Note: Descriptions are shown in the official language in which they were submitted.
212 ~ o so
PREPARATION OF LIGNAN ANALOGUES
The present invention relates to a novel method of
preparing compounds of lignan series and to intermediates thereof.
More particularly, it relates to a method of efficiently preparing
compounds of lignan series having an alkyl ketone chain, which
comprises utilizing Michael addition reaction which provides
regioselective reaction.
The compounds of lignan series which are prepared by the
process of the present invention are useful for treating
arteriosclerosis, particularly atherosclerosis, and various
compounds were disclosed by the present inventors (Japanese
Patent Publication Kokai No. 310634/1993, International Patent
Publication W093/08155).
The present invention relates to a process for preparing a
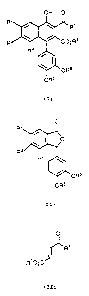
compound represented by the formula (I)
R'
/ I \ , R1
R3 \ / C02R~
2 o R4 / 1
\ ORs
ORS
(s)
in which R1 is alkyl, cycloalkyl, cycloalkyl-lower alkyl, or aralkyl;
each of R2 and R3 is lower alkoxy or R2 and R3 are combined
together to form an alkylenedioxy;
- 212 ~ o so
-2
R4 is lower alkoxy or hydrogen;
each of R5 and R6 is lower alkyl; and
R ~ is lower alkyl;
which is characterized by that a lactone compound represented by
the formula (1l)
R2.
0
%~
R3~
R4
OR°
OR5
(xz>
in which R2, R3, R4, R5, and R6 are as defined above, is allowed
to react with a compound of a formula (III):
0
~R~
R~02C
(=xx)
in which R~ and R~ are as defined above in the presence of a base,
and then the resulting compound is subjected to dehydration. In the
present invention, the aimed compound can be prepared efficiently.
Accordingly, the process of the present invention is a suitable
process for a large scale synthesis of lignan analogues.
In other aspects, the present invention relates to the above
compounds (II) and (III), which are useful in the process of the
212 Los
0
present invention.
In the specification, the term "alkyl" for R1 means linear
or branched C1 to C1p alkyl, while "lower alkyl" means linear or
branched C1 to Cg alkyl such as methyl, ethyl, n-propyl, isopropyl,
n-butyl, isobutyl, sec-butyl, n-pentyl, 1-ethylpropyl and
2-ethylbutyl, etc.. The term "cycloalkyl" means C5 to C~ cycloalkyl
such as cyclopentyl, cyclohexyl and cycloheptyl. The term
"cycloalkyl lower alkyl" means a group in which the above-defined
lower alkyl has the above-defined cycloalkyl substituent and its
examples are cyclohexylmethyl and cyclopentylethyl, etc.. The term
"aralkyl" means a group in which the above-defined alkyl is
substituted with an aryl and its examples are benzyl,
p-methoxybenzyl, phenylethyl, phenylpropyl and naphthylmethyl.
"Lower alkoxy" means a group in which oxo is substituted
with the above-defined lower alkyl and its example are methoxy,
ethoxy and propoxy, etc.
The term "alkylenedioxy" for R2 and R3 means C1 to C3
alkylenedioxy such as methylenedioxy, ethylenedioxy and
propylenedioxy.
The process for preparation of the present invention is
composed of two steps, which are provided below:
z~2~oso
Step 1
R2 O O HO O
/ I R2
O R1 / I \ ~R~
R3 + ~ R \
R4 I \ R~02C Base ~ a HO C02R7
~ R /
/ I
OR6
(III) \ OR6
ORS
OR5
(II)
(IV}
Step 2
HO O OH O
R2 R2
/ I \ wR, / I \ ,R,
R \ C02R~ Acid R3 \ / C02R~
Ra HO ~ Ra
/I /
\ I
~OR6 \ ORs
1 5 OR5 ORS
(IV) (I)
The first step is a step in which a lactone (II) and an
unsaturated ketone (III) are reacted in the presence of a base to
yield a compound (IV).
The second step is a step in which the compound (IV) is
subjected to dehydration by treatment with an acid to yield the
desired compound (I).
Reaction Conditions
There is no particular limitation as to the ratio of the
-s- ~ 212 1 0 6 0
compounds (II) to (III) used in the first step, but the compound (III)
is usually used in an excess amount to the compound (II), preferably
from 1:1 to 1:2.
Examples of the base used in this step are ordinary dialkyl
metal amides such as lithium diisopropylamide and sodium
dicyclohexylamide, etc., and bis(trialkylsilyl) metal amides such as
lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl) -
amide and sodium bis(triethylsilyl)amide, etc.. Preferably, lithium
bis(trimethylsilyl)amide is used.
With regard to the solvent for the reaction, ethers such as
tetrahydrofuran, diethyl ether and dioxane; hydrocarbons such as
n-hexane and n-pentane; aromatic hydrocarbons such as benzene and
toluene; halogenated hydrocarbons such as methylene chloride; and
amides such as N,N-dimethylformamide and hexamethylphosphoric
triamide, may be used either solely or in combination. Preferred
solvents are tetrahydrofuran, methylene chloride, N,N -
dimethylformamide, and hexamethylphophoric triamide.
The reaction of this step is carried out usually at -100°C
to 100°C (preferably from -80°C to the room temperature) and
completed within several minutes to several hours.
Examples of the acid used in the second step are organic or
inorganic acids such as hydrochloric acid, sulfuric acid,
trifluoroacetic acid, sulfonic acids (methanesulfonic acid, p -
toluenesulfonic acid), or Lewis acids (boron trifluoride or titanium
tetrachloride, etc.). Preferably, boron trifluoride or
methanesulfonic acid is used. There is no particular limitation as
_6_ 212106p
to the amount of the acid, but preferably it is used in 1 to 2
equivalents.
With regard to the solvent for the reaction, aromatic
hydrocarbons such as benzene and toluene; halogenated hydrocarbons
such as methylene chloride; and nitrites such as acetonitrile may be
used.
The reaction of this step is usually completed at -70°C to
100°C (preferably -20°C to the room temperature) within several
minutes to several hours.
The lactone (II) and the unsaturated ketone (III) of the
present invention, which are also starting materials in the present
process, can be prepared from the known compounds, for example, by
methods of the following preparations.
The present invention will be further illustrated by way of
the following Preparations and working examples which, however,
are not intended to limit the scope of the present invention.
Synthetic Examples of the Compound II).
Preparation 1.
Synthesis of 3-(3,4-dimethoxyphenyl)-4,5,6-trimethoxy-1 (3H)-
isobenzofuranone: II-1.
Step 1 . Synthesis of 4,4-dimethyl-2-(3,4,5-trimethoxyphenyl)-2 -
oxazoline: 2.
Under cooling in an ice-bath, a solution of 500 g (2.17
moles) of 3,4,5-trimethoxybenzoyl chloride (compound 1 ) in 1 liter
of dry methylene chloride was added dropwise to a solution of 386 g
(4.34 moles) of 2-amino-2-methyl-1-propanol in 1 liter of dry
-'- 2121pgo
methylene chloride over 2 hours. After completion of the addition,
the mixture was stirred for additional 1 hour and the reaction
solution was filtered through a glass filter. The filtered cake was
washed with 500 ml of methylene chloride, and the filtrate was
combined with the washing, which was followed by concentration in
vacuo. The residue was suspended in a mixture of 900 ml of dry
toluene and 100 ml of dry methylene chloride, and 206 ml (2.82
mmoles) of thionyl chloride was added dropwise thereto under
cooling in an ice-bath. The mixture was allowed to warm to room
temperature while being stirred for additional 30 minutes. Then,
the mixture was cooled again by using an ice-bath, and was added
200 ml of water and aqueous sodium hydroxide (560 g of NaOH in 1.8
liter of water). And then the mixture was extracted with toluene.
The extract was washed with water and brine, and dried over
anhydrous magnesium sulfate. After concentration in vacuo, the
residue was crystallized from methylene chloride - n-hexane (200
ml - 2.0 liter) to give 495 g of the desired oxazoline as the first
crystals, and 61 g as the second crystals. Total yield: 556 g (96.5
from compound 1 ). Melting point: 87-89°C.
