Note: Descriptions are shown in the official language in which they were submitted.
WO 93/l112g PC~/EP9~/0269/
212~14
ANTIPARASITIC AGENTS
This invention relates to antiparasitic agents,
related to the milbemycins and to processes for their
preparation and compositions thereof.
The avermectins and milbemycins form an important
group of broad spectrum antiparasitic agents possessing
anthelmintic, ectoparasiticidal, insecticidal,
antibacterial and antifungal activity with application in
the areas of animal and human health, agriculture and
horticulture. The avermectins are a group of macrolide
compounds (previously referred to as C-076) isolated from
the fermentation broth of an avermectin producing-~6train
of Stre~tomyces avermitilis such as those deposlted
strains ATCC 31267, ATCC 31271, ATCC 31272 as described
in US patents 4310519 and 4429042. ~uropean patents
214731 and 276103 disclose a related series of compounds
differing from the C-076 compounds in the nature of the
substituent at C-25. The milbemycins form another group
of related macro1ides which are distinguished from the
avermectins in lacking a sugar residue attached at C-13.
Examples of such compounds are the B-41 series as
described in UK patent 1390336, the LL-F28249 series- - -
described in European patent EP 170006, the E225
compounds described in European patent 254583 and the
N787-182 compounds d~scribed in European patent ` ~-
application 334484. In addition to these fermentation
products a large number of publications describe
compounds derived semi-synthetically from these
fermentation products many of which possess useful
antiparasitic activities. Some of this chemistry is~
reviewed by Davies, H.G. and Green, R.H. in Natura~
Product Reports (1986), 3, 87-121.
~'093J11129 PCT/EP9~/0269,
~122614
Our European Patent Application 410615 describes a
compound, herein designated UK-86956 of formula:
CH~
~ 0~0
(~CH3
OH
It may be made by fermentation of a micro~organism
deposited at the American Type Culture Collection under
accession number ATCC 53928.
It has been found that certain semi-synthetic :
derivatives of the above-mentioned compound UK-86956,-
also have antiparasitic activity. Thus one aspect of :
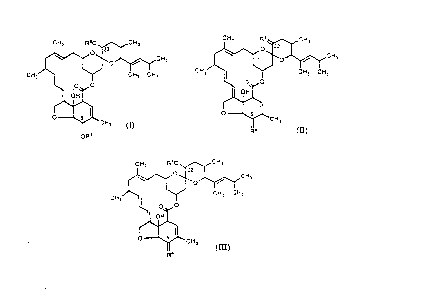
this invention provides compound~ of formula (I): .
CH,~,CH,
;~CH, (I) -
OR~
where Rl and R2 are independently selected from H,
C~-C8 alkyl, alkenyl, alkynyl, Cz-C8 alkanoyl, alkenoyl and
alkynoyl, arylalkanoyl, arylcarbonyl and C~-C8
alkylsulphonyl an~ arylsulphon~ gro~ps, wherein all
these groups other than H may optionally be substituted
with at least one hydroxy, C~-C8 alkoxy, carboxy or halo
group with the pro~iso that at least one of Rl and R2 are
other than hydrogen. The alkane, alkene and alkyne
WO 93/11129 ~ 1 ~ 2 6 1 4 PCT/EP92/0269,
groups may be straight or branched-chain. Halo means F,
Cl, Br or I.
Compounds within this aspect of the invention
include those in which Rl is methyl and R2 is H or methyl.
Another aspect of this invention provides compounds
of formula (II~:
CH3 R ~ ~C~3
CH3~ C~3 C~3
'- 0~5~
CH3
~ (rr) ' '
where R3 is oxo, optionally O-substituted oximino or
methylene optionally substituted with cyano or a Cl-C8
alkyl, cycloalkyl, aryl, aralkyl or C~-C8 alkoxycarbonyl
group which groups may optionally be substituted by a
halo, carboxy and/or other groups; R4 is O-Rs where Rs is
as defined for Rl in formula (I) and the oxygen is
attached at C-5 by a single bond or R4 is oxo, optionally
substituted imino or optionally O-substituted oximino,
the broken line representing an double bond.
