Note: Descriptions are shown in the official language in which they were submitted.
- ~093/102~ - 212 2 7 ~ 4 PCT/US92/09606
RESTRICTION AMPLIFICATION ASSAY
~ACKGROUND O~ THE INVENTION
Field of the Invention:
The present invention relates to methods,
reagents and kits for the determination o~ the
presence of target nucleotide sequences. In
particular, the present invention relates to a method
for conducting an assay for the presence of a
specific target nucleotide sequence in a sample
containing an unknown guantity of said specific
target nucleotide sequence and to probes for use in
such an assay.
pescription of the Prior Art: ~
In the technology of manipulating genetic
lS material or in evaluating the genetic character of an
organi~m, it is often desirable to ascertain if a
particular gene or part of a gene is present in an
organism or in an extracellular extract of genetic
material from that organism. Since any gene or gene
portion i6, in essence, a specific sequence of
nucleotide bases forming all or part of a
polynucleotide molecule, it is possible to directly
t~st the sample polynucleotide to discover if the
sp~cific sequence of nucleotide bases forming the
gQne i8 presQnt in the cample.
Interest in specific sequences of
nucleotide bases may involve detecting the presence
of pathogens, determining the presence of alleles,
detecting the presence of lesions in a host genome
and detecting a particular mRNA or the modification
of a cellular host, to name only a few illustrative
examples. Genetic diseases such as Huntington
Chorea, muscular dystrophy, phenylketonuria,
thalassemias and sickle cell anemia can be diagnosed
by the analysis of an individual's DNA. Furthermore,
,
WO93/10264 PCT/US92/09~6
~122734 ~ `~
diagnosis or identification of viruses, viroids,
bacteria, fungi, protozoa or any other plant or
animal life form can be determined by hybridization
assays with nucleotide probes.
Nucleic acid detection assays of various
types have been documented in the literature. These
types of assays, and in particular those requiring
detection of polynucleotides, are based on the
purine-pyrimidine base pairing properties of
complamentary nucleic acid strands in DNA-DNA or
DNA-RNA duplexes. This base-pairing process most
frequently occurs through formation of hydrogen bonds
in the pairing of adenosine-thymine (A-T) and
guanosine-cytosine (G-C) bases in double-stranded
DNA; adenosine-uracil base pairs may additionally be
formed by hydrogen bonding in DNA-RNA hybrid
molecules. Base pairing of nucleic acid strands for
determination of the presence or absence of a given
nucleotide sequence involving sample nucleotide
sequences and a probe nucleotide sequence is commoniy
r~ferred to as nucleic acid hybridization or simply
hybridization.
One of the most powerful tools of molecular
biology is the ability to fractionate nucleic acids
and to determine which nucleic acids have sequences
complementary to an array of DNA or RNA molecules.
The Southern blot is a well known method for
transferring electrophoretically fractioned DNA from
a gel matrix to a nitrocellulose solid support by
passive diffusion, followed by hybridization to a
labeled probe. Similar procedures are used for
detecting RNA with minor modifications and this
method is known in the art as the Northern blot. The
use of dried agarose gels as the immobilized phase is
known as the Unblot method. All of these assay
techniques are valuable tools for analyzing mRNA's,
- WO93/102~ - PCT/US92/09606
21~3 l
-3-
clones, genes, fragments, flanking sequences,
repetitive elements and the like.
U.S. Patent No. 4,358,535 describes a
method for detecting pathogens using a target nucleic
acid sequence. The method involves fixing a target
nucleic acid se~uence to an inert support before
hybridization with a radioactively labeled nucleotide
probe. The target nucleic acid sequence is then
determined by detecting the presence of any label on
the inert support.
European Patent Application No. 0 117 440
discloses non-radioactive chemically labeled
.~ polynucleotide probes and methods of using the
probes. The target nucleic acid sequence is also
fixed to a solid support.
U.S. Patent Nos. 4,767,699 and 4,795,701
disclose nucleic acid displacement assays. These
assays utilize two polynucleotides; one
polynucleotide is labeled, and the other
polynucleotide is used to displace the labeled probe
from the target sequence, thereby allowing detection
of the target molecule. These assays use ATP to
detoct whether hybridization has occurred with the
target molecule.
Many of the assays using nucleotide probes
have problems in the detection systems. Sensitivity
o~ the labeled probe and background levels that are
generated during the assay often lead to erroneous
results.
To facilitate more efficient detection of a
nucleic acid sequence from a given sequence of DNA or
RNA a target amplification method may be utilized.
This method, known as PCR, is described in U.S.
Patent No. 4,683,195 and uses a set of primers and a
DNA polymerase to extend the nucleic acid sequence of
the target nucleotide and amplify it for future probe
.
WO 93/10264 PCl`/US92/Og~n~
212273'1
--4--
detection. By amplifying the DNA sequence, the
target nucleotide can be more efficiently detected
with the nucleotide probe. One of the problems
encount~red in this probe assay is contamination of
the reaction medium.
ClassII restriction enzymes are known in
the art for makinq double-stranded scissions at
specific sites within a DNA molecule. These enzymes
are prevalent in bacteria, contain only one type of
subunit and Mg2+ alone is required for DNA cleavage.
DNA cleavage or scission occurs at specific sites
within or adjacent to the enzyme's recognition site.
~' More than 500 restriction enzymes have been isolated
from bacterial strains to date. These restriction
enzymes have been characterized primarily with
respect to their recognition sequences and cleavage
specificity. The majority of restriction enzymes or
endonucleases recognize sequences 4-6 nucleotides in
length, but some have been found with 7-8 base
recognition sites. Most, but not all, reco~nition
sites contain a dyad axis of symmetry and in most
cases all the bases within the site are uniquely
specified. Recognition of the symmetrical sequence
of the hybridized sequences or palindromes is made by
endonucleases. Endonucleases with symmetrical
recognition sites generally cleave symmetrically.
The use of restriction enzymes with their
specific cleavage sites is well recognized in the
art. Usually restriction enzymes are used for the
specific mapping, cloning and characterization of DNA
sequences. However, they have been used in various
nucleotide probe assays. For instance, U.S. Patent
No. 4,683,194 discloses a method for detecting the
presence or absence of a specific restriction site in
a nucleic acid sequence by hybridization with a
nucleic acid probe that is complementary to one
~ WO93/102~ PCT/US92/096~
212273 l
.
--5--
strand of the nucleic acid sequence spanning the
restriction site. The hybridized sequence is then
cleaved with a restriction enzyme and the resulting
cut and uncut oligomers are separated and detected
based on the type of probe label.
A similar concept for detecting a target
nucleotide havinq a half-restriction site is set
forth in U.S. Patent No. 4,725,537. This patent
discloses the use of a restriction endonuclease in a
displacement-type of assay.
Another type of assay that uses the concept
of cleaving a nucleic acid sequence in a nucleotide
.~ probe is disclosed in U.S. Patent No. 4,876,187.
This method is used to detect DNA or RNA sequences by
specifically cleaving the nucleic acid sequence of
the probe in at least one point thereby removing any
reporter molecules not bound to a complementary
target DNA sequence. This assay improves the signal
to noise ratio of the detection system and is a
highly sensitive assay.
