Note: Descriptions are shown in the official language in which they were submitted.
WO 93/10767 ~ ~ ~ ~~ ~ PCT/DS92/01009
Ors1 administration form for peptide pharmaceutical
substances, in particular insulin
The invention relates to an oral administration
form for peptide medicaments, which contains at least one
peptide pharmaceutical substance, dispersed in a matrix
of gelatin or a gelatin derivative, in addition to
pharmaceutically customary excipients and auxiliaries.
The invention furthermore relates to a process for the
preparation of such an oral administration form.
In the highly industrialized countries, it can be
assumed that about 2-3% of the population exhibit the
diabetes syndrome. For the effective treatment of this
disorder with its very important symptoms hyperglycemia,
polyuria, glucosuria, as well as hyperlipidemia, we have
to rely today, as before, on the exogenous supply of
insulin, in spite of the enormous variety of pharma-
ceutical developments. Even the oral antidiabetics of the
sulfonylurea type, which are only indicated if the
endogenous production of insulin is at least still
partially maintained, at most offer a limited width of
application.
The administration of insulin is carried out to
the greatest possible extent by injection (parenteral
administration). Other administration routes, e.g. nasal,
pulmonary, rectal or, especially, oral administration are
presently under test. However, it has still not become
public that an appropriate preparation could be ready to
be put on the market. On the contrary, we find ourselves
still at the stage of informative investigations. As is
known, injections are associated with disadvantages.
Thus, for example, lipodystrophy or other foreign body
reactions can occur at the administration site. Problems
with the handling of injection syringes are particularly
to be expected with very young and relatively old
patients. In these groups of patients, a regularly
required injection must often be carried out by a person
looking after them. It is therefore obvious that this
effort does not particularly promote patient compliance.
212~~84
- 2 -
The optimum, simplest and safest use of pharma-
ceutical substances, however, is oral administration, for
example of tablets, capsules or beverage solutions. In
the case of peptide pharmaceutical substances, such as
e.g. insulin, marked difficulties result, however,
because these are inactivated to the greatest part by
enzymatic degradation after release in the gastro-
intestinal tract (GIT; stomach or small intestine) even
before absorption. Enzymatic degradation in the stomach
or small intestinal fluid or on the mucosa threatens to
lower the bioavailability of peptide pharmaceutical
substances, particularly insulin, to a minimum. Addi-
tionally, the mechanism of absorption by means of passive
transport is largely lacking for peptide pharmaceutical
substances. This is based, on the one hand, on the
molecular size, because the exclusion limit for passive
transport is assumed to be about 500 Daltons. On the
other hand, substance-specific properties, such as
hydrophilicity (low distribution coefficient), self-
association to form larger units or binding to
constituents of the gastro-intestinal tract make
absorption difficult. In addition, absorption is also
made difficult if negative charge formed as a result of
dissociation of functional active compound groups leads
to electrostatic repulsion at the glycocalyx, the
negatively charged glycoprotein layer on which the lipid
double layer lies. Absorption of peptide pharamaceutical
substances, however, is of extraordinary importance
despite this if it is wished to manage a parenteral
supply successfully.
It has already been proposed to administer
insulin encapsulated in liposomes. In these investiga-
tions, however, it did not appear possible to determine
the amount of insulin absorbed quantitatively. These
experiments can therefore probably only offer rough
guiding values. The use of liposomes is moreover accom-
panied, as is known, by difficulties both in the prepara-
tion and in the storage of appropriate pharmaceutical
forms.
z~2~~~
- 3 -
More recently, useful starts were reported in
order to be able to administer insulin orally. Of
particular interest in this case are pharmaceutical forms
which are stomach- and small intestine-resistant, and
only release the insulin after reaching the colon, which
is low in peptidase.
It has likewise already been proposed to
introduce insulin into a soft gelatin capsule together
with an absorption accelerator (EP Appl. 0 225 189), the
capsule being provided with a coating which is intended
to dissolve only in the colon, and the insulin being
released together with said absorption accelerator. The
use of absorption accelerators (e. g. certain surfactants
or salicylic acid derivatives) in the GIT, however,
appears only to have limited effectiveness because of the
high dilution which takes place there. The very large
amount which is employed for this reason, which makes up
up to 50~ of the capsule contents, can even cause harmful
side effects. Additionally, the toxic side effects of
surfactants under certain circumstances, particularly in
the action on mucous membranes, are adequately known.
Legitimate doubts, however, may be attached to the use of
salicylic acid derivatives as pharmaceutically utilizable
auxiliaries.
US Patent 4,849,405 proposes the embedding of
insulin in a liquid, aqueous two-phase system based on a
coacervate. As is known, however, coacervates do not
behave in an uncritical manner during preparation.
Accurate monitoring of the process parameters is unavoid-
able. The reproducibility of the process is therefore to
be put in question. The insulin embedded in this
coacervate should be a rapidly releasing pharmaceutical
form, the preparation being present in liquid form
(emulsion). Legitimate doubts, however, may be registered
for the storage stability of this system. The coacervate
can be converted into a storage-stable, delayed-release
pharmaceutical form by heat treatment (curing) or by
crosslinking with aldehydes (e.g. glutaraldehyde), and
subsequent separation of the microcapsules by filtration
2~2~~8~
- 4 -
and drying. In these processes, however, a loss of
activity of the insulin as a result of chemical changes
is not to be excluded. It is known that insulin is both
sensitive to heat and is presumably hardly inert to
aldehydes. Additionally, in the process indicated in US
Patent 4,849,405 a high loss of insulin during encap-
sulation is generally to be expected, which is reflected
with certainty in the preparation costs. Nothing is
reported about the yield of the encapsulated insulin.