The oximino groups may be O-substituted by a wide
variety of substituents including linear or branched CI-C~
alkyl, alkenyl, alkynyl, trialkylsilyl and a~alkyl~groups
which may themselves be substituted by halo, carboxy
and/or other groups.
Compounds within this aspect of the invention
include those in which R3 is methylene and R4 is OH or :.:
oximino and those in which R3 is oxo and R4 is~hy~roxy,
methoxy or oximino.
A further aspect of the invention provides compounds
of formula (III):
WO93/11129 212 2 61 1 PCT/EP92/0269,
CH3 R1o~ ,~CH3
/ CH3 C'3
O~CH3
I l ~ (III)
where R1 is as defined for formula (I) and R4 is oxo or
oximino optionally s~ubstituted as for compounds of
formula (II).
Compounds within this aspect of the invention
include that in which R1 is -OH and R4 is oximino.
The compounds of the invention have several ¦ :
asymmetric centres, the number of such centres depending
on the nature of substituents R1-R4, and accordingly may
exist as several pairs of stereoisomers. Compound UK
86956, when prepared by fermentation of micro-organism
ATCC 53928 is believed to have the following relative
stereochemistry.
CH3 HO,,~.,~CH3
~ J~CH~
CH ""~ : ~ CH3 CH3
~CH,
OH
The compounds of the invention may be prepared by .
replacement of one-or more of the hydroxy ~roups at the 5 :
and 22 positions of compound UK 86956 by the appropriate
~'093/1t129 ~ 1~ 2 6 1 4 PCT/EP92/0269-
substituents. These substitutions may be carried out by
means of reactions which are known in the art. However,
in preparing these compounds it is necessary to react
selectively at one of the 5 and 22 hydroxy groups. In
some instances this may be achieved by choice of
appropriate reagent, for example manganese dioxide which
selectively converts the 5-hydroxy group to a 5-oxo
group. Alternatively selective reaction may be achieved
using suitable protecting groups such as acyl or silyl
derivatives. A particularly preferred protecting group
is the tert-butyldimethylsilyl group (TBDMS) which is
readily removed when required using either an acid in a
protic solvent such as methanol or with a fluoride ion
source such as tetrabutylammonium fluoride dissolved in a
suitable inert solvent such as tetrahydrofuran.
Alkylation of compound UK-86,956 (or a derivative
thereof retaining a hydroxy group at position 5 or 22)
may be achieved using conventional methodology such as
treatment with an alkyl halide, preferably an alkyl
iodide, in the presence of a silver salt or silver oxide.
Alternatively other reactive alkylating agents known to
those skilled in the art such as alkyl triflates or ~-
diazoalkanes may be employed and may show differing_ ',
tendencies to react at the free hydroxy groups at the 5 ~,
and 22 positions. Similarly acylation or sulphonylation `
of UK-86,956 or its derivatives can be achieved-using-
standard procedures for example by using an acyl or
sulphonyl halide or anhydride in the presence of a base
such as triethylamine, pyridine or diisopropylami-ne-.
Oxidation of the hydroxy group at C-22 may be
carried, out using a suitable oxidising agent such as ¦
dimethylsulphoxide/oxalyl chloride. Using, t~ese~ ~-
conditions the C-S hydroxy group will also be oxidised if !
WO 93/11129 PCT/EP9~/0269,
~122614
not protected for example using the TBDMS group. The oxo
group at C-5 or C-22 may be reduced to a hydroxy group
although the stereochemical outcome of this reaction will
depend on the choice of reducing agent. For example
sodium borohydride will reduce either a C-5 or C-22 oxo
group to a hydroxy group possessing substantially the
same stereo~hemistry as that found in the UK-86,~56
starting compound. However, other reducing agents such
as lithium tri-sec-bu~ylborohydride (L-selectride,
Aldrich Chemical Company) will reduce the
C-22 oxo group to give a hydroxy group with the opposite
stereochemistry to that found in the starting compound.
