Note: Descriptions are shown in the official language in which they were submitted.
D 9
. - 1 -
Tricyclic Quinoxalinedione Derivatives
This invention relates to a new class of tricyclic quinoxalinedione
derivatives which are selective antagonists of glycine binding site of the NMDA
(N-methyl-D-aspartate) receptor. Particularly, ~he compounds provided by the
present invention show in viYo antagonism against the excitation of the NMDA
receptors under systemic administrations and therefore, are especially useful
for minlmizing damage of central nervous system induced by ischemic or
hypoxic conditions such as stroke, hypoglycemia, cardiac arrest, and physical
trauma, (see, J. McCulloch, Br. J. clin. Pharmacol., 34, 106 (1992)). The
compounds are also useful in treatment of a number of neurodegenerative
disorders including epilepsy, Huntington's chorea, Parkinson's disease, and : -
Alzheimer's disease (reviews: G. Johnson, Annu. Rep. Med. Chem., 24, 41
(1989) and C,. Johson and C. F. Bigge, ibid., 26, 11, (1991)). The present
1~ compounds may also have analgesic, antidepressant, anxiolytic, and anti -
schizophrenic activities, by virtue of these NMDA-glycine antagonism, as
indicated by recent reports, e.g. A. H. ~ickenson and E. Aydar, Neuroscience
Lett., 121, 263 (1991), R. Trullas and P. Skolnick, Eur. J. Pharmacol., 185, 1
(1990), J. H. Kehne, et al., Eur. J. Pharmacol., 193, 283 (1991), P. H. Hutson, et
al., Br. J. Pharmacol., 103, 2037 (1991), in which the reagents affecting glycine -
binding site of NMDA receptors have shown such aGtivities. Excessive release
of glutamic acid and/or glycine from neuronal and glial cells results in
overexcitation of NMDA receptor-Ca2+ channel complexes and successive
massive amount of Ca2-~ influx into the cell, which leads to neuronal cell death.
NMDA-glycine antagonist described in the present invention would obviously
regulate the amount of Ca2+ influx from the glycine modulatory site of NMDA
receptor-channel complex to maintain normal activities of neuronal cell.
Therefore, the compounds of the present invention may be potential
therapeutic agents for any diseases of animals including human caused by
excessive glutamic acid and/or glycine release in addition to the diseases
indicated above.
Tricyclic quinoxalinediones, 6,7-dihydro-1 H, ~H-pyrido[1 ,2,3,-de]-
2121~
- 2 -
quinoxaline-2,3-diones and 5,6-dihydro-1H-pyrrolo[1,2,3-de]quinoxaline-2,3-
diones are disclosed in WO 93/08188, published after the priority date of this
application, as selective antagonists of glutamate receptors such as NMDA
receptors and AMPA (2-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid)
5 receptors. The compounds of the present invention, however, exhibit much
higher in vivo activities under systemic administrations compared to the
compounds of examples in WO 93/08188.
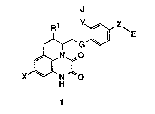
The present invention provides novel tricyclic quinoxalinedione
derivatives depicted by formula 1 and pharmaceutically acceptable salts
1 0 thereof:
J
~1 ~ ~E
,~N ~o
wherein X represents hydrogen, alkyl, halogen, cyano, trifluoromethyl, or
nitro;
R1 represents hydrogen, alkyl, cycloalkyl, or cycloalkylalkyl;
G represents -CONR2- or-NR2CO-, wherein R2 represents hydrogen or
alkyl;
J represents an acidic group or a group which is convertible thereto In
vivo;
E represents an basic group or a group which is convertible thereto in
25 vivo;
Y represents a single bond, alkylene, alkenylene, substituted alkylene,
or y1_Q_y2, wherein y1 represents a single bond or alkylene, y2 represents
alkylene, and Q represents a heteroatom selected from oxygen or sulfur;
~ represents alkylene.
The compounds of the present invention possess both the groups
represented by J and E simultaneously in the same molecule and provide
much higher in vivo activities compared to the compounds which possess one
of the groups represented by J and E in the molecule.
212~6~
This invention further relates to aniline derivatives depicted by formula
6, which are useful intermediates for preparation of compounds 1:
,E
~ Jo
R2NH
wherein R2, Y and Z are as defined above;
J represents a protected carboxyl group;
E represents -NHLl or-NHC(=NL1)NHL1~ wherein L1 represents a
protecting group for amino or guanidino function.
The term "protected carboxyl group" includes -CO2R6, wherein R6
represents alkyl, substituted alkyi, cycloalkyl, cycloalkylalkyl, arylalkyl,
substituted arylalkyl, or alkenyl. A preferable example is -CO2Me.
The term "protecting group for amino or guanidino function" as used
herein includes -CO2R, wherein R represents alkyl, cycloalkyl,
cycloalkylalkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, or alkenyl.
A preferable example is t-butoxycarbonyl.
The term "arylalkyl" as used herein includes straight-chained or
branched alkyl groups attached with aryl group, which contains up to 15 carbon
atoms. Typical examples are benzyl, phenylethyl, 1- or 2-naphthylmethyl, and
1- or 2-naphthylpropyl.
The term "aryl" as used herein includes aryl groups containing up to 10
carbon atoms. Typical examples are phenyl, and naphthyl.
The number of the substituents of substituted aryl or substituted arylalkyl
as used herein rnay be permitted to be up to 3, and the substituents include
alkyl, halogen, trifluoromethyl, and alkoxy.
The number of the substituents of substituted alkyl as used herein may
be permitted to be up to 3, and the substituents include alkoxy, halogen and
trimethylsilyl.
The term "alkyl" as used herein includes straight-chained or branched
alkyl groups containing from 1 to 6 carbon atoms. Typical examples are
methyl, ethyl, n-propyl, isopropyl, sec-butyl, tert-butyl, neopentyl, n-pentyl, and
212~09
- 4 -
n-hexyl.
The term "halogen" as used herein includes fluorine, chlorine, bromine,
and iodine. Typical examples are chlorine and bromine.
The term "alkoxy" as used herein includes straight-chained or branched
alkoxy groups containing from 1 to 6 carbon atoms. Typical examples are
methoxy, ethoxy, propoxy, isopropoxy, sec-butoxy, te~-butoxy, neopentoxy,
pentoxy, and hexoxy.
The term "cycloalkyl" as used herein includes cycloalkyl groups
containing from 3 to 7 carbon atoms. Typical examples are cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl.
The term "cycloalkylalkyl" as used herein includes straight-chained or
branched alkyl groups attached with cycloalkyl groups, which contains up to 13
carbon atoms. Typical examples are cyclopropylmethyl, 2-cyclopentylethyl,
cyclohexylmethyl, and 3-cyclohexylpropyl.
The term "basic group" as used herein means the group which is readily
protonated in v~vo to provide cation. Typical examples are -NH2, -NHR3E, -
NR3ER4E, -NH-C(=NH)-NH2, -NH-C(=NH)-NHR3E, and -NH-C(=NH)-NR3ER4E.
Herein, R3E and R4E independently represent alkyl, cycloalkyl, alkenyl, or
cycloalkylalkyl, or R3E and R4E are joined to form a cyclic amine.
The term "alkenyl" as used herein includes straight-chained or branched
alkenyl groups containing from 3 to 6 carbon atoms, of which an olefinic carbon
atom may not be connected directly with nitrogen atom or oxygen atom.
Typical examples are allyl, 2-butenyl, and 3-butenyl.
The term "group which is convertible to a basic group in vivo" as used
herein includes-NHL, -NLR3E, -NH-C(=NL)-NH2, -NH-C(=NL)-NHR3E, and -
NH-C(=NL)-NR3ER4E. Herein, L means a hydrolyzable group in vivo, such as
alkanoyl group or alkoxycarbonyl group.
The term "alkanoyl" as used herein includes straight-chained or
branched alkanoyl groups containing from 1 to 6 carbon atoms. Typical
examples are formyl, acetyl, propanoyl, n-butanoyl, and pivaloyl.
The term "alkoxycarbonyl" as used herein includes straight-chained or
branched alkoxycarbonyl groups containing from 2 to 6 carbon atoms. Typical
examples are methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl,
- 5 -
isopropoxycarbonyl, sec-butoxycarbonyl, and terf-butoxycarbonyl.
The terrn "acidic group" as used herein means the group which is readily
deprotonated in vivo to provide anion. Typical examples are carboxyl and
tetrazolyl.
The term "group which is convertible to an acidic group in vivo" as used
herein means the group which generates the acidic group in vivo by hydrolysis.
Typica! examples are -CooR3J, -CONH2, -CON(OH)H, -CoNHR3J,
-CoN(OH)R3J, -CON(OR5J)R3J, or-CONR3JR4J, wherein R3J and R4J
independently represent alkyl, cycloalkyl, alkenyl, arylalkyl, substituted
arylalkyl, or cycloalkylalkyl, or R3J and R4J are joined to form a cyclic amine,and R5J represents alkyl.
The term "cyclic amine" which R3E and R4E, or R3J and R4J are joined to
form includes 3 to 7 membered cyclic amine such as azetidine, pyrrolidine, or
piperidine, and 5 to 7 membered cyclic amine containing oxygen or nitrogen
atom wherein the oxygen or nitrogen atom is always bonded to the adjacent
alkylene group, such as piperazine, N-methylpiperazine, or morpholine.
The term "alkylene" as used herein includes straight-chained or
branched alkylene groups containing from 1 to 6 carbon atoms. Typical
examples are methylene, dimethylene, trimethylene, tetramethylene, 2 -
methyltrimethylene, 3-methyltrimethylene, 1,1-dimethylmethylene,
pentamethylene, and hexamethylene.
The term "alkenylene" as used herein includes straight-chained or
branched alkenylene groups containing from 2 to 6 carbon atoms. Typical
examples are vinylene, 1-propenylene, 2-propenylene, 3-butenylene, 2-ethyl-3 -
butenylene, 4-pentenylene, 3-methyl-4-pentenylene, and 1-hexenylene.
The substituent of the term "substituted alkylene" includes hydroxy,
-OR3S, -oCoR3S, amino, -NHCOR3S, -NHCo2R3s~ carboxyl, and Co2R3S,
wherein R3S represents alkyl, cycloalkyl, alkenyl or cycloalkylalkyl. Typical
exampies of the "substituted alkylene" are -CH(OH)-, -CH(OAc)-, -CH(C02-tert-
Bu)-, and -CH2-CH2-CH(CO2Et)-. Preferably, the substituent and J group may
be attached to the same carbon atom.
Typical examples of y1 Q y2 are -O-CH2-, -S-CH2-, -CH2-O-CH2-, -CH2 -
S-CH2-, and-CH2CH2-O-CH(CH3)-.
- 6 -
The expression "pharmaceutically acceptable salts thereof" represents
either non-toxic acid addition salts or base addition salts.
The acid which forms non-toxic salts with the compounds provided by
formula 1 include inorganic acid such as hydrochloric, hydrobromic, sulfuric,
5 and phosphoric acid or organic acid such as acetic, oxalic, citric, lactic, tartaric,
malonic, fumaric, maleic acid, and methansulfonic acid. On the other hand, the
non-toxic base addition salts include inorganic metal salt such as lithium,
sodium, potassium, magnesium, aluminum, and barium salt or organic
quaternary ammonium salt such as ammonium, triethylammonium,
10 tetrabutylammonium, pyridinium, pyrrolidinium, and piperidinium salts.
The compounds provided by the present invention have an asymmetric
center a~t C-5 position. Although the enantio-mixtures of the compounds
regarding the C-5 position are encompassed in the scope of the present
invention, the preferable configuration of the C-5 position may be "S". When
15 the compounds according to the invention have more than two asymmetric
centers, they additionally exist as diastereomers. Such diastereomeric pure
compounds and diastereo-mixtures of these compounds are also
encompassed within the scope of the present invention.
The tricyclic quinoxalinedione derivatives of the present invention can
20 be formulated to conventional pharmaceutical preparations such as tablets,
pills, capsules, powders, granules, suspensions, or emulsions all for oral
administration, and such as sterile parenteral solutions or suppositories for
parenteral or rectal administration, respectively. The solid compositions such
as tablets can be routinely prepared by mixing the active ingredient with
conventional pharmaceutical carrier or diluent such as lactose, sucrose or
cornstarch, binder such as hydroxypropylcellulose, polyvinylpyrrolidone or
hydroxypropylmethylcellulose, disintegrating agent such as sodium
carboxymethylcellulose or sodium starch glycolate, lubricants such as stearic
acid and magnesium stearate, or preservatives. For parenteral administration,
30 the active compound is dissolved or suspended in a physiologically acceptablepharmaceutical carrier such as water, saline, oil or dextrose solution, which
may contain auxiliary agent such as emulsifier, stabilizer, salt for influencingosmotic pressure or buffer, if desired. The dosage range can be varied widely
- ` ' ~: ~.. ~ ' ' ' ' : ,
7 -
depending on the severity of the particular disease, age, weight, and sex of thepatient, and the route of administration. Typically, effective dosages are in the
range of 1 to 1000 mg/day, or preferably of 10 to 500 mg/day orally for adult
patients, which may be given in a single dose or in multiple doses. For
parenteral administration, the dosage range of 0.1 to 500 mg/day, or more
suitably of 3 to 100 mg/day/patient can be employed with a single dose or with
multiple doses.
Examples of compounds within the scope of the invention include:
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]q~inoxaline-2,3 -
dione;
(S)-9-chloro-5-(p-tert-butoxycarbonylaminomethyl-~carboxyphenyl -
carbamoylmethyl)-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione;
(S)-9-chloro-5-(p-aminomethyl-~carboxyphenylcarbamoylmethyl)-6,7 -
dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3 -
dione;
(S)-9-bromo-5-(p-tert-butoxycarbonylaminomethyl-o-carboxyphenyl -
carbamoylmethyl)-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione;
(S)-9-bromo-5-(p-aminomethyl-~carboxyphenylcarbamoylmethyl)-6,7 -
dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride;
(+)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3 -
dione;
(~)-9-bromo-5-[p-aminomethyl-~(methoxycarbonyl)phenylcarbamoyl -
methyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione;
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl -
methyl)phenylcarbamoylmethyl]-6,7-dihydro-1tJ, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-chloro-5-[p-ter~-butoxycarbonylaminomethyl-~(carboxymethyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione;
(S)-9-chloro-5-[p-aminomethyl-~(carboxymethyl)phenylcarbamoyl -
methyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
hydrochloride;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl -
methyl)phenylcarbamoylmethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3~de] -
quinoxaline-2,3-dione;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-o-(carboxymethyl) -
phenylcarbamoylmethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione;
(S)-9-bromo-5-[p-aminomethyl-o-(carboxymethyl)phenylcarbamoyl-
methyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione
hydrochloride
(+)-9-bromo-~-[p-(2,3-di-tert-butoxycarbonylguanidinomethyl)-o -
(methoxycarbonylmethyl)phenyicarbamoylmethyl]-6,7-dihydro-1H, 5H-
pyrido[1,2,3-de]quinoxaline-2,3-dione;
(+)-9-bromo-5-~p-(2,3-di-tert-butoxycarbonylguanidinomethyl)-o -
(carboxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3 -
de]quinoxaline-2,3-dione;
(+)-9-bromo-5 [p-guanidinomethyl-o-(carboxymethyl)phenylcarbamoyl -
methyl]-6,7-dihydro 1 H, SH-pyrido[1 ,2,3-de]quinoxaline-2,3-dione;
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(2-methoxycarbonyl-
ethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline -
2,3-dione;
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(2-carboxyethyl) -
phenylcarbamoylmethyl~-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione; :
(S)-9-chloro-5-[p^aminomethyl- ~(2-carboxyethyl)phenylcarbamoyl -
methyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione
hydrochloride
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3-methoxycarbonyl-
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~?3-carboxypropyl)-
.... . ~ , . , . ~ ,. . . ...... . . . .
-, -: , ~: . - :
- . . - - . -
-~-` 2:12~6~3
.
g
phenylcarbamoylmethyl]-6,7-dihydro-l H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3 -
dione;
(S)-9-bromo-5-[arninomethyl-~(3-carboxypropyl)phenylcarbamoyl -
methyl]-6,7-dihydro-lH, 5H-pyrido~1,2,3-de]quinoxaline-2,3-dione
5 hydrochloride;
(5S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(1 -methoxy -
carbonyl-1-acetoxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-
pyrido[1 ,2,3-de]quinoxaline-2,3-dione;
(5S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(1 -carboxy-1 -
hydroxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione,
(5S)-9-bromo-5-[p-aminomethyl-~(1 -carboxy-1 -hydroxymethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
hydrochloride;
(5S)-9-chloro 5-[p-tert-butoxycarbonylaminomethyl-~(1-methoxy-
carbonyl-1 -acetoxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H -
pyrido[1 ,2,3-de]quinoxaline-2,3-dione;
(5S)-9-chloro-5-~p-tert-butoxycarbonylaminomethyl-~(1 -carboxy-1
hydroxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione;
(5S)-9-chloro-5-[p-aminomethyl-~(1 -carboxy-1 -hydroxymethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione
hydrochloride;
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(tert-butoxycarbonyl -
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-chloro-5-[p-aminomethyl-~(carboxymethoxy)phenylcarbamoyl-
methyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione
hydrochloride;
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(ethoxycarbonyl-
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-chloro-5-[~aminomethyl-~(ethoxycarbonylmethoxy)phenyl-
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione
hydrochloride;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(tert-butoxycarbonyl -
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de] -
5 quinoxaline-2,3-dione;
(S)-9-bromo-5-[p-aminomethyl-~(carboxymethoxy)phenylcarbamoyl -
methyl]~6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione
hydrochloride;
(S~-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(4-ethoxycarbonyl -
butyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline -
2,3-dione;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(4-carboxybutyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de~quinoxaline-2,3 -
dione;
(S)-9-bromo-5-[p-aminomethyl-~(4-carboxybutyl)phenylcarbamoyl-
methyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
hydrochloride;
(S)-9-chloro-5-~p-tert-butoxycarbonylaminomethyl-~(3,3-diethoxy -
carbonylpropyl)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-dicarboxy-
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-chloro-5-[aminomethyl-~(3,3-dicarboxypropyl)phenylcarbamoyl-
methyl]-~,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
hydrochl~ride;
(S)-9-chloro-5-[aminomethyl-~(3-carboxypropyl)phenylcarbamoyl -
methyl]-~,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3^dione
hydrochloride;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-diethoxy-
carbonylpropyl)phenylcarbamoylmethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-dicarboxy-
2~21~9
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione;
(S)-9-bromo-5-~aminomethyl-o-(3,3-dicarboxypropyl)phenylcarbamoyl -
methyl]-6,7-dihydro-1 H, 5H-pyrido~1 ,2,3-de]quinoxaline-2,3-dione
5 hydrochloride;
and salts thereof;
wherein the numbering used for the tricyclic quinoxalinedione system is as
shown in the following figure.
_Igure
~ S
15 Process A-1
Compounds of formula 1 may generally be derived from key
intermediates 3, which themselves are comprised of the invention;
Jo J
R1 Y~Z~ E R1 Y~ E
2 0 ~ G~ ~ GJ~
X ~ N~ O X J~ N ~ O .
25 wherein X, R1, G, Y, Z, J, E, E and J are as defined above.
Process A-2
The key intermediates of formula 3a may be prepared by condensation
of 4 with 6;
Q ~
- 12-
R~ C2H , ~ E R' G1~ E
5XJ~N ~O ~r XJ~ N~ O
H R2NH H
4 6 3a
wherein X, R1, Z, E, Y, J and R2 are as defined above, and G1 is -CONR2-.
The condensation may be carried out in the presence of condensation
1 0 reagent such as 1 -ethyl-3-(3'-dimethylaminopropyl)carboximide-hydroxy -
benztriazole, isobutyric anhydride-triethylamine, and N,N-bis(2-oxo-3 -
oxazolidinyl)-phosphinic chloride-triethylamine in an inert solvent such as
DMF, THF, or dichloromethane at ambient temperature.
Process A-3
Compounds 4 may be prepared starting from compounds of formula 62
as illustrated in the following scheme;
~X R1 0~ R1 ~ R1 ~:
H CO2Me H OH 1~1~ N~CN .
H
62 64 65
G~ CO2Me 3~ CO2Me X ~ CO2Me
COCO2Et COCO2Et
66 67 68
R1 R1
~ CO2Me ~ CO2H
X~CO2Me XJ~N O XJ~N~o
N2 COC02Et H H
69 70 4
.-,, , ",. . : : ,: -,, :.,. . , :. - , :,
21~g~
- 13-
whereln X and R1 ar~ as defined abov~.
Compounds of formula 62 may be readily prepared by a method
doscribed in litera~ures. For ~xample, tetrahydroquinollne-2-carboxyllc acid
methyl ester may conveniently be prepared by hydrogenation of quinaldinic
5 acid over PtO2 in methanol followed by treatment of thionyl chloride In
methanol at ambient to refluxing temperature, or by direc~ hydrogenation of
quinaldinic acid rn~thyl ester in acetic acid. Alternatively, tetrahydroquinoline-2 -
carboxylic acid methyl ester may be prepared by rsduction of quinaidinic acid
methyl ester with combination of NiC12 and sodium borohydrlde in methanol at
10 0C to room temperature. R1 sub.stituted tetrahydroquinoline-2-carboxylic acid
methyl esters comprised in compounds ~2 can be prepared by following
sequence: a) Carboxyl group can be introduced into C 2 position of
R1substituted corresponding quinolines by using Reissert reaction followed by
hydrolysis ~W. E. McEwen and R. L. Cobb, Chem. Rev., 5~, 511 (1955)): and b)
15 the resulUng substituted quinoline-2-carboxylic acids were methylated, and
then reduc0d to tha corresponding tetrahydroquinoline-2-carboxylic acid
methyl esters as mentioned above. The R1 substituted quinolines may be
commercially available or readily prepared by using Skraup reaction ~R. H. F.
Manske and M. Kulka, Org. React., 7, 59 (19~3)). When R1 is not hydrogen,
20 compounds 62 may exist as a diastereomixture. Such a diastereomer can be
convenientiy separated to a pure isomer by a conventional column
chromatography technique.