The present invention is now based on the object
of preparing a medicament for oral administration of
peptide pharmaceutical substances, in particular insulin,
which overcomes the problems in this type of administra-
tion outlined in the prior art and thus makes possible a
safe and effective treatment.
This object is achieved according to the
invention by a medicament which contains peptide
pharmaceutical substances, in particular insulin, in a
gelatin matrix in addition to customary pharmaceutical
excipients and auxiliaries. This object is furthermore
achieved by the peptide pharmaceutical substance, in
particular insulin, being associated, as a charged
molecule, with an oppositely charged gelatin by
adsorptive charge compensation (pseudocoacervate).
Finally, the object is also achieved by the use of a
system of peptide medicaments, in particular insulin,
associated by adsorptive charge compensation (pseudoco-
acervate) to an oppositely charged gelatin, for the
production of medicaments which are suitable for the safe
and effective treatment of the diabetes syndrome.
According to the present invention, for the first
time an oral administration foran for peptide medicaments
is proposed which can be prepared and used in practice.
An advantage is that the release system according to the
invention is suitable both for rapid release and for
delayed release or a combination of rapid release and
delayed release. Furthermore, the known low absorption
rate of peptide pharmaceutical substances, in particular
insulin, is only significantly increased in the GIT by
2~~~~8~
_ - 5 -
the present invention.
In particular, the present invention makes
available an oral administration form for peptide
medicaments, containing at least one peptide
pharmaceutical substance in a matrix which, in addition
to pharmaceutically customary excipients and auxiliaries,
contains at least one hydrophilic macromolecule, selected
from the group consisting of: gelatin, fractionated
gelatin, collagen hydrolyzates and gelatin derivatives;
and also their mixtures.
Inter alia, the present invention additionally
makes available a process for the production of an oral
administration form for peptide medicaments, a powdered
macromolecule/pharmaceutical substance mixture being
prepared using at least one hydrophilic macromolecule
selected from the group consisting of: gelatin,
fractionated gelatin, collagen hydrolyzates and gelatin
derivatives; and also their mixtures, and the peptide
medicament and the mixture being compressed.
Moreover, the present invention provides a
process for the preparation of a slowly dissolving oral
administration form for peptide medicaments, which
comprises
a) selecting a hydrophilic macromolecule selected
from the group consisting of: gelatin,
fractionated gelatin and gelatin derivatives; and
also their mixtures, with a maximum in the
molecular weight distribution in the range from
about 9.5 x 10' - 106 D,
b) converting the hydrophilic macromolecule into the
sol form with water at a temperature below the
inactivation temperature of the peptide,
c) adjusting the pH of the sol to a value between
that of the IEP of the hydrophilic macromolecule
and that of the peptide,
d) adding the peptide to the macromolecule sol in
dissolved or undissolved form,
e) removing the water,
f) pressing the powder obtained to give the
21 25284
- 6 -
administration form by customary processes.
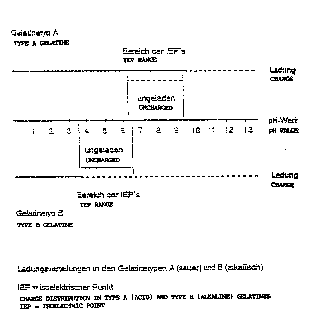
Fig. 1 shows a schematic representation of the
adjustable states of charge of gelatin as a function of the
pH and IEP, it being possible for the IEP to be between 3.5
and 9.5, depending on the manner of preparation. Below pH
3.5, nearly all types of gelatin are positively charged. In
the basic range above pH 9.5, all types of gelatin are
negatively charged.
The International (PCT) Patent Applications W093/10768
and W093/10764 by the same applicant of the same date
...
disclose related subject mater.
Other International (PCT) patent applications of
ALFATEC Pharma GmbH, where appropriate also of the PAZ
Arzneimittelentwicklungsgesellschaft mbH, of the same date
relate to the immediate-effect form of 2-arylpropionic acid
derivatives (WO 93/10761), the sustained-release form of
dihydropyridine derivatives (WO 93/10770), the immediate-
effect form of S- and R-ibuprofen (WO 93/10762), the
sustained-release form of S- and R-ibuprofen (WO 93/10760),
the immediate-effect form of S- and R-flurbiprofen (WO
93/10766), the sustained-release form of S- and R-
flurbiprofen (WO 93/10771) and the sustained-release form
of indolylacetic acid derivatives (WO 93/10769).
a
y21 25284
In the International (PCT) Patent Applications WO 93/10768, the
preparation of colloidally disperse systems (nanosols) containing
gelatin is described, in WO 93/10769 the preparation of
delayed and constant (0 order) active compound-releasing,
oral pharmaceutical forms based on gelatin. Peptide
pharmaceutical substances, in particular insulin, can be
brought into an oral administration form according to the
processes described in the patent applications mentioned.
The combination of these administration forms, however,
can be looked at as particularly advantageous in
principle.