1 H-NMR: 8 (CDC13) 1.39 (6H,s) 3.88 (3H,s) 3.91 (6H,s) 4.10 (2H,s)
7.20 (2H,s).
Step 2. Synthesis of Compound II-1.
Under nitrogen flow, a 1.66 N hexane solution of n-butyl
lithium (800 ml; 1.33 moles) was added dropwise over 1 hour and 15
minutes to a solution of 335 g (1.26 moles) of the above oxazoline
(compound 2) obtained above in 1.7 liter of dry THF, which was
_s_ 2121060
cooled with a refrigerant at -35 to -40 °C. After completion of the
addition, the mixture was stirred for additional 1 hour at -20 to -
35 °C, then cooled to -78°C, and a solution of 231 g (1.39
moles,
1.10 eq.) of 3,4-dimethoxybenzaldehyde in 500 ml of dry THF was
.added dropwise. After completion of the addition, the cooling bath
was replaced by an ice-bath, and the mixture was stirred for 1 hour.
Then, 400 ml of saturated aqueous ammonium chloride and 400 ml
of water were added thereto, and the mixture was extracted with
ethyl acetate. The extract was washed with water and brine, dried
over anhydrous magnesium sulfate and concentrated in vacuo. The
residue was dissolved in 1.4 liter of 10% sulfuric acid and the
mixture was heated under reflux for 40 minutes. Ice water was
added to the reaction solution and the precipitated crystals were
collected by filtration. The crystals were dissolved in 1.5 liter of
methylene chloride, washed with water, dried over anhydrous
magnesium sulfate, and then concentrated in vacuo. The residue
was recrystallized from methylene chloride - methanol to give 390
g (85.7 % from compound 2) of the desired lactone II-1. Melting
point: 141-142°C.
1 H-NMR: b (CDC13) 3.52 (3H,s) 3.83 (3H,s) 3.89 (3H,s) 3.92 (3H,s)
3.95 (3H,s) 6.32 (1 H,s) 6.72 (1 H,s) 6.86 (2H,s) 7.21 (1 H,s).
The reaction of Preparation 1 is expressed by the following
scheme.
_g_
212 ~ o so
Me0 ~ COCI 1 ) H2N~OH Me0
CH2C12 \ I N
Me0 '~ Me0
Me0 2 ) SOC12 Me0
PhMe
1 CH2C12
2
1 ) n - BuLi / THF
Me0 ~ ~ CHO Me0
O
Me0 Me0
Me0
2 ) 10% HpS04
OMe
OMe
II-1
Preparation 2.
Synthesis of 3-(3,4-dimethoxyphenyl)-5,6-methylenedioxy-1 (3H) -
isobenzofuranone: II-2.
Step 1. Synthesis of 2-(3,4-dimethoxy-a-hydroxybenzyl)-4,5 -
methylenedioxybenzaldehyde ethylenedioxyacetal: 4.
i) To a solution of 28.0 g (122 mmoles) of 2-bromo-4,5 -
methylenedioxybenzaldehyde (compound 3) in 250 ml of benzene
were added ethylene glycol (14 ml) and 465 mg of p-toluenesulfonic
acid and the mixture was heated under reflux for 3 hours to effect
dehydration by using a Dean-StarkT"" trap. After cooling in an ice-
bath, saturated aqueous sodium bicarbonate was added to the
reaction solution and the mixture was extracted with ethyl acetate.
The extract was washed with water and brine, dried over anhydrous
magnesium sulfate and concentrated in vacuo to give 33.2 g of a
crude product of the acetal as crystals., This was used in the next
b~. . .,........ ...w.....,.".. . ..... ..... ,..,..~. ,.,n. .,.., ..
-,o- 2121pgp
reaction without further purification.
ii) Under nitrogen flow, a 1.64 N solution of n-butyl lithium in
n-hexane (80 ml) {131 mmoles) was added dropwise to a solution of
33.2 g of the crude acetal obtained above in 300 ml of dry THF at -
78°C. After additional stirring for 30 minutes at the same
temperature, a solution of 20.3 g (122 mmoles) of 3,4-
dimethoxybenzaldehyde in 85 ml of dry THF was added and the
mixture was stirred for additional 30 minutes. Saturated aqueous
ammonium chloride was added to the reaction solution and the
mixture was extracted with ethyl acetate. The extract was washed
with water and brine, dried over anhydrous magnesium sulfate and
concentrated in vacuo. The residue was purified by a medium -
pressure silica gel column chromatography (600 g of Si02; ethyl
acetate : n-hexane = 1:2 to 1:1 ) to give 35.6 g (80.9% from compound
3) of the desired compound 4 as an oil.
1 H-NMR: 8 (CDgOD) 3.77 (3H, s) 3.80 (3H, s) 3.90 - 4.17 (4H, m)
5.91 (1 H, d, J = 1.2Hz) 5.93 (1 H, d, J = 1.2Hz) 5.97 (1 H, s) 6.13 (1 H, s)
6.85 (1 H, s) 6.87 (1 H, s) 6.88 (1 H, s) 6.98 (1 H, s) 7.02 (1 H, s).
St_ ep 2. Synthesis of 2-(3,4-dimethoxy-a-acetoxybenzyl)-4,5 -
methylenedioxybenzaldehyde: 5.
i) Under nitrogen flow, acetic anhydride (12.1 ml; 128 mmoles)
was added to a solution of 35.6 g (98.3 mmoles) of compound 4
obtained above, 360 mg of N,N-dimethylaminopyridine and 21 ml of
triethylamine in 170 ml of dry THF under cooling in an ice-bath. The
mixture was allowed to warm to room temperature while being
~212~os0
-" -
stirred for 50 minutes. Methanol (4.4 ml) was added thereto, the
mixture was stirred for 20 minutes and concentrated in vacuo.
Water was added to the residue and the mixture was extracted with
ethyl acetate. The extract was washed with water and brine, dried
over anhydrous magnesium sulfate and concentrated in vacuo to give
39.6 g of a crude product of the desired acetate as an oil. This was
used in the next reaction without further purification.
ii) To a solution of 39.6 g of the crude product of the acetate
obtained above in 350 ml of acetone was added 35 ml of 1 N
hydrochloric acid under cooling in an ice-bath. The mixture was
allowed to warm to room temperature, while being stirred for 1
hour. The reaction mixture was neutralized by addition of saturated
aqueous sodium bicarbonate and concentrated in vacuo. Water was
added to the residue and the mixture was extracted with ethyl
acetate. The extract was washed with water and brine, dried over
anhydrous magnesium sulfate and concentrated in vacuo to give 39.0
g of a crude product of the desired aldehyde 5 as an oil. This was
used in the next reaction without further purification.
1 H-NMR: b (CDC13) 2.16 (3H, s) 3.85 (6H, s) 6.09 (2H, s) 6.80 -
6.89 (3H, m) 7.09 (1 H, s) 7.32 (1 H, s) 7.58 (1 H, s).
Step 3. Synthesis of Compound II-2.
The crude product (39.0 g) of aldehyde 5 obtained above
was dissolved in a mixture of 500 ml of methanol and 250 ml of
dioxane and then 130 ml of 2-methyl-2-butene was added thereto.
Then, 250 ml of aqueous solution of 44 g (486 mmoles) of sodium
chlorite and 57 g (365 mmoles) of sodium dihydrogenphosphate
212 ~ o so
-12-
dehydrate was added and the mixture was stirred at room
temperature for 20 minutes. After addition of 5 N aqueous sodium
hydroxide (160 ml) to the reaction solution, the mixture was stirred
for 25 minutes, and then 160 ml of 6 N hydrochloric acid was added,
which was followed by stirring for additional 20 minutes. Ice
water was added to the reaction solution and the mixture was
extracted with methylene chloride. The extract was washed with
water, saturated aqueous sodium bicarbonate and brine, and dried
over anhydrous magnesium sulfate. After concentration in vacuo,
the crystalline crude residue was washed with ether and then
recrystallized from methanol to give 27.0 g (86.2% from the
compound 4) of the desired lactone II-2 as crystals. Melting point:
176-178°C.