Although the aforementioned assays do
provide a method for detecting nucleic acid seguences
in a target molecule, the need still exists for an
assay system that provides very high sensitivity,
ease of detection, less contamination in the assay
medium and ease of operation, while avoiding false
positive results.
The present invention overcomes the
disadvantages associated with the techniques
discussed above by introducing a highly sensitive
detection method for detecting a nucleic acid
sequence through the use of a novel form of
restriction amplification. A second oligonucleotide
may be used in the assay to recycle the cleaved
target sequence of interest thereby amplifying the
labeled and cleaved probe oligonucleotide.
WO93/10264 21 2 2 7 ~ 4 - pcT/us92/os~n6
SUMMARY OF THE INVENTION
Accordingly, an object of the present
invention is to provide a highly sensitive nucleic
acid recycling or probe amplification assay for
detecting a nucleic acid sequence.
Another object of the present invention is
to provide an assay that recycles the target sequence
of interest and thereby amplifies the labeled probe.
The present invention provides a method for
detecting the presence of a nucleic acid seguence which
contains a scissile linkage, that is cleavable by a
cleaving enzyme, in a biological sample, said method
.- comprising the steps of:
(a) providing an oligonucleotide which
comprises a nucleic acid sequence and a scissile
linkage that is substantially complementary to a
nucleic acid target sequence, said oligonucleotide
having a detectable marker attached thereto;
(b) adding a cleaving enzyme to said
reaction mixture which is able to cleave the scissile
linkage if said target sequence and said
oligonucleotide hybridize;
(c) hybridizing said reaction mixture; and
(d) detecting the cleaved detectable
marker in the presence of the uncleaved
oligonucleotide having a detectable marker attached
thereto.
Another embodiment of the present invention
recites:
A method of detecting in a sample the
presence of a nucleic acid sequence which contains a
scissile linkage that is cleavable by a cleaving
enzyme, said method comprising the steps of:
(a) forming a reaction mixture by
denaturing a target nucleic acid sequence cohtaining
a scissile linkage in the presence of an
~0 93/10264 PCI~/US~2/09606
21227~
oligonucleotide which contains a scissile linkage and
is substantially complementary to the nucleic acid
sequence of the target molecule;
(b) adding a cleaving enzyme to said
mixture;
(c) permitting.said reaction mixture to
hybridize whereby the cleaving enzyme will release
the detectable marker from said oligonucleotide
sequence; and
(d) detecting the cleaved detectable marker
in the presence of the uncleaved oligonucleotide
having a detectable marker attached thereto.
~' Still another embodiment of the present .
invention recites: ~
A method for detecting the presence of a
nucleic acid sequence which contains a scissile
linkage that is cleavable by a cleaving enzyme, in a
biological sample, said method comprising the steps
of:
(a) hybridizing a target molecule having a
scissile linkage to a labeled oligonucleotide probe
that has a complementary sequence to said target and
a detectable marker to provide a probe:target duplex;
(b) cleaving said duplex at the scissile
linkage;
(c) recycling the target molecule;
(d) reconstituting a scissile linkage site
on said oligonucleotide; and
(e) detecting said cleaved oligonucleotide
probe in the presence of the uncleaved
oligonucleotide having a detectable marker attached
thereto.
Another embodiment of the present invention
recites:
W093/l026~ PCT/US92io9~"~
2122734
-8-
A method for cleaving a nucleic acid
sequence said method comprising providing an
oligonucleotide ~aving a partially double-stranded
recognition and cleavage site and adding to said
olig~nucleotide a sufficient amount of a ClassII
restriction enzyme in a solvent that alters the
dielectric constant of a reaction mixture and/or
enhances cleavage.
Another object of the present invention is
~0 the use of a partial hairpin oligonucleotide sequence
as the labeled oligonucleotide in the assay of the
present invention using ClassII and ClassIIS
restriction enzymes.
Yet another object of the presence
invention is to provide kits for diagnosis of various
diseases using the above-described method.
BRIEF DESCRIPTION OF THE DRAWINGS
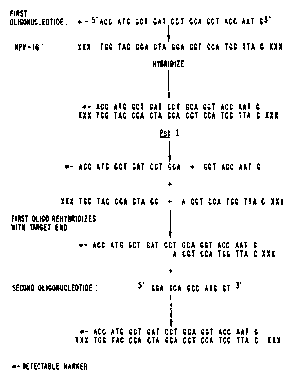
Fig 1. is a schematic representation of the
method of the present invention with a complementary
target sequence.
Fig. 2 i5 a schematic representation of the
method of the present invention with a
non-complementary non-target sequence.
Fig. 3 is an autoradiograph of æeveral
samples run using the second oligonucleotide
according to the present invention.
Fig. 4 is a schematic representation of the
method of the present invention using a partial
hairpin oligonucleotide as the first oligonucleotide
with a ClassIIS restriction enzyme.
Fig. 5 is a schematic rèpresentation of the
method of the present invention using a labeled first
oligonucleotide.
Fig. 6 is an autoradiograph of several
samples run without using the second oligonucleotide
WO93/10264 ~1227~ 1 PCT/US92/09606
according to the present invention.
Fig. 7 is a schematic representation of the
method of the present in~ention using a partial
hairpin oligonucleotide and a ClassII restriction
enzyme.
~ETAILED DESCRIPTION OF THE
PREFERRED EMBOI:)IMENTS OF THE INVENTION
More particularly, the present invention
relates to a method for detecting nucleic acid
sequences of interest in a target molecule. This
assay may or may not use a second oligonucleotide
~, that is able to reconstitute th~ scissile linkage of
~aid target molecule, permitting cleavage of the
first oligonucleotide, thereby recycling the target
sequence and amplifying the amount of the cleaved
first oligonucleotide probe. If the second
oligonucleotide is not used to reconstitute the
scissile linkage, the labeled oligonucleotide is
still available for recycling the target sequence.
It is advantageous to run this method without the use
of the second oligonucleotide, since it is more cost
e~fective.
The term "oligonuc}eotide" as used herein
refers to a compound made up of thé condensation of a
sm~ll number of nucleotides. The exact size of the
oligonucleotide will depend on many factors including
the ultimate function of the use of the
oligonucleotide.
As used herein, the term "scissile linkage"
refers to a site-specific recognition nucleotide
sequence that is cleavable by the use of restriction
enzymes or restriction endonucleases.
As used herein, the terms "restriction
endonucleases" and "restriction enzymes" refer to
enzymes each of which cut double-stranded DNA at or
near a specific recognition nucleotide site.
WO93/10264 PCT/US92/09~
2122734
--10--
As used herein, the term "partially double-
stranded recognition and cleavage site" refers to a
nucleotide sequence that is not fully double-stranded
at the scissile linkage.
As used herein, the term "partial hairpin
sequence" refers to an oligonucleotide sequence that
is able to partially hybridize to itself internally
and has additional single-stranded nucleotide
sequences that are complementary to the target
sequence to be detected.
