Note: Descriptions are shown in the official language in which they were submitted.
P30264
- 1 -
2128 -~3~
COMPOIJNI~S
5 The present invention relates to novel azabicyclic derivatives, processes for their
preparation, pharmaceutical compositions containing them and their use in therapy, in
par~icular as calcium channel antagonists.
Gerrnan OLS 41 16582 describes azabicyclic compounds of the formula:
= (C~t2)n-X-R
A`\N~C
wherein A, B and C independently represent -CH2- or a single bond; n is zero, 1 Qr 2; X is
oxygen or sulphur and R is inter ~ phenylalkyl, diphenylallyl, heterocyclicalkyl, phenyl,
15 diphenyl or a heterocycle, each of which may be optionally substituted. These compounds
are said to be useful as muscarinic agonists.
Chem Abs. Vol 111, 1989, No. ?32473v reports the attempted synthesis of 3-phenyl-1-
azabicyclo[l.l.l.~pentane and 3-phenyl-1-azabicyclo[2.1.1]hexane. Chem Abs. Vol 84,
20 1976, no. 121623y describes~ 4-phenylquimlclidine and Chem Abs. Vol 71, 1969,No. 22026f desc~ibes the synthesis of 4-aminoquinuclidine and its denvatives~ including
the ~ben~ylamino deriva~ive. No therapeutic use is ascribed to any of these compounds.
International ~plication WO91/13885 descnbes a class of bridged azabicyclic compounds
25 subs~ituted at the bridgehead carbon atom by a triazinyl group. The compounds are said to
act at muscarinic receptors and to be of potential use in the treatment and/or prophyla~cis of
dementia.
We have now found novel azabicyclic compounds, substitued at the bridgehead carbon
30 atom, which have activity as calcium channel antagonists.
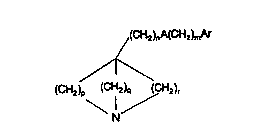
In a first aspect, the present invention provides, a compound of fonnula (I):
S~EET
P30264 ~ 1 2 8 ~ ;!
- 2 -
/(CH2)n A(CH2)~nAr
,f\
(CH2~p (~H2)q ,~CH2)r
\ I /
Formula (I)
in which
s
p, q and r each independently repr~sent an integer from 1 to 4;
A is a bond, -CH=CH-, -C~C-, oxygen, sulphur or NR1; where
Rl iS hydrogen, Cl galkyl or phenylCl 4alkyl;
n is 0 to 6, and m is 0 to 6, such that the length o~ the chain (CH~nA(CH2)m is at least
10 two atoms; and
Ar is aryl or heteroaryl, each o~ which may be optionally substituted;
with the proviso that when p, q and r each represent 2, n is zero, m is 1 and A i~ then
Ar is not unsubstituted phenyl,
5 and s~ts thereo
p and r are preferably 2 or 3.
.,
q is preferably 1 or 2.
llle values of n, m and A should be chosen such that the length of the chain
-(CH~nA(CH2~m is at least two atoms. In general ths length of the chain
-~CH2)nA(CH2)m is ~rom 2 to 6 e.g. 2 to 5 atorns. Preferred values for n and m depend
on the group A. Thus for example, when A is oxygen the sum of n+m is from 1 to 5, for
25 example n may be zero, 1 or 2 and m may be zero or 1 to 5.
A is preferably oxygen or a bond, most preferably oxygen.
When Ar represents aryl~ suitable groups include, for example, unsaturated monocyclic
30 and unsaturated or partially saturated bicyclic or tricyclic ring systems of up to 15 carbon
atoms, such as, for exarnple, phenyl, naphthyl, tetrahydronaphthyl, fluorene, fluorenone,
dibenzosuberene and dibenzosuberenone . Preferred are optionally substituted phenyl
rings.
~il)E3STlT~TE S~E~,
p3()26~ 2 ~
- 2a -
An aryl group may be substituted, for example, by a (: 1 2alkylenedioxy group (e.g. phenyl
substituted by a 3,4-methylenedioxy group) or by 1 to 3 substituents selected from
halogen, Cl 4alkoxy, ni~o, SCl 4alkyl, NR2aR2b (in which R2a and R2b can be
independently H or Cl 4alkyl), OCF3, Cl 6alkyl, tIifluoromethyl, CN, optionally
S substituted phenyl, optionally substituted phenoxy, optionally substituted phenylCl 4allcyl
and ophonally substituted phenylCI 4alkoxy.
Suitable op~ionally substituted phenylCl 4alkyl groups include, for example benzyl.
Suitable optionally substituted phenylCl 4alkoxy groups includs, for example benzyloxy
10 groups.
Suitable subst;tuents for said optionally substituted
phenyl, phenoxy, phenylCl 4alkyl and phenylCl 4alkoxy groups include for examplehalogen, Cl 4alkyl, Cl 4alkoxy, nitro and tlifluoromethyl groups.
S~ TUTESH ~7-
~0 93/15080 ~ pcr/GB93/oo174
Preferably the aryl group Ar is a phenyl ring substituted by one or two substituents, in
particular, by a phenyl, phenyl(Cl 4)alkyl e.g. benzyl, phenoxy or phenylCl 4alkoxy, e.g.
benzyloxy group; or by two chloro atoms especially in the 3- and 4-positions of the phenyl
s ring.
When Ar represents heteroaryl suitable groups include, for example, unsaturated or
par~ially saturated bicyclic and tricyclic ring systems containing at least one heteroatom.
A bicyclic ring systeml preferably contains 8 to 10 nng members, such as quinolinyl,
10 tetrahydroquinolinyl or benzofuranyl. A tricyclic ring system preferably contains from 11
to 15 ring members, and most preferably has the structure:
~Z~
wherein Yl represents Y(CH2)t, Y is O, S or NR3 (wher~ R3 is hydr~gen or Cl:4alkyl), Z
is (C~12)S or -C~=CH-, s is 0, 1 or 2 and t is 0 or 1 or is a corresponding dehydro ring
system. Examples of tricyclic heteroaryl groups include dibenzofuranyl, dibenzothienyl,
carbazole, N-methylcarbazole, acridine and dibenzoxepine . The heteroaryl ring can be
20 linked to the remainder of formula ~I) via any suitable ring atom.
Suitable substituents for said heteroaryl rings include, for example, 1 to 3 subsdtuents
selected from halogen, ~rifluoromethyl, Cl 4allcyl, Cl 4alkoxy, phenyl, phenylCl 4alkyl,
andphenylCl 4alkoxy.
ALkyl groups present in the compounds of formula (I), alone or as part of another group,
can be s~aight or branched. Thus a C l 4allcyl group may be for exarnple methyl, ethyl,
n-propyl, n-butyl or any branched isomer thereof such as isopropyl or t-butyl.
30 It will be appreciated ~hat for use in medicine a salt of a compound ~l) should be
phamlaceutically acceptable. Examples of pharmaceutically acceptable salts include
inorganic and organic acid addition salts such as hydrochloride, hydrobrornide, sulphate,
phosphate, acetate, fumarate, maleate, ci~ate, lactate, tartrate, oxalate, methanesulphonate
or similar pharmaceutically acceptable inorganic or organic acid addition salts. Other non-
35 pharmaceutically acceptable salts may be used for example in the isola~ion of the ~lnalproduct and are included within the scope of this invention.
Wo93/1~08~ ~ 2~ ~ i 4 PCI/GB93tO0174
Particular compounds of the invention include:
4-[2-(3,4-dichlorophenoxy)ethyl]- 1 -azabicyclo~2.2. 1 ]heptane hydrochloride,
4-[2-(4-benzyloxyphenoxy)ethyl]-1-azabicyclol2.2.1]hep~ane hydrochloride,
4-12-(2-dibenzofuranyloxy)ethyl]-1-azabicyclo[2.2.1]heptane hydrochloride,
(i)5-(4-benzyloxyphenoxymethyl)- l-azabicyclo[3.2. l~octane hydrochloride,
(i)5-(4-benzylphenoxymethyl)- 1-azabicyclo[3.2. 1 ]octane hydrochloride,
(+)5-~2-dibenzofuranyloxy)methyl- 1 -azaibicyclo[3.2. 130ctane hydrochloride,
(~ ) 5-[2-(2-dibenzofuranyloxy~ethyl]- 1 -azabicyclo[3.2. 1 ]octane hydrochloride,
4-[2-(4-phenoxyphenoxy)ethyl]-1-azabicyclo[2.2.1]heptane hydrochloride,
4-[3-(4-benzyloxyphenyl)propyloxyme~hyl]- 1 -azabicylo[2.2. 1 ]heptane hydrochloride,
4-[5-(~phenoxyphenyl)pentyloxy]- 1 -azabicyclo[2.2. 1 ~heptane hydrochloride,
4-~4-benzyloxyphenoxymethyl)- 1 -a~abicyclo[2.2. 1 ~heptane hydrochloride,
15 4-[4-(4-phenoxyphenyl)butyloxy]-1-azabicyclo[2.2.1]heptane hydroehlonde,
~3-(4-benzyloxyphenyl)propyloxy]-1-azabicyclo[2.2.1~hep~ane hydrochlonde, and
4-[5-(4-benzyloxyphenyl)pentyloxy]-1-azabicyclo[2.2.1]heptane hydrochlonde.
lt will be appreciated that certain compounds of formula (I) may contain one or more
20 asymmetric centres, for example where p, q and r all have different vall~es. Such
compounds will exist as optical isomers (enan~iomers). Both the pure enantiomers,
racemic mixtures (50% of each enantiomer~ and unequal mixtures of the two are included
within the scope of the invendon. Fur~hert all diastereomeric forrns possible (pure
enantiomers and mixtures thereof) are within the scope of the invention. In addition, when
2s A represents -~H=CH- the compounds will exist as geometric isomers, and the invention
encompasses all such isomers and mixtures thereof.