The oxo group at either C-5 or C-22 is a useful
functional group for performing furthçr semi-synthetic
modification to the compounds. For example reaction
between a phosphor~ne of the general formula XYZP=CHR6,
where X, Y and Z are substituents such as phenyl,
phenoxy, alkoxy or oxo and a 22-oxo derivative of UK-
86,956 in an inert solvent yields a 22-methylene or when
R6 is other than H, a mono-substituted methylene
deri~ative. R6 is ~ or a substituent such as an alkyl,
cyano, phenyl, alkanoyl or alkoxycarbonyl group.
Alternatively the oxo group at C-5 and~or C-22 may be
converted to an oximino group or O-substituted oximino
group by reaction with an optionally O-substituted
hydroxylamine or salt thereof in a suitable solvent such
as methanol, dioxane or water or combination thereof. O-
substituted oximino derivatives may alternatively be
prepared by reaction of the oximino derivative with a
suitable alkylating or acylating agent.
Oximino and substituted methylene derivatives may exist
as E or Z isomers and both stereoisomers are within the
scope of this invention. In addition those derivatives
o~ UK-86,956 which are singly bonded at C-22 may be
prepared with two possible geometries and both are within
the scope of this invention (including the epi-hydroxy
compound).
WO93/11129 ~ l~ 2 6 1 ~ PCT/EP9~/0269-
The compounds of the invention are highly active
antiparasitic agents having particular utility as
anthelmintics, ectoparasiticides, insecticides and
acaricides.
Thus the compounds are effective in treating a
variety of conditions caused by endoparasites including,
in particular, helminthiasis which is most frequently
caused by a group of parasitic worms described as
nematodes and which can cause severe economic losses in
- swine, sheep, horses and cattle as well as affecting
domestic animals and poultry. The compounds are also
effective against oth`er nematodes which affect various
species of animals including, for exa~ple, Dirofilaria
and various parasites of dogs and cats which can infect
humans including gastro-intestinal parasites such as
AncYlostoma, Necator, Ascaris, StronqYloides,
Trichinella, CaPillaria, Trichuris, Enterobius and ~;~
parasites which are found in the blood or other tissues .
and organs such as filiarial worms and the extra ¦
intestinal stages of Strona~loides and Trichinella.
The compounds are also of value in treating ;
ectoparasite infections including in particular arthropod
ectoparasites of animals and birds such as ticks, mites,-
lice, fleas, blowfly, biting insects and migrating
dipterous larvae whlch can affect swine, sheep, cattle
and horses as well as affecting domestic animals- an* -~
poultry.
The compounds are also insecticides active against
household pests such as the cockroach, clothes moth,
carpet beetle and the housefIy as well as being us~eful
against insect pests of stored grain and of agricultural
plants such as spider mites, aphids, caterpillar&~and
against migratory orthopterans such as locusts.
WV93/11129 ~ 6 ~ l PCT/EP92/0269
The compounds of the invention possess a number of
beneficial properties compared to similar compounds such
as the natural product UK-86,956 in terms of their
efficacy, pharmacokinetics and toleration. The benefits ~-
~that arise from this unexpected combination of properties
include efficacy against the important parasitic wcrms or
arthropods afflicting livestock, domesticated animals or
humans at lower doses than are currently employed for
related çompounds and, in addition, the ability to treat
animals previously regarded as ~ensitive to this class of
macrolide with a greater margin of safety.
