Note: Descriptions are shown in the official language in which they were submitted.
W O 93/14026 ~ 4 PC~r/US93/00319
, 1 '.
METHOD AND APPARATUS FOR DESULFURIZATION OF A GAS
Field of the Invention ~ `-
This invention relates to the treatment of gases for the
removal of sulfur therefrom and more particularly to a
continuous method for the treatment of flue gases and the like
5to remove sulphur dioxide.
Back~round of the Invention
The combustion of fossil fuels, particularly coal and
high sulfur petroleum, leads to many ~environmental problems
due to the generation of sulphur oxide products during the
10combustion. These sulphur compounds, generally referred to as
SOx are the primary precursors to environmental problems such
as acid rain which has recently gained much attention.
Various methods have been utilized in the prior art for
the treatment of the effluent from the combustion of such
lS~ ~ fossil; fuel~s to remove the sulphur combustion products. For
example, one method involves the creation of a w`ater solution
of sodium sulfite which is then contacted with the flue gas to
produce acid sodium sulfite. The acid sodium sulfite is then
treated with calcium carbonate or ca}cium hydroxide to
~20generate calcium sulfite crystals which are subsequently
- ~oxidized;;~to gypsum. A similar method involves a slurry of
~calcium hydroxide which is then contacted with the flue gas to
-; produce calcium sulfite which is subsequently oxidized to
gypsum. These methods present a major problem relating to the
25disposal of the large quantities of gypsum produced. Also, in
the sodium sulfite process,disposal of the caustic soda
produced by the process presents an additional environmental
problem. i`
.~ i!
-~Another~method involves contacting the flue gas with a ~`
;~0~slurry of magnesium oxide to form magnesium sulfite which may j
- ~ then be thermally decomposed to regenerate SO2 and MgO. These
operations are of low efficiency because the insolubility of !
~the magnesium oxide only permits contact between the flue gas
.~,
; ' '
W093/14026 PCT/US93/00319
212'~ 88~
and the surfaces of the magnesium oxide particles contained in
the slurry. Once magnesium sulfite is formed on the surface
of a particle, the inside of the particle is prevented from
contact with the flue gas so that the ultimate reaction
efficiency is substantially reduced. Because of the relative
inefficiency of the desulphu-rization process, large amounts of
reactant are required for the SO2 removal and a substantial
amount of unreacted MgO plus the MgSO3 produced by the process
results in a large bulk of material for thermal decomposition.
It becomes necessary to use large indirect thermal heaters to
carry out the decomposition process. These heaters are
expensive to purchase and operate yet, without thermal
decomposition, the consumption of magnesium oxide and the
byproduct magnesium sulfite is large rendering this method for
the treatment of flue gasses uneconomical. For instance,
desulphùrization of gas from a 500 ton steam boiler burning
heavy oil would require about 1,310 tons of magnesium oxide
per month and would produce about 3,400 tons of magnesium
sulfite per month. It is highly desirable to substantially
improve both the desulphurization efficiency and the thermal
efficiency of desulphurization processes in order to render
~ the treatment of flue gas to remove sulphur combustion
j products economically feasible.
To avoid the foregoing problems, methods have been
125 developed which take advantage of the solubility of magnesium
¦~ bicarbonate to more intimately mix with the flue gas to remove
the SO2. Although more efficient than the processes utilizing
the less soluble magnesium compounds, recovery of magnesium
oxide for subseguent reuse in the process and recovery of S2
~30 is reduced by the formation of soluble magnesium sulfate which
is not recovered in the process and by inefficient methods for
converting magnesium sulfite to magnesium oxide.
Su D arv of the Invention
Accordingly, it is an object of the present invention to
provide an improved method for the treatment of combustion gas
to remove SO2. ~
Another object of the present invention is to provide a
: ~
~ W093/14026 PCT/US93/00319
~ ~ 1 2 ~ 5~
method for the treatment of flue gases and the like which
substantially reduces the consumption of the reactant
products.
Yet another object of the present invention is to p~ovide
a method for the treatment of flue gases and the like in which
the efficiency of the sulphur removal is substantially
enhanced.
Another object of the invention is to provide improved
apparatus for carrying out the treatment of sulfur containing
gases.
' Still another object of the present invention is to
provide improved apparatus for the thermal decomposition of
,, sulfite produced in order to efficiently regenerate the
i reactants and substantially reduce the consumption of the
'~ 15 reactants.
j The foregoing objects as well as other advantages and
features are achieved by the present invention in which an
alkaline earth bicarbonate is solubilized in water and
,~ contacted by a flue gas. The solubilized alkaline earth
bicarbonate reacts with the S2 in the flue gas to form an
alkaline~earth sulfite which readily precipitates from the
water solution and is efficiently separated therefrom.