The compounds of the present invention can be prepared by p~cesses analogous to ~hose
known in the art. The present invention therefore provides in a further aspect, a process
30 ~or the preparation of a compound of formula (1) which comprises:
(a) for compounds of formula (I) in which A is 0, S or NR1, reac~ilon of a compound
of formula (II):
WO 93/1~080 5 P~tGB93/00174
.~. .t 2 ~3 i
(CH2)nA H
~,
(CH2)p (C~2)q ,~CH2).
\ I /
Formula (II)
in which p, q, r and n are as described for formula (I) and A 1 is O~ S or NR 1, with a
compound of fonnula L(CH2)mAr in which m and Ar are as described for ~ormula (I), and
L is a leaving group;
10 (b) for compounds of formula (I) in which A is O, S or NR 1, reaction of a compound
of formula (III):
/(CH2)nL
~ .~
(CH2)p (CH2)q ,~;H2),
\ 1/
Formula ~III)
in which p, q, r and n are as descnbed for forrnula (I) and Ll is a group displaceable by a
nucleophile, with a compound of formula HA1~CH2)mAr where m and ~r are as
describe~ for formula (I) and A~ is as dcscribed for formula (II); or
(c) for compounds of fo~mula ~I) in which A is NR1, reduenon of a compound of
formula (IV):
(( H2)p (CH2)q ,~CH2)r
\ 1/
Formula ~IV)
WO 93/15080 ~ 6 - PCI/GB93/00174
in which R4 represents the group
-(cH2)nN(Rl)c(o)(cH2)m lAror-(CH2)n 1C(O)N(R )(CH2)mAr,
and p, q, r, n, m, and Ar are as described for formula (I);
(d) for compounds of formula (I) in which A is a bond, reaction of a compound offormula (V):
(CH2)n~mL
~CH2)p (CH2)q ,5ÇH2)r
\ I /
Formula (V)
.,
15 (wherein Ll, p, q, r, m and n are as hereinbefore defined).
with a compound of formula XlAr in which Ar is as described for formula (I), and Xl is
an aLkali metal;
2~ (e) For compounds wherein A is -~I=CH- reaction of a compound of forsnula ~VI):
(CH2 ~n ~HO
(CH2)p ~CH2)q ,~CH2),
\ I /
Formula ~VI)
(wherein n, p, q and r are as hereinbefore de~lned) with a reagent serving to intrGduce ~he
group Ar;
WO 93/15080 - 7 - 2 l 9 j ~ ~ Pcr/GBg3/00174
(f) lnterconversion of one compound of for ~ O a different compound of
formula (I) e.g. the reduclion of a compound wherein A is -CH=CH- to a compound
wherein A is -CH2CH2-;
and optionally thereafter forming a salt.
In process (a) the reaction between a compound ~; mula (II) and a compound
L(CH2)mAr can take place under conditions wh ~ ~end on the nature of the group Land the value of m. For example, when L is ha' ~r a sulphonic acid residue such as a
tosylate or mesylate and m is other than zero, the ~ on is carried oul under standard
10 conditions in a solvent, op~ionally in ~e presence i ~ase. When a fluoro-substituted
aryl compound F-Ar is employed in process (a), (to prepare cornpounds where m is zero)
the reaction is effected in the presence of a strong base such as sodium hydride, and in an
inert organic solvent such as dimethylformamide. Preferably the aryl group is substituted
by an activating group such as CE;3 or N02. If necesary, the azabicyclic nitrogen atom
ls may be protected during the reaction by methods well known in the art, e.~s. by prior
fonnation of a quaternary derivat; t:uch as an N-benzyl derivative. Protection may also
be effected by formation of a bor BH3) complex. lt will be appreciated that the N-
protecting group should be chose; uch that it can be removed without affectin~ other
moieties in ~he molecule. Thus ~or example a benzyl protecting group may not be
20 appropriate when the side chain (CH2~nA(CH2)mAr also contains a benzyl moiety such as
a benzyloxy group. ln general, N-protection is prefe~ed when the leaving group Lrepresents halogen, e.g. bromine, but when L is a sulphonic acid residue e.g. a tosylate, N-
protection may not be necessary.
2s l~e reaction between a compound of formula (III) and a compound of forrnula
HAl(CH2)mAr (process (b)) can take place under conditions which depend on the nature
of L1 and A. For example when Ll is hydroxy, m is 0 and Al is oxygen or sulphur the
reaction is carried out in the presence of diethyl azodicarboxylate and tnphenyl phosphine.
Such a reaction is 3cnown as the Mitsunobu reaction (as described in Synthesis 198l, 1).
30 Alternatively the leaving group Ll may be for example a halogen atom or a sulphonyloxy
group eg. methane-sulphonyloxy or p-toluene sulphonyloxy in which case the compound
(III) may preferably be protected, e.g. as an acid salt such as a hydrochloride salt.
Reaction may be effected in the presence or absence of solvent, at a temperature in the
range 0 to 200C, and may preferably be carried out in the presence of a base.
The reduction of a compound of formula (IV) according to process (c) can be effected by
methods known in the art, for example using a reducing agent such as lithium aluminium
hydride. Conveniently a compound ~ _ula ~ ~/ca bcp--l~a ~ ~fm ~:am~e ~
Wo93tl50X0 ~ t ~ 8 PCI/GB93/00174
described below) and reduced in a 'one-pot' reaction, without isolation of compound (IV)
itself.
Ihe reaction between a compound of fonnula (V) and a compound of fonnula XlAr ins process (d) can take place under standa~d conditions known to those skilled in the art for
the formation of carbon-carbon bonds.
Process (e) may be effected using a Wadsworth-Emmons reagent for example of the
formula Ar(CH2)m+lP(O)(OAlk)2, such as a diethylphosphonate, or a Wittig reagent of
10 the formula Ar(CH2)m+lPPh3X (where X is an anion) which compounds are available
commercially or can be prepared by known methods. The reaction may be carried out in a
solvent such as tetrahydrofuran, optionally containing a crown ether such as 1 5-crown-5,
or l 8-crown-6, and in the presence of a strong base such as sodium hydride, or potassium
t-butoxide.
Interconversion reactions according to process (f) may be effected by methods well known
in the art. Thus for example conversion of a compound (1~ wherein A represents
=CH- into a compound (i) wherein A represents-CH2-CH2- may be effectet by
satalytic reduction.
Compounds of formula ~Il) wherein n is l-6 and Al is oxygen can be prepared by
reduction of the corresponding ester of formula (VII): -
(CH2~n.l C02Alk
~f~ `'
(C;H2)p (CH2)q ,~CH2~,
\ 1/
Formula (VlI3
wherein p, q and r are as hereinbefore defined and Alk is a Cl 6alkyl group e.g. ethyl.
The reduction may be effected using a reducing agent such as lithium aluminium hydride
30 in a solvent such as diethyl ether or tetrahydrofuran. Esters of formula ~VIl) wherein n is
l are described for example in European Patent Applications 287356 and 3928V3 and by
Eckhardt ~ al Helv. Chim Acta, 1972, 55, 2432, and B.S. Orlek et al., J. Med. Chem.,
1991,34,2726. Esters wherein n is greater than l may be prepared by conversion of an
esler wherein n is I to the corresponding N-methyl-N-methoxycarboxamide (e.g. by
WO 93/15080 2 :b 2 ~ ~' ` PCI/GB93/00174
hydrolysis of the ester followed by reaction with thionyl chlonde and
- N,O-dimethylhydr~xylamine hydrochloride), which is then reduced to the aldehyde using
diisobutylaluminium hydride. The aldehyde is further converted to the cyanomethyl
deriva~ive for example as described in EPA 363085, followed by acid hydrolysis, and
5 esterification to form an ester wherein n is 2. The sequence may be repeated to form
higher homologues.