Tha compounds of formula (I), (II) or (III~ are
administered as a formulation appropriate to the specific
use envisaged and to the particular species of host
animal being treated and the parasite or insect involved. j
For use as an anthelmintic the compounds are preferably
administered ~y injection, either subcutaneously or
intramuscularly, alternatively they may be administered
orally in the form of a capsule, bolus, tablet, chewable
tablet or liquid drench, or they may be administered as a
pour-on formulation or as an implant. Such formulations
are prepared in a conventional manner in accordance with
standard veterinary practice.- Thus capsules, boluses or
tablets may be prepared by mixing the active ingredient
with a suitable finely divided diluent or carrier,
additionally containing a aisi-ntegrating agent and/or
binder such as starch, lactose, talc or magnesium
stearate. A drench formulation may be prepared by
dispersing the active ingredient in an aqueous solution
together with dispersing or wetting agents and injectable
formulations may be prepared_in the form of a sterile
solution or emulsion. These_formulations will vary with
regard to the weight of active compound depending on the
species of host animal to be treated, the severity and
type of infection and the body weight of the host.
WO93/11129 PCT/EP9~0269-
~ ~2~611
g
Generally for oral administration a dose of from about
O.OO1 to 10 mg per Kg of animal body weight given as a
single dose or in divided doses for a period of from 1 to
5 days will be satisfactory but of course there can be
instances where higher or lower dosage ranges are
indicated and such are within the scope of this
invention.
As an alternative the compounds may be administered
with the animal feedstuff and for this purpose a
concentrated feed additive or premix may be prepared for
mixing with the normal animal feed.
For use as an insecticide and for treating
agricultural pests the compounds are applied as sprays, ~;
dusts, pour-on formulations, emulsions and the like in
accordance with standard agricultural practice. I :
For human use the compounds may be administered as a ¦~-
pharmaceutically acceptable formulation of conventional
type. ~-
, .
WO 93/11129 PCT/EP92/0269-
7 122~1 l
The preparation of compounds according to the
invention and certain intermediate compounds is
illustrated by the following Examples. "Silica" refers
to Kieselgel 60, 230-400 mesh, Merck, Art. 9385. The
reaction steps described in the Examples are illustrated
by accompanying Figure 1.
EXAMPLE 1
5-Keto-UK-86 95
UK-86,956 (500mg) was dissolved in diethyl ether
(anhydrous, 50ml) and manganese dioxide (activated,
Aldrich Chemical Co. Ltd., 2.0g) was added. The
suspension was stirred at room temperature for 18h and a
further aliquot of manganese diox~de ~1.0g~ was added.
After a further 8h the mixture was filtered, the solid
washed with et~yl acetate and the com~ined filtrate
evaporated under vacuum to give an off-white powder which
was used directly in the following Example 2.
EXAMPLE 2
5-Oximino-UK-86,956
5-Keto-UK-86,9S6 (300mg) was dissolved in methanol
(7.5ml) at room temperat~ and~then was added, in
sequence, dioxan (7.5ml), hydroxylamine hydrochloride
(300mg) and water (7.5ml). After stlrring for 18h the
mixture was added to diethyl-ether and water, the upper
layer was separated and washed twice with water. The
lower layer was extracted with diethyl ether and the
combined organic layers were dried over anhydrous sodium
sulphate and evaporated under vacuum to give a colourless
glass (460mg). Final puri~ication was achieved by column .
chromatography on silica (Klesèlgel 60, 230-400 mesh,
Merck) (50g) eluting with methylene chloride-methanol
50:1 to give the pure titl~ compound. Mass and NMR
spectra were fully consistent with the structur~ shown in
Figure 1.
W093/11129 ~ 1 ~ 2 ~1~ PCT/EP92/0269
EXAMPLE 3
5-MethoxY-UK-86 956
To a solution of UK-86,956 (500mg) in anhydrous
diethyl ether (125ml) was added iodomethane (lOml) and
freshly prepared silver (I) oxide (2.5g). The mixture
was stirred for 60h at a room temperature, filtered and
evaporated under vacuum to give the crude product
(600mg). This was purified by high pressure liquid
chromatography on a Dynamax column (41.4 x 300mm, 8~m ;~
ODS-silica, Rainin) eluting with methanol-water 87:13 at ~-~
70ml per minute to give the title compound (198mg) and
5,22-dimet~oxy-UX-86,956 (200mg) from later fractions.