1) Compounds 82 are reduced to the corresponding alcohols 64 by
using lithium aluminum hydride in an iner~ solvent such as diethyi ether or T~iF25 at 0C to refluxing temperature.
2) Compounds 64 can be converted into 65 by two step sequences: a)
treatment with triphenyl phosphine imidazole-iodine in toluene or in a mixed
solvent oF toluene-acstonitrile at 0C to ambient temperature to form the
corresponding iodides, and b) replacement ef the iodide to the Gyanide with
30 sodium cyanlde in DMF at amblent tempsra~ure to around 80~C to provide 6~.
3) Hydrolysis of compounds 65 with a strong acid such as 12 N
hydrochloric acid at elevated temperatur~ followed by methylation of the
resulting carboxylic acid with thionyl chloride in methanol gives compounds
; 2~2~9
66.
4) Compounds of formula 66 are transformed to compounds 67 by
reaction with ethyl chloroglyoxalate in an inert solvent such as methylene
chloride, chloroform, tetrahydrofuran (THF), and ethyl acetate, at temperature
range of - 10 to 30C.
5) Compounds 67 are converted to compounds 68 by using
conventional aromatic electrophilic substitution technique (see, for example
~dvanced Organic Chemistry, Jerry March ed., Chapter 13, 576). In the case
of X = Br, compounds 67 are reacted with bromine in halogenated solvent such
as methylene chloride in the presence or absence of a catalyst such as Fe
powder to give the brominated compounds comprised in 68. In the case of X =
Cl, compounds 67 are reacted with N-chlorosuccinimide in dimethylformamide
(DMF) at ambient temperature to give the chlorinated compounds comprised in
68. When X is Br, compounds 68 can also be synthesized by direct
1~ bromination of compounds 66 with N-bromosuccinimide in dimethylformamide
(DMF) followed by acylation with ethyl chloroglyoxalate.
~) Compounds 6~ can be transformed to compounds 69 by
conventional nitration conditions including treatment with fuming nitric acid at0C to ambient temperature, nitric acid or isopropyl nitrate in concentrated
sulfuric acid at 0C to ambient temperature, a mixed reagent of trifluoroacetic
anhydride and ammonium nitrate in halogenated solvent such as chloroform
and methylene chloride at ambient to refluxing temperature, and nitronium
tetrafluoroborate in halogenated solvent such as chloroform and methylene
chloride at ambient temperature.
7) Reductive ring closure of compounds &~ to 70 are effected by
aqueous titanium trichloride in protic solvent such as methanol, ethanol, and
acetic acid or in aprotic solvent such as acetone, THF, or DMF at 0C to
ambient temperature. Other reducing reagents including stannous dichloride,
zinc, iron powder, and formate-palladium on carbon may be utilizable for the
ring closure. The compounds of formula 70 wherein X is Br may be especially
useful, since the Br substituent of X can be readily displaced to various
substituents such as Cl, I, CN, alkyl, and CF3 under conditions such as CuCI -
dimethyl sulfoxide-1 50C, Cul-KI-hexamethylphosphorous triamide-1 50C,
~ t ~
- 15 -
CuCN-dimethyl sulfoxide-150C, alkyl-copper reagent-TllF-0C, and
CF3CO2Na-Cul-N-methylpyrrolidone-1 60C, respectively. Compounds of
formula 70 wherein X is hydrogen may be obtained by catalytic hydrogenation
of compounds of formula 70 wherein X is Br by using Pd/C in methanol.
5 Compounds of formula 70 wherein X is nitro may be obtained by standard
nitration of compounds of formula 7û wherein X is hydrogen.
8) Carboxylic acids 4 can be prepared by hydrolysis of 70. The
hydrolytic conditions include treatment with alkaline metal hydroxide or
carbonate such as lithium hydroxide, sodium hydroxide, potassium carbonate
10 in a mixed solvent of water and protic or aprotic solvent such as methanol,
ethanol, or THF at temperature range of 0 to 60C, or treatment with aqueous
strong acid such as 1 N ~ 12 N hydrochloric acid, or 5 ~ 48% hydrobromic acid
in protic or aprotic solvent such as acetic acid or dioxane at temperature rangeof ambient temperature to 1 00C.
Similarly, Optically active 4 wherein R1 is hydrogen may be prepared, if
desired, starting from optically active 62 wherein R1 is hydrogen of which
synthesis is described in J. Chem. Soc. Perkin 1 1977, ~96.
Process A-4
Compounds 6 (6a and 6b) may be prepared as illustrated in the
20 following scheme;
Z, E z~ E E
~,J ~Jo ~ Jo
NO2 NH2 R2aNH
2~ 7 6a 6b
wherein Z, E, Y and J are as defined above, and R2a is alkyl.
Compounds 6a may be obtained by reduction of the corresponding
nitrobenzene derivatives 7. The reduction may be performed with conventional
catalytic hydrogenation on palladium/charcoal or palladium hydroxide in an
30 inert solvent such as ethyl acetate, methanol, or ethanol. Compounds 6b may
be prepared by alkylation of compounds 6a with R2al in the presence of a base
such as potassium carbonate or sodium hydride. Reductive amination using
an appropriate aldehydes or ketones in the presence of sodium borohydride or
... .... .. . ~ . . . ,.. . . . . -- . . . i . . . .
~ -
~ 2~2.t~09
- 16-
sodium cyanoborohydride in an alcoholic solvent such as methanol at ambient
temperature may also be utilized for the alkylation.
Process B
Compounds of formula 7a may be prepared starting from readily
5 available 8;
a s
- 17-
CO2Me ~ CO2Me ~ CO2Me
N02 N~2 NO2 ~----\
8 / 1 15
R7 OH R~f; o
CO2Me ~ CO2Me
N02 NO2
/ ~1 16 1 17
J o~
q-~ ~ z
r N3 r N~ R7~r N~o ~ R
[~CO2Me ~ CO2Me ~CO2Me ~CO2Me
NO2 NO2 NO2 NO2
10a ~ 10b 10c
20 [$`Co2R6 A A
11 l ~ / ~
H N~ NH
~' 1 ! ,N` Ll ,NH
N~" N~ Ll N~ R7 N~ R7 N~ Rs~ R9 zl
$` C~2R6 ~ CO R6 ~ CO2R6 ~ CO2R6 ~ CO2R6 ~ C02R
NO2 N02 NO2 NO2 NO2 N02
7c 7b 7d 7e 7t 79
, E
CO2R6
7a
212-~ ~as
- 18-
wherein Z, E, L1 and R6 are the same as defined above, R7 is alkyl, Ra is
hydrogen or alkyl, R9 is hydrogen or alkyl, and Z1 is a single bond or alkylene.Process B-1
Compounds ~ may be converted into compounds of formula 7b and 7c,
5 as outlined below.
1) Compounds ~ may be brominated with N-bromosuccinimide in the
presence of azobisisobutyronitrile (AIBN) or benzoyl peroxide ~BPO) in an inert
solvent such as carbon tetrachloride or chlorobenzene under reflux to provide
9.
2) Treatment of 9 with potassium phthalimide in an aprotic solvent such
as DMF at temperature ran3e of 50 to 80C may provide 1 Oa.
3) Hydrolysis of ~Oa under acidic conditions followed by re-esterification
with R60H in the presence of acid promoter such as thionyl chloride or
hydrogen chloride may afford compounds 11. The hydrolytic conditions
15 include treatment with aqueous strong acid such as 6 N ~ 12 N hydrochloric
acid, or 25 ~ 48% hydrobromic acid in protic or aprotic solvent such as acetic
acid or dioxane at temperature range of 50 to 1 00C. Hydrolysis of phthalimide
group of 10a may be utilized by hydrazine or methylhydrazine in protic solvent
such as methanol at temperature range of O to 60C. Compounds 11 may also
20 be obtaine~ by reaction of 9 with sodium azide in an aprotic solvent such as
DMF at temperature range of 20 to 80C followed by hydrogenation over
palladium/charcoal in an inert solvent such as ethyl acetate.
4) Free amines 11 may be protected with L1 group to afford compounds
of formula 7~. For example, reaction of 11 with di-t-butyl-dicarbonate in the
25 presence of an organic base such as triethylamlne in an inert solvent such asdichloromethane or ethyl acetate at ambient temperature affords compounds of
formula 7b wherein L1 is t-butoxycarbonyl. Treatment of 11 with
MeSC(=NL1)NHL1 in the presence of an organic base such as triethylamine in
an inert solvent such as dichloromethane or ethyl acetate at ambient
30 temperature may afford compounds of formula 7c.
Process B-2
Compounds of formula 7d and 7e may be prepared as outlined below.
1) Bromides 9 can be oxidized to aldehydes 15 by an appropriate
2~ 2~6~
`~
- 19-
oxidation conditions. The conditions include reaction with dimethyl sulfoxide
and trimethylamine N-oxide in dichloromethane at temperature range of 20 to
40C~
2) Treatment of 15 with an alkylating reagent such as R7M~Br or R7Li in
5 diethyl ether or THF at temperature range of -20 to 0C provides alcohols 16.
3) Mitsunobu reaction of 16 by using phthalimide, triphenylphosphine,
and diethyl azodicarboxylate in THF at ambient temperature may afford
compounds 10b, which may be converted into compounds of formula 7d and
7e, as described in the conversion of 1 Oa into the compounds of formula 7b
10 and 7c.
Process B-3
Compounds of formula 7f and 7g, may be prepared as outlined below.
1) Alcohols 16 may be oxidized to the corresponding ketones 17 by an
appropriate oxidation conditions. The conditions include treatment with
15 manganese oxide in dichloromethane at room temperature, treatment with
DMSO-ClCOCOCI-triethylamine in dichloromethane at -70C, treatment with
pyridinium chlorocromate in dichloromethane at ambient temperature, and
treatment with Dess-Martin reagent.
2) ~eaction of aldehydes 15 or ketones 17 with Wittig reagent
20 PPh3=CR9-Z1-phthalimide in an inert solvent such as Ttl~ at temperature
range of - 70 to 60C followed by reduction with diimine generated from
reaction of potassium azodicarboxylate with acetic acid in acetonitrile at
temperature range of - 20 to 0C affords compounds 10c, which may be
converted into compounds of formula 7~ and 79 as described in the
25 conversion 1~a into the compounds of formula 7b and 7c.
Process C-1 and C-2
Compounds of formula 7h and 7i, may be prepared as illustrated in the
following scheme;
:: :
, ....
. ~ ~ . .-. . - - . - - - - ~.
. . .. . - . . . .
,- . ~ -- - . ~ . - ~. -
2~ ~6~9
- 20 -
z.E z.E z.E
CO2R6 ~OH ~
NO2 NO2 NO2 H
7a 18 ~ 19
//
/
z,E z, E ,E
~OH ~O
NO2 Rla NO2 R10a N2 R10
21 ~ 7h
,E
Me Me J0 Z J
CO2Me ~ ~
NO2 NO2 R10 ~ NO2 Rl
2~ 8 1 25 / 7i
i~
Me J0 :
'
W`R11
N2 Rl
26
.. - , . ... , . -, . .. -
~ 2:~216~9
- 21 -
wherein R10 and R11 are independently hydrogen or alkyl, Y3 is a single bond
or alkylene, Rla is alkyl, and Z, E and J are as defined above.
Process C-1
1) Compounds 7a may be reduced to alcohols 1~ by using sodium
5 borohydride-methanol in THF at ambient to refluxing temperature.
2) Alcohols 18 may be oxidized to the corresponding aldehydes 19
under the conditions described in the conversion of 16 to 17.
3) Treatment of 19 with alkylating reagent such as R10aMgBr or R10aLi in
diethyl ether or THF at temperature range of - 20 to 0C provides alcohols 20.
4) Alcohols 20 may be oxidized to the corresponding ketones ~1 by an
appropriate oxidation conditions. The conditions include treatment with DMSO -
ClCOCOCI-triethylamine in dichloromethane at -70C, treatment with
pyridinium chlorocromate in dichloromethane at ambient temperature, and
treatment with Dess-Martin reagent.
5) Reaction of aldehydes 19 or ketones 21 with Wittig reagent
PPh3=CR1 1-Y3-J0 in an inert solvent such as THF at temperature range of - 70
to 60C affords compounds of formula 7h, which may be selectively reduced to
compounds of formula 7i, by catalytic hydrogenation using palladium/charcoal
or by diimine reduction.
Process G-2
Alternatively, compounds of formula 7h and 7i may be prepared s~arting
from compounds 8 as described below.
1 ) Compounds 8 may be converted into compounds 25 and 26
according to a sequence analogous to that used in the conversion of
compounds 7a into compounds 7h and 7i, respectively (process C-1). ..
2) Compounds 25 and 26 may be converted into the corresponding
compounds 7h and 7i, respectively, according to a sequence analogous to
that used in the conversion of 8 into compounds 7a (process B).
Process C-3
Convenient methods for making a -Y-J part of formula 7 may be a use
of malonate anion.
Compounds of formula 32 and 7j, may be prepared as illustrated in the
following scheme;
~- - , , ,
2 ~ n s
- - 22 -
M~ Me M~ Mq
~ CO2Me~~ ~L ~C02FI~
~2 NO2 R12 NO2 R12 NOz R12
B 29 30 31
6 ~/
, E ~
~3 OH \ ~b~3~Co3~8
,E / NO2 R12 NO2 R12
IJ~ ~ 32 7J
b~OH
NO2 R~
1~ 2
wherein Z, E, R6 and R10 are as ciefinad above, R12 is hydrogen or alkyl, R
is hydrogen or alkyl, and L4 is a 10aving group such as Cl, Br, I, OSO2Ph, and
OS02CF3.
Compounds 8 may be convsrted into alcohols 29 by a sequence
20 analogous to that used in the convcrsion of compounds 7a into 18 and 20
(process C-1, 1) - 3)). Compounds 29 may be converted to 30. Herein, Cl
and Br may be introduced by reaction with SOCI2 or SO~r2, and CCI4-PPh3 or
C:Brq-PPh3, respectively and I may be introduced by reaction of 12-PPh3 -
imida~ole in an in~rt solvent at tamperature range of 0 to ~0C. PhSO2O- and
25 CF3SO2O- groups may be prepared by reaction with PhSO2Cl-triethylamine ~ ~:
and (CF3SO2)2O-triethylamina, respectively in an iner~ solvent such as
dichlorome~hane at 0C to ambient temperature. Treatment of 30 with
R6OCOCHR13CO2R6 in the presenca of a base such as sodium hydride,
potassium t-butoxide, and lithium hexamethyldisilæide in an aprotic solvent
30 such as Tl IF, DMF, or DMSO at temparature range of 0 to 60C provid~ 31.
Compounds 3 i may be converted into compounds 32, which are comprised of
compounds of formula 7, according to a sequence analogous ~o that us~d in
the converslon of 8 into compounds of formula 7a (procass B). !n sorne cases,
, .
, ~ . . .. . ;
" 2~2:~09
- 23 -
an alkoxycarbonyl group of 32 may be simultaneously decarboxylated during
the hydrolysis step of the phthalimide group to afford compounds of formula 7j.
Alternatively, 18 and 20 may be converted into compounds 32 by a sequence
similar to that described in the conversion of 29 to 31.
5 Process C-4
Compounds of formula 36 and 7k, may be prepared as Illustrated in the
following scheme;
E E E
Z' ;Z' Z'
~O or ~O -- ~ ~14
NO2 H No~ R10a NO~ R12
19 21 3~
,E~ ,E
~CO2R6 ~1R13
NO2 R12 CO2R6N02 Rl2 CO2R6
36 7k
wherein Z, E, R6, R10a~ R12 and R13 are as defined above, R14 and R15 are . -
independently hydrogen or alkyl.
Compounds 19 and 21 may be converted into 35 by Wittig reaction with
PPh3=CR14R15 in an inert solvent such as THF at temperature range of - 70 to
60C. Reaction of 35 with anion of R6OCOCHR13CO2R6 under conditions
similar to those mentioned in the conversion of 30 into 31 may provide
compounds 36 which are comprised of compounds of formula 7. An
alkoxycarbonyl group of compounds 36 may be decarboxylated to hydrogen, if
necessary, to provide compounds of formula 7k, under certain conditions such
as heating in DMSO at elevated temperature in the presence of a salt such as
sodium chloride.
; . - . . ~ .. - - ~ - ~ .
:; ; - . .
- . .
` 21~6~
- 24 -
Process C-5~
Compounds of formula 7m and 7n, may be prepared as illustrated in
the following scheme;
o~ o~
Me Me z,N O z,N O
OH ~ L5 ~ Ls ~ CO2R6
NO2 N02 NO2 NO2CO2R6
41 37 38 39
E ~/ E
Z' Z'
~,1co R6 b~,R13
NO2 CO2R6NO2 CO2R
7m 7n
wherein Z, E, R6 and R13 are as defined above, and L5 iS a leaving group
such as OSO2Ph or OSO2CF3. :
Compounds 37 readily prepared from 41 as described in the
conversion of 29 to 30 (process C-3), may be converted into 38 by a method
similar to that described in the conversion of 8 into 10a,b,c (process B).
Reaction of 38 with anion of R6OCOCHR13CO2R6 as mentioned in synthesis of
3t (process C-3) may provide compounds 39 which may be converted into -
compounds of formula 7m and 7n, according to a sequence analogous to that
used in the conversion of 10a,b,c into the corresponding compounds of
formula 7a (process B).
Process D-1
Compounds of formula 7p, may be prepared as illustrated in the
following scheme;
2 ~ 21609
- 25 -
Me Me Z
b y2 ~ y2
OH ~ O CO2R~ ~ O CO2R6
N02 NO2 NO2
41 42 7p
wherein y2 iS alkylene, and Z, E and R6 are as defined above.
Compounds of formula 7p may be prepared from compound 41.
Reaction of 41 with BrY2CO2R6 in the presence of base such as potassium
carbonate, potassium t-butoxide, or sodium hydride in an inert solvent such as
10 acetonitrile, THF, DMF, or DMSO at temperature range of 0 to 60C may
provide compounds 42. Conversion of 42 into compounds of formula 7p may
be performed as described in the conversion of 8 into compounds of formula
7a (process B). :
Process D-2 -
l ~ Compounds of formula 7q, may be prepared as illustrated in the
following scheme;
o~ o~ ~ -
N_~' N_~ z E
~S CO2R6 ¢lS CO2R6
NO2 N02 N02
38 44 7q
wherein Z, F, y2, L5 and R6 are as defined above.
Compounds of formula 7q may be prepared from compounds 38.
Reaction of 38 with HS-Y2CO2R6 in the presence of a base such as sodium
hydride, potassium t-butoxide, and lithium hexamethyldisilazide in an aprotic
solvent such as THF, DMF, or DMSO at temperature range of 0 to 60C may
provide 44. Conversion of 44 into compounds of formula 7q may be
performed as described in the conversion of 10a,b,c into the corresponding
compounds of formula 7a (process B).
; ~ - .. - .; . ~. -
.. .. ~
. ~ . ~- . . . :
- . .
2 1 ~ 9
- 26 -
Process E-1
Compounds of formula 7r, may be prepared as illustrated in the
following scheme;
Me Me
(~, L4 1~, C02R6
NO2 Rl2 NO2 Rl2
~0 51
Me Me / Me
CO2R6 ~Ys OH (~Y; O co2r~
N02 ¦ NO2 N2
15 ~ y3 I E
26 ) ~y510' `CO R6
7r :-
~0 wherein Z, E, JQ, L4, y3, y2, R12 and R6 are as defined above, and Y5 is a
single bond or alkylene.
Reaction of 30 with sodium cyanide in an aprotic solvent such as THF,
DMF, or DMSO at ambient temperature to 60C followed by acid hydrolysis and
esterification may provide compounds 51. Compounds 8, 26, and 51 may
25 readily be converted into compounds of formula 48 as described in the
conversion of 8 into 29 (process C-3). Reaction of 48 with BrY2CO2R6 as
described in the conversion of 41 to 42 (process D-l ) followed by
transformation similar to that described in the conversion of 8 into compounds
of formula 7a (process B) may provide compounds of formula 7r.
- 27 -
Process E-2
Compounds of formula 7s, may be prepared as illustrated in the
following scheme;
`~
Me 7 o
~y OH ¢~yS L
NO2 N2
48 /52
~ ~C2~ Z
NO2 NO2
7s
wherein Z E L5 y5 y2 R12 and R6 are as defined above.
Compounds 48 may be converted into compounds 52, as described in
the conversion of 41 to 38 (process C-5). Reaction of ~2 with HSY2CO2R6 as
described in the conversion of 38 to 44 (process D-2) followed by
transformation similar to that described in the conversion of 10a,b,c into the
corresponding compounds of formula 7a (process B) may provide compounds
of formula 7s.
Process F-1
Compounds of formula 7t, may be prepared as illustrated in the
following scheme;
Me Me Me Z
~Y5 R12 ~yS~Y 12 ~ X2CO r~S
NQ2 N02 NO2 NO2
48 49 50 7t
- : ~..... -.. ,
.: - ~ -::: . :, ,,: . :- ::: .:., :: : . . : :
.~, ~:: ~. .
:: ~ : -:: ~. -:. - . . .:: .:: -- .. : - - .
-` 2 ~
- 2~ -
wherein Z, E, J, R3S, R6, Y5 and R12 are as defined above, and X2 is R3So-,
R3SCO2, R3SCoN~-, or R3SoCoNH-.
Compounds 48 may readily be oxidized into compounds of formula 49,
as described in the conversion of 16 to 17 (process B). Treatment of 49 with
5 trimethylsilyl cyanide in the presence of zinc iodide in dichloromethane at
temperature range of 0 to 40C followed by acid hydrolysis and successive
protection of the resulting carboxylic acid into J and the alcohol into x2 may
provide compounds of forrnula 50 wherein X2 represents R3So- or R3SCOO-
wherein R3S is as defined above. The protection of the alcohol to -oR3S may
10 be carried out by treatment with R3SI in the presence of a base such as sodium
hydride, potassium carbonate, and lithium hexamethyldisilazide in an aprotic
solvent such as DMF, THF, or DMSO at temperature range of 40 to 80C. The
protection of the alcohols to -OCoR3S may be carried out by treatment with
R3SCOCl or (R3SCO)2O in the presence of an organic base such as
15 triethylamine and pyridine in an inert solvent such as dichloromethane at
ambient temperature.