During the desulfurization step, a~significant portion of
the magnesium sulfite formed undergoes further reaction with
~-25 the SO2 in the flue gas to form magnesium bisulfite which is
'~ soluble in the aqueous phase of the slurry and is normaIly
~ ~ lost in the a,queous pha~se. In accordance with the invention,
¦ the slurry exiting the desulfurization step is subjected to a
heating step which causes the soluble bisulfite to be
converted to the insoluble sulfite. Following the heating
step, the slurry is then subjected to a separation step to
recover the solid phase of the slurry from the aqueous phase.
The aqueous phase is then preferably recycled to the
,~ desulfurization step or may be disposed of.
',35 The solids are subjected to thermal degradation to
recover the alkaline earth oxide and SO2. The alkaline earth
- oxide can be recirculated and reused in the desulfurization
'
~ ,
~r WO93/14026 PCT/US93/00319
21278~4 ;`
- 4
process to reduce the consumption of the alkaline earth
reactant while the So2, which has practical uses as a
precursor in various chemical processes and therefor is of
commercial value, is liquified.
Thermal degradation of the solids from the
desulfurization step is carried out in apparatus which
includes a preheater zone, an ignition and heating chamber and
a degradation zone. The thermal degradation is carried out in
the presence of heated pellets which are themselves inert to
the degradation reaction. Preferably, the pellets are heated
in the ignition and heating chamber prior to contact with the
solids from the desulfurization step.
It is been found that the alkaline earth sulfite which is
relatively insoluble in water is more readily precipitated and
~l5 more easily separated when an alkali metal sulfite is also
present. Consequently it is highly preferred that the
reactant solution contain an alkall metal bicarbonate which
will also be converted into an alkali metal sulfite and will
enhance precipitation of the alkaline earth sulfite via the
common ion effect.
The solids separated in the process are essentially the
sulfites of both the alkaline earth metal or the alkali metal
and thus the bulk of material is substantially decreased and
~ less expensive equipment can be used for thermally decomposing
25~ the sul~fites.
The method of the present invention is advantageously
carried out in a continuous counter current fashion in one or
more reaction columns. In the preferred embodiment, the
-~ columns are divided into a bicarbonation zone and a
desulphurization zone and the reactants introduced at the
bicarbonation zone travel countercurrent to the flue gas so
that the treated gas exits a~ one end of the column and a
liquid phase and sulfite produced by contact with the flue gas
~ exit as a slurry at the opposite end of the column. In a
-35~ multi-column configuration the zones may be defined in
~ separate columns.
,
The slurry is conveyed to a heated vessel for conversion
",
i~; W093/14026 PCT/US93/00319
2127~38
r~3
of soluble bisulfate to insoluble sulfite. Since the solid
byproduct is e~sentially alkaline earth or alkali metal
-1 sulfite, the bulk of the material relative to the volume of
flue gas being treated is less than with conventional me~hods
and can be more efficiently thermally decomposçd than is the
case for conventional methods in which only the surface area
i of an oxide particle reacts with the So2 to form a sulfite
`~ coating on the particle.
It has also been found highly advantageous to provide
~lO antioxidants in the reactant in order to prevent the oxidation
of sulfite to sulfate which is both water soluble and thus
hard to separate and which is not thermally decomposable and
thus increases consumption of the reactant products.
.
These and other objects and features of the present
invention will become apparent from the following detailed
description of the invention taken in conjunction with the
drawings.
:
`~ Brief Description of the Drawinqs
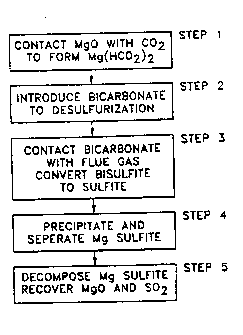
~ig. l is a b}ock diagram of the steps of the method of
the present invention;
Fig. 2 is a schematic diagram of a single column
apparatus for carrying out the method of the present
i m ention;
Fig. 3 is a schematic diagram showing an apparatus for
`~25 ~ thermally decomposing magnesium sulfite;
Fig. 4 is another embodiment illustrating a use of two
columns for thermally decomposing magnesium sulfite; and
Detailed DescriDtion of the Invention
' The present invention provides an improved method for the
O stripping of SO2 from gases produced by the combustion of
- suIphur containing fossil fuels such as high sulphur oil, coal
and the like. Sulphur in such exhaust gases, hereinafter
referred to as flue gas, is oxidized to S2 and if released to
~ the atmosphere in will produce a harmful complex of sulfur
- 35~ compounds, commonly referred to as SOx, which react with
~::
,~
WO93f14026 PCT/US93/00319
21278~3~ -
moisture in the air to form the so called acid rain which is
so harmful to the environment as well as contributing to smoq
which is common in urban locations throughout the world. The
invention is illustrated hereinafter in connection wi~h the
use of magnesium reactants although it will be clearly
- understood that the other members of the alkaline earth group
can be utilized in the present invention.