1 H-NMR: b (CDC13) 3.83 (3H, s) 3.89 (3H, s) 6.12 (2H, ABtype, J =
1.2 Hz) 6.20 (1 H, s) 6.66 (2H, d, J = 1.2 Hz) 6.86 (1 H, s) 6.87 (1 H, s)
7.25 (1 H, s).
The above reaction of Preparation 2 is expressed by the
following scheme.
2121060
-1 3-
1 ) HO ( CH2 )20H O
O CHO Ph~H ( cat. ) O i 0O
i
off
0
2)n-BuLi/THF
Me0 ~ ~ CHO ~ I OMe
Me0 OMe
4
O ~ CHO
1 ) AcpO, Et3N ~ ~ I OAc
DMAP/THF O
2 ) 1 N HCI / acetone
OMe p
OMe O
5 ~ i I v0
O
NaCl02, NaH2P04, ~ i
aq. MeOH - dioxane,
OMe
aq. NaOH; 6N HCI OMe
I I-2
The compound (III) may be prepared by any one of three
procedures, which are provided below.
First procedure for ,preparing the compound (,III
Preparation 3
Synthesis of methyl (E)-4-cyclohexyl-4-oxo-2-butenoate: III-a
Step 1 . Synthesis of 3-cyclohexyl-5-(methoxycarbonyl)-2
isoxazoiine: 8a
i) Under cooling in an ice-bath, a solution of 224 g (2.00 moles)
of cyclohexanecarboxyaldehyde (compound 6a) in 133 ml of 99%
ethanol was added dropwise to 800 ml of an aqueous solution of 154
g (2.20 moles) of hydroxylamine hydrochloride and 166 g (1.20
4
-, 4- 212 1 0 g p
moles) of potassium carbonate. After completion of the addition,
the mixture was allowed to warm to room temperature while being
stirred for 1 hour, and then extracted with 900 ml of ethyl acetate.
The extract was washed with water and brine to give a solution of
the desired oxime in ethyl acetate.
ii) Under cooling in an ice-bath, the solution of the oxime
obtained above in ethyl acetate, containing 220 ml (2.40 moles) of
methyl acrylate was added dropwise to a mixture of 3 liter of about
10% aqueous solution of sodium hypochlorite (about 4 moles) and 28
ml (200 mmoles) of triethylamine with vigorous stirring over 2
hours. After completion of the addition, the mixture was allowed to
warm to room temperature, while being stirred for 1 hour, and then
extracted with ethyl acetate. The extract was washed with water
and brine, dried over anhydrous magnesium sulfate, and concen -
trated in vacuo to give 369 g of a crude product of isoxazoline
(compound 8a) as an oil (88% from compound 6a).
~ H-NMR: 8 (CDC13) 1.10-1.45 (5H, m) 1.48-1.90 (5H, m)
2.35-2.50 (1 H, m) 3.19 (1 H, d, J = 8.2 Hz) 3.20 (1 H, d, J = 9.4 Hz) 3.79
(3H, s) 4.96 (1 H, dd, J = 8.2 Hz, 9.4 Hz).
Step 2. Synthesis of Compound Ill-a
i) The crude product (369 g) of the isoxazoline obtained above
(compound 8a) was dissolved in a mixture of 1.5 liter of methanol,
316 ml of water and 474 ml of acetic acid, and the mixture was
stirred for 6 hours at room temperature under hydrogen atmosphere
in the presence of 11.1 g of 10 % palladium-carbon. The palladium -
carbon was filtered off, and the filtrate was concentrated in vacuo.
212 ~ 0 6p
To the residue was added water, and the mixture was extracted
with ethyl acetate. The extract was washed with water, saturated
aqueous sodium bicarbonate and brine, dried over anhydrous
magnesium sulfate, and concentrated in vacuo to yield 280 g of a
crude product of the desired hydroxyketone as an oil.
ii) Under cooling in an ice-bath, 163 ml (2.10 moles) of
methanesulfonyl chloride was added dropwise to a solution of 280 g
of the crude hydroxyketone obtained above and 819 ml (5.89 moles)
of triethylamine in 1.4 liter of dry ethyl acetate over 1 hour. The
mixture was allowedg to warm to room temperature while being
stirred for 1 hour. After addition of ice-water, the mixture was
acidified with conc. hydrochloric acid, and extracted with ethyl
acetate. The extract was washed with water, saturated aqueous
solution of sodium bicarbonate and brine, dried over anhydrous
magnesium sulfate and concentrated in vacuo. The residue was
crystallized from 80% aqueous ethanol to give 158 g of unsaturated
ketoester as crystals (62% from compound 8a). Melting point: 53-56
°C
~ H-NMR: 8 (CDCIg) 1.10-1.50 (5H, m) 1.60-2.00 (5H, m)
2.50-2.63 (1 H, m) 3.81 (3H, s) 6.69 (1 H, d, J = 15.6 Hz) 7.17 (1 H, d, J
= 15.6 Hz).
Preparation 4
Synthesis of Methyl (E)-6-ethyl-4-oxo-2-octenoate: III-b
Step 1 . Synthesis of 3-(2-ethylbutyl)-5-(methoxycarbonyl)-2 -
isoxazoline: 8b
212 X060
i) Under nitrogen flow, a solution of 399 g (2.42 moles) of 1 -
bromo-2-ethylbutane (compound 7b) in 650 ml of dry THF was added
dropwise over 2 hours to a suspension of 61.7 g (2.54 moles) of
magnesium in 1 liter of dry THF while the reaction was kept under
mild reflux. After completion of the addition, the mixture was
allowed to warm to room temperature while being stirred for 1.5
hours. The mixture was cooled by using an ice-bath, and to the
mixture was added dropwise a solution of 269 ml (2.42 moles) of 1 -
formylpiperidine in 450 ml of dry THF. After additional stirring for
1 hour, 300 ml of an aqueous solution of 202 g (2.9 moles) of
hydroxylamine hydrochloride was added, and the mixture was
allowed to warm to room temperature while being stirred for 1 hour
and 20 minutes. The reaction mixture was decanted, and the residue
was washed with ether. The combined organic phase was washed
with water and brine, dried over anhydrous magnesium sulfate, and
concentrated in vacuo to give 362 g of a crude product of oxime as
an oil. This was used in the next reaction without further
purification.
ii) Under cooling in an ice-bath, a solution of 362 g of crude
oxime obtained above in 850 ml of toluene was added dropwise to a
mixture of 2.55 liter of about 10% aqueous solution of sodium
hypochlorite (about 3.8 moles) and a solution of 306 ml (3.39 moles)
of methyl acrylate and 51 ml (363 mmoles) of triethylamine in 850
ml of toluene with vigorous stirring. After completion of the
addition, the mixture was allowed to warm to room temperature
while being stirred for 2 hours. To the reaction mixture was added
.
2121psp
ice water, and the mixture was extracted with ethyl acetate. The
extract was washed with water, an aqueous sodium thiosulfate and
brine, dried over anhydrous magnesium sulfate, and concentrated in
vacuo. The residue was distilled in vacuo to yield 255 g of the
desired isoxazoline (compound 8b) as an oil (49.5% from compound
7b). Boiling point: 130-135 °C (5 mmHg)
~ H-NMR: 8 (CDC13) 0.88 {3H, t, J = 7.2 Hz) 0.89 (3H, t, J = 7.2 Hz)
1.27-1.61 (5H, m) 2.32 (2H, d, J = 7.0 Hz) 3.21 (2H, d, J = 9.0 Hz) 3.79
(3H, s) 4.99 (2H, d, J = 9.0 Hz).
Step 2. Synthesis of Compound III-b
The reactions were conducted in a procedure similar to
that of step 2 in Preparation 3 starting from the isoxazoline
(compound 8) to give the desired unsaturated ketoester III-b.