Similarly, compounds 49 may be converted into compounds of formula
50 wherein x2 represents R3SCoNH- and R3SoCoNH- by conventional
Strecker or Bucherer synthesis followed by protection. The protection of the
20 resulting amine may be carried out by treatment with R3SCOCI, (R3SCO)2o~
R3SoCoCi, or (R3Soco)2o in the presence of an organic base such as
triethylamine or pyridine in an inert solvent such as dichloromethane at
ambient temperature.
Compounds 50 may be transformed into compounds of formula 7t, by a
25 route similar to that described in the conversion of 8 into compounds 7a
(process B).
Process F-2
Compounds of formula 7u, may be prepared as illustrated in the
following scheme;
~; i' -' ~ ':. :: ' -. ' :
','": :~ ' :'. ',
, . . . . .
-` 2121~09
- 29 -
Me / Me \ Z
~Y R (~ySJ~ ) No2 ~ C02RS
wherein Z, E, J, Y5, R12 and R6 are as defined above, y6 iS a single bond or
alkylene, R16 is hydrogen or alkyl.
Reaction of compounds of formula 49 with Wittig reagent PPh3=CR16-Y6 -
J in an inert solvent such as THF at temperature range of - 70 to 60C followed10 by transformation similar to that described in the conversion of 8 into
compounds 7a (process B) affords compounds of formula 7u.
Process G
Compounds of formula 3b, may be prepared as illustrated in the
following scheme;
o ~
R~ NHR2 z/E RlR2NJ~3~ ,E
XJ~N Xo ~ X~N O
2 0 H C02H H
57 3b
wherein X, R1, R2, Z, E, Y and J are as defined above.
Compounds of formula 1 wherein G is -NR2C0- may also be derived
from the key intermediates 3b, which themselves are comprised of the
25 invention. The key intermediates 3b may be prepared by condensation of 55
with 57, as described in that of 4 with 6 (process A-2).
Process H-1
Compounds of formula 55 (55a and 55b), may be prepared starting
from compounds 64 as illustrated in the following scheme;
30 -
,~ .
o N
Rl ¦
~ ~
OH ~ ~ N~ x~ ~
64 73 / 74
~ R1
~f NH2 ~ NHRza
~X C02M~ ~ X
~2 i5a 55b
wherein X, R1 and R2a are as defined above.
1) Compounds 64 can be converted into 73 by ~NO steps sequence: a)treatmsnt with triphenyl phosphine-imidazola-iodine in toluene or in a mixed
solYent of toluane-acetonitrite at 0C to ambient temparature to form the
20 correspondin~ iodide, and b) replacement of the iodide to the phthaiimide with
potassium phthalimide in DMF at ambient t~mperature to around 80C to
provide 73. Alternatively, compounds 73 are prepared from 62 by t'nres steps
sequence: a) treatment of 62 with ammonia in methanoi to give the
corresponding amides, b) eonYersion of the amides to the corresponding
25 diamines by using lithium aluminum hydride in THF at reflux tamperature, and
c) condensation of the diamines with phthalic anhydride in toluene under
azeotropic conditions.
2) Transformation of 73 into the compounds 74 can be carried out as
described in the conversion of 66 into 70. The compounds of formula 74
~0 wherein X is ~r may be especially useful, since the Br substituent of X can be
r~adily displaced to various substitu0nts such as Cl, I, CN, alkyl, and Cf3 under
conditions such as CuCI-dimethyl sulfoxide-150C, Cul-KI-hexamethyl -
phosphorous triamida-150C, CuCN-dimethyl sulfoxide-150C, alkyl-copper
_ .... ... . .. .
- P. 6/7
2 ~
-31 -
raagent-THF-0C, and CF3CO2Na-Cul-N-methylpyrrolidone-1 60C,
respectively. Compounds of formula 74 wharein X is hy~rogen may be
obtainod by catalytic hydrogenation of compounds of ~orrnula 74 wherein X is
Br by using Pd/C in mathanol. Compounds of formula 74 wharaln X Is nitro
5 may be obtained by standard nitration of compounds of formula 74 wherein X
is hydrogen~
3) Acid hydrolysis of 74 may afford the compounds of formula 5~a. The
acid hydrolysis conditions include treatmant with aqueous strong acid such as
6 N - 12 N hydrochlorlc acid, or 2~ ~ 48% hydrobromic acid in protic or aprotic
solvent such as acetic acid or dioxane at temperature range of 50 to 1 00C.
Compounds 55b may be prepared by alkylation of compounds 5~a with
~2al in the presenca of a base such as potassium carbonate or sodium hydrida.
Reductive amination using an appropriate aldehydes or ketones in the
presence of sodium borohydride or sodium cyanoborohydride in an alcoholic
1~ solv~nt such as methanol at ambient temperature may also bs utilized for the
alkylatlon.
Similarly, Optically active 5~ whor3in R1 is hydrogen may be prepared
starting from optically acffve 62 wherein R1 is hydrogen of which synthesis is
described in J~ Chem. Soc. Perkin 1 1g77, 596.
20 pr~cess H-
~
The key intermediates 57 may ba prepared from compounds 6a;
E E
~Y~J ~ J
NH2 CO2H
6a 57
wherein Z, E, Y and J are as defined abova.
Reaction of 6a with alkyl nitrite such as isopropyl nitrite in the presence- of copper cyanide in an inert solYent such as acetonitrile followed ~y acid or
30 alkaline hydrolysis of the resulting cyano group and selecti\/e ra-protection of
the carboxylic acid group of tha side chain into ~i and tha arnino or guanidinogroup (if necessary~ into E may provida 57~ The hydrolytic conditions includ3
treatrnent with alkaline metal hydroxide or carbonate such as lithium hydroxide, ~ -
... , ,.. ... .,.. ,.,;,.,.".,.,,,..,., ,., .,,....; ....,.., ,.. " , , , , . ,,, ,~
:. .
~-~2~6a~ ~
- 32 -
sodium hydroxide, potassium carbonate in a mixed solvent of water and protic
or aprotic solvent such as methanol, ethanol, or THF at temperature range of 0
to 50C, or treatment with aqueous strong acid such as 1 N ~ 12 N hydrochloric
acid, or 5 ~ 48% hydrobromic acid in protic or aprotic solvent such as acetic
5 acid or dioxane at temperature range of ambient temperature to 1 00C.
Process J
Compounds 3 of the invention may be hydrolyzed to compounds of
formula 1 wherein E is E and J is CO2H by treatment of aqueous alkaline
hydroxide such as lithium hydroxide and sodium hydroxide in a mixed solvent
10 of methanol and THF at ambient temperature. Compounds of formula 1
wherein E is E and J is CO2H may be condensed with NH3, NH2R3J,
HNR3J~4J, HN(OH)R3J, HN(oR5J)R3J, H2NOH and HOR3J to give compounds
of formula 1 wherein J is CONH2, CoNHR3J, CONR3JR4J, CoN(oH)R3J~
CoN(oR5J)R3J, CON(OH)H and CO2R3J, respectively, wherein R3J, R4J and
15 R5J are as defined above and E is E. The condensation may be carried out in
the presence of condensation reagent such as 1-ethyl-3-(3'-dimethylamino -
propyl)carboximide-hydroxybenztriazole, isobutyric anhydride-triethylamine,
and N,N-bis(2-oxo-3-oxazolidinyl)-phosphinic chloride-triethylamine in an inert
solvent such as DMF, THF, or dichloromethane at temperature range of 0C to
20 ambient temperature.
Similar condensation of compounds of formula 1 wherein E is E and J
is CO2H with 3-aminopropionitorile followed by treatment of triphenyl -
phosphine, diethyl azodicarboxylate, and trimethylsilyl cyanide in THF at
ambient temperature and alkaline hydrolysis may provide compounds of
25 formula 1 wherein J is tetrazolyl and E is E.
Compounds of formula 1 wherein E is E may be selectively deprotected
to compounds of formula 1 wherein E is -NH2 or -NHC(=NH)NH2 by mild acid
hydrolysis. The hydrolytic conditions includes treatment with 0.1 ~ 4 N
hydro~en chloride in an inert solvent such as 1,4-dioxane and ethyl acetate at
30 ambient temperature.
Compounds of formula 1 wherein E is NHR3E or -NHC(=NH)NHR3E may
be prepared by alkylation of compounds of formula 1 wherein E is -NH2 or
-NHC(=NH)NH2 with R3EI wherein R3E is as defined above in the presence of
:- : . .. : ~ ` ` - '
' ?~
a base such as potassium carbonata or sodium hydride. Compounds of
formula ti wh~rein E is NR3ER4E or NHC~=NH)N~3ER4E may also be
pr~pared by further alkylation of compounds of formula 1 wherein E is NHR3E
or -NHC(=NH~NHR3~ with R4~1 as described above, wherein R3E and R4E are
5 as defined above. Compounds of formula 1 wharain E is NR3ER4E or
-NHC(=NH)NR3ER4E wherein R3E and R4E are join~d to forrn a cyclic amine
may aiso be prapared by alkylation of compounds of formula 1 wherein E is
NH2 or -NHC(=NH)NH2 with l t;~2 1 as descri~ed above, wherein Q2 represents
aikylene containing 2 to 6 carbon atoms which may be a straight chained or
10 alk~lene containing 4 to 6 carbon atoms and an oxygen or nitrogen atom which
may be straight-chained. The oxygen or nitrogen atom of Q2 is always bonded
to tne adjacent all~lsns group. Reductive aminaUon using an appropriate
aldehydes or ketanes in the presencs of sodium borohydride or sodium
cyanoborohydrids In an alcoholic solvent such as methanol at ambient
15 temperature may also be utilized for introducing ~3E and R4E groups.
Compounds of formula 1 wherein E is NH2, -NHR3E, -NH-C(=NH)-NH2, -NH -
C(=NH)-NHR3E, and -NH-C(=NH)-NR3~R4E may be converted into compounds
- of formula 1 wherein E is -NHL, -NLR3E, -NH-C(=NL)-NH2, -NH-C(=NL) -
NHR3E, and -NH~C(=NL)-NR3ER4E wherein L is R17CO or R170CO wherein
20 R17 is alkyl, and R3E and R4E are as defined above. The conversion may be
carried out by treatment with R17COCI, (R17CO)20, R170COCI, or ~R170CO)20
in tha presence of an organic base such as triethylamine or pyridine in an inertsolvent such as dichloromethane at ambient temperatur0.
According to the methods as described above, the compounds of the
2~ invention may be prepared as racemic form. However, the compounds of tha
invention may be obtained as enantlomeric pure form by resolving an
appropriate racemic intermediate during the synthesis, or the compounds of the
invention themselves. The resolutlon includes salt-formation of the compound
having a basic moiety with optically pure acid such as (+)-tartaric acid, and also
30 salt-format~on of the compound having an acidic moiety with optically pure
amine such as quinine or quinidine, followed by fractional recrystalll2ation andregeneration of the parent compound. The resolution technique also includes
amide or ester formation of the compound having carboxylats, amine, or
,
. . - :. -
,. . ;.. .
- . - -
2~216~9
- 34 -
alcohol with chiral-auxiliary, followed by chromatographic separation and
removal of the auxiliary. Alternative resolution technique includes enzymatic
hydrolysis of the ester or amide of the intermediate during the synthesis or thecompound in the invention.
A certain compound in the invention may be obtained by using
conventional protection-deprotection techniques, if necessary or desirable,
during synthesis as described above. Such techniques are described in T. W.
Greene, Protective Groups in Organic Synthesis, John Wiley ~ Sons, 1981.
The compounds of the present invention strongly inhibit both [3H] 5,7 -
dichlorokynurenic acid (DCKA) and [3H] glycine binding to the rat brain
synaptic membrane preparation, implying that these compounds possess the
potent affinities for strychnine-insensitive glycine modulatory site of NMDA (N -
methyl D-aspartate) receptors (see, for example, Y. Yoneda, et al., J.
Neurochem., ~0, 634 (1993)). The activities of the compounds were measured
by [3H] DCKA and [3H] glycine binding inhibition studies as illustrated below.
[3H] glycine binding studies
A crude rat brain synaptic membrane preparation was washed three
times by centrifugation at 50,000 x g for 30 min with 50 mM tris acetate (pH 7.4).
The pellets obtained were suspended in 0.23 M sucrose solution and stored at
-80C. For binding studies, the frozen suspension was thawed, treated with
0.08% triton X-100 at 2C for 10 min, and washed twice by the centrifugation as
mentioned above. The synaptic membrane thus prepared (ca. 150 - 200 ~19 of
protein) was incubated with 10 nM [3H] glycine (1.11 TBq/mmol) and the test
compound (10 ng/mL ~ 0.1 nglmL) at 2C for 10 min in 50 mM tris acetate (pH
7.4). The incubation was terminated by suction filtration using Whatman GF/B
glass filter. The radioactivities bound to the membrane on the filter was
measured by scintillation counting. Non-specific binding was calculated by the
radioactivities measured under the incubations in the presence of 0.1 mM D -
serine. The ~3H] glycine binding was not inhibited by addition of 0.1 mM
strychnine.
[3H] DCKA binding studies
A crude rat brain synaptic membrane preparation was washed three
times by centrifugation at 50,000 x g for 30 min with 50 mM tris acetate (pH 7.4).
.
2~2~ 9
- 35 -
The pellets obtained were suspended in 0.23 M sucrose solution and stored at
-80C. For binding studies, the frozen suspension was thawed, treated with
0.08% triton X-100 at 2C for 10 min, and washed twice by the centrifugation as
mentioned above. The synaptic membrane thus prepared (ca. 100 lly of
protein) was incubated with 10 nM [3H] (DCKA) (603 GBq/mmol) and the test
compour.d (10 ng/mL ~ 0.1 ng/mL) at 2C for 10 min in 50 mM tris acetate (pH
7.4). The incubation was terminated by suction filtration using Whatman GF/B
glass filter. The radioactivities bound to the membrane on the filter was
measured by scintillation counting. Non-specific binding was calculated by the
radioactivities measured under the incubations in the presence of 0.1 mM
glycine.
The compounds of the present invention attenuated strongly NMDA -
induced seizure under systemic administrations in the following in vivo model.
NMDA-induced seizure model
Thirty min later following intraperitoneal administration of the test
compound (0.3 ~ 30 mg/kg) into each of ten mice tested, NMDA (5 nmol) was
administered intracerebroventricularly (i.c.v.). Under the conditions without
pretreatment of the test compound, all of the mice exhibit tonic seizures. The
number of mice which did not exhibit tonic seizures after i.c.v. administration of
NMDA was counted as considered to be protected. The activity of the test
compound may be shown by the E~50 value. As favourable examples, the
compounds of Example 31, 35, and 11 inhibited the seizures with ED50 values
of 1.0, 2.1 and 3.1 mg/kg, respectively.
Reference Example 1
9-Bromo-5-methoxycarbonylmethyl-6,7-dihydro-1H, SH-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
1) 2-Hydroxymethyltetrahydroquinoline
To a suspension of LiAlH4 (8.3 g, 0.22 mol) in THF (200 mL) was added
dropwise 2-methoxycarbonyltetrahydroquinoline (42 g, 0.22 mol) in Tl IF (200
mL) at 0C. The mixture was stirred for 3 h at room temperature and refluxed
for 0.5 h. The excess reagent was decomposed by addition of aqueous sodium
hydroxide in THF. To the mixture was added 1 N aqueous NaOH, water, and
diethyl ether, successively. The organic layer was separated, washed with
- . .. , ~.. . . ... . ........ . . . .
. ~ , ~ . - . - .
2~ 21~0')
- 36 -
brine, dried over magnesium sulfate, and cocentrated to give 38.4 9 of 2 -
hydroxymethyltetrahydroquinoline (quant).
1H NMR (270 MHz, CDGI3) ~ 6.95 ~ 7.00 (m, 2 H), 6.63 (t, 1 H, J = 7.4 Hz), 6.54
(d,1 H,J=7.4Hz),3.74(dd,1 H,J=10.2,3.6Hz),3.56(dd,1 H,J=10.2,8.6
Hz), 3.41 ~ 3.49 (m, 1 H), 2.70 ~ 2.85 (m, 2 H), 1.85 ~ 1.90 (m, 1 H), 1.68 ~ 1.77
(m, 1 H).
2) 2-Cyanomethyltetrahydroquinoline
To a solution of 2-hydroxymethyltetrahydroquinoline (35.9 9, 0.22 mol),
imidazole (35~9~ g, 0.528 mol), and triphenylphosphine (69.24 g, 0.264 mol) in
1 0 a mixed solvent of 5: 1 toluene/acetonitrile (750 mL) was added iodine (61.42
g, 0.242 mol) at 0C. The mixture was stirred for 15 min at 0C and for 30 min
at room temperature. Aqueous sodium thiosulfate solution (200 mL) was
added. The organic layer was separated, washed with brine, dried over
magnesium sulfate, and concentrated. The residue was triturated with diethyl
1 5 ether and the insoluble materials were removed by filtration. The filtrate was
concentrated and the residual oil was dissolved in DMF (200 mL). To the
solution was added sodium cyanide (43.2 g, 0.881 mol) and the mixture was
heated at 80C for 10 h. The resulting mixture was poured into ice-water and
extracted with a mixture of toluene and ethyl acetate. The organic layer was
washed with brine, dried over magnesium sulfate, and concentrated. The
residue was purified by silica gel column chromatography with 1: 1
hexane/dichloromethane to 1 00% dichloromethane as eluents to give 31.6 9 of
the title compound (94%).
1H NMR (270 MHz, CDCI3) ~ 6.97 ~ 7.04 (m, 2 H), 6.68 (t, 1 H, J = 7.4 Hz), 6.54
(d, 1 H, J = 7.4 Hz), 4.03 (br,1 H), 3.70 (m, 1 H), 2.70 ~ 2.86 (m, 2 H), 2.54 (d, 1
H, J = 6.6 Hz), 2.02 ~ 2.13 (m, 1 H), 1.78 ~ 1.91 (m, 1 H).
3) 2-Methoxycarbonylmethyltetrahydroquinoline
2-Cyanomethyltetrahydroquinoline (28.0 g, 0.163 mol) was dissolved in
concentrated hydrochloric acid (200 mL) and the mixture was refluxed for 4 h.
The resulting mixture was concentrated and the residue was dissolved in
methanol (500 mL). Thionyl ch!oride (36 mL, 0.49 mol) was added slowly at
0C. The mixture was refluxed for 5 h and concentrated. To the residual solid
~12~ ~09
- 37 -
was added slowly saturated sodium bicarbonate (1 L) and the mixture was
extracted with ethyl acetate. The organic layer was washed with brine, dried
over magnesium sulfate, and concentrated to give 31.6 g of the title compound
(94%). 2-Methoxycarbonylmethyltetrahydroquinoline hydrochlorida.
lH NMR (270 MHz, ~::DCI3) ~ 7.69 ~ 7.72 (m, 1 H), 7.23 ~ 7.38 (m, 3 H), 3.96 ~
4.05 (m, 1 H), 3.75 (s, 3 H), 3.48 ~ 3.55 (m, 1 H), 2.96 ~ 3.12 (m, 3 H), 2.19 ~2.41 (m, 1 H).
4) 6-Bromo-2-methoxycarbonylmethyltetrahydroquinoline
To a solution of 2-methoxycarbonylmethyltetrahydroquinoline (31.5 9,
1 0 0.153 mol) in DMF (750 mL) was added dropwise a solution of N-bromo -succinimide (27.41 g, 0.154 mol) in DMF (550 mL) at 0C. The mixture was
stirred for 2 h at the same temperature, poured into water (2 L), and extracted
with a mixture of toluene and ethyl acetate. The organic layer was washed with
water, dried over magnesium sulfate, and concentrated to give 44.72 g of the
1 5 title compound (quant).
1H NMR (270 MHz, CDCI3) ~ 7.02 ~ 7.06 (m, 2 H), 6.38 ~dd, 1 H, J = 1.7, 7.3
Hz), 4.53 (br,1 H), 3.75 (s, 3 H), 3.72 ~ 3.75 (m, 4 H), 2.70 ~ 2.85 (m, 2 H), 2.49
~ 2.53 (m, 1 H), 1.89 ~ 1.99 (m, 1 H), 1.61 ~ 1.75 (m, 1 H).
5) 6-Bromo-2-methoxycarbonylmethyl-N-ethoxalyltetrahydroquinoline
To a solution of 6-bromo-2-methoxycarbonylmethyltetrahydroquinoline
(43.8 g, 0.153 mol) and triethylamine (37.5 9, 0.371 mol) in dichloromethane
(700 mL) was added slowly ethyl chlorooxalate (25.5 9, 0.187 mol) at 0C. The
mixture was stirred for 3 h at 0C, poured into 0.3 N hydrochloric acid (750 mL)and extracted with dichloromethane. The organic layer was washed with brine,
dried over magnesiwm sulfate, and concentrated. The residue was purified by ~ -
silica gel column chromatography with 4: 1 to 3: 1 hexane/ethyl acetate to give
56.9 g of the title compound (97%).
1 H NMR (270 MHz, CDCI3) ~ 7.36 (s, 1 H), 7.30 (d, 1 H, J = 8.3 Hz), 6.92 (d, 1
H,J=8.3Hz),4.94~5.01 (m,1 H),4.13-4.16(m,2H),3.~4(s,3H),2.43
2.75 (m, 6 H), 1.11 - 1.26 (m, 3 H).
6) 6-Bromo-2-methoxycarbonylmethyl-8-nitro-N-ethoxalyltetrahydroquinoline
A solution of 6-bromo-2-methoxycarbonylmethyl-N-ethoxalyltetrahydro-
- .. - ~ . ~ - ~ . . . - . - .
.. . . ~ . . - . . .-
--~ 2 ~
- 38 -
quinoline (56.0 9, 0.146 mol) in dichloromethane (500 mL) was added
dropwise to a suspension of nitronium tetrafluoroborate (25.0 9, 0.179 mol) in
dichloromethane (500 mL) at 0C. The mixture was stirred for 3 h at 0C,
poured into ice-water and extracted with dichloromethane. The organic layer
5 was washed with brine, dried over magnesium sulfate, and concentrated. The
residue was purified by silica gel column chromatography with 3: 1 to 2: 1
hexanelethyl acetate to give 52.0 g of the title compound (83%).