Insulin is a peptide pharmaceutical substance
which consists of 51 amino acids which are arranged in
two chains (A and B chains) . Insulin is very sensitive
with respect to external influences. Thus heat- and
alkali-sensitivity, sensitivity to oxidizing and reducing
agents, and also strongly acid-reacting substances is
known. On account of its isoelectric point (IEP) of 5.3 -
5.4, however, insulin is adequately soluble and also
adequately stable in weakly acidi:e medium at pH 3-4, and
also in weakly alkaline uiedium at pH 7-8. In the pH
ranges indicated, the molecule, however, is positively
charged (pH less than IEP) or negatively charged (pH
greater than IEP).
In a particular embodiment of the present
invention which is claimed in the dependent claims,
peptide pharmaceutical substances, in particular insulin,
are present in a form in which the peptide pharmaceutical
substance is associated in charged and simultaneously
dissolved form with an oppositely charged gelatin or a
gelatin derivative by adsorptive charge compensation
(pseudocoacervate).
In the acidic range below pH 5.3 - 5.4, where the
insulin molecule is positively charged, only negatively
charged gelatin is suitable for this purpose. Apart from
type B gelatin, certain molecular fractions of this
gelatin, so-called fractionated gelatin, and also gelatin
derivatives, in particular succinylated gelatin, can also
t
- g -
be used. In the pH range indicated, these exhibit
identical behavior to Type B gelatin. As one, the Type B
is suitable which must have an IEP of less than 5.3 - 5.4
and is thus negatively charged at pHs above its IEP.
Conversely, insulin is negatively charged at pHs of
greater than 5.3 - 5.4. This negative charge can be
analogously compensated only by type A gelatin which is
positively charged at pHs of greater than 5.3 - 5.4 up to
pH about 9.5.
With a pharmaceutical form prepared according to
this principle, the type B gelatin is particularly to be
preferred. To be precise, the following has surprisingly
been shown:
After reaching the large intestine or colon,
where physiological pHs of about 6 - 7.5 prevail and the
release of insulin from the pharmaceutical form begins,
the covering gelatin particles protect the insulin
molecule effectively from enzymatic degradation by
peptidases. At the same time, an additional effect of the
gelatin further makes itself advantageously noticeable.
The high molecular weight components of the gelatin
(preferably from a molecular weight of about 10' D) form
spherically shaped networks. Diffusion of the degrading
enzymes through these networks is further additionally
made difficult so that the insulin molecule is even
better protected. On the other hand, these gelatin
particles or networks show a good adhesion to mucous
membrane surfaces, which ensures optimum conditions for
absorption. As a result of the pH shift to pHs of greater
than 6, the insulin is now no longer present in
positively charged form, but its charge is reversed and
it can thus escape from the "complex" (pseudocoacervate)
with the gelatin, whose charge shifts more and more into
the negative range. This "charge inversion process" can
additionally be accelerated according to the invention by
buffer substances (e. g. disodium hydrogen phosphate)
being present in the gelatin matrix whose buffer capacity
maximum happens to be at pHs of greater than 6. However,
it is to be stressed that in this case it is not a true
2125284
_ g -
inclusion complex, such as, for example, in the case of
cyclodextrins. The release of insulin in each case takes
place without the customary advance equilibrium, for
example, with cyclodextrin compounds. Optimum conditions
for the absorption of insulin in the gastrointestinal
tract are thus provided.
In order to utilize this principle for an oral
pharmaceutical form of insulin or alternatively other
pharmaceutical substances more effectively, the pharma-
ceutical form described in the present invention can
preferably be a two-layer tablet, or even better a
layered tablet. The tablet is enteric-coated using
suitable film coatings, e.g. Eudragits~ (Rohm-Pharma,
Germany). Eudragit S, mixtures of Eudragit S and
Eudragit RS types, or mixtures of Eudragit S, Eudragit L
and Eudragit RS types have proven particularly suitable.
These film coatings have the advantage that they are
water-impermeable up to dissolution and only begin to
dissolve at pHs from about 7, i.e. after the
pharmaceutical form is already in the lower intestinal
sections or already in the colon. Up to this point in
time, the pharmaceutical form and the active compound
contained (insulin) are thus additionally effectively
protected from enzymatic degradation by the enzymes of
the digestive fluid.
The first layer or the coat of said pharma-
ceutical form is then built up such that a relatively
slow (delayed) release of active compound takes place
within about 4 h. The second layer, on the other hand. or
the core of the layered tablet is constructed such that
a rapid (non-delayed) release of active compound takes
place. This combination of ia~ediate-effect and delayed-
release form in a single tablet has the advantage that
the rapid release of insulin only takes place in any case
after reaching the colon, where, as is known, only a low-
peptidase medium is to be found.
Thus a continuous supply of the body with insulin
is always provided so that an adjustment to the insulin
requirement of a patient after food absorption can be
- 10 -
easily performed. In this manner, according to the
invention independence from the injection of insulin can
be achieved and patient compliance can be decisively
increased.