The æample nucleic acid which may be
employed herein may be derived from àny source(s),
'' including organisms, provided that it contain either
the particular restriction site of interest within a
given nucleic acid sequence when using a ClassII
restriction enzyme or can be cleaved downstream using
a ClassIIS restriction enzyme. Thus, the process may
employ pure DNA which is single or double stranded or
a cDNA or a mixture of nucleic acids. Sources
include, for example, plasmids such as pBR322, cloned
DNA and genomic DNA from any sourc~. Typical sources
can be ~rom biological samples including bacteria,
yeasts, viruses, and higher organisms such as plants,
birds, reptiles and mammals.
Genomic DNA may be prepared from blood,
urine, tissue material such as chorionic villi or
amniotic cells by a variety of techniques tsuch as
those described by Maniatis et al., in Molecular
Clonina, pp. 280-281 (1982). If necessary or desired
to lower the viscosity, the sample of prepared human
DNA to be analyzed may be physically sheared or
digested using a specific restriction endonuclease.
The first oligonucleotide used in the
present invention is a single-stranded
oligonucleotide and has a structure complementary to
the nucleic acid sequence being detected. The probe
~ W093/102~ 2 1 2 2 7 3 ~ - PCT/US92/09606
is usually DNA and may contain an unlimited number of
bases. However, it is preferable that the probe
contains up to about 100 bases, more preferably
between about 10 to 40 bases. The probe may be
obtained from messenger RNA, from a complementary
strand of cDNA obtained by reverse transcription of
messenger RNA with reverse transcriptase or by
cleavage of the genome, conveniently by endonuclease
digestion, followed by cloning of the gene or gene
fragment in accordance with known techniques. See,
for example, Kornberg, DNA Replication, W.H. Freeman
and Co., San FranGisco, (198~) pp. 670-679; So et
al., Infect. Immun.,_21:pp.405-411 (1978). After
isolation and characterization of the desired gene or
DNA fragment, the gene or DNA fragment may be used
for preparation of the probe. The probe may also be
chemically synthesized using an automated synthesizer
such as a MILLIGEN synthesizer. Chemical synthesis
of the probes is the preferred method to obtain the
desired probe for use in the present invention.
For the most part, the oligonucleotide
probe will be labeled with a detectable marker using
an atom, an inorganic radical, radionucleotide, heavy
mQtal, antibody, enzyme, biotin, immunobiotin and the
like. Conveniently, a radioactive label may be
employed. Radioactive labels include 32p, 3H, 14C and
the like. Any radioactive label may be employed
which provides for an adequate signal and has
sufficient half-life. Other labels include ligands,
which can serve as a specific binding pair member to
a labeled antibody, fluorescers, chemiluminescers,
enzymes, antibodies which can serve as a specific
binding pair member for a labeled ligand and the
like. A wide variety of labels have been employed in
immunoassays which can be readily employed in the
present assay. The choice of label will be governed
WO 93~102~ PCT/US92/096n6
21~734
--12--
by the effect of the label on the rate of
hybridization and binding of the probe to the genetic
DNA. It will be necessary that the label provide
sufficient sensitivity to detect the amount of DNA
s available for hybridization. Other considerations
include ease of synthesis of the probe, readily
available instrumentation, the ability to automate,
convenience and the like.
The manner in which the label is bound to
the probe will vary depending upon the nature of the
label. For a radioactive label, a wide variety of
techniques can be employed. Co~monly employed is end
~- labeling with a ~-32P-NTP and T4 polynucleotide
kinase. Alternatively, nucleotides can be
synthesized where one or more of the elements present
are replaced with a radioactive isotope, e.g.,
hydrogen with tritium.
Where other radionucleotide labels are
involved, various linking groups can be employed. A
terminal hydroxyl can be esterified with inorganic
acids. For example, 32p phosphate or 14C organic acids
can be esterified via the terminal hydroxy or
esteri~ied to provide linking groups to the label.
Alternatively, intermediate bases may be substituted
with activatable linking groups which can be linked
to the label.
Ligands and antiligands may be varied
widely. Where a ligand has a natural receptor such
as biotin, thyroxine and cortisol, the ligand can be
used in conjunction with labeled naturally occurring
receptors. Any compound can be used, either haptenic
or antigenic, in combination with an antibody.
Enzymes of interest as labels will
primarily be hydrolases, particularly-esterases and
glycosidases, phosphatases, or oxidoreductases,
particularly peroxidases. Fluorescent compounds
-`WO93/10264 ~ 1 2 2 7 3 4 PCT/US92/09606
-13-
include fluorescein and its derivatives, rhodamine
and its derivatives, dansyl, umbelliferone and the
like. Chemiluminescers include luciferin and
2,3-dihydrophthalazinediones, i.e., luminol.
Yet another method for labeling and
detecting the nucleotide probe is disclosed in U.S.
Patent No. 4,868,103, in which an energy transfer
results in the generation of bathochromic and/or
delayed fluorescence emission. Fluorescence
radiation, emitted from a first energy emitter (El)
i8 absorbed by a second energy emitter (~). The
second emitter emits fluorescence radiation of a
~' longer wavelength than the first energy emitter. (E~)
and (~) must be within a proximate distance of each
other 50 that the energy emitted by (E~) can be
absorbed by (~) and (~) emits fluorescent energy at
a longer wavelength.
Any means for labeling and detecting the
labeled probe can be used in the present invention.
It i8 preferable, however, that the label is bound at
a distance away from the scissile linkage and is
situated at the 3 prime or 5 prime end of the first
oligonucleotide.
Besides having a label on the first
oligonucleotide the first oligonucleotide must also
contain a scissile linkage that is complementary to
the target molecule of interest. The scissile
linkage must be readily cleavable by restriction
enzymes or other means after hybridization has
occurred when ClassII restriction enzymes are used.
However, the use of CIassIIS restriction enzymes
requires the recognition sequence, but cleaves
downstream from this sequence.
In a preferred embodiment of the present
invention, the first oligonucleotide is immobilized
to a solid support. Any solid support in which an
.
W093/102~ PCT/US92/09~
2122734
-14-
oligonucleotide can be affixed may be utilized in the
present invention. Examples of these supports
include glass, test tubes, microtiter plates, nylon,
beads, agarose beads, magnetic beads, glass beads,
teflon, polystyrene beads, photodetectable chips and
the like. It is well known within the art how t~
attach the oligonucleotides to a solid support by
derivatizing one of the ends of the oligonucleotide
with a carboxyl or amino group. Another example o~
attachment is taking an avidin-bound bead and attach
the oligonucleotide via biotin. If a solid support
i8 used, it is advantageous to have the label at the
~' opposite end of the oligonucleotide to which the
support is attached.
The restriction enzymes are well known in
the art and have specific recognition sites. The
restriction enzymes that can be used in the present
invention are ClassII restriction enzymes and include
Aat II, Acc II, Acc III, Aha III, Alu I, Aoc I, Apa
I, ApaL I, Ava I, Ava II, Bal I, Bam HI, Bcl I, Bgl
II, BssH II, BstE lI, Cla I, Dra I, Eco52 I, Eco RI,
Eco RII, Eco RV, Fsp I, Nae II, Hha I, Hind III, Hp~ :
I, Hpa II, Rpn I, Rsp I, Nci I, Mst II, Nae I, Pst I,
Pvu I, Xba I and the like. There are more than 500
different restriction endonucleases which can be used
and the present invention contemplates the use of any
of these enzymes. The criteria for choosing the
restriction enzyme is based upon t~e recognition site
of the target nucleotide molecule, the first
oligonucleotide probe, and the properties of the
enzyme. The first oligonucleotide probe containing
the scissile linkage must be complementary to the
target nucleotide's recognition or scissile linkage
site which upon hybridization of these
oligonucleotides and addition of the restriction
enzyme, the duplex will be cleaved.
