Note: Descriptions are shown in the official language in which they were submitted.
WO 93/15208 - .f ` ~ 2 7 1 2 ~ ~ P(~/GB93/00204
CHIRAL SYNTHESIS WITH MODIFIED ENZYMES.
This invention relates to chiral synthesis; mDre
particularly, it relates to the modification of enzym~-s to
facilitate such synthesis.
Enzymes are biological catalysts which are specific
~oth in ter~s of chemical activity and substrate structure, and
it is ~his specifici~y which has been exploited in a variety of
co~merci~l applications. Although many such activities are
known, it may be desirable to change the range of substr~tes that
are suitable for catalysis and/or to change the efficiency of a
given catalysis for a particular type of enzyme. Given a type
of enzyme with known key elements vis-à-vis substrate preference
and hence activi~y, it may be possible purposefully to change
those elements to bring about desired modifications and hence to
expand thPi poten~ial industrial utility of a particular enzyme.
~nzyme activity lS primarily controlled by the amino
acid composition especiaIly in certain important functional areas
of the enzyme, altering these amino acids is known to change
activity and may be achieved by the use of either specific or
non-specific techniques. For example, the introduction of a
neutralising amino acid may facilitate the cata ly5is of a
substrate with an altered charge and this could be regarded as
a predi~table altexation, although no result may ~ver be
p~edicted with total certainty, especially where the tertiary
structures of enzymes are~not as precisely known as would be
necessary for complete confidence. However, while it is possible
to make individual changes by known means, this would prove an
almost infinite task and so it is often convenient initially to
make a "macro-change" and then to "fine tune" with discrete
changes. Of course, in a given case, a macro-change may prove
to be sufficient, or, indeed, discrete changes may be all that
are required.
WO93/1~208 ~ PCT/GB93/00204~
Although alteration of the enzyme structure has been
describedt this is not achieved by any direct effect on the amino
acid components, but ~y known techni~ues on the DNA encoding for
the enzyme prior to protein transcription. Taking as an example
the enzyme lactate dehydrogenase (natural substrate pyruvate),
when acting on the carboxylic acid analogue of pyruvate, oxalo
acetic acid, it would have substantially reduced activity due to
the negative charge introduced into the active site. In this
case, site-direc~ed mutagenesis involving the introduction of a
~10 neutralizing charge into the correct region of the active site
alters substrate specificity allowing the enzyme to take on the
activity that would be expected of a malate dehydrogenase. Such
spe~cific mutations may be considered predictable in gross terms,
but are very unlikely to be the ultimate refinement in increasing
specificity towards such a substrate. For alternative
substrates, such as those with increased alkyl chain lengths,
phenyl residues or heterocyclic additions, predictions of site-
specific cha~ges are unlikely to be reliable. It is probable
that the changes necessary to accommodate such "~nnatural
substrates" are most likely to be required adjacent to or in the
active site region of the enzyme, which in many enzymes may
involve up to 20 amino acids, which may be derived from many
disparate parts of the primary sequence. Clearly, if one tried
to proceed by altera~lons in individual amino acids, the scale
~ of the undertaking would be impractical even with modern
techniques.
In order to achieve the desired objective while
circumventing the above disadvantages, it is possible in the case
~ 30 of lactate dehydrogenase, for example, to make use of the knswn
; loop region forming part of the active site~ As a convenient
first step, at least a portion of the loop region may be
~ exchanged for a larger or smaller section of loop region from a
;~ similar enzyme. This may be expected to allow some variation in
substrate specificity and relative catalytic efficiency, while
retaining the typical activity. Having chosen the most promising
loop region for a desired substrate, which could indeed be the
W093/15208 ~12 7 ~ PCT/GB93/00204
; '
starting wild-type loop, specific amino acid residues may be
targeted for further change. In order to secure the best
possible option, it is necessary to survey all possible amino
acid combinations in the positions of interest. This is done by
S generating random nucleotides in the region coding for the amino
acids targeted. Following routine cloning, it becomes necessary
to select for a ~esired modification from amongst the numerous
alternatives produced. Such screens are in common use. This
approach to enzyme engineering is fac~litated by the introduction
of unique endonuclease restriction sites into the coding DNA, if
such are not already present, at desired points. Such changes
may often be achieved by alteration in the bases without altering
the amino acid encoded due to code degeneracy or alternatively
they are achieved by the introduction of codes as far as possible
for similar amino acids. This allows the region of particular
interest to be handled independently of the remainder.
As will be appreciated from the foregoing, the present
invention relates to a method for modifying the specificity
and/or efficiency of an enzyme, while retaining its catalytic
activity, characterised in that it comprises: selecting an
enzyme, the tertiary structure of which is substantially known
or deduced; i~entifying at least one specificity and/or
efficiency-related region; identifying or constructing unique
~25 restriction sites bounding the identified region in the DNA
coding therefor; ~enerating a DNA sequence which corresponds to
at least a portion of the identified region, except that the
nucleotides of at least one codon are randomized, or selecting
as a substitute for at least a portion of the identified region
an alternative such region, which may itself be similarly
randomized; using the generated or substitute DNA sequence to
replace the original such sequence; expressing the DNA including
the generated or substitute DNA sequence; and selecting for a
desired modification so that the DNA coding therefor may be
isolated.
It will be described in more detail below, but the
WO93/1~208 . P~T/GB93J00204
~ 4
present method may be illustrated by reference to a
dehydrogenase, in particular an ~-hydroxy a~id dehydrogenase,
such as lacta~e de~ydrogenase. In this illustration, it is the
loop region of the enzyme which is identified initially as being
specificity and/or efficiency-related. Generally, the randomized
DNA is ~enerated by means of an inosine triphosphate PCR method
or a spiked oligonucleotide method or a PCR assembly method, all
of which will be discussed in more detail below. If a substitute
is to be selected for at least a portion of the region of
interest, it is often based on a corresponding sequence from a
similar enzyme. Once the original DNA sequence has been replaced
by the generated or substitute DNA sequence, it is cloned into
a plasmid or phage vec~or and transformed into a bacterium or
virus for expression. Thereafter, a screen may be used to select
for a desired modification. Taking L-lactate dehydrogenase as
an example, positions l0l and lQ2 are particularly appropriate
for randomization.
The present invention also relates to the use of such
modified enzymes particularly i~ the production of chiral
products. Often, such processes involve the use of a cofactor
recycling system. One example is the reduction of 2-oxo-4-
phenyl-propanoîc acid characterised in that it comprises the use
of L-lactate dehydrogenase which has been modified in the loop
region by the present method and another is the reduction of 4-
methyl-2-oxo-3-pentenoic acid characterised in that it comprises
the use of MVS/GG obtainable by the present method.
Having outlined the present invention, it will now be
described more fully.