Apart from insulin, under which regular insulin,
insulin complexed with zinc or alternatively globin-zinc-
insulin is understood, other peptide pharmaceutical
substances which can be enzymatically inactivated in the
gastrointestinal tract are also suitable for the present
invention, such as octreotide, desmopressin, vasopressin,
triptorelin, endogenous peptide hormones, such as
gonadotropin-releasing hormone, somatotropin-releasing
hormone, corticotropin-releasing hormone or thyrotropin-
releasing hormone, polypeptide antibiotics, cyclosporin,
buserelin, calcitonin, gonadorelin, lysoprenin, oxytocin,
protirelin, hirudin, glucagon, encephalin or adreno-
corticotropic hormone. Substances for the treatment of
AIDS (renin antagonists), treatment of hypertension
(renin antagonists, enalapril, captopril, antibiotics
which are derived from amino acids, penicillins
(ampicillin), cephalosporins, (cefalexin), carbapenems
(thienamycin), interferons (alpha-interferon) and
vaccines.
The present invention additionally proposes a
simple process for the production of the pharanaceutical
forms described.
According to International (PCT) Application
81AL2739, a relatively viscous gelatin having an appro-
priate bloom number is first selected, with a maximum in
the molecular weight distribution in the range
9.5 x 10' - 106, preferably type B with an IEP in the
range from 3.5 to about 5.3, which is completely freed
from foreign ions. The gelatin, which can be used for the
delaying layer or the coat of the pharmaceutical form
according to the invention, is first converted into the
sol form with water at a temperature which is above 37°C
and below the temperature at which the insulin is already
"inactivated". The gelatin concentrations are customarily
0.1-20% (percentages by weight), but preferably 0.1-5%.
~12~~8~
_. - 11 -
The pH of the sol is adjusted by addition of acid or base
to a value which is above the IEP of the gelatin used and
below the IEP of the insulin employed. By this means,
sufficient negative charge is generated on the gelatin
molecules in order to cause the adsorptive charge compen-
sation (pseudocoacervate) with the insulin molecules.
Customarily, the insulin, e.g. 50-500 I.U., can be added
directly to the gelatin sol and dissolved therein with
stirring or added to the gelatin sol already in dissolved
form. The progressive charge compensation (pseudoco
acervate formation) can be monitored here, for example,
by a simple conductivity measurement of the system. It
may be necessary to readjust the pH of the system to the
prespecified value if this should alter during the
preparation process.
The water can then be removed by known processes,
such as e.g. spray- or freeze-drying, the required state
of the system being fixed in dry form.
Completely analogously to this, a second, dry
system is to be prepared which forms the basis for the
second layer or the core of the pharmaceutical form
according to the invention. The gelatin of the same type
and with identical IEP used here preferably possesses a
maximum in the molecular weight distribution of below
105, such that a non-delayed release can be guaranteed.
The dried powders can then be pressed to give
customary tablets or to give two-layer or layered tablets
with the addition of customary pharmaceutical auxili-
aries, such as, for example, fillers, buffer substances,
flow regulators, lubricants and mold release agents.
Surprisingly, the tablets according to the invention are
distinguished by high breaking strength and low
friability.
That layer of the two-layer tablet which is not
intended to be delayed-release can be separately prepared
and preisolated by coating with one of the abovementioned
film-forming agents.
The customary tablets, two-layer or layered
tablets prepared according to the invention are then
- 12 -
coated by customary coating processes (for example in a
fluidized bed, in a coating pan or the like) with the
film-forming agents mentioned. Eudragit S is particularly
advantageously used, or mixtures of Eudragit S with
Eudragit RS, e.g. in a mixing ratio of 3:2.
In principle, the procedures and process variants
mentioned in the abovementioned German Patent Application
P 41 40 195.6 of ALFATEC-Pharma GmbH "Pharmazeutisch
applizierbares Nanosol and Verfahren zu seiner Herstel-
lung" (Pharmaceutically administrable nanosol and process
for its preparation), which are referred to once more in
the following, are also particularly suitable for the
preparation of the administration form according to the
invention:
Several processes for the preparation of the
nanosols are proposed. These are an exemplary, incomplete
list. The person skilled in the art can independently
work out further variants in the context of the present
invention on the basis of his expert knowledge:
Process I
This can be used if the pharmaceutical substance
is soluble in a mixture of:
a water-miscible organic solvent and water, or
several water-miscible organic solvents and water:
a) a gelatin selected in the preliminary tests is
converted into sol form with water;
b) the pH of the solution found in the preliminary
tests is adjusted;
c) one or more water-miscible, organic solvent(s),
preferably ethanol, isopropanol or methanol, is/are
added to this solution;
d) the pharmaceutical substance is added to the solu-
tion in solid form and dissolved;
~12~284
.. - 13 -
e) the organic solvent (s) is/are removed, preferably by
evaporating in vacuo; the nanosol is formed during
the course of this;
f) the colloidally disperse solution is then dried,
preferably by spray- or freeze-drying.
The organic solvent has the aim of dissolving the
pharmaceutical substance and also changes the hydration
shell of the gelatin molecules.
Process II
This embodiment can be used if the pharmaceutical
substance is an acid or a base whose salt is soluble in
water:
a) a gelatin selected in the preliminary tests is
converted into the sol form with HsO;
b) a pH is set which enables formation of the salt of
the pharmaceutical substance;
c) the pharmaceutical substance is dissolved in the
gelatin sol with salt formation;
d) by addition of alcohol or similar organic solvents,
the hydration shell of the gelatin molecules can be
loosened;
e) by addition of a suitable amount of acid or base,
the pH is set which leads to the formation of the
isoionic point (IIP) and the nanosol results;
f) the colloidally disperse solution is dried as in
process I.