. :
~-WO 93/10264 2 1 ~ 2 7 3 4 PCI~US92/09606
--15--
In a preferred embodiment of the present
invention, an oligonucleotide containing a partial
hairpin sequence may be utilized as the
oligonucleotide, provided that this oligonucleotide
5 is labeled. By definition, the partial hairpin
oligonucleotide sequence should be able to hybridize
internally to itself thereby forming a loop and
contains additional single-stranded oligonucleotide
sequences that can hybridize to the target sequence
10 c~f interest. The hairpin loop can reside either at
the 5 prime or 3 prime end of the oligonucleotide and
is labeled appropriately as discussed above. In
con~unction with the use of a partial hairpin
oligonucleotide sequence, ClassIIS restriction
15 endonucleases may be used to cleave double-stranded
DNA at precise distances from their recognition
sites. Examples of ClassIIS restriction
endonucleases include BbvI, BbvII, BinI, FokI, HgaI,
HphI, MboII, MnlI, SfaNI, TaqII, TthlllII, HinGuI and
20 the like. In particular, see for example, Szybalski,
Gene, 40:169-173 (1985) and Podhajska et al Gene,
40:175-182 (1985). ClassIIS restriction enzymes may
also be used without partial hairpin oligonucleotide
~equences in the practice of the present invention.
25 Figures 4 and 7 are illustrative of how the hairpin
oligonucleotide can be used in the method of the
present invention.
Restriction enzymes or endonucleases are
relatively stable proteins. Their purification to
30 homoqeneity is often not necessary. All restriction
enzymes require a cofactor for cleavage such as Mg2+
or Mn2+ and are most active in the pH range of 7.2 to
7.6. Typically for enzyme cleavage an appropriate
buffer system is used. These buffer systems vary
35 among the restriction enzymes used to cleave a
hybridized duplex. Therefore, in addition to the DNA
2122734
PCT/US92/09606
-16-
substrate and restriction enzyme, most reaction solutions
will contain TRIS, i.e., tris(hydroxymethyl)aminomethane
buffer, Mg2+, NaCl, 2-mercaptoethanol and bovine serum
albumin (ssA). The predominant difference among the
restriction enzymes is their dependence on ionic strength.
To maximize cleavage efficiencies the buffering systems are
varied among the restriction enzymes. For instance, if a
Hlnd III restriction enzyme is used in the assay, a buffer
containing 50 mM NaCl, 25 mM TRIS-HCl, pH 7.7, 10 mM MgCl2,
10 mM ~-mercaptoethanol and 100 ~tml ssA is used to
maximize the cleavage efficiency. If an Eco RI enzyme is
~' used the buffering system contains 50 mM NaCl, 100 mM
TRIS-HCl, pH 7.5, 5 mM MgCl2 and 100 ~ml BSA. Therefore,
the present invention contemplates the use of different
buffering sysSems, which vary according to the restriction
enzyme used in the assay.
Restriction enzymes may also vary in temperature
optima. Most cleavages are performed at 37C, but a few
endonucleases such as Sma I prefer lower incubation
temperatures, and several, mainly those isolated from
thermophiles such as Tag I, require much higher
temperatures. Therefore, the reaction temperature in the
`present invention is chosen taking into consideration the
re~triction enzyme used in the assay.
Be~ides cleaving the double-stranded molecule formed
after hybridization with restriction enzymes, any other
method available can be used in the cleavage process. For
instance, certain chemicals may be used to cleave
doublestranded complexes at specific sites.
The present invention may also use a second
oligonucleotide which reconstitutes with the cleaved target
molecule of interest such that hybridization
.
~iJB~TlTUTE SHEET
WO93/1026~ 2 1 2 2 7 ~ ~I PCT~US92/09606
-17-
with the probe oligonucleotide occurs within the
assay. The second oligonucleotide acts to recycle
the target sequence of interest and thereby amplifies
the amount of cleaved probe for detection purposes.
This second oligonucleotide is a single-stranded
nucleotide that is aomplementary to an end of the
first probe oligonucleotide such as the 3 prime end
or 5 prime end. The second oligonucleotide may be
derived via processes similar to the synthesis of the
first oligonucleotide, as described above.
Besides being complementary to the 3 prime
or 5 prime end of the first oligonucleotide, the
econd oligonucleotide should contain bases such as a
3 prime or 5 prime base that will reconstitute the
lS target molecule at the site that was cleaved by the
restriction enzyme. After reconstituting with the
cleaved end of the target molecule, hybridization
once again occurs with the first labeled
oligonucleotide and the process "cycles" once again
and releases the labeled probe into the reaction
media. The second oligonucleotide may contain up to
lO0 bases, preferably between about lO to 20 bases,
more preferably about 14 bases. Figures l and 2 are
illustrative examples of how the present invention
works.
In a preferred embodiment of the present
~nvention, the second oligonucleotide is not utilized
and therefore reconstitution of the cleaved end of
the target molecule does not occur. Even though a
second oligonucleotide may not be used in the
preferred embodiment of the present invention,
nonetheless, the cleaved detectable marker does
accumulate. Thus, positive target sequences are
easily detectable. Contrary to teachin~s in the
prior art, it has been discovered by the present
inventor that the use of ClassII restriction enzymes
WO93/10264 PCr/US9~/09~
21~273~
-18-
requires only a partially double-stranded recognition
and cleavage site in order to cleave the hybridized
oligonucleotide under varying chemical conditions.
Examples of the various chemicals (which are
generally diluted in appropriate solvents) that can
be used to enhance the cleavage of the partially
double-stranded recognition and cleavage site in the
present invention include divalent cations such as
cobalt, magnesium, zinc and the like, glycerine,
dimethylsulfoxide (DMSO), dimethylformamide (GMF),
formamide, ethylene glycol and the like. See, George
et al, Journal of Bioloaical ChemistrY, 55, pp.
~- 6521-6524 ~1980). The concentrations of these diluted
chemicals, that may be used to vary the dielectric
constant and/or enhance cleavage of the restriction
site, may vary depending on the assay conditions.
Usually between 10% to 50% by volume of the
aforementioned chemicals may be used; preferably
between 10% to 30% by volume. Figures 5 and 6 are
illustrative examples of how the present invention
works without the use of the second oligonucleotide.
The assay is initiated by denaturing the
sample target molecule to form a single-stranded
molQcule. The denaturation of the target molecule or
substrate is generally performed by boiling. In
addition, the substrate, first oligonucleotide,
second oligonucleotide buffer and distilled water may
be mixed together and boiled for 5 to 10 minutes.
The mixture is then allowed to cool to room
temperature. After cooling, the restriction enzyme
may then be added to initiate the reaction.