Boiling point: 85-90 °C (0.5 mmHg)
1 H-NMR: 8 (CDC13) 0.86 (6H, t, J = 7.2 Hz) 1.19-1.50 (4H, m)
1.79-1.96 (1 H, m) 2.54 (2H, d, J = 6.6 Hz) 3.82 (3H, s) 6.67 (1 H, d, J =
15.8 Hz) 7.09 (1 H, d, J = 15.8 Hz).
Preparation 5
Synthesis of methyl (E)-5-ethyl-4-oxo-2-heptenoate: III-c
Step 1. Synthesis of 3-(1-ethylpropyl)-5-(methoxycarbonyl)-2 -
isoxazoline: 8c
The reactions were conducted in a procedure similar to
that of step 1 in Preparation 3 starting from (2-ethyl)butylaldehyde
(compound 6c) to give the desired isoxazoline (compound 8c).
Boiling point: 100-110 °C (2 mmHg)
~212~oso
-, 8-
1 H-NMR: 8 (CDC13) 0.88 (3H, t, J = 7.4 Hz) 0.89 (3H, t, -J = 7.4 Hz)
1.33-1.71 (4H, m) 2.36-2.53 (1 H, m) 3.13 (1 H, d,J = 9.6 Hz) 3.14 (1 H,
d, J = 7.8 Hz) 3.79 (3H, s) 4.99 (1 H, dd, J = 9.6 Hz, 7.8 Hz).
Step 2. : Synthesis of Compound III-c
The reactions were conducted in a procedure similar to
that of step 2 in Preparation 3 starting from the isoxazoline
(compound 8c) obtained above to give the unsaturated ketoester III -
c. Boiling point: 68-72 °C (0.8 mmHg)
~ H-NMR: 8 (CDC13) 0.86 (6H, t, J = 7.4 Hz) 1.42-1.80 (4H, m)
2.51-2.70 (1 H, m) 3.82 (3H, s) 6.72 (1 H, d, J = 15.8 Hz) 7.19 (1 H, d, J
= 15.8 Hz).
Preparation 6
Synthesis of methyl (E)-5-methyl-4-oxo-2-hexenoate: III-d
Step 1 . Synthesis of 5-(Methoxycarbonyl)-3-(1-methylethyl)-2 -
isoxazoline: 8d
The reactions were conducted in a procedure similar to
that of step 1 in Preparation 3 starting from isobutylaldehyde
(compound 6d) to give the desired isoxazoline (compound 8d).
Boiling point: 84-88 °C (1 mmHg)
1 H-NMR: 8 (CDCIg) 1.19 (6H, d, J = 7.0 Hz) 2.65-2.82 (1 H, m) 3.23
(2H, d, J = 9.4 Hz) 3.80 (3H, s) 4.99 (1 H, t like, J = 9.4 Hz).
Step 2. Synthesis of Compound III-d
The reactions were conducted in a procedure similar to
that of step 2 in Preparation 3 starting from the isoxazoline
(compound 8d) obtained above to give the unsaturated ketoester III-
.2121060
-19-
d. Boiling point: 76-80 °C (6 mmHg)
1 H-NMR: 8 (CDC13) 1.16 (6H, d, J = 7.0 Hz) 2.76-2.94 (1 H, m) 3.82
(3H, s) 6.74 (1 H, d, J = 15.8 Hz) 7.20 (1 H, d, J = 15.8 Hz).
The above reactions of Preparations 3 to 6 are described in
the following reaction scheme.
212~o6p
-20-
1 st Procedure
1 ) Mg I THF; N - CHO;
R' - Br
aq. H2NOH~HC1
7b; R' _ - CH2CHEt2
2 ) aq. NaClO
Et3N / PhMe
~C02Me
1 ) H2NOH~HCI \
K2C03 / aq. EtOH N-O
R' - CHO R1 i~C02Me
2 ) aq. NaClO / EtOAc
Et3N ~C02Me
6a; R' _
8a; R'=-
c; R' _ - CHEt2
b; R' =-CH2CHEt2
d; R' _ - CHMe2
c; R' =-CHEt2
d; R' _ - CHMe2
O
1 )H2/Pd-C
MeOH - aq. AcOH R~
Me0 C
2 ) MsCI / EtOAc
Et3 N
n1- a; R' _ (Preparation 3)
b; R' _ - CH2CHEt2 (Preparation 4)
c; R' _ - CHEt2 (Preparation 5)
d; R' =-CHMe2 (Preparation 6)
2121060
-21-
Second procedure for preparing the compound jlll,)
Preparation 7
Synthesis of methyl (E)-4-cyclohexyl-4-oxo-2-butenoate: III-a
St_ ep 1. Synthesis of (~)-3-acetoxy-3-(methoxycarbonyl) propionic
acid : 10
A mixture of 201 g (1.50 moles) of DL-malic acid
(compound 9) and 750 ml of acetyl chloride was stirred at room
temperature for 68 hours, and at 50 °C for additional 2 hours. The
reaction solution was concentrated in vacuo and 300 ml of toluene
was added to the resulting residue. The mixture was concentrated
in vacuo again, and then the residue was subjected to such addition -
concentration operation once more. The resulting acid anhydride
was crystallized from 100 ml of toluene. To the crystalline residue
obtained by evaporation of toluene in vacuo, was added 1.5 liters of
methanol under cooling in an ice-bath. The mixture was stirred at
room temperature for one hour, and then allowed to stand for 15
hours. After concentration in vacuo, 300 ml of toluene was added to
the residues, and the mixture was concentrated in vacuo again. The
residue was crystallized from 150 ml of toluene, and the crystals
were washed with petroleum ether to give 279 g (98%) of the
desired carboxylic acid (compound 10) as crystals. Melting point: --
63 °C
1 H-NMR: b (CDC13) 2.16 (3H, s), 2.96 (2H, d, J = 6.0 Hz), 3.78 (3H,
s), 5.49 (1 H, t, J = 6.0 Hz ).
St_ ep 2. Synthesis of Compound III-a
i) A mixture of 196 g (1.10 moles) of the carboxylic acid
-22-
2121060
(compound 10) obtained above and 196 ml (1.65 moles) of thionyl
chloride was stirred at 30 °C for 30 minutes, and at 50 °C for
an
additional 1 hour. The reaction solution was concentrated in vacuo
and 250 ml of toluene was added to the resulting residue. The
mixture was concentrated in vacuo again, and then the residue was
subjected to such addition-concentration operation once more to
give the desired acid chloride. This was used in the next reaction
without further purification.
ii) Under nitrogen flow, 1.17 liters (1.19 moles) of 1.02 M
solution of cyclohexylmagnesium bromide in THF was added
dropwise to a suspension of the crude acid chloride obtained above
and 9.52 g (5.00 mmoles) of cuprous iodide in 1 liter of dry THF at -
25 °C over 2.5 hours. After completion of the addition, the mixture
was stirred for an additional 30 minutes, and the reaction was
quenched by addition of 1 liter of 1.3 N hydrochloric acid and ice.
The reaction solution was extracted with ethyl acetate, the extract
was washed with brine, saturated aqueous sodium bicarbonate,
aqueous sodium thiosulfate and brine, and dried over anhydrous
magnesium sulfate. The solution was concentrated in vacuo to give
a crude product of the desired ketone. This was used in the next
reaction without further purification.
iii) To a solution of the crude ketone obtained above in 550 ml
of dry acetonitrile was added 230 ml (1.65 moles) of triethylamine,
and the mixture was heated under reflux for 1 hour. To the reaction
solution was added 1 liter of 1.3 N hydrochloric acid under cooling
in an ice-bath, and the mixture was extracted with ethyl acetate.
A
272 10 60
-23-
The extract was washed with brine, and dried over anhydrous
magnesium sulfate. After concentration in vacuo, the residue was
crystallized by addition of 1 liter of petroleum ether. The crystals
were recrystallized from 85% aqueous methanol to give 76.8 g of
the desired unsaturated ketoester III-a as crystals. Further, mother
liquids of crystallization and recrystallization were combined and
distilled in vacuo, and then the distillate was recrystallized from
85% aqueous methanol to give additional 32.5 g of the desired
compound. Total yield: 109 g (51 % from compound 10). Melting
point: 56-57 °C.
1 H-NMR spectrum of this compound is identical with that
of the compound prepared in Preparation 3.