Stage d) is optional, but preferred.
_.. - 14 -
Process III
This embodiment can be used if the pharmaceutical
substance is a neutral substance:
a) a gelatin sol is prepared as described in (1) a) and
b) .
b) a second solution is prepared from a water-miscible
organic solvent, preferably ethanol, methanol,
isopropanol or acetone and the pharmaceutical
substance.
c) the two solutions are combined.
d) the organic solvent is removed and the colloidally
disperse solution is dried.
Process IV
a) As described in (I) a) and b).
b) A colloidally disperse system is briefly formed with
the pharmaceutical substance, but without gelatin,
in a second solution.
c) The solution obtained in (b) is continuously
combined with the gelatin solution.
In step (IV) c), the continuous mixing of the
solutions described in (IV) a) and b) can be controlled
in a time-dependent manner by on-line measurement of the
particle size using a suitable process, such as e.g. by
laser light scattering (BI-FOQELS On-line Particle
Sizer) . It is thus possible to continuously set a desired
particle size.
All processes mentioned are also suitable for
collagen hydrolyzates and gelatin derivatives and can be
applied without problems on an industrial scale.
2125284
- 15 -
The essential steps can largely run in an auto-
mated manner, it also being possible to carry out
processes I to III continuously. In the case of the
immediate-effect form for 2-arylpropionic acid
derivatives, variants No. II and III may be mentioned as
preferably suitable processes.
All gelatins, gelatin derivatives, collagen
hydrolyzates and fractionated gelatin, and also their
mixtures are suitable for the medicaments according to
the invention. Types of gelatin which have an isoelectric
point (IEP) described according to the invention which is
not commercially available can be prepared according to
Examples I to III from the abovementioned German Patent
Application.
Compared with commercially available products,
the use of gelatin which has been prepared in a special
manner leads to nanosols described according to the
invention having increased stability.
Examples of the preparation of grades of gelatin
particularly suitable according to the invention are
given below.
Examples of the preparation of particularly suitable
types of gelatin according to the iaventioa with
isoelectric points of 3.5 to 9.5
Example I:
Process for obtaining an IEP of 7.5 to 9.5
Collagen-containing starting material such as
e.g. pig skins is treated for 12 to 20 hours with an
aqueous solution of a 0.45 N mineral acid, preferably
sulfuric acid, in a liquor ratio of 1 . 1. The excess of
acid is then removed by washing several times, it being
possible to use sodium hydrogen carbonate to shorten the
process. The extraction of the stock-rich material is
carried out using hot water at 55 - 80°C at a pH of 2.5
to 4.5. At pHs below 3.5, an IEP of 8.5 to 9.5 can be
achieved, at pHs above 3.5, the IEP is 7 to 8.5. In this
manner, various IEPs from 7 to 9.5 can be achieved as a
2125284
- 16 -
direct function of the pH during the extraction.
After the extraction process step, the aqueous
solution is neutralized and worked up as customary.
Depending on the temperature selected during the
extraction, types of gelatin having high to medium
molecular weight distributions can furthermore be
obtained by this process.
At temperatures of 50 - 55°C, particularly highly
viscous and high-bloom grades are obtained. Types of
gelatin having low molecular weight or cold water-soluble
gelatins can be obtained by controlled degradation with
collagenases.
Example II:
Process for achieving an I$P of 4 to 7.5
The collagen-containing starting material is
first washed to remove foreign substances. and comminuted,
and then homogeneously rendered alkaline by addition of
magnesite, sodium hydroxide solution or calcium hydroxide
by thorough mixing in the liquor ratio 1 . 1.2. The
material pretreated in this way is briefly hydrolyzed by
pressure hydrolysis at 1.01 x 105 to 2.02 x 105 Pa and a
pH of the aqueous solution of 8 - 14. After hydrolysis,
it is immediately neutralized and the still hot aqueous
gelatin solution is filtered, deionized, concentrated and
dried in the usual manner.
If a weakly basic hydrolyzing agent such as
magnesite is taken, an IEP of 6 to 7.5 is obtained if the
reaction is carried out at 1.01 x 105 Pa. IEPs of 5 to 6
are obtained when using a dilute milk of lime suspension,
and when using 0.005 to 0.1 N sodium hydroxide solution
IEPs of 4 to 5 can be achieved.
Types of gelatin having a low degree of
racemization and a low peptide content can be obtained
with pressure ratios of 1.01 x 105 Pa and residence times
of at most 10 min.
Medium to low molecular weight types to cold
water-soluble types are afforded by correspondingly
longer residence times.
2125284
- 17 -
Example III:
P=ocess for achieving as I$P of 3.5 to 6
Collagen-containing starting material, preferably
split or ossein, is subjected after the starting wash to
treatment with a high-speed usher. In this case, two
process variants in the liquor ratio 1 . 1.3 offer
themselves, which either use a saturated milk of lime
suspension or a 0.1 to 1 N sodium hydroxide solution.
then using a milk of lime suspension, the raw
material is hydrolyzed for a maximum of 3 to 4 weeks with
continuous agitation. The material is then neutralized by
addition of acid and washed several times. Further
working up follows in the usual manner. IEPs of 4 to 6
can be set in this manner.