1 H NMR (270 MHz, CDC13) ~ 8.11 and 7.99 (d and d, 1 H, J = 2 Hz), 7.66 and
7.61 (d and d,1 H, J = 2 Hz), 5.03 ~ 5.16 and 4.74 ~ 4.85 (m and m, 1 H), 4.37
1 0 ~ 4.49 and 4.13 (m and q, 2 H, J = 7.2 Hz), 3.72 and 3.62 (s and s, 3 H), 2.44 ~
3.02 (m, 5 H), 1.65 ~ 1.80 and 1.50 ~ 1.60 (m and m, 1 H), 1.42 and 1.23 (t and
t,3H,J=7.2and7.2Hz).
7) 9-Bromo-5-methoxycarbonylmethyl-6,7-dihydro-1H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
1 5 To a mixture of 20% aqueous titanium trichloride (670 g, 0.867 mol),
water (500 mL), and acetone (500 mL) was added dropwise a solution of 6 -
bromo-2-methoxycarbonylmethyl-8-nitro-N-ethoxalyltetrahydroquinoline (52.0
g, 0.121 mol) in acetone (600 mL) at 0C. The mixture was stirred overnight at
room temperature, concentrated to ca. 1 L and diluted with water (1 L). The
20 precipitates formed were collected by filtration, washed with water, and dried in
vacuo to give 35.~ 9 of the title compound. The filtrate was extracted with ethyl
acetate. The organic layer was washed with brine, dried over magnesium
sulfate, and concentrated. The residue was purified by silica gel column
chromatography with ethyl acetate to give 6.0 9 of the titie compound (total
25 89%).
mp 185~ 187C
1H NMR (270 MHz, DMSO-d6) â 12.04 (bs, 1 H), 7.20 (d, 1 H, J = 2 Hz), 7.15 (d,
1 H,J=2Hz), 5.04~5.13(m, 1 H),3.62(s,3H),2.94(ddd, 1 H,J=17.1, 13.5,
4.5 Hz), 2.78 (dm,1 H, J = 17.1 Hz), 2.63 (dd, 1 H, J = 18, 7.2 Hz~, 2.57 (dd, 1 H,
J = 18, 3.6 Hz), 2.09 (dm, 1 H, J = 13.5 Hz), 1.80 ~ 1.95 (m, 1 H).
8) 9-Bromo-5-carboxymethyl-6,7-dihydro-1H, 5H-pyrido[1,2,3-de~quinoxaline-
2,3-dione
. . - ~
. . ~. .
.. ~ .... ~ , ................. . .
2~21~9
- 39 -
To a solution of 9-bromo-5-methoxycarbonylmethyl-6,7-dihydro-1H, 5H-
pyrido~1,2,3-de]quinoxaline-2,3-dione (25.0 9, 0.071 mol) in a mixture of THF
(350 mL) and methanol (350 mL) was added aqueous 1 N NaOH (440 mL).
The mixture was stirred for 2 h at room temperature, concentrated to ca. 500
S mL, and acidified by addition of aqueous 1 N HCI. The precipitates formed
were collected by filtration, washed with distilled water, and dried in vacuo togive 22.9 g of the title compound (95%).
mp > 27ûC
1H NMR (270 MHz, DMSO-d6) â 12.06 (bs,1 H), 7.20 (d,1 H, J = 2 Hz), 7.15 (d,
1 H, J = 2 Hz), 5.02 ~ 5.12 (m,1 H), 2.95 (ddd,1 H, J = 17.1,13.5, 4.5 Hz), 2.79(dm,1 H, J = 17~1 Hz), 2.43 ~ 2.61 (m, 2 H), 2.12 (dm,1 H, J = 13.5 Hz),1.78 ~
1.96 (m,1 H).
Reference Example 2
9-Chloro-S-carboxymethyl-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione
1) 9-Chloro-5-methoxycarbonylmethyl-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione
A mixture of 9-bromo-5-methoxycarbonylmethyl-6,7-dihydro-1 H, 5H -
pyrido[1,2,3-de]quinoxaline-2,3-dione (530 mg, 1.50 mmol) and cuprous
chloride (1.0 g,10.1 mmol) in dimethyl sulfoxide (5 mL) was heated at 160C -
for 4.5 h and poured into 1 N aqueous ammonium chloride (200 mL). The ~ ~
mixture was extracted with a mixed solvent of THF and ethyl acetate (600 mL).
The extract was washed with 1 N aqueous ammonium chloride (200 mL x 2)
and brine (200 mL), dried over magnesium sulfate, and concentrated. The
residue was recrystalized from ethanol to give 125 mg of the title compound
(27%).
mp 218 ~ 220C (dec)
1H NMR (270 MHz, DMSO-d6) ~ 12.08 (bs,1 H), 7.08 (d,1 H, J = 2 Hz), 7.02 (d,
1 H, J = 2 Hz), 5.04 ~ 5.13 (m,1 H), 3.62 (s, 3 H), 2.94 (ddd,1 H, J = 17.1,13.5,
4.5Hz),2.78(dm,1 H,J=17.1 Hz),2.63(dd,1 H,J=18,7.2Hz),2.57(dd,1 H,
J = 18, 3.6 Hz), 2.09 (dm,1 H, J= 13.5 Hz),1.80 - 1.95 (m,1 H).
2) 9-Chloro-~-carboxymethyl-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-
. ~ . .
r
- 40 -
2,3-dione
Hydrolysis of 9-chloro-5-methoxycarbonylmethyl-6,7-dihydro-1 H, 5H -
pyrido[1,2,3-de]quinoxaline-2,3-dione (246 mg, 0.8 mmol) was carried out as
described in Reference Example 1-8) to give 210 mg of the title compound
5 (89%)~
mp > 280C
1H NMR (270 MHz, DMSO-d6) ~ 12.06 (bs,1 H), 7.08 (d,1 H, J = 2 Hz), 7.02 (d,
1 H, J = 2 Hz), 5.02 ~ 5.13 (m,1 H), 2.95 (ddd,1 H, J = 17.1,13.5, 4.5 Hz), 2.78(dm,1 H,J=17.1 Hz),2.41 ~2.60(m,2H),2.14(dm,1 H,J=13.5Hz),1.88~
1.95 (m,1 H).
Example 1~
(S)-9-Chloro-5-[~tert-butoxycarbonylaminomethyl-o-(methoxycarbonyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
H CO2Me
1 5 ~,~N~,NHCO2t-Bu
CI~H~O
1) (S)-9-Chloro-5-carboxyrnethyl-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione
The ~itle compound was prepared starting from (S)-2-methoxycarbonyl -
tetrahydroquinoline ([Cl]D = ~ 41.4 ) according to the method described in
Reference Example 1 and 2. [~]D= -126.1 (c= 0.1, MeOH).
2) Methyl 5-azidomethyl-2-nitrobenzoate
A mixture of methyl 5-methyl-2-nitrobenzoate (5.5 9, 30 mmol), N -
bromosuccinimide (NBS, 5.87 g, 33 mmol), and azobisisobutyronitrile (AIBN,
200 mg) in carbon tetrachloride (60 mL) was refluxed for 3 h. The insoluble
material formed was removed by filtration and the filtrate was concentrated.
The residue was dissolved in DMF (10 mL) and sodium azide (2.92 g, 45
mmol) was added. The mixture was stirred for 2 h at 50C, poured into brine,
and extracted with a 1: 1 mixture of toluene and ethyl acetate. The organic
212 ~ ~9
- 41 -
layers were washed with brine, dried over magnesium sulfate, and
concentrated. The residue was purified by silica gel column chromatography
with 4: 1 hexane/ethyl acetate to give 3.97 g of the title compound (56%).
1H NMR (CDCI3) ~ 7.95 (d, 1 H, J = 8~1 Hz), 7.69 (d, 1 H, J = 2 Hz), 7.57 (dd, 1H,J=8.1,2Hz),4.52(s,2H),3.96(s,3H).
3) Methyl 5-tert-butoxycarbonylaminomethylanthranylate
A solution of methyl 5-azidomethyl-2-nitrobenzoate (8.19 g, 34.7 mmol)
in ethyl acetate (300 mL) in the presence of di-tert-butyl dicarbonate (8.3 g,
38.1 mmol) and 10% Pd/C (1 g) was hydrogenated for 10 h under atmospheric
1 0 pressure of hydrogen at room temperature. The catalyst was removed by
filtration through celite and the filtrate was concentrated. The residue was
puriFied by silica gel column chromatography with 4: 1 hexane/ethyl acetate to
give 6.5 g of the title compound (67%).
1H NMR (CDCI3) â 7.75 (d,1 H, J = 2 Hz), 7.21 (dd, 1 H, J = 8.1, 2 Hz), 6.64 (d,1 H,J=8.1 Hz),5.64~5.76(br,2H),4.66~4.72(br,1 H),4.17(bd,2H,J=6.3
Hz),3.88(s,3H),1.47(s,9H). ~-
4) (S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl)-
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione
A mixture of methyl 5-tert-butoxycarbonylaminomethylanthranylate (802
mg, 2.85 mmol), (S)-9-chloro-5-carboxymethyl-6,7-dihydro-1H, 5t~-pyrido[1,2,3 -
de]quinoxaline-2,3-dione (756 mg, 2.57 mmol), triethylamine (0.86 mL, 6.16
mmol), and N,N-bis(2-oxo-3-oxazolidinyl)-phosphinic chloride (Bop-CI, 726
mg, 2.85 mmol) in dichloromethane (8 mL) was stirred for 3.5 h at room
temperature. Bop-CI (726 mg) was added and the mixture was stirred further
for 2 h. The mixture was diluted with ethyl acetate (800 mL). The mixture was
washed with 5% potassium hydrogen sulfate, water, 1/15 phosphate buffer (pH
7.5), water, and brine, successively, dried over magnesium sulFate, and
concentrated. The residue was recrystallized from acetone-ethyl acetate to
give 445 mg of the title compound. The mother liquid was concentrated and
the residue was purified by silica gel column chromatography with ethyl acetate
to 1 00% THF to give additionally 63 mg of the title compound (36%).
2~ 21~
,. .
- 42 -
1H NMR (DMSO-d6) ~ 12.0 ~ 12.5 (br, 1 H), 10.46 (s, 1 H), 8.03 (d, 1 H, J = 8.6
Hz),7.75(d, 1 H,J=2Hz),7.45(bt, 1 H,J=5.9Hz),7.45(dd, 1 H,J=8.6,2
Hz),7.11 (d,1 H,J=2Hz),7.04(d,1 H,J=2Hz),5.18(m,1 H),4.11 (d,2H,J
=5.9Hz),3.84(s,3H),3.0~3.15(m, 1 H),2.83(dm, 1 H,J=17.1 Hz),2.60~
2.78 (m, 2 H), 2.17 (dm, 1 H, J = 14 Hz),1.80 ~ 2.00 (m, 1 H), 1.40 (s, 9 H).
Exampie 2
(S)-9-Chloro-5-(p-tert-butoxycarbonylaminomethyl-~carboxyphenylcarbamoyl -
methyl)-6,7-dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
A solution of (S)-9-chloro-5-~tert-butoxycarbonylaminomethyl-o -
1 0 (methoxycarbonyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3 -
de]quinoxaline-2,3-dione (445 mg) in a mixture of 1 N sodium hydroxide (5
mL), THF (5 mL), and methanol (5 mL) was stirred for 3.5 h at room temperature
and the solvent was concentrated to ca. 5 mL. To the residue was added 5%
potassium hydrogen sulfate and the precipitates formed were collected by
1 5 filtration, washed with water, and dried to give 453 mg of the title compound.
Example 3
(S)-9-Chloro-5-(p-aminomethyl-Gcarboxyphenylcarbamoylmethyl)-6,7-dihydro -
lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride
A suspension of (S)-9-chloro-5-(~tert-butoxycarbonylaminomethyl-o -
carboxyphenylcarbamoylmethyl)-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione (430 mg) in 2 N hydrogen chloride in 1,4-dioxane (14
mL) was stirred overnight at room temperature and diluted with diethyl ether.
The precipitates were collected by filtration and recrystallized from water to give
350 mg of the title compound.
1H NMR (DMSO-d6) ~ 12.0 ~ 13.0 (br, 1 H), 12.12 (bs, 1 H), 11.06 (bs, 1 H),
8.36 (d, 1 H, J = 8.6 Hz), 8.20 ~ 8.40 (br, 3 H), 8.10 (d, 1 H, J = 2.3 Hz), 7.69 (dd,
1 H,J=8.6,2.3Hz),7.10(d,1 H,J=2.3Hz),7.07(d,1 H,J=2.3Hz),5.20(m,
1 H), 4.03 (bd, 2 H, J = 6.6 Hz), 3.00 ~ 3.15 (m, 1 H), 2.78 ~ 2.90 (dm, 1 H,
J=14.0Hz),2.71 (bd,2H,J=7.3Hz),2.15~2.28(dm, 1 H,J=14.0Hz), 1.82~
2.00 (m, 1 H).
Example 4
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl)phenyl-
2:~216~9
- 43 -
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxallne-2,3-dione
1) (S)-g-Bromo-5-carboxymethyl-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
The title compound was prepared starting from (S)-2-methoxycarbonyl -
5 tetrahydroquinoline ([a]D = + 41.4 ) according to the method described in
Reference Example 1. [O~]D = - 108.3 (c = 0.1, MeOH).
2) (S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl)-
phenylcarbamoylmethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3~de]quinoxaline-2,3 -
dlone
1 0 A procedure similar to that described in Example 1-4) was performed
with (S)-9-bromo-5-carboxymethyl-6,7-dihydro-1H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione (760 mg, 2.24 mmol) and methyl 5-tert-butoxycarbonyl -
aminomethylanthranylate (700 mg, 2.49 mmol) to give 640 mg oF the title
compound (48%).
1H NMR (DMSO-d6) â 12.06 (bs,1 H),10.44 (s,1 H), 8.01 (d,1 H, J = 8.1 Hz),
7.76 (d,1 H, J - 2 Hz), 7.42 ~ 7.50 (m, 2 H), 7.24 (d, l H, J = 2 Hz), 7.17 (d,1 H,
J=2Hz),5.11~5.27(m,1 H),4.12(d,2H,J=6.3Hz),3.84(s,3H),3.05(ddd,
1 H, J = 17.1,13.5, 4.5 Hz), 2.83 (dm,1 H, J = 17.1 Hz), 2.60 ~ 2.76 (m, 2 H),
2.17 (dm,1 H, J = 13.5 Hz),1.78 ~ 1.97 (m,1 H),1.39 (s, 9 H).
20 Example 5
(S)-9-Bromo-5-(p-tert-butoxycarbonylaminomethyl-~carboxyphenylcarbamoyl -
methyl)-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
A solution of (S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-o -
(methoxycarbonyl)phenylcarbamoyimethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3 -
2~ de]quinoxaline-2,3-dione (680 mg, 1.06 mmol) in a mixture of 1 N sodium
hydroxide (6 mL), THF (6 mL), and methanol (6 mL) was stirred For 3.5 h at
room temperature and the solvent was concentrated to ca. 6 mL. To the
residue was added 5% potassium hydrogen sulfate and extracted with ethyl
acetate. The organic layer was washed with water, dried over magnesium
30 sulfate, and concentrated to give 608 mg of the title compound.
1H NMR (DMSO-d6) ~ 12.06 (bs,1 H),11.93 (s,1 H), 8.31 (d,1 H, J = 8.6 Hz),
-`` 2~2~6~9
- 44 -
7.85 (d,1 H, J = 2 Hz), 7.30 ~ 7.46 (m, 2 H), 7.10 ~ 7.30 (m, 4 H), 7.17 (d, 1 H, J
=2Hz),5.11 ~5.27(m,1 H),4.08(d,2H,J=6.3Hz),3.05(ddd,1 H,J=17.1,
13.5, 4.5 Hz), 2.80 (dm,1 H, J = 17.1 Hz), 2.60 ~ 2.76 (m, 2 H),2.17 (dm,1 H, J
= 13.5 Hz),1.78 ~ 1.97 (m,1 H),1.39 (s, 9 H).
5 Example 6
(S)-9-Brorno-5-(p-aminomethyl-~carboxyphenylcarbamoylmethyl)-6,7-dihydro -
1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride
A procedure similar to that described in Example 3 was performed with
(S)-9-bromo-5-(p-tert-butoxycarbonylaminomethyl-~carboxyphenylcarbamoyl -
methyl)-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (350 mg) to
give 275 mg of the title compound.
1H NMR (CD30D) ~ 8.58 (d,1 H, J = 8.1 Hz), 8.20 (d,1 H, J = 2 Hz), 7.64 (dd,1
H,J=8.1,2Hz),7.24(bs,1 H),7.22(bs,1 H),5.35~5.46(m,1 H),4.13(s,2
H), 3.15 (ddd,1 H, J = 17.1,13.5, 4.5 Hz), 2.92 (dm,1 H, J = 17.1 Hz), 2.84 (dd,1 H, J = 6.3,13.5 Hz), 2.75 (dd,1 H, J = 8.1,13.5 Hz), 2.34 (dm,1 H, J = 13.5
Hz),1.97 ~ 2.15 (m,1 H). [CC]D = - 37.6 (c = 0.1, MeOH).
Example 7
(+)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione
The ti~le compound was prepared from (+)-9-bromo-5-carboxymethyl-6,7 -
dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (300 mg, 1.17 mmol)
and methyl 5-tert-butoxycarbonylaminomethylanthranylate (361 mg, 1.28
mmol) according to a method described in Example 1-4.
Exampie 8
(+)-9-Bromo-5-[p-aminomethyl-~(methoxycarbonyl)phenylcarbamoylmethyl] -
6,7-dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
A procedure similar to that described in Example 3 was performed with
(+)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
(35 mg) to give 31 mg of the title compound (89%).
1H NMR (CD30D) ~ 8.50 (d,1 H, J = 8.1 Hz),8.16 (d,1 H, J = 2 Hz), 7.66 (dd,1
. ~ . .... . .. . . .
2~2~6~
- 45 -
H, J -- 2, 8.1 Hz), 7.25 (bs,1 H), 7.23 (bs,1 H),5.37 ~ 5.47 (m,1 H), 4.16 (s,2
H), 3.95 (s,3 H),3.16 (ddd,1 H, J = 17.1,13.5, 4.5 Hz), 2.91 (dm,1 H, J = 17.1
Hz), 2.86 (dd,1 H, J - 6.3,13.5 Hz), 2.75 (dd,1 H, J = 8.1,13.5 Hz), 2.39 (dm,1
H, J = 13.5 Hz),1.99 ~ 2.14 (m,1 H).
5 Example 9
(S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonylmethyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione
CO2Me
10 ~,N~ ,NHCO2t-Bu
CIJ~N~O
15 1) 4-Nitro-3-trifluoromethanesulfonyloxytoluene
To a solution of 3-methyl-6-nitrophenol (3.06 g, 20 mmol) and 2,4,6 -
colidine (4.0 mL, 30 mmol) in dichloromethane (100 mL) was added slowly
trifluoromethanesulfonic anhydride (7.05 g, 25 mmol) at room temperature.
The mixture was stirred overnight at the sarne temperature, poured into water,
20 and extracted with ethyl acetate. The organic layer was washed successively
with 0.2 N hydrochloric acid, water, saturated aqueous sodium bicarbonate,
and brine, dried over magnesium sulfate, and concentrated to give 5.0 9 of the
title compound (B8%).
1HNMR(CDCI3)â8.03(d,1 H,J=8.3Hz),7.36(d,1 H,J=8.3Hz),7.24(s,1
H),2.52(s,3H).
2) 1-Nitro-4-phthalimidomethyl-2-trifluoromethanesulfonyloxybenzene
A mixture of 4-nitro-3-trifluoromethanesulfonyloxytoluene (5.7 9, 20
mmol), N-bromosuccinimide ( 5.7 9, 32 mmol), and benzoyl peroxide (1 9) in
carbon tetrachloride (75 mL) was refluxed for 18 h. The insoluble material
30 formed was removed by filtration and the filtrate was concentrated. The residue
was dissolved in DMF (40 mL) and potassium phthalimide (2.6 9, 14 mmol)
was added. The mixture was stirred for ~ h at room temperature, poured into
2 ~ 9
- 46 -
brine, and extracted with a 1: 1 mixture of toluene and ethyl acetate. The
organic layers were washed with brine, dried over magnesium sulfate, and
concentrated. The residue was purified by silica gel column chromatography
with 9: 1 to 1: 1 hexane/ethyl acetate to give 3.4 g of the title compound (39%).
lHNMR(CDCI3)~8.14(d,1 H,J-8.3Hz),7.86~7.91 (m,2H),7.75~7.79
(m,2H),7.63(dd, 1 H,J=8.3, 1.6Hz),7.54(d, 1 H,J= 1.6Hz),4.93(s,2H).
3) Diethyl 2-nitro-5-phthalimidomethylphenylmalonate
To a suspension of 60% sodium hydride (5.8 g, 145 mmol) in DMF (150
mL) was added diethyl malonate (26.4 mL, 175 mmol) at room temperature,
1 0 while the sodium hydride was washed with dry hexane before use. The
mixture was heated at 40C for 1.5 h, allowed to cool at room temperature and
1-nitro-4-phthalimidomethyi-2-trifluoromethanesulfonyloxybenzene (25 g, 58
mmol) was added. The mixture was stirred overnight at room temperature,
poured into 3% potassium hydrogen sulfate, and extracted with a 1: 1
1 5 toluene/ethyl acetate. The extract was washed successively with 5%
potassium hydrogen sulfate, water, saturated aqueous sodium bicarbonate,
and brine, dried over magnesium sulfate, and concentrated. The unreacted
diethyl malonate was distilled out in vacuo and the residual solid was washed
with 1: 1 diethyl ether/hexane to give 24.5 g of the title compound.