Preparation 8
Synthesis of methyl (E)-6-ethyl-4-oxo-2-octenoate: III-b
The reactions were conducted in a procedure similar to
that of step 2 in Preparation 7 starting from compound 10 (Step 1
of Preparation 7) and (2-ethyl)butylmagnesium bromide to give the
desired unsaturated ketoester III-b. The physical properties of the
resulting compound are completely identical with those of the
compound prepared in Preparation 4.
Preparation 9
Synthesis of methyl (E)-5-ethyl-4-oxo-2-heptenoate: III-c
The reactions were conducted in a procedure similar to
that of step 2 in Preparation 7 starting from compound 10 (Step 1
of Preparation 7) and (1-ethyl)propylmagnesium bromide to give the
desired unsaturated ketoester III-c. The physical properties of the
-24- Z12~osp
resulting compound are completely identical with those of the
compound prepared in Preparation 5.
Preparation 10
Synthesis of methyl (E)-4-oxo-2-hexenoate: III-a
The reactions were conducted in a procedure similar to
that of step 2 in Preparation 7 starting from compound 10 (Step 1
of Preparation 7) and ethylmagnesium bromide to give the desired
unsaturated ketoester III-e. Boiling point: 87-90 °C (8 mmHg)
1 H-NMR: 8 (CDCIg) 1.14 (3H, t, J = 7.2 Hz) 2.67 (2H, q, J~ = 7.2 Hz)
3.81 (3H, s) 6.69 (1 H, d, J = 16.0 Hz) 7.09 (1 H, d, J = 16.0 Hz).
The above reactions of Preparations 7 to 10 are described
in the following reaction scheme.
2nd Procedure
1 ) AcCI Ac O
HO
~.,~ ~ ~C02H
H02C~C02H ~' Me02C
2 ) MeOH
O
R'
1 ) SOC12
Me0 C
2 ) R - MgBr / THF
Cul ( cat. )
3 ) Et3N / MeCN III- a; R' _ ---~ (Preparation 7)
b; R' =-CH2CHEt2 (Preparatione 8)
c; R' =-CHEt2 (Preparation 9)
e; R' _ - Et (Preparation 10)
_25_ . 212 1 0 6 0
Third procedure for pr~arin4 the compound (III)
Preparation 11
Synthesis of methyl (E)-6-ethyl-4-oxo-2-octenoate: III-b
Step 1. Synthesis of (E)-3-(methoxycarbonyl) acryloyl chloride: 12
i) To a suspension of 300 g (3.06 moles) of malefic anhydride in
900 ml of dry toluene were added 136 ml (3.37 moles) of methanol
and 3.0 ml of thionyl chloride, and the mixture was heated under
reflex for 2 hours. The reaction solution was cooled by using an ice -
bath, and the precipitated crystals were collected to give crude
crystals of the desired compound. The cystals were recrystallized
from THF (500 ml) -isopropyl ether (1 liter) to give 210 g (52.7%) of
the desired mono-methyl ester. Melting point: 145-146 °C.
ii) To a suspension of 97.5 g (750 mmoles) of the methyl ester
obtained above in 250 ml of dry benzene were added 82.2 ml (1.13
moles) of thionyl chloride and 1.0 ml of DMF, and the mixture was
stirred at 70 °C for 2.5 hours. The reaction mixture was
concentrated and distilled in vacuo to give 95.3 g (85.3%) of the
desired acid chloride (compound 12) as an oil. Boiling point: 72-73
°C (15 mmHg)
1 H-NMR: 8 (CDC13) 3.86 (3H,s) 6.99 (1 H, ABtype, J = 15.4 Hz) 7.00
(1 H, ABtype, J = 15.4 Hz).
Step 2. Synthesis of Compound III-b
Under nitrogen flow, 728 ml (837 mmoles) of 1.15 M
solution of zinc chloride in THF was added dropwise over 55
minutes to a 0.97 M THF solution of (2-ethyl)butylmagnesium
bromide (860 ml; 837 mmoles) under cooling in an ice-bath. After
212 1 0 60
-26-
completion of the addition, the mixture was stirred for additional
50 minutes, and 14.8 g (12.8 mmoles) of tetrakis(triphenylphophine) -
palladium was added thereto. Subsequently, a solution of 95.3 g
(639 mmoles) of the above acid chloride (compound 12) obtained
above in 250 ml of dry THF was added dropwise over 1.5 hours.
After completion of the additon, the mixture was stirred for
additional 30 minutes, 600 ml of 3N hydrochloric acid was added
thereto, and the mixture was extracted with ethyl acetate. The
extract was washed with water and brine, and dried over anhydrous
magnesium sulfate. The residue after concentration in vacuo was
dissolved in 200 ml of a mixture of ether and n-hexane (1:4), and
filtered through a pad of silica gel (300 g). The silica gel was
washed with 1 liter of the same mixed solvent described above. The
filtrate and the washing were combined together, and then the
mixture was concentrated in vacuo. The residue was distilled in
vacuo to give 67.6 g (53.4%) of the desired unsaturated ketoester III -
b as an oil.
The physical properties of the resulting compound are
completely identical with those of the compound prepared in
Preparation 4.
The above reactions of Preparation 11 is described in the
following reaction scheme.
.2121060
-27-
3rd Procedure
O 1 ) MeOH / PhMe O
SOC12 ( cat. )
'CI
2) SOC12 / PhH Me0 C
O 2
12
11
O
R' - MgBr / THF
ZnCl2 ~ ~ R1
~ phsP lapd (cat. ) Me02C
III-b; R1 =-CH2CHEt2
Examples for preparing the Compounds {I) are provided
below.
Example 1
Synthesis of 3-(cyclohexanecarbonyl)-1-(3,4
dimethoxyphenyl)-4-hydroxy-2-(methoxycarbonyl)-6,7,8 -
trimethoxynaphthalene: I-1 a.
Step 1. Synthesis of 3-(cyclohexanecarbonyl)-1-(3,4
dimethoxyphenyl)-1 ,2-dihydro-1 ,4-dihydoxy-2-(methoxycarbonyl) -
6,7,8-trimethoxynaphthalene: IV-1 a.
Under nitrogen flow, 1.64 M solution of n-butyl lithium in n -
hexane (700 ml; 1.15 moles) was added dropwise to a solution of
256 ml (1.15 mmoles) of (TMS)2NH in 1.15 liter of dry THF under
cooling in an ice-bath. A solution of 207 g (575 mmoles) of lactone
i1-1 (Preparation 1 ) in 1.15 liter of dry DMF was added dropwise
~212106p
-28-
over 1 hour to the reaction solution which was cooled by using a dry
ice-acetone bath. After the mixture was stirred for additional 45
minutes, a solution of 134 g (684 mmoles) of unsaturated ketoester
III-a (Preparation 3) in 456 ml of dry THF was added dropwise over
45 minutes. After completion of the addition, the mixture was
stirred for 15 minutes, allowed to warm to 0 °C, and stirred for 3
hours at the same temperature. To the reaction solution was added
1.8 liter of 2 N hydrochloric acid, and the mixture was extracted
with ethyl acetate. The extract was washed with water and brine,
dried over anhydrous magnesium sulfate and concentrated in vacuo.
The residue was crystallized from 1 liter of methanol to give 180 g
of crude crystals of the desired compound IV-1 a. The crystals were
further recrystallized from methylene chloride - methanol to give
148 g of the desired compound IV-1 a. Then, the mother liquid of
recrystallization was concentrated, and the residue was
recrystallized twice to give additional 7.0 g of compound IV-1 a.
Total yield: 155 g (49% from compound I I-1 ).
Melting point: 154-155 °C.
~ H-NMR: 8 (CDCIg) 0.90-1.83 (10H, m) 2.40-2.58 (1 H, m) 3.45
(3H, s) 3.67 (3H, s) 3.82 (3H, s) 3.85 (3H, s) 3.93 (1 H, s) 3.94 (3H, s)
3.99 (3H, s) 5.75 (1 H, s) 6.42 (1 H, dd, J = 8.4 Hz, 2.4 Hz) 6.64 (1 H, d,
J = 8.4 Hz) 7.14 (1 H, d, J = 2.4 Hz) 7.51 (1 H, s).