When using sodium hydroxide solution, the usher
process can be shortened again, the material, depending
on the degree of coamninution, being hydrolyzed even after
6 - 12 hours at concentrations of 1 N sodium hydroxide
solution. Neutralization is carried out using equimolar
amounts of mineral acid and the neutral salts are removed
by washing several times or by deionizing the aqueous
gelatin solution obtained in the extraction. In this
process variant, IEPs of 3.5 to 5 can be obtained.
Particularly low-peptide types of gelatin are
obtained with a short residence time in the usher. Types
of gelatin with high to average molecular weight distri
bution (M = 10' - 10' D) can thus be obtained.
Low molecular weight to cold water-soluble types
of gelatin can be obtained by thermal degradation or
enzymatically.
Depending on the gelatin preparation procedure
(extent of depredation of native collagen and acidic or
alkaline hydrolysis process), gelatin of Type A or Type B
has a characteristic molecular weight spectrum or
molecular weight distribution. Table 1 indicates the
molecular weight distributions of various types of
gelatin or of collagen hydrolyzates, and the percentage
content (frequency) of individual molecular weight
ranges.
2125284
- 18 -
Table 1
Molecular weight distribution of various known
types of gelatin or of known collagen hydrolyzates
MolecularNativeG)slatia(~elatiaCollagenCollagenCollagen8laetia
Mare CollagenType Type hydroly:atehydroly:atehydrolysatehydrolysate
Die- H a
tributiont t ~k Cielita fielita Oelita G)alita
(kD) CollagelCollagelSol fislastia
71 H C
>360 100 18.0 18.0 0 0 0 0
285 0 7.0 9.0 0 0 0 0
1 145-237 0 20.0 34.0 1.0 1.5 0 0
0
95 0 26.0 11.0 0 0 0 0
95-50 0 16.3 13.4 2.6 4.0 1.1 0
50-20 0 7.4 9.1 18.0 14.5 0.3 0
20-10 0 3.9 3.8 43.0 31.5 3.7 0.2
1 10-5 0 3.0 3.0 15.4 20.0 12.2 5.2
5
5-2 0 0 0 6.0 14.0 26.0 93.9
2-1 0 0 0 7.0 8.0 23.0 0
c 1 0 0 0 6.5 7.0 34.0 0
MW 360 165 185 12-18 12-18 3 2-3
20 The predominance of an individual range compared
with the other molecular weight ranges of the same
gelatin can be seen clearly in the individual columns.
This range is thus the maximum in the molecular weight
distribution (it is 95 kD e.g. for the Type B gelatin
25 shown in the figure). The concept of the "maximum of the
molecular weight distribution", however, is to be
separated strictly from the concept of the "average mean
molecular weight". This mean value is 165 kD for the
gelatin of Type B mentioned.
30 Customary pharmaceutical auxiliaries and/or other
macromolecules, if they are technologically necessary,
can be added to the nanosols according to the invention
in the liquid or dry state.
For example, an addition of polyvinylpyrrolidone
35 in the quantitative ratio gelatin to polyvinylpyrrolidone
in the range from 5 . 1 to 500 . 1 may be suitable.
The technological processing properties of an
immediate-effect form within the meaning of the inven-
tion, which is processed e.g. to give tablets or is to be
40 lyophilized, can be improved by addition of low molecular
weight types of polyvinylpyrrolidone in the range 10:1 to
50:1 without the stability of the nanosols being
2125284
- 19 -
adversely affected.
The preferred preparation processes, procedures
and names in the following examples relate as follows to
the German Patent Application "Pharmazeutisch applizier-
bares Nanosol and Verfahren zu seiner Herstellung"
(Pharmaceutically administrable nanosol and process for
its preparation) (P 41 40 195.6) or the abovementioned
processes and examples:
Nanosol preparation . Processes II and III
Gelatin preparation . Examples I to III
Preliminary test . see the following description:
Preliminary test:
As already mentioned at the beginning and as is
evident from Fig. l, the absolute, maximum possible net
charge of an individual gelatin molecule depends mainly
on the number of free COOH and NH, groups and the pH of
the solution. As Type A, B, collagen hydrolyzates or
gelatin derivatives differ in the number of free COON
groups, their maximum possible net charge is thus also
different. With gelatin derivatives, the state of charge
can additionally depend on the type of modification.
When carrying out the process according to the
invention, the suitable gelatin and the suitable pH are
selected in a preliminary test.
First, a working pH range suited to the physico-
chemical properties of the pharmaceutical substance is
selected. Physicochemical properties of the pharmaceu-
tical substance to be taken into account in particular
are: the solubility (in organic solvents or water), its
properties as an acid, base or neutral substance and its
stability to acids and alkali solutions.
In a first rapid test, it is determined what
charge the precipitated particles have. This results,
taking into account the working pH range, in the choice
of a suitable type of gelatin. If the particles are, for
example, negatively charged, a gelatin is picked which is
positively charged under the given pH conditions. This
?125284
- 20 -
rapid test for the determination of the particle charge
has the advantages that it can be carried out without a
great outlay in terms of apparatus and time. A time-
consuming and inaccurate zeta potential measurement can
thus be dispensed with entirely.