Macs and NMR spectra were fully consistent with the ~
proposed structures. `
i ''
EXAMPLE 4
5-Tertbutyldimethylsilyoxy~UK-86 956
To a solution of UK-86,956 (4.9g) in anhydrous
methylene chloride (lOOml) was added tertbutyl-
dimethylsilyl chloride (3.62g) and imidazole (3.2g~ and
the mixture was neated under reflux for 2h. Water (50ml)
was then added and the organic layer separated. The -
aqueous layer was extracted once with methylene chloride
(20ml) and the combined organic solution was washed with
dilute hydrochloric acid, water and then dried over
anhydrous sodium sulphate before evaporating to-dry~ess~
under vacuum to give the crude product (7.58g) as~a
yellow foam. This material was purified by column
chromatography twice on silica (Kieselgel 60, 230-400-
mesh, Merck) (50g) eluting with methylene chloride to -
give the title compound, 3.77g as a white foam. The N~R
spectrum of this material was fully consistent wi~h- ~he
proposed structure. !-
WO 93/11129PCT/EP92~0269ï
~122614
12
EXAMPLE 5
5-TertbutYldimethYlsilYloxY-22-keto-UK-86,956
To a solution of oxalyl chloride (0.5ml) in
anhydrous methylene chloride (5ml) under nitrogen at
~78C was added a solution of dimethylsulphoxide (0.4ml)
in anhydrous methylene chloride (lOml) at such a rate
that the temperature did not rise above -60C. After 10
minutes gas evolution had ceased and a solution of
5-tertbutyldimethylsilyloxy-UK-86,956 (150mg) o~tained
from Example 4 in anhydrous methylene chloride (Sml) was
added quickly. After stirring for lh at -78C a solution
of diisopropylethylamine (2.Oml) in anhydrous methylene
chloride (5ml) was added. The yellow solution was then
allowed to warm to room temperature. After 30 minutes
diethyl ether (50ml) was added and the solution was
washed quickly with ice cold dilute hydrochloric acid
(SOml). The aqueous layer was extracted with diethyl
ether (20ml) and the combined organic solutions were
washed with water (4 x 20ml), dried over anhydrous sodium
sulphate and evaporated under vacuum to give the crude
product (292mg) as a yellow oil. This material was
purified by column chromatography on silica (Kieselgel
60, 230-400 mesh, Merck)- (30g) eluting with methylene
chloride to give the pure title compound (125mg). The
NMR spectrum was fully consistent with the proposed
structure. ~~ ~~- -
EXAMPLE 6
22-Keto-UK-86,956
5-Tertbutyldimethylsilyloxy-22-keto-UK-86,956
(116mg) from Example S-was dissolved in methanol and p-
toluenesulphonic acid--(S8mg3 was added. The mixture was
stirred at room temperature for lh then a mixture of
saturated aqueous p~tassium hydrogen carbonate solution
and diethyl ether
WO 93/1 l 129 ~ 1 2 2 S 1 4 PCT/EP92/0269,
--13--
was added. The organic layer was separated and the ;~
aqueous layer was extracted with diethyl ether. The
combined organic solution was washed successively with
water and saturated sodium chloride solution, dried over
anhydrous sodium sulphate solution and evaporated under
vacuum to give the crude product as a white solid
(120mg). Final purification was achieved by column ~
chromatography on silica (12g) eluting initially with `-
methylene chloride followed by methylene chloride:
methanol }00:1 to give the pure title compound (lOOmg).
Mass and NMR spectra were fully consistent with the
proposed structure. ~ ;
- EXAMPLE 7
5-MethoxY--22-keto-UK 86 956
To a solution of 22-keto-UK-86,956 from Example 6 1 ~-
(lOOmg) in anhydrous ether (25ml) was added iodomethane -
(2.Oml) and silver (I) oxide (0.5g). The mixture was
stirred at room temperature for 18h then filtered and
evaporated to dryness under vacuum. The crude product -~
was purified by column chromatography on silica (12g) `
eluting initially with methylene chloride followed by
methylene~chloride: methanol 100:1 to give the desired -
product (77.9mg). Final purlfication was achieved by
reverse~phase high pressure liquid chromatography on a
Zorbax- column (21.2 x 250mm, 8~m ODS-silica, Dupont)~
eluting with acetonitrile:water 85:15 to give the pure -~
title compound (30mg). Mass and NMR spectra were fully ~-
consistent with the proposed structure.