The use of enzymes in chemical synthesis has gained
increasing acceptance as an academic possibility, while its
introduction into industrial chemical procedures is rare. The
potential advantages of enzymes as catalysts, such as obtaining
stereospecificity and regiospecificity under mild conditions,
have initiated many attempts to obtain enzymes suitable for
WO93/15208 i~4 ~ P~r/GB93/002
.
particular chemical conversions.
Several approaches to selection of the enzyme are
possible. Experimentation with currently-availa~le enzymes may
yield surprising results in terms of breadth of substrate
specificity not predictable from the literature. It is thus
possible to utilise commercially-available enzymes, which may
have a low catalytic efficiency, but, ~ecause of cost, may form
the basis of an industrial process. A second approach is to
screen large numbers of environmental micro-organisms in an
attempt to effect a particular transformation. Shoul~ such an
activity be obtained, it is often required that the enzyme be
obtained in a purer form than whole microbial cells or crude
preparations thereof. To obtain enzymes frcm such a screen in
sufficient quantity and at a reasonable cost for an industrial
process requires extensive development often with the involvement
of clonin~J and over expression of the gene. Another approach for
ob~aining suitable enzyme catalysts is to modify the structure
of an existing enzyme to improve its catalysis for a particular
~20 subs~rateO This approach of so-called "enzyme engineeriny",
which is in its very early~stages has great potential for the
preparation of catalysts for the synthesis of homochiral
moleculesO The importance~of these molecules in the synthesis
of single isomer pharmaceuticals and agrochemicals is well
25~ recognised.
Despite the obvious attraction of enzyme engineering,
the results of amino acid changes are often, at best, only of
limited predictability due to the structural complexity of
enzyme~O At present, it is not possible to predict the effect
of certain amino acid changes on the finer points of substrate
recognition and catalytic performance where the substrate is
altered in size and additional functionalities introduced from
the natural substrate. It is generallv easy to predict the
removal of activity by the elimination of one of the
catalytically-vital amino acids which are generally well known
from the classical studies of enzyme mechanism and function. To
~
.
WO93/15208 PCT/~B93/00204
f3, ~
enhance the activity towards an unnatural substrate remains a
challenge.
The opportunity for enzyme engineering may be
calculated for a 300 residue protein of 20 amino acids as 1039
possi~le sequences. The vast majority of these sequences cannot
have been explored for biological function. It may be suggested
that a typical large protein of 300 amino acids residues cannot
represent a global optimum for any biological function, but at
best is an assembly of empirically optimised 25-35 amino acid
domains. Thus, enzyme engineering should be capable of improving
a large frame-work for any particular target function.
Recently, methods have ~een developed to express random
sequences of DNA as protein fused to phage M13 coat protein and
it has been suggested that it will be possible to mimic the
process of evolution by suitable affinity chromatography to
isolate both the required protein sequences and its gene (Xang,
PNAS, 88, 1991, 4363~. However, just as e~olution has bPen
unable ~o sample all possible sequences, so too the protein
engineer will be limited to ~he num~er of M13 phage that may pe
screened (1 ol5 plaque-forming units are produced per litre
culture of E. coli cells containing the phage M13). With 1015
variants, the length of DNA which may be optimised is o~tained
from 4w = lol5~ i.e.~N = 24 bases or 8 amino acids. The other
problem encountered is that a phage display system determines
binding not catalysis and thus is not designed to obtain enzymes
with new chemical potential.
Random mutagenesis of existing proteins is also limited
in i~s ~bi}ity to produce radically altered proteins by problems
of sampling all the possible variants. In addition, the genetic
~; code is very resistant to change. Not only are codons redundant
at the third position, but also amino acid residues with similar
properties are coded by similar sequences and thus resistant to
sparse mutagenesis. For example: (i) a codon having a T at the
second position always codes for an amino acid residue having 2
WO93/1~208 PCT/GB93/002~
~ hl 7 1 ~
hydrophobic side chain; (ii) the codons for aspartate and
glutamate differ only at the third position. Therefore,
strategies, such as use of thioate nucleotides (Holm, Prot Eng,
3, l99O, 181), which create randomly dispersed mutations (in
which only one mutation is likely to be present in any codon) are
unlikely to yield new proteins having dramatically different
properties to those of the parent proteins.
Although it should be possible to engineer any designed
property into any protein framework, only those which have been
well characterised are likely to be redesigned successfully.
In order to obtain the fundamental knowledge required
for rational redesign, a combination of crystallography, site-
directed mutagenesis and transient kinetic techni~ues was usedto relate function to structure in the NAD-dependent lactate
dehydrogenases from both prokaryotes and eukaryotes. That
knowledge not only revealed those amino acids required for the
catalytic pathway, but also mapped those amino acids which are
~O par~ of a major rearrangement of shape which is induced when tpe
negatively-charged substrate acid enters the acti~e site and
causes the protein to se~uester the substrate in an internal
~acuole which is sensitive to the size of the substrate and which
contains exactly balanced charge. Using this knowledge, it has
been possible to design specific new enzymatic properties with
respect to charged substrates and so avoid the low statistical
pro~abilities associated with random mutagenesis. It should, of
course, be appreciated that the present invention is more
generally applicable than to this particular illustration.
-
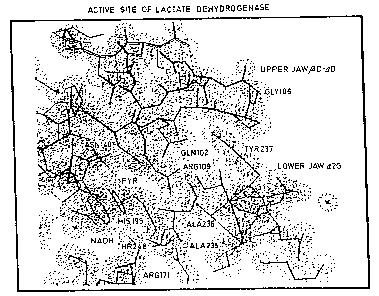
Accordingly Fig. 1 depicts the active site of lactate
dehydrogenase. In this illustration, some of the residues which
determine substrate specificity are carried on the under-surface
of the "upper jaw". The rate-limiting step in lactate
dehydrogenase catalysis is the rate at which this loop may sweep
through a viscous solvent to close onto the upper surface of
helix ~2G. The rate-limiting step is largely independent of the
WO93/15208 ,~ 6 PCT/GB93~00204
~, 8
se~uence of amino acids on the "upper jaw" and since the chemical
step is much faster than the shape change, the lactate
dehydrogenase system has the advantage that the loop sequence may
be easily varied to achieve different substrate specificities
without much danger that the chemical step will become rate-
limiting~ Thus, in order to obtain enzymes improved by
engineering towards particular substrates, a combination of
techniques may be preferentially employed. Specific residues may
be changed to accommodate functional groups, such as an altered
charge to that of the natural substrate, but to perfect the
enzyme for acti~ity towards a different substrate, elements of
the infinite variability of random amino acid changes may be
required. This may be applied to a particular area of the enzyme
and selected for usIng screening techniques.