Referring to Fig. 1, the first step in the method is the
bicarbonization step in which an aqueous slurry of magnesium
in the form of an oxide, carbonate or hydroxide is reacted
with carbon dioxide to produce magnesium bicarbonate which is
water soluble in accordance with the following:
MgO + 2 CO2 + HzO - Mg(HCO3) 2 ( 1 )
MgCO3 + H2O + CO2 - Mg(HCO3)z (2)
Mg(OH)2 + 2CO2 - Mg(HCO3)2
In preparing the reactant for the bicarbonization step
the magnesium oxide, hydroxide or carbonate or mixtures
thereof, are added to water to form a slurry of the
~, ~
-~20 essentially water insoluble magnesium compounds. The amount
-~- of the oxygen containing magnesium compound added in the
initial slurry is not critical although it will be seen from
reaction (4) that essentially one mole of the magnesium
- bicarbonate will react with one mole of the SO2.
~25~ Consequently, sufficient magnesium oxide, hydroxide or
carbonate muæt be added to provide sufficient bicarbonate for
the reaction with the SO2 present in the flue gas.
Preferably, however, the oxygen containing magnesium compound
is added in excess to ensure the production of sufficient
bicarbonate in the reactant to strip the SO2 in the flue gas.
The slurry is contacted with CO2 to produce the magnesium
bicarbonate for the reaction in accordance with the reactions
~ set out (1), (2~ and (3) above. After contact with the CO2
^~ ~the solids portion of the reactant slurry becomes
~35 substantially solubilized in the aqueous phase as the
insoluble oxygen containing magnesium compounds react with the
C2 to form the soluble magnesium bicarbonate. Since no such
:
:
~:
W093/14026 PCT/US93/00319
215 i
reaction is 100% efficient, it will be understood that after
contact with the C02 the reactant still will contain some
solids although far less than in the original slurry. These
solids will comprise unreacted oxygen containing magn~sium
compounds as well as magnesium bicarbonate which is not been
solubilized in the water base. These solids may be separated
from the reactant at this point, although such separation is
not required.
The second step in the method is the desulfurization step
in which at least the aqueous phase of the slurry containing
the solubilized magnesium bicarbonate is contacted with the
S02 of the flue gas to form magnesium sulfite in accordance
with the following:
Mg(HC03)2 + S02 - MgS03 ~ 2 C02 + H2O (4)
As is apparent from reaction 4, there is a mole to mole
reaction between the solubilized bicarbonate and the S02 and
intimate contact between the bicarbonate in solution and the
S02 of the gas being treated provides a highly efficient
~ desulfurization operation. The magnesium sulfite produced by
20~ the reaction of the bicarbonate and S2 is relatively
insoluble in water and is thus readily separated for recovery
of the magnesium sulfite using conventional liquid/solid
separation eguipment. The carbon dioxide formed in the
desulfurization step is preferably recirculated back to the
-25 bicarbonization step to provide the C02 for the
¦~ bicarbonization of the magnesium oxide, carbonate or
¦~ hydroxide.
It will also be noted that the magnesium sulfite formed
during the desulfurization step will react with the S02 in the
flue gas to form magnesium bisulfite in accordance with the
following:
MgS03 + S02 + H20 ~ Mg(HS03)2 ( )
Although this reaction is not undesirable in that it removes
an additional mol of S02, magnesium bisulfite is soluble in
the aqueous phase of the reactant and, unless treated, will be
lost in the aqueous phase. It has been found that from between
about 0.2 to about 0.6 mols of the bisulfite per mol of
,~
::
~ WO93/14026 PCT/US93/00319
~ ~12~8~
sulfite will be formed in the reactant during the
desulfurization step.
The third step of the process is to heat the liquid phase
of the reactant to a temperat~re sufficient to convert~ the
bisulfite back to the insoluble sulfite. It has been found
that heating the liquid phas~ to between about 60 C and about
140-C will effect the conversion in accordance with the
following:
Mg(HSO3)2 - MgSO3 + SO2 (6)
The MgSO3 is then subsequently separated from the aqueous
- phase and recovered as described below~ Although the liquid
phase of the reactant slurry can be heated as described to
effect the conversion of the soluble bisulf~ites to insoluble
sulfites, it is preferred to subject the entire slurry exiting
S from the reaction column to the heating step prior to
separating solids from the acqueous phase. It will be apparent
that, unless treated as described herein, as much as one half
of the magnesium compound may be lost in the process as
soluble bisulfite.