Step 2. Synthesis of Compound I-1 a
Under nitrogen flow, a solution of 44.8 ml (364 mmoles) of
BF3~OEt2 in 90 ml of dry methylene chloride was added to a solution
CA 02121060 2003-07-25
of 155 g (279 mmoles) of the compound IV-1 a obtained above in 750
ml of dry methylene chloride over 30 minutes under cooling in an ice -
bath, and the mixture was stirred at the same temperature for
additional 50 minutes. To the reaction solution was added 58.4 ml
(420 mmoles) of triethylamine, and the mixture was stirred at room
temperature for 30 minutes. After concentration in vacu.o, water
was added to the residue and the mixture was extracted with ethyl
acetate. The extract was washed with 1 N hydrochloric acid, water
and brine, and dried over anhydrous magnesium sulfate. After
concentration iii vacuo. the residue was crystallized from 500 ml of
methanol, and the crystals were recrystallized from methylene
chloride-methanol twice to give 140 g (93%) of the desired
compound I-1 a.
Melting point: 150-151 °C.
~H-NMR: 8 (CDC13) 1.15-7.90 {10H, m) 2.70-2.90 (1H, m) 3.24 (3H, s)
3.44 (3H, s) 3.86 {3H, s) 3.89 {3H, s) 3.93 (3H, s) 4.03 (3H, s)
6.81-6.87 (3H, m) 7.71 {1H, s) 13.99 {1H, s).
1R: v (Nujol)M1714, 1606, 1580, 1516, 1489, 1410, 1240, 1197,
1142, 1107, 1063, 1027, 1004 cm-i .
Analysis calculated for C3GH34~9- C 66.90%, H 6.36%,
Found: C 66.92°/p, H 6.39%
Example 2
Synthesis of 1-{3,4-dimethoxyphenyl)-3-{3-ethyl-1 -
oxopentyl)-4-hydroxy-2-(methoxycarbonyl)-6,7,8 -
trimethoxynaphthaiene: I- i b.
~212~os~
-3 0-
Step 1 . Synthesis of 1-(3,4-dimethoxyphenyl)-3-(3-ethyl-1
oxopentyl)-1 ,2-dihydro-1 ,4-dihydroxy-2-(methoxycarbonyl)-6,7,8 -
trimethoxynaphthalene: IV-1 b
Under nitrogen flow, a solution of 180 g (0.05 moles) of
lactone II-1 (Preparation 1 ) in 500 ml of dry methylene chloride
was added dropwise to 1 liter of 1.0 M LiN(TMS)2-THF solution (1.0
mole, 2.0 eq.) at -78°C over 1 hour. After the mixture was stirred
for additional 40 minutes, a solution of 109 g (0.55 moles, 1.1 eq.)
of unsaturated ketoester III-b (Preparation 4) in 300 ml of dry THF
was added dropwise over 40 minutes. After completion of the
addition, the mixture was allowed to warm up to 0 °C, and stirred
for 3 hours under cooling in an ice-bath. To the reaction solution
were added 1 liter of 2 N hydrochloric acid and ice, and the mixture
was extracted with ethyl acetate. The extract was washed with
water and brine, dried over anhydrous magnesium sulfate and
concentrated in vacuo. The residue was crystallized from 700 ml of
methanol to give crystals. The crystals were further recrystallized
from 600 ml of methanol twice to give 194 g (70%) of the desired
compound IV-1 b.
Melting point: 132-133 °C.
1 H-NMR: b {CDCIg) 0.65 (3H, t, J = 7.2Hz) 0.69 (3H, t, J = 7.2Hz)
0.80-1.29 (5H, m) 2.03 (1 H, dd, J = 14.0 Hz, 7.0 Hz) 2.31 (1 H, dd, J =
14.0 Hz, 6.4 Hz) 3.40 (3H, s) 3.67 (3H, s) 3.81 (3H, s) 3.86 (3H, s)
3.88 (1 H, s) 3.94 (3H, s) 3.99 (3H, s) 5.73 (1 H, s) 6.43 (1 H, dd, J = 8.4
Hz, 2.2 Hz) 6.64 (1 H, d, J = 8.4 Hz) 7.14 {1 H, d, J = 2.2 Hz) 7.54 (1 H,
2121060
-31-
s).
Step 2. Synthesis of Compound I-1 b.
The reaction was conducted in a procedure similar to that
of step 2 in Example 1 starting from the compound IV-1 b obtained
above to give the desired compound I-1 b.
Melting point: 128.5-129.5°C (methylene chloride-methanol).
1 H-NMR: 8 (CDC13) 0.83 (6H, t, J = 7 Hz) 1.20-1.42 (4H, m) 1.96-2.12
(1 H, m) 2.73 (2H, d, J = 6Hz) 3.25 (3H, s) 3.44 (3H, s) 3.86 (3H, s)
3.89 (3H, s) 3.93 (3H, s) 4.04 (3H, s) 6.78-6.90 (3H, m) 7.73 (1 H, s)
14.38 (1 H, s)
IR: v (CHC13) 2968, 1729, 1606, 1576, 1514, 1489, 1464, 1412,
1139, 1064, 1028 cm-1
Analysis calculated for C3pH3609: C 66.65%, H 6.71 %;
Found: C 66.72%, H 6.69%
Example 3
Synthesis of 1-(3,4-dimethoxyphenyl)-3-(2-ethyl-1 -
oxobutyl)-4-hydroxy-2-(methoxycarbonyl)-6,7,8 -
trimethoxynaphthalene: I-1 c.
Step 1 . Synthesis of 1-(3,4-dimethoxyphenyl)-3-(2-ethyl-1 -
oxobutyl)-1 ,2-dihyro-1 ,4-dihydroxy-2-(methoxycarbonyl)-6,7,8 -
trimethoxynaphthalene: IV-1 c.
The same reaction was conducted in a procedure similar to
that of step 1 in Example 1 starting from lactone II-1 (Preparation
1 ) and unsaturated ketoester I I I-c (Preparation 5) to give the
desired compound IV-1 c.
212~os0
-32-
Melting point: 154.5-156 °C (methylene chloride-methanol)
1H-NMR: 8 (CDC13) 0.19 (3H, t, J = 7.4 Hz) 0.75 (3H, t, J = 7.4 Hz)
1.13-1.72 (4H, m) 2.35- 2.51 (1 H, m) 3.42 (3H, s) 3.65 (3H, s) 3.79
(3H, s) 3.86 (3H, s) 3.94 (3H, s) 3.96 (1 H, s) 3.99 (3H, s) 5.76 (1 H, s)
6.42(lH,dd,J=8.4Hz,2.2Hz)6.64(lH,d,J=8.4Hz)7.14(lH,d,J
= 2.2 Hz) 7.56 (1 H, s).
Step 2. Synthesis of Compound I-1 c.
The reaction was conducted in a procedure similar to that
of step 2 in Example 1 starting from the compound IV-1 c obtained
above to give the desired compound I-1 c.
Melting point : i 13-115 °C (acetone-n-hexane)
1 H-NMR: 8 (CDC13) 0.82 (3H, t, J = 8 Hz) 0.83 (3H, t, J = 8 Hz)
1.42-1.60 (2H, m) 1.64-1.81 (2H, m) 2.78-2.90 (1 H, m) 3.24 (3H, s)
3.42 (3H, s) 3.86 (3H, s) 3.89 (3H, s) 3.93 (3H, s) 4.03 (3H, s)
6.81-6.87 (3H, m) 7.72 (1 H, s) 14.18 (1 H, s)
IR: a (CHCIg) 1730, 1606, 1575, 1523, 1490, 1463, 1412, 1137,
1062, 1029 cm-1
Analysis calculated for C29H3409: C 66.14%, H 6.51%;
Found: C 66.09%, H 6.64%
Example 4
Synthesis of 1-(3,4-dimethoxyphenyl)-4-hydroxy-2 -
(methoxycarbonyl)-3-(2-methyl-1-oxopropyl)-6,7,8 -
trimethoxynaphthalene: I-1 d.