An object of the present invention was to modify an
already useful, but substrate-restricted enzyme, S lactate
:: :
dehydrogenase, to provide an improved catalyst for reduction of
the ~-keto group in acids larger than the natural substrate,
pyruvate. In particular, the substrates of interest contain
bulky aromatic groups.
The natural enzyme used as the basis for engineering
~Z5~ was the thermophilic lactate dehydrogenase (LDH) isolated from
Bacillus stearothermo~hilus, which has been cloned and expressed
~: `
~; in Escherichia coli.
~:: : :
This enzyme has been one of the most thoroughly
characterised protein frameworks (Dunn, C. R., et al, Philos.
Trans. R. Soc. London Ser. B, l99l, 332, 184), including the
;~ study of inhibition, substrate interaction and genetic
manipulation. The physical stability of the enzyme, especially
to thermal dena~urationj makes it an ideal candidate for
demonstrating the features of redesign which would be generally
applicable to ~-hydroxy acid dehydrogen-ses, for example.
:
W~93/15208 PCT~GBg3/00204
~.2~
The modification of wild-type enzymes presents a
significant challenge because, even in the case of a protein with
considerable literature knowledge, the results may be unexpected
and surprising. Thus, redesign of even well- studied enzymes is
of limited predictability.
Changes in the amino acid composition of enzymes and
thus effects on kinetics and substrate specificity have occurred
throughout nature and various methods have been developed in
order to potentiate the natural divergence of enzyme structure.
Random mutations may be produced in genetic information [and thus
in the protein coded for) by the use of classical mutagenesis.
Lately, the technique of site directed mutagenesis has allowed
the alteration of specific bases in genes, thus produring
l~ directed amino acid changes in the target protein at a known
p~sition. Using similar techniques, it has been possible to
achieve the replacement of slgnificant amino acid sequonces in
8 functionally important area of the enzyme.
Detailed knowledg2 of the proteinl such as primary
sequence~and tertiary structure from X-ray analysis, along with
molecular mode~ling allow the identification of the position of
varioug amino acids in what are known as ronserved regions. This
is illustrated with the nomenclature of the amino acids of
various lactate dehydrogenase enzymes. Thus, any structure in
the protein whi~h is retained between species is regarded as
conserved and probably essential for the enzyme's function. This
~information will allow any change in a particular enzyme to be
pinpointed for all other homologous enzymes across all general
substrate types; if this were not possible the enzymes would not
fulfil the same biochemical function. The enzymes of particular
interest at present are ~-hydroxy acid dehydroyenases, which
catalyse the NADH/NADPH dependent reduction of a keto group in
an ~-position to a carboxylic acid, or, alternatively, the
reverse reaction where the ~-hydroxy group is oxidised to the
ketone.
WO93/15208 ~ PCT~GB93/00204
q ~ 3 ~.
~ '- 10
Attempts to modify ~he enzyme lactate dehydrogenase to
expand the natural substrate specificity to allow an increased
reaction rate with larger substrates with various functional
groups has led to the present unpredictable observations.
Although it may be possible to prepare substrates and
corresponding chiral products of interest by chemical synthesis,
followed by wild-type enzyme reduction, such an approach may not
be attractive and it may be that preparation via a redesigned
protein framework may provide a more rational and ~ost effective
approach. Additionally, the alteration of the enzyme has
demonstrated that the activity towards the natural substrate may
be so dramatically reduced that completely different substrate
selecti~ity is produced. This may not be a re~uirement of a
biotransformation catalyst, where the enzyme is presented with
only one substrate species for reduction, but, when a mixture of
potential substrates is presentl such as may occur in a
biological sample, this may be essential for achievement of
~ selective conversion or the determination of one particular
`; chemical species. This alteration in substrate specificity could
a~so be advantageous in a biotransformation using whole cells
where the intended;su~strate is necessarily contaminated with
other entities which could also be transformed.
In the work of Wilks et al (Biorhemistry, 1990, 27,
~587) a mutation strategy is described for the production of NAD-
~;~ dependent dehydrogenases which have altered substrate
specificity. The dlsclosed enzymes catalyse the reduction of
homologues of pyruvic acid corresponding to the general formula:
CnH2n~l CO COOH, which may include straight- and ~ranched-chain
alkyl residues. The initial intention of the present work was
to con~inue the design method for substrates with an aromatic
function, in addition to extended alkyl residues and hydroxyl and
keto substitution associated with the same base structure of ~-
oxoacids.
Enzymes capable of reducing such substrates would be
of particular value in the field of synthetic chemistry where an
I .1~
WO93/15208 ~ 71 ~' i~ PCT/GB93/~204
11
~-keto compound could be conver~ed stereospecifically to the
corresponding secondary alcohol. The production of individual
optical isomers of secondary alcohols is especially valuable in
the manufacture of optical isomers of pharmaceuticals and drug
intermediaries. The feature of thermophilicity which may be
obtained with some ~-hydroxy acid dehydrogenases is valuable as
it enables the enzymic reactions to be carried out at relatively
high temperature where a rate acceleration may exist and the
enzymes are inherently stable. These enzymes may also be
suitable for incorporation into determinations of the levels of
particular substrates obtained in biological samples under
certain disease states.
A num~ering convention has evolved in the field of
NAD-dependent dehydrogenases, which was originally based on an
X-ray structure of dogfish muscle lactate dehydrogenase. This
system numbers amlno acids ln ascending order extending from the
N terminus. This system identifies conserved residues, such as
glycine at positions 30 and 33, tyrosine at position 85, arginine
at posi~ion lO9, serlne at position 163 and aspartic acid at
; position 168. ~ ~
Thus, in any given NAD dependant dehydrogenase, natural
or~subject to mutation,~ there are regions of sequence whlch are
:
2S homologous with the amino acid sequence of the numbering
convention. An important aspect of this convention is that any
amino acid change in an NAD dependent dehydrogenase may ~e
~accùrately described.
-~ 30In Table 1 below,~an alignment of amino acid sequences
is shown for three NAD dependent lactate dehydrogenases: the M4
isoenzyme of pig, the testis isoenzyme of man and the Bacillus
~;~stearothermophiIus enzyme. (The symbols " - " do not signify
breaks in the continuous polypeptide chains, instead they are
conventional representation of discontinuities of numbering which
allow alignment with sequences of other enzymes to give maximum
homology.)