2~0 ~ From the reaction it is seen that as a result of heating
~; ~ the slurry to convert the bisulfite to insoluble sulfite, a
mol~e of~SO2 is also formed. By maintaining an excess of MgO in
the~slurry or~by introducing MgO prior to the heating step the
52 thus~formed is converted to the insoluble sulfite in
accordance with the following: ~
Mg(HSO3) 2 + MgO ~ 2MgSO3 + H2O (7)
Also~ during the desulfurizatioh step, some magnesium
sulfite mày~be~oxid~ized to magnesium su}fate (MgSO~) which is
also soluble in water and which represents additional loss of
l~O magnesium in the process because it is~not easily separated
from the }iquid phase. This reaction represents a potential
loss of magnesium from the process and must be made up by the
; ; addition of fresh magnesium oxide, carbonate or hydroxide at
the bicarbonization step. The loss of magnesium due to
~5~ oxidation of the sulfite to sulfate can be reduced by the
addition of anti-oxidants to the reactant either at the
bicarbonization step or just prior to desulfurization. The
'~
,:.
::~ :
W093/14026 PCT/US93/00319
~127 ~ 8~1
.
. g
anti-oxidant serves to prevent the oxidation of magnesium
sulfite to magnesium sulfate in the presence of oxygen in the
flue gas. The anti-oxidant should be soluble in water and
have a low vapor pressure so as to maintain its anti-ox~idant
effect over a substantial period of time in the presence of
relatively high temperature~. Among the anti-oxidants which
have been found useful in the present invention are hydrazine
and hydrazine salts. Also aryl- and alkyl- hydroxylamine
containing materials such as p-, o-, or m-amino phenol are
~10 also suitable anti-oxidants. Carboxylic acid such as tartaric
and citric can serve as anti-oxidants in the method of this
invention as well as aromatic polyamines such as ortho-, meta-
or para-diaminobenzene. Aromatic hydroxy compounds such as
pyrocatechol, pyrogallol and 1,2,4, trioxybenzene have also
been found suitable as anti-oxidants in the method of this
invention. The anti-oxidant is preferably added in
concentrations of between about 50ppm to about 500ppm with the
-~ exact amouht being a matter of choice depending upon the
oxygen content of the flue gas being treated. As previously
mentioned, magnesium sulfite is insoluble in water and thus
precipitates out of the liquid phase in the desulfurization
step.
The fourth step of the process involves the separation of
1~ the insoluble sulfite from the liquid phase of the reactant.
i~25 The separation of the magnesium sulfite precipitate is carried
out using any conventional liquid solid separation apparatus.
Good results have been achieved using conventional
quid/solid separation means such as for example,
centrifugation, filtration, and screening. Apparatus utilized
for carrying out such operations such as for example
centrifuges, filters, settling bowls and the like are well
known in the art of solid/liquid separation and do not per se
~ ~ form a part of this invention.
- It has been found that the precipitation of magnesium
~35 sulfite from the liquid phase of the reactant is greatly
enhanced in the presence of an alkali metal sulfite- such as
for example sodium sulfite. The alkali metal sulphite is
.
W093/14026 PCT/US93/00319
'~l'Z7~
:
prepared by the addition of an alkali bicarbonate to the
liquid phase of the reactant slurry or can be added as the
carbonate, oxide or hydroxide to the slurry of oxygen
containing magnesium compounds for reaction in~ the
bicarbonization step in the same manner as the magnesium
compounds. The alkali bicarbonate reacts with the SO2 in the
flue gas accordingly to the following:
2NaHCO3 + SO2 - Na2SO3 + H20 + 2C02 (8)
In addition, one mole of alkali sulfite will further
react with a mole of S02 in accordance with the following:
Na2SO3 ~ So2 + H20 2NaHSo3 (9)
Thus, in addition t~ promoting the precipitation of
magnesium sulfite from the liquid phase, the alkali sulfite
also helps to remove the SO2 from the flue gas. -
As mentioned, it is preferred that the alkali metal be
added in the form of an oxide, carbonate or hydroxide for
reaction in the bicarbonation step to form the bicarbonate of
the alkali~metal. In this connection excellent results have
~ ~ i
been achieved using natural alkali minerals sùch as nahcolite,
;~20~ trona, and natron. Nahcolite is a mineral whose major
component is sodium bicarbonate and which is found in saline
mineral deposits. Trona (Na3H(co3) 2. 2H20 iS also a natural
soda. Nahcolite is a mineral comprising hydrated sodium
;bicarbonate.
2S;~ The~quantity of alkali metal bicarbonate which is present
in the re~actant should be sufficient to provide between about
O.l to about 3 moles of alkali sulfite per mole of magnesium
sulfite present after desulfurization. A highly preferred
range is about 0.4 mole to about 1 mole of alkali sulfite per
mole of magnesium sulfite.