1H NMR (CDCI3) ~ 8.04 (d, 1 H, J = 8.9 Hz), 7.85 ~ 7.90 (m, 2 H), 7.72 ~ 7.78
(m,2H),7.56(d,1 H,J=8.9Hz),7.54(s,1 H),5.24(s,1 H),4.90(s,2H),4.27
(q,4H,J=7.3Hz), 1.28(t,6H,J=7.3Hz).
4) Methyl 5-aminomethyl-~-nitrophenylacetate hydrochloride
A solution of diethyl 2-nitro-5-phthalimidomethylphenylmalonate in a
mixture of concentrated hydrochloric acid (150 mL) and 1,4-dioxane (150 mL)
was heated at 120C for 24 h. The solvents was removed in vacuo and the
residual solid was dissolved in methanol (1 00 mL). To the solution was added
thionyl chloride (11.8 g) dropwise at 0C. The mixture was stirred for 2 h at
40C and the solvent and the excess reagent was removed in vacuo. The
residue was washed with diethyl ether and dried to give 6.5 g of the title ~ `
compound (quant).
lH NMR (CD30D) â 8.18 (d,1 H, J = 8.6 Hz), 7.63 (dd, 1 H, J = 8.6, 1.6 Hz),
-~ ~ .. , . -
.- ~ ... . . .
. .
~1$`~9
- 47 -
7.56(d,1 H,J-1~6Hz),4.23(s,2H),4.09(s,2H),3.69(s,3H).
5) Methyl 5-tert-~utoxycarbonylaminomethyl-2-nitrophenylacetate
To a solution of methyl 5-aminomethyl-2-nitrophenylacetate
hydrochloride (6.80 g, 26.1 mmol) and triethylamine (12 mL) in
dichloromethane (100 mL) was added di-tert-butyl dicarbonate (9 mL, 39.2
mmol) at room temperature. The mixture was stirred for 1.5 h and diluted with
ethyl acetate. The mixture was washed successively with 5% potassium
hydrogen sulfate, water, saturated aquaous sodium bicarbonate, and brine,
dried over magnesium sulfate, and concentrated. The residue was purified by
1 0 silica gel column chromatography with 6: 1 to 2: 1 hexane/ethyl acetate to give
8.55 g of the title compound (quant).
1H NMR (CDCI3) ~ ~.11 (d, 1 H, J = 8.3 Hz), 7.38 (d, 1 H, J = 8.3 Hz), 7.25 (s, 1
H),4.98(br, 1 H),4.39(d,2H,J=6.3Hz),4.02(s,2H), 1.49(s,9H).
6) 4-tert-Butcxycarbonylaminomethyl-2-methoxycarbonylmethylaniline
1 5 A solution of methyl 5-tert-butoxycarbonylaminomethyl-2-nitrophenyl -
acetate (6.8 g, 21 mmol) in methanol (250 mL) in the presence of 10% Pd/C
was hydrogenated under atmospheric pressure of hydrogen at room
temperature. The catalyst was removed by filtration through celite and the
filtrate was concentrated to give 5.8 g of the title compound.
1H NMR (CDCI3) ~ 7.02 (d, 1 H, J = 7.6 Hz), 7.00 (s, 1 H), 6.67 (d, 1 H, J = 7.6Hz), 4.72 (br, 1 H), 4.18 (d, 2 H, J = 5.7 Hz), 4.05 (br, 2 H), 3.55 (s, 2 H), 1.46 (s,
9 H).
7) (S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonyl-
methyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
A mixture of methyl 4-tert-butoxycarbonylaminomethyl-2-methoxy -
carbonylmethylaniline (1.20 g, 4.27 mmol), (S)-9-chloro-5-carboxymethyl-6,7 -
dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (1.26 g, 4.27 mmol),
triethylamine (1.49 mL, 10.7 mmol), and Bop-CI (1.19 g, 4.69 mmol) in dichloro -methane (26 mL) was stirred for 4.5 h at 0C ~o room temperature and diluted
with ethyl acetate. The mixture was washed successively with 5% potassium
hydrogen sulfate, water, 1/15 phosphate buffer (pH 7.5), water, and brine, dried
~ 21~6~9
- 48 -
over magnesium sulfa~e, and concentrated. The residual solid was washed
with a 1: 1 mixture of diethyl ether and dichloromethane, and dried to give 1.82g of the title cornpound (74%).
1H NMR (DMSO-d6) ~ 12.06 (s,1 H), 9.48 (s,1 H), 7.39 (t,1 H, J = 7.2 Hz), 7.26
(d, lH, J = 9.0 Hz), 7.15 ~ 7.05 (m, 3H), 7.04 (bs,1H), 5.26 ~ 5.14 (m,1H), 4.08(d,2H,J=7.2Hz),3.66(s,2H),3.57(s,3H),3.06(ddd,1H,J=17.1,13.5,4.5
Hz), 2.83 (dm, lH, J = 17.1 Hz), 2.66 ~ 2.52 (m, 2H), 2.11 (dm,1H, J = 13.5 Hz),1.97 ~ 1.77 (m,1H),1.41 (s, 9H).
Example 10
(S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(carboxymethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
A solution of (S)-9-chloro-5-l~tert-butoxycarbonylaminomethyl-o -
(methoxycarbonylmethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H -
pyrido[1,2,3-de]quinoxaline-2,3-dione (1.82 g, 3.18 mmol) in a mixture of 1 N
aqueous sodium sulfate (20 mL), THF (20 mL), and methanol (20 mL) was
stirred for 4 h at room temperature. The mixture was acidified to pH 3 by
addition of 5% potassium hydrogen sulfate and extracted with a 1: 1 mixture of
ethyl acetate and THF. The organic layers were washed with brine, dried over
magnesium sulfate, and concentrated . The residual solid was washed with
dichloromethane, and dried in vacuo to give 1.499 g of the title compound.
Example 11
(S)-9-Chloro-5-[p-aminomethyl-~(carboxymethyl)phenylcarbamoylmethyl]-6,7-
dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride ~ -
A suspension of (S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-o -
(carboxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]
quinoxaline-2,3-dione (1.499 g) in 2 N hydrogen chloride in 1,4-dioxane (30
mL) was stirred overnight at room temperature and diluted with diethyl ether.
The precipitates were collected and recrystallized from water to give 1.247 g ofthe title compound (79%).
1H NMR (DMSO-d6) ~ 12.40 (bs,1 H),12.12 (s,1 H), 9.63 (s, lH), 8.25 (br, 3H),
7.46 (d, l H, J = 9Hz), 7.36 (d,1 H, J = 9.0 Hz), 7.34 (s,1 H), 7.13 (s,1 H), 7.06 (s,
1H), 5.28 ~ 5.14 (m,1H), 4.05 ~ 3.93 (m, 2H), 3.64 (d,1H, J = 16Hz), 3.62 (d,
5`09
. ~
- 49 -
1H, J = 16Hz), 3.08 (ddd, 1H, J = 17.1, 13.5, 4.5Hz), 2.82 (dm,1H, J = 17.1Hz),
2.72 ~ 2.55 (m, 2H), 2.11(dm, 1H, J = 17.1Hz), 1.96 ~1.76 (m, 1H).
Example 12
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(methoxycarbonylmethyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione
A procedure similar to that described in Example 9-7) was performed
with methyl 4-tert-butoxycarbonylaminomethyl-2-methoxycarbonylmethyl -
aniline (7.63 g, 22.51 mmol) and (S)-9-bromo-5-carboxymethyl-6,7-dihydro-1H,
1 0 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (6.03 9, 21.43 mmol) to give 8.02 g of
the title compound (61%).
1H NMR (DMSO-d6) ~ 12.06 (bs, 1 H), 9.48 (s, 1 H), 8.01 (d, 1 H, J = 8.1 Hz),
7.38 (t,1 H, J = 7.2 Hz), 7.20 ~ 7.30 (m, 2 H), 7.09 ~ 7.20 (m, 3 H), 5.13 ~ 5.23
(m, 1 H),4.08(d,2H,J=7.2Hz),3.67(s,2H),3.60(s,3H),2.97~3.14(m, 1
H),2.82(dm, 1 H,J=17.1 Hz),2.56~2.64(m,2H),2.04~2.14(m, 1 H), 1.79~
1.93 (m, 1 H).
Example 13
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(carboxymethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
A procedure similar to that described in Example 10 was performed with
(S)-9-bromo-5-~p-tert-butoxycarbonylaminomethyl-~(methoxycarbonylmethyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione (8.02 g, 13.03 mmol) to give 6.52 g of the title compound.
Example 14
(S)-9-Bromo-5-[p-aminomethyl-~(carboxymethyl)phenylcarbamoylmethyl]-6,7-
dihydro-1H, 5.'J-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride
A procedure similar to that described in Example 11 was performed with
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(carboxymethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
(6.5 g) to give ~.438 g of the title compound (80%).
1H NMR (CD30D) ~ 7.54 (d, 1 H, J = 9 Hz), 7.40 (bs, 1 H), 7.37 (bd,1 H, J = 9
Hz), 7.25 (bs, 1 H), 7.22 (bs,1 H), 5.38 ~ 5.50 (m, 1 H), 4.11 (s, 2 H), 3.71 (s, 2
~ ~ ~3
- 50 -
H), 3~16 (ddd,1 H, J = 17.1,13.5, 4.5 Hz), 2.91 (dm,1 H, J = 17.1 Hz), 2.79 (dd,1 H, J = 5.4,13.5 Hz), 2.71 (dd,1 H, J = 8.1,13.5 Hz), 2.34 (dm,1 H, J = 13.5
Hz),1.95 ~ 2.12 (m,1 H). ~c~]D = - 60.0 (C = 0.1, MeOH).
Example 15
5 (+)-9-Bromo-5-[p-(2,3-di-tert-butoxycarbonylguanidinomethyl)-~(methoxy-
carbonylmethyl)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-
de]quinoxaline-2,3-dione
CO2Me
H
1 0 ~ N ~, N ~ NCO2t-Bu
Br ~N ~O NHCO2t-Bu
1) Methyl 3-(2,3-di-tert-butoxycarbonylguanidinomethyl)-6-nitrophenylacetate
A solution of methyl 3-(aminomethyl)-6-nitrophenylacetate hydrochloride
(652 mg, 2.5 mmol), 1,3-di-tert-butoxycarbonyl-2-methylisothiourea (850 mg,
2.9 mmol), and triethylamine (708 mg, 7.9 mmol) in DMF (10 mL) was stirred for
7 h at 50 - 55C. The mixture was diluted with water and extracted with a 1: 1
mixture of toluene/ethyl acetate. The organic layer was washed with brine,
dried over magnesium sulfate, and concentrated. The residue was purified
with silica gel column chromatography with 6: 1 to 4: 1 hexane/ethyl acetate to
give 705 mg of the title compound (6~%).
lH NMR (CDCI3) ~ 11.54 (bs,1 H), 8.74 (bt,1 H, J = 5.6 Hz), 8.12 (d,1 H, J =
8.6Hz),7.41 (dd,1 H,J=8.6,1.7Hz),7.27(d,1 H,J=1.7Hz),4.72(d,1 H,J=
5.6 Hz), 4.03 (s,2 H), 3.72 (s, 3 H),1.50 (s,18 H).
2) 4-~2,3-Di-tert-butoxycarbonylguanidinomethyl)-2-methoxycarbonyl-
methylaniline
A procedure similar to that described in Example 9-6) was performed
with methyl 3-(2,3-di-tert-butoxycarbonylguanidinomethyl)-6-nitrophenylacetate
(400 mg, 0.92 mmol) to give 362 mg of the title compound (97%~.
3) (+)-9-Bromo-~-[p-(2,3-di-tert-butoxycarbonylguanidinomethyl)-~(methoxy-
carbonylmethyl)phenylcarbamoylmethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3-de]-
.,, ~ - - . -
~ : .. . .
., ~ - - ~, - : - : - :
~ -. - - . . - . . .
~ ~2~.9
- 51 -
quinoxaline-2,3-dione
A procedure similar to that described in Example 9-7) was performed
with 4-(2,3-di-tert-butoxycarbonylguanidinomethyl)-2-methoxycarbonyl-
methylaniline (220 mg, 0.54 mmol) and (+)-9-Bromo-5-carboxymethyl-6,7-
dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (190 mg, 0.56 mmol) to
give 185 mg of the title compound after silica gel column chromatography with
0.3% acetic acid/ethyl acetate (47%).
1H NMR (DMSO-d6) â 1 2.07 (br, 1 H), 11.53 (br, 1 H), 9.50 (br, 1 H), 8.66 (bt, 1
H,J=5.9Hz),7.32(d,1 H,J=8.6Hz),7.13~7.28(m,4H),5.13~5.28(m,1
H), 4.48 (bd, 2 H, J = 8.6 Hz), 3.68 (bs, 2 H), 3.59 (s, 3 H), 3.00 ~ 3.18 (m, 1 H),
2.78 ~ ~.89 (dm, 1 H, J = 14.0 Hz), 2.59 (bd, 2 H, J = 7.3 Hz), 2.05 ~ 2.17 (dm, 1
H, J = 14.0 Hz),1.79 ~ 1.95 (m, 1 H), 1.48 (s, 9 H), 1.39 ~s, 9 H).
Example 16
(+)-9-Bromo-5-[p-(2,3-di-tert-butoxycarbonylguanidinomethyl)-~(carboxy -
1 5 methyl)phenylcarbamoylmethyl~-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
A procedure similar to that described in Example 10 was performed with
(+)-9-Bromo-5-[p-(2,3-di-tert-butoxycarbonylguanidinomethyl)-~(methoxy -
carbonylmethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3 -
de]quinoxaline-2,3-dione (1 60 mg, 0.22 mmol) to give 129 mg of the title
compound (82%). .:
Example 1 7
(+)-9-Bromo-5-[p-guanidinomethyl-~(carboxymethyl)phenylcarbamoylmethyl] -
6,7-dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
A procedure similar to that described in Example 11 was performed with
(+)-9-bromo-5-[p-(2,3-di-tert-butoxycarbonylguanidinomethyl)-~(methoxy -
carbonylmethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3 -
de]quinoxaline-2,3-dione (125 mg, 0.176 mmol) to give 88 mg of the title -
compound (90%).
1H NMR (DMSO-d6) ~ 12.09 (bs, 1 H), 9.55 (bs, 1 H), 7.93 (bt, 1 H, J = 5.9 Hz),
7.41 (d, 1 H, J = 8.9 Hz), 7.05 ~ 7.39 (m, 8 H), 5.18 ~ 5.27 (m, 1 H), 4.33 (bd, 2
H, J = 5.9 Hz), 3.61 (d, 2 H, J = 2.6 Hz), 3.00 ~ 3.18 (m, 1 H), 2.77 ~ 2.90 (dm, 1
, . - ., - .
. ,- . . .
2 ~21~
- 52 -
H, J = 14 Hz), 1.78 ~ 1.95 (m, 1 H).
Example 18
(S)-9-Chloro-5-~p-tert-butoxycarbonylaminomethyl-~(2-methoxycarbonylethyl) -
phenylcarbamoylrnethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
5 dione
1) 3-Methyl-2-nitrobenzylalcohol
To a refluxed solution of methyl 5-methyl-2-nitrobenzoate (50 g, 0.256
mol) and sodium borohydride (29 9, 0.768 mol) in THF (400 mL) was added
dropwise methanol (60 mL) over 2.5 h. After the addition was completed, the
1 0 mixture was refluxed for 1 h and allowed to cool at room temperature. The
excess reagent was decomposed by addition of diluted hydrochloric acid (300
mL) and the mixture was extracted with ethyl acetate. The organic layer was
washed with brine, dried over magnesium sulfate, and concentrated to give
43.0 9 of the title compound (100%).
1 5 1H NMR (CDCI3) ~ 8.03 (d, 1 H, J = 8.6 Hz), 7.51 (s,1 H), 7.26 (d, 1 H, J = 8.6
Hz), 4.94 (bs, 2 H), 2.63 (br, 1 H), 2.47 ~s, 3 H).
2) Diethyl 3-methyl-6-nitrobenzylmalonate
A mixture of 3-methyl-2-nitrobenzylalcohol (30.0 g, 0.18 mol) and thionyl
chloride (30 mL, 0.42 mol) in diethyl ether (200 mL) was refluxed for 4 h and
concentrated. The residual reagent and hydrogen chloride was removed by
azeotropic evaporation with toluene to give 37.0 9 of the crude benzylic
chloride. To a solution of sodium diethyl malonate (0.36 mol) prepared from
diethyl malonate (0.46 mol) and 60% sodium hydride (14.5 g, 0.36 mol) in DMF
(300 mL) was added the crude benzylic chloride (37 g) in toluene (60 mL) at
room temperature. The mixture was stirred for 2 h at 5 ~ 60C, poured into 0.2
N hydrochloric acid (1.5 L), and extracted with a 1: 1 mixture of toluene and
ethyl acetate. The organic layèrs were washed with brine, dried over
magnesium sulfate, and concentrated. The residue was purified by silica gel
column chromatography with toluene to 50: 1 toluene/ethyl acetate to give 25.4
9 of the title compound (46%).
1H NMR (CDCI3) â 7.95 (d, 1 H, J = 8.3 Hz), 7.19 (d, 1 H, J = 8.3 Hz), 7.17 (bs, 1
H),4.17(q,2H,J=7.3Hz),4.16(q,2H,J=7.3Hz),3.86(t,1 H,J=7.6Hz),
., . ~, .~ . , ;, .. ... , .. . . ., - . -
. - , .
.
~:, ~ -
.- ~ - ~, . . -.
2-~ 2~6~
- 53 -
3.49(d,2H,J=7.6Hz),2.39(s,3H), 1.2~ (t, 6H,J=7.3Hz).
3) Diethyl 3-phthalimidomethyl-6-nitrobenzylmalonate
A mixture of diethyl 3-methyl-6-nitrobenzylmalonate (25.0 g, 80.8 mmol),
N-bromosuccinimide (18.5 g, 103.9 mmol), and azobisisobutyronitrile (AI~N,
1.1 g) in carbon tetrachloride (400 mL) was refluxed for 11 h, while 3 x 100 mg
of AIBN were added every 3 h during the reaction. The insoluble material
formed was removed by filtration and the filtrate was concentrated. The residue
was dissolved in DMF (100 mL) and potassium phthalimide (13.5 g, 72.9 mmol)
was added. The mixture was stirred for 3 h at 5 ~ 60C, poured into saturated
1 0 sodium bicarbonate, and extracted with a 1: 1 mixture oF toluene and ethyl
acetate. The organic layers were washed with brine, dried over magnesium
sulfate, and concentrated. The residue was purified by silica gel column
chrornatography with 4: 1 hexane/ethyl acetate to give 18.66 g of the title
compound (51%).
1 5 1 H NMR (CDCI3) ~ 7.96 (d, 1 H, J = 8.3 Hz), 7.83 ~ 7.90 (m, 2 H), 7.71 ~ 7.78
(m,2H),7.43(dd, 1 H,J-8.3,~.0Hz),7.40(d, 1 H,J=2.0Hz),4.86(s,2H),
.14(q,2H,J=7.3Hz),4.13(q,2H,J=7.3Hz),3.82(t, 1 H,J=7.6Hz),3.48
(d,2H,J=7.6Hz),1.20(t,6H,J=7.3Hz).
4) Methyl 3-(3-tert-butoxycarbonylaminomethyl-6-nitrophenyl)propionate
A solution of diethyl 3-phthalimidomethyl-6-nitrobenzylmalonate (1 8.0 g,
39.6 mmol) in a mixture of conc. hydrochloric acid (100 mL) and dioxane (100
mL) was refluxed for 30 h and concentrated. The residual water and
hydrochloric acid were azeotropically distilled out with toluene. The residue
was dissolved in methanol (250 mL) and thionyl chloride (50 mL) was added.
The mixture was refluxed for 3 h and concentrated. The residual reagent and
hydrogen chloride were azeotropically distilled out with toluene. The residue
was dissolved in dichloromethane (10 mL) and di-tert-butyl dicarbonate (0.35
mL) and triethylamine (0.5 mL) were added. The mixture was stirred for 1 h at
room temperature, diluted with ethyl acetate, and washed with 5% potassium
hydrogen sulfate, water, saturated sodium bicarbonate, water, and brine,
successively. The organic layer was dried over magnesium sulfate and
concentrated. The residue was purified by siiica gel column chromatography
,.. , . . :, . .. ., .. . - .:
:.:. . . ~ . - , .
212~6~
- 54 -
with 7: 1 to 4: 1 hexane/ethyl acetate to give 8.03 9 of the title compound
(60%).
1H NMR (CDCI3) ~ 7.94 (d, 1 H, J = 8.9 Hz), 7.29 (d, 1 H, J = 2.0 Hz), 7.28 (dd, 1
H, J = 8.9, 2.0 Hz), 4.90 ~ 5.10 (br, 1 H), 4.36 (bd, 2 H, J = 5.9 Hz), 3.68 (s, 3 H),
3.22(t,2H,J=7.6Hz),2.71 (t,2H,J=7.6Hz),1.47(s,9H).
5) 2-(2-Methoxycarbonylethyl)-4-tert-butoxycarbonylaminomethylaniline
A solution of methyl 3-(3-tert-butoxycarbonylaminome~hyl-6-nitrophenyl) -
propionate (3.2 g, 9.5 mmol) in ethyl acetate (35 mL) in the presence of 10%
Pd/C was hydrogenated under atmospheric pressure of hydrogen at room
1 0 temperature. The catalyst was removed by filtration by using celite and the -
filtrate was concentrated to give 3.4 g of the title compound.
1HNMR(CDCI3)~6.95(dd, 1 H,J=8.3,2.0Hz),6.94(d, 1 H,J=2.0Hz),6.40
(d, 1 H, J = 8.3 Hz), 4.70 ~ 4.80 (br, 1 H), 4.17 (bd, 2 H, J = 5.3 Hz), 3.68 (s, 3 H),
2.81 (t,2H,J=7.3Hz),2.62(t,2H,J=7.3Hz), 1.46(s,9H).