Alternatively compounds of formula (II) may be prepared by reaction of an aldehyde of
formula (VI) with triethylphosphonoacetate or triethylphosphonocrotonate, followed by
o catalytic hydrogenation to give an ethoxycarbonylaL~cyl derivative which is further reduced
e.g. using lithium aluminium hydride, to the desired hydroxyalkyl compound. It will be
appreciated that use of trie~ylphosphonoacetate results in a 2-carbon homologation whilst
triethylphosphonocrotonate gives a 4-carbon homologation.
S Compounds of fo~mula (II) wherein n is zero and p and r are both 2 may be prepared from
l-benzyl4-piperidone, by a vanety of methods. For example, a compound (lI) wherein r
is 1 may be prepaIed according to the following reaction scheme:
(CH3)3SO+I ~O ~1) HCO2H OHXCH2GH
(2) NaHC03 ~ J
N NaHJDMSO l CH2Ph
CH2Ph CH2Ph MeOH-H20
.,
(1) tosyl
choIide/
py~dine ~ r
(2) refluxholuene
C)H H2110% Pd-C OH
Ethanol ~
CH2 Ph
Alternatively the 1-benzyl-4-piperidone may be converted to the 4-hydroxy-4-
hydroxymethyl compound by the method described in EPA 188~55, via coIresponding
cyano and ester derivatives
WO 93/15080 ~ - 10 PCl/GB93/00174
A compound of formula (II) wherein n is zero, and p, q and r are each 2 may be prepared
by the method of C.A. Grob and P. Brenneisen (Helv. Chim. Acta.~ 41, 1184, 1958) in
which 1-benzyl-4-piperidone is reacted with zinc and ethylbromoacetate to give the
corresponding 1-hydroxy-1-ethoxycarbonylmethyl derivative which is reduced usingS lithium aluminium hydride to the 1 -hydroxy- 1-hydroxyethyl compound and then treated as
above to effect cyclisation and deprotection.
Compounds of formula (II) wherein n is zero, p is 3, r is 2 and q is 1, may be prepared as
described in EPA 2~73S6.
Compounds of formula (II) wherein A1 is S or NR1 may be prepared from the
corresponding hydroxy compound by standard methods, for example via formation of an
aL~cyl halide followed by reaction with an appropriate amine or thiol.
15 Compounds of forrnula (III) wherein Ll is OH can be prepared as described forcompounds of formula (Il), and compounds of formula (III) wherein Ll is a halogen atom,
or a mesyloxy or tosyloxy group can be prepared from the corresponding alcohol in
conventional manner. ~
20 The compounds of formula L(CH2)mAr and HA 1 (CH2)mAr may be prepared by standard
methods well known in the art. For example compounds L~CH2)mAr wherein Ar is a
substituted phenyl group and L is halo, e.g. bromo, may be prepared by Friedel-Crafts
acylation of lhe corresponding substituted benzene derivative, using an appropriate acid
chloride and catalysed by aluminium ~ichloride, followed by reduction in situ with
2s ~iethylsilane.
When Ar represents a phenyl group subs~itued by benzyloxy compounds L(CH2)mAr may
be prepared according to the following scheme:
OH OCH2Ph OCH2Ph
cH2sr ~1~ losylehloride ,~
K2C03 ~ pyridine
(CH2)n-OH ~CH2)nOH (CH2)nOTs
3~
The starting materials are available commercially or may be prepared by standardmethods, e.g. by reaction of 4-benzyloxybenzaldehyde with triethylphnsphonocrotonate in
a similar manner to that described for the preparation of compounds (II).
WO 93~15080 2 ~ 2 ~3 ~ . PCI /GB93/00174
Compounds of formula (IV) wherein R4 is a group
-(CH2)nN(Rl)C(O)(CH2)m 1Ar can be prepared by reacting a compound of folmula (II)
wherein Al represents NR1 with an acylating agent corresponding to the group -
5 (CH2)mAr, for example an acid chloride ClOC(CH2)m 1 Ar.
Compounds of formula (IV) wherein R4 is a group
-(CH2)n lC(O)N(Rl)(CH2)mAr may be prepared for example by reaction of a
corresponding compound wherein R4 represents -(CH2)n I C02H or an activated
lo derivative thereof such as an acid halide, ester or anhydride, with an amine of formula
HN(Rl)(CH2)m Ar. It will be appreciated that when the acid itself is employed, reaction
with the amine should be effected in the presence of a coupling agent. The carboxylic acid
may itself be prepared for example by oxidation of the corresponding alcohol? ie. a
compound of formula (II) wherein A 1 is oxygen.
Compounds of formula (V) may be prepared in analogous manner to compounds of
formula (III); where necessary the chain length may be increased using methods well
known in the art.
20 Compounds of formula (VI) may be prepared by conventional methods, for example the
oxidation of a compound of formula (II) wherân A 1 is oxygen, or conversion of the
corresponding ester, e.g. via the corresponding N-methyl-N-methoxycarboxamide and
reduction with diisobutylaluminium hydride, as described hereinabove. Compounds of
fonnula ~VI) wherein n is 1 may be prepared from the corresponding compound wherein n
2~ is zero by various methods. For example the aldehyde wherein n is zero may be ~eated
with (methoxyrnethyl) triphenylphosphonium chloride and potassium t-butoxide, followed
by a strong acid, e.g. concentrated sulphuric acid, resulting in the aldehyde wherein n is 1.
Altçrnatively the aldehyde may be converted to the corresponding cyanomethyl derivative
as described in EPA 363085 followed by acid hydrolysis, convçrsion tO the N-methyl-
30 N-methoxycarboxamide and reduction. These procedures may also be used to fo~n higher
homologues.
When a compound of formula (1) is obtained as a mixture of enantiomers, these may be
separated by conventional methods such as crystallisation in the presence of a resolving
35 agent, or chromatography. for example using a chiral HPLC column.
The compounds of the invention have been found to exhibit high calcium influx blocking
activity for example in neuronal cells. As such the compounds are expected tO be of use in
WO 93/15080 ~ 12 pcr/GB93/oo174
therapy in treating conditions and diseases related to an accumulalion of calcium in ~he
brain cells of mammals, in panicular humans. For example, the compounds are expected
to be of use in the treatment of anoxia, ischaemia including for example stroke, migraine,
epilepsy, traumatic head inju~y, AIDS-related dementia, neurodegenerative diseases such
5 as Alzheimer's disease and age-related memory disorders, and drug addiction withdrawal
such as ethanol addiction withdrawal.
In a further aspect of the invention there is therefore provided a method of treatment of
conditions or diseases related to (e.g. caused or exacerbated by) the accumula~on of
10 calcium in the brain cells of marnmals (e.g. humans) which comprises administering to a
subject in need thereof an effective amount of a compound of formula (I) or a
pharmaceu~ically acceptable salt thereof. Thus for example, the present invention
provides a method of trea~ment of anoxia, ischaemia including for example stroke,
migraine, epilepsy, traumatic head injury, AIDS-related dementia, neurodegenerative
15 diseases such as Alzheimer's disease and age-related memory disorders, and drug addiction
withdrawal such as ethanol addiction withdrawal, which comprises administering to a
subject in need thereof, an effective amount of a compound of formula (I) or a
pharmaceutically acceptable salt thereof. ..
20 In a yet further aspect the invention also provides the use of a compound of fortnula (I) or
a phannaceutically acceptable salt thereof in the manufacture of a medicamen~ for the
treatment of a condition or a disease related to ~e.g. caused or exacerbated by) the
accumulation of calcium in the brain cells of a mammal ~e.g. a human).
2s For use in medicine, the com~ounds of the present invention are usually administered in a
standard pharmaceutical composition. The present invention therefore provides in a
further aspect pharrnaceutical compositions compnsing a compound of formula (1) or a
pharmaceutically accep~able salt thereof and a phannaceutically acceptable carrier or
excipient.
The compounds of the inven~ion may be administered by any convenient method, forexarnple by oral, parenteral, buccal, rectal or transdennal administration, and the
pharmaceutical compositions adapted accordingly.
3~ The compounds of formula (I) and their pharmaceutically acceptable salts which are active
when given orally can be formulated as liquids, for example syrups, suspensions or
emulsions, tablets, capsules and lozenges.
WO 93/15080 2 ~ 2 8 ~ X ~ PCl/GB93/00174
A liquid formulation will generally consist of a suspension or solution of the compound or
pharmaceutically acceptable salt in a suitable liquid calTier(s) for example, ethanol~
glycerine, non-aqueous solvent, for example polyethylene glycol, oils, or water with a
suspending agent, preservative, flavouring or colouring agent.
s
A composition in the fo~n of a tablet can be prepared using any suitablç pharmaceutical
carrier(s) routinely used for preparing solid formulations. Examples of such carr~ers
include magnesium stearate, starch, lactose, sucrose and cellulose.