The fifth step in the process involves the thermal
degradation of the sulfite (both the magnesium and alkali
metal) for recovery of MgO, alkali earth oxide, if present,
and S02. Since magnesium oxide is decomposed at a temperature
of between about 800-C and 1200-C which is well above the
decomposition of any of the alkaline earth metal sulfites or
al~ali metal sulfites, the degradation temperature is carried
'
,
, ~:
W O 93/14026 P ~ /~S93/00319
?127'~ f"'9.
~ 11
out below the degradation temperature of Mg
Oo and above that of the magnesium sulfite. In this fashion
the alkaline earth and alkali metal sulfites are decomposed to
' their oxides and recirculated to the bicarbonization for reuse
i 5 in the process. SO2 of high purity is formed as a byproduct
¦ of the sulfite decompositio~ and is separated and liquified
for subsequent use for the production of sulphur containing
products such as, for example, sulfuric acid production.
I The method of the present invention is preferably carried
¦10 out as an essentially continuous process in which the
¦ bicarbonate containing reactant is caused to flow
¦ countercurrent to the flue gas being treated. In this manner,
intimate contact between the reactant and the flue gas is
achieved.
Referring to Fig. 2 there is schematically illustrated
¦ apparatus for carrying out the method of the present
invention. A reaction column 12 is provided with an inlet 14
at its lower end for flue gas and an outlet 16 at its top for
treated gas. A spray head 18 is disposed in the top of the
reaction column 12 and is connected by a line 20 and a line 22
to a mixing tank 24 in which the alkali metal oxide containing
sIurry is prepared. The spray head 18 serves to evenly
~istribute the slurry in the reaction column 12. The reaction
column 12 is divided into zones 26 and 28 by a partition 30
including an open ended cylinder 32 which defines a passage 34
through the column 12 for communication between the zones, 26
and 28 respectively. The cylinder 32 is partially closed off
by a member 36 which permits gaseous communication between the
zones but essentially prohibits liquid çommunication
therebetween. Bicarbonation of the alkali and alkaline earth
oxide, carbonate and hydroxide occurs in the upper zone or
bicarbonation zone 26 of the reaction column 12 while the
desulfurization of the gas occurs in the lower or
desulfurization zone 28. Liquid containing bicarbonate
~35 collects at the partition 30 and is pumped through a line 38
¦~ by a pump 40 to a spray head 18' which is disposed in the
upper portion of the desulfurization zone 28. The precipitated
!
W093/l4026 PCT/US93/00319
2 1 2 ~1 ~ 8 '1
sulfites and the liquid phase of the reactant are collected in
a reservoir 29 in the lower portion of the desulfurization
zone 28 of the reaction column 12. These products are pumped
out in the form of a slurry by a pump 42 through a line ~4 and
a line 46 to a heating tank 48 where the slurry is heated to
convert soluble bisulfites in the liquid phase to insoluble
sulfites which are precipitated and combined with the slurry
solids. So2 formed d~ring the heating step is returned to
inlet 14 for processing. Preferably, magnesium oxide is added
at the heating tank 48 to ensure an excess of the oxide for
- reaction with So2 formed during bisulfite conversion to reduce
or eliminate the necessity of processing S02 formed during the
bisulfite conversion step. From the heating tank 48, the
slurry is sent to a liquid/solid separator 50 for separation
of the solids from the liquid phase. The solids are moved by
a line 52 to a thermal degradation unit 54 as will be
described hereinafter as will be described in detail in
connection with Fig.3. The liquid phase is conveyed by a pump
5S through a line 56 and line 20 back to the bicarbonation
zone 26.
The reaction column 12 is further subdivided by fluid
- ~ permeable packing supports 60, for example movable trays,
- screens or the like. Each of the packing supports support
packing materials 62 which serve to diffuse both the flue gas
and the reactant to ensure intimate contact therebetween.
Permeability is provided by openings in the supports 60. The
size of the openings is not critical so long as packing
material 62 can be retained by the supports 60 The packing
material 62 may comprise any of the conventionally used
materials such as Raschig rings or Berl saddles. Preferably,
however, the packing material 62 comprises a plurality of
hollow balls 64 which, as illustrated in FIG.5 are provided
with openings 66 and edge portions of which describe inwardly
extending contoured baffles 68. The design and function of
-35 the hollow balls 64 is set forth in Japanese patent
publication 54-37586, dated Nov 15, 1979. The use of the
hollow balls 64 as the packing material of choice is preferred
- W093/14026 PCT/US93/00319
2~278~ l
since the hollow balls 64 are constantly agitated and moving
by the action of the flue gas and the counterflowing reactant
in the openings 66 and against the baffles 68 to caus~ the
balls to oscillate and vibrate so that the build up of sulfite
on the surface of the ball is avoided. The separated magnesium
sulfite is pumped to a heating unit 70 for thermal degradation
at a temperature of between about 8000C to about 1200C. The
thermal degradation is achieved using a column heater in which
the magnesium sulfite is heated in the presence of alumina
pellets. The apparatus may comprise a single unit or multiple
units.