Steh 1 . Synthesis of 1-(3,4-dimethoxyphenyl)-1,2-dihydro-1,4-
212~ogo
-33-
dihydroxy-2-(methoxycarbonyl)-3-(2-methyl-1-oxopropyl)-6,7,8 -
trimethoxynaphthalene: I V-1 d.
The reaction was conducted in a procedure similar to that
of step 1 in Example 1 starting from lactone l1-1 (Preparation 1 )
and unsaturated ketoester III-d (Preparation 6) to give the desired
compound IV-1 d.
Melting point: 155-156 °C (methylene chloride-methanol)
1 H-NMR: 8 (CDCIg) 0.67 (3H, d, J = 7.0 Hz) 1.04 (3H, d, J = 6.8 Hz)
2.73-2.88 (1 H, m) 3.43 (3H, s) 3.67 (3H, s) 3.81 (3H, s) 3.86 (3H, s)
3.91 (1 H, s) 3.94 (3H, s) 3.99 (3H, s) 5.74 (1 H, s) 6.44 (1 H, dd, J = 8.4
Hz, 2.0 Hz) 6.65 (1 H, d, J = 8.4 Hz) 7.14 (1 H, d, J = 2.0 Hz) 7.52 (1 H,
s).
St, ep 2. Synthesis of Compound I-1 d.
The reaction was conducted in a procedure similar to that
of step 2 in Example 1 starting from the compound IV-1 d obtained
above to give the desired compound I-1 d.
Melting point: 108-110°C (90% aqueous methanol).
1 H-NMR: 8 (CDC13) 1.15 (3H, d, J = 7 Hz) 1.16 (3H, d, J = 7 Hz)
3.09-3.20 (1 H, m) 3.24 (3H, s) 3.43 (3H, s) 3.86 (3H, s) 3.89 (3H, s)
3.93 (3H, s) 4.03 (3H, s) 6.79-6.90 (3H, m) 7.71 (1 H, s) 13.76 (1 H, s)
IR: a (nujol) 1724, 1604, 1579, 1510, 1410, 1195, 1133, 1104,
1024, 988, 959, 843 cm' 1
Analysis calculated for C2~H3QOg: C 65.05%, H 6.07%;
Found: C 65.16%, H 6.08%.
Example 5
~~21060
-3 4-
Synthesis of 1-(3,4-dimethoxyphenyl)-4-hydroxy-2 -
{methoxycarbonyl)-3-(1-oxopropyl)-6,7,8-trimethoxynaphthalene: I -
1 e.
Step 1 . Synthesis of 1-(3,4-dimethoxyphenyl)-1,2-dihydro-1,4 -
dihydroxy-2-(methoxycarbonyl)-3-{1-oxopropyl)-6,7,8 -
trimethoxynaphthalene: IV-1 e.
The reaction was conducted in a procedure similar to that
of step 1 in Example 2 starting from lactone II-1 (Preparation 1 )
and unsaturated ketoester III-a (Preparation 10) to give the desired
compound IV-1 e.
Melting point: 143-145 °C (methanol)
1H-NMR: 8 (CDC13) 0.89 (3H, t, J = 7.4 Hz) 2.31 (2H, q, J = 7.4 Hz) 3.41
(3H, s) 3.68 (3H, s) 3.82 (3H, s) 3.86 (3H, s) 3.87 (1 H, s) 3.94 (3H, s)
3.99 (3H, s) 5.73 (1 H, s} 6.44 (1 H, dd, J = 8.4 Hz, 2.2 Hz) 6.65 (1 H, d,
J = 8.4 Hz} 7.12 (1 H, d, J = 2.2 Hz) 7.51 (1 H, s).
St. ep 2. Synthesis of the Compound I-1 e.
The reaction was conducted in a procedure similar to that
of step 2 in the Example 1 starting from the compound IV-1 a
obtained above to give the desired compound I-1 e.
Melting point: 145-146 °C (ethyl acetate-diisopropyl ether).
1 H-NMR: 8 (CDC13) 1.18 (3H, t, J = 7.2 Hz) 2.81-2.91 (2H, m) 3.25 (3H,
s) 3.45 (3H, s) 3.86 (3H, s) 3.89 (3H, s) 3.93 (3H, s) 4.03 (3H, s)
6.80-6.85 (3H, m) 7.74 (1 H, s) 14.60 (1 H, s)
(R: v (nujol} 1730, 1604, 1577, 1202, 1117, 1023 cm-1
Analysis calculated for C26H2809:C 64.46%, H 5.83%;
212060
-3 S-
Found:C 64.53%, H 5.80%.
Example 6
Synthesis of 3-(cyclohexanecarbonyl)-1-(3,4 -
dimethoxyphenyl)-4-hydroxy-2-(methoxycarbonyl)-6,7 -
methylenedioxynaphthalene: I-2a.
Step 1. Synthesis of 3-(cyclohexanecarbonyl)-1-(3,4 -
dimethoxyphenyl)-1,2-dihydro-1,4-dihydroxy-2-(methoxycarbonyl) -
6,7-methylenedioxynaphthalene: IV-2a.
The reaction was conducted in a procedure similar to that
of step 1 in Example 1 starting from lactone Il-2 (Preparation 2)
and unsaturated ketoester Ill-a (Preparation 3) to give the desired
compound IV-2a.
Melting point: 193-195 °C (methylene chloride-methanol)
1 H-NMR: 8 (CDCIg) 0.90-1.88 (10H, m) 2.39-2.57 (1 H, m) 3.65 (3H, s)
3.80 (3H, s) 3.82 (3H, s) 4.02 (1 H, s) 4.96 (1 H, br. s) 6.07 (2H, s) 6.33
(1 H, dd, J = 8.4 Hz, 2.2 Hz) 6.62 (1 H, d, J = 8.4 Hz) 6.98 (1 H, d, J = 2.2
Hz) 7.20 (1 H, s) 7.45 (1 H, s).
Steep 2. Synthesis of Compound I-2a.
The reaction was conducted in a procedure similar to that
of step 2 in Example 1 starting from the compound IV-2a obtained
above to give the desired compound I-2a.
Melting point : 177-178 °C (methylene chloride-methanol)
1 H-NMR: 8 (CDC13) 1.06-1.92 (1 OH, m) 2.70-2.88 (1 H, m) 3.52 (3H, s)
3.86 (3H, s) 3.96 (3H, s) 6.06 (2H, s) 6.77 (1 H, s) 6.79 (1 H, d, J = 2.0
Hz) 6.82 (1 H, dd, J = 8.0 Hz, 2.0 Hz) 6.94 (1 H, d, J = 8.0 Hz) 7.79 (1 H,
212~oso
-3 6-
s) 13.74 (1 H, s).
I R:v (CHC13) 1724, 1618, 1583, 1514, 1450, 1241, 1169, 1039 cm-1
Analysis calculated for C28H2808: C 68.28%, H 5.73%;
Found: C 68.05%, H 5.77%.
Example 7
Synthesis of 1-(3,4-dimethoxyphenyl)-3-(3-ethyl-1 -
oxopentyl)-4-hydroxy-2-(methoxycarbonyl)-6,7 -
methylenedioxynaphthalene: I-2b.
Step 1 . Synthesis of 3-(3-ethyl-1-oxopentyl)-1-(3,4 -
dimethoxyphenyl)-1,2-dihydro-1,4-dihydroxy-2-(methoxycarbonyl) -
6,7-methylenedioxynaphthalene: IV-2b.
The reaction was conducted in a procedure similar to that
of step 1 in Example 2 starting from lactone II-2 (Preparation 2)
and unsaturated ketoester III-b (Preparation 4) to give the desired
compound IV-2b.
Melting point: 144.5-146.5 °C (methanol)
1 H-NMR: b (CDCIg) 0.60-1.41 (11 H, m) 2.13 (1 H, dd, J = 14.2 Hz, 7.0
Hz) 2.28 (1 H, dd, J = 14.2 Hz, 7.0 Hz) 3.66 (3H, s) 3.80 (3H, s) 3.82
(3H, s) 3.90 (1 H, s) 4.88 (1 H, s) 6.07 (2H, s) 6.39 (1 H, dd, J = 8.4 Hz,
2.2 Hz) 6.63 (1 H, d, J = 8.4 Hz) 6.98 (1 H, d, J = 2.2 Hz) 7.18 (1 H, s)
7.48 (1 H, s).