10 A composit~on in the form of a capsule can be prepared using routine encapsulation
procedures. For example, pellets containing the active ingredient can be prepared using
stand~rd carriers and then filled into a hard gelatin capsule; alternatively, a dispersion or
suspension can be prepared using any suitable pharmaceutical can~er(s), for example
aqueous gums, celluloses, silicates or oils and the dispersion or suspension then filled into
lS a soft gelatin capsule.
Compounds of the invention may also be administered parenterally, by bolus injection or
continuous in~usion. Typical parenteral compositions consist of a solution or su~pension
of the compound or pha~naceutically acceptable salt in a sterile aqueous carrier or
20 parenterally acceptable oil, for example polyethylene glycol, polyvinyl pyrrolidone,
lecithin, arachis oil or sesame oil. Alterna~ively, the solution can be Iyophilised and then
reconstituted with a sui~ble solvent just prior to administration.
Both liquid and solid compositions msy contain other excipients known in the
2~ pharrnaceutical art, such as cyclodex~ins.
Preferably the cvmposition is in unit dose form such as a tablet, capsule Qr ampoule.
Each dosage unit for oral adminis~ation contains preferably from 1 to 25S) mg (and for
30 parenteral administration contains pre~erably from 0.1 to 60 mg) of a compound of the
formula (I) or a phannaceutically acceptable salt thereof calculated as the free base.
~e daily dosage regimen for an adult patient may be, for example, an oral dose of
between 1 mg and 500 mg, preferably between 1 mg and 250 mg, eg. 5 to 200 mg or an
35 in~ravenous, subcutaneous, or intramuscular dose of between 0.1 mg and l O0 mg,
preferably between 0.1 mg and 60 mg, eg. I to 40 mg of the compound of the formula (1)
or a phartnaceutically acceptable salt thereof calculated as the free base, the compound
being administered 1 to 4 times per day. Alternatively the compounds of the invention
wo 93/15080 ~ ~ ?~ ~ - 14 - pcr/GB93/ool74
may be administered by continuous intravenous infusion, preferably at a dose of up to
400 mg per day. Thus the total daily dosage by oral administration will be in the range l
to 2000 mg and the total daily dosage by parenteral administration will be in the range 0.1
to 400 mg. Suitably the compounds will be administered for a period of continuous
5 therapy, for example for a week or more.
BIOLOGICAL DATA
Ca2+ Current Measurement
Cell preparations
Sensory neurons from dorsal root ganglia were dissociated from 1 day old rat pups (Forda
et al, Developmental Brain Research, 22 (1985), S5-65). Cells were plated out onto glass
coverslips and used within 3 days to pennit effective vollage clamp of Ca2+ currents.
Solutions
The pipette (internal solution) contained in mM: CsCl, 130; HEPES, l(); EGTA, l0;
MgCL2, 4; ATP, 2; buffered to pH 7.~ with CsOH. Cells were bathed in a normal
Tyrodes solution before esta~lishment of whole cell recording when the bathing solution
20 was changed to one allowing isolation of Ca2+ currents. The external solution for
recording Ca2+ channel currents contained in mM: EaCL2, l0; TEA-CI, 130; glucose,
10; HEPES, l0; MgCL2, 1; buffered to pH 7.3 with TEA-OH. Barium was used as the
charge carrier as this assists in current isolation and caIcium dependent inactivation of
current is avoided. Compounds were dissolved in DMSO to make a 2Q mM stock
2s solu~ion. At the drug concentra~ion used the vehicle (0. l %) had nv significant effect on
Ca2+ currents. All experiments were perfo~med at 2l to 24C. Whole cell currents were
recorded using List EPC-7 amplifiers and stored, digitised for la~er analysis using PC
based software similar to that described previously (Benham & Tsien, Journal of
Physiology (l9~8), 404, 767-784).
3~
Ca2~ currents
Peak voltage gated Ca2+ channel currents of up to l0 nA from dorsal root ganglion
neurons were recorded using 10 mM Ba2+ as charge carrier. Currents were evolced frorn
a holding potential of -80 mV to a test potential of 0 or +10 mV every lS seconds. This
3~ test potential was at the peak of the cu~ent voltage relationship and assessing block at this
point reduced any elTors due to drifting holding potential. Some cells showed slow
rundown of current as is commonly seen when recording Ca2+ currents. The rundownrate was measured in control conditions and extrapolated through the time of drug
WO 93/15080 ~ ~ 2 3 ~ 3 ~; pcr/GB93/ool74
application to derive a control value to relate the dlug affected current to. Block by 2()
,uM drug was assessed 3 minutes after drug application.
Compounds of the invention gave percentage inhibi~ion of plateau Ca2+ current in the
S range 71 to 99%.
WO 93/15080 r s~ r~ ~ 16 - pcr/~B93/oo174
8 ~
~, .
PHARMACEUTICAL ~ORMULATIONS
1. Formulation for intravenous infusion
Compound of fonnula (1) 0.1 - 60 mg
Sodium hydroxide/hydrochloric acid to pH ca 7
polyethylene glycol 0 - 30 ml
propyleile glycol 0 - 30 ml
alcohol 0 - 10 ml
water to lOOml
2. Formulation for bolus injection
Compound of fonnula (1) 0.1 - 60 mg
sodium hydroxide or hydrochloric acid to pH ca 7
polyethylene glycol 0 ^ 2.~ ml
alcohol 0 - 2.5 ml
water to S ml
20 A toni~ty adjusting agent eg. sodium chloride, dex~ose or mannitol may also be added.
3. Tablet for oral administration
mg/tablet
2s Compound of formula (I) 25
lactose 153
starch 33
crospovidone 12
microcrystalline cellulose 30
magnesium stearate 2
~ L22~
W093/15080 - 17- PCr/GB93100174
The following non-limiting examples illustrate the preparation of compounds of formul~
(I)
Preparation 1
s 4-(Methoxycarbonylmethyl)-l-azabicyclo[2.2.1]heptane
A solution of 4-(cyanomethyl)-1-azabicyclo[2.2.1~heptane (EP 363085, Description 25)
(1.94g, 14.3mmol) in SN hydrochloric acid (25ml) was heated under reflux for 12h. The
reaction mixture was concentrated in vacllo, then coevaporated with toluene to remove the
tast ~aces of water. The residue was dissoh~ed in methanol (~Oml), treated with lM
ethereal hydrogen chlolide then heated at reflux for 2h. The solution was concentrated
in vacuo, treated with aqueous potassium carbonate (25ml) and extrac~ed into chlorofonn
(3x25ml). The combined organic extracts were dried over sodium sulphate, concentrated
in vacuo and the oil produced was distilled to afford the title compound as a clear oil
(2.1g, 87%) b.p. 125 C, 0.4mm Hg (Kugelrohr).
lH Nmr (CDC13) ~: 1.28-1.42 (2H, m), 1.49-1.64 (2H, m), 2.37 (2H, s), 2.52-2.71 (4H,
m), 2.87-3.02 ~2H, m), 3.67 (3H, s).
Preparation 2
4-(2-Hydroxyethyl)-l-azabicyclo[2.2~1]heptane
A ~olution of 4-(methoxycarbonylmethyl)-1-azabicyclo[2.2.1]heptane (2.1g, 12.4mmol) in
dry diethyl ether (lOml) was added to a stirred suspension of lithium aluminium hydride
- 2s (0.94g, 24.9mmol~ in dry diethyl ether (70m1), under nitrogen. The reaction was stirred at
room temperature ~or 2.5h and then quenched by careful addition of the minimum amount
of water. The reaction was filtered and the precipitate washed thoroughly wi2h 20%
methanol in diethyi ether. The combined ~ ate and washings were concentrated in vacuo
and the r~sidue was dis~illed to afford the title compound as a white sQlid (1.49g, 8~%)
b.p. 1~5 C, 0.3mm Hg (Kugelrohr).
lH Nmr (CDC13) ~: 1.20-1.35 ~2H, m), 1.42-1.57 (2H, m), 1.94 (2H, t, J=7Hz), 2.29 (2H,
m), 2.39-2.80 (3H, m), 2.84-3.00 (2H, m), 3.72 (2H, t, J-7Hz).
wo 93/15080 ~ ? $~ 18 - P~/GB93/00174
jJ
Preparation 3
~+)~-Hydroxymethyl-l azabicyclo[3.2.1]octane
The title compound was prepared in a similar manner to Preparation 2 from (+)ethyl 1-
5 azabicyclo[3.2.1~-oct-5-yl carboxylate (EP 287 356, Example 7) (2g, 10.~mmol) and
lithium aluminium hydride (0.83g, 21.9mmol). This afforded the title compound as a
clear gum (1.24g, 80%) b.p. 150 C, 0.7mm Hg (Kugelrohr).