In many cases, it will be adequate for this rapid
test to convert two commercially available Type A and B
gelatins with an IEP of 9.5 or 3.5 respectively and with
peptide contents of < 30 % and a bloom number of 200,
which are additionally designated as standard gelatins,
into the sol form at a pH of 6 (5 % strength aqueous
solution) and to dissolve the pharmaceutical substance in
a water-miscible solvent, such as e.g. ethanol,
isopropanol or acetone, and in each case to mix
homogeneously with the gelatin solutions. At the same
dose of the pharmaceutical substance, in the case of the
gelatin which is unsuitable in its state of charge a
colloidal system will either not form or immediately
become unstable or the pharmaceutical substance will
flocculate. If the resulting particles are negatively
charged, they are stabilized earlier by the gelatin
solution of Type A, which is positively charged at a pH
of 6, than by the solution containing Type B gelatin; in
contrast, in this case Type H either will form no
colloidal system or the system will immediately
destabilize. The flocculation of the particles can be
monitored e.g. via a simple turbidity measurement.
In this rapid test, the working pH range must be
taken into account in each case. Other gelatins can also
be selected as a standard, but they must be selected in
their IEP such that they carry an opposite net charge at
this pH (see also Fig. 1). In most cases, said standard
Type A and B gelatins are adequate for this rapid test.
Starting from the result of the preliminary
experiment, the optimum conditions for the formation of
the nanosols are determined by stepwise variation of the
IEP by use of appropriate types of gelatin and of the pH
of the solution in relatively small ranges (e.g. 0.1 pH
steps), i.e. the stability optimum which is characterized
- 21 -
by the isoionic point (IIP) must be found in order to
guarantee an adequate stability for the pharmaceutical
applications mentioned.
It can definitely be the case that a stability of
the nanosols which is acceptable within the meaning of
the invention is already found in a relatively narrow pH
range (about 0.5 units) around the isoionic point, so an
adjustment of this point itself is not absolutely
necessary. On the other hand, several gelatins can also
lead to the same, stable results. Thus, for example
(Example 5) with the oral antidiabetic glibenclamide in
the case of a Type B gelatin with an IEP of 5.5 the
stability optimum can be at a pH of 3.2, while in the
case of a Type B gelatin with an IEP of 3.8 the stability
optimum is at a pH of 2.2.
Characterized by a stability maximum, in both
cases the isoionic point was reached (the dependence of
the net charge on the pH and the IEP must be non-linear,
as it is given by the pKa value of the COOH or NH3+ groups
present).
The two systems described for the delayed and
non-delayed release of insulin can also be shaped by
suitable granulation methods to give granules or conven-
tional pellets. Such granules or pellets can be filled,
for example, into hard gelatin capsules. Granules,
pellets and hard gelatin capsules are customarily coated
with the same film-forming agents as indicated for the
tablet according to the invention in order to achieve at
least a resistance to gastric juice. In this manner, for
example, mixtures of rapid- and delayed-release pellets
can be realized in a single pharmaceutical form (hard
gelatin capsules), it being possible for the types of
pellet to be additionally coated with various film-
forming agents. It is thus possible to carry out the
adjustment to the insulin requirement of the body even
more accurately, as is already possible anyway with a
tablet.
Oral pellet pharmaceutical forms are furthermore
distinguished in that in their gastrointestinal transit
2125284
- 22 -
times they are substantially more independent of physio-
logical effect factors, such as e.g. the nature and
amount of food absorbed among other things, as single-
unit pharmaceutical forms such as e.g. tablets.
The oral pharmaceutical forms described in the
present patent application can also be employed advan-
tageously for other administration routes.
Thus a tablet according to the invention, in
particular a simple sustained-release preparation can be
used for the administration of peptide pharmaceutical
substances in the oral cavity (buccal or sublingual) . The
bioadhesive properties of the gelatin in this case cause
adhesion to the oral mucous membrane after contact with
physiological fluid.
According to the invention, spray- or freeze-
dried powders can be advantageously employed for the
development of nasal sprays or nasal gels. (nasal adminis-
tration) . After insufflating into the nasal cavities, the
gelatin/pharmaceutical substance particles adhere to the
nasal mucous membrane as a result of bioadhesive
properties and exhibit a residence period in the nose of
on average 3 to 4 hours.
In order to explain the physiological background
of the absorption of pharmaceutical substances in general
and the improved absorption rate of the nanosols or
pseudocoacervates according to the invention adequately,
first a consideration of the mechanism of physiological
absorption of pharmaceutical substances, as is also
presented in relevant publications, is necessary.
However, the present invention is neither tied to the
following attempt at a scientific explanation of the
phenomena occurring according to the invention nor can it
be restricted by this.
Passive pharmaceutical substance absorption takes
place according to the present state of knowledge (theory
according to Brodie et al.), if the following conditions
exist:
a) the gastrointestinal membrane acts as a lipid
2125284
- 23 -
barrier,
b) the pharmaceutical substance is only absorbed in
dissolved and uncharged, i.e. nonionized form,
c) acidic pharmaceutical substances are preferably
absorbed in the stomach and basic pharmaceutical
substances preferably in the intestine.
After the oral uptake of a pharmaceutical
substance into the body, its absorption, i.e. the cross-
ing into the general circulation (biophase) is prevented
to a great degree by physical barriers (see Fig. 2),
namely
- by the mucus layer and an aqueous layer adhering
thereto
- the cell membranes of the intestinal epithelial
cells with the glycocalyx covalently bonded thereto
and
- the so-called "tight junctions" which connect the
epithelial cells with one another on their apical
side.