WO 93/15208 PCI`/GB93/00204 ~"~
~b~r~
12
Table 1
2 2 2 2 3
o 5 o 3 4 5 0
5 A T L K E K L I A P V A Q Q E T T I P N N K I T V V G V G - Q V G M
S T V K E Q L I E K L I E D D E - - S Q C K I T I V 5 T G - A V G M
M K N N G G A R V V V I G A G - F V G A
10 3 4 4 5 5
0 5 0 5
A C A I S I L G K - - S - L - - - - T D E L A L V D V L - - E D K
A C A I S ~ L L K - - D - L ~ A D E L A IJ V D V A - ~ L D K
S Y V F A L M N Q - - G ~ - A D E I V L I D A N - - E S K
6 6 7 78 8 9
0 5 o 50 5 0
I. K G E M M D L Q H G S L F L Q T P K I V A N X D Y - S V T A - N
20 L K G E M M D L Q H G S L F F S T S K V T S G K D Y - S V S A - N
A I G D A M D F N H G K V F A P K P V D I W H G D Y - D D C R - D
2 5 9 o 0 1 1 2
5 0 ~ 5 0 5 0
S K I V V V T A G - V R Q Q E G E S R L N L V Q R N V N V F K F I
S R I ~7 I V T A G - A R Q Q E G E T R L A L V Q R N V A I M K I
A D L V V I C A G - A N Q K P G E T R L D L V D K N I A I F R S
3~ ~
- ~: 1 1 3
~: 2 3 2 2 3 4 4 5
~:: 5. 0 A B 5 0 5
3 5 I P Q I V K Y S P - N C I I I V V S N P V D I L T Y V T W K . L S G
I P A I V H Y S P - D C K I L V V S N P V D I L T Y I Y W K I S G
V E S V ~5 A S G F - Q G L F L V A T N P V D I L T Y A T W K F S G
~,
5 6: 6 7 7 8
;: 5 0 S 0 5 0
- - - - L P K H R V I G S G C N L D S A R F R Y L M A E K L G V H
- - -- - L P V T R V I G S G C N L D S A R F R Y L I G E K L G V H
45 - - - L P H E R V I G S G T I L D T A R F R F L L G E Y F S V A
i ~ ~ 1 1 1 2 2
~: 8 9 3 0 0 9 9 9 9 0
5 0 ~, o 5 o 5 A B C D A
P S S C H G W I L -- G E H G D - S S V A V W S G V N V A G V S - L
P T S C H G W I I - G E H G D - S S V P L W S G V N V A G V A - L
P Q N V H A Y I I -- G E H G D - T E L P V W S Q A Y I G V M P - I
WO93/1~08 . PCT/GB93/00204
~ ~ 2 7 ~ s
Table 1 fcont~
1 2 2 2 2 2 2
5 0 1 2 2 3 3 4
B 5 0 5 0 5 0
Q Q L N P E M G T D N D S E N W K E V H K M V V E S A Y E V I K L
K T L D P K L G T D S D K E H W K N I H K Q V I Q S A Y E I I K L
R K L V E S K G E E A Q K D - L E R I F V N V R D A A Y Q I I E K
2 2 2 2 2 2
: 4 5 5 6 6 7
5 o 5 0 S 0
15 K - G Y T N - W A I G L S V A D L I E S M L K N - - L S R I H P V
K - G Y T S - W A I G L S V M D L V P - - L K N - - L R R V H P V
K - G A T Y - Y G I A M G L A R V T R A I L H N - - E N A I L T V
2 2 2 2 2 ~ 3
7 8 8 9 9 9 o
5 0 5 0 5 9 l
S T M V Q G - - - M Y G I E N E V F L S L P C V L N A R G L T S
S T M V K G - - - L Y G I K E E L F L S I P C V L G R N G V S D
~: 25 S A Y L ~ G ~ L Y G E R D - V Y I G V P A V I N R N G I R E
,
3 ~ 3 3 3 3 3 3 3
0 l 1 2 2 0 0 3
30 ~ : 0 5 0 5 A B 1
V I N Q K L K D D E V A ~ L K N S A D T L W G I Q K D L K D L
V V K I D L S E E E - A L L K K S A E T L W N I Q K N L I - F
- V I E I E L N D D E K N R F H H S A A T L X S V L A R A F T R
Expression cloning of human testis~specific lactate dehydrogenase cDNA.
Millan, J.L., Driscoll, ~.E. and Goldberg E.
Sequence ~rom cDNA - Genbank accession number J~2938 (19~6).
40 The DNA sequence c~ the thermophilic lactate dehydrogenase from
Barstow, D., Clarke, A.R., Wigley, D., Holbrook, J.J. and Atkinson, T.
Gene, 46, ~1986), 47-55
WO93/15208 PCT/GB93/00204
3- h 14
~ Within the conventional numbering system are short
sequences which may be correlated with specific structural
elements in the ~olded polypeptide and which may have specific
functional properties such as the substrate recognition site or
the activation site.
The substrate recognition site is carried in part by
a mobile 190p of polypeptide chain, conventionally numbered 98
to llO. This sequence is contiguous but traditionally omits a
residue 103.
It is known for ~-hydroxy acid dehydrogenases of the
: L type which generate S stereochemistry on reduction to the
hydroxy function that a mobile surface loop exists which changes
conformation after substrate binding. This 1GOP consists of the
amino acid residues 98-llO and contains an arginine at position
lO9 which is impcrtant for catalysis as the positive charge from
: . the amidine group stabi1ises the stretched substrate carbonyl and
:: thus decreases the energy required to obtain the transition state
necessary for hydride transfer.
The loop region is also involved in substrate selection
and for that reason was the particular object for the present
enzyme engineering s~udy.
~25
~: The mechanism by which lactate dehydrogenase
dis~inguishes diff ren~ substrates is the ability of the
substrate to fit into a proton-impermeable, fixed-sized internal
vacuole which is formed when the mobile surface polypeptide loop
closes down onto the protein surface. Not only is.loop closure
only possible over suitably small and singly negatively chargPd
s:ubstrates, but also the loop closure triggers catalysis through
the arginine lO9 residue. The variation in composition and
: length of this mobile loop region is the immediate object. For
the convenience of these experiments, a particular gene for wild-
type Bacillus stearothermoPhilus lactate dehydrogenase was chosen
where the amino acids alanine at positions 235 and 236 had been
WO93/15208 ~ PC~/GBg3/00204
changed for glycines. The effe ts of this particular amino acid
substitution have been presented by Wilks et al. for a limited
range of substrates tBiochemistry, 28, 8587) and generally
increased the activity towards substrates with larger alkyl
groups. Although used to demonstrate the principle of loop
exchange, the technique would not be constrained to this
; particular enzyme, rather it is applicable not only to this
mutant enzyme, but also to all other structurally-related ~-
hydroxy acid dehydrogenases, for example.
The mutation where alanines at 235,236 are replaced by
glycines has been combined with three mutations in the mobile
;~ polypeptide loop (reSldues 98-112), namely glycine 102 by
methionine, lysine 103 by valine and proline 105 by serine
(M~S/GG).