Preparation 4
( ~ ) 5-(2-Hydroxyethyl)-l-azabicyclol3.2.1]octane
The title compound was prepased in a similar manner to Preparation 2 from (~) 5-methoxycarbonylmethyl-1-azabicyclo[3.2.1]octane (EP 363085, Description 11) (0.74g,
4.0 mmol) and lithium aluminium hydride (0.46g, 12.1 mmol). This afforded the title
compound as a clear oil (0.54g, 87%) b.p. 200C, 0.2 mm Hg (Kugelrohr)
lH Nrnr (CDC13) ~: 1.37-1.82 (8H, m, overla~ping signals), 2.43-3.05 (6H, m,
overlapping signals), 3.68 (2H, t, J=7Hz)
Preparation 5
4-Hydroxymethyl~l-azabicyclo[2.2.1]heptane
The title compound was prepared in a similar manner to Preparation 2 from methyl 1-
azabicyclo[2.2.1]hept-4-yl carboxylate (B.S. Orlek et al., J. Med. Chem., 1991, 34, 2J26)
2~ (3.13g, 20.2 mmol) and lithium aluminium hydride ~1.92g, S0.6 mmol). This afforded ~he
title compound as a colourless solid (1.76g, 69%) b.p. 2~0C, 0.3 mrnHg ~Kugelrohr).
lH Nrnr (CD(: 13) ~: 1.27 (2H, m), 1.65 (2H, m), 2.32 (2H, s~, 2.62 (2H, m), 2.94 (2H,
m), 3.72 (lH, br s), 3.87 (2H, s)
Preparation 6
l-Benzyl-4-hydroxymethyl-4-hvdroxypiperidine
To a suspension of sodium hydride (4.76g of an 80% dispersion in mineral oil~ 0.159 mol)
3s in dry dimethyl sulphoxide was added trimethyl sulphoxonium iodide (34.92g, 0.159 mol)
over 1.5h. The mixture was stirred for lh, then cooled in ice and treated with a solution of
1-benzyl-4-piperidone (25g, 0.132 mol) in d~y dimethyl sulphoxide (25 ml). The reaction
was allowed to warm to room ~emperature and stirred for a further I h. The mixture was
wo93/15080 2:12g i ~, Pcr/GBg3/00l74
diluted with wa~cr (500 ml) and extracted into diethyl ether (3x250 ml). The combined
organic extracts were washed with bnne, dried over sodium sulphate and concentrated in
vacuo. The residue was extracted into pentane and purifled by distillation to give a clear
oil (22.48g) b.p. 175C. Ql mm Hg (Kugel~hr). A solution of this cpoxidc in foImic
S acid (100 ml) was stirrcd at soom tcmpcraturc ovcrnight, then conce~trated in vacuo. The
residuc was dissolved in methanol (200 ml), treatcd with a soludon of sodium bicarbonate
(30g) in water (100 ml) and stisred at room tempcrature for 21h. Thc reaction was
concent~ed to approximately onc third thc original volumc, saturated with potassium
carbonate, and extracted cxhaustivcly with chlo~ofoqm. The combined cxtracts wcre dried
0 ovcr sodium sulphatc and concentratcd in vacuo to givc the title compound (24.5 g).
I'reparation 7
l-Benzyl-l-azoniabicyclo[22.1]heptan~d p-toluenesulphonate
5 A solution of l-benzyl~hydroxymcthyl~hydroxypiperidinc (16.8g, 0.076 mol) in
pyddine (150 ml) at 0C was treated portionwise ovcr 10 min with ~tolucnesulphonyl
chlodde (14.86g, 0.078 mol) and the resulting solution was stored ov~night at 8C. The
reaction was concentratcd in vacuo and the residual oil was dissolved in chla¢ofaQm and
washcd with sa~uratcd aqueous potassium car'oonate. After drying over sodium sulphate
20 ~hc solution was concentTatcd in vacuo. The sesidual oil was dissoh/ed in dry toluerlc and
heated undrcflux for 1.5h. Thc product was isolated by filtration and washed with
diethyl ether to give the tille compound as a beige solid (23.3g, 81%)
~:
.
Preparation 8
25 4-Hydroacy-l-azabicyclo[22.1]heptane
A solution of l-benzyl-1-azoniabicyclo[2.2.1]heptan~o1 ~tolucnesulphonate (S.0 g, 13.3
mmol) in ethanol (100 ml) was hyd~genated over 10% Pd-C (1.0 g) at atmosphcric
pressur~ for 18h. The catalyst was removcd by filtration through kieselguhr, and the
30 filtrate was concentrated in vacuo. The residue was partitioned between saturated aqueous
potassium carbonate (20 ml) and chlorofolm (25 ml) and the aqueous layer was extracted
cxhaustivdy with chlorof~m The combined organic extracts were dried over sodium
sulphate. concentrated in vacuo and dis~lled to give the title compound as a colourless
solid (1.2g, 80%), b.p. 17SC (Icugel~hr).
H Nmr C:DC13~ ~: 1.58 (2H, m), 1.78 (2H, m), 2.37 (2H, s), 2.75 (2H, m). 3.17 (2H, m),
~ 3.65 (lH, br s).
`~:
WO 93/15080 ~a5~ - 20 - PC~/GBg3/00174
Preparation 9
3-(4-Benzyloxyphenyl)- 1-propanol
A solution of 3-(4-hydroxyphenyl)-1-propanol (2.5g, 16.4 mmol) in acetone (40 ml)
S containing po~assium carbonate (5.9g, 42.75 mmol) was treated with benzyl bromide (2.54
ml, 21.35 mmol) and refluxed for 3h. The mixture was concentrated in vacuo and
partitioned between water (S0 ml) and chloroform (S0 ml). The aqueous phase was further
extracted with chloroform (2xS0 ml) and the combined organic e~tracts were dried over
sodium sulphate and concentrated in vacuo. Purification by chromatography on silica
using 0-8% methano! in chloroform as eluant afforded the title compound as a colourless
solid (3.g2g, 99%).
1H Nmr (CDC13) ~: 1.36 (lH, s), 1.85 (2H, m), 2.63 (2H, t, J=7Hz), 3.65 (2H, t, J=7Hz),
5.04 (2H, s), 6.91 (2H, d, J=8Hz), 7.11 (2H, d, J=8Hz), 7.27-7.50 (SH, m).
Preparation 10
3-(4-Benzyloxyphenyl)propyl p-toluenesulphonate
A solution of 3-(4-benzyloxyphenyl)-1-propanol (4.15g, 17.1 mmol) in absolute
chloroform (30 ml) was cooled in ice and treated with pyridine (3.92 ml, 48.5 mmol)
followed by p-toluenesulphonyl chloride (6.16g, 32.3 mmol). The mixture was allowed to
warm slowly to room temperature and left overnight. After dilution with diethyl ether (90
ml) the solution was washed with lM orthophosphoric acid (S0 ml) followed by saturated
aqueous potassium hydrogen carbonate~(S0 ml) and water (S0 ml). The organic pha,se was
dried over sodium sulphate and then purified by chromatography on silica using 0-8%
methanol in ~hlorofonn as eluant to give the title compound as a colourless solid (S.lg,
75%)-
lH Nmr (CDC13) ~: 1.92 (2H, q, J=7Hz), 2.45 (3H, s), 2.57 (2H, t, J=7Hz), 4.01 (2H, t,
J=7Hz), 5.02 (2H, s), 6.83 (2H, d, J=8Hz), 6.97 (2H, d, J=8Hz), 7.30-7.47 (7H, m), 7.78
(2H, d, J=8Hz).
Preparation 11
4-(4-Phenoxyphenyl)butyl bromide
To a stirred suspension of aluminium chloride (13.33g, 0.1 mol) in dry dichloromethane
(200 ml) was added dropwise over O.Sh 4-bromobutyryl chloride (11.6 ml, 0.1 mol). The
resulting mixture was filtered and then added to a solution of diphenyl ether (25.5 g, O.lS
WO93/15080 ~ 8~ PCI/C;;B93/00174
mol) in dry dichloromethane (150 ml) over 0.5 h. The mixture was stirred overnight and
then treated with triethylsilane (48 ml, 0.3 mol). After 2 h the reaction was quenched
with ice-water. The organic phase was washed with water (200 ml) followed by brine
(2x100 ml) then dried over sodium sulphate and concentrated in vacuo. The residue was
chromatographed on silica gel using 0-15% diethyl ether in 40-60 petroleum ether as
eluant. Subsequent distillation afforded the title compound as a clear oil (22.12g, 73%)
b.p. 146-160C at 0.1 mm Hg.
lH Nrnr (CDCI3) ~: 1.68-2.00 (4H, m, overlapping signals), 2.60 (2H, t, J=7Hz), 3.42
(2H, t, J=7Hz), 6.86-7.40 (9H, m).