Referring to Fig. 3 there is schematically illustrated a
single column thermal degradation unit 54 which comprises a
hollow column 72 having closed ends defining a top wall 71 and
a bottom wall 78. Magnesium sulfite is introduced to the
column through an inlet port 74. The alumina pellets are
separately added through an inlet port 75 in the top wall 71
opposite the port 74. A depending partition 80 extends across
the diameter of the column 72 in the upper portion thereof and
~;20 terminates intermediate the top and bottom walls 71 and 78 to
define in cooperation with the top wall 71 and side wall of
the column 72 a drying and pre-heating zone 73 and an ignition
and heating zone 76. Fuel and air are introduced to the
ignition zone 76 through inlet 82. Alumina pellets are heated
~ 25 to the degradation temperature by the ignited fuel/air mixture
! ~ in the ignition and heating zone 76. Flue gas generated in the
¦ ignition zone 76 is conveyed by a line 73 to the inlet 14 of
the reaction column 12 for processing. Magnesium sulfite is
pre-heated and dried in the pre-heating zone 73 which is
, 30 heated by radiation from the ignition and heating zone 76
¦ through the partition 80. Both of the zones 73 and 76 are open
at the bottom and the lower portion of the column 72 is
undivided so that the magnesium sulfite and the now heated
pellets come into direct contact. A rotating cone shaped mixer
86 is }ocated in the column 72 and the mixer 86 is driven
-~ through a shaft 88 and motor 90 to mix the pellets and the
~ sulfite solids which are thermally degraded by the heated
'~:
:
~`:
WOg3/14026 PCT/US93/00319
21278~4
pellets to produce finely divided magnesium oxide. The finely
divided magnesium oxide and the alumina pellets are separated
by a vibrating screen 92 located at the bottom of the column
72 and the magnesium oxide is sent to the make up tank ~4 for
reuse in the process. The alumina pellets are returned by a
line 94 to the port 74 and ~eintroduced in the column 72 for
reheating and reuse in the thermal degradation process. SO2
is separated and exits the column 72 at line 96 for
liquification and storage for use in other chemical operations
in accordance with known procedures.
Referring now to Fig. 4, there is illustrated a pair of
reaction columns 98 and 100 designed for the thermal
degradation of larger volumes of magnesium sulfite in large
S02 stripping operations. The columns are paired with the
column 98 serving as the degradation column and the column 1~0
serving as the ignition and heating column. The ignition
column 100 includes a pair of inlets 102 and 103 for receiving
fue} and pellets to be heated, respectively. Flue gas
generated during the ignition of the fuel is conducted out of
the column 100 through an outlet 104. A rotating mixing
element 106 of the type described above in connection with
Fig.3 is provided to intimately mix the pellets and the fuel
which are ignited in the midsection of the column 100 just
~ below the mixing element 106. A vibrating screen 108 is
provided at the bottom of the column lOO for separating ash,
if any, from the heated pellets and the heated pellets are
then transferred by a line 110 to the degradation column 98.
The degradation column 98 is provided with inlet ports 112 and
114 for receiving magnesium sulfite and the heated pellets,
respectively. An outlet port 116 for S02 is provided in the
upper portion of the degradation column 98 and a rotating
power driven mixing element 118 is provided in the column 98.
The magnesium sulfite and the heated pellets are intimately
- mixed by the mixing element 118 to cause the degradation of
the magnesium sulfite to magnesium oxide and So2. A vibrating
screen 120 is provided at the bottom of the column 98 for
separating finely divided magnesium oxide from the alumina
W093/14026 PCT/US93/00319
~127~
j15
pellets. The pellets are then returned by a line 122 to the
ignition column 100 for reuse in the process and the magnesium
oxide is transferred by a line 133 to the make up tan~ 24 for
reuse in the sulfite stripping process.
The following examples are illustrative of specific modes
of practicing the invention-and are not intended as limiting
the scope of the invention as defined by the appended claims.
~:
ExamDle 1
~ The following example illustrates the S2 efficiency of
a single~ reaction column 12 comprlsing a bicarbonation zone
and a desulfurization zon~e.
A glass column 380cm in height and 10 cm in~diameter was
~- partitioned into an upper~zone`l60cm~in length and a lower
zone 220cm in length. The upper zone was further~divided into
two 15cm coaxial sections by~permeable supports consisting of
screen~having 7.25mm openlngs~and the lower zone was divided~
into~three~;15cm ~sections by the~ screens. ~ Each section
contained~`a~plural~ity of hollow polypropylene packing baIls of
lO~mm~ diameter.~ Each ball had a~surface wall thickness of
~2-0~ ~ 0~.03 mm~and~was provided with five through running openings,
each~2mm ~in diameter.~ Each section ~contained sufficient
number~of~packing~bal~ls to~fill it to~a depth of 30 cm.