This new enzyme construction was evaluated f~r activity
towards longer substrates, in particular an unsaturated branched
substrate 4-methyl-2-oxo-3-pentenoic acid, which is reduced to
the f~llowing alcohol:
OH
~ ~ \ CO Na
: 2
;:~ 25 :-~
;~: Steady state kinetic measurements indicated that
reduction of this compound by the wild-type enzyme proceeded
:; slowly, o~taining an estimate for turnover of 0.03S-l in contrast
~ to that obtained with the mutant enzyme of 1.2S-1. The Km
.~ 30 de~ermined under similar conditions of substrate concentration
(1-20mM) in the presence of 5mM fructose 1,6-bisphosphate was
22mM. This observation regarding the specificity alteration
towards a less flexible substrate indicates that the loop region
~: has importance in substrate reduction.
The method used to make new loop constructions was to
insert restriction enzyme sites at either end of the DNA coding
WO93/15208 PCT/CB93/00204
~ 3 ~6
for the loop region. These new restriction sites which are
unique within the DNA coding for the enzyme, are cleaved and then
religated with synthetic DNA designed to code for the required
new loop region. One of the restriction sites introduced was for
SacII near amino acid 97. The construction of the Sac II
restriction site required that the wild type coding sequence for
cysteine g7 was changed to threonine. The Xbal site retained the
wild-type amino acid sequence with arginine at l09, but did
result in the creation of an MluI site close to threonine 108.
The new MluI site was used to advantage as it was destroyed in
transformants and thus enabled easy distinction thereof from the
wild-type gene.
To illustrate~the utility of the loop design approach
to enzyme engineering, novel loops were introduced, two shorter
by 3 amino acids and one lon~er by 4 amino acids. The new
enzymes generated~in-this manner were evaluated against a range
of experimental substrates to determine the effect of the loop
` exchanges.
;~ .
It was clearly demonstrated that the new loops altered
the pro:perties of the enzyme~from that of the framework used in
th~e construction ~thereof.~ The results also illustrate the
difference obtained~with~ the ~alanine - glycine alteration at
amino acids 235 and~236 and the introduction of the threonine in
place of cysteine at amino acid 97.
- ~ The incre~se 1n turnover of ~-ketocaproate and ~-
ketoisocaproate with ~the alanine - glycine double mutation was
consistent with the results of Wilks et al. ~Biochemistry, 29,
; l990, 8587). The increase in turnover for the aromatic substrate
~ 2-oxo-4-phenyl propanoic acid:
;~
;~
- ~:~
::
~:
:
:
WO~3/1~20~ PCT/~Bg3/0~04
Ph I ~
~ C02H
along with increases in the Km for both was not obvious and
indicates useful improvement with respect to the use of the
mutant enzyme in the synthesis of the chiral ~-hydroxy group of
this aromatic substrate.
The exchange of threonine for cysteine at amino acid
97 maintained the beneficial Km effect for 2-oxo-4-phenyl
butanoic acid:
o
15 J~
Ph ~ / C~2H over the wild-type enzyme.
: The effect of these individual mutations on the
reduction of ~he aromatic substrates is of clear interest as the
homochir~l hydroxyacids produced form useful chiral building
; blocks for the synthesis of bioactive compounds.
::: The introduction of the new loop sequences further
alters the substrate specificity of the enzyme reducing the
turnover ~f the na~ural substrate from that. of the wild type
enzyme. The t~ree new loop enzymes retaine~ most of the wild
: type catalytic potential t~wards the 2-oxo-4-phenyl propanoic
ac~d as shown by turnover and Km and, in the example of the
longer loop and second shorter loop version, resulted in an
increase in turnover.
These examples serve to illustrate that the activity
of the enzyme may be dramatically altered by chanyes in the loop
sequences, both towards the natural substrate and larger
unnatural substrates.
In the large loop, it is o~served that the Kcat/Km for
WO93/15208 PCT/GB93/002
~,~A~ j ` 18
2-oxo-4 phenyl propanoic acid was 1700 times better than for
pyruvate compared to the wild type enzyme which is conversely 23Q
times better for pyruvate, representing a switch in specificity
of 391,000 fold.
The alteration in specificity of the enzyme from
pyruvate to 2-oxo~4-phenyl propanoic acid renders the new enzyme
suitable for the determination of the concentration of 2-oxo-4-
phenyl propanoic acid, often termed phenyl pyruvate in clinical
chemistry nomenclature, especially from body fluids, such as
blood and urine.
Phenyl pyruvate levels are normally low, but rise to
significant levels with the increase in phenylalanine
1~ concentration, which is associated with the genetic disease
phenylketonuria (Langenbeck et al., J. Inher. Metab. Dis. 4,
~; 1981~ 69) o It is~ also possible that the phenyl pyru~ate
reductase or phenyl lactate dehydrogenase enzyme could be used
in conjugation with phenylalanine dehydrogenase, a current method
~20 of determining the phenylketonuria level such that interference
from phenyl pyruvate could be negated, thereby enhancing the
sensitivity of the phenylalanine-based method.
; , :
The construct having the restriction sites at either
end of the loop region may be used to produce a series of
dehydrogenases having ~loops of variable length and variable
:
sequence. Thus, by restricting random mutagenesis to the region
of lactate dehydrogenase which has been identified as being
important for substrate recognition, it is possible to isolate
enzymes which may carry out a desired chiral reduction. The
random mutagenesis may be generated by use of spiked
oligonucleotides at specific positions and on different length
loops or, alternatively, by the incorporation of inosine
triphosphate in a polymerase chain rea~tion (PCR) that randomises
3~ either the entire loop region or specific residues. Both of
these techniques have been employed to prepare mutant libraries
using the restriction sites engineered into the DNA coding for
WOg3/15208 ~ PCT/GB93/002~4
19
the loop region of LDH.~ A further PCR method was used to
generate a random combinational DNA library of spe~ific positions
of the loop region. This technique was specifically targeted to
positions 101 and 102 as these are involved in defining enzyme
substrate specificity.
The PCR was initially used to generate 300 & 800 base
pair fragments that had complementary overlapping ends. These
primary products which had random sequences incorporated in the
overlap, were then primed on each other and extended to yield an
LDH hybrid gene. A second PCR with two outer primers annealing
at non-overlapping ends was finally used to amplify the LDH
product.
Previous manipulation of the Bacillus
~tearothermophilus LDH gene involved cloning an EcoRI/PstI
digested gene in to PKK 233-2, or Ml3 plasmid vectors. Where,
as now, it ls possible to clone the PCR product into any one of
a number of vectors, because one of the outer primers (2), which
~; 20 anneals past the codiny region, was designed with an additional
EcoRI site incorporated. For example, in order to verify that
there is a representative library with random sequences in ~he
desired positions, it is possible to clone the gene with unique
EcoRI sites into PUC18, which produces a high yield of DNA from
~25 mlni-preps, and subsequently the PCR product may be cloned into
plasmid or phage expression vectors, such as PKK 233-2. (See
accompanying illustrative Fig. 2.) Yf
The following advantages are obtained with the PCR
method: -
1. High yield of PCR product obtained.
2. The ability to identify product as mutant DNA and select
against wild-type sequences via MluI digestion.