Preparation 12
~-(4-Phenoxyphenyl)pentyl bromide
15 The title compound was prepared in a similar manner to Preparation 11 from 5-~romovaleryl chloride (13.39 ml, 0.1 mol), aluminum chloride (13.33g, 0.1 mol), diphenyl
ether (25.5g, 0.15mol) and ~iethylsilane (48 ml, 0.3 mol). This afforded the title
compound as a clear oil (17.56g, 55%) b.p. 165-180C at 0.2 mm Hg.
lH Nmr (CDC13) ~: 1.36-1.75 (4H, m, overlapping signals), 1.88 (2H, m), 2.60 (2H, t,
J=7Hz), 3.40 (2H, t, J=7Hz), 6.8~7.38 (9H, m).
Preparation 13
S-(4-Hydroxyphenyl3-1-pentanol
A solution of 4-benzyloxybenzaldehyde (5.0g, 23.56 mmol) and triethyl 4-phosphono-
c~otonate (7.07g, 28.25 mmol) in dry tetrahyd~furan (50 ml) was added dropwise to a
stirred ice cold slurry of sodium hydride (0.78g of an 80% dispersion in mineral oil, 26.0
mmol) in dry tetrahydrofuran (50 ml) containing 15-crown-5 (0.18g). The mixture was
allowed to warrn tO room temperature. After 1 h the reaction was quenched with glacial
acetic acid (Sml) and concentrated in vacuo. The residue was partitioned between water
(50 ml~ and chloroform (100 ml). The aqueous layer was further extracted with
chloroform (2x50 ml) and the combined organic extracts were dried over sodium sulphate
and concentrated in vacuo. Crystallisation from ethyl acetate-pentane afforded a pale
yellow solid (5.77g) which was dissolved in ethanol (100 ml) and hydrogenated atatmospheric pressure over 10% Pd-C (lg). After 3 h the reaction mixture was filtered
through kieselguhr and then concentrated in vacuo to give ethyl 5-(4-
hydroxyphenyl)pentanoate as a clear oil (4.08g). A solution of this ester in dry diethyl
wo 93/1~080 2 i 2 ~ i 3 ~J - 22 - PCI/GB93/00174
ether (lOml) was added dropwise to a stined suspension of lithium aluminum hydride
(2.08g, 54.8 mmol) in dry diethyl ether (150 ml). After 2.5h the reaction was quenched
with wet diethyl ether followed by a minimum amount of water. The reaction was filtered,
and the precipitate was washed with methanol. The combined filtrate and washings were
5 concentrated in vacuo. The residue was dissolved in water, acidified with SM
hydrochloric acid. saturated with sodium chloride and extracted exhaustively with
chloroform. The combined chloroforrn extracts were dried over sodium sulphate and
concentrated in vacuo to give the title compound as a colourless solid. The precipitate
was dissolved in SM hydrochloric acid, and the solu~ion was saturated with sodium
0 chloride and filtered through kieselguhr. Exhaustive extraction with chloroform yielded
additional product, bringing the to~al recove~y of the title compound tO 3.04g (92%).-
lH Nmr (CDC13) ~: 1.25 -1.80 (7H, m, overlapping signals), 2.52 (2H, t, J=7Hz), 3.62
(2H, t, J~7Hz), 4.90 (lH, br s), 6.74 (2H, d, J=8Hz), 7.02 (2H, d, J=8Hz).
Preparation 14
5-(4-Benzyloxyphenyl)-l -pentanol
A solution of 5-(4-hydroxyphenyl)-1-pentanol (3.04g, 16.98 mmol) in ethanol (40 ml)
20 containing potassium carbonate (6.1g, 44.2 mmol) was treated with benzyl bromide
(2.63ml, 22.07 mmol) and refluxed for lh. The reaction mixture was concentrated in
vacuo and the residue was partitioned between water (SO ml) and chloroform (SO ml). The
aqueous phase was further extracted with chloroform (2x50 ml) and the combined organic
extracts were dried over sodium sulphate and concentrated in vacuo. Purification by
25 chromatography on silica using 0-8% methanol in chloroform as .oluant afforded the title
compound as a colourless solid (4.31g, 94%)
lH Nmr (CDC13)~: 1.15-1.70 (7H, m, overlapping signals3, 2.55 (2H, t, J=7Hz), 3.62
(2H, t, J=7Hz), S.O1 (2H, s), 6.87 (2H, d, J=8Hz), 7.07 (2H, d, J=7Hz~, 7.23-7.50 (SH, m).
3û
Preparation 1~
5-~4-Ben~yloxyphenyl)pentyl p-toluenesulphonate
35 The ~itle compound was obtained using the procedure described in Prepara~ion 10 from 5-
(4-benzyloxyphenyl)-1-pentanol (3.3g, 12.2 mmol), pyridine (3.0 ml, 37.2 mmol) and p-
toluenesulphonyl chloride (4.66g, 24.4 mmol). After a reaction time of 4h at room
WO 93/1~080 23 9 l 2~5 3 ~ PCr/GB93/00l74
temperature the mixture was worked up as previously described to give the title
compound as a colourless solid (3.64 g, 70~).
1H Nrnr (CDC13) ~: 1.20-1.76 (6H, m, overlapping signals), 2.33-2.60 (5H, m,
s overlapping signals), 4.0 (2H, t, J=6Hz), 5.0 (2H, s), 6.83 (2H, d, J=8Hz,), 7.02 (2H, d,
J=8Hz), 7.23-7.50 (7H, m), 7.77 (2H, d, J=8Hz).
wO 93/15080 ~ 24 - pcr~GB93too174
9 ~
`J
Example 1 ~ ~
4-[2-(3,4-Dichlorophenoxy)ethyll-l-azabicyclo[2.2.1]heptane hydrochloride (El)
A solution of 4-(2-hydroxyethyl)-1-azabicyclo[2.2.1]heptane (O.Sg, 3.55mmol) in dry
te~ahydrofuran (40ml), under nitrogen, was ~eated with 3,4-dichlorophenol (0.87g,
5.32mmol) in dry tetrahydrofuran (2ml), followed by ~phenylphosphine (1.21g,
4.61mmol) in tetrahydrofuran (2ml). Diethyl azodicarboxylate (0.80g, 4.61mmol) was
added to the r~action over O.Sh and the mixture was stilTed ovemight at room temperature~
The reac~on was concentrated in vacuo, treated with saturated aqueous potassium
carbonate (25ml) then extracted into chloroform (3x25 ml). The combined organic
extracts were dried over sodium sulphate, concentrated in vacuo and the residue
chromatographed on neutral alumina in a gradient of 0-2% methanol in chloroform. The
gum produced was converted into ~e HCl salt, which was washed thoroughly with diethyl
ether then crystallised to afford the title compound as a white solid (0.65g), m.p. 188-
190 C (methanol-diethyl ether).
1H Nmr (DMSO-d6) ~: 1.70-2.09 (4H, m), 2.18 (2H, t, J=7Hz), 3.12 (2H, s), 3.23-3.56
(4H, m), 4.20 ~2H, t, J=7Hz), 7.06 (lH, dd, J=3, 10Hz), 7.35 (lH, d, J=3Hz3. 7.62 (lH, d,
20 J=lOHz).
Example 2
4-[2-(4-Benzyloxyphenoxy)ethyl]-l-azabicyclo~2.2.1]heptane hydrochloride (E2)
The ~tle compound was prepared in a similar manner to Example 1 from 4-(2-
hydroxyethyl)-l-azabicyclo~2.2.1~heptane ~O.Sg, 3.55mmol), 4-benzyloxyphenol (1.06g,
5.32mmol), ~iphenylphosphine (1.21g, 4.61mmol) and diethyl azodicarboxylate (0.80g,
4.61mmol). This afforded the title compound as a white solid (0.24g~, m.p. 209-212 C
30 (from rnethanol-diethyl ether.
H Nmr (DMSO-d63 ~: 1.70-2.09 (4H, m), 2.16 (2H, t,(J=6Hz~, 3.13 (2Ht s), 323-3.66
~4H, m), 4.10 (2H, t, J=6Hz), 5.12 (2H, s), 6.89-7.08 (4H, m), 7.36-7.S7 (SH, m3.
35 Example 3
4-12-(2-Dibenzofuranyloxy3ethyl]-1-azabicyclol2.2.1]heptane hydrochloride (E3)
WO 93/15080 - 25 - PCl /GB93/00174
~ 2 ~;..'