These barriers presuppose that absorption of
pharmaceutical substances takes place through the lipid
double layers fundamentally independently of their
distribution mechanism and state of charge (so-called
passive diffusion).
The epithelial cells of the entire gastro-
intestinal tract are covered with a mucus layer which
consists of mucins (glycoproteins), electrolytes,
proteins and nucleic acids. In particular, the glyco-
proteins form with the main component of mucus, namely
water, a viscous gel structure which primarily performs
protective functions for the underlying epithelial layer.
The mucus layer is bound to the apical surface of the
epithelial cells via the glycocalyx. The glycocalyx
likewise has a glycoprotein structure which is covalently
z~2~zs4
- 24 -
bonded to components of the membrane double layer of the
epithelial cells. The branched polysaccharides of the
glycocalyx, which are either directly covalently bonded
to amphiphilic molecules of the double membrane or to the
proteins incorporated in the double membrane, possess
charged N-acetylneuraminic acid and sulfate radicals and
are therefore negatively charged, which can lead to an
electrostatic bond or repulsion of charged pharmaceutical
substance molecules or of electrostatically charged
particles respectively. The epithelial cell membranes
consist of phospholipid double layers in which proteins
are anchored via their hydrophobic regions. The
phospholipid double layers with their lipophilic content
represent a further barrier for the transport of the
pharmaceutical substances to be absorbed.
From this description, it clearly follows that
charged pharmaceutical substance molecules or electro-
statically charged particles therefore only have a very
low chance of being absorbed via the oral administration
route.
The nanosols according to the invention for the
first time provide the technical teaching to form a
system with which these abovementioned obstacles to
absorption can be overcome. As the active compound
nanoparticles are stabilized in neutrally charged form by
the gelatin according to the invention, they can be
transported through the negatively charged glycocalyx
without relatively great obstructions, in contrast to
other described nanoparticles of the prior art, which are
not or cannot be stabilized in neutrally charged form.
According to the invention, the adjustment of the iso-
ionic state of charge can additionally be effected in
coordination with the physiological conditions.
As the active compound nanosols or pseudo
coacervates according to the invention can pass through
the glycocalyx without hindrance, without being bonded or
repelled by electrostatic effects, they thus also reach
the surface of the epithelial cells and are available
there in a high concentration.
~12~~84
- 25 -
Active, carrier-mediated transport mechanisms or
phagocytosis can then also make a substantial
contribution to the absorption of the active compound
nanosols.
The following examples are intended to illustrate
the invention in greater detail, but without making a
claim to completeness:
Example l:
Active compound: normal insulin (German Pharmacopeia 9)
Gelatin: Type B, completely freed from foreign ions.
IEP: 3.5
Characteristic bloom number: 280 ,for delayed release
30 for rapid release
500 g in each case of the gelatins specified
above are converted into the sol form with distilled
water at 40° such that a 5% strength solution results. A
pH of 3 . 9 is adjusted in each sol. 30, 000 I .U. each of
insulin of the described specification are then dissolved
in both batches. The adsorptive charge compensation
(pseudo-coacervate foranation) taking place is monitored
by conductivity testing (e. g. using a microprocessor-
controlled high performance conductometer from WTW) until
a change in the total conductivity no longer occurs.
Both solutions are than converted into the dry
form by separate spray-drying at an outlet temperature of
the spray stream of about 45 - 50°C.
Layered tablets which, as the core, possess the
insulin in non-delayed form are produced in a tablet
press with admixture of customary pharmaceutical
auxiliaries. The unfinished tablet blanks are then coated
in a coating pan by spraying on a solution of Eudragit S
and Eudragit RS in the ratio 3:2 in acetone.
Example 2:
Analogously to Example 1, the dried powders are
first prepared. Each powder is granulated per se with
admixture of customary pharmaceutical auxiliaries and
shaped to give pellets.
The pellets are then coated in a coating pan for
rapid release of insulin by spraying on an acetone
~~.25284
- 26 -
solution of Eudragit S and Eudragit RS in the ratio 3:2
aad the pellets for sustained insulin release analogously
with Eudragit S.
Both types of pellet are filled in a mixture
ratio of 1:1 into hard gelatin capsules which are coated
with Eudragit S after sealing.
Example 3:
Active compound: Corticotropin, IEP in the weakly
alkaline range at about 8
Gelatin: Type A, completely freed from
foreign ions.
IEP: 9.0
Characteristic
bloom number: 320 for delayed release
30 for rapid release
300 g in each case of the gelatins specified
above are converted into the sol form with distilled
water at 40° such that a 3% strength solution results. By
means of hydrochloric acid (2%), a pH of 8.5 is adjusted
in each sol. 200 mg each of corticotropin of the
described specification are then dissolved in both
batches.
Both solutions are then converted into the dry
form by separate spray-drying at an outlet temperature of
the spray stream of about 45 - 50°C.
Layered tablets which, as the core, possess the
corticotropin in non-delayed form are produced in a
tablet press with admixture of customary pharmaceutical
auxiliaries.
The unfinished tablet blanks are then coated in
a coating pan by spraying on a solution of Eudragit S in
acetone.