid. 216C m.p.
WO 93/08166 2 1 1 g ~ ~ Q PCl/US92/07314
-45-
Preparation 133(b), Ex~nple 17, (-,1-10-Methylsulfonamido-1,2,3,4,4a,5,6,10b-trans-
octahydrobenzo[tlisoquino-line hydrochloride, see CHART C, STEP 133, Compound C134, R
CH3. (-)-7-Chloro- I O-methylsulfonamido- 1,2~3 ,4,4a,5,6, 1 Ob-trans-octahydro-3N-
benzylbenzolf~isoquinoline (0.78 g, 1.93mmol), ethanol (50 mL). and palladium hydroxide. 20%
5 on carbon (0.69 g) are shaken under hydrogen (43 PSI) for 21 hours. The mixture is filtered
through diatomaceous earth and solvents removed under vacuum to yield a pale foam, m.p. 95C.
a]2558~ -148.1 (c= 0.96).
STEP 134
Preparation 134(a), Example l 8, (-)-10-Sulfonan~ido-1,2,3,4,4a,~36,l0btrans-octahydro-
3N-propylbenzolfJiso-quinoline, see CHART C, STEP 134, Compound C135, R = H.
Bromopropane (0.02 mL, 0.25 mmol) is added to (-)-10-sulfonamido-1,2,3,4,4a,5,6,10b-Jrans-
octahydrobenzo[f~isoquinoline hydrochloride (0.077 g, 0.25 mmol) and triethylamine (0.08 mL, 0.56
mmol) in a~etonitrile (5 mL3 and DMF (2 mL). After heating at 80C for 4 hours, e~her and
15 aqueous sodium carbonate are added. The organic layer is washed with water and brine and dried
over sodiurn sulfate to yield an amber oil.
Preparation 134(b), Example 19, ~ 10-Methylsulfonan~ido-1,2,3,4,4a,5,6,10b-~ns-
octahydro-3N-propyl-benzo[flisQquinoline, see CHART C, STEP 134, Compound C135, R =
20 CH3. Bromopropane (0.05 mL, 0.50 mmol) is added to ~-)-10-methylsulfonamido-
1,2,3,4,4a,5,6,10b-trans-octahyd~benzo[flisoquinoline hydrochloride (0.15 g, 0.50 mmol) and
triethylarnine (0.14 mL, 1.00 rnmol) in acetonitrile (4 mL) and DMF ( I mL). After heating at 50C
for 4 hours, ether and aqueous sodium carbonate are added. The organic layer is washed with water
and brine and dried over sodium sulfate to yield a white foam, m.p. g4- 100C. la]25589= -187.6
25 (c= 0.47). This foam is dissolved in methanol and ether, then hydrochloric acid in ether is added.
The solid is recrystallized from methanol and e~er to yield gray crystals, m.p. 299-301 C.
CHART C begins next page.
2 1 ~ 2 ~
WO 93/08166 PCI`/US92/07314
46-
G~RT C (6-6-6 ring system) page 1
~c"
~2 0
Step 1 l C
Rl
x2 oa c a
Step2
~,
R2 ~r
c 3
~tep3
(c~ntinued)
WO 93/08166 2 1 1 ~ ~ 2 û Pcr/uS9~/07314
-47-
CHART C page 2
Step3
~2 C 4
step 4
Rl~
R2 c 5
StepS
Rl~ `C
~2
C 6
Step 6
....
(continued)
'2 1 ~
WO 93/08166 Pcr/US92/~7314 .
-48-
C~ART C page 3
Step 6
Rl~OE C 7
Step~ I
~C~ ~ C~3
~ O
R2 C 8
Step8
R
~0
C9
Step 9
(continued)
. WO 93/08166 2 ~ ?
Pcr/US92/07314
-49-
CHQRT C page 4
Step 9
R~CN O~\c33
Sl;ep 10 c 10
R2 `N~C~O~ CH3
Stepll 1
R3
R2 c 12
Step 12
(co~tinued)
WO 93/08166 PCT/US92/07314
-50-
CHART C page 5
Step 12
1ll~) C 13
R2
Step 130
Step13 /
c~ c
N'C~3 Step 131
R~ C 14 ¦~
step 14 Cl
c 132
~ r Step 132 r
Rz N~c~3 R~N
C 15
C 133
Step 15 Step 133 r
.. R~IN
~ ~N~
(co~tinued) ¢~.~cl
C 134
WO 93/08166 2 1 1 ~ !1 2 ~) PCr/US92/07314
-51 -
C~ART C page 6
Step 15
~ 3~&N C ~
,
~ N~c~3
R
R2 A 16
Step 16 ~ \
R~ C ~D Cll3 Step 17~
R2 Rl~&N~cll3
Step 18
~c~tlnued)
2 ~
WO 93/08166 PCI`/US92/07314
-52-
C~RT C page 7
Step 18
~~C~;