The title compound was prepared in a similar manner to Example 1 from 4-(2-
hydroxyethyl)-1-azabicyclo~2.2.1]heptane (0.39g, 2.77mmol), 2-hydroxydibenzofuran
(0.76g, 4.1~mmol), triphenylphosphine (0.94g, 3.60mmol) and diethyl azodicarboxylate
(0.63g, 3.60mmol). The crude product was chromatographed on silica in a gradient of 5-
5 20% methanol in chlorofrom. Pooling of ~ractions containing the major slower runningcomponent afforded an oil which was treated with ethereal hydrogen chloride tO give the
title compound as a white solid (0.12g), m.p 206-209 C (from methanol/acetonetdiethyl
ether).
lH Nmr (DMSO-d~ 1.75-2.13 (4H~ m), ~.26 (2H, t, J=6Hz), 3.20 (2H, s), 3.26-3.~7
(4H, m), 4.30 (2H, d, J=6Hz), 7.19 tlH, dd, J=2, 7Hz), 7.40-7.79 (SH, m), 8.22 (lH, d,
J=7Hz).
Example 4
(+)S-(4-Benzyloxyphenoxymethyl)-l-azabicyclo[3.2.11octane hydrochloride (E4)
The ~itle compound was prepared in a similar manner to Example 1 from ~+)5-
hydroxymethyl-l-azabicyclo[3.2.1]octane (O.Sg, 3.55mmol~, 4-benzyloxyphenol (1.065g,
5.32mmol), mphenylphosphine (1.21g, 4.61mmol) diethyl azodicarboxylate (0.80g,
4.61mmol). l~his afforded the title compound as a white solid (0.1 lg), m.p. 188-191 C
(from methanol-acetone-diethyl ether).
1H Nmr (DMSO-d6) ~: 1.48-1.55 (lH, m), 1.60-1.97 (SH, m), 3.02-3.16 (4H, m), 3.25-
3.37 (2H, m), 3.80 (2H9 s~, 4.95 (2H, s), 6.74 6.98 (4H, m), 7.18-7.35 (SH, m).
Example 5
~ 35-(4-l~en~ylphenoxymethyl)-1-azabicyclo(3.2.1]octarle hydrochloride (E~)
The title compound was prepared in a similar manner to Example 1 from (*)~-
hydroxymethyl-l-azabicyclo~3.2.1]octane (O.~g, 3.55mmol), 4-hydroxydiphenylmethane
(0.98g, 5.32mmol), triphenylphosphine (1.21g, 4.61mmol~ and diethyl azodicarboxylate
(0.80g, 4.61mmol). This afforded the title compound as a white solid (O.lSg), m.p.
35 168 C (dec) (~;rom methanol-ether).
wo 93/15080 c~ S~ 26 - pcr/GBs3/oo174
lH Nmr (DMSO-d6) ~: 1.62-1.70 (lH, m), 1.7~2.11 (5H, m), 3.16-3.30 (4H, m), 3.38-
3.51 (2H, m), 3.92 (2H, s), 3.97 (2H, s) 6.88-6.93 (2H, m), 7.14-7.25 (SH, m), 7.27-7.34
(2H, m).
E~cample 6
~i)5~(2-Dibenzofuranyloxy)methyl-l-azabicyclol3.2.11octane bydrochloride (E6)
The dde compound was prepared n a similar manner to Example 1 f~m (i)5-
o hydroxymethyl-1-azabicyclo[3.2.1]octane (Q53g, 3.76mmol), 2-hydroxydibenzofuran
(1.04g, 5.64mmol) triphenylphosphine (1.28g, 4.89mmol) and diethyl azodicarboxylate
(0.8~g, 4.89mmol). The crude product was chromatographed on neut~al alumina in agradicnt of 0.5-2% methanol in toluene. Pooling of fractions containing the faster running
component and conversion of the res~ting gum into the hydrochlo~ide salt afforded the
title compound as a white solid (Q98g), m.p. 22~225 C (from methanol-acetone-diethyl
ether).
lH Nmr (I)MSO~ 1.62-2.22 (6H, m), 3.15-3.59 (6H, m), 4.10 (2H, s), 7,08-7.17
(lH, m), 7.33-7.87 (SH, m), 8.09-8.18 (lH, m).
E~ample 7
( t ) 5-[2-(2-Dibenzofuranyloxy)ethyl]-l-azabicyclo[3.2.1~oc~ne hydrochloride ~E7)
2~ The ~de compound was pr~par~ in a similar manner to Example 1 ~m ( ~ ) S-(2-
hydroxyethyl~l-axabicyclo[3.2.1]oc~ne (0.53g, 3.42 mmol), 2-hydroxydibenzofu~an
(09Sg, 5.13 mmol~, triphenylphosphine (1.17g, 4.45 mmol) and di~dlyl awdi~arboxylate
(0.54ml, 3.42 mmol3 emplo~g a reac~on ~me of 2h. The crude p~odua was purified by
chromatography on neutral al~na using 0.5-2% methanol in toluene as eluan~ and
conveTted into the hydrochlonde salt to give the title compound as a coulourless solid
(0.7g), m.p. 137-138C (from methanol-acetone-die~yl ether).
lH Nmr (DMSad6) ~: 1.60-2.08 (8H, m), 3.10-3.50 (6H~ m), 4.14 ~, t, J=5Hz), 7.12,
(lH, dd, J=8Hz and 2Hz), 7.40 ~lH, t, J=8Hz), 7.52 (lH, t, J=8Hz), 7.63 (lH, d, J=8Hz),
3s 7.67 (lH, d, J=8Hz), 7.77 (lH, d, J=2Hz), 8.14 (lH, d, J=8Hz), 10.93 ~lH, br s).
wo 93/1~080 2t~ ~ 2 ~ ? pcr/GB93/ool74
Example 8
4-[2-(4-Phenoxypheno~y)ethyl]-l-azabicyclo[2.2.1]heptane hydrochloride (E8)
s The dde compound was prepared in a similar manner to Example 1 from 4-(2-
hydroxyethyl)-l-azabicyclo~2.2.1]heptane (0.62g, 4.4 mmol), ~phenoxyphenol (1.23g,
6.61 mmol), triphenylphosphine (1.5g, 5.72 mmol) and diethyl azodicarboxylate ~û.9 ml,
5.72 mmol). The reacdon mixture was concentrated in vacu~, and the residue was
convcrted into the hydrochloride salt with éthereal hydrogen chloride. Aftcr tritu~ation
0 with cther, the salt was treated with sa~urated aqueous potassium carbonate (20 nil) and
extracted into chlorofonn (3x25 ml). The combined cxt~acts were d~ied over sodium
sulpbate, concentrated ~n vacuo and then purified on neutral alumina using ~2% methanol
in cbloroform as eluant. i~fter pooling pure fracdons the product was treated with ethereal
hydrogen chlolide to give the title compound as a colourless solid (0.85g), m p. 209-
5 211C (methanol - acetone - diethyl edler)
lH Nmr a)MSO-d6) ~: 1.73 (2H, m), 1.93 (2H, m), 2.12 (2~I, t, J~lz), 3.07 (2H, s),
3.25 (4Hr m), 4.12 (2H, t, J=6Hz), 6.88-7.13 (7H, m), 7.38 (2H, t, J=7Hz).
20 E~mple 9
4-[3-(4-Benzyloxyphenyl)propylo~cymethyl]~ zabicylol2.2.1]heptane hydrochloride
~E9)
A solution of ~hydroxymethyl-l-azabicyclo[2.2.1]he~ e (0.2g, 1.57 mmol) in ~ N,N-
dimed~ylformamide (Snil) wa~ treated ~i~ sodium hydride (SOmg ~f an 80% di~ersion in
nnn~al Qil, 1.66 mmol) and the mix~ure was s~d at 50C unde~ nitrogen u~l hydrogen
evolution had ceased. Por~on~nse add~non of 3-(4-benzyloxyphenyl)I~ropyl
toluenesulphonate (û.58g, 1.57 ~1) was casried out over lh, and d~e mixt~Me was st~
for a further lh. The reac~on was concen~ated in vacuo and dle residue was co
evapo~a~ed wi~h successive ~ons of toluene. The residue was treated w~th samrated
aqueous postassium carbonate (lOml) and extIacted into chlorofo~m (3x15 ml). Thecombined extracts were dried over sodium sulphate and dlen concentrated in vacuo. The
crude product was pusified by flash chromatography on neu~al alumina using ~15%
3s methanol in chloroform as eluant. Pooled ~ac~ons containing dle fastellunningcomponent were converted into the hydrogen chloride salt to give the title oompound as a
colourless solid (0.1 lg), m.p. 16~168C (f~om methanol-diethyl edler).
wO93/15080 ~ 9 ~5 ~J ~ 28 - pcr/Gss3/ool74
lH Nmr (DMSO-d6) ~: 1.72 (2H, m), 1.87(2H, m), 2.03 (2H, m), 2.65 (2H, t, J=8Hz),
3.12 (2H, s), 3.2~-3.55 (6H, m, overlapping signals), 3.68 (2H, s), ~.16 (2H, s), 7.01 (2H,
d, J=9Hz), 7.20 (2H, d, J=9Hz), 7.37-7.63 (SH, m).