A gas~comprising 0.2% S02, 15% C02~, ~84.8% N2 and no oxygen
was~introduced~into the~;column~at a f~low~rate of~I.18 nm3/min.
~ A reàction slurry compri~sing; 4.5 gr of magnesium
~bicarbonate ~ànd ~5.7~;qr ~of~ehe ~ mineral trona in water was
charged~to~the coIumn~at~an~initial flow rate of about 8.4
liters per~minute. 0.6~gr~of magnes~ium carbonate and 0.7gr of
trona were added on an~ ~hourly basis to the reactant to
~30 ~ compensate for any lost magnesium or sodium bicarbonate.
-~ The influent and~effluent gases were analyzed every two
: hours over a ten hour period and the results are set forth in
T~ble 1.
S,'' ~
W093/14026 PCT/US93/00319
2127~84
16
TABLE l
Elapsed Influent Effluent Desulfurization
time (SO2 PPm) (SO2PPm) Efficiençy
2 2000 2.2 99.89
4 2000 1.0 99.95
6 200-0 0.8 99.96
8 2000 2.0 99.90
2000 1.6 99.92
` 10
The exiting material from the reaction column was
filtered to separate magnesium sulfite from the liquid phase
and the liquid phase analyzed for its bisulfite content. The
I ratio of magnesium bisulfite in the liquid phase to magnesiumsulfite solids was found to be in the mole ratio of between
0.2:1 to 0.6:1. The liquid phase was heated to about 65 C to
convert the magnesium bisulfite salt to lnsoluble magnesium
sulfite and refiltered to recover the magnesium sulfite.
The magnesium sulfite was thermally decomposed in the
-20~ presence of alumina pellets heated to approximately 800-C and
the magnesium oxide in finely divided form was separated from
the alumina pellets. Recovery of the magnesium oxide was on
the order of 99.5% of the magnesium oxide introduced to the
-
~ thermal degradation.
: :
Exam~le 2
,~
A series of operations were performed to determine
recovery efficiency of MgO in the thermal degradation
operation using a pellet heater of the type described herein.
The heater was 1 meter in height and had an inside diameter of
i::
~30 2 meters. The inner surface of the heater was lined with
,,~
:::
~:
:
W093/l4026 PCT/US93/00319
2 1 ~ 4
17
magnesium chrome bricks and the outside was lined with
insulating brick, 25 mm in thickness. A pellet hopper was
provided at the top of the apparatus for feeding pelle~s to
the interior. The inner wall of the hopper was lined with a
mortar consisting of a mixture of magnesium oxide and alumina
to avoid contaminating the thermal degradation unit with
metal particles from the sidewalls of the hopper. An
electrically powered vibrating screen (3.9mm openings) was
installed at the outlet of the apparatus for separating the
pellets and the magnesium oxide.
A test material comprising magnesium sulphite (58.14%
moisture) was supplied to the apparatus through the feeding
hopper at the rate of 10.37 kilograms an `hour. Alumina
1~ pellets (about Smm in diameter and length) were fed to the
!~ 15 thermal degradation unit at the rate of about SOkg/hr.
Temperatures inside the apparatus were measured at 700-C. at
I the top, 990-C. at the midsection, and 850-C. at the bottom of
j the apparatus. The fuel used was propane which was fed at the
¦ rate of 4 kilograms per hour. The amount of magnesium oxide
recovered was l.S9 kg. per hour which represented a mol
percent yield of 94.84% of the magnesium sulfite introduced.
In a second operation magnesium sulfite (68.28% moisture)
was introduced at the rate of 10.08 kg/hour while the pellets
were introduced at 5Okg/hr . Temperatures inside the
apparatus were measured as at 788-C. at the top, 998^C. in the
midsection and 845-C at the bottom of the apparatus. Propane
; fuel was fed at the rate of 4 kil. per hour. The amount of
,
- ma~nesium oxide recovered was l.S kil. per hour with a mol
~ .
-,
,
W O 93/14026 PC~r/US93/00319
21278$4 ` `
i 18
yield efficiency of 92.99%.
A third run was made in the same manner as described
above using magnesium sulfite test material (66.57% mois~ure)
fed at the rate of 13.87 kg/hr. The pellets were fed at the
S rate of about 70 kg/hr. ~emperatures measured inside the
apparatus were 775~C. at the top, 998C. at the midsection and
827-C at the bottom. Propane was utillzed at the rate of
4kg/hr as the fuel. The amount of magnesium recovered was
1.76 kil. representing a mol yield efficiency of 9~.28%.