When using the ClassII restriction enzymes
in conjunction with the partial hairpin loop
oligonucleotide, the procedure differs from that
described above. Basically, the reaction requires
that the ClassII restriction enzyme be cut initially
~093/10264 PCT/US92/09606
212273 l
--19--
in the reaction mixture comprising the labeled
oligonucleotide, then the reaction mixture is
denatured, hybridized and then recut again. Figure 7
illustrates the use of ClassII xestriction enzymes
using a partial hairpin oligonucleotide.
The temperature at which the assay is run
may vary according to the restriction enzyme used,
the length of the oligonucleotide probe and the G + C
content of the oligonucleotides present in the assay.
Suggs et al, in "Developmental Biology Using Purified
Genes," (D.D. Brown, ed.), p.683. Academic Press,
New York, 1981., developed an empirical formula to
determine the appropriate hybridization temperature
based on the temperature at which an oligonucleotide
DNA complex is dissociated. The formula derived was:
T~ = 2 (number of A+T residues) + 4 (number of G+C
residues). This empirical formula can be used to
estimate the reaction temperature, but one must also
take into account the restriction enzyme used in the
assay. As discussed above, many restriction enzymes
are active at 37C, bu~ others may require higher or
lower temperatures. The tem~erature of the reaction
is chosen such that optimal rates of hybridization,
as well as cleavage of the restriction enzyme occurs.
The reaction time may vary depending upon
the concentration of the sequence of interest, the
stringency, the length of the complementary first
oligonucleotide probe sequence, the restriction
enzyme used and the like. Enough time should be
provided to permit amplification of the probe by
recycling the target sequence of interest. Usually
thP assay is run from 1 to 3 hours, more preferably
for two hours.
After the reaction has run, it is stopped by
placing an aliquot of the reaction mixture into a
polyacrylamide gel loading buffer. The sample is
W093/102~ - PCT/US92/09~
212273~
-20-
then electrophoresed and an autoradiograph i~ taken
of the electrophoresed gel. One can then proceed to
quantitate the amount of probe generated on a
scanning densitometer, if desired.
In order to further illustrate the present
invention and the advantages thereof, the following
specific examples are given it being understood that
same are intended as illustrative and in nowise
limitative.
~ ~Ynthesis of Oliqonucleotides
Two oligonucleotides may be used in the
~' restriction amplification process. The synthesis of
the first oligonucleotide having the sequence 5ACC
ATG GCT GAT CCT GCA GGT ACC AAT G3 (28 mer) and the
second oligonualeotide having the sequence 5GGA TCA
GCC ATG G~ (18mer) were prepared by phosphoramidite
chemistry and were subsequently purified by anion
exchange HPLC. The oligonucleotides synthesized by
the phosphoramidite approach contained a free 5'-OH.
The partial hairpin oligonucleotide can be
synthesized in the same manner as described above.
Labelina of First Oliaonucleotide
The first oligonucleotide contained a 5'-OH
group which was labeled by transfer of the
[32P]phosphate from t~2P]ATP using polynucleotide
kinase. The labeling reaction was carried out by
dissolving each oligonucleotide in distilled water.
The concentration of each oligonucleotide dissolved
in the water was 0.5 x 1o-5 molar. The reaction was
performed using 1 ~1 of the first oligonucleotide
(0.5 x 105 molar), 5 ~1 of kinase buffer which
contained O.5 M TRIS-HCI at pH 7.6, 1 mM spermidine,
50 mM dithiothreitol, 1 mM EDTA and 0.1 M MgCl2, 30.5
ml distilled water, 12.5 ~1 32P-ATP (0.125 millicuFies
-`VO93/102~ PCT/US92/09606
212273ll
-21-
or 3000 curiPs/~ mole) and 1 ~l of polynucleotide
kinase (10 units). The abové-mentioned reaction
ingredients were combined and incubated at 37C for 1
hour. The reaction was terminated by heating the
reacted mixture to 80C for 10 minutes.
The partial hairpin oligonucleotide can be
labeled in the same manner as the first
oligonucleotide.
The labeled first oligonucleotide and
partial hairpin oligonucleotide prepared by this
method are stable for at least one week when stored
at -20C.
~,
HPv 16 Substra~e
The HPV16-DNA virus was isolated from a
cervical lesion by methods known in the art and
cloned into pBR322 using standard techniques. The
cloned DNA was isolated by a modified procedure
described by H.C. ~irnboim and J. Daly, Nucleic Acids
Res., 7, 1513 (1979).
The cells were grown overnight in 5 ml of
LB broth containing 100 ~g/ml ampicillin at 37C with
vigorous shaking. 1.5 ml of the culture was
transferred to a 1.5 ml centrifuge tube and
centrifuged at 10,000 X g for 1 minute. The
supernatant was then carefully removed leaving the
pellet as dry as possible. The cells were
resuspended by vortexing in 100 ~l of an ice-cold
solution containing 50 mM glucose, 10 mM EDTA and 25
mM TRIS-HCL at pH 8Ø The resuspended cells were
allowed to incubate for 5 minutes at room
temperature. 20 ~l of a freshly prepared solution
containing 0.2 N NAOH, 1% sodium dodecyl sulfate
(SDS) was added to the cells and mixed by inversion.
The cells were further incubated for 5 minutes at
OC.
WO 93~10264 PCI`/US92/09~'`.
21~2734
-22-
After incubation, 150 ~1 of ice-cold
potassium acetate at pH 4-8 (prepared by adding 11.5
ml glacial acetic acid and 28.5 ml of water to 60 ml
of 5 M potassium acetate) was added and the mixture
s was inverted for lo seccnds and incubated at ooc for
5 minutes.
The mixture was then centrifuged for 5
minutes at 10,000 X g, and the supernatant was
transferred to another tube. The supernatant was
centrifuged again at 10,000 X g for 5 minutes, and
the supernatant was transferred to another fresh test
tube. RNase A was then added to the supernatant,
,- ha~ing a final concentration of 20 ~g/ml. The
reaction was incubated at 37C for 20 minutes.
After incubation, an equal volume of
phenol: chloroform (1:1, saturated with 50 mM
TRIS-HCL at pH 8.0, 100 mM NaCl, 1 mM EDTA) was added
and the mixture was vortexed for 30 seconds and
centrifuged at 10,000 g for 30 seconds. The aqueous
phase was then transferred to a fresh test tube.
2.5 volumes of ethanol was then added to
the aqueous phase and mixed. The mixture was
incubated at -70C for 5 minutes. The mixture was
centrifuged at lO,OOO g for 5 minutes and the
supernatant was removed. The pellet was then rinsed
by Adding 1 ml of prechilled 70% ethanol and mixed
briefly. The mixture was then centrifuged for 1
minute. The supernatant was then removed, and the
pellet was dried under vacuum.
The DNA obtained from this procedure was
dissolved in 20 ~1 of deionized water.
Restriction Am~lification Assay Usina Two
Oliaonucleotides
The restriction amplification assay was
initiated by combining the HPV 16 substrate, the
W093/102~ - PCT/US92tO9606
212273'1
-23-
first oligonucleotide labeled probe, the first
unlabeled oligonucleotide, a Pst I buffer, and
deionized water. In this RAMP assay two controls
were simultaneously run with the samples. Two
different temperatures of 32C and 37C were utilized
in this example.