`` ` ' - . `
EXAMPLE 8 -
22-MethYlene-UK-86 956 - -
n-Butyllithium (1.6M in hexanes, 0.375ml) was added
dropwise to a stirred suspension of triphenylmethyl-
phosphonium bromide in anhydrous tetrahydrofuran (lOml)
W093/11129 ' 1 2 2 5 1 I PCT/EP9~/0269,
under nitrogen at 0C. After 15 minutes a solution of 5-
tertbutyldimethylsilyloxy-22-keto-UK-86,956 (Example 5)
in anhydrous tetrahydrofuran (5ml) was added. The
mixture was allowed to warm to room temperature and
~stirred for lh before adding saturated ammonium chloride
solution (50ml) and methylene chloride (20ml). The
organic layer was separated and the aqueous layer
extracted with methylene chloride. The combined organic
solution was dried over anhydrous sodium sulphate and
evaporated to dryness under vacuum to give crude 5-
tertbutyldimethylsilyloxy-22-methylene-UK-86,956 (216mg)
which was purified by column chromatography on silica
(20g) eluting with hexane: diethyl ether 2:1. This
material (60mg) was dissolved in methanol (1.5ml)
containing p-toluenesulphonic acid (30mg) and stirred at
40C for 30 minutes. Potassium hydrogen carbonate
solution was then added followed by diethyl ether and the
organic layer was separated, washed with water and
saturated sodium chloride solution and dried over
anhydrous sodium sulphate. The crude product was
purified by column chromatography on silica (lOg) eluting
with methylene chloride:methanol 50:1 to give the pure
title compound (47.5mg). The mass and NMR spectra were
fully consistent with the proposed structure.
The product of Fxample 8 may be converted to the
corresponding 5-oxo and 5-oxi~i~o-compounds by the
methods described in Examples 1 and 2.
,.: ,; -" ,, : -
WO 93/1 1 1 29 212 2 S 1~ PCI /EP92/0269 1
Figure I
a~, "~ " C~ o c
o,o Cll, a~, 2 Clt, ~ C1~, Cl(, .
Il o~ O,~r
o~ clll (~c", .,
o ~I~ -'
I ' ....
Ci1, ItO~ ~C11, CH, HO~ , ,,CII, ~
~ ~ ;o~CI~, 1'~ ~o,~C11, ''
ci~, cl~ 3 CH~ 1I Cl~, Cl~
O 0~, ~:
~a~, O~-CH, ~-~
O~t OCI~
- 14,S ' ' ' ",~,,.
al~ ~CHJ CH, H,C;~,~ ,Cii, --:
~/~o~CI~, =o~CK, I ,
CH, ~ CH, Cit, 7, 6 l~ o O Cit, CH, 1
~$CI~, ~C~
OSiMcz'Bu OH
¦ G . ~i ¦ 1, 2
CH, ~,Cii, CH, 11zC Ci.,
jol~CI~ ~G ~Ci~
C i ~ C ~ ~ ! Ch,~ ~ C!!, C1;,
~c~, ~;J`c~
OCIi, ~Oi~ : - - - --
Reagents
l. Manganese dioxide, diethyl ether.
2. Hydroxylamine hydrochloride, methanol, dioxane, water. - ~-
3. Methyl iodide, silver (I) oxide, diethyl ether. ~ -
4. tert-Butyltimethylsilyl chloride, imidazole, methylene chloride. --~-~ -
5. Oxalyl chloride, dimethyl sulphoxide, diisopropylethylamine,methylene chloride.
6. p-Toluenesulphonic acid, me~hanol.
7. Triphenylphosphonium bromide, n-butyl lithium, tetrahydrofuran.