6) (S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(2-methoxycarbonyl-
ethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline -
2,3-dione
A mixture of 2-(2-methoxycarbonylethyl)-4-tert-butoxycarbonylamino-
methylaniline (3.4 g), (S)-9-chloro-5-carboxymethyl-6,7-dihydro-1 H, 5H-pyrido -[1,2,3-de]quinoxaline-2,3-dione (2.80 9, 9.5 mmol), triethylamine (2.43 g, 24
mmol), and Bop-CI (2.42 9, 9.5 mmol) in dichloromethane (40 mL) was stirred
for 2 h at room temperature and diluted with ethyl acetate (800 mL). The
mixture was washed with 5% potassium hydrogen sulfate, water, 1/15
phosphate buffer (pH 7.5), water, and brine, successively, dried ovar
25 magnesium sulfate, and concentrated. The residue was recrystallized from
acetone-ethyl acetate to give 3.22 9 of the title compound. The mother liquid
was concentrated and the residue was purified ~y silica gel column
chromatography with ethyl acetate to 1 % AcOH/ethyl acetate to give
additionally 0.8 9 of the title compound (72%).
1H NMR (DMSO-d6) ~ 12.08 (bs, 1 H), 9.45 (bs, 1 H), 7.35 (bt, 1 H, J = 6.3 Hz),
7.24(d, 1 H,J=7.9Hz),7.00-7.18(m,4H),5.18~5.30(m, 1 H),4.07(d,2H,
J=6.3Hz),3.61 (s,3H),3.03-3.20(m,1 H),2.74~2.90(m,3H),2.63(d,2H,
. ~ ~
~ . . .. -
~ . 7 ~) ~ 9
- 55-
J = 7.3 Hz), 2.55 (d, 2 H, J = 7.3 Hz), 2.05 ~ 2.20 (dm, 1 H, J = 14.0 Hz), 1.80 ~
2.00 (m, l H), 1.40 (s, 9 H).
Example 19
(S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(2-carboxyethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-l H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
A procedure similar to that described in Example 2 was performed with
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(2-methoxycarbonylethyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione (3.05 g) to give 2.86 g of the title compound (96%).
1H NMR (DMSO-d6) ~ 11.50 ~ 12.50 (br, 2 H), 9.52 (bs, 1 H), 7.34 (bt, 1 H, J =
5.9 Hz), 7.26 (d,1 H, J = 8.3 Hz), 7.03 ~ 7.09 (m, 4 H), 5.19 ~ 5.29 (m, 1 H), 4.07
(d,2H,J=6.3Hz),3.03~3.20(m,1 H),2.74~2.89(m,3H),2.63(d,2H,J=
7.3 Hz), 2.42 (d, 2 H, J = 7.3 Hz), 2.07 ~ 2.20 (dm, 1 H, J = 14.0 Hz), 1.80 ~ 1.98
(m, 1 H), 1.40 (s, 9 H).
1 5 Example 20
(S)-9-Chloro-5-[p-aminomethyl-~(2-carboxyethyl)phenylcarbamoylmethyl]-6,7 -
dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride
A procedure similar to that described in Example 11 was performed with
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(2-carboxyethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
(2.86 g) to give 2.50 g of the title compound (98%).
1H NMR (DMSO-d6) ~ 11.60 ~ 12.~0 (br, 1 H), 12.13 (bs, 1 H), 9.61 (bs, 1 H)l
8.10 ~ 8.45 (br, 3 H), 7.41 ~d, 1 H, J = 7.9 Hz), 7.37 (d, 1 H, J = 2.0 Hz), 7.30 (dd,
1 H,J=7.9,2.0Hz),7.10(d,1 H,J=2.0Hz),7.10(d,1 H,J=2.0Hz),5.20~
5.30 (m,1 H), 3.97 (d, 2 H, J = 5.6 Hz), 3.05 ~ 3.20 (m, 1 H), 2.74 ~ 2.91 (m, 3H), 2.61 ~ 2.73 (m, 2 H~, 2.10 ~ 2.20 (dm, 1 H, J = 14.0 Hz), 1.82 ~ 1.98 (m, 1 H).
Example 21
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3-methoxycarbonyt -
propyl)phenylcarbamoylmethyl3-6,7-dihydro-1H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
1) 3-Methyl-6-nitrobenzaldehyde
A mixture of 3-methyl-6-nitroben~ylalcohol (10 9, 59.8 mmol) and
- :.. - . -, ~ ~ . -. . :
212i~
- 56 -
manganese dioxide (80 g) in dichloromethane (100 mL) was stirred for 9 h and
passed through a celite short column. The eluent was concentrated and the
residue was purified by silica gel column chromatography with 8: 1
hexane/ethyl acetate to give ~.25 g of the title compound (84%).
1HNMR(CDCI3)~10.44(s, l H),8.05(d, 1 H,J-8.3Hz),7.72(d, 1 H,J=1.7
Hz),7.53(dd,1 H,J=8.3,1.7Hz),2.53(s,3H).
2) 3-Methyl-6-nitrostyrene
To a suspension of methyltriphenylphosphnium bromide (18.86 g, 52.8
mmol) in THF (120 mL) was added 0.5 M potassium hexamethyldisilazide in -
1 0 toluene (106 mL, 53.0 mmol) at - 10C. The mixture was stirrad for 40 min at
the same temperature and 3-methyl-6-nitrobenzaldehyde (8.0 g, 48 mmol) in
THF (60 mL) was added. The mixture was stirred for 1 h at - 10C and 0.2 N
hydrochloric acid (400 mL) was added. The mixture was extracted with ethyl
acetate, washed with brine, dried over magnesium sulfate, and concentrated.
The residue was purified by silica gel column chromatography with 15: 1
hexane/ethyl acetate to give 5.10 g of the title compound (65%).
1H NMR (CDCI3) ~ 7.88 (d, 1 H, J = 8.3 Hz), 7.39 (d, 1 H, J = 1.7 Hz), 7.21 (dd, 1
H, J = 17.2, 11.2 Hz), 7.19 (dd, 1 H, J = 8.3, 1.7 Hz), 5.71 (dd, 1 ~1, J = 17.2, 1.0
Hz), 5.46 (dd, 1 H, J = 11.2 Hz), 2.46 (s, 3 H).
3) Diethyl 2-(3-methyl-6-nitrophenyl)ethylmalonate
To a suspension of 60% sodium hydride (1.36 g, 34 mmol) in DMF (35
mL) was added diethyl malonate (7.00 g, 43.7 mmol). The mixture was heated
at 60C for 1.5 h, allowed to cool at room temperature and 3-methyl-6-nitro -
styrene (4.80 9, 29.4 mmol) in DMF (10 mL) was added dropwise. The mixture
was again heated at 4 ~ 50C for 4 h, poured into 0.1 N hydrochloric acid, and
extracted with a 1: 1 toluene/ethyl acetate. The extract was washed with brine,
dried over magnesium sulfate, and concentrated. The residue was purified by
silica gel column chromatography with 10: 1 hexane/ethyl acetate to give 2.70
g of the title compound (28%).
1HNMR(CDCI3)~7.89(d, 1 H,J=8.9Hz),7.16(s, 1 H),7.15(d, 1 H,J=8.9
Hz),4.22(q,4H,J=7.3Hz),3.42(t,1 H,J=7.3Hz),2.94(t,2H,J=7.9Hz),
2.41 (s,3H),2.25(q,2H,J=7.9Hz),1.29(t,6H,J=7.3Hz).
. ~- - ~ -- -; ; - -- - - :. -
.
2~1609
- 57 -
4) Diethyl 2-(3-phthalimidomethyl-6-nitrophenyl)ethylmalonate
A mixture of diethyl 2-(3-methyl-6-nitrophenyl)ethylmalonate (2.7 9, 8.35
mmol), N-bromosuccinimide t1.63 9, 9.19 mmol), and azobisisobutyronitrile
(100 mg) in carbon tetrachloricle (35 mL) was refluxed for 14 h, while 3 x 50 mg5 of AIBN were added every 3 h during the reaction. The insoluble material
formed was removed by filtration and the filtrate was concen~rated. The residue
was dissolved in DMF (100 mL) and potassium phthalimide (1.3 g, 7.02 mmol)
was added. The mixture was stirred for 5 h at 4 ~ 50C, poured into saturated
sodium bicarbonate, and extracted with a 1: 1 mixture of toluene and ethyl
1 0 acetate. The organic layers were washed with brine, dried over magnesium
sulfate, and concentrated. The residue was purified by silica gel column
chromatography with 4: 1 hexane/ethyl acetate to give 580 mg of the title
compound (15%).
5) Methyl 4-(3-tert-butoxycarbonylaminomethyl-6-nitrophenyl)butanoate
A solution of diethyl 2-(3-phthalimidomethyl-6-nitrophenyl)ethylmalonate
(580 mg, 1.24 mmol) in a mixture of dioxane (10 mL) and concentrated
hydrochloric acid (10 mL) was refluxed for 30 h and concentrated. The residual
water and hydrochloric acid were azeotropically distilled out with toluene. The
residue was dissolved in methanol (10 mL) and thionyl chloride (2 mL) was
added dropwise. The mixture was refluxed for 2 h and concentrated. The
residual reagent and hydrogen chloride were azeotropically distilled out with
toluene. The residue was dissolved in dichloromathane (150 mL) and di-tert -
butyl dicarbonate (11 mL, 48 mmol) and triethylamine (15 mL) were add0d.
The mixture was stirred for 5 h at room temperature, diluted with ethyl acetate,and washed with 5% potassium hydrogen sulfate, water, saturated sodium
bicarbonate, water, and brine, successively. The organic layer was dried over
magnesium sulfate and concentrated. The residue was purified by silica gel
column chromatography with 7: 1 to 5: 1 hexane/ethyl acetate to give 8.03 g of
the title compound (60%).
1H NMR (CDCI3) ~ 7.90 (d, 1 H, J = 8.9 Hz), 7.26 (dd, 1 H, J = 8.9,1.7 Hz), 7.25(d,1 H,J=1.7Hz),4.92~5.13(br,1 H),4.36(bd,2H,J=6.3Hz),3.68(s,3H).
2.92 (t, 2 H, J = 7.3 Hz), 2.41 (t, 2 H, J = 7.3 Hz),1.99 (5et, 2 H, J = 7.3 Hz), 1.47
~.
.. ~ ,, , .- - , -
~ . - - , -
2 ~ g
- 58 -
(s, 9 H).
6) 4-tert-Butoxycarbonylaminomethyl-2-(3-methoxycarbonylpropyl)aniline
A procedure similar to that described in Example 9-6) was performed
with methyl 4-(3-tert-butoxycarbonylaminomethyl-6-nitrophenyl)butanoate (140
mg, 0.4 mmol) to give the title compound.
1H NMR (CDCI3) ~ 6.94 (dd,1 H, J = 8.3, 2.0 Hz), 6.92 (d, l H, J = 2.0 Hz), 6.61
(d,1 H,J=8.3Hz),4.65~4.77(br,1 H),4.15~bd,2H,J=5.3Hz),3.77(s,3H),
2.50 (t,2 H, J = 7.3 Hz), 2.40 (t, 2 H, J = 7.3 Hz),1.90 (5et,2 H, J = 7.3 Hz),1.45
(s, 9 H)-
7) (S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3-methoxycarbonyl-
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione
A procedure similar to that described in Example 9-7) was performed
with 4-tert-butoxycarbonylaminomethyl-2-(3-methoxycarbonylpropyl)aniline
1 5 (130 mg, 0.4 mmol) and (S)-9-bromo-5-carboxymethyl-6,7-dihydro-1 H, 5H -
pyrido[1,2,3-de]quinoxaline-2,3-dione (136 mg, 0.4 mmol) to give 118 mg of the
title compound (62%).
1H NMR (DMSO-d~) ~ 12.07 (bs,1 H), 9.37 (bs,1 H), 7.36 (bt,1 H, J = 1.7 Hz),
7.24 - 7.31 (m,2 H), 7.17 (d,1 H, J = 2.0 Hz), 7.07 (d,1 H, J = 2.0 Hz), 7.02 ~
7.10 (m,1 H), 5.19 ~ 5.30 (m,1 H), 4.07 (d, 2 H, J =2.0 Hz), 3.58 (s, 3 H), 3.00 ~
3.20 (m,1 H), 2.89 ~ 2.91 (dm,1 H, J = 14.0 Hz), 2.65 ~ 2.70 (m, 2 H), 2.32 (t, 2
H, J = 7.3 Hz), 2.08 ~ 2.19 (dm, ~ H, J = 14.0 Hz),1.80 ~ 1.98 (m,1 H),1.73
(5et, 2 H, J = 7.3 Hz),1.40 (s, 9 H).
Example 22
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3-carboxypropyl)phenyl-
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3- de]quinoxaline-2,3-dione
A solution of (S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3 -
methoxycarbonylpropyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido -
[1,2,3-de]quinoxaline-2,3-dione ~110 mg, 0.17 mmol) in a mixture of 1N
aqueous sodium hydroxide (1.5 mL), THF (1.5 mL), and methano~ (1.5 mL) was
stirred for 3 h at room temperature. The mixture was concentrated to ca. 2 mL
and acidified with 5% aqueous potassium hydrogen sulfate. The precipitates
., - ~. ~. - . ~ .
, -. .
,. .
$ ~ ~3
- 59 -
formed were collected and dried in vacuo to give 103 mg of the title compound
(96%).
Example 23
(S)-9-Bromo-5-[p-aminomethyl-o-(3-carboxypropyl)phenylcarbamoylmethyl] -
6,7-dihydro-1H,S~l-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride
To a solution of (S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3 -
carboxypropyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione (103 mg, 0.164 mmol) in 1,4-dioxane (5 mL) was added
4 N hydrogen chloride in 1,4-dioxane (5 mL) at room temperature. The mixture
1 0 was stirred for 20 h at room temperature and concentrated. The residual solid
was dried in vacuo to give 90 mg of the title compound (97%).
1H NMR (DMSO-d6) ~ 12.12 (br, 1 H), 11.40 ~ 11.90 (br, 1 H), 9.51 (br, 1 H),
8.05~8.50(br,3H),7.43(d,1 H,J=7.6Hz),7.33(d,1 H,J=2.0Hz),5.18~
5.30 (m, 1 H), 3.98 (bs, 2 H), 3.03 ~ 3.21 (m, 1 H), 2.81 ~ 2.90 (dm, 1 H, J = 14.0
Hz),2.50~2.75(m,4H),2.27(t,211,J=7.3Hz),2.06~2.18(dm, 1 H,J=14.0
Hz), 1.80 ~ 1.95 (m, 1 H), 1.73 (Set, 2 H, J = 7.3 Hz).
Example 24
(5S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(1 -methoxycarbonyl-1 -
acetoxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
AcO C02Me
f ~ ~, NHCO2t-Bu
Br J~N ~0
1) 1-(3-Methyl-6-nitrophenyl)-1-trimethylsiloxyacetonitrile
To a solution of 3-methyl-6-nitrobenzaldehyde (4.3 g, 26.0 mmol) in
dichloromethane (40 mL) in the presence of zinc iodide (200 mg) was added
trimethylsilyl cyanide (5.2 9, 52.0 mmol) at room temperature. The mixture was
stirred for 2 h at room temperature, diluted with ethyl acetate, washed with
brine, dried over magnesium sulfate, and concentrated to give 7.38 9 of the title
. ~ .- : -. :
2~2:~09
- 60 -
compound.
~) Methyl 1-(3-methyl-6-nitrophenyl)-1-hydroxyacetate
A solution of 1-(3-methyl-6-nitrophenyl)-1-trimethylsiloxyacetnitrile (7.3
g) in concentrated hydrochloric acid (60 mL) was heated at 80C for 2.5 h and
5 a mixed solution of methanol and toluene was added. The solvents and
hydrogen chloride were removed azeotropically by evaporation. The residue
was dissolved in ethyl acetate, washed with brine, dried over magnesium
sulfate, and concentrated to give 5.80 g of the title compound (99%).
1H NMR (CDCI3) ~ 7.95 (d, 1 H, J = 8.3 Hz), 7.46 (d, 1 H, J = 1.7 Hz), 7.29 (dd, 1
H,J=8.3,1.7Hz),5.81 (s,1 H),3.76(s,3H),2.46(s,3H).
3) Methyl 1-(3-methyl-6-nitrophenyl)-1-acetoxyacetate
A mixture of methyl 1-(3-methyl-6-nitrophenyl)-1-hydroxyacetate (5.70 g,
~5.3 mmol), acetic anhydride (3.62 g, 35.5 mmol), triethylamine (3.60 g, 35.5
mmol), and 4-dimethylaminopyridine (500 mg) in dichloromethane (50 mL) was
1 5 stirred for 1 h at room temperature and concentrated. The residue was
dissolved in a mixture of ethyl acetate and 0.5 N hydrochloric acid. The organiclayer was separated, washed with brine, dried over magnesium sulfate, and
concentrated. The residue was purified by column chromatography with 4: 1 to
3: 1 hexane/ethyl acetate to give 5.7 9 of the title compound (84%).
1H NMR (CDCI3) ~ 7.99 (d, 1 H, J = 8.6 ~Iz), 7.40 (d, 1 H, J = 1.3 Hz), 7.33 (dd, 1
H, J = 8.6, 1.3 ~Iz), 6.86 (s, 1 H), 3.76 (s, 3 H), 2.47 (s, 3 H), 2.21 (s, 3 H).
4) Methyl 1-(3-azidomethyl-6-nitrophenyl)-1-acetoxyacetate
A mixture of methyl 1-(3-methyl-6-nitrophenyl)-1-acetoxyacetate (3.5 g,
13.8 mmol), N-bromosuccinimide (NBS, 2.70 9, 15.2 mmol), and
azobisisobutyronitrile (AIBN, 100 mg) in carbon tetrachloride (65 mL) was
refluxed for 13 h, while 3 x 50 mg of AIBN ware added every 3 h during the
reaction. NBS (1.35 9, 7.6 mmol) and AIBN (100 mg) were added further and
the mixture was refluxed additionally for 7h. The insoluble material formed was
removed by filtration and the filtrate was concentrated. The residue was
dissolved in DMF (50 mL) and sodium azide (1.0 g, 15.4 mmol) was added.
The mixture was stirred for 1 h at room temperature, poured into a mixture of
toluerie, ethyl acetate, and 5% aqueous potassium hydrogen sulfate. The
. .~ -. - . . . ~ ~ -
-
2:~2:~0'~
- 61 -
organic layer was separated, washed with brine, dried over magnesium sulfate,
and concentrated. The residue was purified by silica gel column
chromatography with 5: 1 to 4: 1 hexane/ethyl acetate to give 1.97 g of the title
compound (46%).
1H NMR (CDCI3) ~ 8~09 (d, 1 H, J = 8.3 Hz), 7.57 (d, 1 H, J = 2.0 Hz), 7.51 (dd, 1
H,J=8.3,2.0Hz),6.88(s,1 H),4.52(s,2H),3.77(s,3H),2.23(s,3H).
5) 4-tert-Butoxycarbonylaminomethyl-2-(1-methoxycarbonyl-1-acetoxy-
methyl)aniline
A solution of methyl 1-(3-azidomethyl-6-nitrophenyl)-1-acetoxyacetate
(1.9 ~, 6.16 mmol) in ethyl acetate (35 mL) in the presence of di-tert-butyl
dicarbonate (1.48 g, 6.78 mmol) and 1 0% Pd/C was hydrogenated for 4 h
under atmospheric pressure of hydrogen at room temperature. The catalyst
was removed by filtration by using celite and the filtrate was concentrated to
give 1.1 9 of the crude title compound. The product was used for the next step
1 5 without further purification.
1H NMR (CDCI3) ~ 7.15 (d, 1 H, J = 2.0 Hz), 7.11 (dd, 1 H, J = 8.3, 2.0 Hz), 6.67
(d, 1 H, J = 8.3 Hz), 6.02 (s, 1 H), 4.68 ~ 4.82 (br, 1 H), 4.20 (bd, 2 H, J = 5.6 Hz),
4.10~4.20(br,2H),3.74(s,3H),2.20(s,3H), 1.46(s,9H).
6) (5S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-o-(1-methoxycarbonyl-1 -
acetoxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de~ -
quinoxaline-2,3-dione
A mixture of 4-tert-butoxycarbonylaminomethyl-2-(1-methoxycarbonyl-1 -
acetoxymethyl)aniline (500 mg, 1 mmol), (S)-9-bromo-5-carboxymethyl-6,7-
dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (475 mg, 1.4 mmol),
triethylamine (0.55 mL), and Bop-CI (360 mg, 1.4 mmol) in dichloromethane
(15 mL) was stirred for 48 h at rcom temperature and diluted with ethyl acetate
(200 mL). The mixture was washed with 5% potassium hydrogen sulfate,
water, 1/15 phosphate buffer (pH 7.5), water, and brine, successively, dried
over magnesium sulfate, and concentrated. The residue was purified by silica
gel column chromatography with 0.3% acetic acid/ethyl acetate to give 340 mg
of the title compound (50%).
1H NMR (DMSO-d6) ~ 12.07 (bs, 1 H), 9.67 (bs, 1 H), 7.44 (bt, 1 H, J = 5.6 Hz),
.. .. . .
. ..
2121~9
,
- 62 -
7.36(d,1 H,J=8.9Hz),7.27(d,1 H,J=8.9Hz),7.26(s,1 H),7.25(d,1 H,J=
2.0Hz),7.18(d,1 H,J=~.OHz),6.20,6.17(s,1 H),5.15~5.28(m,1 H),4.11
(bd,2H,J=5,6Hz),3.65(s,3H),2.99~3.17(m,1 H),2.76~2.90(dm,1 H,J
= 14.0 Hz), 2.55 ~ 2.68 (m, 2 H), 2.04 ~ 2.20 (m, 1 H), 2.11, 2.12 (s, 3 H), 1.80 ~
1.99 (m, 1 H), 1.40 (s, 9 H).
Example 25_
(5S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(1 -carboxy-1 -hydroxy -
methyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyridol1,2,3-de] -
quinoxaline-2,3-dione
1 0 A solution of (5S)-9-bromo-5-[~tert-butoxycarbonylaminomethyl-o-(1 -
methoxycarbonyl-1-acetoxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H,
5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (310 mg, 0.46 mmol) in a mixture of
1 N aqueous sodium hydroxide (6 mL), THF (5 mL), and methanol (5 mL) was
stirred for 6 h at room temperature. The mixture was concentrated to ca. 6 mL
1 5 and acidified with 5% aqueous potassium hydrogen sulfate. The precipitatesformed were collected and dried to give 276 mg of the title compound (97%).