Stock solutions of the reaction components
were first prepared. The oligonucleotides were
diluted to 0.5 x 105 molar solutions. Similarly, the
second oligonucleotide was also diluted to create a
0.5 x 10-5 sto~k solution. The HPV 16 stock was
diluted with distilled water to form a 100 ~g/ml
stock solution. The Pst I restriction enzyme stock
#olution contained 50 units/~l.
~5 A specific aliquot was taken from these
stock solutions for each assay. The total reaction
volume was SO ~1. Each reaction assay contained 1 ~1
(100 ng) of HPV 16, 2 ~1 of the labeled first
oligonucleotide, 2 ~1 of unlabeled first
oligonucleotide, 4 ~1 of the second oligonucleotide,
5 ~1 of Pst I buffer containing 100 mM NaCl, 10 mM
TRIS-HCl at pH 7.7, 10 mM MgCl2, 1 mM DTT and 100
~g/ml BSA. 5 ~1 of Pst I was used. Distilled water
wa8 ~dded in varying quantities such that the final
volume in each assay tube was equal to 50 ~1.
Five assays were run in separate tubes.
The first tube (#1) contained 2 ~1 of the first
oligonucleotide labeled with 32p, 2 ~1 of cold
unlabeled first oligonucleotide, 4 ~1 of the second
oligonucleotide, 5 ~1 Pst I buffer and 32 ~1
distilled water. No HPV 16 was added to the first
assay mixture.
The second assay tube (#2) contained 1 ~1
(100 ~g) HPV-16, 2 ~1 of the first oligonucleotide
labeled with 32p, 2 ~1 of cold unlabeled first
nucleotide, 4 ~1 of the second oligonucleotide, 5 ~1
WO93/102~ PCT/US92/09~^~
21~2734
-24-
of Pst I buffer and 31 ~l of distilled water~
The third assay tube (#3) contained 1 ~l of
HPV 16 (100 ~g), 2 ~1 of the first 32p
oligonucleotide, 2 ~l of cold first oligonucleotide,
5 ~l of Pst I buffer, and 35 ~l of distilled water.
The fourth assay tube (#4) contained 1 ~1
of HPV 16 (100 ~g), 2 ~l of the first 32p
oligonucleotide, 2 ~1 of cold unlabeled first
oligonucleotide, 5 ~l of Pst I buffer, and
35 ~l of distilled water.
The fifth assay tube (#5) contained 1 ~1 of
HPV 16 (lO0 ~g), 2 ~l of labeled32P first
oligonucleotide, 2 ~l of cold unlabeled first
oligonucleotide, 4 ~l of second oligonucleotide, 5 ~1
lS of Pst I buffer, and 31 ~l of distilled water.
Prior to addition of Pst I, the five assay
tubes described above were boiIed for S to lO minutes
and then cooled at room temperature for approximately
10 minutes.
After cooling the samples, the samples were
placed in an incubator; assay tube numbers 1, 4, and
S, discussed above, were incubated at 37C. Assay
tubes numbers 2 and 3 were incubatéd at 32C. To
initiate the assay, S ~1 of Pst I enzyme was added to
each assay tube. The assay was run for
2 hours.
After incubation, the reaction was stopped
by adding 10 ~l of a solution containing 80%
formamide, 15 mM TRIS-HCl at pH 7.6, 1 mM EDTA, 0.1%
w/v bromophenol blue and 0.1% w/v xylene cyanoIe FF
to each assay tube. Each tube was then heated for 5
minutes at 65C and cooled rapidly.
Polyacrylamide gels were prepared by making
a stock solution containing 30% acrylamide (l9:1
acrylamide:bis), l9 grams of acrylamide, l gram of
N,N'-methylenebisacrylamide and enough deionized
~093/10264 PCT/US92/09606
~1227~ 1
-25-
water to dilute the solution to 67 ml total volume.
A concentrated solution of TBE buffer was prepared by
diluting to 1 liter, 108 grams of TRIS base,
55 grams of boric acid and 9.3 grams of Disodium
EDTA-2H20. The pH of the concentrated TBE buffer
should be adjusted to 8.3, if appropriate. 48 grams
of ultra-pure grade urea (8 M) was added to the stock
solution containing the 30~ acrylamide. lO ml of TBE
buffer was added and the urea was dissolved in this
solution. 50 ~1 (per 100 ml of acryla~ide solution)
of N,N,N',N'-tetramethylethylenediamine(TEMED) and 1
ml (per 100 ml) of 10% ammonium persulfate was added
and the solution was mixed well. The gel was poured
between two glass plates into the electrophoresis
apparatus and the comb was inserted immediately.
20 ~1 of each sample was loaded into the
polymerized gel and a running buffer was added that
contained 0.089 M TRIS-borate, pH 8.3 and 0.025 M
Disodium EDTA. The samples were electrophoresed at
80 volts for 4 hours.
The gels were then wrapped in a plastic
folder placed next to Xodak X-OMAT AR film and
exposed for 10 minutes.
Figure 3 is an illustrati~e example of the
25 autoradiograph of the present invention. From the ;
autoradiograph, one can easily determine the
formation o~ the substrate by the presence of the 28
mer oligonucleotide band and the 18 mer
oligonucleotide band. Lanes 2 and 5 illustrate a
doublet pattern when the substrate is present in the
reaction media at two different temperatures. Lanes
3 and 4 are indicative of the presence of only the
labeled first oligonucleotides without the second
oligonucleotide used to recycle the substrate. Lane 1
is indicative of the pattern obtained when both the
first and second oIigonucleotides are present in the
WO g3/10264 PCl /US92/09~ ~
2122734
-26-
assay mixture, but no substrate (i.e., HPV 16 ) was
present in the reaction.
Restriction Am~lification Assay usinq
One Oli~onucleotide
The restriction amplification assay was
initiated by combining the HPV 16 substrate, the
first oligonucleotide labeled probe, Pst I buffer,
deionized water and varied concentrations of
glycerine. In this RAMP assay, a control was run
with 5% glycerine.
Stock solutions of the reaction co~ponents
were first prepared. The oligonucleotide was diluted
' to a 0.5 x 105 molar solution. The HPV 16 stock w~s
diluted with distilled water to form a loo ~g/ml
stock solution. The Pst I restriction enzyme stock
solution contained 50 units/~l.
A specific aliquot was taken from these
stock solutions for each assay. The total reaction
volume was 50 ~1. Each reaction assay contained 1 ~1
`~ 20 (100 ng) of HPV 16, 2 ~1 of the labeled first
oligonucleotide, 2 ~1 o~ unlabeled first
. ~
oligonucleotide, 5 ~1 of Pst I buffer containing 100
mM NaCl, 10 mM TRIS-HCl at pH 7.7, 10 mM MgCl2, 1 mM
DTT and 100 ~g/ml BSA. 5 ~1 of Pst I was used.
Distilled water was added in varying quantities such
that the final volume in each assay tube was equal to
50 ~1, after addition of the glycerine in varied
quantities.