3. Ease of handling and monitoring a lkb product compared to
previous attempts which involved designing restriction sites
either side of the loop region, such that a 40 base pair wild-
type sequence may be replaced with a mutant sequence.
WO93/1~208 ~t~Jb 20 PCT/GB93/00204
4. Speed of method.
5. The design of primer 2 with an EcoRI site enables the
cloning of gene product in~o a number of vectors.
6. Use of double-stranded template for mutagenesis.
7. Application of method to manipulate other areas of the LDH
gene and the ease by which interesting mutations in different
regions may be brought together in one molecule using this splice
overlap extension method.
8. Having mutant oligos with a high region of complementarity
to the templ~te at the 3'-end ensures that annealing of oligos
to the vector is highly efficient.
In order successfully to utilise a directed random
mutagenesis method that generates a library of mutants covering
the loop region of the enzyme, or indeed any specific region of
any target enzymet requires a suitable screen for clones which
express mutant enzymes of the desired specificity. For the
dehydrogenases, this is simply provided by coupling NADH
production with phenazine metasulphate to formation of insoluble
blue formazan dye.
The screen is based on the work of Katzen and Schimkel
PNAS, 54, 1218) and relies on the ability of a colony expressing
an enzyme with specificity to oxidise the required substrate and
2~5~ to reduce NAD+ to NADH. The reduced coenzyme then reduces
phenazine metasulphate which in turn reduces nitroblue
tetrazolium to form an insoluble blue dye.
The mutant DNA is transformed into competent E. coli
cells and is stored on agar plates containing 15% glycerol and
ampicillin at -80C. Obtaining electro-competent cells with high
transformation rates has produced rates of 106 per ~g of DNA, a
rate which pr~duces a sufficiently representative population of
mutant colonies for screening. Copies of this plate are made
using a velvet replicator and the copies grown up overnight.
(The E. coli LDH activity is removed by incubation of the filter
paper at 67C for 30 minutes, the activity of the wild-type
W093/15208 PCT/GB93/00204
~27~
21
enzyme is not lost until 45 minutes at this temperature.) The
copies are then screened against a range of substrates and
individual colonies may be compared. Each master plate is
scre~ned at least three times to ensure conditions are ideal in
each case.
Using this technique demonstrates differential rates
of staining have been shown between filter copies of wild-type
colonies and those containing the malate dehydrogenase activity
mutan~ enzyme (Q}02R) with lactate and malate as su~strates,
respectively, confirming the validity of the screen to identify
individual colonies.
The following illustrates the present invention:
Mutaqenesis of lactate dehydroqenase
Mutants ~f lactate dehydrogenase from Bacillus
stearothermophilus were generated by the oligonucleotide mismat h
~20 procedure of Winter et al. (Nature, 1982, 299, 756) in M13 with
the mutagenic oligonucleotide as the prlmer for in vitro chain
extensions. The double~alanine replacement at 235 and 236 by
glycine was obtained us;ing the oligonucleotide sequence
3'CGCGCTACCGCCGATGTTTA5~'. The~wild type and mutant enzymes were
~25 expressed in the PKK223-3 plasmid in E. coli (Barstow et al.,
Gene,~1986, 46, 47). ~ ~
; Mutaaenesis to construct Sac II and XbaI sites at either end of the qene codinq for wild t~pe active site loo~.
~30
A 54-mer oligonucleotide was used to direct mutagenesis
to introduce unique restriction sites (SacII and X~aI) at either
end of the active site loop (amino acids 98-110) using the wild-
type template (Barstow loc. cit~. The mutagenic oligonucleotide
was:
S'GTCCACAAGGTCTAGACGCGTCTCGCCCGGTTTTTGGTTGGCGCCCGCGGTAATGACAAC3',
~:
~ ' .
W093/15208 PC~/GB93/002~ ~
,~ ,
~lt~ ~ 22
the annealing, chain extension and cloning were as described by
Clarke et al. ~Nature, 1986, 329, ~99).
.
: 5 Mutants were identified by making mini-preps and
restricting with SacII and XbaI. Mutant mini-preps were
restricted with EcoRI and XhoI and the small fragment was sub-
: cloned into PKK223-3 containing Ala235Gly, Ala236Gly mutant LDH
from which the small EcoRI/XhoI fragment had been removed (Wilks
et al. Biochemistry, 19so, 29, 8587). The resulting plasmid
(pLDHrs) was transformed into competent E. .coli TG2 cells. The
whole sequence was redetermined using a "Dupont Genesis 2000"
automatic sequencer and showed the correct loop sequence had been
; inserted. The partial DNA sequences of the wild type gene and
the mutant with inserted restriction sites are shown in Table 2
below.
~:
l ~
.~
.: : ~
1 ~ ~
~ :
::~ :
;~
:
~.~093/15208 PCT/G W3/00204
. 23 ~ ~ 2 7 ~ ~ ~
Table 2 comparison of the protein and DNA sequences of B.
stearothermophilus lactate dehydrogenase in the loop (93-ltlJ region
of wild-type and the mutant with SacII and XbaI restriction sites at
either end of the loop, and the variable loop sequences derived from
5 them.
Wild-type DNA sequence in loop region (Cys changed to Thr is shown
bold):
: `
LeuValValIleCysAlaGlyAlaAsnGlnLysProGlyGluThrArgLeuAsp
5'TTGGTTGCTATTTGCGCCGGCGCCAACCAAAAACCGGGCGAGACGCGGCTTGAT3'
3'AACCAACGATAAACGCGGCCGCGGTTGGTTTTTGGCCCGCTCTGCGCCGAACTAS'
Mutant DNA (p~DHrs) sequence in loop region:
~:15
~:;:LeuValValIleThrAlaGlyAlaAsnGlnLysProGlyGluThrArgLeuAsp
5~TTGGTTGcTATTAccGcGGGcGccAAccAAAAAccGGGcGAGAcGcGTcTAGAc3
~:~ 3~AAccAAcGATAATGGcGcccGcGGTTGGTTTTTGGcccGcTcTGcGcAGATcTG5
~ SacII XbaI
:::20 MluI
: :
Two oligonucleotides (~LA and LLB) used to synthesise the big loop by
PCR:
~25~5'TACCGC GGCAACATTAAATTGCAACAAGATAA3' ~LLA ?