Example 10
4-15-(4-Phenoxyphenyl)pentyloxy~ azabicyclol2.2.1]heptane hydrochloride (13:10)
A solution of l-benzyl-l-azoniabicyclol2.2.13heptan~ol ~toluenesulphonate (0.5g, 1.33
lo mmol) in dry N,N-dimethylforma~de ~7ml) was treated with sodium hydride (80 mg of
an 80% dispersion in mineral oil, 2.66 mmol) and then stirred under nitrogen at room
te~perature f~r lh. A soludon of 5-(~phenoxyphenyl)pentyl bromide (0.85g, 2.66 mmol)
in dry N, N-dimethylformamide (2ml) was added, and dle mi~cture was sdrred for 3h. The
rcaction was quenched with glacial acetic acid (O.Sml) and then concen~rated in vacuo.
After c~evaporadon with successive pordons of tolllene, the residue was disolved in
- ~ ~ ethanol (lO ml) and hydrogenated at atmospheric pressure over 10% Pd-C (O.lg) for 2h.
The reaction n~ix~re was filtered through a pad of kieKlguhr and the filtrate was
conclu~ed in vacuo. The residue was treated with saturated aqueous potassiym
carl~nr~ (~0 ml) and ex~d into chlorof~m (3x20ml). The combined extracts were
dried~over sodium sulphate and conc~ntratcd in vacuo. The crudc p~duct was purified by
flash chn~tography on neu~al alumina using 0-15% mcthanol in cblo~ofo~m as eluant.
Afte~ pooling pure fractions the product was ~eated with ethereal hydrogen chlo~ide to
give ~e title oompound as a col~less solid ~0.14g), m.p. 129-131C (~om acetone--~ diedlyl edler).
: 25
lH Nmr (DMS~d6) ~: 1.33 (2P~ m), 155 (4H, m), 1.8~2.10 (4H, m), 2.53 (~, t,
J=7Hz), 3.18 ~2H, s), 3.3~3.58 (~iH, m, overlapping signals), 6.93 (4H, m~, 7.11(1H, t,
J=8Hz), 7.22 (lH, d, J-8H~, 7.38 (2~ t, J=8~1z).
30 Example 11
,
4~ Benzylo~ypheno~methyl)-l-azabicyclol2.2.1]heptane hydrochloride (Ell)
The title compound was prepared in a similar manner to Example 1 from 4
3s hydroxymethyl-1-azabicyclo[2.2.1]heptane (0.5g, 3.94 mmol), ~benzyloxyphenol (1.18g,
~; 5.91 mmol), tnphenylphosphine (1.55g, 5.91 mmol) and diethyl azodicarboxylate (0.93ml,
5.91 mmol). After a re~cd(m period of Sh the mixture was worked up as pre~riously
described for E~xample 1. Purification on neut~al alumina using 5% ethanol in chlorofo~m
~'
WO 93/150X0 - 29 2 ~ ? ~ Pcr/GB93/00l74
as eluant afforded a pale yellow oil which was treated with ethereal hydrogen chloride to
give the title compound as a colourless solid (0.09g), m.p. 20~208C (~om methanol-
ace~ne-diethyl ether).
s lH Nmr (DMS~d6) ~: 1.73 (2H, m), 2.02 (2H, m), 3.12 (2H, s~, 3.20-3.50 ~4H, m,
overlapping signals), 4.17 (2H, s), 5.03 (2H, s), 6.93 (4H, m), 7.2~7.48 (SH, m).
E~ample 12
'I l4-(4-Phenoxyphenyl)butyloxy]-l-azabicyclo[2æ.1]heptane hydrochloride (E12)
The title compound was prepared in a similar manner to Example 10 from l-benzyl-l-
azoniabicyclo[2.2.1]heptan-~ol p-toluenesuphonate ~0.5g, 1.33 mmol), sodium hyd~ide
(80 mg of an 80% dispersion in mineral oil, 2.66 mmol) and ~(~phenoxyphenyl)butyl
b~omide (0.89g, ~.91 mmol). The crude product was purified by chromatography on silica
using 5-10% edlanol in chloroform as duant, and ~eated with ethe~eal hydrogen chloride
to givc the title compound as a ~olouriess solid nLp. 115-117.5C (~om acetonc-died yl
e~h~).
lH Nmr (DMS~d6) ~: 1.46-1.67 (4H, m), 1.85-2.06 (4H, m~, 2.57 (2H, t, J--7Hz), 3.17
(2~I, s), 3.3~3.60 (6H, m, ove~ ping signals), 6.93 (4H, m, overlapping signals), 7.11
(lH, t, J=7Hz), 7.21 (2H, d, J=8Hz), 7.38 (2H, t, J=8Hz).
Example 13
4.~3-(4-Benzyloxyphenyl)pr~pyloY~y]-l~azabicyclo[2.2.1~heptane hydrochloride (E13)
A solu~on of ~hydroxy-l-azabicylo~2.2.13heptane (0.38g, 3.36 mmol) in dry N, N-
dimedlylfoImamide (l~ml) was treated under ni¢ogen with sodium hydride ~120 mg of an
~0% dispersion in ~eral oil, 4.0 mmol). The mixture was warmed to 4~50C and
sti~red for 2h. The temperature was raised to 60C and a solu~ion of 3-(4-
benzyloxyphenyl)pr~pyl tosylate (0.9Og, 2.69 mmol) in dry N, N-dimethylfo~de
(8ml) was added dropwise over 2h. After a further lh at 60C ~e reac~ion was cooled and
quenched with glacial acetic acid (0.23 ml). The reaction was concentratecl in vacuo and
~en paltitioned between cbloroform (20 ml) and satarated a~ueous potassium carbonate
(20 ml). The aqueous phase was extracted with chlorofoIm (2x20 ml) and the combined
organic layers were dried oYer sodium sulphate and concen~ated in vacuo. Purification on
neutral alumina using 1-2% ethanol in chloroform as eluant afforded a colourless
wo 93/1~080 ~" ~ 30 - PCI`/GB93/00174
crystalline solid which was treated with ethereal nydrogen chloride to give the title
compound as a colourless solid (0.30g) m.p. 180-181 C (from methanol-acetone-diethyl
ether).
s lH Nmr (DMSO-d6) ~: 1.78 (2H, m); 1.92 (2H, m); 2.02 (2H, m); 2.56 (2H, t, J= 7Hz);
3.18 (2H, s); 3.25-3.60 (6H, m); 5.07 (2H, s~, 6.92 (2H, d, J=8Hz); 7.12 (2H, d, J=8Hz);
7.28-7.50 (SH, m).
Example 14
4-15-(4-Benzyloxyphenyl)pentyloxy~ azabicyclol2.2.1~heptane hydrochloride (E14)
A solution of 4-hydroxy-1-a~abicyclo 12.2.1] hep~ane (0.38g, 3.36 mmol) in dry N, N-
dimethylformamide (lSml) was treated under nitrogen with sodium hydride (120mg of an
80% dispersion in mineral oil, 4.0 mmol). The mixture was warmed tO SOC and stilTed
for 2h. The temperature was raised to 60C and a solution of 5-(4-benzyloxyphenyl)
pentyl p-toluenesulphonate (1.07g, 2.52 mmol) an dry N, N-dimethylfonnamide (9ml) was
added dropwise over a period of 3h. After a further 45min at 60C the mixtur~ was cooled
and quenched with glacial ace~ic acid (0.23ml). The reaction was concenlrated in vacuo
then par~tioned between chloroform (75ml) and satura~ed aqueous potassium carbonate
(75ml). The aqueous pha~e was exhaustively extracted with chlorofonn. The combined
organic layers were dried over sodium sulphate and concentrated in vacuo. Purification on
neu~ral alumina using 1% ethanol in chloroform as eluanl followed by treatment with
ethereal hydrogen chloride afforded Ihe title compound as a colourless so~id (0.30g) m.p.
15~151.5C (acetone-diethyl ether).
lH Nmr (DMSO-d6) ~: 1.32 (2H, m); 1.53 (4H, m~; 1.82-2.08 (4H, m), 2.S0 (2H, t,
J=7Hz), 3.17 (2H, s); 3.25-3.60 (6H, m); 5.06 (2H, s); 6.92 (2H, d, J=8Hz), 7.10 (2H, d,
J=8Hz); 7.28-7.50 (5H, m).