Six assays were run in separate tubes. The
first tube (#1) contained 2 ~1 of the first
oligonucleotide labeled with 32p, 2 ~1 of c~ld
unlabeled first oligonucleotide, 5 ~1 Pst I buffer, 1
~1 HPV 16, 2.5 ~1 glycerine (5% by volume gIycerine)
and 32.5 ~1 distilled water. Tube #l was used as a
control.
The second assay tube (#2) contained 1 ~1
.
W093/102~4 2 1 2 2 7 3 I PCT/US92/09606
-27-
(100 ~g) HPV-16, 2 ~1 of the first oligonucleotide
labeled with 32p ~ 2 ~1 of cold unlabeled first
nucleotide, 5 ~l of Pst I buffer, 5 ~1 glycerine (10%
by volume glycerine) and 30 ~1 of distilled water.
The third assay tube (#3) contained l ~l of
HPV 16 (100 ~g), 2 ~l of the first 32p
oligonucleotide, 2 ~1 of cold first oligonucleotide,
5 ~1 of Pst I buffer, 10 ~1 glycerine (20% by volume
glycerine) and 25 ~1 of distilled water.
The fourth assay tube (#4) contained 1 ~1
of HPV 16 (lOo ~g), 2 ~1 of the first 32p
oligonucleotide, 2 ~1 of cold unlabeled first
oligonucleotide, S ~1 of Pst I buffer,
15 ~1 glycerine (30% by volume glycerine) and 20 ~1
of distilled water.
The fifth assay tube (#5) contained 1 ~1 of
HPV 16 (100 ~g), 2 ~1 of labeled32P first
oligonucleotide, 2 ~1 of cold unlabeled first
ol~gonucleotide, 5 ~1 of Pst I buffer, 20 ~1
glycerine (40% by volume glycerine) and 15 ~1 of
distilled water.
The sixth assay tube (#6) contained 1 ~1 of
HPV 16 (100 ~g), 2 ~1 of the first 32p
oligonucleotide, 2 ~1 of cold unlabeled first
oligonucleotide, 5 ~1 of Pst I buffer,
25 ~1 glycerine (50% by volume glycerine) and 10 ~1
of d~stilled water.
Prior to addition of Pst I, the six assay
tubes described above were boiled for 5 to 10 minutes
and then cooled at room temperature for approximately
10 minutes.
After cooling the samples, the samples were
placed in an incubator at 37C. To initiate the
assay, 5 ~1 of Pst I enzyme was added to each assay
tube. The assay was run for 2 hours.
After incubation, the reaction was stopped
w093/10~ PCT/US92/09~<;
2122734
-28-
by adding lo ~1 of a solution containing 80%
formamide, 15 mM TRIS-HCl at pH 7.6, 1 mM EDTA, 0.1%
w/v bromophenol blue and 0.1% w/v xylene cyanole FF
to each assay tube. Each tube was then heated for 5
minutes at 65C and cooled rapidly.
Polyacrylamide gels were prepared by making
a stock solution containing 30% acrylamide (19:1
acrylamide:bis), 19 grams of acrylamide, 1 gram of
N,N'-methylenebisacryl&mide and enough deionized
water to dilute the solution to 67 ml total volume.
A concentrated solution of TBE buffer was prepared by
diluting to 1 liter, 108 grams of TRIS base, 55 grams
~' of boric acid and 9.3 grams of Disodium EDTA-2H20.
The pH of the concentrated TBE buffer should be
adjusted to 8.3, if appropriate. 48 grams of
ultra-pure grade urea (8 M) was added to the stock
solution containing the 30% acrylamide.
10 ml of TBE buffer was added and the urea was
dissolved in this solution. 50 ~1 (per 100 ml of
acrylamide solution) of N,N,N',N'-tetramethylethyl-
enediamine(TEMED) and 1 ml (per 100 ml) of 10%
ammonium persulfate was added and the solution was
mixed well. The gel was poured between two glass
plates into the electrophoresis apparatus and the
comb was inserted immediately.
20 ~1 of each sample was loaded into the
polymerized gel and a running buffer was added that
contained 0.089 M TRIS-borate, pH 8.3 and 0.025 M
Disodium EDTA. The samples were electrophoresed at
80 volts for 4 hours.
The gels were then wrapped in a plastic
folder placed next to Kodak X-OMAT AR film and
exposed for 10 minutes.
Figure 6 is an illustrative example of the
autoradiograph of the present invention using the
met~od of the present invention. From the autoradio-
WO93/10264 PCT/US92/09606
212273~
-29-
graph, one can easily determine the presence of the
target sequence by the appearance of the 18 mer band
derived from the 28 mer oligonucleotide. It can be seen
that the use of glycerine in the assay with only one
labeled oligonucleotide probe is feasible in the range
between 10% to 40% by volume glycerine. A more
preferred range is about 40% by volume glycerine.
Restriction Am~lification Assay
USING A PARTIAL HAIRPIN LOOP OLIGONUCLEOTI~E
The restriction amplification assay is
initiated by combining the HPV 16 substrate, the
partially hairpin oligonucleotide labeled probe, ~st I
buffer and deionized water. Stock solutions of -the
reaction components are first prepared. The
oligonucleotide is diluted to a 0.5 x 10'5 molar
solution. The HPV 16 stock is diluted with distilled
water to form a 100 ~g/ml stock solution. The Pst I
restriction enzyme stock solution contains 50 units/~l.
A specific aliquot is taken from these stock
solutions for each assay. The total reaction volume is
50 ~1. Each reaction assay contained 1 ~1 (100 ng) of
HPV 16, 2 ~1 of the labeled partial hairpin
~ligonucleotide, 2 ~1 of unlabeled partial hairpin
oligonucleotide, 5 ~1 of Pst I buffer containing 100 mM
NaCl, 10 mM TRIS-HCl at pH 7.7, 10 mM MgCl2, 1 mM DTT
and 100 ~g/ml BSA. 5 ~1 of Pst I is used. Distilled
water was added in varying quantities such that the
final volume in each assay tube is equal to 50 ~1.
Four assays are run in separate tubes. The
first tube (#1) contains 2 ~1 of the partial hairpin
oligonucleotide labeled with 32p, 2 ~1 of cold unlabeled
partial hairpin oligonucleotide, 5 ~1 Pst I buffer, 0
~1 HPV 16 and 36 ~1 distilled water.
The second tube (#2) contains 2 ~1 of the
partial hairpin oligonucleotide labeled with 32p, 2 ~1
of cold unlabeled partial hairpin oligonucleotide, 5 ~1
W093/102~ PCT/US92/09~,~
2I 22 734
--30--
Pst I buffer, 1 ~1 of a control DNA which does not
contain the HPV 16 sequence and 35 ~1 distilled water.
The third tube (#3) contains 2 ~1 of the
partial hairpin oligonucleotide labeled with 32p, 2 ~1
of cold unlabeled partial hairpin oligonucleotide, 5 ~1
Pst I buffer, 1 ~1 of HPV 16 and 35 ~1 distilled water.
The fourth tube (#4) contains 2 ~1 of the
partial hairpin oligonucleotide labeled with 32p, 2 ~1
of cold unlabeled partial hairpin oligonucleotide, 5 ~1
lOPst I buffer, 5 ~1 of HPV 16 and 31 ~1 distilled water.