5'GGTCTAGACGATCGCCCGTCGGGTTATCTTGTT3' (LLB)
~:;3:0: Big }oop sequence in~ the 97-110 region ~note the MluI site is
:: destroyed)~
` CysAlaGlyA~aAsnGlnLys~ -ProGlyGluThrArgLeuAsp (wild-type)
ThrA~aGlyAs~IleLysLeuGlnGlnAsDAsnProThrGlvAs~ArgLeuAsp (big loop)
3;s~:s~TAccGcGGG:cAAcATTAAATTGcAAcAAGATAAcccGAcGGGcGATcGTcTAGAcc3~
3'ATGGCGCCCGTTGTAATTTAACGTTGTTCTATTGGGCTGCCCGCTAGCAGATCTGGS'
SacII ~ XbaI
Oligonucleotides for PCR synthesis of LeuLysGly and SerLysGly short
~^40:1Oops: ~ :
SLA :5'T~CCGCGGGCGCCAACT3'~::
::SLB 5'GGTCTAGACGGCCTTTCAAGTTGGCGCC3'
~: SLC S'GGTCTAGACGGCCTTTGGAGTTGGCGCC3'
: 4S
: Short loop sequence in the original 97-111 region (MluI site is again
destroyed):
:Gl~GluThr
50 CysAlaGlyAlaAsnGlnLysProArgLeuAsp (wild-type)
ThrAla~lyAlaAsnLeuL~sGlvArgLeuAsp (SLl)
5'TACCGCGGGCGCCAACTTGAAAGGCCGTCTAGACC3'
~3'ATGGCGCCCGCGGTTGAACTTTCCGGCAGATCTGG5'
:~55 ThrAlaGlyAlaAsnSerLYsGlv~rgLeuAsp (SL2)
5'TACCGCGGGCGCCAACTCCAAAGGCCGTCTAGACC3'
: 3'ATGGCGCCCGCGGTTGAGGTTTCCGGCAGATCTGG5'
SacII : XbaI
WO93/15208 ~ 24 PCT/GB93/00204 ~
PCR assembly method for generation of random
combinational library ~of the loop region of the B.
stearothermophilus LDH gene:
l. Single-stranded oligos were made such that the oligos were
only differen~ to the wild-type sequence at positions encoding
amino acids l0l and 102 where each one of the bases A, T, C, G
has an equal chance of being inserted. (Oligo mix l0l,102
forward.)
2. An MluI restriction site which is present in the wild-type
lQ template i5 destroyed by change of the third codon position of
~; amino acid 108 from an ACG to an ACT without altering threonine
as the amino acid being coded. The absence of the MluI site
enables verification that the mutants have been generated and to
select against wild-type sequences.
3. A DNA primer which has 14 base homology to olio mix l0l,1~2
forward was used to make the complementary strand toligo mix
l0l,102 reverse) using a Klenow reaction.
4. Single-stranded library oligos were used with primer l and
5ng of wild-type ~emplate in order to generate a 300 base pair
product with 25 cycles of PC~ (94C, for l minute, 55C ~or l
minute, 72C for 2 minutes).
5~ Double-stranded Klenow oligos were used with primer 2 and
~.
; 5ng of wild-type template to gener~te an 800 base pair product
which overlaps the 300 base pair product. (PCR conditions as in
4-~)
The use of double-stranded oligo as primer in 5 is very important
in ensuring that both the 300 and 800 base pair products are made
;and primed using mutant oligos and that the wild-type sequence
at position l0I and 102 is not copied.
6. After gel purification, 20ng of the 300 base pair product
and 60 ng of the 800 base pair product were mixed without primers
and thermocycled seven times in order to join the fragments ~94C
for 2 minutes, 55C for l minute, 72C for 4 minutes).
7. After seven cycles, primers l and 2 were added, and the
product amplified for twen~y cycles (94OC for 1.5 minutes, 55C
for l minute, 72C for 2.5 minutes).
8. The l kb PCR product was then gel purified, digested with
EcoRI, and gel purified again before ligation into EcoRI-cut
PUCl8 plasmid vector and transformation into E. coli.
WO93/15208 . ~ ~ 7 ~ ~ ~ PCT/GB93/00204
9. Recombinant colonies were selected for by IPTG and X-Gal
insertional inactivation.
lO. Of the nine white colonies picked, seven were verified for
the presence of the LDH gene and to resistance to MluI digestion
via gel and restriction analysis. The other two did not have
inserts.
ll~ Six of the mutants were sequenced using a Dupont 2000
sequencer and con~irm that the random mutagenesis approach had
been achieved.
~: : See Table 3 below:
~ : .
~ :::
. ~
.
~:
.
, ~
:
:~;: :
: .
WO 93/1520~ ,, rl ~ ;` 26 PCI`/GB93/00204 ,~.,
~ .~ o ~ .~ .~ .~ ~ _ _ _ c e c c c c c
~ .~ ~ ~ ~t ~ ~ ~ v o . . . . . ~ . , ,
o ~' ~ ~ s ~: ~ ~ ~ ~
o ~ ~ ~ ~ o ~ _ _ _ _ _ _ _ _
e~ ~ ~ ~ ~ ~1 ~t O ~ r _ ~ ~ r ~ _ _
V ~ ~ ~ V t~ ~ ~ O ~ _~ ~ ~ ~ ~ _
V ~ V O O V S~ V V O _ 17 _ ~ ~ ~ ~ O
~ '1: ~ V 1~ C ~ ~ o C C O :. :- J C C :-
~ ~1~ ~ ~ ~ ~ -0 ~ a~ ~ ~ ~
i9~ ~ I ~ ~ ~ o ~ ~ ~q9~
U G V ~ ~ IJ 1.) 1~ _. O _ ~ _ _ ~t _ ~ ~
Cl ~O~ O ~.) O~ (.~ O ~ O~ Cl ~:1 _ ~; _ ~ ~) C~ C~
O O 1~ O Cl C~ O ~ ~ ~ e <~ ~e ~~ ~ ~ ~ ~ ~
~ V ~ ) ~t , ~ L~ ~ V V I~ _ r _ S _ _ _ _ (~A)
l_ l_ ~_ ~_ ~ l_ : 1~- _ _ _ _ _ _ _ _ _
: ~ ~ ~ ~ ~ ~ ~ v ~ u~ ~: ~ ~ ~: ~ _ ~ ~ ~
~ lo ~ 1- 1-- ~- lo ~
~ ~ ~ ~ ~ ~ ~7 ~ ~ ~ ~ a~ _ ~ ~ :~ _ ~ ~ ~ _
E~ _ ~ ~ U) r- co ~ 3a 3 _ ~ ~ ~ co ~ U3~ 3
~ .