Exampie 26
(5S)-9-Bromo-5-[p-aminomethyi-~(1 -carboxy-1 -hydroxymethyl)phenyl -
carbamoylmethyl]-6,7-dihydro-l H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
hydrochloride
To a solution of (5S)-9-bromo-5-[~tert-butoxycarbonylaminomethyl-~(1
carboxy-1-hydroxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido
[1,2,3-de]quinoxaline-2,3-dione (260 mg, 0.42 mmol) in 1,4-dioxane (10 mL)
was added 4 N hydrogen chloride in 1,4-dioxane (10 mL) at room temperature.
The mixture was stirred for 14 h at room temperature and concentrated. The
residual solid was washed with diethyl ether and dried in vacuo to give 240 mg
of the title compound (100%).
1H NMR (DMSO-d6) â 12.13 (br, 1 H), 9.55, 9.58 (bs, 1 H), 8.25 ~ 8.45 (br, 3 H),7.66,7.71 (d,1 H,J=8.3Hz),7.52(d,1 H,J=1.7Hz),7.42(dd,1 H,J=8.3, -
1.7 Hz), 7.21 (bs, 2 H), 5.28, 5.30 (s, 1 H), 5.15 ~ 5.25 (m, 1 H), 3.99 (bd, 2 H, J
= 5.6 Hz), 3.01 ~ 3.21 (m, 1 H), 2.65 ~ 2.95 (m, 3 H), 2.10 ~ 2.20 (dm, 1 H, J =14.0 Hz),1.80~ 1.95 (m, 1 H).
. = . . , . . .. , . = .. ,, , , .... ~, ... , . , , .,, . - . .
2 ~ 2 ~ 9
- 63-
Example 27
(5S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(1 -methoxycarbonyl-1 -
acetoxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3 -
de]quinoxaline-2,3-dione
A procedure similar to that described in Example 9-7) was performed
with 4-tert-butoxycarbonylaminomethyl-2-(1-methoxycarbonyl-1-acetoxy-
methyl)aniline (500 mg, 1 mmol) and (S)-9-chloro-5-carboxymethyl-6,7-dihydro -
1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (413 mg, 1.4 mmol) to give 275
mg of the title compound (43.7%).
1H NMR (DMSO-d6) â 12.08 (bs, 1 H), 9.67 (bs, 1 H), 7.44 (bt, 1 H, J = 5.9 Hz),
7.36(d>1 H,J=8.3Hz),7.26(d,1 H,J=8.3Hz),7.22(s, 1 H),7.11 (d,1 H, J =
2.0 Hz), 7.03 (d, 1 H, J = 2.0 Hz), 6.20, 6.17 (s, 1 H), 5.15 ~ 5.28 (m, 1 H)l 4.10
(bd, 2 H, J = 5.9 Hz), 3.65 (s, 3 H), 2.98 ~ 3.18 (m, 1 H), 2.75 ~ 2.89 (dm, 1 H, J
= 14.0 Hz), 2.56 ~ 2.67 (m, 2 H), 2.05 ~ 2.21 (m, 1 H), 2.11, 2.12 (s, 3 H), 1.80 ~
1.99 (m, 1 H), 1.40 (s, 9 H).
Example 28
(5S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(1 -carboxy-1 -hydroxy -
methyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione
A procedure similar to that described in Example 25 was performed with
(5S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~1 -methoxycarbonyl-1 -
acetoxymethyl)phenylcarbamoylmethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3 -
de]quinoxaline-2,3-dione (260 mg, 0.41 mmol) to give 210 mg of the title
compound (89%).
Example 29
(5S)-9-Chloro-5-[p-aminomethyl~ carboxy-1-hydroxymethyl)phenyl-
carbamoylmethyl]-6,7-dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
hydrochloride
A procedure similar to that described in Example 26 was performed with
(5S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(1-carboxy-1-hydroxy-
methyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3~de] -
quinoxaline-2,3-dione (195 mg, 0.34 mmol) to give 180 mg of the title
, . ~ -. . -
g ~
- 64 -
compound (100%).
1H NMR (DMSO-d6) ~ 12.14 (br, 1 H), 9.54, 9.57 (bs, 1 H), 8.20 8.45 (br, 3 H),
7~66,7.71 (d,1 H,J=8.3Hz),7.52(d,1 H,J=1.7Hz),7.41 (dd,1 H,J=8.3,
1.7 Hz), 7.11 (bs, ~ H), 5.27, 5.30 (s, 1 H), 5.15 ~ ~.30 (m, 1 H), 3.99 (bd, 2 H, J
= 5.6 Hz), 3.03 ~ 3.20 (m, 1 H), 2.60 ~ 2.90 (m, 3 H), 2.10 ~ 2.20 (dm, 1 H, J =14.0 Hz), 1.80 ~ 1.95 (m, 1 H).
Example 30
(S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(tert-bu toxycarbonyl -
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H pyrido[1 ,2,3-de] -
1 0 quinoxaline-2,3-dione
H C02t-Bu
~,~, N ~, NHC02t-Bu
~N~o
Cl N o
1) 3-tert-Butoxycarbonylmethoxy-4-nitrotoluene
A mixture of 3-methyl-6-nitrophenol (30.62 9, 200 mmol) and tert-butyl
bromacetate (46.8 g, 240 mmol) in acetonitrile (700 mL) in the presence of
potassium carbonate (69.1 g, 500 mmol) was refluxed for 2 h. Inorganic
materials were removed by filtration and the filtrate was concentrated. The
residue was diluted with ethyl acetate, washed with brine, dried over
magnesium sulfate, and extensively concentrated in vacuo to give 54.2 9 of the
title compound (quant).
1H NMR (CDCI3) ~ 7.82 (d, 1 H, J = 8.3 Hz), 6.87 (d, 1 H, J = 8.3 Hz), 6.74 (s, 1
H), 4.65 (s, 2 H), 2.40 (s, 3 H), 1.47 (s, 9 H).
2) 4-Azidomethyl-2-~ert-butoxycarbonylmethoxynitrobenzene
A mixture of 3-tert-butoxycarbonylmethoxy-4-nitrotoluene (36.1 g, i20
mmol), N-bromosuccinimide (21.3 g, 120 mmol), and benzoyl peroxide (4 g) in
carbon tetrachloride (500 mL) was refluxed for 18 h. The insoluble material
formed was removed by filtration and the filtrate was concentrated. The residue
was dissolved in DMF (1 0 mL) and sodium azide (5.2 g, 80 mmol) was added.
. ... .
2 ~ 21S~
- 65 -
The mixture was stirred for 2 h at 50C, poured into brine, and extracted with a1: 1 mixture of toluene and ethyl acetate. The organic layers were washed
with brine, dried over magnesium sulfate, and concentrated. The residue was
purified by silica gel column chromatography with 9: 1 to 7: 3 hexane/ethyl
acetate to give 13.4 g of the title compound (36%).
1H NMR (CDCI3) â 7.89 (d, 1 H, J = 8.3 Hz), 7.01 (dd, 1 H, J = 8.3, 1.7 Hz), 6.93
(d,1 H,J=1.7Hz),4.70(s,2H),4.43(s,2H),1.48(s,9H).
3) 4-tert-Butoxycarbonylaminomethyl-2-tert-butoxycarbonylmethoxyaniline
A solution of 4-azidomethyl-2-tert-butoxycarbonylmethoxynitrobenzene
1 0 (2.96 g, 10 mmol) in ethyl acetate (50 mL) in the presence of di-tert-butyl
dicarbonate (2.40 9, 11 mmol) and 10% Pd/C (1 g) was hydrogenated for 12 h
under atmospheric pressure of hydrogen at room temperature. The catalyst
was removed by filtration through celite and the filtrate was concentrated. The
residue was purified by silica gel column chromatography with 3: 1 to 1: 1
1 5 hexane/ethyl acetate to give 1.75 9 of the title compound (50%).
1H NMR (CDCI3) ~ 6.73 (dd, 1 H, J = 8.3 Hz), 6.6~ (d, 1 H, J = 8.3, 1.7 Hz), 6.66
(d,1 H,J=8.3Hz),4.65~4.75(br,1 H),~.52(s,2H),4.16(d,2H,J=5.6Hz),
3.90 ~ 4.00 (br, 2 H), 1.49 (s, 9 H), 1.46 (s, 9 H).
4) (S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(tert-butoxycarbonyl-
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione
A mixture of 4-tert-butoxycarbonylaminomethyl-2-tert-butoxy -
carbonylmethoxyaniline (93û mg, 2.61 mmol), (S)-9-chloro-5-carboxymethyl -
6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione (807 mg, 2.74
mmol), Bop-CI (770 mg, 2.99 mmol), and triethylamine (2 mL) in dichloro -
methane (20 mL) was stirred for 4 days at room temperature. The mixture was
diluted with ethyl acetate (500 mL) and washed with 5% aqueous potassium
hydrogen sulfate, water, phosphate buffer (pH 7.5), water, and brine,
successively, dried over magnesium sulfate, and concentrated. The residual
solid was recrystalli~ed from acetone-ethyl acetate to give 1.04 g of the title
compound. The mother liquid was concentrated and the residue was purified
by silica gel column chromatography with 0.3% acetic acid/ethyl acetate to give
~ . . .. ..
;`~`: : ` : `
- 66 -
additionally 360 rng of the title compound. A total amount of 1.40 g of the title
compound was obtained (85%).
1H NMR (DMSO-d6) ~ 12.08 (bs, 1 H), 9.30 (bs, 1 H), 7.79 (d, 1 ~I, J = 8.3 Hz),
7.32 (bt, 1 H, J = 5.9 Hz), 7.10 (d, 1 H, J = 2.0 Hz), 7.03 (d, 1 H, J = 8.3 Hz), 6.80
(dd, 1 H, J = 8.3,1.7 Hz), 6.78 (d, 1 H, J = 1.7 Hz), 5.15 ~ 5.25 (m, 1 H), 4.64 (s,
2 H), 4.05 (bd, 2 H, J = 5.9 Hz), 3.05 ~ 3.20 (m, 1 H), 2.70 - 2.88 (m, 2 H), 2.55 ~
2.70 (bd,1 H, J = 14.0 H~), 2.03 ~ 2.20 (dm, 1 H, J = 14.0 Hz), 1.80 ~ 1.95 (m~ 1
H), 1.43 (s, 9H), 1.39 (s, 9 H).
Example 31
1 0 (S)-9-Chloro-5-[p-aminomethyl-~(carboxymethoxy)phenylcarbamoylmethyl] -
6,7-dihydro-lH, 5H-pyrido[1,2,3-de]quinoxaline-2,3-diona hydrochloride
A suspension of (S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-o -
(tert-butoxycarbonylmethoxy)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H -
pyrido[1,2,3-de]quinoxaline-2,3-dione (1.30 g) in 4 N hydrogen chloride in 1,4 -1 5 dioxane (50 mL) was stirred for 20 h at room temperature and 2 N hydrochloric
acid (35 mL) was added. The mixture was stirred further for 1 h and
concentrated in vacuo. The residue was recrystallized from water to give 900
mg of the title compound.
1H NMR (DMS~-d6) ~ 12.50 ~ 13.50 (br, 1 H), 12.13 (bs, 1 H), 9.44 (bs, 1 H),
8.15~8.45(br,3H),8.10(d,1 H,J=8.3Hz),7.19(d,1 H,J=1.7Hz),7.10(d,
1 H,J=1.7Hz),7.07(d,1 H,J=1.7Hz),7.05(dd,1 H,J=8.3,1.7Hz),5.15~
5.30 (m, 1 H), 4.73 (s, 2 H), 3.95 (bd, 1 H, J = 5.3 Hz), 3.05 ~ 3.20 (m, 1 H), 2.75
~ 2.88 (m, 2 H), 2.63 (dd, 1 H, J = 5.3, 14 Hz), 2.09 ~ 2.20 (dm, 1 H, J = 14 Hz),
1.80 ~ 1.95 (m, 1 H).
Example 32
(S)-9-Chloro-5-[~tert-butoxycarbonylaminomethyl-~(ethoxycarbonyl-
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione
A mixture of (S)-9-chloro-5-[p-aminomethyl-~(carboxymethoxy) -
phenylcarbamoylmethyl]-6,7-dihydro-lH) 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione hydrochloride (1.50 g, 2.86 mmol), triethylamine (2 mL), and di-tert-butyldicarbonate (750 mg, 3.44 mmol) in dichloromethane was stirred for 6 h at
`~- 2~2~
- 67-
room temperature and concentrated. To the residue was added 2% aqueous
potassium hydrogen sulfate (200 mL) and the precipitates were collected by
filtration. The precipitates were dried in vacuo and suspended in dichloro -
m~thane (20 mL). Bop-CI (884 mg, 3.43 mmol), ethanol (264 mg, 5.72 mmol),
and triethylamine (2.5 mL) were added and the mixture was stirred for 20 h at
room temperature. The mixture was diluted with ethyl acetate, washed
successively with 5% potassium hydrogen sulfate and brine, dried over
magnesium sulfate, and concentrated. The residue was purified by silica gel
coiumn chromatography with 5: 1 ethyl acetate/hexane to 5: 1 0.3% acetic
1 0 acid in ethyl acetate/hexane to give 310 mg of the title compound (18%).
1H NMR (DMSO~d6) ~ 11.90 ~ 12.00 (br, 1 H), 9.31 (bs, 1 H), 7.78 (d, 1 H, J =
8.3Hz),7.33(bt,1 H,J=5.9Hz),7.10(d,1 H,J=2.0Hz),7.03(d,1 H,J=8.3
Hz), 6.81 (dd, 1 H, J = 8.3, 1.7 Hz), 6.80 (d, 1 H, J = 1.7 Hz), 5.18 ~ 5.28 (m, 1
H),4.77(bs,2H),4.17(q,2H,J=7.3Hz),4.05(bd,2H,J=6.3Hz),3.02~
3.20 (m, ~ H), 2.70 ~ 2.90 (m, 2 H), 2.55 ~ 2.70 (dd, 1 H, J = 14.0, 4.0 Hz), 2.05
~ 2.18 (dm,1 H, J = 14.0 Hz), 1.78 ~ 1.95 (m, i H), 1.39 ( s, 9 H),1.22 (t, 3 H, J =
7.3 Hz).
Example 33
(S)-9-Chloro-5-[p-aminomethyl-~(ethoxycarbonylmethox~)phenylcarbamoyl -
methyl]-6,7-dihydro-1 H, 5H-~yrido[1,2,3-de]quinoxaline-2,3-dione
hydrochlorid~
A suspension of (S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-o -
(ethoxycarbonylmethoxy)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-
pyrido[1,2,3-de]quinoxaline-2,3-dione (150 mg) in ethyl acetate (4 mL) was
added 4 N hydrogen chloride in 1,4-dioxane (2 mL). The mixture was stirred
for 3 h at room temperature, and concentrated in vacuo to give 135 mg of the
title compound.
1H NMR (DMSO-d6) ~ 12.13 (bs, 1 H), 12.13 (bs, 1 H), 9.42 (bs, 1 H), 8.10 ~
8.50(br,3H),7.92(d,1 H,J=7.92Hz),7.17(d,1 H,J=2.0Hz),7.11 (d,1 H,J
= 2.0 Hz), 7.07 (d, 1 H, J = 2.0 Hz), 7.05 (dd, 1 H, J = 7.9, 2.0 Hz), 5.18 - 5.28
(m, 1 H), 4.82 (bs, 2 H), 4.18 (q, 2 H, J = 7.3 Hz), 3.95 (bd,1 H, J = 5.0 Hz), 3.02
~ 3.20 (m, 1 H), 2.72 ~ 2.89 (m, 2 H), 2.64 (dd, 1 H, J = 5.0,14 Hz), 2.05 ~ 2 20
~ ~ 9
- 68 -
(dm, 1 H, J = 14Hz), 1.80 ~ 1.98 tm, 1 H), 1.22 (t, 3 H, J =7.3 Hz).
Example 3~
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(tert-butoxycarbonyl -
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-l H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
A procedure similar to that described in Example 18-6) was performed
with 4-tert-butoxycarbonylaminomethyl-2-tert-butoxycarbonylmethoxyaniline
(990 mg, 2.61 mmol) and (S)-9-bromo-5-carboxymethyl-6,7-dihydro-1H, 5H-
pyrido[1,2,3-de]quinoxaline-2,3-dione (1.06 g, 3.12 mmol) to give a total
1 0 amount of 1.42 9 of the title compound (81 %). - -
1H NMR (DMSO-d6) ~ 12.04 (bs, 1 H), 9.29 (bs, 1 H), 7.79 (d, 1 H, J = 8.3 Hz),
7.31 (bt, 1 H, J = 5.9 Hz), 7.22 (d,1 H, J = 1.7 Hz), 7.16 (d, 1 H, J = 1.7 Hz), 6.80
(dd, 1 H, J = 8.3,1.7 Hz), 6.78 (d, 1 H, J = 8.3 Hz), 5.18 ~ 5.28 (m, 1 H), 4.64 (s,
2 H), 4.05 (bd, 2 H, J = 5.9 Hz), 3.05 ~ 3.20 (m, 1 H), 2.70 ~ 2.95 (m, 2 H), 2.58 ~
2.68 (bd, 1 H, J = 14.0 Hz), 2.08 ~ 2.18 (dm, 1 H, J = 14.0 Hz), 1.75 ~ 1.90 (m, 1
H), 1.43 (s, 9 H),1.39 ( s, 9 H).
Example 35
(S)-9-Bromo-5-[p-aminomethyl-~(carboxymethoxy)phenylcarbamoylmethyl]-
6,7-dihydro-1 H, 5H-pyrido~1,2,3-de]quinoxaline-2,3-dione hydrochloride
A procedure similar to that described in Example 31 was performed with
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(tert-butoxycarbonyl-
methoxy)phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione (1.35 g, 2.0 mmol) to give 997 mg of the title compound.
1H NMR (DMSO-d6) ~ 11.50 ~ 12.50 (br, 1 H), 12.11 (bs, 1 H), 9.43 (bs, 1 H),
8.10~8.40(br,3H),7.92(d,1 H,J=8.3Hz),7.21 (d,1 H,J=1.7Hz),7.18(bs,
1 H), 7.16 (d, 1 H, J = 2.0 Hz), 7.04 (d, 1 H, J = 8.3 Hz), 5.20 ~ 5.30 (m, 1 H),
4.73(s,2H),3.96(d,1 H,J=5.3Hz),3.05~3.20(m,1 H),2.73~2.88(m,2H),
2.60 ~ 2.69 (bd,1 H, J = 14 Hz), 2.08 ~ 2.20 (dm, 1 H, J = 14 Hz), 1.82 ~ 1.98
(m, 1 H).
Example 36
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(4-ethoxycarbonylbutyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-
.. , . .. . - . : ~-
., .. , ,. - , .-
-.,. :- : : - ... . ..
2~2~6~9
- 69 -
dlone
1) Methyl 5-phthalimidomethyl-2-nitrobenzoate
A mixture of methyl 5-methyl-2-nitrobenzoate (35.0 g, 179 mmol), N -
bromosuccinimide (33.5 g, 188 mmol), and azobisisobutyronitrile (500 mg) in
5 carbon tetrachloride (450 mL) was refluxed for 14 h~ The insoluble material
formed was removed by filtration and the filtrate was concentrated. The residue
was dissolved in DMF (300 mL) and potassium phthalimide (19.41 9, 45 mmol)
was added. The mixture was stirred for 2 h at 50C, poured into brine, and
extracted with a 1: 1 mixture of toluene and ethyl acetate. The organic layers
10 were washed with brine, dried over magnesium sulfate, and concentrated. The
residue was purified by silica gel column chromatography with 4: 1 to 2: 1
hexane/ethyl acetate to give 20.0 9 of the title compound (33%).
1H NMR (GDC~13) ~ 7.85 ~ 7.92 (m, 3 H), 7.73 ~ 7.80 (m, 3 H), 7.68 (dd, 1 H, J =8.2,2.0Hz),4.92(s,2H),3.91 (s,3H?.
15 2) Methyl 5-aminomethyl-2-nitrobenzoate hydrochloride
A solution of methyl 5-phthalimidomethyl-2-nitrobenzoate (10.0 g, 29.4
mmol) in a mix~ure of 1,4-dioxane (50 mL) and concentrated hydrochloric acid
(50 mL) was refluxed for 25 h and concentrated in vacuo. The residue was
washed with a mixture of toluene/ethyl acetate and dried in vacuo to give 8.16
20 g of a solid. The residual solid was dissolved in methanol (100 mL) and thionyl
chloride (12 mL) was added slowly. The mixture was refluxed for 1 h and
concentrated. The residual solid was dispersed in toluene, collected by
filtration, and dried in vacuo to give 6.60 g of the title compound (91%).
1H NMR (DMSO-d6) ~ 8.40 ~ 9.90 (br, 3 H), 8.15 (d, 1 H, J = 8.3 H7), 8.02 (d, 1
H, J = 2.0 Hz), 7.95 (dd, 1 H, J = 8.3, 2.0 Hz), 4.20 (bs, 2 H), 3.87 (s, 3 H).
3) Methyl 5-tert-butoxycarbonylaminomethyl-2-nitrobenzoate
A mixture o~ methyl 5-aminomethyl-2-nitrobenzoate hydrochloride (6.50
g, 26.4 mmol~, di-tert-butyl dicarbonate (6.8 g, 31.1 mmol), and triethylamine (8
mL) in dichloromethane (150 mL) was stirred for 5 h at room temperature. After
30 being concentrated, the residue was dispersed between ethyl acetate and 5%
aqueous potassium hydrogen sulfate and the organic layer was washed
successively with saturated aqueous sodium bicarbonate, water, and brine,
~ .
~: . . - : . - :::
.. ~ ., ~ .