Pst I is then added to the four assay tubes
described above and the tubes are bQiled for 5 to lo
minutes and then cooled at room temperature for
approximately 10 minutes.
15After cooling the samples, the samples are
placed in an incubator at 37C. To initiate the assay,
5 ~1 of Pst I enzyme is added to each assay tube. The
assay is run for 2 hours.
After incubation, the reaction is stopped by
20adding 10 ~1 of a solution containing 80% formamide, 15
mM TRIS-HCl at pH 7.6, 1 mM EDTA, 0.1% w/v bromophenol
blue and 0.1~ w/v xylene cyanole FF to each assay tube.
Each tube i8 then heated for 5 minutes at 65C and
cooled rapidly.
25Polyacrylamide gels are prepared by making a
stock solution containing 30% acrylamide (19:1
acrylamide:bis), 19 grams of acrylamide, 1 gram of
N,N'-methylenebisacrylamide and enough deionized water
to dilute the solution to 67 ml total volume. A
concentrated solution of TBE buffer is prepared by
diluting to 1 liter, 108 grams of TRIS base, 55 grams
of boric acid and 9.3 grams of Disodium EDTA-2H20. The
pH of the concentrated TBE buffer should be adjusted to
8.3, if appropriate. 48 grams of ultra-pure grade urea
(8 M) is added to the stock solution containing the 30%
acrylamide. 10 ml of TBE buffer is added and the urea
WO93/102~ PCT/US92/09606
212 2 ~ 3 4
-31-
is dissolved in this solution. 50 ~1 (per 100 ml of
acrylamide solution) of N,N,N',N'-tetramethylethylene-
diamine(TEMED) and 1 ml (per 100 ml) of 10% ammonium
persulfate is added and the solution is mixed well.
The gel is poured between two glass plates into the
electrophoresis apparatus and the comb is inserted im-
mediately.
20 ~1 of each sample is loaded into the
polymerized gel and a running buffer is added that
contained 0.089 M TRIS-borate, pH 8.3 and 0.025 M
D~sodium EDTA. The samples ar~ electrophoresed at 80
volts for 4 hours.
~` The gels are then wrapped in a plastic folder
placed next to Kodak X-OMAT AR film and exposed for 10
minutes.
Restriction Amplification Assav usina FokI
The restriction -amplification assay is
initiated by combining the HPV 16 substrate, the
partial hairpin oligonucleotide labeled pr~be, FokI
buffer containing 10 mM TRIS-HCl, pH 7.4; 50 mM NaCl;
1 mM EDTA; and S mM MgCli and deionized water. Stock
solutions of the reaction components are first
prepared. The oligonucleotide is diluted to a 0.5 x 105
molar solution. The HPV 16 stock is diluted with
distilled water to form a 100 ~g/ml stock solution.
The FokI restriction enzyme stock solution contains 50
units/~l.
A specific aliquot is taken from these stock
solutions for each assay. The total reaction volume is
50 ~1. Each reaction assay contained 1 ~1 (100 ng) of
HPV 16, 2 ~1 of the labeled partial hairpin
oligonucleotide, 2 ~1 of unlabeled partial hairpin
~ligonucleotide, 5 ~1 of FokI buffer, 5 ~1 of FokI is
used. Distilled water was added in varying quantities
such that the final volume in each assay tube is equal
to 50 ~1.
WO 93~10264 PCr/US92/09~
2122~34
--32--
Four assays are run in separate tubes. The
first tube (#l) contains 2 ~1 of the partial hairpin
oligonucleotide labeled with 32p, 2 ~1 of cold unlabeled
partial hairpin oligonucleotide, 5 ~1 FokI buffer, 0 ~1
5 HPV 16 and 36 ~1 distilled water.
The second tube (#2) contains 2 ~1 of the
partial hairpin oligonucleotide labeled with 32p, 2 ~1
of cold unlabeled partial hairpin oligonucleotide, 5 ~1
FokI buffer, 1 ~1 of a control DNA which does not
10contain the HPV 16 sequence and 35 ~1 disti}led water.
The third tube (#3) contains 2 ~1 of the
partial hairpin oligonucleotide labeled with 32p, 2 ~1
~' of cold unlabeled partial hairpin oligonucleotide, 5 ~1
FokI buffer, 1 ~1 of HPV 16 and 35 ~1 distilled water.
lSThe fourth tube (#4) contains 2 ~1 of the
partial hairpin oligonucleotide labeled with 32p, 2 ~1
of cold unlabeled partial hairpin oligonucleotide, 5 ~1
FokI buffer, S ~1 of HPV 16 and 31 ~1 distilled water.
The four assay tubes described above are
boiled for 5 to 10 minutes and then cooled at room
temperature for approximately 10 minutes.
After cooling the samples, the samples are
placed in an incubator at 37C. To initiate the assay,
5 ~1 of FokI enzyme is added to each assay tube. The
assay is run for 2 hours.
After incubation, the reaction is stopped by
adding 10 ~1 of a solution containing 80% formamide, lS
mM TRIS-HCl at pH 7.6, 1 mM EDTA, 0.1% w/v bromophenol
blue and 0.1% w/v xylene cyanole FF to each assay tube.
Each tube is then heated for S minutes at 65OC and
cooled rapidly.
Polyacrylamide gels are prepared by making a
stock solution containing 30% acrylamide (19:1
acrylamide:bis), 19 grams of acrylamide, 1 gram of
N,N'-methylenebisacrylamide and enough deionized water
to dilute the solution to 67 ml total volume. A
~093/10264 2 1 2 ~ 7 3 I PCT/US9~/09606
-33-
concentrated solution of TBE buffer is prepared by
diluting to l liter, 108 grams of TRIS base, 55 grams
of boric acid and g.3 grams of Disodium EDTA 2H~0. The
pH of the concentrated TBE buffer should be adjusted to
5 8.3, if appropriate. 48 grams of ultra-pure grade urea
(8 M) is added to the stock solution containing the 30%
acrylamide. lO ml of TBE buffer is added and the urea
is dissolved in this solution. 50 ~l (per lO0 ml of
acrylamide solution) of N, N, N ', N '-tetramethylethylene-
diamine(TEMED) and 1 ml (per lO0 ml) of 10% ammoniumpersulfate is added and the solution is mixed well.
The gel i~ poured between two glass plates into the
~' electrophoresis apparatus and the comb is inserted im-
mediately.
~5 20 ~l of each sample is loaded into the
polymerized gel and a running buffer is added that
aontained 0.089 M TRIS-borate, pH 8.3 and 0.025 M
Disodium EDTA. The samples are electrophoresed at 80
volts for 4 hours.
The gels are then wrapped in a plastic folder
placed next to Kodak X-OMAT AR film and exposed for lO
minut~s.
While the invention has been described in
terms of various preferred embodiments, the skilled
artisan will appreciate that various mo~ifications,
substitutions, omissions, and changes may be made
without departing from the spirit thereof.
Accordingly, it is intended that the scope of the
present invention be limited solely by the scope of the
following claims, including equivalents thereof.