Rc~T~ ITF ~HEET
WO93/1~208 PCT/GB93/00204
~7 ~
27
Generation of double-stranded DNA looP _fraoments by
oliaonucleotide-overlaP
Each pair of overlapping oligonucleotides (20~M of
each) were subjected to 30 cycles of annealing and extension
(~4C for 1 minute, cool to 45~C for 2 mînutes, 45C for 1
minute, heat to ~2C in 1 minute, 72C for 1 minute in 50~1
containing 0.05 M KCl, 10mM Tris pH 8.3, 1.5 mM MgC12, 0.01%
gelatin), 200~M of each dNTP and 2.5 units TAQ DNA polymerase).
The double-stranded DNA product was purified and then cut with
SacII and XbaI before ligating it into the plasmid pLDHrs cut
with the same enzymes. The ligated products were restricted with
MluI to cleave wild-type plasmid pLDHrs.
The DNA was purified, microdialysed and used to
transform E. coli TG2 cells by electroporation. Transformed
cells were selected for ampicillin resista~ce. Ten such colonies
were picked and plasmid DNA purified from overnight cultures.
:The presence of mu ant loops was confirmed by resistance to MluI
digestlon.
The expression of the enzymes was obtained as de~criked
above.
Purification of lactate dehYdroaenase and mutants
Overnight cultures ~ were centrifuged and the packed
cells were resuspended in 50 mM triethanolamine, pH 6Ø The
cells were sonicated and the debris was removed by
centrifugation. The protein in the supernatant was precipitated
by the addition of 65% ammonium sulphate. The precipitate was
spun down and resuspended in 50 mM triethanolamine, pH 6.0 and
dialysed against the same buffer. After dialysis, NADH and FBP
were added to the protein to final concentrations of 5mM and 10mM
before loading onto an oxamate Sepharose column which had been
WO93~1~208 ~4 ~ & PCT/GB93/00204
28
pre-equilibrated with 50 mM triethanoloamine, pH 6.0, 3 mM NADH
and 5 mM FBP. After washing off unbound protein with column
buffer mutant LDH was eluted with 50 mM triethanolamine, pH 9.o,
0.3 M NaCl. The elutant was precipitated with 65% ammonium
sulphate and then resuspended in and dialysed against 50 mM
triethanolamine, pH 7.5. The protein was then loaded onto a Q-
Sepharose Fas~ Flow column and eluted with a salt gradient. LDH
eluted at a concentration of 0.25 M N~Cl. For the double glycine
mutant enzyme, the first chromatography procedure with oxamate
Sepharose was replaced by chromatography on Blue Sepharose -F3GA,
otherwise the procedure was essentially the same. All proteins
were judged to ~e greater than 98% pure from the intensity of
Coomassie blue staining .on an SDS Phast gel tPharmacia). The
yield of protein was usually 0.2g/1 of original broth.
St.eady-State Kinetics
Steady-state measurements were made by following the
~; reduction in absorbance at 340nm in the NADH/NAD+ conversion.
All assays were at 25C in the buffer Bis-Tris, pH 6, (20mM~,
con~aining KCl (50mM) and when used fructose-1,6- bisphosphate
at 5mM. Protein concentration was determined from ~he absorbance
at 280nm using ~he value of 0.91 for lmg/ml protein in lcm path
and an Mr of 33,000.
.
The results from these determinations ~re shown in
Table 4 below~
WO 93~152Q8 ;~ s~ S PCr/GB93/00204
Cl, ~ ~ V OD V ~ ~rl
a:~ o ~ o ~ o . o o . o . o o ~
~ o C~ V ~ . ~
o o o o
U~ `
~D
P~ I` o ~ o 7
o o ,~ D ~ O O
U~ ~ o o ~ ~ o
C~ o r ~ o
m ,~ . o O o O o O O
U~ ~ ~ I ~ I o ~ ' o ~ I ~ '
o o
.
~ a~
.~~ OL~ O Ln 1` U~ ~ U~ 00
~ ~ ~ , . a:~ ~.D O ~ ' ~ ' ~
XU~ ~ ~ o ~ S O O U
~ P~ Ul ~ o o
a: o ~ ,~ o ,~ , O u~
~,8 ~ o ~ ~ o ~ ~ o ~ ~ o ~ I Q
O ~q ~ . o 0 ~ t` J~
c~:l ~ o ~ o ~ o
E ~ :~
m ~ o ~ ' o ~ I ~ '`3
O ~ ~ r
U~
u3~_~ In r co ~ ~, o In ~D _
.~ O CO U~ CD -
3 + ~ ~:
E P~ O
h m
V
~ D~ ~ W ~r
:~ -- ~ t` ~ o ~ ~ . ~ 0 .
~ ~ ~ ~ ~ ~ ~ . ,
_~~ ~ ~1 ~ ~ r
x ~
o ~r U .
P~ ~ In W
~s~ I ~ ~ In
~ ~ ~o
u~ l ~ ~ ~r u
a P. ~ a~
~,~ ~ o o ~
H ~ Lt~ o
t~:l 3 + ~ O er ~ O ~ U~ ~D O ~ ~ -1 ~1
S~
_ 1~
U~ v ~ V ~ U~ V ~ U~ ;q O
~ ~ u~
Z t~ E ~ ~ E ~ ) e ~ E
~ :~ X ~ X X ~ X ~ ~ X 1~ ~
:~: ~ ~ ~ ~: O
: ~:; X X Z X Z Q
~ a W ~ u I ~ t~ ~n
E~~ ~ x O ~ x ~ X ~:r O ~C
I O I Z
:> 0 1~ 0 0 Z 0 .~3:
~:1 ~ O p~ O ~ X ~: X
m ~ O ~ O O
~ ~ hl F~ E
.~ P~ ~ ~ ~
W093/15208 PCT/GBg3/flV2~ ~.
~ ~i<3 3~
Reduct~o~ of 4-methyl-2-oxo-3-pentenoic acid using MVS/GG:
MVS/GG (6 units (~ moles/minuteJ30C)) and yeast
formate dehydrogenase ~5 units~ were added to a solution of 4-
methyl-2-oxo-3-pentenoic acid ~1.0 mM) in deoxygenated Tris
buffer (5mM:pH 6.0; ~Oml) containing NADH (0.02 mM), sodium
formate (3.1 mM), fructose-1,6 bisphosphate (0.4 mM) and
dithiothreitol ~O~08 mM). The solution was stirred at room
temperature (~20C) under nitrogen for 5 days with periodic
addition of 0.2 mM HCl to ma:intain p~ in the range of 6.0 ~ 6.2.
Acidification to pH 2.0 and extractive work-up with ethyl acetate
gave (S)-2-hydroxy-4-methyl-3-pentenoic acid in 91% isolated
yield. Analysis of the (R)-MTPA derivative and comparison to a
racemic standard gave a value of at least 99% for entantiomeric
excess.
: : :
.
::
::
~:
.
l ~ :
I ~
.