Step 2. Synthesis of Compound I-2b.
The reaction was conducted in a procedure similar to that
of step 2 in the Example 1 starting from the compound IV-2b
obtained above to give the desired compound I-2b.
2121060
-37-
Melting point : 126-128 °C (methanol)
1 H-NMR: 8 (CDC13) 0.83 (6H, t, J = 7.3 Hz) 1.20-1.42 (4H, m)
1 .96-2.16 (1 H, m) 2.73 (2H, d, J = 6.6 Hz) 3.51 (3H, s) 3.86 (3H, s)
3.96 (3H, s) 6.06 (2H, s) 6.72-6.98 (4H, m) 7.81 (1 H, s) 14.17 (1 H, s)
IR: v (CHCIg) 1730, 1623, 1610, 1586, 1517, 1463, 1242, 1176,
1043 cm-1
Analysis calculated for C2gHgpOg: C 68.00%, H 6.11 %;
Found: C 67.88%, H 6.15%.
Exam~ale 8
Synthesis of 1-(3,4-dimethoxyphenyl)-4-hydroxy-2 -
(methoxycarbonyl)-6,7-methylenedioxy-3-(1-oxopropyl) -
naphthalene: I-2e.
Step 1. Synthesis of 1-(3,4-dimethoxyphenyl)-1,2-dihydro-1,4 -
dihydroxy-2-(methoxycarbonyl)-6,7-methylenedioxy-3-(1 -
oxopropyl)naphthalene: IV-2e.
The reaction was conducted in a procedure similar to that
of step 1 in Example 2 starting from lactone II-2 (Preparation 2)
and unsaturated ketoester III-a (Preparation 10) to give the desired
compound IV-2e.
Melting point: 138-140 °C (methanol)
1 H-NMR: S (CDC13) 0.93 (3H, t, J = 7.4 Hz) 2.21-2.50 (2H, m) 3.66 (3H,
s) 3.82 (6H, s) 3.94 (1 H, s) 4.86 (1 H, br. s) 6.08 (2H, s) 6.42 (1 H, dd, J
= 8.4 Hz, 2.2 Hz) 6.65 (1 H, d, J = 8.4 Hz) 6.93 (1 H, d, J = 2.2 Hz) 7.20
(1 H, s) 7.45 (1 H, s).
Step 2. Synthesis of Compound I-2e.
2~2 ~ o.so
-38-
The reaction was conducted in a procedure similar to that
of step 2 in the Example 1 starting from the compound IV-2e
obtained above to give the desired compound I-2e.
Melting point: 169-170 °C (methylene chloride-methanol)
1 H-NMR: 8 (CDC13) 1.19 (3H, t, J = 7.2 Hz) 2.81-2.91 (2H, m) 3.52 (3H,
s) 3.86 (3H, s) 3.95 (3H, s) 6.06 (2H, s) 6.72 (1 H, s) 6.79-6.92 (3H,
m) 7.81 (1 H, s) 14.36 (1 H, s).
1R: a (CHCIg) 1724, 1619, 1582, 1459, 1175, 1038, 1025 cm-1
Analysis calculated for C24H22~s~ C 65.75%, H 5.06%;
Found: C 65.55%, H 5.14%.
Example 9.
Synthesis of compound I-1 b of Example 2 using
methanesulfonic acid as a dehydrating agent
Synthesis of compound I-1 b.
Methanesulfonic acid (1.15 g) was added to a suspension of
5.58 g {10.0 mmoles) of compound IV-1 b, which was prepared in the
step 1 of Example 2, in 23 ml of dry acetonitrile, and the mixture
was stirred at room temperature for 1 hour and 15 minutes. To the
reaction solution was added 24 ml of water and the mixture was
stirred under cooling in an ice-bath for 30 minutes. The
precipitated crystals were collected by filtration. The crystals
were recrystallized from acetone-methanol twice to give 5.04 g
(93%) of the desired compound I-1 b.
Melting point: 128-129 °C.
2121oso
-39-
All of the other physical properties of this compound are
identical with those of the compound of Example 2.
The compounds prepared in the above examples are listed
in the following tables.
~212~os0
-40-
Table 1
OH
O
Me0 / I
\
R'
\ /
Me0 ~ ~C02Me
Me0
home
OMe
(Example 1 ) I -1
a;
R'
(Example 2) b;
R'
=-CH2CHEt2
(Example 3) c;
R'
_
-
CHEt2
(Example 4) d;
R~
=-CHMe2
(Example 5) e;
R~
_
-
Et
~ 5 Table 2
OH O
/ \ R,
o \~ /
C02Me
\ OMe
OMe
(Example 6) I - 2a; R' _
(Example 7) b; R~ =-CH2CHEt2
(Example 8) e; R' _ - Et
2121060
-41-
Effect of the invention
Pharmacological experiments were conducted with the use
of the compounds obtained in Examples 1 and 2.
Experiment 1
Inhibitory Action against Oxidative Modification of LDL
Testing and Evaluatin4 Methods
Experiments were conducted as follows according to the
method of Kita, et al. described in Proceedings of the National
Academy of Sciences, USA, volume 84, pages 5928 (1987).
First, LDL was separated from the blood of New Zealand
White rabbits fed with a feed containing 0.5% of cholesterol for
three weeks and dissolved in phosphate-buffered physiological
saline (final LDL concentration: 0.2 mg protein/ml). To this was
added an ethanolic solution of each test compounds, then cupric
sulfate was added thereto (final Cu2+ concentration: 0.5 ~M) and the
mixture was incubated at 37°C for 24 hours.
The amount of lipid peroxides in each of the incubated
solutions was measured as thiobarbituric acid reacting substances
(TBA reactive substances) and, from the regression line of the
inhibitory rate for oxidative modification of LDL and the
concentration of the compound, the 50% inhibiting concentration
(ICSp) was calculated. Quantitative determination of the TBA
reactive substances was conducted by measuring the TBA reactive
substances in the supernatant (prepared by removing protein from
the incubated solution) by means of TBA method.
The result is shown in Table 3 below.
212'060
-42-
The ICSO values of the compounds obtained by the method
of the present invention were not more than 10 ~.M and, accordingly,
the compounds are understood to exhibit strong antioxidative action
against LDL.
Experiment 2
Cholesterol-lowering Action
Testing and Evaluating Methods
Male ICR mice (body weight: 30-40 grams) were freely fed
for seven days with a feed containing 1 % of cholesterol and 0.5% of
sodium cholate to which 0.12% of the test compound was added (no
such a compound was added for the control group), then blood was
collected from the mice and the total cholesterol in serum was
measured by the method of Allain described in Clinical Chemistry,
volume 20, page 470 (1974).
Total amount of VLDL cholesterol and LDL cholesterol was
calculated by subtracting the amount of HDL cholesterol from the
amount of total cholesterol. The amount of the HDL cholesterol was
measured by the method of Ash and Hentschel described in Clinical
Chemistry, volume 24, page 2180 (1978).
Cholesterol lowering action of the test compound was
evaluated from the cholesterol lowering rate calculated from the
following expressions.
X12 1 0 60
-43-
Total cholesterol lowering rate =
{1-[(total cholesterol in the group with the test compound) /
(total cholesterol in the control)]} x 100
(VLDL + LDL) cholesterol lowering rate =
{1-[(VLDL + LDL cholesterol in the group with the test compound) /
(VLDL + LDL cholesterol in the control)]} x 100
The result is given in Table 3.
Table 3
Example LDL-Oxidation Total Cholesterol (VLDL+LDL)
No. Inhibiting Lowering Rate Cholesterol Lowering
IC~~.,~.M) (%) Rate l%)
1 0.46 22 61
2 0.40 35 72
Both compounds tested exhibited excellent lowering action
for (VLDL + LDL) cholesterol and do not show a decrease in HDL
cholesterol and, therefore, the compounds obtained by the present
invention are understood to exhibit strong and selective cholesterol
lowering action.