.
:
Exam~le 3
The desulfurization process of the present invention was
utilized on a flue gas from~a boiler utilizing the apparatus
illustra~ted in Fig. 1. The reactio~ column was 12 meters in
~height and had an inside diameter of 1.5 meters. The column
was divided into a carbonization zone and a desulfurization
.
zone with~each zone being $urther divided in sections by
;screens~ hav~ing ~38mm openings, each section included hollow
plastic ba~lls to~a depth of 50 centimeters. Each plastic ball
was 45mm in diameter, had a surface wall thickness of O.lmm,
-~20 ~ was provided with fourteen through running holes of 4 mm in
;diameter and provided with inwardly extending contoured
baffles as illustratéd in Fig. 5.
Fuel gas from a boiler having an average composition of
2000ppm so2, 14%-15% co? and 3%~5~ 2 with the remainder being
~-2~5~nitrogen was introduced into the reaction column at a rate of
,
17,000 nm3/hr. A reactant slurry comprising 3.2 kg/l of
~magnesium carbonate and 3.6 kg/l of the mineral trona (C0
! ~ .
. . . . . . . . . . . .. .... . .. ..
W093~4026 '~1 ~7,Q~'~ PCT/US93/00319
, 19
23%, HC03 17%) was prepared in water to a salt concentration
of 0.0759 kmols/Xl. The slurry was heated to a temperature of
about 50 C. and introduced through a spray head intQ the
bicarbonation zone of the reaction column. Following
bicarbonization, the reactant, in the form of a thin slurry
was led into the desulfurization zone of the reaction column
through a spray head as shown in FIG. 2. The slurry was
introduced at a rate of about 2 kl/min which resulted in a
magnesium compound supply of about 1.6 mols per hour and a
trona supply of about 0.8 mols per hour.
The influent and effluent flue gas was measured every two
hours and the results are set forth in Table 4 below.
.
:
~ .
~, ' ' ` '
WO93/14026 PCT/US~3/00319
~12788~
.
TABLE_2
Elapsed time Inlet SO~ Outlet S02 Desulphur-
(hours) ~ppm! (ppm) ization
_ _ __ __ Efficiency~c
2 20~0 1.78 99.91
4 1945 19.64 98.99
6 2020 6.46 99.68
8 1980 6.34 98.68
2016 1.00 99.95
12 2113 0.92 99.96
14 1976 0.69 99.97
,
Example 4
Utillzing the apparatus of the foregoing example~ the
flue gas was modified by the addition of oxygen to provide an
oxygen contene of 10%. The desulfurization operation was run
as described in Example 4 above and the process was operated
for two-hours. The sulphate content of the liquid phase after
separation of the magnesium sulphite was measured and reported
,
as a percent of magnesium sulphite recovered. After the first
two hours of operation antioxidant (100 ppm~ was added prior
to introduction of the slurry to the bicarbonization zone and
after 2 hours of operation the sulphate ion concentration of
the liquid phase was determined~as a percent of the magnesium
sulphite recovered. A second 2 hour run was then conducted
using a different antioxidant. Runs were repeated as described
above for a total of six different antioxidant compositions.
The antioxidants tested were hydrazine, hydroxylamine, sodium
~30 polythionate and diaminodiphenyl, p-phenylenediamine and o-
-- phenylenediamine. At the end of each run of each of the
antioxidants the sulphaté ion content was of the liquid phase
was measured and reported as a percentage of the magnesium
sulphite recovered. The results are set forth in Table 3
.35 below.
~'
SUBSTIT~JT~ SHEET
`~
f
W O 93/14026 PC~r/US93/00319
21278~4
TABLE 3
Antioxidation Inhibitor .o,,M~,S,O
None ~ 8.1
S Hydrazine 0.2?
Hydoxylene 0.41
Sodium Polvthionate 1.15
Diaminediphen~l 0.',9
P-phenvlenediamine 0.73
O-phenvlenediamine 0.65
The foregoing results show the effectiveness of the use
of antioxidant in preventing the formation of the sulphate of
magnesium sulphate and sodium sulphate which are soluble in
1~ the liquid phase and which represent a loss of magnesium and
sodium since the sulphate salts are not recovered.
While the foregoing invention has been described~in
` connection~with certain preferred embodiments thereof~ various
embodlments other than those described in detail in this
20~ speci;f~ication will occur to those persons skilled in the art,
which arrangements lie within the spirit and scope of the
invention. It is therefore to be understood that the
invention is to be limited only to the claims appended hereto.
Having described the inven~tion~ we claim:
~ ~ :
:
.
.
~,
:; S~JBSTIT~JTE~ S~ET
.. , . ,, ,,, . .. .. . . .. ... , . . . . .. , ,, . . , .. ~ .. .. . . ..