~ ~ 2 ~
~`
- 70 -
-
dried over magnesium sulfate, and concentrated. The residue was purified by
silica gel column chromatography with 6: 1 to 3: 1 hexane/ethyl acetate to give
7.6 g of the title compound (93%)~
lH NMR (CDCI3) ~ 7.91 (d, 1 H, J = 8.6 Hz), 7.61 (d, 1 H, J = 2.0 Hz), 7.54 (dd, 1
H,J=8.6,2.0Hz),4.90~5.12(br,1 H),4.41 (bd,1 H,J=5.9Hz),3.92(s,3H),
1.46 (s, 9 H~.
4) 5-tert-3utoxycarbonylaminomethyl-2-nitrobenzylalcohol
To a solution of methyl 5-tert-butoxycarbonylaminomethyl-2-nitro -
benzoate (2.0 9, 6.45 mmol) and sodium borohydride (740 mg, 19.56 mmol) in
THF (11 mL) was added dropwise methanol (1.5 mL) over 1.5 h under reflux.
After the addition was completed, the excess reagent was clecomposed with
5% aqueous potassium hydrogen sulfate. The mixture was extracted with ethyl
acetate. The organic layer was washed with brine, dried over magnesium
sulfate, and concentrated. The residue was purified by silica gel column
chromatography with 3: 1 to 1: 1 hexane/ethyl acetate to give 1.16 9 of the title
compound (64%).
1 H NMR (CDCI3) ~ 8.09 (d, 1 H, J = 8.3 Hz), 7.65 (d, 1 H, J = 2.0 Hz), 7.38 (dd, 1
H,J=8.3,2.0Hz),4.95~5.15(br,1 H),4.98(d,2H,J=6.3Hz),4.41 (d,2H,J
= 5.9 Hz), 2.65 (t, 1 H, J = 6.3 Hz), 1.47 (s, 9 H).
5) 5-tert-Butoxycarbonylaminomethyl-2-nitrobenzylaldehyde
A mixture of 2-nitro-5-tert-butoxycarbonylaminomethylbenzylalcohol
(700 mg, 2.48 mmol) and manganese oxide (7.0 g) in dichloromethane (20 mL)
was stirred at room temperature for 5 h. The reagent was removed by filtration
through celite and the ~iltrate was concentrated to give 580 mg of the title
compound (84%).
1H NMR (CDCI3) ~ 10.43 (s, 1 H), 8.11 (d, 1 H, J = 8.6 Hz), 7.83 (d, 1 H, J = 2.0
Hz), 7.68 (dd, 1 H, J = 8.6, 2.0 Hz), 5.05 ~ 5.25 (br, 1 H), 4.45 (bd, 2 H, J = 5.9
Hz), 1.47 (s, 9 H).
6) Ethyl 5-(5-tert-butoxycarbonylaminomethyl-2-nitrophenyl)pent-4-enoate
To a suspension of 3-ethoxycarbonylpropyltriphenylphosphonium
bromide (940 mg, 2.05 mmol) in THF (7 mL) at -78C was added a 0.5 N
solution of potassium hexamethyldisilazide (4 mL, 2.0 mmol). The mixture was
. - ~ . .
-- .
`:
- 71 -
stirred for 1 h at -78C and a solution of 5-tert-butoxycarbonylaminomethyl-2 -
nitrobenzylaldehyde (580 mg, 2.07 mmol) in THF (10 mL) was added slowly.
The mixture was allowed to warm to room temperature, poured into 1%
aqueous potassium hydrogen sulfate (100 mL), and extracted with ethyl
5 acetata. The organic layers were washed with brine, dried over magnesium
sulfate, and concentrated. The residue was purified by silica gel column
chromatography with 3: 1 hexane/ethyl acetate to give 590 mg of the title
compound (78%) as a 3: 1 mixture of cis and trans isomers.
Cis isomer: 1H NMR (CDCI3) ~ 8.02 (d, 1 H, J = 8.3 Hz), 7.33 (dd, 1 H, J = 8.3,
1 0 2.0 Hz), 7.29 (d, 1 H, J = 2.0 Hz), 6.76 (d,1 H, J = 11.5 Hz), 5.80 (dt, 1 H, J =
11.5, 7.0 Hz), 5.08 ~ 5.22 (br, 1 H), 4.39 (bd, 2 H, J = 6.6 Hz), 4.11 (q, 2 H, J =
7.3 Hz), 2.30 ~ 2.65 (m, 4 H), 1.46 (s, 9 H),1.24 (t, 3 H, J = 7.3 Hz).
Trans isomer: 1H NMR (CDC13) ~ 7.89 (d, 1 H, J = 8.6 Hz), 7.44 (d, 1 H, J = 2.0
Hz), 7.27 (dd, 1 H, J = 8.6, 2.0 Hz), 6.92 (d, 1 H, J = 17.2 Hz), 6.23 (dt, 1 H, J =
17.2,7.0H7),4.90~5.10(br, 1 H),4.37(bd,2H,J=6.~Hz),4.17(q,2H,J=
7.3 Hz), 2.30 ~ 2.65 (m, 4 H), 1.46 (s, 9 H), 1.27 (t, 3 H, J = 7.3 Hz).
7) 2-(4-Ethoxycarbonylbutyl)-4-tert-butoxycarbonylaminomethylaniline
A solution of ethyl 5-(5-tert-butoxycarbonylaminomethyl-2-nitrophenyl) -
pent-4-enoate (560 mg, 1.48 mmol) in ethyl acetate (20 mL) was hydrogenated
over platinum oxide (100 mg) under atmospheric pressure of hydrogen at room
temperature for 2.5 h. The mixture was passed through celite and the filtrate
was concentrated to give 560 mg of the title compound (100%).
1H NMR (CDCI3) ~ 6.90 ~ 7.00 (m, 2 H), 6.62 (d, 1 H, J = 8.6 Hz), 4.60 ~ 4.80
(br,1 H),4.17(bd,2H,J=8.6Hz),4.13(q,2H,J=7.3Hz),3.30~3.88(br,2
H),2.49(t,2H,~=7.3Hz),2.35~t,2H,J=7.3Hz),1.58~1.80(m,4H),1.49
(s,9H), 1.26(t,3H,J=7.3Hz).
8) (S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(4-ethoxycarbonylbutyl)-
phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione
A procedure similar to that descrlbed in Example 18-6) was performed
with 2-(4-ethoxycarbonylbutyl)-4-tert-butoxycarbonylaminomethylaniline (256
mg, 0.73 mmol) and (S)-9-bromo-5-carboxymethyl-6,7-dihydro-1H, 5H-
- . ~: . . . . . .
-~` 2121~
- 72 -
pyrido~1,2,3-de]quinoxaline-2,3-dione (297 mg, 0.88 mmol) to give 365 mg of
the title compound after silica gel column chromatography with 0.3% acetic
acid/ethyl acetate (74%).
1H NMR (DMSO-d6) ~ 12.08 (bs, 1 H), 9.37 (bs, 1 H), 7.37 (bt, 1 H, J = 5.3 Hz),
7.25(dd,1 H,J=8.3,2.0Hz),7.24(d,1 H,J=2.0Hz),7.17(d,1 H,J=2.0Hz),
7.03 (d, 1 H, J = 8.3 Hz), 5.18 ~ 5.30 (m,1 H), 4.05 (bd, 2 H, J = 5.3 Hz), 4.03 (q,
2 H, J = 7.3 Hz), 3.05 ~ 3.21 (m, 1 H), 2.80 ~ 2.90 (dm, 1 H, J = 14 Hz), 2.51 ~2.69(m,4H),2.30(t,2H,J=7.3Hz),2.05~2.18(dm, 1 H,J=14.0Hz), 1.80~
1.98 (m, 1 H), 1.40 ~ 1.65 (m, 4 H), 1.39 (s, 9 H),1.16 (t, 3 H, J =7.3 Hz).
1 0 Example 37
(S)-9-Bromo-5-~p-tert-butoxycarbonylaminomethyl-~(4-carboxybutyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1H, 5H-pyridol1,2,3-de]quinoxaline-2,3-dione
A procedure similar to that described in Example 10 was performed with
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(4-ethoxycarbonylbutyl) -
1 5 phenylcarbamoylmethyl]-6,7-dihydro-1 f 1, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione (300 mg, 0.446 mmol) to give the title compound (quant).
Example 38
(S)-9-Bromo-5-~p-aminomethyl-G(4-carboxybutyl)phenylcarbamoylmethyl]-6,7
dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride
A procedure similar to that described in Example 11 was performed with
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(4-carboxybutyl)phenyl -
carbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
(0.446 mmol) to give 250 mg of the title compound (97%).
lH NMR (DMSO-d6) â 11.0 ~ 13.0 (br, 2 H), 9.53 (bs, 1 H), 7.80 - 8.80 (br, 3 H),7.40(d,1 H,J=8.3Hz),7.32(d,1 H,J=2.0Hz),7.27(dd,1 H,J=8.3,2.0Hz),
7.25 (d, 1 H, J = 2.0 Hz), 7.19 (d,1 H, J = 2.0 Hz), 5.20 ~ 5.30 (m,1 H), 3.98 (bs,
2 H), 3.05 ~ 3.22 (m,1 H), 2.80 ~ 2.91 (dm, 1 H, J = 14.0 Hz), 2.52 ~ 2.80 (m, 4H),2.24(t,2H,J=7.3Hz),206~2.16(dm,1 H,J=14.0Hz),1.80~1.98(m,1
H), 1.42~1.62(m,4H).
2:l2~6~
- 73 -
Example 39
(S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-diethoxycarbonyl -
propyl)phenylcarbamoylmethyl]-6,7-dihydro-l H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione
CO2Et
O q~ N ~2 Et
~1 ~, NHCO2t-Bu
1 0 Cl~H~O
1) 3-tert-Butoxycarbonylaminomethyl-5-nitrostyrene
To a suspension of methyltriphenylphosphonium bromide (4.1 9, 11.5
mmol) in THF (40 mL) at -78C was added 0.5 N potassium hexamethyl -
disilazide (24 mL, 12 mmol) in toluene. The mixture was stirred for 3 h at -78Cand a solution of 5-tert-butoxycarbonylaminomethyl-2-nitrobenzaldehyde (3.2
g, 11.4 mmol) in THF (40 mL) was added slowly. The mixture was allowed to
warm to room temperature, poured into 2% aqueous potassium hydrogen
sulfate (250 mL), and extracted with ethyl acetate. The organic layers were
washed with brine, dried over magnesium sulfate, and concentrated. The
residue was purified by silica gel column chromatography with 7: 1 to 5: 1
hexane/ethyl acetate to give 2.4 9 of the title compound (76%).
1 H N M R (C D Cl3) ~ 7.93 (d, 1 H, J = 8.3 Hz), 7.50 (d, 1 H, J = 2.0 Hz), 7.32 (dd, 1
H, J = 8.3, 2.0 Hz), 7.1 9 (dd, 1 H, J = 1 7.2, 11.2 Hz), 5.73 (dd, 1 I l, J = 17.2, 1.0
Hz), 5.49 (dd, 1 H, J = 11.2, 1.0 Hz), 4.90 - 5.11 (br, 1 H), 4.3g (bd, 2 H, J = 5.9
Hz), 1.47 (s, 9 H).
2) Ethyl 4-(5-tert-butoxycarbonylaminomethyl-2-nitrophenyl)-2-ethoxycarbonyl -
butanoate
To a suspension of 60% sodium hydride (757 mg, 18.9 mmol) in D M F
(80 mL) at room temperature was added slowly diethyl malonate (7.6 9, 47.3
mmol). The mixture was stirred for 2 h at 40 ~ 50C and a solution of 3-tert -
butoxycarbonylaminomethyl-5-nitrostyrene (2.4 g, 8.6 mmol) in D M F (20 mL)
2i~:~6~
- 74 -
was added slowly. The mixture was allowed to warm to room temperature,
poured into 2% aqueous potassium hydrogen sulfate (500 mL), and extracted
with a 1: 1 mixture of toluene and ethyl acetate. The organic layers were
washed with brine, dried over magnesium sulfate, and concentrated. The
5 residue was purified by silica gel column chromatography with 8: 1 to 3: 1
hexane/e~hyl acetate to give 3~24 g of the title compound (86%).
1H NMR (CDC13) â 7.93 (d, 1 H, J = 8.3 Hz), 7.28 (dd, 1 H, J = 8.3, 2.0 Hz), 7.26
(d,1 H,J=2.0Hz),4.90~5.10(br,1 H),~.36(bd,2H,J=6.3Hz),4.22(q,4H,
J=6.9Hz),3.41 (t, 1 H,J=7.3Hz),2.94(t,2H,J=7.6Hz),2.25(dt,2H,J=
7.3,7.6Hz),1.47(s,9H),1.29(t,6H,J=6.9Hz).
3) 2-(3,3-Diethoxycarbonylpropyl)-4-tert-butoxycarbonylaminomethylaniline
A solution of ethyl 4-(5-tert-butoxycarbonylaminomethyl-2-nitrophenyl)-2 -
ethoxycarbonylbutanoate (3.2 g, 7.3 mmol) in ethyl acetate (30 mL) was
hydrogenated over palladiumlcharcoal (500 mg) under atmospheric pressure
1 5 of hydrogen at room temperature for 2.5 h. The mixture was passed through
celite and the filtrate was concentrated to give 2.8 9 of the title compound
(94%).
1H NMR (CDCI3) ~ 6.96 (dd, 1 H, J = 7.9, 2.0 Hz), 6.93 (d, 1 H, J = 2.0 Hz), 6.63
(d,1 H,J=7.9Hz),4.58~4.80(br,1 H),4.30~4.60(br,2H),4.22(q,4H,J=
7.3 Hz), 4.16 (bd, 2 H, J = 5.9 Hz), 3.41 (t,1 H, J = 7.3 Hz), 2.53 (dt, 2 H, J = 7.6
Hz),2.12(q,2H,J=7.3Hz),1.45(s,9H),1.28(t,6H,J=6.9Hz).
4) (S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-diethoxycarbonyl-
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
A procedure similar to that described in Example 18-6) was performed
with 2-(3,3-diethoxycarbonylpropyl)-4-tert-butoxycarbonylaminomethylaniline
(559 mg, 1.37 mmol) and (S)-9-chloro-5-carboxymethyl-6,7-dihydro-1 H, 5H -
pyrido[1,2,3-d2]quinoxaline-2,3-dione (445 mg, 1.51 mmol) to give 735 mg of
the title compound after silica gel column chromatography with 0.3% acetic
acid/ethyl acetate (78%).
1H NMR (DMSO-d6) ~ 11.80 ~ 12.30 (br,1H), 9.41 (bs, 1 H), 7.39 (bt, 1 H, J =
6.3Hz),7.25(d,1 H,J=8.3Hz),7.10(d,1 H,J=2.0Hz),7.09(dd,1 H,J=8.3,
., . , .. .,, ... . . ~ . .... . . .. . . .. .
2:l21~
- 75 -
2.0 Hz), 7.07 (d,1 H, J = 2.0 Hz), 7.04 (d, 1 H, J = 2.0 Hz), 5.15 ~ 5.28 (m, 1 H),
4.13(q,4H,J-7.3Hz),4.0~(bd,2H,J=6.3Hz),3.47(t,1 H,J=7.3Hz),3.02
~ 3.18 (m, 1 H), 2.76 ~ 2.88 (dm,1 H, J = 14.0 Hz), 2.48 ~ 2.67 (m, 4 H), 2.06 ~2.18(dm,1 H,J=14.0Hz),1.75~1.98(m,3H),1.40(s,9H),1.18(t,6H.J=
5 7.3 Hz).
Example 40
(S)-9-Chloro-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-dicarboxypropyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione
1 0 A procedure similar to that described in Example 10 was per~ormed with
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-diethoxycarbonyl -
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de] -
quinoxaline-2,3-dione (700 mg, 1.02 mmol) to give the title compound (quant).
Exarnple 41
1 5 (S)-9-Chloro-5-[p-aminomethyl-~(3,3-dicarboxypropyl)phenylcarbamoyl -
methyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione
hydrochl~ride
A procedure similar to that described in Example 11 was performed with
(S)-9-chloro-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-dicarboxypropyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione (1.02 mmol) to give 530 mg of the title compound (96%).
1H NMR (DMSO-d6) â 12.50 ~ 13.50 (br, 2 H), 12.15 (bs, 1 H), 9.56 (bs, 1 H),
8.10 ~ 8.40 (br, 3 11), 7.41 (d, 1 H, J = 8.3 Hz), 7.33 (d, 1 H, J = 2.0 Hz), 7.30 (dd,
1 H,J=8.3,2.0H~),7.12(d,1 H,J=2.0Hz),7.08(d,1 H,J=2.0Hz),5.15~
5.30 (m, 1 H), 3.90 ~ 4.02 (m, 2 H), 3.29 (t, 1 H, J = 7.3 Hz), 3.03 ~ 3.20 (m, 1 H),
2.80 ~ 2.90 (dm,1 H, J = 14.0 Hz), 2.50 ~ 2.70 (m, 4 H), 2.03 ~ 2.18 (dm, 1 H, J= 14.0 Hz), 1.78 ~ 1.98 (m, 3 H).
Example 42
(S)-9-Chloro-5-[p-aminomethyl-~(3-carboxypropyl)phenylcarbamoylmethyl] -
6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-dione hydrochloride
A suspension of (S)-9-chloro-5-[p-aminomethyl-~(3,3-dicarboxypropyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3-
.. - - - . .. - - ~ - -
`~t2lGa9
- 76 -
dione hydrochloride (550 mg, 0.98 mmol) in acetic acid (300 mL) was refluxed
for 5 h~ To the mixture was added 0.5 N hydrochloric acid and the solvent was
removed in vacuo. The residual solid was recrystallized from wat0r to give 450
mg of the title compound (89%).
1H NMR (DMSO-d6) ~ 11.50 ~ 12.~0 (br, 1 H), 12.17 (bs, 1 H), 9.57 (bs, 1 H),
8.10 ~ 8.50 (br, 3 H), 7.42 (d, 1 H, J = 8.3 Hz), 7.33 (d, 1 H, J = 2.0 Hz), 7.30 (dd,
1 H,J=3.3,2.0Hz),7.12(d,1 H,J=2.0Hz),7.09(d,1 H,J-2.0Hz),5.18~
5.30 (m, 1 H), 3.97 (bs, 2 H), 3.05 ~ 3.25 (m, 1 H), 2.80 ~ 2.91 (dm, 1 H, J = 14.0
Hz),2.50~2.80(m,4H),2.27(t,2H,J=73Hz),205~218(dm,1 H,J=14.0
1 0 Hz), 1.82 ~ 1.98 (m, 1 H), 1.74 (5et, 2 H, J = 7.3 Hz).
Example 43
(S)-9-Bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-diethoxycarbonyl -
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de] -
quinoxaline-2,3-dione
1 5 A procedure similar to that described in Example 18-6) was performed
with 2-(3,3-diethoxycarbonylpropyl)-4-tert-butoxycarbonylaminomethylaniline
(1.5 9, 3.67 mmol) and (S)-9-bromo-5-carboxymethyl-6,7-dihydro-1 H, 5H -
pyrido[1,2,3-de]quinoxaline-2,3-dione (1.37 g, 4.04 mmol) to give 1.60 g of the
title compound after silica gel column chromatography with 0.3% acetic
acid/ethyl acetate (60%).
1H NMR (DMSO-d6) ~ 11.00 ~ 13.00 (br, 1H), 9.41 (bs, 1 H), 7.39 (bt, 1 H, J =
6.3Hz),7.25(d,1 H,J=8.3Hz),7.19(d,1 H,J=2.0Hz),7.16(d,1 H,J=2.0
Hz),7.07(d,1 H,J=2.0Hz),7.06(dd,1 H,J=8.3,2.9Hz),5.18~5.30(m,1
H),4.13(q,4H,J=7.3Hz),4.08(bd,2H,J=5.6Hz),3.47(t,1 H,J=7.6Hz),
3.02 ~ 3.20 (m,1 H), 2.78 ~ 2.90 (dm, 1 H, J = 14.0 Hz), 2.50 ~ 2.70 (m, 4 H),
2.07~2.18(dm,1 H,J=14.0Hz),1.80~2.00(m,3H),1.40(s,9H),1.18(t,6
H,J=7.3Hz).
Example 44
(S)-9-Bromo-5-~p-tert-butoxycarbonylaminomethyl-~(3,3-dicarboxypropyl)- ~ -
phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 - -
dione
A procedure similar to that described in Example 10 was performed with
~: :
~ l 21 ~ Q~
- 77 -
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-diethoxycarbonyl -
propyl)phenylcarbamoylmethyl]-6,7-dihydro-1 H, 5H-pyrido~1 ,2,3-de] -
quinoxaline-2,3-dione (1.50 g, 2.06 mmol) to give the titl0 compound (quant).
Example 45
5 (S)-9-Bromo-5-[p-aminomethyl-~(3,3-dicarboxypropyl)phenylcarbamoyl-
methyl]-6,7-dihydro-1 H, 5H-pyrido[1 ,2,3-de]quinoxaline-2,3-dione
hydrochloride
A procedure similar to that described in Example 11 was performed with
(S)-9-bromo-5-[p-tert-butoxycarbonylaminomethyl-~(3,3-dicarboxypropyl) -
phenylcarbamoylmethyl]-6,7-dihydro-1H, 5H-pyrido[1,2,3-de]quinoxaline-2,3 -
dione (2.06 mmol) to give 1.45 g of the title compound (quant).
1H NMR (DMSO-d6) ~11.00 ~ 13.00 (br, 2 H), 12.14 (bs, 1 H), 9.55 (bs, 1 H),
8.15 ~ 8.40 (br, 3 H), 7.40 (d, 1 H, J = 7.9 Hz), 7.33 (d, 1 H, J = 2.0 Hz), 7.30 (dd,
1 H,J=7.9,2.0Hz),7.24(d,1 H,J=2.0Hz),7.20~d,1 H,J=2.0Hz),5.18~
5.30 (m, 1 H), 3.98 (m, 2 H), 3.29 (t, 1 H, J = 7.3 Hz), 3.04 ~ 3.21 (m, 1 H), 2.80 ~
2.91 (dm, 1 H, J = 14.0 Hz), 2.50 ~ 2.70 (m, 4 H), 2.05 ~ 2.18 (dm, 1 H, J = 14.0
Hz), 1.7~ ~ 2.00 (m, 3 H).
,: ` `'' , . ~ '